
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Is copper a new target to counteract the progression of chronic diseases?
Clara
Balsano
 *,
Cristiana
Porcu
and
Silvia
Sideri
*,
Cristiana
Porcu
and
Silvia
Sideri
F. Balsano Foundation, Via Giovanni Battista Martini 6, 00198, Rome, Italy. E-mail: clara.balsano@cc.univaq.it; Tel: +39 0649970894
First published on 19th October 2018
Abstract
Altered regulation of copper (Cu) homeostasis may contribute to the development of many pathologies, such as metabolic, cardiovascular, neurodegenerative and cancerous diseases. Cu serum concentrations are strictly related to oxidative stress. During the past decade, it has been well demonstrated that even marginal deficits of this element contribute to development and progression of a number of chronic diseases. On the other hand, an excess of Cu may become a potent oxidant causing the generation of reactive oxygen species (ROS) and leading to the formation of macromolecules dangerous to health. In this review we will describe several chronic inflammatory diseases pathogenically related to the alteration of serum copper levels. Some considerations on present and future perspectives for use of natural antioxidants as effective drugs for the treatment of chronic diseases will be made to shed a glimmer of light on some of the mechanisms employed by natural antioxidants in controlling the distribution and concentration of biometals.
Introduction
Oxidative stress can be a physiological protective mechanism against different toxic stimuli, but if this process is not adequately controlled it may play a major part in the development of chronic and degenerative illnesses.Cu is an essential trace metal. This ion is crucial due to its pleiotropic effects on biological systems. Cu is a key element for a wide range of physiological cellular functions: aerobic respiration, oxidative stress protection, cardiovascular functionality, neuropeptide biogenesis, lipid, iron and potassium metabolism, erythropoiesis and angiogenesis.1–6 Cu in metabolic processes is associated with its ability to fluctuate between oxidized (Cu2+) and reduced (Cu+) states, as a result of accepting and donating single electrons in oxido-reductive cellular reactions.7 Thus, Cu is essential as a structural component for different proteins, as well as a catalytic cofactor for the activation of several metallo-enzymes, such as superoxide dismutase (SOD) and cytochrome c oxidase (or mitochondrial complex IV),8,9 as seen in Fig. 1. Due to this, the intra- and extra-cellular levels of Cu undergo stringent regulation. The imbalance of Cu homeostasis, in fact, can induce oxidative damage. This contributes to the onset and progression of inflammation and oxidative stress in several pathologies.10 In particular, an increase in unbound Cu promotes pro-oxidant effects that, in turn, determine tissue damage. In fact, during the transition between the reduced and oxidized states, free Cu ions induce the overproduction of ROS, which is a common feature of several pathologies.11 This event stimulates the production of advanced glycation end-products (AGEs), advanced lipoxidation end-products (ALEs) and protein oxidation products (PrOPs).10,12 The latter molecules activate pro-inflammatory cascades, participating in tissue damage and inducing stress-related responses. Thus, the interplay between Cu, oxidative stress and inflammation represents a crucial regulatory mechanism to prevent or induce tissue damage during chronic inflammatory diseases,13 as seen in Fig. 2.
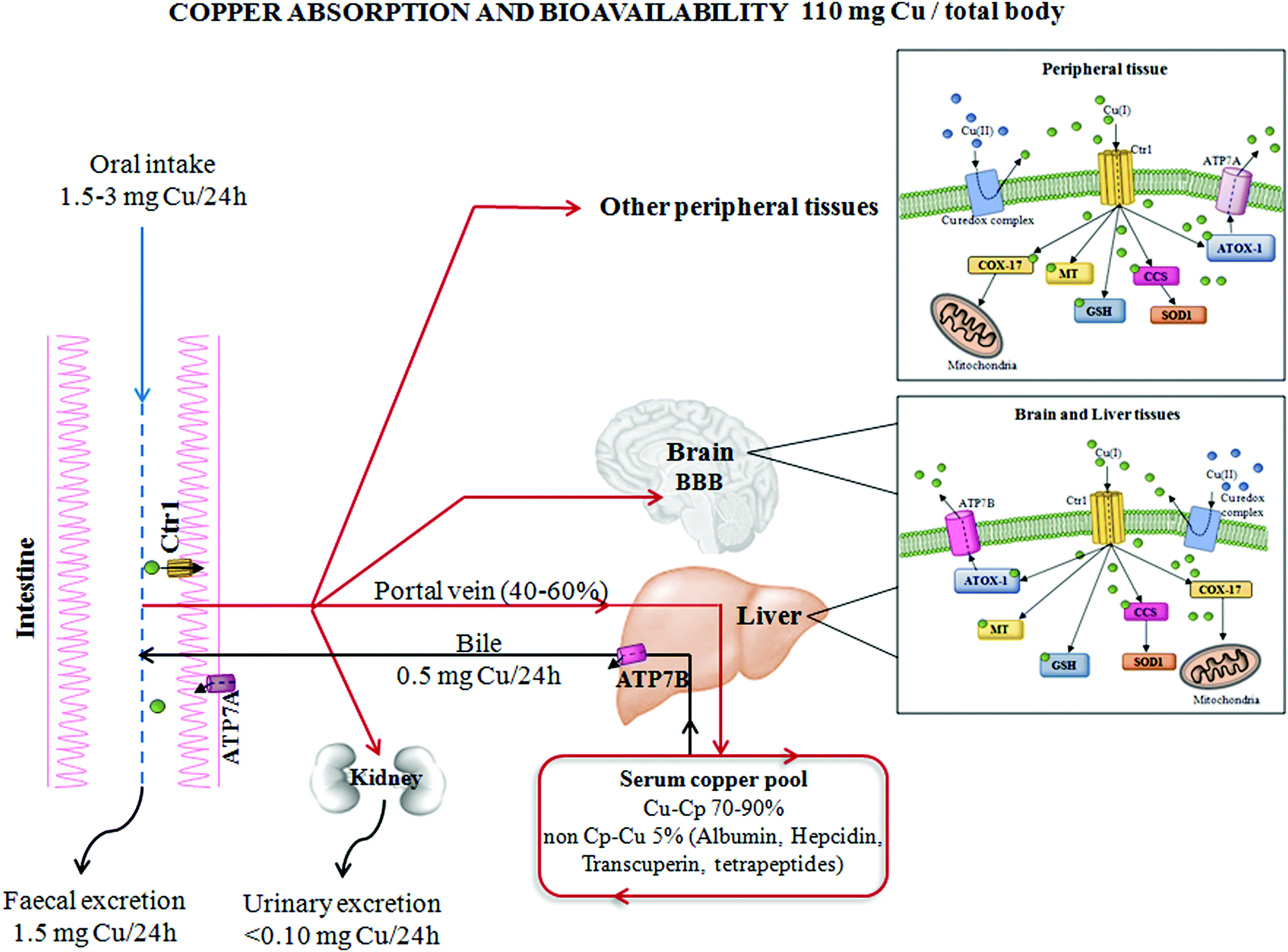 | ||
| Fig. 1 Copper absorption and bioavailability. The absorption and distribution of copper from the human gastrointestinal system to peripheral tissues. The main proteins involved in copper homeostasis have been reported, as well as total body copper amount and specific distribution in different body compartments. Abbreviations: Cu, copper; Ctr1, high affinity copper uptake protein 1; ATP7A/B, copper-transporting ATPase (ATP7); Cp, ceruloplasmin; ATOX1, antioxidant 1 copper chaperone; MT, metallothionein; GSH, glutathione; SOD1, superoxide dismutase 1; CCS, copper chaperone for superoxide dismutase; COX-17, cytochrome c oxidase copper chaperone; and BBB, blood–brain barrier. | ||
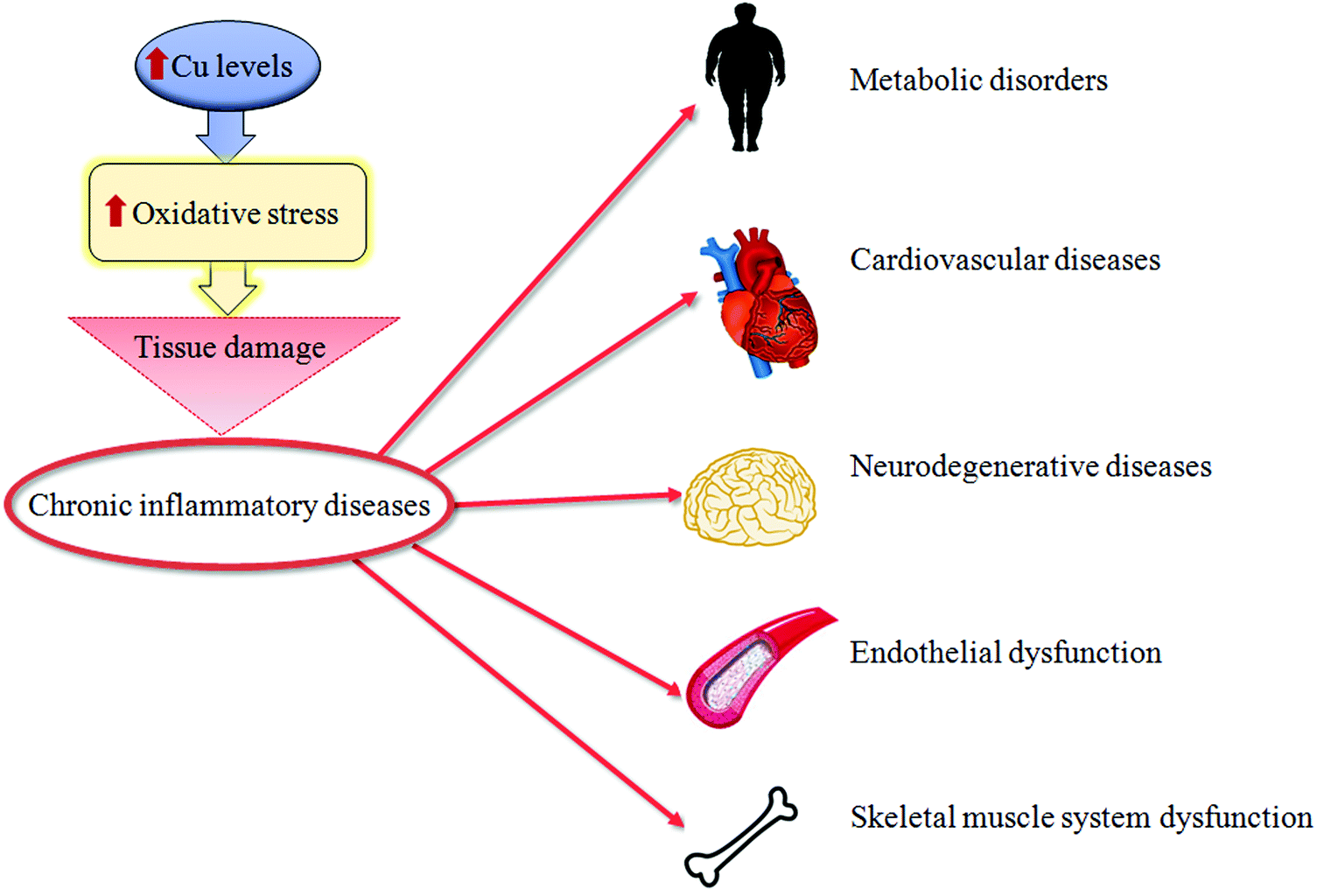 | ||
| Fig. 2 Copper and tissue damage. A synthetic representation of how high copper (Cu) levels might cause tissue damage of different organs during chronic inflammatory diseases. | ||
In this review, we will highlight the importance of a Cu imbalance in the pathogenesis of several chronic inflammatory diseases.
All of the information reported herein leads us to give a brief opinion on issues related to use of natural antioxidants that are able to target Cu. We are convinced that these drugs, if used properly, may give important opportunities in the future to develop innovative and effective personalized treatments.
Cu homeostasis
Cells have evolved specific and complex trafficking systems that participate in Cu binding and compartmentalization, to ensure both the delivery and detoxification of any Cu excess. The amount of this biometal in the human body may be influenced by dietary intake, efficiency of absorption and excretion, as well as by the presence of nutritional components that might promote or impair its absorption (e.g. high levels of zinc).14 The recommended daily Cu intake is between 1 and 3 mg, and the main sources of this micronutrient in food are grains, nuts, beans, seeds, shellfish and liver,15 as seen in Fig. 1.The majority of dietary Cu is absorbed in the duodenum through high-affinity Cu transporter 1 (CTR1), encoded by the SLC31A1 gene. CTR1 mediates the influx of reduced Cu ions (Cu+) into human cells.16 In intestinal epithelial cells, this metal is carried across the basolateral membrane by a Cu-transporting P-type ATPase ATP7A in the portal system up to the liver. From the liver, which is the principal organ for Cu storage, the ion is incorporated into ceruloplasmin (Cp–Cu, which is responsible for chelating between 70 and 90% of Cu in the blood) and is subsequently distributed to other peripheral tissues. Less than 5% of the total Cu concentration circulates as “free Cu” independent of binding with ceruloplasmin (non-Cp–Cu)17 and is the main factor in clinically significant oxidative stress. “Free Cu” binds to several proteins, such as albumin, hepcidin, transcuperin and low-molecular-weight complexes or tetrapeptides.18,19 The excess Cu returns to the liver to be excreted into the bile duct via Cu-transporting ATPase ATP7B. Moreover, ATP7A and B participate in intracellular routing of Cu between cytosol and cellular organelles16 (Fig. 1).
