
Remote stereocontrolled asymmetric 1,6-addition/1,4-addition cascade reactions between cyclic dienones and 2-indolinethiones†
Xiaohua Sun,
Jie Fei,
Chuncheng Zou,
Min Lu and
Jinxing Ye*
Engineering Research Center of Pharmaceutical Process Chemistry, Ministry of Education, Shanghai Key Laboratory of New Drug Design, School of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: yejx@ecust.edu.cn
First published on 31st October 2016
Abstract
Highly enantioselective 1,6-addition/1,4-addition cascade reactions between cyclic dienones and 2-indolnethiones have been realized by employing a cinchona-based primary amine catalyst. This method displays a broad substrate scope and provides a simple and highly effective way to achieve the chiral spiro[thiopyranoindole-cyclohexanone] scaffold.
Carbon–sulfur bond formation,1 has attracted intense attention in organic synthesis due to organosulfur compounds' wide presence in many synthetic drugs and bioactive natural products.2 To date, significant progress has been achieved for the construction of carbon–sulfur bonds via organocatalytic methods.3 However, it remains challenging to attempt to build a carbon–sulfur bond in which the sulfur atom participates in the construction of a quaternary spirocyclic center to obtain chiral thio-spirocyclic compounds.4
Organocatalytic cascade strategy5 in asymmetric transformation, especially in accessing complex chiral architectures, allows the construction of multiple fresh chiral bonds in one single process.6,7 Recently, the application of vinylogous cascade reaction affords a new way to construct stereochemically dense molecules simply and effectively.8 Key to the success of this tactics is to control the regioselectivity and stereoselectivity of reactive sites far from the catalytic center which is still a great challenge for modern chemical methodology. Although this strategy appeals to synthetic chemists greatly, existing examples in this field are rare.8 In 2013, Melchiorre and co-workers firstly reported a vinylogous 1,6-addition/Aldol cascade reaction in which cyclic dienones acted as an electron donor–acceptor to access spirocyclic framework.9 After that, Melchiorre and Jørgensen managed to develop a 1,6-addition/1,4-addition reaction between dienals and dinucleophiles via enolization intermediates catalyzed by diphenylprolinolsilyl ether for the construction of tetrahydrofuran spirooxindole derivatives and oxa-containing heterocyclic compounds respectively.10 In 2015, we successfully reported an unprecedented doubly vinylogous Michael addition/vinylogous Michael addition/isomerization cascade reaction of α,β-unsaturated γ-butyrolactams to cyclic dienones, generating chiral fused tricyclic γ-lactams with four newly formed stereocenters.11 Recent studies on 2-indolinethiones, benefiting from the nucleophilicities of both thiol and indole C3, have shown their potential reactivities in the cascade reactions as dinucleophiles.4c,12 To the best of our knowledge, the remote stereocontrolled cascade reactions of dienone substrates as dielectrophiles possessing a conjugated π-system and 2-indolinethiones as dinucleophiles have not been explored yet. Herein, we envisaged that a 1,6-addition/1,4-addition cascade reaction between cyclic dienones and 2-indolinethiones could be feasible for the construction of chiral thio-spirocyclic compounds through a remote stereocontrol driven by vinylogous iminium ion activation by employing primary amine catalysis.
Initially, we conducted the reaction between 2-indolinethione 2a and cyclic dienone 3a utilizing L-tert-leucine derived primary amine 1a11,13 which had been used as a powerful catalyst in our previous Michael addition/cascade works. For the convenience of chiral separation, a further step of N-protection was carried out. Delightfully, the desired adduct 4a was successfully delivered with full conversion and moderate stereoselectivity (Table 1, entry 1, 3/1 dr, 57% ee). Changing the bulky substituent adjacent to the amino group in 1a to diphenylmethoxymethyl group increased the enantioselectivity slightly (entry 2). However, the reaction became drastically suppressed when the secondary amine moiety was replaced by a tertiary amine substituent (entry 3). Meanwhile, the reaction hardly conducted when the cyclohexyl in 1a was substituted by piperidine (entry 4) while relatively low enantioselectivity was obtained by changing the bulky substituent adjacent to the amino group in 1d to diphenylmethyl substituent (entry 5), which meant the bulky tert-butyl substituent was adverse to the reaction. Then, we turned to test the cinchona alkaloid derived primary amine system. Delightfully, amine 1f14,15 in cooperation with a Brønsted acid additive strongly improved the enantioselectivity albeit affording a lower diastereoselectivity (entry 6, 2/1 dr, 86% ee). Without the catalyst, the reaction didn't proceed. Thus, amine 1f was appointed as the optimized catalyst for this cascade reaction.
