DOI:
10.1039/C6RA17898G
(Paper)
RSC Adv., 2016,
6, 86468-86476
Green synthesis, optical and magnetic properties of a MnII metal–organic framework (MOF) that exhibits high heat of H2 adsorption†
Received
13th July 2016
, Accepted 3rd September 2016
First published on 5th September 2016
Abstract
Green synthesis of a 3D, Mn(II) metal–organic framework (MOF) composed of a trimeric Mn(II) cluster and NDC linker formulated as [Mn3(NDC)3(DMA)4]n (1) (where, NDC = 2,6 napthalene dicarboxylic acid, DMA = N,N-dimethylacetamide) has been achieved by employing mechanochemical and sonochemical routes. Interestingly, MOF 1 undergoes reversible structural transformation upon desolvation and solvation of DMA molecules. The desolvated framework, 1′ containing two unsaturated Mn(II) sites exhibits interesting H2 and CO2 uptake properties with isosteric heats of adsorption (Qst) for H2 and CO2 of 11.8 and 29.2 kJ mol−1, respectively. Remarkably, the Qst of H2 estimated for 1′ is found to be the highest value amongst the Mn(II) MOFs reported so far and the high value has been attributed to the stronger interaction of H2 with the unsaturated Mn(II) centers. Further, variable temperature magnetic susceptibility measurements of 1 revealed weak antiferromagnetic coupling interactions between the adjacent Mn(II) ions. The thermal stability of 1 has also been examined and was found to be highly stable. Photoluminescence investigation revealed that the emission from MOF 1 is owed to ligand based charge transfer transitions. Furthermore, compound 1 undergoes temperature induced solid-state conversion into phase pure MnO nanocrystals of about 10–18 nm in size embedded in a carbonaceous layer to form MnO–C nanocomposite (NC). The MnO–C NC has been characterized by PXRD, FE-SEM, EDAX and TEM analyses.
Introduction
Metal–organic frameworks (MOFs) are a new class of crystalline materials which are attracting growing interest not only due to their intriguing network topologies1–4 but also due to their potential applications in sensing,5–10 catalysis,11–14 separation,15–17 carbon dioxide capture,18–21 magnetism,22–24 drug delivery25–27 and so on. Particularly, MOFs are gaining considerable attention as prospective candidates for hydrogen storage applications.28–33 Since hydrogen has been considered as an eco-friendly alternative to fossil fuels, the U.S. department of energy (DOE) has set an ultimate target of volumetric storage capacity of 70 g L−1 and gravimetric storage capacity of 7.5 wt% for onboard applications of H2.34 Several MOFs exhibiting very high H2 uptake properties at low temperatures (77 K) have been reported but the maximum uptake achieved at ambient conditions is still close to 2–3%.35 The low uptake at ambient conditions has been attributed due to weak interaction of H2 (Qst < 10 kJ mol−1) with the framework. To overcome this limitation, several strategies such as, doping of noble metal (Pd) nanoparticles,36,37 pore size modulation,38,39 introduction of open metal centers, doping of alkali metal ions,40,41 and etc., have been investigated to enhance the H2 uptake properties of MOFs. Furthermore, MOFs with smaller pores (3–4 Å) are found to be appropriate candidates for application as hydrogen storage materials due to high interaction energy resulting from an increased van der Waals contact area.28–33 In this context, we have recently reported construction of Zn(II) MOF constituted by partially fluorinated ligand exhibiting isosteric heat of adsorption (Qst) of −8.8 kJ mol−1.42
It is worth to note that incorporation of multiple components such as, porosity and magnetism in MOFs is not common as magnetism operates over short distances whereas porosity generation needs large organic linkers. Further, magnetic properties strongly depend on the nature of metal ion or clusters and can be tuned by altering the length of organic linkers. Therefore for establishing magnetic ordering, use of paramagnetic metal ions as spin carriers and multicarboxylate ligands capable of facilitating magnetic coupling between the metal ions are essential. In this direction, MOFs exhibiting interesting magnetic properties like, ferro-,43 antiferro-,45,46 meta-magnetic,47,48 single chain magnetism,49–51 spin canting52,53 and spin glass behavior54 have been reported.
Towards rapid synthesis of MOFs which can provide access to unsaturated metal centers for stronger interaction with H2 herein we have employed green synthetic routes55–57 for synthesis of 3D MOF, [Mn3(NDC)3(DMA)4]n (1) constituted by [Mn3(CO2)6] secondary building units (SBUs) which are bridged by NDC ligands. Interestingly, MOF 1 undergoes reversible structural transformation upon desolvation and solvation of DMA. The desolvation generates a microporous framework, 1′ containing two unsaturated Mn(II) sites which exhibits an interesting H2 and CO2 uptake properties with isosteric heat of adsorption (Qst) of 11.8 and 29.2 kJ mol−1, respectively. Remarkably, the Qst value of H2 for 1′ is the highest amongst the Mn(II) MOFs reported so far and the high value has been attributed to the stronger interaction of H2 with coordinatively unsaturated Mn(II) centers. Further, variable temperature magnetic susceptibility measurements of 1 revealed weak antiferromagnetic coupling interaction between the adjacent Mn(II) ions. Photoluminescence investigation show emission from 1 owing to ligand based charge transfer transitions. Moreover, compound 1 undergoes temperature induced solid-state conversion into phase pure MnO nanocrystals embedded in carbonaceous layer to form MnO–C nanocomposite (NC). The MnO–C NC has been characterized by PXRD, FE-SEM, EDAX and TEM analyses. The present study demonstrates the application of MOFs with unsaturated metal centers for stronger interaction with H2 and its utility as a clean source for phase pure formation of MnO–C nanocomposite.
Experimental details
Materials
All the reagents employed were commercially available and used as provided without further purification. Mn(OAc)2·4H2O and 2,6-napthalene dicarboxylic acid (H2NDC) were obtained from Sigma Aldrich Chemical Company. N,N-Dimethylacetamide (DMA) was obtained from Loba Chemie, India.
Physical measurements
Elemental analysis was carried out using a Thermo Fischer Flash 2000 Elemental Analyzer. IR spectra were recorded on a Thermo Scientific Nicolet iS10 FT-IR spectrometer in the 4000–400 cm−1 region. Thermogravimetric analysis (TGA) was carried out using a Mettler Toledo thermogravimetric analyzer in a nitrogen atmosphere (flow rate = 40 mL min−1) in the temperature range 25–1000 °C (heating rate = 10 °C min−1). Powder X-ray diffraction (PXRD) patterns of the compounds were recorded using Cu Kα radiation (k = 1.542 Å; 40 kV, 20 Ma) with PANalytical's X'PERT PRO diffractometer. Gas adsorption analyses were carried out using surface area analyzer Quadrasorb SI (Quantachrome) with high purity gases (99.995%). UV-Vis spectra of the solid samples of the compounds were recorded on Shimadzu UV-2600 240 V spectrophotometer using the solid state absorbance mode using BaSO4 as reference material. Photoluminescence spectra were recorded at room temperature on a PerkinElmer LS55 fluorescence spectrophotometer. DC magnetic susceptibility measurements were carried out on a vibrating sample magnetometer, PPMS (Physical Property Measurement System, Quantum Design, USA) in the temperature range 2.5–300 K with applied field of 100 Oe. Field variation (−5 kOe to 5 kOe) magnetization measurements were carried out at 3 K. Diamagnetic corrections were applied using Pascal's constants. Selected-area electron diffraction (SAED) and TEM measurements were performed with a FEG TEM 300 kV field-emission transmission electron microscope.