In the cytoplasm, Cu may be chelated by metallothionein for storage bound to Cu chaperones for delivery to specific proteins or to reduced glutathione. In the presence of an excess of intracellular Cu, these proteins move to intracellular vesicles contributing to an export of Cu.20 When extracellular Cu is increased, CTR1 undergoes Cu-dependent endocytosis, in peripheral tissues, to protect these tissues against an excess of Cu accumulation.21 Contrary to other tissues, in the liver, CTR1 expression is elevated under high Cu conditions to facilitate detoxification from circulation and to counteract Cu overload in other Cu-susceptible peripheral tissues.22
Note that, in response to high levels of Cu concentrations, cell systems displace ATP7A to post-Golgi vesicles and to the plasma membrane and increase the expression of this protein in order to enhance the export of Cu to restore intracellular Cu homeostasis.22
In our opinion, the crucial role of ATP7A in maintaining Cu homeostasis should be better studied, because its altered expression may even sensitize cells toward transformation.
Cu and human chronic diseases
Inflammation and oxidative stress are characteristic of chronic diseases. Excess non-Cp–Cu becomes a potent oxidant by generation of ROS, implicated in the decay of biological macromolecules.11 For the reasons described above, Cu levels are fine-tuned and Cu imbalance has important implications in the development and progression of several chronic inflammatory diseases, such as metabolic, cardiovascular, neurodegenerative and cancerous diseases.2,3,23,24 Thus, currently the main open issue of a correlation between altered copper regulation and chronic inflammatory diseases should be more deeply and better studied.Cu and metabolic diseases
Recent results indicate that altered serum Cu homeostasis has an important role in diabetes, obesity, metabolic syndrome and non-alcoholic fatty-liver disease (NAFLD).25,26 In fact, diabetes mellitus and obesity are known to correlate with an imbalance of transition metals, including Cu.27 Oxidative stress, insulin resistance, end-product molecules, adipokines, pro-inflammatory cytokines and mitochondrial dysfunction are all related to higher Cu serum levels and are implicated in the development and progression of several chronic inflammatory diseases.28 Accordingly, down-regulation of CTR1, Cu-responsive transition-metal-binding metallothionein proteins (MT1/MT2), antioxidant-protein-1 (ATOX1) and ATP7B was observed in the left-ventricular myocardium of diabetic rats and has been associated with Cu deficiency.29 Moreover, in obese ob/ob mice and in high-fat diet-induced (HFD) mice, pathogenic lipid accumulation promotes a hepatic marginal Cu deficiency, as well as changes in the mitochondrial morphology and function.30,31 Interestingly, our recent data highlighted a high Cu serum concentration in NAFLD-cirrhotic patients, which became even more evident in patients with NAFLD-related hepatocellular carcinoma (HCC).32All of the data reported above suggest that it will be an important agenda in research to elucidate details of the pathogenic role of altered Cu serum levels (especially “free Cu”) and the expression of the Cu-related proteins (e.g. CTR1, ATP7A, ATP7B, MT1, MT2, ceruloplasmin, albumin, transferrin, hepcidin, transcuperin, etc.) during chronic inflammatory diseases. In fact, despite studies on redox-active tracers being quite advanced, Cu-related pathological features in metabolic diseases have been poorly studied until now.
Cu and heart diseases
The heart has the highest resting energy expenditure compared to any other organ in the body and has a constant high demand for bioenergy to maintain its workload. Transition metals (e.g. Fe, Cu, Zn, etc.) are particularly important in heart mitochondrial respiration, where they play essential roles in the production of ATP and detoxification of ROS. Unfortunately, these transition metals can also promote the formation of dangerous radicals,33,34 and so meticulous control of their concentration and subcellular compartmentalization is needed.Cu deficiency has been described in heart attacks and hypertension and causes a decrease in the activity of several important Cu-related enzymes, including Cu,Zn SOD, the potent cellular anti-oxidant enzyme that prevents the formation of free radicals and increases peroxidation of lipids and proteins.35 The pathogenic role of Cu,Zn SOD seems to be mainly related to the over-production of hydrogen peroxide during the transfer of an electron from one molecule of a superoxide anion to another. Although hydrogen peroxide is not a ROS, it is a potent toxic oxidizing agent, playing, in this way, an important role in inducing oxygen toxicity.36
In addition, human cardiac hypertrophy has been described as result of dietary Cu deficiency,35 but, on the other hand, heart-specific deletion of CTR1 allowing increased serum Cu levels together with an increased expression of ATP7A, correlate with the appearance of cardiac hypertrophy.22 Thus, to date, the importance of dietary Cu intake on the onset of cardiac hypertrophy is a question that has yet to be answered. In fact, even if several Cu trafficking proteins have been reported to be implicated in cardiovascular diseases, the related-pathogenic mechanisms are not completely understood and should be better studied in the near future.