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. 1 | Additive | Conv.b (%) | drc | eed (%) |
| a Reactions were performed in toluene at room temperature using 1.0 equiv. of 2a (0.2 mmol, 0.2 M), 1.5 equiv. of 3a, 0.2 equiv. of cat. 1, and 0.2 equiv. of additive. The products, purified on silica gel in the first step, were treated with 0.2 equiv. DMAP (4-dimethylaminopyridine) and 1.5 equiv. (Boc)2O (di-tert-butyl dicarbonate) in THF (0.2 M) at room temperature for 30 min.b Determined by GC analysis.c Determined by 1H NMR analysis of the crude reaction mixture.d The main diastereoisomer was determined by HPLC analysis on chiral stationary phase.e The reaction was performed at 4 °C for 24 h.f Reaction temperature = −10 °C, reaction time = 24 h.g 0.6 equiv. of 5a was used and the reaction was performed at 4 °C for 24 h.h In toluene (0.4 M).i In toluene (0.1 M). | |||||
| 1 | 1a | C6H5CO2H | Full | 3/1 | 57 |
| 2 | 1b | C6H5CO2H | Full | 2/1 | 67 |
| 3 | 1c | C6H5CO2H | Trace | — | — |
| 4 | 1d | C6H5CO2H | Trace | — | — |
| 5 | 1e | C6H5CO2H | Full | 2/1 | −31 |
| 6 | 1f | C6H5CO2H | Full | 2/1 | 86 |
| 7 | 1f | — | — | — | — |
| 8 | 1f | 5a | Full | 4/1 | 92 |
| 9 | 1f | 5b | 87 | 3/1 | 89 |
| 10e | 1f | 5a | Full | 4/1 | 96 |
| 11f | 1f | 5a | 85 | 4/1 | 93 |
| 12g | 1f | 5a | Full | 5/1 | >99 |
| 13h | 1f | 5a | Full | 3/1 | 85 |
| 14i | 1f | 5a | Full | 3/1 | 88 |
Then, different additives and solvents were tested (shown in ESI†).16 It was worth to note that the reaction didn't occur in the absence of an acid additive (entry 7). Switching the additive to (S)-mandelic acid17 (5a) greatly enhanced the stereoselectivity (entry 8, 92% ee). In contrast, its (R)-enantiomer4b,17 gave a little lower result (entry 9). Amazingly, the adduct was formed as a single enantiomer with a better dr by lowering the reaction temperature and increasing the amount of 5a (entry 12, 5/1 dr, >99% ee). Further examination of reaction concentration failed to improve the stereochemical outcome (entries 13–14).