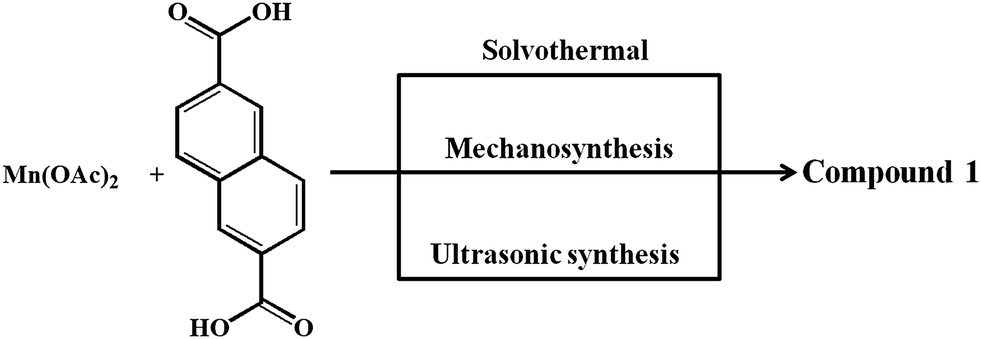
Synthesis of 1 by solvothermal method
Compound 1 was synthesized under solvothermal conditions at 100 °C. Mn(OAc)2·4H2O (0.20 mmol, 0.049 g) was dissolved in a mixture of 2 mL ethanol and 4 mL of DMA to which DMA solution (4 mL) of H2NDC (0.20 mmol, 0.043 g) was added with constant stirring. To this solution 0.5 mL of formic acid was added to maintain the pH of the solution at 3 and the contents were stirred for 5 min and then taken in a 30 mL glass vial sealed with parafilm and teflon and heated at 100 °C for 3 days. After being cooled to room temperature, pale yellow block shaped crystals of 1 were isolated. Yield calculated 72% based on MnII ions and the phase purity of as-synthesized sample was confirmed by PXRD analysis (Fig. S1†). IR (cm−1) (Fig. S2†): ν(CH-Ar), 2977(w); ν(C–N), 1612(s); ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1477–1347(s); ν(C–O), 1287–1189(s).
C), 1477–1347(s); ν(C–O), 1287–1189(s).
Synthesis of 1 by sonochemical route
1 was synthesized by ultrasonication using bath sonicator at room temperature and atmospheric pressure for half an hour. An ethanolic solution (2 mL) of Mn(OAc)2·6H2O (0.1 mmol, 0.029 g) was added drop wise to the DMA solution (8 mL) of H2NDC (0.1 mmol, 0.039 g) in a 30 mL glass vial, and the mixture was sonicated in an bath sonicator. After 30 min, a pale yellow product was obtained by centrifugation followed by washing with methanol and water two to three times and dried under vacuum. Yield calculated 55% based on MnII ions and the phase purity of as-synthesized sample was confirmed by PXRD analysis (Fig. S1†). IR (cm−1) (Fig. S3†) ν(CH-Ar), 2988(w); ν(C–N), 1608(s); ν(CC), 1482–1325(s); ν(C–O), 1254–1194(s).
Synthesis of 1 by solvent-assisted mechanochemical route
Synthesis of 1 was carried out by grinding vigorously the mixture of Mn(OAc)2·6H2O (0.049 g, 0.20 mmol) and H2NDC (0.043 g, 0.20 mmol) in a mortar with a pestle for 5 min. Then 2–3 drops of DMA was added to the mixture and grinded for 15 min. The color of the mixture changes to pale yellow indicating the formation of the product. The pale yellow powder was washed with minimal amount of DMA, distilled water and methanol to eliminate any unreacted starting materials and vacuum dried. Yield calculated 91% based on MnII ions and the phase purity of as-synthesized sample was confirmed by PXRD analysis (Fig. S1†). IR (cm−1) (Fig. S4†) ν(CH-Ar), 2977(w); ν(C–N), 1608(s); ν(CC), 1466–1314(s); ν(C–O), 1265–1172(s).
Adsorption measurements
The N2 adsorption–desorption measurements were carried out at 77 K, while H2 adsorption–desorption measurements were carried out at 77 and 87 K, whereas CO2 adsorption–desorption experiments were conducted at 273 and 298 K using Quadrasorb SI surface area analyzer, (Quantachrome) volumetric instrument. Ultrapure (99.995%) N2, H2, He and CO2 gases were used for the adsorption experiment. Before starting the adsorption measurements, the sample (∼0.1 g) was activated at 523 K under vacuum (18 mTorr) for 20 h. Then the activated MOF sample was introduced to the surface area analyzer. 87 K and 77 K were attained using liquid argon and liquid nitrogen respectively. Dead volume of sample cell was measured using helium gas.
Results and discussion
Syntheses and structure
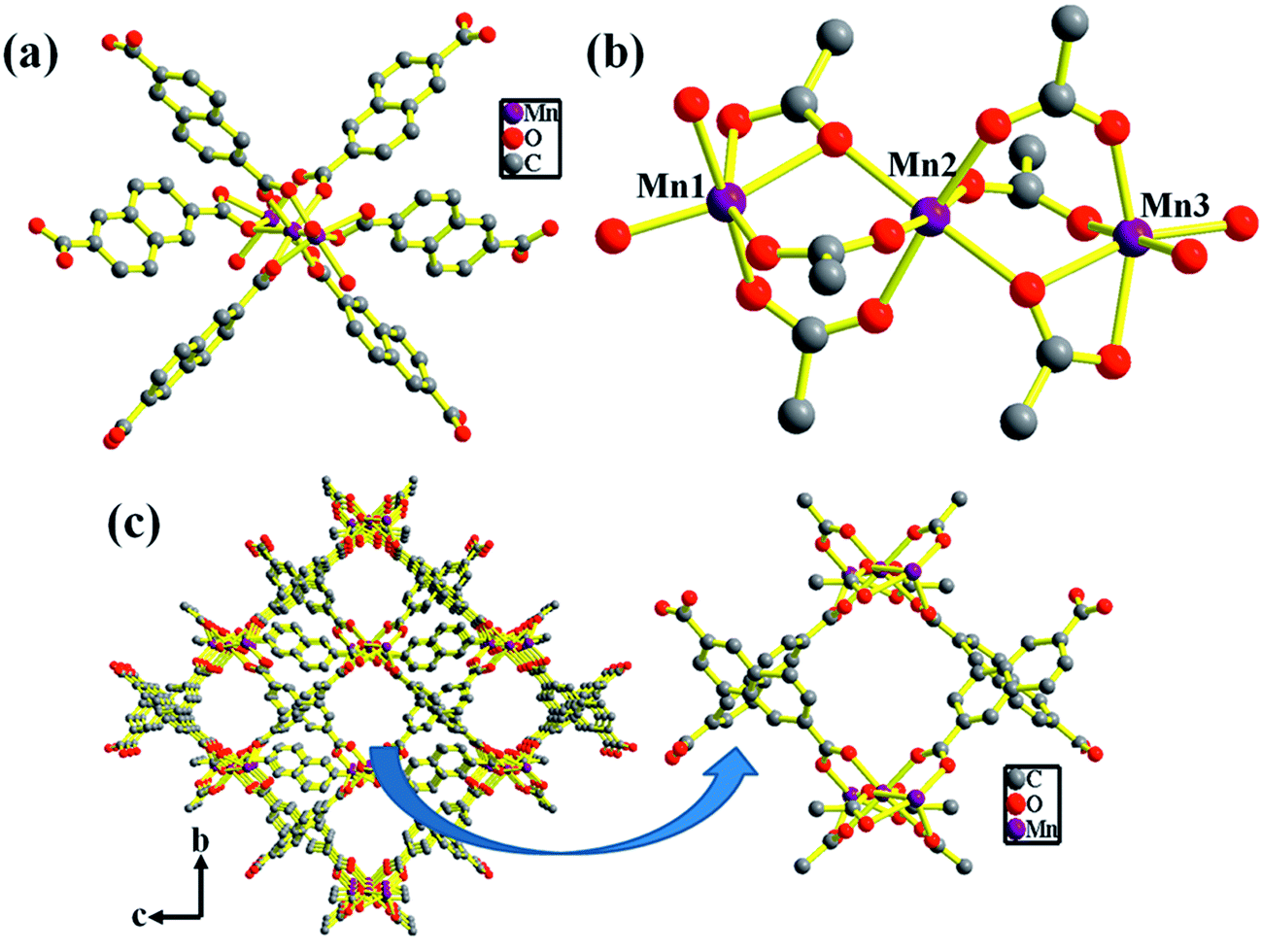
The structure of compound 1 has been reported earlier by S.-Y. Yang et al.58 It has a 3D framework structure constituted by Mn3 clusters which are connected through six carboxylate groups of three NDC ligands including four coordinated DMA molecules (Fig. 1). The carboxylate groups of NDC ligand adopt bidentate and tridentate coordination modes to bridge Mn(II) nodes (Scheme S1†).