To give an example: ceruloplasmin is involved as ferroxidase in the conversion of iron from the ferrous state (Fe2+) to the ferric state (Fe3+) mediating transferrin iron uptake.37 Thus the lack of the ferric state of iron can cause anemia that, in turn, is involved in cardiac hypertrophy and/or ischemia. Thus, oversimplification should be avoided and further studies should be dedicated to finely understanding metal ion regulation in cardiovascular diseases.
Cu and neurodegenerative diseases
After the liver, the brain is the organ containing the highest Cu concentration.38 In a normal brain, Cu has a heterogeneous distribution with a higher concentration in grey matter (e.g. Substantia nigra, dentate nucleus and cortex and basal ganglia) rather than in white matter.39 Currently, it is known that Cu is transported to the brain through the blood–brain barrier (BBB). The BBB regulates the influx of Cu into the brain, and together with the blood–cerebrospinal fluid contributes to the maintenance of Cu homeostasis in the brain’s extracellular fluids by specific Cu transporters and chaperones.38 One pathogenic mechanism seems to be related to the altered ability of Cu in preventing excessive activity of N-methyl-D-aspartate receptors (NMDARs), which determine synaptic plasticity and are excitatory neurotransmitter receptors.40 Hypo- or hyper-activation of NMDARs seems to contribute to the onset and progression of depression, strokes and Alzheimer's disease.41In genetic and neurodegenerative diseases, a Cu deficiency or overload may appear for ineffective Cu-related homeostatic mechanisms. Following on from this, anomalous Cu transport and aberrant Cu–protein interactions have been described in genetic diseases, as well as in numerous human neurological disorders, confirming the critical relevance of this trace metal for proper neuro-development and neurological functions.42
Menkes' disease is an X-linked fatal disorder characterized by the accumulation of Cu in the intestine and kidney, while Cu is low in the liver, brain and other tissues, such as bones, skin and hair.43 Patients with Menkes’ disease lack functional ATP7A protein, causing a severe Cu deficiency that is often fatal in early childhood.44 To date, the molecular mechanisms at the basis of the pathogenic effects of ATP7A inactivation are yet to be clarified, even if it is known that this condition leads to altered Cu concentrations in cytosol, nuclei and mitochondria.45
Although most mutations in the ATP7A gene cause Menkes’ disease, some missense mutations induce a weaker disease called Occipital Horn Syndrome.46,47 Patients with Occipital Horn Syndrome suffer from connective tissue abnormalities as with Menkes’ disease patients, but do not usually present neurological symptoms and have a considerably longer average lifespan.47
Genetic Wilson's disease is an autosomal recessive disorder characterized by a lack of fully functional ATP7B protein, thus Cu accumulation in the liver and subsequently in the brain and kidney occurs. In addition, Wilson's disease patients typically develop severe liver disease and may present additional neurological symptoms as well.44 In particular, the excess of Cu in the liver is caused by its defective excretion by bile, that, in turn, increases oxidative stress, leading cells to apoptosis and fat accumulation. At the same time, the incorporation of Cu into ceruloplasmin is impaired, thus elevated levels of free Cu are released into the bloodstream exerting extrahepatic Cu toxicity.45
Interestingly enough, polymorphism in the ATP7B gene has also been associated with an increased risk of Alzheimer's disease (AD) and may be useful to stratify specific metabolic subtypes of AD.48 AD is a late-onset multifactorial disease and is related to multiple risk factors. AD is the most common form of dementia worldwide and AD patients have higher Cu serum levels49,50 that seem to be involved in the formation of the β-sheet form of β-amyloid peptides.51 β-Amyloid in turn becomes redox-active in the presence of high levels of Cu.52 In particular, Cu, when complexed with β-amyloid, catalyzes the production of ROS. Among the different ROS generated by Cu and β-amyloid interactions, it is worth mentioning that hydroxyl radicals are able to induce oxidative damage, not only on the β-amyloid peptide, but also on other molecules, such as lipids and proteins.53 Accordingly, it has been demonstrated that serum non-Cp–Cu (“free Cu”) levels predict the progression of mild cognitive impairment during AD.48,49
Besides AD, a large number of studies have described the dysregulation of Cu homeostasis in diseases of the central nervous system, such as Parkinson's disease (PD), Huntington's disease (HD) and amyotrophic lateral syndrome (ALS).38,42
PD is the second most common neurodegenerative disease, and oxidative stress is one of the most common causes of its etiology and pathogenesis.54 PD is characterized by gradual loss of dopaminergic neurons, and by the existence of aggregates of misfolded proteins (mainly α-synuclein), known as Lewy bodies.54 Cu, as an oxidative stress promoter, plays a key role in the pathogenesis of PD by its ability to form a complex with α-synuclein, inducing the formation of neurotoxic masses.55 Accordingly, the reduction of cellular Cu levels results in a significant decrease in aggregate formation.56 On the other hand, it has been reported that the presence of Cu induces the formation of structurally different and less-damaging α-synuclein aggregates, suggesting that protein aggregation could not be a primary cause of cytotoxicity.57 It has been reported that post-mortem Substantia nigra derived from PD patients shows increased levels of iron and decreased levels of Cu.58 Moreover, in the Substantia nigra of a PD mouse model, the ceruloplasmin protein shows severely reduced ferroxidase activity.59 Finally, in PD, CTR1 is decreased in the Substantia nigra, and mutations of ATP7B have been indicated to be involved in the pathogenesis of this disease.60 Given this, depending on the interplay between genetic and micro-environmental conditions, some groups of patients may benefit from Cu supplementation, whereas others need to be treated with iron or Cu chelators.61,62
HD is a neurodegenerative disorder, caused by a dominant polyglutamine expansion within the N-terminus of the huntingtin protein and is characterized by oxidative stress and striatal degeneration.63 In the striatum of HD brains, high Cu and iron concentrations have been detected.64 Studies by Fox and colleagues (2007) have demonstrated the ability of Cu2+ to directly interact with the N-terminal of the huntingtin protein through histidine residues, making them redox active. Interestingly, metal chelation inhibits the formation of huntingtin aggregates.