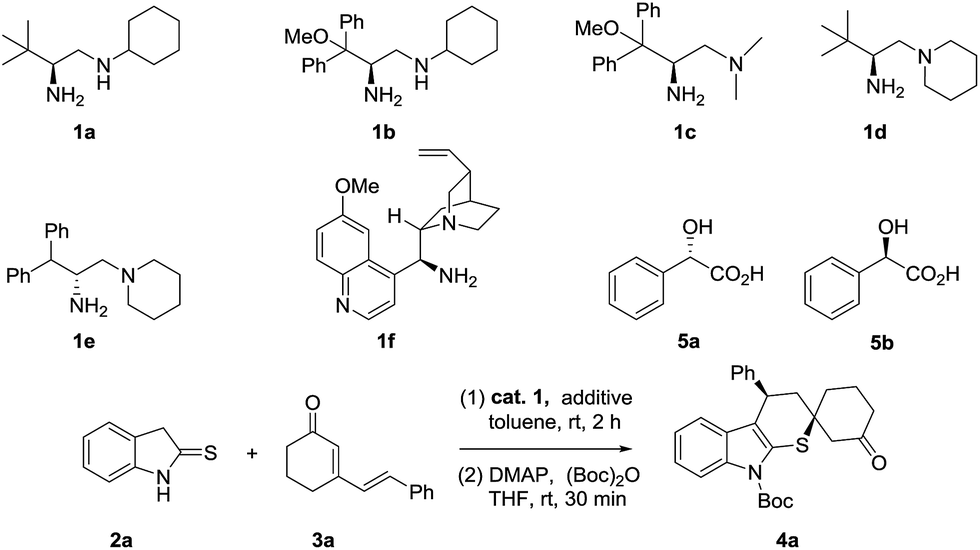
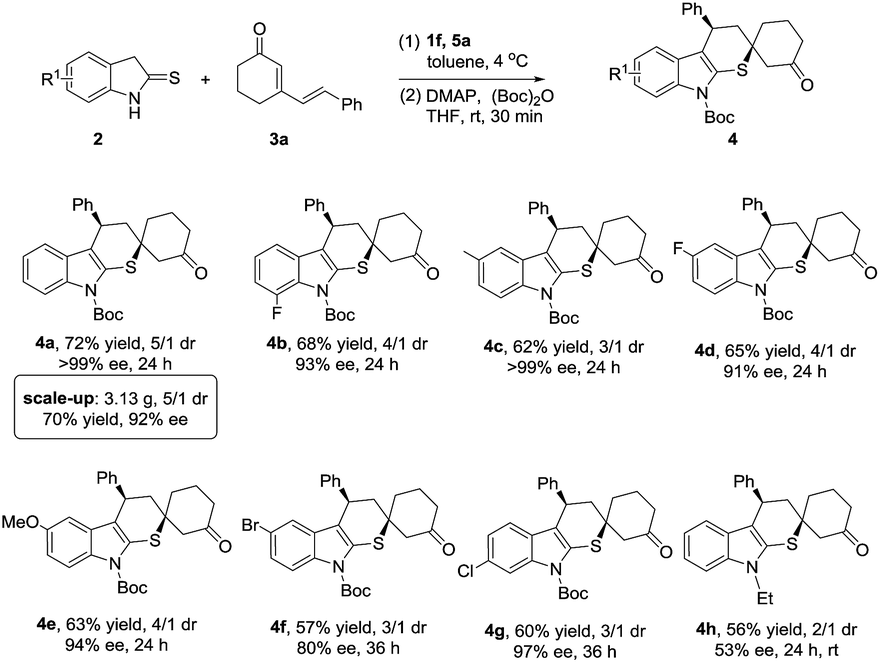
After establishing the optimized reaction conditions, the generality of this cascade reaction was studied. As presented in Schemes 1 and 2, it seemed that this method was amenable to the structural and electronic variations of both dinucleophilic and dielectrophilic substrates and yielded a variety of complex spirocompounds with high enantioselectivities. Except for 5-bromo substituted 4f, a series of spirocyclohexanones were prepared with good regioselectivities and excellent enantioselectivities regardless of the position and electronic features (Scheme 1, 4a–4e, 4g, 60–72% yield, 91–99% ee). Although the relatively lower dr were obtained, the chemical yields were still satisfied. Unexpectedly, N-ethyl substituted 2-indolinethione strongly suppressed the reaction in terms of reaction rate and stereoselectivity (Scheme 1, 4h, 56% yield, 2/1 dr, 53% ee) (Scheme 3).