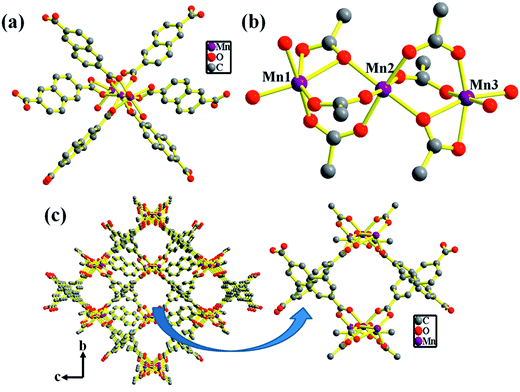 |
| | Fig. 1 (a) Polyhedral representation of 1 showing the coordination environment around Mn(II) (hydrogen atoms are removed for clarity) (b) distorted octahedral SBU's of Mn3 cluster. (c) View of the rectangular 1D channels along the crystallographic a-axis of 3D framework of 1′ (coordinated DMA molecules are omitted for clarity). | |
Considering the presence of four DMA molecules which are coordinating to two Mn(II) centers, we envisaged that the DMA-free MOF with two unsaturated Mn(II) centers would be a suitable candidate to study H2 storage properties. In this regard, we investigated the rapid synthesis of phase pure sample of 1 using green synthetic techniques such as solvent-assisted mechano- and sonochemical routes. Manual grinding of Mn(II) salt and H2NDC ligand in pestle and mortar for 20 min yielded 1 in phase pure form (Fig. S1†). On the other hand, ultrasonication of a mixture of Mn(II) salt and deprotonated NDC2− ligand in DMA/EtOH (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 30 min yielded 1 in phase pure form (Scheme 1). The phase purity of the isolated samples obtained by different synthetic routes was confirmed by powder XRD patterns which are in good agreement with the simulated pattern from the crystal structure (Fig. S1†). The DMA-free framework, 1′ was obtained by evacuating 1 at an elevated temperature of 523 K under vacuum (18 mTorr) for 20 h. Interestingly, upon removal of coordinated DMA molecules the color of the sample changed from pale yellow to grey. PXRD analysis of the desolvated sample revealed change in the diffraction pattern upon removal of coordinated DMA molecules (Fig. S5†). Further, upon soaking the desolvated sample 1′ in DMA for 3 days regenerated the original framework 1, as revealed from PXRD analysis suggesting the reversible structural change upon desolvation and solvation (Fig. S5†).
:1) for 30 min yielded 1 in phase pure form (Scheme 1). The phase purity of the isolated samples obtained by different synthetic routes was confirmed by powder XRD patterns which are in good agreement with the simulated pattern from the crystal structure (Fig. S1†). The DMA-free framework, 1′ was obtained by evacuating 1 at an elevated temperature of 523 K under vacuum (18 mTorr) for 20 h. Interestingly, upon removal of coordinated DMA molecules the color of the sample changed from pale yellow to grey. PXRD analysis of the desolvated sample revealed change in the diffraction pattern upon removal of coordinated DMA molecules (Fig. S5†). Further, upon soaking the desolvated sample 1′ in DMA for 3 days regenerated the original framework 1, as revealed from PXRD analysis suggesting the reversible structural change upon desolvation and solvation (Fig. S5†).
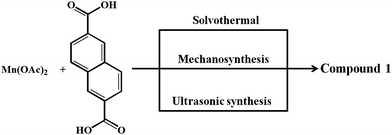 |
| | Scheme 1 Various synthesis routes for 1. | |
Gas adsorption studies
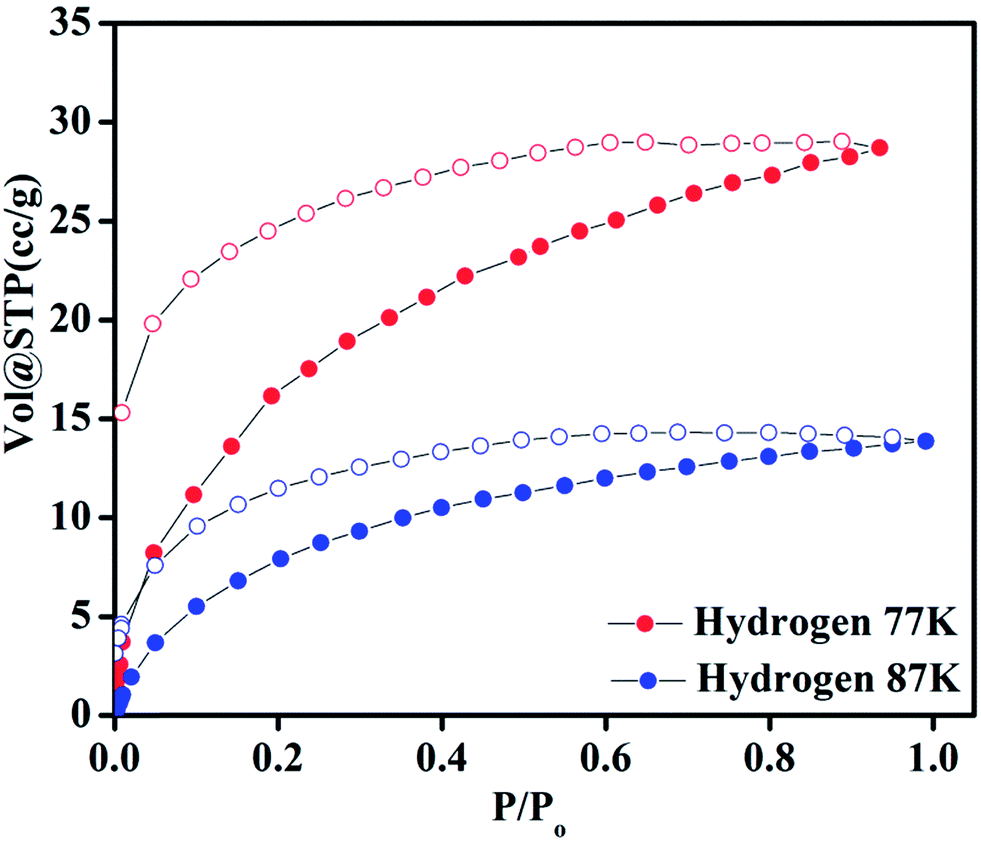
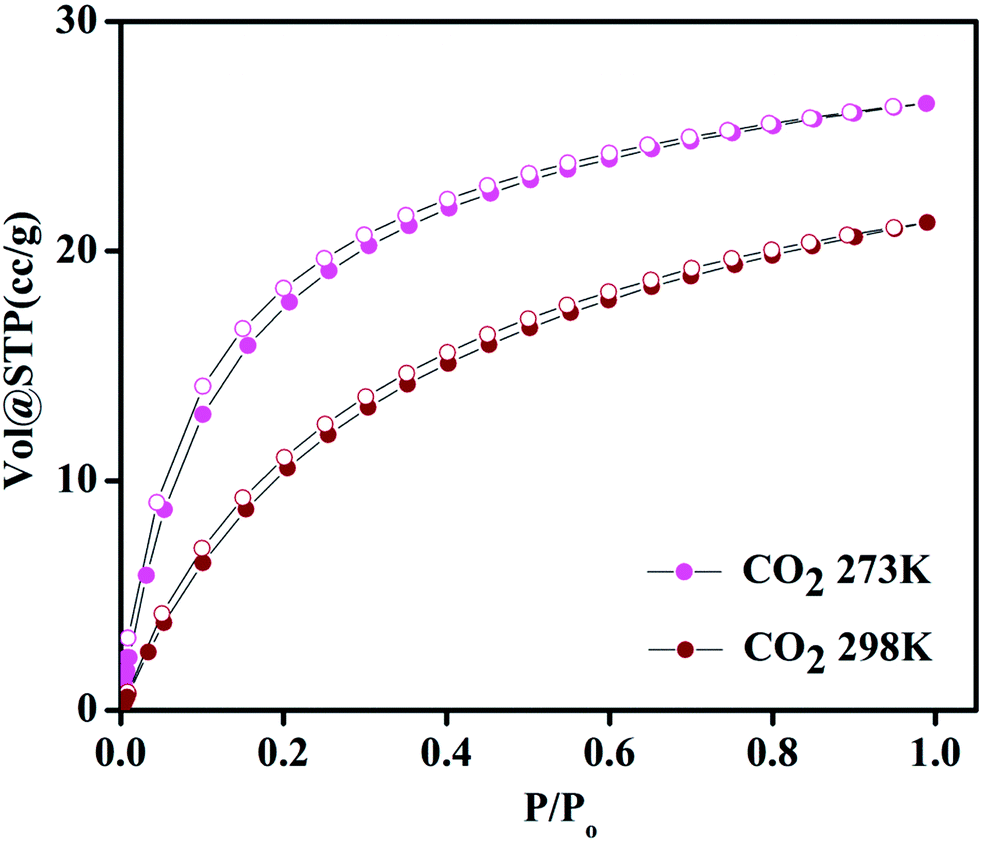