ALS is a sporadic disease. Only 10% of this malignancy is familial with a known genetic background.65,66 ALS is a fatal neurodegenerative disorder affecting upper and lower motor pathways of the central nervous system, characterized by muscle atrophy, paralysis and death within a few years after disease onset.67 Currently, the ALS pathogenic mechanisms are still unclear, but the involvement of several altered intracellular signaling pathways have been described, controlling Cu homeostasis,68 oxidative stress, inflammation and dysfunction of mitochondrial respiration (Fig. 3). Among several genes responsible for familial ALS, mutations in the Cu,Zn SOD1 gene have been deeply studied and over 180 different mutations have been recognized as causes of ALS.69 SOD1 is a major Cu-binding protein and regulates Cu homeostasis in cells; therefore, toxicity of mutant SOD1 could arise from the disruption of Cu homeostasis.45
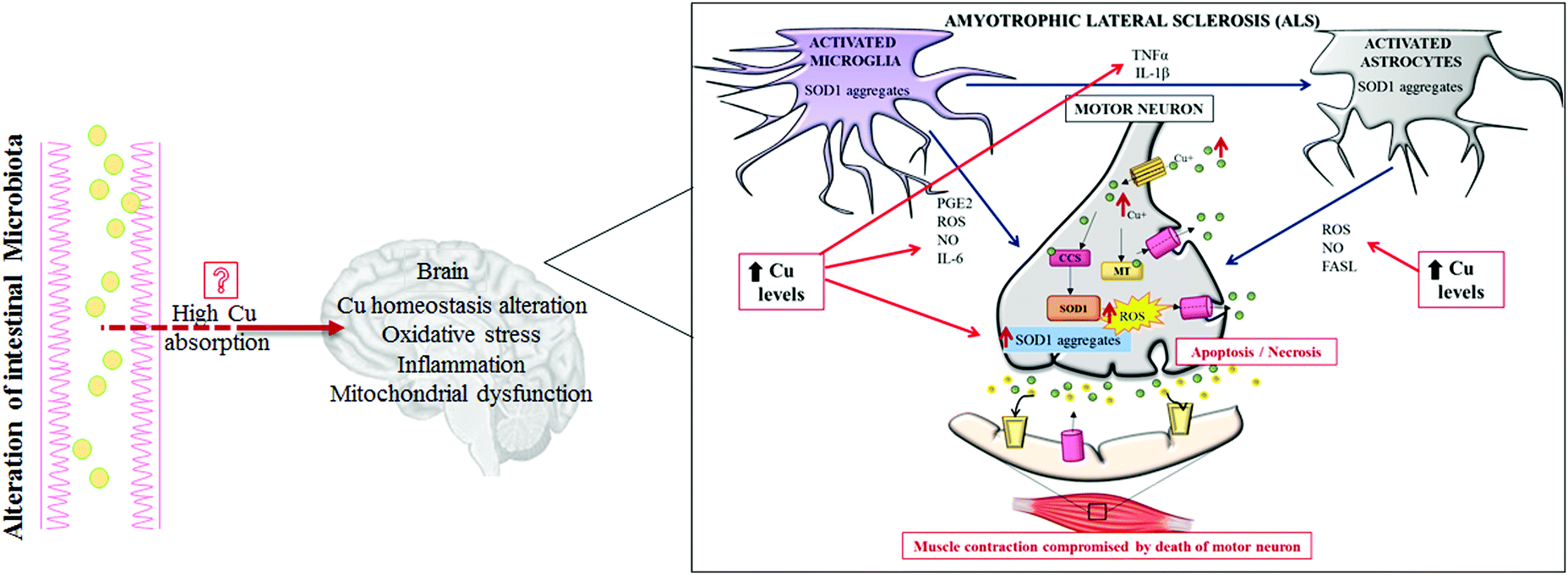 | ||
| Fig. 3 Pathogenic role of copper in amyotrophic lateral sclerosis (ALS). A representation of how high copper levels may directly contribute to the worsening of all of the most important pathogenic mechanisms described to be involved in ALS. Abbreviations: Cu, copper; MT, metallothionein; SOD1, superoxide dismutase 1; CCS, copper chaperone for superoxide dismutase; PGE2, prostaglandin E2; IL-6, interleukin-6; IL-1β, interleukin-1β; TNFα, tumor necrosis factor α; FASL, ligands to cell surface death receptors; ROS, reactive oxygen species; and NO, nitric oxide. | ||
Interestingly, increased levels of Cu have been evidenced in the ventral areas of the spinal cords of sporadic ASL patients, as well as other metals (lead and zinc), but mutations in the SOD1 gene have not been highlighted in those cases. To note, even over-expression of SOD1 alone, still able to bind Cu, can function as an efficient trigger for increasing Cu spinal levels; this effect is probably due to the high Cu affinity of this enzyme.70 To date, a consensus does not exist on which Cu-related proteins (SOD1, CCS, ATP7A, MT etc.) are really implicated in the pathogenesis of ALS, and how.
Given this, restoration of Cu homeostasis could be a good therapeutic approach for ALS. Accordingly, administration of Cu chelator drugs (e.g. ammonium tetrathiomolybdate, TM) has significantly improved and/or slowed down disease progression in severe phenotypes of ALS transgenic mice recently.71
We think it should be found that the most efficient way to correct intracellular Cu homeostasis is by choosing drugs able to chelate and stabilize abnormal accumulation of Cu ions. However, to achieve this aim in ASL patients, we should beforehand finely characterize how, when and where Cu accumulates in the spinal cord.
Cu and cell transformation
An important mechanism for the development of several cancers is the deregulation of oxidative stress that impairs cell DNA repair mechanisms due to the overproduction of ROS.72 Some lifestyle habits, such as smoking or drinking alcohol, may promote oxidative stress, whereas consumption of nutrients, rich in dietary antioxidants, may protect against oxidative stress.73High Cu levels, both in serum and in tumor tissue, have been reported in different tumor types, including cancers of the lung, liver, breast, gastrointestinal tract and gynecological malignancy.74–77 A persistent status of Cu-related oxidative stress contributes to tumor development and progression by inducing cell damage and impairing cellular repair processes.78 Furthermore, it has been recently reported that a high Cu/Zn ratio is positively associated with colorectal cancer.79 In addition, recently, it has been demonstrated that increased levels of exogenous Cu push hepatocytes towards transformation through the Cu-dependent over-expression of c-MYC oncogene,32 as seen in Fig. 4.
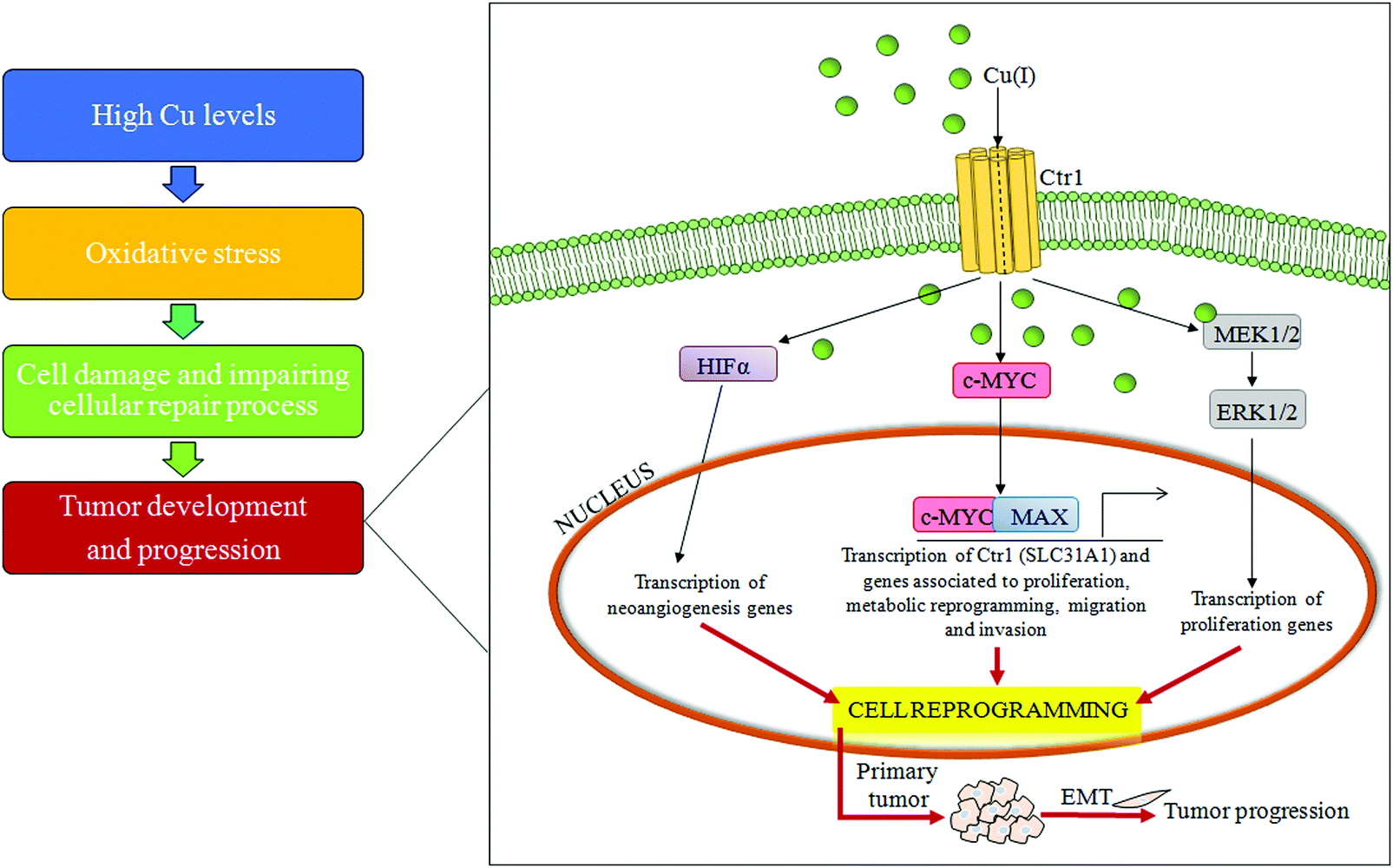 | ||
| Fig. 4 Copper and cell transformation. A schematic representation of the copper-related pathogenic liver transformation. In particular, high serum copper levels have the ability to induce over-expression of the c-MYC oncogene, reprogramming differentiated cells. Abbreviations: Cu, copper; Ctr1, high affinity copper uptake protein 1; MAX, MYC associated factor X; HIF-1α, hypoxia inducible factor 1 subunit alpha; MEK1/2, MAP kinse-ERK kinase; ERK1/2, extracellular regulated MAP kinase; and EMT, epithelial–mesenchymal transition. | ||
Moreover, several studies have highlighted the role of Cu in tumor growth and neo-angiogenesis, and so Cu seems to be a promising target for anticancer therapy.80,81 Accordingly, Cu chelators act as anti-angiogenic agents reducing the secretion of many positive regulators of angiogenesis, such as vascular endothelial growth factors (VEGFs) and microvascular density, in animal models.82–84 Cu stabilizes the transcription factor promoting tumor angiogenesis, hypoxia-inducible factor-alpha (HIFα), under normoxic conditions, inducing hypoxia-response element (HRE)-dependent reporter gene expression.80,85 Furthermore, Cu is required for oncogenic BRAF signaling and its related-tumorigenesis.86 Mutated BRAF kinase (V600E) phosphorylates and activates MEK1 and MEK2 kinases, which in turn phosphorylate the ERK1 and ERK2 kinases, thus promoting cell transformation. If treated with TM, tumor-carrying mice, derived from mouse embryonic fibroblasts expressing mutated BRAFV600E (Val600 → Glu), have a significant decrease in tumor size.87 In line with these findings, TM efficacy is increased by association with a low Cu diet.88