 | ||
| Scheme 1 Scope of the 2-indolinethiones. | ||
 | ||
| Scheme 2 Scope of the cyclic dienones. | ||
 | ||
| Scheme 3 The absolute configuration of 4s′-a and 4s′-b. | ||
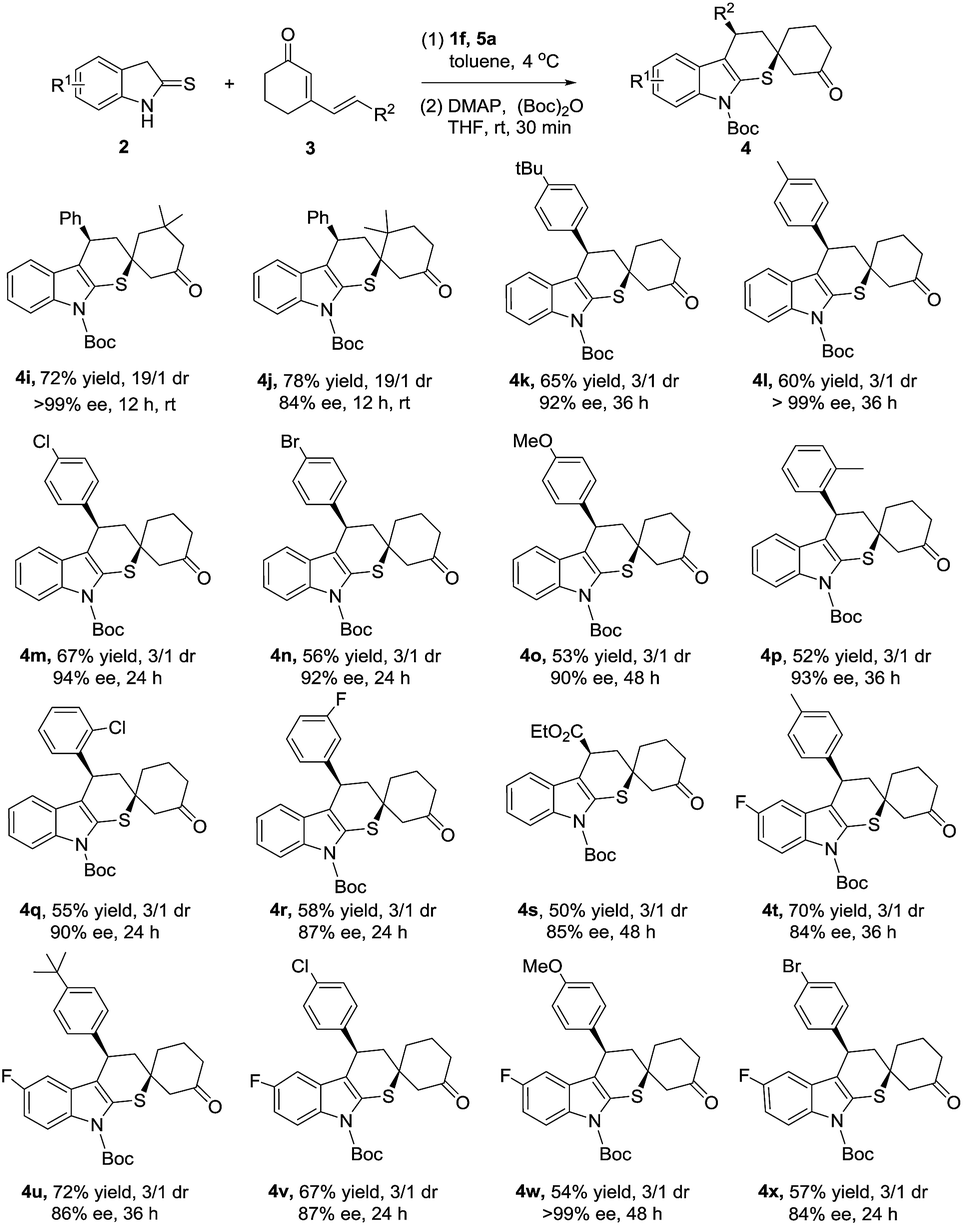
Subsequently, the investigation of dienone substrates involving substituents at the phenyl ring and cyclohexanone ring was carried out. As summarized in Scheme 2, all the cyclized products were delivered in good yields and excellent enantioselectivities (Scheme 2, 4k–4r, 52–67% yield, 87–99% ee). Excitingly, the dienones possessing dimethyl substituent at the cyclohexanone ring successfully converted to the corresponding cycloadducts as a single diastereoisomer although a higher reaction temperature was required. This was mainly attributed to the steric effect of the dimethyl substituent helping the stereocontrol in the second Michael addition step (Scheme 2, 4i, 4j: >19/1 dr). Meanwhile, dienone containing ester functionality was also compatible to this method, affording 4s in 50% yield and 3/1 dr with 85% ee. In order to explore the tolerance of our reaction, cross reactions were proceeded, achieving the target products with good yields and enantioselectivities (Scheme 2. 4t–4x, 54–72% yield, 84–99% ee).