Compound 1 possesses 1D channels in which the DMA molecules are protruded (Fig. 1) and the DMA-free framework obtained by heat treatment of 1 possess microporous 1D channels decorated with unsaturated Mn(II) sites (Fig. 1c). Such MOFs have been found to be suitable for selective adsorption of H2 and CO2.44 Therefore, we investigated the gas adsorption properties of the desolvated sample 1′ to establish the permanent porosity of the framework. N2 (kinetic diameter, 3.64 Å) adsorption isotherms of compound 1′ revealed a type II isotherm with hysteresis indicative of mainly surface adsorption (Fig. S6†). The hysteresis behaviour observed upon desorption can be attributed to the flexibility of framework and hindered diffusion through narrow pore apertures.59 The BET surface area of 1′ estimated from N2 adsorption isotherm carried out at 77 K was found to be 31.65 m2 g−1. Further, H2 adsorption studies of 1′ carried out at cryogenic temperatures show an irreversible type-I isotherm with large hysteresis at 77 and 87 K (Fig. 2) with uptake of 0.26 and 0.13 wt%, respectively as the pressure approaches to 1 bar. The observation of large hysteresis is an indicative of stronger interaction of H2 with the framework owing to presence of unsaturated Mn(II) centers.44 On the other hand, 1′ shows reversible type I isotherms for CO2 at 273 and 298 K suggesting the microporous nature of the compound with uptake of 5.19 wt% and 4.29 wt%, respectively, as the pressure approaches to 1 bar (Fig. 3). The isosteric heat of adsorption (Qst) for H2 was calculated (Fig. S9†) using the Clausius–Clapeyron equation.60,61 Adsorption branches of isotherms measured at 77 and 87 K were fitted to Freundlich–Langmuir equation62 (shown in Fig. S7 and S8†) to obtain a precise prediction over the quantity of gas adsorbed at saturation. The Qst for hydrogen was estimated to be 11.8 kJ mol−1 (Fig. S9†) at lower coverage which is found to be the highest amongst the Mn(II) MOFs and higher or comparable to other robust microporous MOFs reported in literature (Table 1). The considerably high value of Qst suggests stronger interaction of H2 with the framework which can be attributed due to presence of coordinatively unsaturated Mn(II) centers decorated in the 1D channels of 1′ (Fig. 1) and also due to the aromatic π-electron clouds of NDC ligand. Further, the Qst value for H2 decreases at higher coverage which can be explained as the adsorbed H2 molecules blocking the favorable sites of binding for the next coming gas molecule (Fig. S9†). By following the similar procedure discussed above the Qst value for CO2 was also determined and the value was found to be 29.2 kJ mol−1 (Fig. S10–S12†). The moderate value of Qst for CO2 can be ascribed due to its interaction with the electric field generated by aromatic π-electron clouds of NDC ligand and also with the unsaturated Mn(II) centers owing to its significant quadrupole moment (−1.4 × 10−39 C m2) and polarizability.