Future perspectives for alternative treatment of chronic diseases
A good knowledge of Cu regulatory mechanisms and related redox reactions will give additional indications of how oxidative stress should be counteracted to prevent tissue damage during chronic diseases. In this context, in our opinion, it is very important to better explore metal-binding specificity and selectivity of antioxidants. In fact, the excess of antioxidant activity could be, in particular conditions, even dangerous and not associated with an increased life span.It has been reported that some enzymatic and non-enzymatic antioxidants (Fig. 5), such as oleuropein, epigallocatechin gallate, gallic acid and curcumin, display metal chelating activity, becoming interesting drugs to counteract the progression of several chronic diseases.89,90
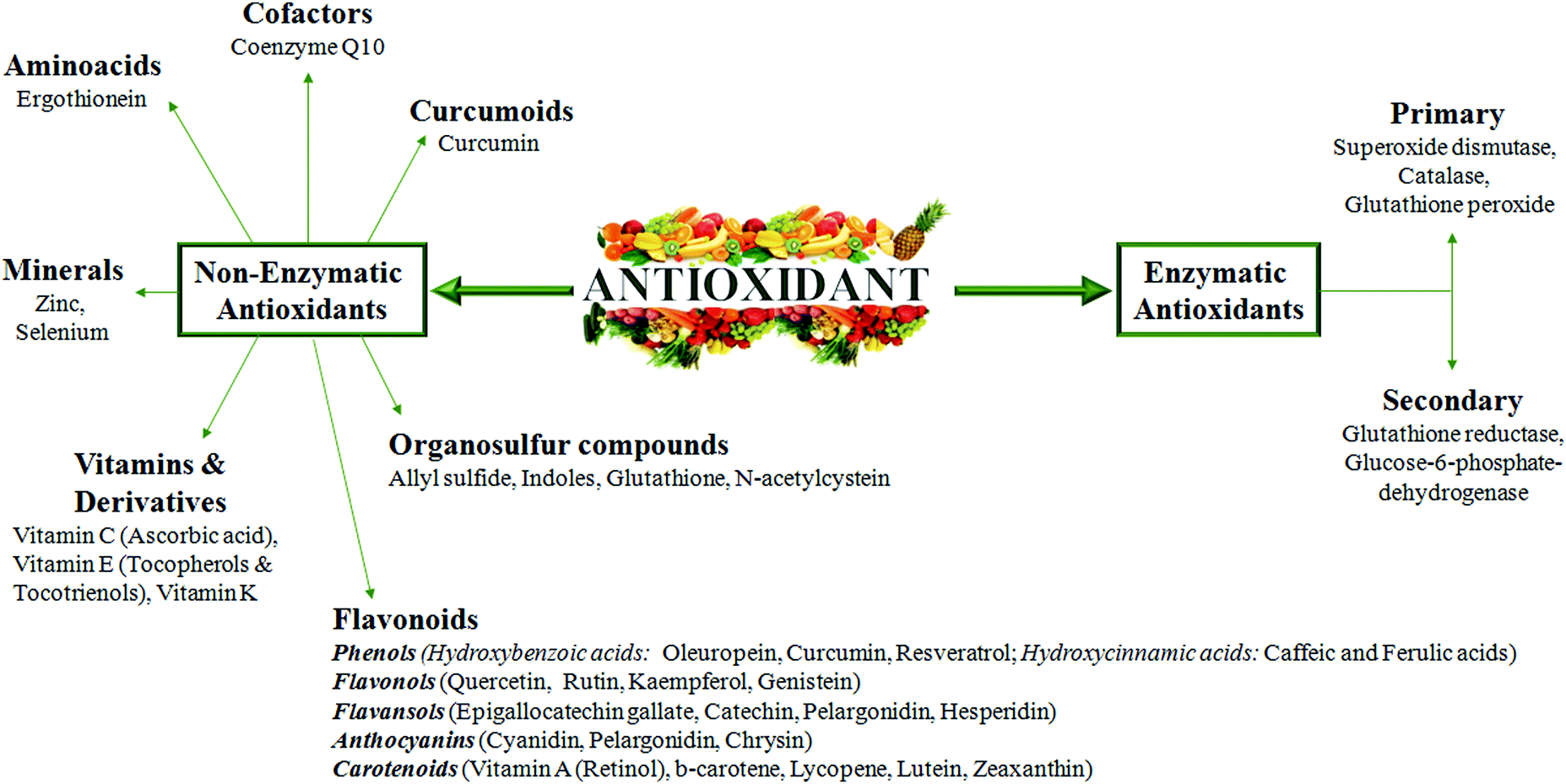 | ||
| Fig. 5 Non enzymatic and enzymatic antioxidant compounds. The figure represents all of the antioxidant molecules reported to be able to counteract oxidative stress and inflammation in chronic diseases. | ||
Taking into account the Cu-related pathogenic mechanisms described above, natural antioxidants, with an ability to chelate metals, could hopefully be an adjuvant therapeutic strategy for chronic inflammatory diseases. However, an antioxidant compound, to be safe, should have the ability, after scavenging the radical, to form a new radical that must be stable through intramolecular hydrogen bonding upon further oxidation.91 Therefore, it is clear that these drugs should be modified in order to create selective, efficient and safe molecules.
In this context, it is very important to point out again that natural antioxidants may not be effective in all patients even if they are affected by the same pathology, because often different pathogenic mechanisms underlie the onset and the progression of oxidative stress-related damage. Emblematic of this is a study on 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 772 post-menopausal women, who regularly supplemented their diet with vitamins and micronutrients such as Cu, Zn, Fe, Mg, folic acid, vitamin E etc., to avoid chronic disease. These women presented an increased total mortality risk.92 This is sobering work, because it clearly highlights that antioxidants, in the form of vitamins or other supplements, can sometimes cause damage and not benefits in healthy individuals.93 Thus, it is urgent to improve our knowledge on the mechanisms of action of natural antioxidants for a conscious use of them.
772 post-menopausal women, who regularly supplemented their diet with vitamins and micronutrients such as Cu, Zn, Fe, Mg, folic acid, vitamin E etc., to avoid chronic disease. These women presented an increased total mortality risk.92 This is sobering work, because it clearly highlights that antioxidants, in the form of vitamins or other supplements, can sometimes cause damage and not benefits in healthy individuals.93 Thus, it is urgent to improve our knowledge on the mechanisms of action of natural antioxidants for a conscious use of them.
Even if at present, most of our knowledge is limited to in vitro and preclinical studies, in the near future, we are convinced that ongoing clinical trials will yield very crucial indications to what is necessary to obtain positive effects by using antioxidants.
Conclusion
In the light of what has been said, metals need extensive screening, mainly for recent evidence of their physiological correlation to cardiovascular diseases, metabolic and degenerative diseases. Recent developments in imaging techniques (magnetic resonance imaging) for metal detection might elucidate the role of copper in the pathogenesis of chronic inflammatory diseases. Interestingly, advanced MRI techniques and susceptibility weighted neuroimaging can detect metal deposition in tissues with high sensitivity.94,95In this context, there is no doubt that Cu plays an important role in a huge number of chronic inflammatory diseases, thus it might represent a promising target for the treatment of these diseases and their complications.
In this review, we highlighted how a Cu imbalance may contribute to the clinical expression of chronic inflammatory diseases, although a full understanding of the biological basis of the imbalance of Cu homeostasis has yet to be achieved. In fact, the specific chemical details of Cu-shunting mechanisms, point by point from its cell entry to its functional targets, should evolve over the next few years through genetic, biochemical, structural and functional studies.
Major comprehension of complex mechanisms in the regulation of Cu signaling pathways in chronic inflammation may be useful in the rationale for developing new promising and personalized therapeutic strategies.