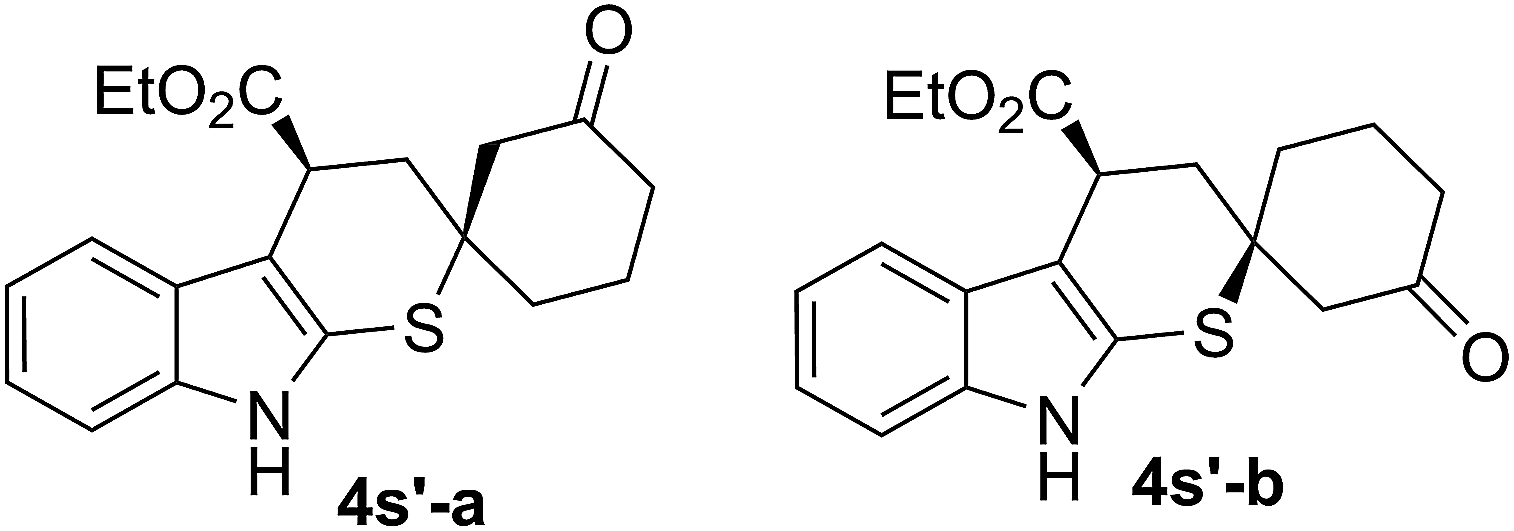
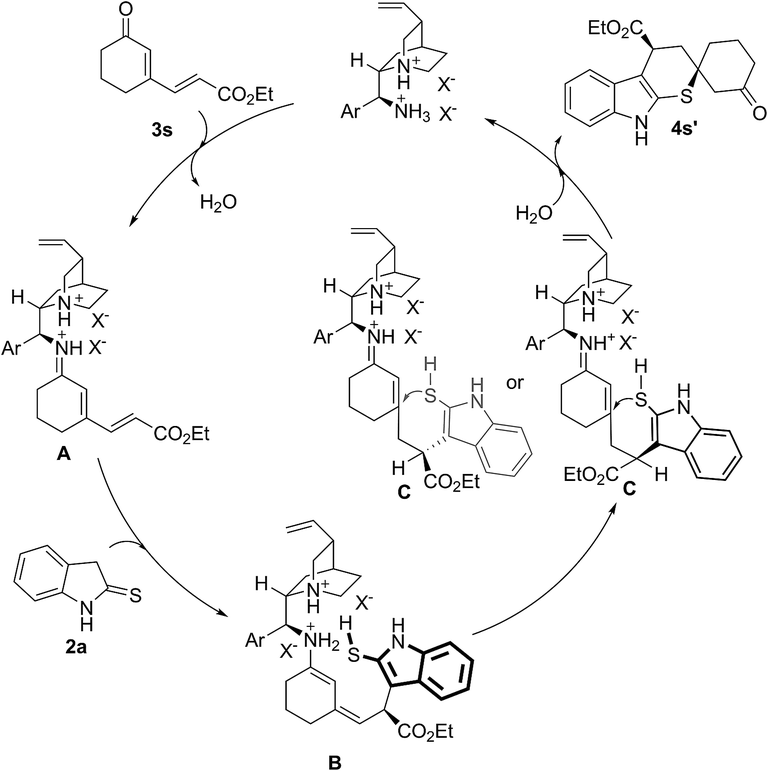
In addition, the model reaction could be easily scaled up to prepare 3.13 grams of 4a (Scheme 1). The absolute configuration of the major product 4a (more polar) was confirmed by single crystal X-ray crystallography, and each one in the unit that included four molecules was (R, R) (shown in ESI†).18 In order to investigate the reaction pathway further, the minor product 4s′-a (less polar) and the major product 4s′-b (more polar), two diastereomers of 4s′ which was the direct product of cyclic dienones possessing ester functional group and 2-indolinethione without the process of N-Boc protection, were selected to conduct the single crystal X-ray analysis due to their preferable crystallinity.18 As indicated in Scheme 4, a proposed reaction pathway was given for the 1,6-addition/1,4-addition cascade reaction based on the absolute configuration of 4s′-a and 4s′-b. First, a remote controlled asymmetric 1,6-addition reaction proceeded between indole-2-thiol resulted from the isomerization of 2-indolinethione and conjugated iminium intermediate A which was formed through the combination of cyclic dienone possessing ester substituent and catalyst 1f. Then, the resulting dienamine intermediate B from A went through a tautomeric process, and yielded the iminium intermediate C. A rotation of the carbon–carbon bond, due to the possible steric effect of amine salt formed by the tertiary amino group in catalyst 1f and (S)-mandelic acid, was conducted, producing the other conformer of intermediate C. Subsequently, the intramolecular thio-Michael addition of C proceeded with indole-2-thiol motif as a highly reactive nucleophile, affording 1,6-addition/1,4-addition cascade product. The 1,4-addition should account for the unsatisfactory diastereoselectivity based on the steric outcome of cascade products.