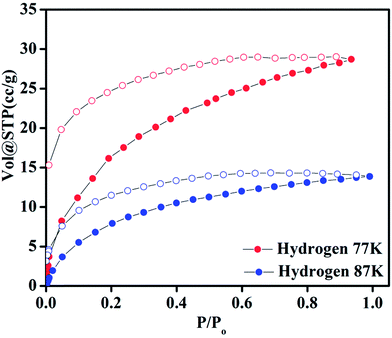 |
| | Fig. 2 Hydrogen adsorption–desorption isotherms of 1′ at 77 and 87 K. | |
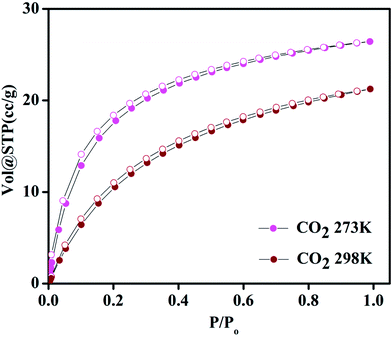 |
| | Fig. 3 Carbon dioxide adsorption–desorption isotherms of 1′ at 273 and 298 K. | |
Table 1 Isosteric heat of adsorption (Qst) values for H2 of various MOFsa
| Material |
Pressure (bar) |
Qst for H2 |
Reference |
| Where, btt = 1,3,5-benzenetristetrazolate, bdt = 1,4-benzeneditetrazolate, ndc = 2,6-napthalenedicarboxylate, tpt-3tz = 2,4,6-tris(p-phenyltetrazolate)-s-triazine, dmf = N,N′-dimethylformamide, bipy = 4,4′-bipyridine, DMA = N,N-dimethylacetamide, MeIM = 2-methylimidazole, BDC = 1,4-benzenedicarboxylate, BTC = 1,3,5-benzenetricarboxylic acid, bptc = biphenyl-3,3′,5,5′-tetracarboxylic acid, dhtp = 2,5-dihydroxyterephthalic acid, TATB = 4,4′,4′′-s-triazine-2,4,6-triyl-tribenzoic acid, dobdc = 2,5-dihydroxybenzenedicarboxylic acid, NOTT = Nottingham, PCN = Porous Coordination Network, MIL = Materials from Institut Lavoisier, IRMOF = isoreticular metal organic framework, ZIF = Zeolitic Imidazolate Framework. |
| Zn(MeIM)2 (ZIF-8) |
1 |
4.5 |
77 |
| Zn4O(BDC)3 (MOF-5) |
1 |
4.8 |
78 and 79 |
| Zn4O(NDC)3 (IRMOF-8) |
1 |
6.1 |
80 and 81 |
| Cr3OF(BTC)2 (MIL-100) |
90 |
6.3 |
82 |
| Cu2(bptc) (NOTT-100) |
1 |
6.3 |
83 |
| Zn4O(BDC)3 interpenetrated (MOF-5) |
1 |
7.6 |
84 |
| Mn3[(Mn4Cl)3(tpt-3tz)8(dmf)12]2 |
80 |
7.6 |
76 |
| Zn2(dhtp) (MOF-74) |
1 |
8.3 |
79 |
| Mn3(bdt)3 |
1.2 |
8.4 |
75 |
| Cu3[(Cu2.9Mn1.1Cl)3(btt)8]2·2CuCl2 |
1.2 |
8.5 |
74 |
| Mn2(bdt)Cl2 |
1.2 |
8.8 |
75 |
| Li3.2Mn1.4[(Mn4Cl)3(btt)8]2·0.4LiCl |
1.2 |
8.9 |
74 |
| Ni2.75Mn0.25[(Mn4Cl)3(btt)8]2 |
1.2 |
9.1 |
74 |
| Zn3[(Zn0.7Mn3.3Cl)3(btt)8]2·2ZnCl2 |
1.2 |
9.6 |
74 |
| Mn3[(Mn4Cl)3(btt)8]2·0.75CuPF6 |
1.2 |
9.9 |
74 |
| Cr3OF(BDC)3 (MIL-101) |
60 |
10.0 |
82 |
| Mn3[(Mn4Cl)3(btt)8(CH3OH)10]2 |
1.2 |
10.1 |
74 |
| Mn3[(Mn4Cl)3(BTT)8]2 (Mn-BTT) |
1.2 |
10.1 |
85 |
| H2[Co4O(TATB)8/3] (PCN-9) |
1 |
10.1 |
86 |
| Mg2(dobdc) (MOF-74Mg) |
1 |
10.3 |
87 |
| Co3[(Mn4Cl)3(btt)8]2·1.7CoCl2 |
1.2 |
10.5 |
74 |
| {[Mn3(bipy)3][Cr(CN)6]2} |
45 |
11.5 |
44 |
| Mn3(NDC)3(DMA)4 |
1 |
11.8 |
This work |
| CPO-27-Ni |
— |
13.5 |
88 |
Magnetic studies
Compound 1 contains tri-nuclear Mn(II) metal clusters connected with each other by NDC ligand.63 The inter-nuclear distance between the two adjacent and terminal Mn⋯Mn centers in the [Mn3(COO)6] SBU are 3.665 and 7.320 Å and the Mn–O–Mn bond angle is of 108.35°. Thus one can anticipate stronger magnetic exchange coupling between the adjacent Mn centers in comparison to the terminal ones. The variable temperature zero-field-cooled (ZFC) and field-cooled (FC) magnetic susceptibility (χ) data of a powder sample of 1 recorded at 100 Oe are shown in Fig. 4. At 300 K, the value of χMT is 12.8186 cm3 mol−1 K with an effective magnetic moment of 10.16 μB per formula units, which is slightly lower than the spin-only value of 10.95 μB for two high spin Mn(II) ions. The high temperature (75–200 K) inverse susceptibility data obeys the Curie–Weiss law with C = 13.1978 cm3 mol−1 K and θ = −7.97 K indicating a weak antiferromagnetic exchange interaction between the Mn(II) centers (Fig. S13†). On lowering the temperature, both the FC and ZFC susceptibilities gradually increase following each other and the value of χM at around 2 K is about 2.77 emu mol−1 (Fig. 4). Further, the variable temperature χMT plot shows a continuous decrease to a minimum value of 4.51 cm3 mol−1 K at 2 K supporting the weak antiferromagnetic interaction between the Mn(II) centers (inset of Fig. 4). The M vs. H plot recorded at 2 K shows no hysteresis loop with a saturation magnetization of 5.2 μB per formula unit at 50 kOe which is much lower than the saturation value of 7.5 μB expected for spin-only Mn3 ions (Fig. S14†). This behavior also supports the antiferromagnetic coupling interaction between the Mn(II) ions in 1.
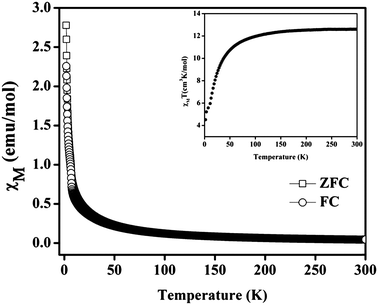 |
| | Fig. 4 Temperature dependence of the magnetic susceptibility (χM) of 1 at 100 Oe under field-cooled (FC) and zero-field-cooled (ZFC) conditions. Inset shows temperature dependence of χT for compound. | |
Optical and photoluminescence properties
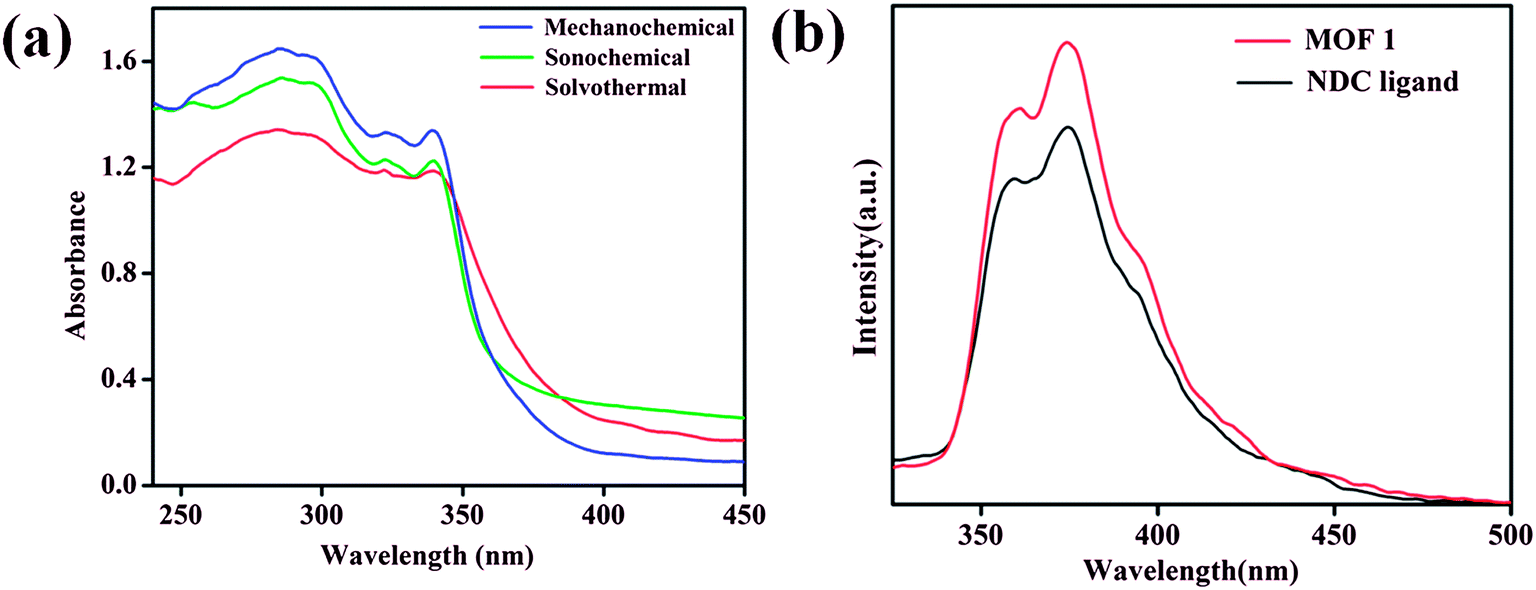
The room temperature solid state UV-vis absorption spectra of 1 synthesized by different approaches are shown in Fig. 5a. The spectra shows sharp absorption band in the UV region with absorption maxima around 285 nm.64 From the UV-vis plot the value of band gap energy (Eg) of 1 was estimated from a plot of (αhν)2 vs. photon energy (hν) following the Tauc relationship,65–67 αhν = A(hν − Eg)n, where hν = photon energy, A = constant, α = absorption coefficient, α = 4πk/λ; k is the absorption index, λ is the wavelength and n depends on the type of transition, n = 1/2 for the allowed direct band gap.68–70 The estimated value of band gap by extrapolating the absorption edge by a linear fitting method was found to be around 3.50 eV for 1 synthesized by different approaches (Fig. S15–S17†). The band gap value of 1 is found lower than the expected value for MnO (3.6–4.2 eV)71 and the lower value can be attributed due to the influence of ligand environment around metal centers. Further, the luminescence properties of 1 along with NDC ligand dispersed in DMA were investigated at room temperature. MOF 1 exhibits intense emission bands at 361 and 374 nm upon excitation at 285 nm which are appearing at similar positions as those of NDC ligand (Fig. 5b). Therefore, the emission bands observed for 1 can be attributed due to ligand based charge transfer (π → π* and n → π*) transitions.