Conflicts of interest
The authors have no conflicts of interest to disclose.Acknowledgements
The authors responsibilities were as follows: C. B. wrote the manuscript; the authors C. P. and S. S. edited the manuscript, and read and approved the final version of the manuscript. The authors thank Dr Simona Tavolaro for her critical suggestions.References
- Z. N. Baker, P. A. Cobine and S. C. Leary, The mitochondrion: a central architect of copper homeostasis, Metallomics, 2017, 9, 1501–1512 RSC.
- T. Fukai, M. Ushio-Fukai and J. H. Kaplan, Copper transporters and copper chaperones: roles in cardiovascular physiology and disease, Am. J. Physiol.: Cell Physiol., 2018, 315, C186–C201 CrossRef PubMed.
- A. Morrell, S. Tallino, L. Yu and J. L. Burkhead, The role of insufficient copper in lipid synthesis and fatty-liver disease, IUBMB Life, 2017, 69, 263–270 CrossRef CAS PubMed.
- G. Tarantino, C. Porcu, M. Arciello, P. Andreozzi and C. Balsano, Serum copper bioavailability predicts carotid intima-media tickness in obese patients with low prevalence of co-morbidities, J. Gastroenterol. Hepatol., 2018, 33, 1511–1517 CrossRef CAS PubMed.
- E. Urso and M. Maffia, Behind the Link between Copper and Angiogenesis: Established Mechanisms and an Overview on the Role of Vascular Copper Transport Systems, J. Vasc. Res., 2015, 52, 172–196 CrossRef CAS PubMed.
- X. Wu, H. Kim, J. Seravalli, J. J. Barycki, P. J. Hart, D. W. Gohara, E. Di Cera, W. H. Jung, D. J. Kosman and J. Lee, Potassium and the K +/H+ Exchanger Kha1p Promote Binding of Copper to ApoFet3p Multi-copper Ferroxidase, J. Biol. Chem., 2016, 291, 9796–9806 CrossRef CAS PubMed.
- L. M. Gaetke and C. K. Chow, Copper toxicity, oxidative stress, and antioxidant nutrients, Toxicology, 2003, 189, 147–163 CrossRef CAS PubMed.
- M. Fukuoka, E. Tokuda, K. Nakagome, Z. Wu, I. Nagano and Y. Furukawa, An essential role of N-terminal domain of copper chaperone in the enzymatic activation of Cu/Zn-superoxide dismutase, J. Inorg. Biochem., 2017, 175, 208–216 CrossRef CAS PubMed.
- A. Aich, C. Wang, A. Chowdhury, C. Ronsör, D. Pacheu-Grau, R. Richter-Dennerlein, S. Dennerlein and P. Rehling, COX16 promotes COX2 metallation and assembly during respiratory complex IV biogenesis, eLife, 2018, 7 DOI:10.7554/eLife.32572.
- B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine (4th Ed.), Free Radical Biol. Med., 1992, 12, 93–95 CrossRef.
- A. Hordyjewska, L. Popiołek and J. Kocot, The many “faces” of copper in medicine and treatment, Biometals, 2014, 27, 611–621 CrossRef CAS PubMed.
- C. M. S. Marques, E. A. Nunes, L. Lago, C. N. Pedron, T. M. Manieri, R. H. Sato, V. X. Oliveira Junior and G. Cerchiaro, Generation of Advanced Glycation End-Products (AGEs) by glycoxidation mediated by copper and ROS in a human serum albumin (HSA) model peptide: reaction mechanism and damage in motor neuron cells, Mutat. Res., 2017, 824, 42–51 CAS.
- T. C. Pereira, M. M. Campos and M. R. Bogo, Copper toxicology, oxidative stress and inflammation using zebrafish as experimental model, J. Appl. Toxicol., 2016, 36, 876–885 CrossRef CAS PubMed.
- J. Osredkar and N. Sustar, Copper and Zinc, Biological Role and Significance of Copper/Zinc Imbalance, J. Clin. Toxicol., 2011, S3, 001 Search PubMed.
- L. Antonucci, C. Porcu, G. Iannucci, C. Balsano and B. Barbaro, Non-Alcoholic Fatty Liver Disease and Nutritional Implications: Special Focus on Copper, Nutrients, 2017, 9, E1137 CrossRef PubMed.
- H. Öhrvik and D. J. Thiele, How copper traverses cellular membranes through the mammalian copper transporter 1, Ctr1, Ann. N. Y. Acad. Sci., 2014, 1314, 32–41 CrossRef PubMed.
- R. Squitti, M. Siotto, E. Cassetta, I. G. El Idrissi and N. A. Colabufo, Measurements of serum non ceruloplasmin bound copper by a direct fluorescent method specific to Cu(II), Clin. Chem. Lab. Med., 2017, 55, 1360–1367 CAS.
- K. Kulprachakarn, Y. L. Chen, X. Kong, M. C. Arno, R. C. Hider, S. Srichairatanakool and S. S. Bansal, Copper(II) binding properties of hepcidin, J. Biol. Inorg. Chem., 2016, 21, 329–338 CrossRef CAS PubMed.
- M. C. Linder, Ceruloplasmin and other copper binding components of blood plasma and their functions: an update, Metallomics, 2016, 8, 887–905 RSC.
- F. Tadini-Buoninsegni and S. Smeazzetto, Mechanisms of charge transfer in human copper ATPases ATP7A and ATP7B, IUBMB Life, 2017, 69, 218–225 CrossRef CAS PubMed.
- R. J. Clifford, E. B. Maryon and J. H. Kaplan, Dynamic internalization and recycling of a metal ion transporter: Cu homeostasis and CTR1, the human Cu+ uptake system, J. Cell Sci., 2016, 129, 1711–1721 CrossRef CAS PubMed.
- H. Chun, T. Catterton, H. Kim, J. Lee and B. E. Kim, Organ-specific regulation of ATP7A abundance is coordinated with systemic copper homeostasis, Sci. Rep., 2017, 7, 12001 CrossRef PubMed.
- C. M. Ackerman and C. J. Chang, Copper signaling in the brain and beyond, J. Biol. Chem., 2018, 293, 4628–4635 CrossRef CAS PubMed.
- D. Denoyer, S. Masaldan, S. La Fontaine and M. A. Cater, Targeting copper in cancer therapy: 'Copper That Cancer', Metallomics, 2015, 7, 1459–1476 RSC.
- E. Aigner, G. Weiss and C. Datz, Dysregulation of iron and copper homeostasis in nonalcoholic fatty liver, World J. Hepatol., 2015, 7, 177–188 CrossRef PubMed.
- J. Lowe, R. Taveira-da-Silva and E. Hilário-Souza, Dissecting copper homeostasis in diabetes mellitus, IUBMB Life, 2017, 69, 255–262 CrossRef CAS PubMed.
- M. Arciello, A. Longo, C. Viscomi, C. Capo, A. Angeloni, L. Rossi and C. Balsano, Core domain mutant Y220C of p53 protein has a key role in copper homeostasis in case of free fatty acids overload, Biometals, 2015, 28, 1017–1029 CrossRef CAS PubMed.
- U. J. Jung and M. S. Choi, Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease, Int. J. Mol. Sci., 2014, 15, 6184–6223 CrossRef PubMed.
- S. Zhang, H. Liu, G. V. Amarsingh, C. C. Cheung, S. Hogl, U. Narayanan, L. Zhang, S. McHarg, J. Xu, D. Gong, J. Kennedy, B. Barry, Y. S. Choong, A. R. Phillips and G. J. Cooper, Diabetic cardiomyopathy is associated with defective myocellular copper regulation and both defects are rectified by divalent copper chelation, Cardiovasc. Diabetol., 2014, 13, 100 CrossRef PubMed.
- A. F. Stättermayer, S. Traussnigg, E. Aigner, C. Kienbacher, U. Huber-Schönauer, P. Steindl-Munda, A. Stadlmayr, F. Wrba, M. Trauner, C. Datz and P. Ferenci, Low hepatic copper content and PNPLA3 polymorphism in non-alcoholic fatty liver disease in patients without metabolic syndrome, J. Trace Elem. Med. Biol., 2017, 39, 100–107 CrossRef PubMed.
- K. Y. Peng, M. J. Watt, S. Rensen, J. W. Greve, K. Huynh, K. S. Jayawardana, P. J. Meikle and R. C. R. Meex, Mitochondrial dysfunction-related lipid changes occur in non-alcoholic fatty liver disease progression, J. Lipid Res., 2018, 59, 1977–1986 CrossRef CAS PubMed.
- C. Porcu, L. Antonucci, B. Barbaro, B. Illi, S. Nasi, M. Martini, A. Licata, L. Miele, A. Grieco and C. Balsano, Copper/MYC/CTR1 interplay: a dangerous relationship in hepatocellular carcinoma, Oncotarget, 2018, 9, 9325–9343 CrossRef PubMed.
- J. Xu, J. T. F. Wise, L. Wang, K. Schumann, Z. Zhang and X. Shi, Dual Roles of Oxidative Stress in Metal Carcinogenesis, J. Environ. Pathol., Toxicol. Oncol., 2017, 36, 345–376 CrossRef PubMed.