 | ||
| Scheme 4 Proposed Reaction Pathway. | ||
Conclusions
In summary, a facile and desirable method to construct chiral spiro[thiopyranoindole-cyclohexanone] scaffold has been developed between cyclic dienones and 2-indolinethiones via vinylogous iminium ion activating 1,6-addition/1,4-addition cascade reaction. The reaction proceeded with good yield and excellent enantioselectivity. We successfully realized the remote transmission of the stereochemical information through the conjugated π-system of β-substituted cyclic dienones, and this strategy was expected to open a new avenue to prepared the highly enantioenriched spiro scaffold in one-step process.Acknowledgements
This work was partially supported by National Natural Science Foundation of China (21272068, 21572056), Program for New Century Excellent Talents in University (NCET-13-0800) and the Fundamental Research Funds for the Central Universities.Notes and references
- For selected reviews on the formation of carbon–sulfur bond, see: (a) T. Kondo and T.-A. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS PubMed; (b) M. Fontecave, S. Ollagnier-de-Choudens and E. Mulliez, Chem. Rev., 2003, 103, 2149–2166 CrossRef CAS PubMed; (c) P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807–8864 CrossRef CAS PubMed; (d) J.-S. Yu, H.-M. Huang, P.-G. Ding, X.-S. Hu, F. Zhou and J. Zhou, ACS Catal., 2016, 6, 5319–5344 CrossRef.