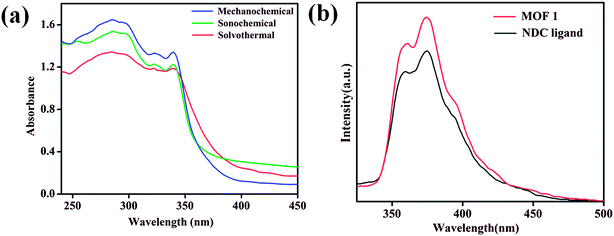 |
| | Fig. 5 (a) Room temperature solid state UV-vis absorption spectra of 1 synthesized by different routes (b) room temperature emission spectra (λex = 285 nm) of 1 and NDC ligand. | |
Thermal stability of 1
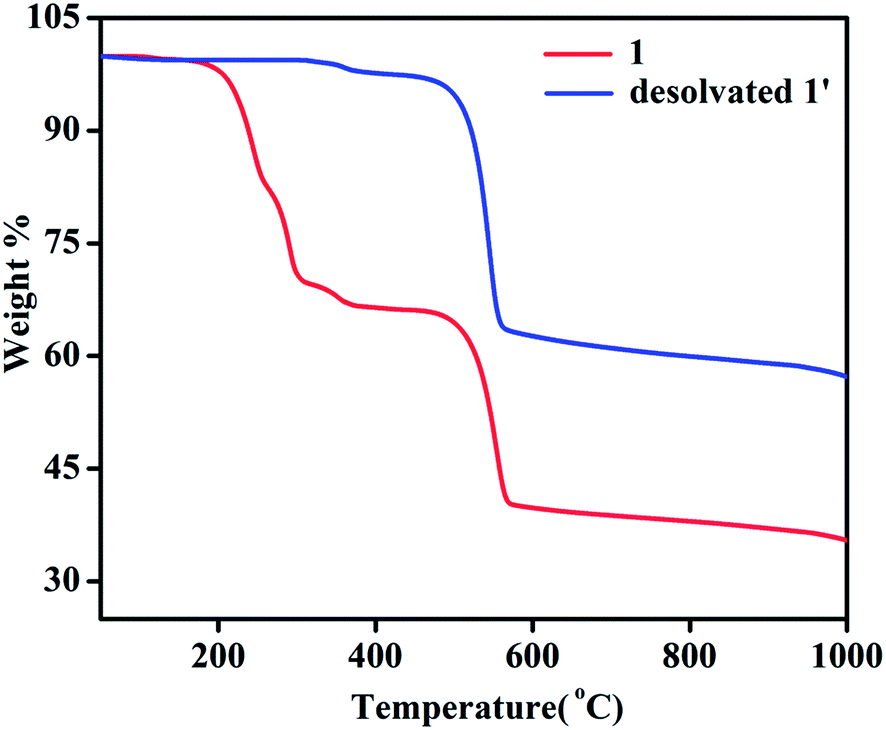
TGA of 1 shows a weight loss of ∼30.5% in the temperature regime (50–315 °C) corresponding to the loss of 4 molecules of DMA (calc. wt% 30.2) (Fig. 6). The second weight loss of ∼28.6% in the temperature range of 315 to 565 °C corresponds to the loss of 1.5 molecules of NDC (calc. wt% 27.8) and further increase of temperature leads to decomposition of the compound yielding a black solid. Whereas, TGA analysis of desolvated sample, 1′ shows absence of weight loss corresponding to four DMA molecules upto the temperature of 315 °C (Fig. 6). The first weight loss of ∼36.90% in the temperature regime (315–560 °C) corresponds to the loss of nearly 2 molecules of NDC (calc. wt% 37.06) and further increase of temperature leads to decomposition of the compound.