- A. K. Rines and H. Ardehali, Transition metals and mitochondrial metabolism in the heart, J. Mol. Cell. Cardiol., 2013, 55, 50–57 CrossRef CAS PubMed.
- D. M. Medeiros, Perspectives on the Role and Relevance of Copper in Cardiac Disease, Biol. Trace Elem. Res., 2017, 176, 10–19 CrossRef CAS PubMed.
- A. Lumb, Oxygen Toxicity and Hyperoxia, Nunn's Applied Respiratory Physiology, 8th edn, 2017 Search PubMed.
- C. Eid, M. Hémadi, N. T. Ha-Duong and J. M. El Hage Chahine, Iron uptake and transfer from ceruloplasmin to transferrin, Biochim. Biophys. Acta, 2014, 1840, 1771–1781 CrossRef CAS PubMed.
- I. F. Scheiber, J. F. Mercer and R. Dringen, Metabolism and functions of copper in brain, Prog. Neurobiol., 2014, 116, 33–57 CrossRef CAS PubMed.
- N. Krebs, C. Langkammer, W. Goessler, S. Ropele, F. Fazekas, K. Yen and E. Scheurer, Assessment of trace elements in human brain using inductively coupled plasma mass spectrometry, J. Trace Elem. Med. Biol., 2014, 28, 1–7 CrossRef CAS PubMed.
- P. Paoletti, C. Bellone and Q. Zhou, NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease, Nat. Rev. Neurosci., 2013, 14, 383–400 CrossRef CAS PubMed.
- L. Yao and Q. Zhou, Enhancing NMDA Receptor Function: Recent Progress on Allosteric Modulators, Neural Plast., 2017, 2017, 2875904 Search PubMed.
- O. Bandmann, K. H. Weiss and S. G. Kaler, Wilson's disease and other neurological copper disorders, Lancet Neurol., 2015, 14, 103–113 CrossRef CAS PubMed.
- S. Zlatic, H. S. Comstra, A. Gokhale, M. J. Petris and V. Faundez, Molecular basis of neurodegeneration and neurodevelopmental defects in Menkes disease, Neurobiol. Dis., 2015, 81, 154–161 CrossRef CAS PubMed.
- S. La Fontaine, M. L. Ackland and J. F. Mercer, Mammalian copper-transporting P-type ATPases, ATP7A and ATP7B: emerging roles, Int. J. Biochem. Cell Biol., 2010, 42, 206–209 CrossRef CAS PubMed.
- R. Giampietro, F. Spinelli, M. Contino and N. A. Colabufo, The Pivotal Role of Copper in Neurodegeneration: A New Strategy for the Therapy of Neurodegenerative Disorders, Mol. Pharmaceutics, 2018, 15, 808–820 CrossRef CAS PubMed.
- T. Skjørringe, P. Amstrup Pedersen, S. Salling Thorborg, P. Nissen, P. Gourdon and L. Birk Møller, Characterization of ATP7A missense mutants suggests a correlation between intracellular trafficking and severity of Menkes disease, Sci. Rep., 2017, 7, 757 CrossRef PubMed.
- S. W. Mercer, J. Wang and R. Burke, In Vivo Modeling of the Pathogenic Effect of Copper Transporter Mutations That Cause Menkes and Wilson Diseases, Motor Neuropathy, and Susceptibility to Alzheimer's Disease, J. Biol. Chem., 2017, 292, 4113–4122 CrossRef CAS PubMed.
- R. Squitti, I. Simonelli, M. Ventriglia, M. Siotto, P. Pasqualetti, A. Rembach, J. Doecke and A. I. Bush, Meta-analysis of serum non-ceruloplasmin copper in Alzheimer's disease, J. Alzheimer's Dis., 2014, 38, 809–822 CAS.
- R. Squitti, R. Ghidoni, M. Siotto, M. Ventriglia, L. Benussi, A. Paterlini, M. Magri, G. Binetti, E. Cassetta, D. Caprara, F. Vernieri, P. M. Rossini and P. Pasqualetti, Value of serum nonceruloplasmin copper for prediction of mild cognitive impairment conversion to Alzheimer disease, Ann. Neurol., 2014, 75, 574–580 CrossRef CAS PubMed.
- S. Bucossi, M. Ventriglia, V. Panetta, C. Salustri, P. Pasqualetti, S. Mariani, M. Siotto, P. M. Rossini and R. Squitti, Copper in Alzheimer's disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies, J. Alzheimer's Dis., 2011, 24, 175–185 CAS.
- D. Yugay, D. P. Goronzy, L. M. Kawakami, S. A. Claridge, T. B. Song, Z. Yan, Y. H. Xie, J. Gilles, Y. Yang and P. S. Weiss, Copper Ion Binding Site in β-Amyloid Peptide, Nano Lett., 2016, 16, 6282–6289 CrossRef CAS PubMed.
- A. I. Bush, C. L. Masters and R. E. Tanzi, Copper, beta-amyloid, and Alzheimer's disease: tapping a sensitive connection, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11193–11194 CrossRef CAS PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Oxidative stress and the amyloid beta peptide in Alzheimer's disease, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed.
- J. Blesa, I. Trigo-Damas, A. Quiroga-Varela and V. R. Jackson-Lewis, Oxidative stress and Parkinson's disease, Front. Neuroanat., 2015, 9, 91 Search PubMed.
- D. R. Brown, α-Synuclein as a ferrireductase, Biochem. Soc. Trans., 2013, 41, 1513–1517 CrossRef CAS PubMed.
- X. Wang, D. Moualla, J. A. Wright and D. R. Brown, Copper binding regulates intracellular alpha-synuclein localisation, aggregation and toxicity, J. Neurochem., 2010, 113, 704–714 CrossRef CAS PubMed.
- A. Villar-Piqué, T. Lopes da Fonseca, R. Sant'Anna, É. M. Szegö, L. Fonseca-Ornelas, R. Pinho, A. Carija, E. Gerhardt, C. Masaracchia, E. Abad Gonzalez, G. Rossetti, P. Carloni, C. O. Fernández, D. Foguel, I. Milosevic, M. Zweckstetter, S. Ventura and T. F. Outeiro, Environmental and genetic factors support the dissociation between α-synuclein aggregation and toxicity, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6506–6515 CrossRef PubMed.
- M. P. Yeager and R. A. Coleman, In silico evidence for glutathione- and iron-related pathogeneses in Parkinson's disease, J. Neurosci. Methods, 2010, 188, 151–164 CrossRef CAS PubMed.
- S. Ayton, P. Lei, J. A. Duce, B. X. Wong, A. Sedjahtera, P. A. Adlard, A. I. Bush and D. I. Finkelstein, Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease, Ann. Neurol., 2013, 73, 554–559 CrossRef CAS PubMed.
- S. Montes, S. Rivera-Mancia, A. Diaz-Ruiz, L. Tristan-Lopez and C. Rios, Copper and copper proteins in Parkinson's disease, Oxid. Med. Cell. Longevity, 2014, 2014, 147251 Search PubMed.
- P. Aguirre, O. García-Beltrán, V. Tapia, Y. Muñoz, B. K. Cassels and M. T. Núñez, Neuroprotective Effect of a New 7,8-Dihydroxycoumarin-Based Fe2+/Cu2+ Chelator in Cell and Animal Models of Parkinson's Disease, ACS Chem. Neurosci., 2017, 8, 178–185 CrossRef CAS PubMed.
- B. Das, A. Kandegedara, L. Xu, T. Antonio, T. Stemmler, M. E. A. Reith and A. K. Dutta, A Novel Iron(II) Preferring Dopamine Agonist Chelator as Potential Symptomatic and Neuroprotective Therapeutic Agent for Parkinson's Disease, ACS Chem. Neurosci., 2017, 8, 723–730 CrossRef CAS PubMed.
- J. A. Kama, G. Lieberman, R. Chopra, K. Dorsey, V. Chopra, I. Volitakis, R. A. Cherny, A. I. Bush and S. Hersch, Mechanisms of copper ion mediated Huntington's disease progression, PLoS One, 2007, 2, e334 CrossRef PubMed.
- J. H. Fox, J. A. Kama, G. Lieberman, R. Chopra, K. Dorsey, V. Chopra, I. Volitakis, R. A. Cherny, A. I. Bush and S. Hersch, Mechanisms of copper ion mediated Huntington's disease progression, PLoS One, 2007, 2, e334 CrossRef PubMed.
- A. L. Ji, X. Zhang, W. W. Chen and W. J. Huang, Genetics insight into the amyotrophic lateral sclerosis/frontotemporal dementia spectrum, J. Med. Genet., 2017, 54, 145–154 CrossRef CAS PubMed.