- For selected references on the applications of organosulfur compounds in the biochemistry, see: (a) C. Schöneich, D. Pogocki, G. L. Hug and K. Bobrowski, J. Am. Chem. Soc., 2003, 125, 13700–13713 CrossRef PubMed; (b) S. Adolph, V. Jung, J. Rattke and G. Pohnert, Angew. Chem., Int. Ed., 2005, 44, 2806–2808 CrossRef CAS PubMed.
- For selected references on organocatalytic enantioselective methods to access organosulfur compounds, see: (a) Y. Gao, Q. Ren, H. Wu, M. Li and J. Wang, Chem. Commun., 2010, 46, 9232–9234 RSC; (b) X. Tian, Y. Liu and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 6439–6442 CrossRef CAS PubMed; (c) Z. Zhou, X. Feng, X. Yin and Y.-C. Chen, Org. Lett., 2014, 16, 2370–2373 CrossRef CAS PubMed; (d) N. Fu, L. Zhang, S. Luo and J.-P. Cheng, Org. Lett., 2014, 16, 4626–4629 CrossRef CAS PubMed; (e) Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 5320–5323 CrossRef CAS PubMed; (f) A. J. M. Farley, C. Sandford and D. J. Dixon, J. Am. Chem. Soc., 2015, 137, 15992–15995 CrossRef CAS PubMed.
- For selected examples on sulfur atom participating in the construction for quaternary spirocyclic center, see: (a) W. Wu, H. Huang, X. Yuan, K. Zhu and J. Ye, Chem. Commun., 2012, 48, 9180–9182 RSC; (b) J. Stiller, D. Kowalczyk, H. Jiang, K. A. Jørgensen and L. Albrecht, Chem.–Eur. J., 2014, 20, 13108–13112 CrossRef CAS PubMed; (c) X. Chen, J.-Q. Zhang, S.-J. Yin, H.-Y. Li, W.-Q. Zhou and X.-W. Wang, Org. Lett., 2015, 17, 4188–4191 CrossRef CAS PubMed.
- For the selected reviews on organocatalytic cascade strategy, see: (a) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed; (b) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed; (c) C. M. R. Volla, L. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS PubMed.
- For the selected examples on organocatalytic cascade strategy in total synthesis, see: (a) P. Jakubec, D. M. Cockfield and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 16632–16633 CrossRef CAS PubMed; (b) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed; (c) S. B. Jones, B. Simmons, A. Mastracchio and D. W. C. MacMillan, Nature., 2011, 475, 183–188 CrossRef CAS PubMed; (d) R. Ardkhean, D. F. J. Caputo, S. M. Morrow, H. Shi, Y. Xiong and E. A. Anderson, Chem. Soc. Rev., 2016, 45, 1557–1569 RSC.
- For the key examples on organocatalytic cascade reactions, see: (a) X. Yu and W. Wang, Org. Biomol. Chem., 2008, 6, 2037–2046 RSC; (b) Y.-N. Xuan, Z.-Y. Chen and M. Yan, Chem. Commun., 2014, 50, 10471–10473 RSC; (c) R. Zhou, Q. Wu, M. Guo, W. Huang, X. He, L. Yang, F. Peng, G. He and B. Han, Chem. Commun., 2015, 51, 13113–13116 RSC; (d) H. Huang, S. Konda and J. C. G. Zhao, Angew. Chem., Int. Ed., 2016, 55, 2213–2216 CrossRef CAS PubMed; (e) B.-L. Zhao and D.-M. Du, Chem. Commun., 2016, 52, 6162–6165 RSC.
- (a) R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247–7290 RSC; (b) I. D. Jurberg, I. Chatterjee, R. Tannert and P. Melchiorre, Chem. Commun., 2013, 49, 4869–4883 RSC; (c) H. Jiang, L. Albrecht and K. A. Jørgensen, Chem. Sci., 2013, 4, 2287–2300 RSC; (d) D. Cheng, Y. Ishihara, B. Tan and C. F. Barbas III, ACS Catal., 2014, 4, 743–762 CrossRef CAS; (e) C. V. Gomez, D. C. Cruz, R. Mose and K. A. Jørgensen, Chem. Commun., 2014, 50, 6035–6038 RSC.
- X. Tian and P. Melchiorre, Angew. Chem., Int. Ed., 2013, 52, 5360–5363 CrossRef CAS PubMed.
- (a) M. Silvi, I. Chatterjee, Y. Liu and P. Melchiorre, Angew. Chem., Int. Ed., 2013, 52, 10780–10783 CrossRef CAS PubMed; (b) P. H. Poulsen, K. S. Feu, B. M. Paz, F. Jensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 8203–8207 CrossRef CAS PubMed.
- X. Gu, T. Guo, Y. Dai, A. Franchino, J. Fei, C. Zou, D. J. Dixon and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 10249–10253 CrossRef CAS PubMed.
- (a) L. K. Ransborg, L. Albrecht, C. F. Weise, J. R. Bak and K. A. Jørgensen, Org. Lett., 2012, 14, 724–727 CrossRef CAS PubMed; (b) X. Chen, Z.-H. Qi, S.-Y. Zhang, L.-P. Kong, Y. Wang and X.-W. Wang, Org. Lett., 2015, 17, 42–45 CrossRef CAS PubMed.
- For the review on the enamine catalysis with L-tert-leucine derived catalysts, see: (a) W. Wu, X. Yuan, J. Hu, X. Wu, Y. Wei, Z. Liu, J. Lu and J. Ye, Org. Lett., 2013, 15, 4524–4527 CrossRef CAS PubMed; (b) H. Huang, W. Wu, K. Zhu, J. Hu and J. Ye, Chem.–Eur. J., 2013, 19, 3838–3841 CrossRef CAS PubMed; (c) L. Zhang, N. Fu and S. Luo, Acc. Chem. Res., 2015, 48, 986–997 CrossRef CAS PubMed; (d) J. Fei, Q. Qian, X. Sun, X. Gu, C. Zou and J. Ye, Org. Lett., 2015, 17, 5296–5299 CrossRef CAS PubMed.
- For selected reviews on cinchona alkaloid derived primary amine catalysis, see: (a) L. Jiang and Y.-C. Chen, Catal. Sci. Technol., 2011, 1, 354–365 RSC; (b) P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 9748–9770 CrossRef CAS PubMed.
- For key examples, see: (a) J.-W. Xie, W. Chen, R. Li, M. Zeng, W. Du, L. Yue, Y.-C. Chen, Y. Wu, J. Zhu and J.-G. Deng, Angew. Chem., Int. Ed., 2007, 46, 389–392 CrossRef CAS PubMed; (b) G. Bartoli, M. Bosco, A. Carlone, F. Pesciaioli, L. Sambri and P. Melchiorre, Org. Lett., 2007, 9, 1403–1405 CrossRef CAS PubMed; (c) S. H. McCooey and S. J. Connon, Org. Lett., 2007, 9, 599–602 CrossRef CAS PubMed.
- For more reaction condition optimizing details, see ESI.†.
- (a) A. O. Patil, W. T. Pennington, I. C. Paul, D. Y. Curtin and C. E. Dykstra, J. Am. Chem. Soc., 1987, 109, 1529–1535 CrossRef CAS; (b) J. Lin, Q.-S. Hu, M.-H. Xu and L. Pu, J. Am. Chem. Soc., 2002, 124, 2088–2089 CrossRef CAS PubMed.
- See ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1481742, 1495419 and 1495499. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra19916j |
| This journal is © The Royal Society of Chemistry 2016 |