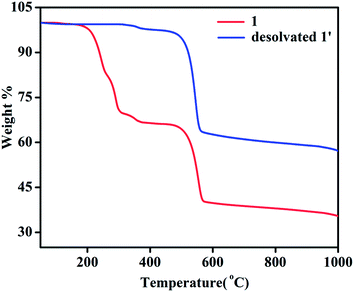 |
| | Fig. 6 Thermogravimetric curves of 1 and desolvated compound 1′. | |
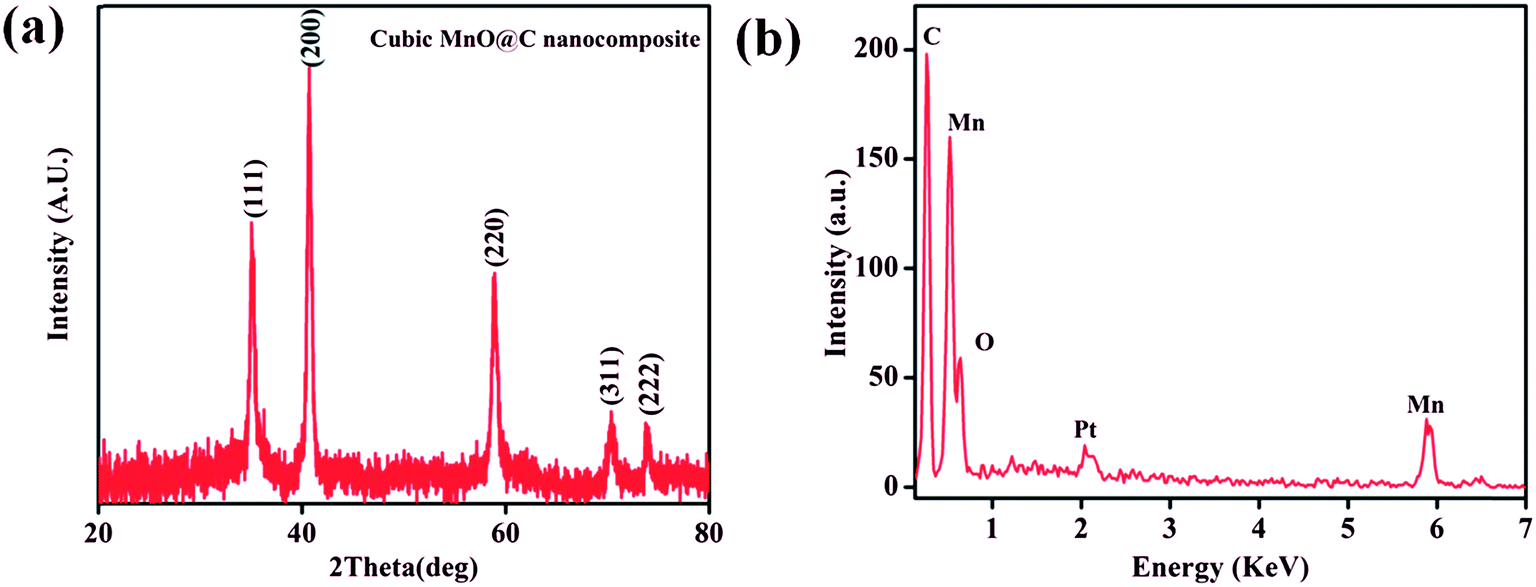
Interestingly, the PXRD pattern of the decomposed black solid showed diffraction peaks at 2θ = 35.1, 40.7, 58.9, 70.4, and 73.9° which can be assigned to (111), (200), (220), (311), and (222) planes of the cubic structure of MnO (JCPDS reference card no. 07-0230) (Fig. 7a). The broadness of the diffraction peaks indicates a finite size of the crystallites.72,73 Further, the mean crystallite size (D) of the MnO nanocrystals calculated using Scherrer equation, D = 0.94λ/Bcosθ (where D = crystallite size, λ = wavelength of X-ray (1.540598 Å), B = value of full width at half maximum (FWHM) and θ is the Bragg's angle) was 15.6 nm. Furthermore, energy dispersive X-ray (EDX) spectrum of the isolated black solid of MnO shows peaks corresponding to manganese, oxygen and carbon and the quantitative analyses confirmed the atom ratio of Mn2+/O2− to be 1:1 (Fig. 7b). FE-SEM analysis of the black solid shows the presence of uniform distribution of MnO nanoparticles embedded on porous carbon matrix (Fig. 8). The low-magnification bright-field TEM image of the MnO–C NC (Fig. 9a) shows the presence of MnO nanocrystals with a crystallite size in the range of 10–18 nm which is in good agreement with the size calculated from PXRD data (15.6 nm). Further, the diffraction planes obtained from the selected-area electron diffraction (SAED) pattern matches very well with the XRD patterns (Fig. 9b).
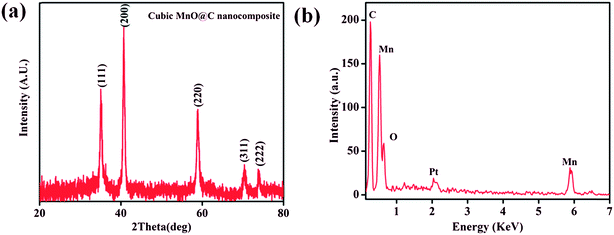 |
| | Fig. 7 (a) PXRD pattern of the cubic MnO–C nanocomposite formed by thermal solid state conversion of 1. (b) EDS spectra of MnO–C nanocomposite (extra peak arising due to electroplating with platinum). | |
 |
| | Fig. 8 FE-SEM image of the MnO–C nanocomposite showing the presence of MnO nanocrystals embedded in porous carbon support. | |
 |
| | Fig. 9 (a) TEM image of the MnO–C nanocomposites showing the presence of MnO nanocrystals (b) selected-area electron diffraction (SAED) pattern of MnO. | |
Conclusions
In summary, we have demonstrated the rapid synthesis of 3D Mn(II)–MOF from a rigid dicarboxylate ligand, H2NDC by employing green synthetic routes such as solvent-assisted mechanochemical and sonochemical methods. MOF 1 undergoes a reversible structural transformation upon desolvation and solvation of DMA molecules. The desolvated framework possesses 1D channels decorated with unsaturated Mn(II) centers and exhibits interesting H2 and CO2 uptake properties with isosteric heat of adsorption (Qst) for H2 and CO2 of 11.8 and 29.2 kJ mol−1, respectively. Remarkably, the Qst of H2 is highest amongst the Mn(II) based MOFs reported so far and the high value has been attributed to stronger interaction of H2 with the unsaturated Mn(II) centers. Compound 1 exhibits luminescence emission owing to ligand based charge transfer (π → π* and n → π*) transitions. Further, 1 undergoes temperature-induced solid state conversion to a phase pure MnO nanocrystals incorporated in carbon matrix to generate MnO–C nanocomposite. This study demonstrates the potential advantage of MOFs with coordinatively unsaturated metal centers for enhanced interaction with H2.
Acknowledgements
Authors gratefully acknowledge the financial support from the Board of Research in Nuclear Sciences (BRNS), DAE, Government of India project reference no. 2013/36/69-BRNS/20091. Authors thank Prof. A. Sundaresan, JNCASR, Bangalore for the magnetic measurements.
References
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466–1496 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. V. Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- D. Singh and C. M. Nagaraja, Dalton Trans., 2014, 43, 17912–17915 RSC.
- D. Singh and C. M. Nagaraja, Cryst. Growth Des., 2015, 15, 3356–3365 CAS.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- A. Corma, H. Garcıa and F. X. LlabrésiXamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- S. C. Xiang, Z. Zhang, C. G. Zhao, K. Hong, X. Zhao, D. R. Ding, M. H. Xie, C. D. Wu, M. C. Das, R. Gill, K. M. Thomas and B. Chen, Nat. Commun., 2011, 2, 204 CrossRef PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- C. M. Nagaraja, R. Haldar, T. K. Maji and C. N. R. Rao, Cryst. Growth Des., 2012, 12, 975–981 CAS.
- B. Wang, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS PubMed.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC.
- Y. F. Zeng, X. Hu, F. C. Liu and X. H. Bu, Chem. Soc. Rev., 2009, 38, 469–480 RSC.
- C. M. Nagaraja, N. Kumar, T. K. Maji and C. N. R. Rao, Eur. J. Inorg. Chem., 2011, 2057–2063 CrossRef CAS.
- P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- T. Kundu, S. Mitra, P. Patra, A. Goswami, D. D. Diaz and R. Banerjee, Chem.–Eur. J., 2014, 20, 10514–10518 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- Y. Kubota, M. Takata, R. Matsuda, R. Kitaura, S. Kitagawa, K. Kato, M. Sakata and T. C. Kobayashi, Angew. Chem., Int. Ed., 2005, 44, 920–923 CrossRef CAS PubMed.
- D. J. Collins and H. C. Zhou, J. Mater. Chem., 2007, 17, 3154–3160 RSC.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- R. B. Getman, Y. S. Bae, C. E. Wilmer and R. Q. Snur, Chem. Rev., 2012, 112, 703–723 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- U.S. Department of Energy Website, https://www1.eere.energy.gov/hydrogenandfuelcells/storage/current_technology.html.
- W. X. Lim, A. W. Thornton, A. J. Hill, B. J. Cox, J. M. Hill and M. R. Hill, Langmuir, 2013, 29, 8524–8533 CrossRef CAS PubMed.
- Y. E. Cheon and M. P. Suh, Angew. Chem., Int. Ed., 2009, 48, 2899–2903 CrossRef CAS PubMed.
- C. Zlotea, R. Campesi, F. Cuevas, E. Leroy, P. Dibandjo, C. Volkringer, T. Loiseau, G. Férey and M. Latroche, J. Am. Chem. Soc., 2010, 132, 2991–2997 CrossRef CAS PubMed.
- M. Dinca and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376–9377 CrossRef CAS PubMed.
- S. H. Jhung, H. K. Kim, J. W. Yoon and J. S. Chang, J. Phys. Chem. B, 2006, 110, 9371–9374 CrossRef CAS PubMed.
- T. A. Maark and S. Pal, Int. J. Hydrogen Energy, 2010, 35, 12846–12857 CrossRef CAS.
- Z. Xiang, Z. Hu, W. Yang and D. Cao, Int. J. Hydrogen Energy, 2012, 37, 946–950 CrossRef CAS.
- S. S. Dhankhar, M. Kaur and C. M. Nagaraja, Eur. J. Inorg. Chem., 2015, 34, 5669–5676 CrossRef.
- E. Pardo, C. Train, G. Gontard, K. Boubekeur, O. Fabelo, H. Liu, B. Dkhil, F. Lioret, K. Nakagawa, H. Tokoro, S. Ohkoshi and M. Verdaguer, J. Am. Chem. Soc., 2011, 133, 15328–15331 CrossRef CAS PubMed.
- A. Hazra, P. Kanoo and T. K. Maji, Chem. Commun., 2011, 47, 538–540 RSC.
- N. Xu, W. Shi, D. Z. Liao, S. P. Yan and P. Cheng, Inorg. Chem., 2008, 47, 8748–8756 CrossRef CAS PubMed.
- M. Kurmoo, H. Kumagai, K. W. Chapman and C. J. Kepert, Chem. Commun., 2005, 3012–3014 RSC.
- P. Lama, A. Aijaz, E. C. Sanudo and P. K. Bharadwaj, Cryst. Growth Des., 2009, 10, 283–290 Search PubMed.
- X. H. Zhang, Z. M. Hao and X. M. Zhang, Chem.–Eur. J., 2011, 17, 5588–5594 CrossRef CAS PubMed.
- Y. Q. Wang, W. W. Sun, Z. D. Wang, Q. X. Jia, E. Q. Gao and Y. Song, Chem. Commun., 2011, 47, 6386–6388 RSC.
- J. T. Brockman, T. C. Stamatatos, W. Wernsdorfer, K. A. Abboud and G. Christou, Inorg. Chem., 2007, 46, 9160–9171 CrossRef CAS PubMed.
- H. B. Xu, B. W. Wang, F. Pan, Z. M. Wang and S. Gao, Angew. Chem., Int. Ed., 2007, 46, 7388–7392 CrossRef CAS PubMed.
- Y. Z. Zheng, W. Xue, M. L. Tong, X. M. Chen, F. Grandjean and G. J. Long, Inorg. Chem., 2008, 47, 4077–4087 CrossRef CAS PubMed.
- X. N. Cheng, W. X. Zhang and X. M. Chen, J. Am. Chem. Soc., 2007, 129, 15738–15739 CrossRef CAS PubMed.
- V. H. Tran and B. Świątek-Tran, Dalton Trans., 2008, 4860–4865 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2011, 112, 933–969 CrossRef PubMed.
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC.
- S. Y. Yang, H. B. Yuan, X. B. Xu and R. B. Huang, Inorg. Chim. Acta, 2013, 403, 53–62 CrossRef CAS.
- Z. Q. Jiang, G. Y. Jiang, F. Wang, Z. Zhao and J. Zhang, Chem. Commun., 2012, 48, 3653–3655 RSC.
- H. Pan, J. A. Ritter and P. B. Balbuena, Langmuir, 1998, 14, 6323–6327 CrossRef CAS.
- N. Sikdar, A. Hazra and T. K. Maji, Inorg. Chem., 2014, 53, 5993–6002 CrossRef CAS PubMed.
- R. T. Yang, Gas Separation by Adsorption Processes, Butterworth, Boston, 1997 Search PubMed.
- B. Liu, R. Q. Zou, R. Q. Zhong, S. Han, H. Shioyama and T. Yamada, Microporous Mesoporous Mater., 2008, 111, 470–477 CrossRef CAS.
- H. R. Moon, N. Kobayashi and M. P. Suh, Inorg. Chem., 2006, 45, 8672–8676 CrossRef CAS PubMed.
- S. Baskoutas, A. F. Terzis and W. Schommers, J. Comput. Theor. Nanosci., 2006, 3, 269–271 CAS.
- N. Bouropoulos, I. Tsiaoussis, P. Poulopoulos, P. Roditis and S. Baskoutas, Mater. Lett., 2008, 62, 3533–3535 CrossRef CAS.
- X. Fang, U. K. Gautam, Y. Bando, B. Dierre, T. Sekiguchi and D. Golberg, J. Phys. Chem. C, 2008, 112, 4735–4742 CAS.
- S. Kar and S. Chaudhuri, J. Phys. Chem. B, 2005, 109, 3298–3302 CrossRef CAS PubMed.
- N. Serpone, D. Lawless and R. Khairutdinov, J. Phys. Chem., 1995, 99, 16646–16654 CrossRef CAS.
- Q. Xiong, G. Chen, J. D. Acord, X. Liu, J. J. Zengel, H. R. Gutierrez, J. M. Redwing, L. C. Lew Yan Voon, B. Lassen and P. C. Eklund, Nano Lett., 2004, 4, 1663–1668 CrossRef CAS.
- M. A. Bryushinin, A. A. Petrov, R. V. Pisarev and I. A. Sokolov, Phys. Solid State, 2015, 57, 907–913 CrossRef CAS.
- M. Kaur and C. M. Nagaraja, RSC Adv., 2014, 4, 18257–18263 RSC.
- M. Kaur, N. K. Gupta and C. M. Nagaraja, CrystEngComm, 2015, 17, 2359–2367 RSC.
- M. Dinca and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172–11176 CrossRef CAS PubMed.
- M. Dinca, A. F. Yu and J. R. Long, J. Am. Chem. Soc., 2006, 128, 8904–8913 CrossRef CAS PubMed.
- M. Dinca, A. Dailly, C. Tsay and J. R. Long, Inorg. Chem., 2008, 47(1), 11–13 CrossRef CAS PubMed.
- W. Zhou, H. Wu, M. R. Hartman and T. Yildirim, J. Phys. Chem. C, 2007, 111, 16131–16137 CAS.
- J. L. C. Rowsell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666–5667 CrossRef CAS PubMed.
- J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304–1315 CrossRef CAS PubMed.
- P. Krawiec, M. Kramer, M. Sabo, R. Kunschke, H. Frode and S. Kaskel, Adv. Eng. Mater., 2006, 8, 293–296 CrossRef CAS.
- B. Panella and M. Hirscher, Adv. Mater., 2005, 17, 538–541 CrossRef CAS.
- M. Latroche, S. Suble, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J.-H. Lee, J. S. Chang, S. H. Jhung and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 8227–8231 CrossRef CAS PubMed.
- X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schreoder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef CAS PubMed.
- H. Kim, S. Das, M. G. Kim, D. N. Dybtsev, Y. Kim and K. Kim, Inorg. Chem., 2011, 50, 3691–3696 CrossRef CAS PubMed.
- M. Dinca, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876–16883 CrossRef CAS PubMed.
- S. Ma and H. C. Zhou, J. Am. Chem. Soc., 2006, 128, 11734–11735 CrossRef CAS PubMed.
- K. Sumida, C. M. Brown, Z. R. Herm, S. Chavan, S. Bordiga and J. R. Long, Chem. Commun., 2011, 47, 1157–1159 RSC.
- J. G. Vitillo, L. Regli, S. Chavan, G. Ricchiardi, G. Spoto, P. D. C. Dietzel, S. Bordiga and A. Zecchina, J. Am. Chem. Soc., 2008, 130, 8386–8396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra17898g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.