- A. Masala, S. Sanna, S. Esposito, M. Rassu, M. Galioto, A. Zinellu, C. Carru, M. T. Carrì, C. Iaccarino and C. Crosio, Epigenetic Changes Associated with the Expression of Amyotrophic Lateral Sclerosis (ALS) Causing Genes, Neuroscience, 2018, 390, 1–11 CrossRef CAS PubMed.
- Q. Q. Tao, Q. Wei and Z. Y. Wu, Sensory nerve disturbance in amyotrophic lateral sclerosis, Life Sci., 2018, 203, 242–245 CrossRef CAS PubMed.
- S. Sheykhansari, K. Kozielski, J. Bill, M. Sitti, D. Gemmati, P. Zamboni and A. V. Singh, Redox metals homeostasis in multiple sclerosis and amyotrophic lateral sclerosis: a review, Cell Death Dis., 2018, 9, 348 CrossRef PubMed.
- O. Abel, J. F. Powell, P. M. Andersen and A. Al-Chalabi, ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics, Hum. Mutat., 2012, 33, 1345–1351 CrossRef CAS PubMed.
- P. A. Jonsson, K. S. Graffmo, T. Brännström, P. Nilsson, P. M. Andersen and S. L. Marklund, Motor neuron disease in mice expressing the wild type-like D90A mutant superoxide dismutase-1, J. Neuropathol. Exp. Neurol., 2006, 65, 1126–1136 CrossRef CAS PubMed.
- E. Tokuda and Y. Furukawa, Copper Homeostasis as a Therapeutic Target in Amyotrophic Lateral Sclerosis with SOD1 Mutations, Int. J. Mol. Sci., 2016, 17, E636 CrossRef PubMed.
- J. Y. Tang, A. A. Farooqi, F. Ou-Yang, M. F. Hou, H. W. Huang, H. R. Wang, K. T. Li, S. Fayyaz, C. W. Shu and H. W. Chang, Oxidative stress-modulating drugs have preferential anticancer effects – involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration, Semin. Cancer Biol., 2018 DOI:10.1016/j.semcancer.2018.08.010.
- G. Bjørklund and S. Chirumbolo, Role of oxidative stress and antioxidants in daily nutrition and human health, Nutrition, 2017, 33, 311–321 CrossRef PubMed.
- A. Gupte and R. J. Mumper, Elevated copper and oxidative stress in cancer cells as a target for cancer treatment, Cancer Treat. Rev., 2009, 35, 32–46 CrossRef CAS PubMed.
- M. Zowczak, M. Iskra, L. Torliński and S. Cofta, Analysis of serum copper and zinc concentrations in cancer patients, Biol. Trace Elem. Res., 2001, 82, 1–8 CrossRef CAS PubMed.
- S. Blockhuys and P. Wittung-Stafshede, Roles of Copper-Binding Proteins in Breast Cancer, Int. J. Mol. Sci., 2017, 18, E871 CrossRef PubMed.
- M. Zhang, M. Shi and Y. Zhao, Association between serum copper levels and cervical cancer risk: A meta-analysis, Biosci. Rep., 2018, 38 DOI:10.1042/BSR20180161.
- D. Denoyer, S. Masaldan, S. La Fontaine and M. A. Cater, Targeting copper in cancer therapy: 'Copper That Cancer', Metallomics, 2015, 7, 1459–1476 RSC.
- M. Stepien, M. Jenab, H. Freisling, N. P. Becker, M. Czuban, A. Tjønneland, A. Olsen, K. Overvad, M. C. Boutron-Ruault, F. R. Mancini, I. Savoye, V. Katzke, T. Kühn, H. Boeing, K. Iqbal, A. Trichopoulou, C. Bamia, P. Orfanos, D. Palli, S. Sieri, R. Tumino, A. Naccarati, S. Panico, H. B. A. Bueno-de-Mesquita, P. H. Peeters, E. Weiderpass, S. Merino, P. Jakszyn, M. J. Sanchez, M. Dorronsoro, J. M. Huerta, A. Barricarte, S. Boden, B. van Guelpen, N. Wareham, K. T. Khaw, K. E. Bradbury, A. J. Cross, L. Schomburg and D. J. Hughes, Pre-diagnostic copper and zinc biomarkers and colorectal cancer risk in the European Prospective Investigation into Cancer and Nutrition cohort, Carcinogenesis, 2017, 38, 699–707 CrossRef CAS PubMed.
- D. C. Rigiracciolo, A. Scarpelli, R. Lappano, A. Pisano, M. F. Santolla, P. De Marco, F. Cirillo, A. R. Cappello, V. Dolce, A. Belfiore, M. Maggiolini and E. M. De Francesco, Copper activates HIF-1α/GPER/VEGF signalling in cancer cells, Oncotarget, 2015, 6, 34158–34177 CrossRef PubMed.
- N. Chan, A. Willis, N. Kornhauser, M. M. Ward, S. B. Lee, E. Nackos, B. R. Seo, E. Chuang, T. Cigler, A. Moore, D. Donovan, M. Vallee Cobham, V. Fitzpatrick, S. Schneider, A. Wiener, J. Guillaume-Abraham, E. Aljom, R. Zelkowitz, J. D. Warren, M. E. Lane, C. Fischbach, V. Mittal and L. Vahdat, Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases, Clin. Cancer Res., 2017, 23, 666–676 CrossRef CAS PubMed.
- C. K. Sen, S. Khanna, M. Venojarvi, P. Trikha, E. C. Ellison, T. K. Hunt and S. Roy, Copper-induced vascular endothelial growth factor expression and wound healing, Am. J. Physiol. Heart Circ. Physiol., 2002, 282, H1821–7 CrossRef CAS PubMed.
- X. Z. Wu, New strategy of antiangiogenic therapy for hepatocellular carcinoma, Neoplasma, 2008, 55, 472–481 CAS.
- S. R. Bharathi Devi, M. A. Dhivya and K. N. Sulochana, Copper transporters and chaperones: Their function on angiogenesis and cellular signalling, J. Biosci., 2016, 41, 487–496 CrossRef CAS PubMed.
- F. Martin, T. Linden, D. M. Katschinski, F. Oehme, I. Flamme, C. K. Mukhopadhyay, K. Eckhardt, J. Tröger, S. Barth, G. Camenisch and R. H. Wenger, Copper-dependent activation of hypoxia-inducible factor (HIF)-1: implications for ceruloplasmin regulation, Blood, 2005, 105, 4613–4619 CrossRef CAS PubMed.
- D. C. Brady, M. S. Crowe, M. L. Turski, G. A. Hobbs, X. Yao, A. Chaikuad, S. Knapp, K. Xiao, S. L. Campbell, D. J. Thiele and C. M. Counter, Copper is required for oncogenic BRAF signalling and tumorigenesis, Nature, 2014, 509, 492–496 CrossRef CAS PubMed.
- R. D. Hall and R. R. Kudchadkar, BRAF mutations: signaling, epidemiology, and clinical experience in multiple malignancies, Cancer Control, 2014, 21, 221–230 CrossRef PubMed.
- I. Lokody, Signalling: Inhibiting oncogenic BRAF signalling by copper depletion, Nat. Rev. Cancer, 2014, 14, 384–385 CrossRef CAS PubMed.
- Q. Y. Eng, P. V. Thanikachalam and S. Ramamurthy, Molecular understanding of Epigallocatechin gallate (EGCG) in cardiovascular and metabolic diseases, J. Ethnopharmacol., 2018, 210, 296–310 CrossRef CAS PubMed.
- S. H. Omar, Oleuropein in olive and its pharmacological effects, Sci. Pharm., 2010, 78, 133–154 CrossRef CAS PubMed.
- S. A. Cherrak, N. Mokhtari-Soulimane, F. Berroukeche, B. Bensenane, A. Cherbonnel, H. Merzouk and M. Elhabiri, In Vitro Antioxidant versus Metal Ion Chelating Properties of Flavonoids: A Structure-Activity Investigation, PLoS One, 2016, 11, e0165575 CrossRef PubMed.
- J. Mursu, K. Robien, L. J. Harnack, K. Park and D. R. Jacobs, Dietary supplements and mortality rate in older women: the Iowa Women's Health Study, Arch. Intern. Med., 2011, 171, 1625–1633 CrossRef CAS PubMed.
- M. W. Moyer, The myth of antioxidants, Sci. Am., 2013, 308, 62–67 CrossRef CAS PubMed.
- L. Finney, S. Vogt, T. Fukai and D. Glesne, Copper and angiogenesis: unravelling a relationship key to cancer progression, Clin. Exp. Pharmacol. Physiol., 2009, 36, 88–94 CrossRef CAS PubMed.
- S. Makino, T. Umemoto, H. Yamada, E. M. Yezdimer and I. Tooyama, In vivo detection of copper ions by magnetic resonance imaging using a prion-based contrast agent, Appl. Biochem. Biotechnol., 2012, 168, 504–518 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |