
Solution chemistry-based nano-structuring of copper dendrites for efficient use in catalysis and superhydrophobic surfaces†
Rangarajan Bakthavatsalama,
Subrata Ghosha,
Ratul Kumar Biswasa,
Aayushi Saxenaa,
Alagar Rajab,
Musthafa Ottakam Thotiylb,
Sandip Wadhaic,
Arun G. Banpurkarc and
Janardan Kundu*a
aPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pashan, Pune, Maharashtra 411008, India. E-mail: j.kundu@ncl.res.in
bDepartment of Chemistry, Indian Institute of Science Education and Research, Dr Homi Bhabha Road, Pashan, Pune, Maharashtra 411008, India
cDepartment of Physics, Savitribai Phule Pune University, Ganeshkhind, Pune, Maharashtra 411007, India
First published on 6th January 2016
Abstract
Despite their performance and economic advantages over Ag and Au, there have been no focused research efforts on the nano-structuring of Cu dendrites with respect to fine-tuning their structure/morphology towards the efficiency enhancement of suitable applications. Reported here is a simple, versatile, environmentally-friendly and galvanic replacement reaction-based solution chemistry methodology to synthesize highly nano-structured copper dendrites targeted towards the efficiency enhancement of desired applications. Herein, copper is deposited galvanically on an Al foil in the presence of NaCl/HCl, wherein the chloride anions augment an uninterrupted replacement reaction. The growth process of Cu dendrites has been probed in detail. The presence of acid, the type of Cu2+ precursor salt, the Cu2+ ion concentration, the surfactant concentration and the reaction temperature are all demonstrated to provide useful means of modulating the surface structure/morphology of the dendrites. Notably, dendrites formed in the presence of acid are found to be highly nano-structured. Moreover, it is also found that the morphology/structure of the obtained Cu deposit depends considerably upon the choice of the Cu2+ precursor salt, a parameter that has been completely overlooked in the past. The acid-induced nano-structuring of the dendrites is exploited for enhancing their efficiency in the catalytic reduction of para-nitrophenol and for fabricating self-cleaning superhydrophobic surfaces. These nano-structured dendrites are demonstrated to have the highest ever normalized rate constant for the catalytic reduction reaction. Superhydrophobic surfaces fabricated using these dendrites demonstrate excellent self-cleaning abilities, showing a high contact angle (159°) with low contact angle hysteresis (2°). This facile synthetic strategy for the fabrication of highly nano-structured Cu dendrites is expected to open up avenues for the production of Cu-based low-cost functional nano/micro-materials.
Introduction
Metallic nanostructures of group 11 elements (namely Cu, Ag and Au) have been extensively exploited for their unique optical, electronic, magnetic and catalytic properties.1–7 Tailoring their intrinsic and unique properties is achievable by tuning the size, shape, morphology and crystal phase composition of the metallic nanostructures.6,8,9 These nanostructures, in comparison to their bulk counterparts, find interesting applications in catalysis, microdevices, electrical and thermal conduction, lubrication, fabrication of superhydrophobic surfaces and plasmonic sensing.6,7,10–13 With the advancement of wet-chemical synthetic techniques, it is now possible to controllably fabricate a variety of metallic nanostructures in solution. An important class of nanostructures that has recently been the focus of interest is the so-called dendritic structures. In simple words, dendritic nanostructures are self-assembled, hierarchical, repetitive structures with a main stem and many side branches resembling fern or bracken-like structures. Consequently, these networked structures have high surface areas, high conductivity and can act as model systems in the field of mathematics for understanding pattern formation. Dendritic nanostructures are believed to be formed under kinetic control, far from thermodynamic equilibrium growth conditions.14,15 Several growth mechanisms, such as the diffusion-limited-aggregation (DLA) model,16,17 oriented attachment,18,19 nanoparticle-aggregated self-assembly crystallization20–22 and anisotropic crystal growth15,23 have been proposed to explain the formation process. However, DLA and nanoparticle aggregated self-assembly crystallization mechanisms have been widely utilized to explain the growth process of these dendritic structures.Amongst the group 11 metals, copper dendrites are receiving current attention due to their low price/earth abundance and the unique enabling properties of metallic copper.15,24–34 Copper offers several economic and performance advantages over precious metals. The conductivity of copper is comparable to that of silver and is 40% better than that of gold.35 The stability of copper at high frequencies is better than gold or silver and it is compatible with most solder materials.35 Although the inherent susceptibility of metallic copper to undergo oxidation has been cited as a difficulty in synthesizing copper nanostructures, many successful efforts have been reported with respect to mitigating this issue by utilizing protective coatings/ligands/surfactants during synthesis of copper nanostructures.36–39 Hence, high-purity, uniformly shaped, nano-structured Cu is highly desirable for use in the electronic industry. Research efforts are now being devoted to the controllable fabrication of Cu-based dendritic nanostructures for the development of functional materials that find practical applications in cheap and effective catalysis, microdevices as nano-joints, electrical and thermal conduction, superhydrophobicity for self-cleaning surfaces and SERS (surface-enhanced Raman scattering)-based biochemical sensing.
Copper dendrites have been fabricated by various routes: traditional electrochemical deposition,24,25 hydrothermal synthesis28,40 and the electroless deposition method (galvanic replacement reaction/GRR).26,27,29,30 Of these synthetic routes, a GRR-based strategy (redox displacement reaction) is the preferred choice for the fabrication of dendritic nanostructures, primarily due to the simplicity and versatility of the GRR process. Although the GRR strategy has been extensively implemented for the synthesis of dendrites of Ag and Au, there have been few reports on Cu dendrites. In many of the existing reports on GRR-based fabrication of dendrites, Cu dendrites were fabricated to showcase the generality of the synthetic strategy.26,30 Reports that specifically focus on the GRR-based fabrication of Cu dendrites, targeted towards particular applications, are scarce.26,27 Most of the reports on the fabrication of Cu dendrites, however, do not implement the various reaction parameters as effective handles to fine-tune the structure/morphology of the dendrites. Interestingly, there are no reports on utilizing different Cu2+ precursor salts during the GRR fabrication of Cu dendrites. The effect of surfactant during the GRR deposition of Cu dendrites has also not been probed extensively. These reaction parameters (alone or in combination) are expected to have a drastic influence on the shape/structure/morphology of the Cu dendrites, which can immensely impact the efficiency of their applications. Hence, the primary objective of this work is to focus comprehensive research efforts on the nano-structuring of Cu dendrites, targeted towards the efficiency enhancement of desired applications.
In a typical GRR process, metal ions (in a bath solution) of higher reduction potential undergo spontaneous reduction at the expense of oxidation of a metal under-layer substrate with a lower reduction potential, without the application of an external current or reducing agents. Generally utilized metal under-layer substrates with low reduction potentials include Mg foil/powder, Al foil and Zn foil. Al foil, being economically viable and having a lower reduction potential (E0 Al/Al3+ = −1.67 V vs. SHE) than most metal ions, is an excellent choice as an under-layer substrate. However, utilizing Al foil for the deposition of metals with higher reduction potentials inevitably fails due to the formation/presence of a thin layer of alumina (Al2O3) that acts as an insulating barrier to the GRR process. To overcome this difficulty, fluoride ions (F−) were demonstrated as a reagent that dissolves the alumina layer, thereby allowing the uninterrupted proceeding of the galvanic displacement reaction.11,30 In particular, Li et al. demonstrated a facile and uninterrupted galvanic replacement route to produce metal dendrites on commercial aluminum foil in the presence of NaF or NH4F as the alumina etchant.30 Undoubtedly, such a strategy of using commonly available/low cost reagents (NaF or NH4F) helps in the large-scale production of nano-materials. Nonetheless, the process involves fluoride anions that can strongly bind to metal dendrites and negatively affect their performance. Fluoride ions are also considered to be highly toxic.41 Moreover, this process30 of fluoride-assisted galvanic replacement synthesis of dendrites on Al foil takes a long time (10 h).26,41
Here, we report a simple yet versatile, rapid, environment-friendly and uninterrupted solution chemistry-based methodology to synthesize nano-structured copper dendrites by the galvanic displacement route on Al foil with the use of NaCl/HCl as the etching reagent for alumina. The chloride anions can dissolve alumina by forming [AlCl4]− anions that can be easily washed off, rendering the process environment-friendly. Here, the GRR proceeds along with the reaction of the Al substrate with the acid, which produces hydrogen gas. The hydrogen gas produced maintains a local oxygen-free environment, thereby suppressing oxide formation. This chloride-assisted methodology is easily scalable with a high yield being obtained in less than 2 hours of total reaction time. Notably, the presence of the acid in the system is found to be an effective handle that can produce a high degree of nano-structuring in the morphology of the fabricated dendrites. We also explore the effect of the choice of Cu2+ precursor salts, such as CuSO4 and Cu(NO3)2 (a parameter that has largely been ignored/overlooked in past reports of copper dendrites),15,24–30 surfactants and reaction temperature on the yield and morphology of the copper dendrites obtained. Surprisingly, we find that the choice of Cu2+ precursor salts has profound effects on the yield/morphology of the copper deposits thus obtained. These acid-induced nano-structured dendrites are demonstrated to be highly useful for applications in: (1) catalytic reduction of 4-nitrophenol and (2) fabricating superhydrophobic surfaces. These dendrites act as efficient catalysts with the highest ever reported rate constant for the catalytic reduction reaction. These dendrites also exhibit superhydrophobicity with excellent self-cleaning abilities. The material properties of these dendrites are largely shaped by solution chemistry-based nano-structuring, which enhances their efficiency in various applications.
Experimental
Al foil (99.5%, thickness: 0.14 mm), CuSO4 (>99.5%), Cu(NO3)2 (>99.5%), CuCl2 (>99.5%), Na2SO4 (>99.5%), NaCl (>99.5%) and HCl (AR) were all purchased from Loba Chemie. Sodium borohydride (>98%), polyvinylpyrrolidone (PVP, 55 K), PDDA (poly(diallyldimethylammonium chloride)) (20 wt% in H2O), poly(4-styrenesulfonic acid) (Mw ∼ 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and n-dodecanethiol (≥98%) were purchased from Sigma Aldrich and NaNO3 (>99.5%) was purchased from Merck. Commercial Cu powder (99.5%, spherical, mesh size: −100) was bought from Alfa Aesar. Milli-Q water (resistivity > 18 MΩ cm) has been used throughout for carrying out the reactions while ethanol (99%) has been used for washing samples.
000) and n-dodecanethiol (≥98%) were purchased from Sigma Aldrich and NaNO3 (>99.5%) was purchased from Merck. Commercial Cu powder (99.5%, spherical, mesh size: −100) was bought from Alfa Aesar. Milli-Q water (resistivity > 18 MΩ cm) has been used throughout for carrying out the reactions while ethanol (99%) has been used for washing samples.
Aluminium foil was cut into 1 × 1 cm2 squares and cleaned by sonicating in ethanol and water sequentially for two minutes each. The copper dendrites were synthesized by immersing the cleaned Al foil in 2 ml solution mixture of a desired concentration of CuSO4/Cu(NO3)2 and NaCl/HCl in a polypropylene reaction vial of 5 ml volume. The reaction proceeded with bubbling and deposition of metallic copper on the Al foil. The reaction was stopped after 2 hours by removing the Al foil from the reaction mixture. After completion of the reaction, the copper deposits were separated from the foil and cleaned thoroughly with deionized water and ethanol. For XRD, XPS and catalytic activity studies, the cleaned copper deposits were dried in a vacuum oven at 60 °C for 4–6 hours. For SEM analysis, the deposits on the Al foil were cleaned by successive washing in water and ethanol before they were directly imaged on the Al foil. For TEM and HRTEM measurements, the foil containing the deposits was sonicated to disperse the copper structures in ethanol before drop-casting on TEM grids (carbon-coated copper grids, 200 mesh size).
Catalytic activity
To test the catalytic activity of the prepared Cu dendrites and commercial Cu powder, the reduction of 4-nitrophenol in excess NaBH4 was studied spectrophotometrically. The as-prepared Cu dendrites were washed successively with water, ethanol, dilute NaOH and again with water and ethanol to remove any remaining salt/acid. To 20 ml of 1 mM 4-nitrophenol, 10 ml of 0.4 M NaBH4 was added and a specified amount of Cu dendrite catalyst was added at the start of the reaction. Once the catalyst was added, the reaction was monitored by spectrophotometry. On addition of NaBH4, 4-nitrophenol (4-NP) shows a strong absorption at 400 nm. The color of the solution turns from pale to bright yellow due to the formation of 4-nitrophenolate ions. In the presence of the catalyst (Cu dendrites), the solution color slowly changes from yellow to colorless. This is monitored (as a function of time) by observing the intensity of the 400 nm peak in UV-Vis spectra and the concomitant increase of the 300 nm peak corresponding to the formation of 4-aminophenol as the product. A catalytic assay of the commercial Cu powder was run as described above, except for using 3 ml of 1 mM 4-NP, 10 ml of 0.4 M NaBH4 and 10 mg of the Cu powder. These catalytic reactions were repeated at least 5 times to ensure reproducibility.Cyclic voltammetry
CV experiments were performed using a BASI Epsilon cyclic voltammeter with N2 purging. The experiment was performed in a typical three-electrode system with glassy carbon as working electrode, Ag/AgCl (Eo = 0.197 vs. SHE) as reference and platinum as counter electrode. For copper electrodeposition, a solution of 50 mM CuSO4 and 100 mM Na2SO4 was used as a background electrolyte to mitigate issues with uncompensated resistance. The scan rate was fixed at 20 mV s−1. CV scanning was done at various switching potentials ranging from −500 to −750 mV vs. Ag/AgCl electrode. For comparison purposes, similar experiments were done with the same parameters but using copper nitrate instead of copper sulfate as the Cu2+ precursor.Superhydrophobic surface preparation
Glass slides were cut into 1 × 1 cm2 squares and immersed in a 3:1 mixture of sulfuric acid and hydrogen peroxide for 4–5 h. They were then washed with copious amounts of DI water and dried in air. To deposit copper dendrites on a glass substrate, a multi-layer of charged polymer was initially coated on the cleaned glass slides. This was achieved through a layer-by-layer deposition process. The cleaned glass substrate was immersed in a 5% PDDA solution for 10 min followed by rinsing with water and drying under a nitrogen stream. To coat a layer of PSS onto this substrate, it was immersed in a solution of 5% PSS for 10 min, washed with DI water and dried. Then, another set of layers of PDDA-PSS was obtained in the same way. This process was repeated 2–3 times to obtain the desired number of layers. The copper dendrites were then drop-casted on these slides and dried in air. The Cu-coated glass slides were then immersed in a 100 mM ethanolic solution of n-dodecanethiol for 24 h. They were then washed with ethanol and dried in air.
Static water contact angle (SCA) and contact angle hysteresis (CAH) measurements
The equilibrium contact angle for a water droplet on several hydrophobic surfaces was determined using an optical contact angle (OCA) goniometer. A CCD camera was attached with a variable magnification microscope (1–5×). A sessile droplet was gently casted on the surface using a precision needle attached to a water delivery system for precise control of the drop volume. A micro-syringe pump (pico-plus, Harvard) was employed for precisely controlling the water drop volume. The sessile drop was back-illuminated using diffused LED lights. The CCD camera, water drop and backlight were kept coaxial and fixed on a rigid stand. Using a syringe pump, a water drop of nearly 8 μL was deposited on the superhydrophobic surface. This was a “sessile-drop needle-in” arrangement and the drop image was constantly monitored and analyzed using SCA 20 software (Dataphysics, Germany). The contact angle was measured using elliptical fitting to the droplet boundary. The experiment was repeated many times on the same sample at different places.Advancing-receding contact-angle (ARCA) goniometry was utilized for the real time measurement of the contact angles of the water drop, characterizing the hysteresis. The water drop volume was controllably increased from 10 μL to 20 μL at a rate of 3 μL min−1 and then restored to its initial volume (10 μL). During this process, the contact angle of the water drop was measured and analyzed using SCA 20 software. The difference between the advancing and the receding contact angle is the measured contact angle hysteresis.
Characterization
The as-prepared products are characterized by X-ray diffraction (XRD) using a PAN analytical X'Pert Pro equipped with Cu Kα radiation (λ = 1.5406 Å). Field emission scanning electron microscope (FE-SEM) images were taken using a ZEISS Ultra Plus instrument. XPS surface analysis was performed using a Multi-Lab, ESCA-3000 (VG Microtech, England). Transmission electron microscope images were obtained from a Tecnai T20 TEM operating at an accelerating voltage of 200 kV. HRTEM images were obtained using an FEI Tecnai G2 30 S-Twin instrument operating at 300 kV. UV-Vis spectroscopy was done using a Shimadzu UV-3600 plus UV-Vis-NIR spectrophotometer. Real time video of water droplets rolling off the superhydrophobic surface was captured using a Sony HDR CX 160 Handycam.Results & discussion
Deposition process
We have performed environment-friendly, rapid, very simple electroless deposition, utilizing uninterrupted galvanic replacement reaction (GRR) strategy, for fabricating Cu dendritic micro/nano-structures on Al foil without using fluoride anions. Dendrites are formed during the galvanic deposition of CuSO4 on Al foil in the presence of NaCl/HCl. This GRR deposition of Cu on Al foil does not proceed at all in the absence of chloride anions (see Fig. S1, ESI†). Driven by the differences in the redox potentials, Cu2+ ions are spontaneously reduced to metallic copper at the expense of oxidation of Al to Al3+ ions in the presence of the chloride anions. The dissolved dioxygen in the aqueous solution can undergo reduction to form hydroxyl anions (E0 O2/OH− = 0.4 V) that can lead to the formation of Al(OH)3, acting as a barrier to the GRR process. However, the presence of Cl− anions helps to complex the released Al3+ ions in the form of soluble [AlCl4]− anions, thereby preventing formation of an undesired alumina barrier layer. Considering the harmful/undesired effects of fluoride anions, as mentioned earlier, we utilized chloride anions for performing uninterrupted GRRs. The presence of acid during the GRR stops the production of hydroxyl ions and produces H2 gas that maintains a local oxygen-free environment, which prevents oxide formation and hence augments the uninterrupted GRR process. A schematic of such processes is shown in Fig. 1a. Simplicity, easiness, eco-friendliness and versatility (high yield >94%, short reaction time <2 hours) are the major attractions of the utilized solution chemistry-based facile methodology for controllable fabrication of Cu dendrites on Al foil.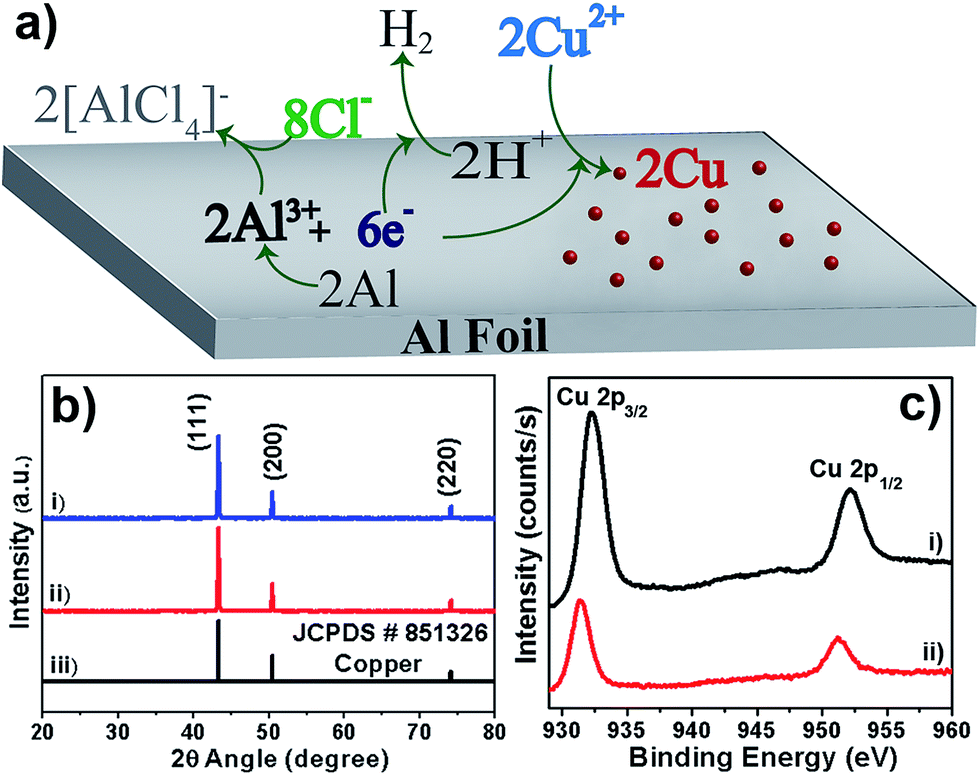 | ||
| Fig. 1 (a) Schematic (not to scale) of the various reactions occurring during the GRR deposition of metallic Cu on an Al foil in the presence of Cl− anions; (b) XRD pattern (stacked) of Cu dendritic structures prepared from (i) CuSO4 (50 mM) + NaCl (100 mM), (ii) CuSO4 (50 mM) + HCl (100 mM) and (iii) Standard JCPDS pattern of Cu; (c) XPS spectra (stacked) of Cu dendrites prepared from (i) CuSO4 (50 mM) + HCl (100 mM) and (ii) CuSO4 (50 mM) + NaCl (100 mM). | ||
Elemental and phase composition
Structure and morphology
The structure and morphology of the prepared products were characterized using SEM and TEM/SAED. Fig. 2 shows SEM images of Cu dendrites fabricated at room temperature using CuSO4 (50 mM) and NaCl/HCl (100 mM), exhibiting typical dendritic structures. The low-magnification SEM images (Fig. 2a and d) reveal that a large quantity of Cu dendrites with good uniformity can be obtained using this approach. The micro/nano-structured dendrites consist of a pronounced central backbone with many symmetrical primary and secondary branches. (Fig. 2b and e). It is also seen that not all the primary branches are fully developed. Coexistence of large and small primary branches is observed because the branch alignment experiences space-limited growth. SEM images (Fig. 2c and f) and TEM images of single dendrites (Fig. S2a and b: ESI†) clearly show that the primary branches are parallel to each other, emerging at 55–60° angles with respect to the central backbone and that primary branches preferentially grow along two definite directions rather than via randomly ramified growth. A closer view of the SEM images clearly reveals that the central backbone and the various branches are composed of aggregates of small nanoparticles. Moreover, the discontinuous concentric ring-like pattern of the selected area electron diffraction pattern (SAED), as shown in the insets in Fig. S2a and b (ESI†), indicates the characteristics of a single crystal structure of cubic Cu. | ||
| Fig. 2 (a)–(c) FESEM images of Cu dendritic structures prepared at room temperature from CuSO4 (50 mM) and NaCl (100 mM) at different magnifications (CuS_Na); (d)–(f) FESEM images of Cu dendritic structures prepared at room temperature from CuSO4 (50 mM) and HCl (100 mM) at different magnifications (CuS_H). | ||
It is interesting to compare and contrast the structure/morphology of the dendrites prepared using NaCl and HCl, prepared with 50 mM CuSO4 and 100 mM NaCl/HCl. The dendrites prepared using NaCl (CuS_Na) are observed to be dense and much longer in dimension, while the dendrites prepared using HCl (CuS_H) are observed to be slender and shorter. The average lengths of the dendrites for CuS_Na are ∼20 μm, compared to ∼10 μm for CuS_H. The secondary branches in the case of CuS_Na are seen to be fused and not crisply divided into individual leaves. However, for Cu_H, secondary branches are clearly seen with no fusion and appear crisply demarcated. Experimentally, the utilization of HCl leads to observed differences in the extent of fusion and surface nano-texturing (surface area), compared to the use of NaCl. Hence, we believe that the presence of the acid during the GRR process is an effective handle that can tailor the surface area and morphology of the fabricated dendrites. The acid present in the reaction system not only helps to mitigate oxidation issues but also acts as a competitive channel working against GRR deposition of Cu due to the reduction of H+ to H2 gas by the Al foil. This acid-based competition, however, is a relatively weaker channel than the GRR-based Cu2+ reduction (EH+/H20 = 0.0 V; ECu2+/Cu0 = 0.33 V). Due to this weaker competition, the GRR deposition rate is expected to slightly decrease for the HCl system when compared to the scenario for NaCl. The relatively higher nucleation rate while using NaCl might lead to a higher growth rate that can result in fusion of the nanoparticles. However, for the HCl system, the lower nucleation and growth rates would provide sufficient time for reorientation and preferred attachment of the nano-particles to each other, as dictated by minimization of surface energy, which might mitigate the random fusion of nanoparticles during the growth of branches in the dendritic structures. This observed increased surface area for the dendrites prepared with HCl compared to NaCl is an excellent feature of this approach as many of the applications of dendrites (such as catalysis, wettability) crucially depend on the available surface area and the trapped air fraction in these micro/nano structures. This difference in the morphology is heavily exploited for demonstrating markedly different efficiencies in applications of these dendrites in catalysis and superhydrophobic surfaces as presented in the later section of this report.
It is notable that the Cu dendrites fabricated here have [Cu2+]:[Cl−] = 1:2. We have also tried ratios of 1:3, 1:5 and 1:10 to obtain Cu deposits on the Al foil. The reaction system was found to be highly robust and tolerant towards small variations in the chloride concentration. Dendritic structures are still the major product at a ratio of 1:5. Only at a much higher [Cu2+]:[Cl−] ratio of 1:10, did we observe changes in the morphology of the Cu deposits. SEM analysis of the Cu deposits, as shown in Fig. S3 (ESI†), shows that along with Cu ‘dendritic-like’ structures, the formation of ‘micro rod’-like structures occurs in modest relative amounts. These faceted rod-like structures are smooth and long (∼20 microns) with progressive tapering at the tips (∼1 micron ‘diameter’). These smooth rod facets are found to be decorated with tiny Cu nanoparticles that act as sites for further growth, resembling “cactus” structures. The seemingly ‘dendritic-like’ structures are found to have a different structure/morphology when compared to the structures shown in Fig. 2. Here, in the case of an increased [Cu2+]:[Cl−] ratio, the dendritic-like structures have less dense primary branching with no signs of secondary branching. Moreover, there is three dimensional growth of the primary branches from the central trunk. Also, these primary branches look more like smooth micro-rods with clearly observable faceting. The observed differences in the structure/morphology of the Cu deposit obtained by varying the Cu2+/Cl− ratio can be attributed to the increased [Cl−]. Shao et al.25 and Wu et al.,42 have previously demonstrated this increased faceting of Cu nanostructures with increased [Cl−]. This chloride-assisted growth with increased faceting has been attributed to preferential binding and stabilization of the chloride anion on the (100) planes of the developing Cu nanostructure.25 We believe that similar chloride-assisted growth might be operative in our case at increased [Cu2+]:[Cl−] ratios.
In order to test this, we have attempted GRR-based deposition of Cu on Al foil, utilizing CuCl2 as the precursor. The obtained morphology of the Cu deposit, shown in Fig. S4 (ESI†), is clearly composed of dendritic-like structures with sparse primary branches and no secondary branches. The growth of the primary branches is three-dimensional and these branches are smooth faceted rods. The obtained morphology utilizing CuCl2 is very similar to structures obtained using CuSO4 with added excess chloride (sulfate: chloride = 1:10; Fig. S3†). We think that in the case of the CuCl2 precursor, the chloride anions, adsorbing preferentially on the 100 facets of the developing Cu nanostructures, dictate the morphology of the dendrites with faceted micro-rod-like structures developing as the primary branches. Here, one might consider the CuCl2 system and the CuSO4 system with added chlorides (Cu2+:Cl− = 1:2) to be similar and to yield similar morphology. However, the structures obtained with these two systems are very different (Fig. S4† vs. Fig. 2a–c, respectively). The only difference in the chemical composition of the system lies in the presence of sulfate anions that are known to bind to developing metallic nuclei.43 Hence we believe that, in the case of CuSO4 with excess chloride (sulfate:chloride = 1:2), the sulfate anions can strongly bind to developing Cu nanostructures even in the presence of chloride anions, leading to the formation of the dendritic structures shown in Fig. 2a–c. Only at much higher chloride amounts (sulfate: chloride > 1:10), does the binding ability of chloride anions surpass that of sulfate anions and the morphology is dictated by the adsorbed chloride anions, leading to the formation of smooth, faceted rod-like structures, as shown in Fig. S3.†
We have also varied the amount of HCl in the reaction system by altering the [Cu2+]:[HCl] ratio to 1:2, 1:5, 1:10 and 1:20. Again, the reaction system is surprisingly found to be highly robust and tolerant towards small variations in the HCl concentration. Dendritic structures are still the major product at a ratio of 1:5. Only at much higher [Cu2+]:[HCl] ratios of 1:10 and above, do we observe changes in the morphology of the Cu deposits. SEM analysis of the Cu deposits, as shown in Fig. S5 (ESI†), shows that, along with Cu dendritic structures, we also observe a modest (∼25%) yield of wire-like structures. The dendritic structures obtained here are smaller in size, measuring ∼5–7 microns with less developed secondary side branches. This subtle morphology change in the dendritic structures, along with the formation of wire-like structures constituted of small aggregated particles, indicates the “slowness” of the GRR reaction when carried out in the presence of a larger amount of acid. The restrained nucleation and growth leads to the formation of incomplete dendrites with the central trunk being composed of aggregated Cu nanoparticles. Cu nanoparticles are formed on the Al foil along with the evolution of hydrogen gas as observed during the experiments. However, the presence of a relatively larger amount of acid (Cu2+:H+ = 1:10), opens up the weaker H+/H2 competitive channel, which leads to decreased nucleation and growth and affects the morphology/structure, thereby forming rough, wire-like Cu deposits. It is worth pointing out here that with this increased amount of acid (Cu2+:HCl = 1:10), there is a concomitant increase of the chloride amount. This increased chloride amount manifests increased faceting only weakly, as observed by SEM analysis. We do not understand why the increased chloride amount would not result in increased faceting but we think that copious evolution of H2 gas might play a role in this.
Growth process of dendrites
In order to gain insight into the growth process of the dendrites, we monitored the time-dependent evolution of the morphology of Cu structures/dendrites through SEM investigation. Fig. 3a–j shows SEM images of products obtained at various reaction times (5 s, 10 s, 25 s, 45 s, 60 s, 2 min, 3 min, 5 min, 9 min and 60 min). It is clearly observed that the initial stage of the reaction (5 s) involves formation of Cu “microparticles” on the surface of the Al foil. It is expected that tiny Cu nanoparticles rapidly nucleate on the surface of the Al foil, followed by further nucleation of Cu on these existing nanoparticles. The Ostwald ripening and/or migration of the nanoparticles and their coalescence leads to the formation of the observed “microparticles” on the Al foil surface. As the reaction time increases (5–25 s), these “microparticles” act as the centers on which 3D growth along the vertical direction occurs selectively. Again here, homogenous nucleation is suppressed and successive nucleation events occur on the existing Cu structures. Interestingly, as depicted in the inset of Fig. 3c, these vertical rod-like structures are seen to be composed of tiny Cu nanoparticles stacked one on top of one another. This also indicates that nanoparticles self-assemble/aggregate after achieving preferred relative orientations. These clusters of rod-like structures, growing vertically outward from the surface, turn into “flowery” structures as can be seen after 45 s of reaction time. The first sign of dendritic-like structures is clearly visible after 45 s of reaction time (Fig. 3d inset). | ||
| Fig. 3 SEM investigation of the morphological evolution of Cu dendrites, as a function of Al foil immersion time, in a solution mixture of CuSO4 (50 mM) and HCl (100 mM) for (a) 5 seconds, (b) 10 seconds, (c) 25 seconds, (d) 45 seconds, (e) 60 seconds, (f) 2 minutes, (g) 3 minutes, (h) 5 minutes, (i) 9 minutes and (j) 60 minutes. The insets in (a)–(e) show higher magnification images of the structures formed. | ||
After about 1 min of reaction time, these flower-like structures have grown both in size and in number and resemble “bush”-like structures. As can be seen from Fig. 3e, these “bushes” have branches all over the surface of the Al foil and are highly interconnected to nearby bushes. The inset in Fig. 3e indicates that the central trunk and its primary branches are forming shapes that will lead to the formation of dendritic structures. It is important to note here that the structure shown in the inset of Fig. 3e is clearly composed of self-assembled/aggregated nanoparticles. By the end of 2 minutes of reaction time, the whole surface of the Al foil is covered with a mat of structures that resembles the basic framework of dendrites. After 2 minutes, the “bush”-like structures are completely invisible due to the heavy growth and huge number of interconnections between branches. The Al surface is now dominated by structures that are composed of a central trunk with growing primary side branches. From here on, we essentially find that the growth of these central trunks and branches continues as the reaction time increases due to further nucleation events. After 3 minutes of reaction time, we can clearly see the central trunk being completely formed and the primary branches starting to lengthen. Interestingly, the incoming copper nanoparticles from fresh nucleation events attach themselves to the tips of these primary branches, leading to the growths shown in Fig. 3g. Further increasing the reaction time results in growth of the primary branches and after 9 minutes, smooth and long primary branches are already formed. With further reaction time, we see the evolution of secondary branches from the primary branches. These secondary branches are observed to be highly separated from each other and the structures obtained at 60 minutes reaction time are fully grown dendrites that are smooth and highly symmetrical with striking self-similarity. One of the common events that occur throughout the whole growth process of the dendrites is nanoparticle aggregation with self-assembly/preferred orientation.
Typically, a DLA mechanism is proposed as the growth mechanism of dendrite formation. However, oriented attachment (OA) and a nanoparticle-aggregated self-assembly crystallization mechanism has also been frequently used to rationalize dendrite formation. It is understandable that structure formation during dendrite growth is a complex, many-body problem where various factors contribute to crystal growth and any single mechanism can only explain a part of the dendrite formation process. In our scheme of reactions where we clearly see evidence of nanoparticle-aggregated self-assembly crystallization, we believe that both DLA and the nanoparticle-aggregated self-assembly crystallization mechanism are relevant. The DLA model includes a single cluster, to which additional particles attach once they reach a site adjacent to the edge of the cluster.44 Tiny Cu nanoparticles that are formed initially on the surface of Al foil undergo surface migration and reorientation to form larger clusters. Concurrently, the growing Cu nuclei tend to deplete precursor ions near the Al foil surface, which sets up concentric diffusion fields around the growing Cu clusters.45 With further reaction time, the quantity of reactants declines, which greatly decreases supersaturation and restrains nucleation. Driven by the need to minimize surface energy, these small Cu clusters undergo reorientation, aggregation and self-assembly into rod-like structures that act as further nucleation sites. These rod-like structures continue to grow during the course of the reaction, whereby successive nucleation occurs heterogeneously and the added nuclei undergo stacked aggregation with preferred orientation, giving the impression of self-assembled nanoparticle aggregates. Further nanoparticle deposition and oriented aggregation through self-assembly leads to the development of directed dendritic structures with central trunks and branches (primary and secondary) as governed by the DLA model. The whole process of dendrite formation is an interplay of the DLA mechanism with the nanoparticle-aggregated self-assembly crystallization mechanism. This proposed mechanism is consistent with the previously reported mechanism of nanoparticle-aggregated self-assembly crystallization.18,46,47 In addition, Ostwald ripening plays an important role in forming the smooth surface and regular shape of the final crystal.48
HRTEM analysis of the completely formed dendritic structure was performed to gain insight into the preferred orientation within the dendrites. Fig. S6 (ESI†) shows images from the highlighted regions of the central trunk and the primary branches of the dendrites. The image (Fig. S6b†) taken from the tip of the central trunk shows obvious fringes with a ∼0.21 nm inter-planar separation corresponding to the {111} planes. This indicates that the growth direction of the central trunk is along 〈111〉. Fig. S6c† shows a typical image of the tip of the primary branch of the dendrites. Interestingly, fringes with inter-planar spacings of ∼0.21 nm are also found here, indicating that the growth direction of the primary branch is also along 〈111〉. This implies that the Cu dendrites grow along a preferential direction with preferred orientation within the dendrites.
:Cl− at 1:2. Fig. 4 below shows the structure/morphology of the Cu dendrites obtained from CuSO4 (25 mM) and NaCl/HCl (50 mM). A direct comparison of the SEM images shown in Fig. 2 and 4 reveals the effect of the amount of CuSO4 on the structure and morphology of the Cu dendrites. Clearly, for the NaCl system, a decrease in the amount of CuSO4 is accompanied by the formation of less-developed dendritic structures (Fig. 4a and b) with a lower overall yield. Unsurprisingly, the structures are shorter and less dense and the growth of the primary and the secondary structures is incomplete compared to the case with a higher CuSO4 concentration (Fig. 2a–c), where the dendrites are longer, denser and fully formed. A reduced amount of CuSO4 led to lowering of the nucleation and growth rate, which led to the formation of less-developed and shorter dendritic-like structures, as found in Fig. 4a and b. On the other hand, a comparison of structures obtained using HCl with different amounts of CuSO4 presents interesting findings. Even for the system with a lower amount of CuSO4, the dendrites that are formed are very similar to the dendrites formed with higher amounts of CuSO4, as revealed by analysis of the low magnification SEM images (Fig. 2 and 4c). However, a closer look at the high-magnification SEM images (Fig. 2e and 4d) reveals differences in the morphology of the Cu dendrites obtained with different amounts of CuSO4. The secondary branches are not developed where a lower amount of CuSO4 is used. The aggregation of smaller nanoparticles to form the primary branches is clearly observable, demonstrating the effect of a lowered amount of CuSO4. The expected lowered nucleation and growth rate for this system is manifested in the form of “slowness” of the reaction where the primary branches are now barely formed with no signs of the evolution of secondary branches. Hence, these structures appear rougher than the structures obtained with higher amounts of CuSO4. Consequently, these dendrites are anticipated to have a higher surface area with increased nano-structuring. Such aspects make them very attractive with respect to a variety of applications (catalysis, wettability, SERS sensors).
 | ||
| Fig. 4 (a), (b) FESEM images of Cu dendritic structures prepared from CuSO4 (25 mM) and NaCl (50 mM) at different magnifications; (c), (d) FESEM images of Cu dendritic structures prepared from CuSO4 (25 mM) and HCl (50 mM) at different magnifications. | ||
In order to demonstrate the feasibility of such surfactant-induced surface morphology engineering, we have performed a GRR of 50 mM CuSO4 with Al foil and 100 mM HCl in the presence of polyvinylpyrrolidone (PVP) at various concentrations (100, 10, 1 and 0.1 mM). Fig. 5a–d below shows SEM images of the structures of Cu deposits obtained at various PVP concentrations. For 100 mM PVP, the Cu deposits are characterized by random aggregates of small, spherical Cu particles (Fig. 5a). At such a high surfactant concentration, the Cu2+ can favorably interact with PVP to form a stable Cu2+–PVP complex, which would lead to a reduced rate of nucleation.49 Moreover, the presence of excess surfactant would coat the freshly-nucleated Cu nanoparticles and would hinder their fusion with preferred orientation, forming only particles that undergo clustering. Since the entire surface of the copper nuclei are covered by PVP, subsequent copper atoms deposit on an exposed Al foil rather than on an existing copper nucleus. Therefore, a uniform layer of copper covers the foil completely, as has been observed experimentally here while using a high concentration of surfactant. This is in stark contrast to the observation of thick red deposits covering only certain regions of the Al foil during a GRR in the absence of PVP. When the amount of PVP is reduced to 10 mM, the obtained Cu deposits clearly show 1-D wire-like growth pointed along the surface normal of the Al foil, as shown in Fig. 5b. Here, the Cu nanoparticles have a modest tendency to fuse through self-assembly with preferred orientation and the structures have a corncob-like morphology. However, the reduced nucleation and growth rates restrict the successful formation of complete dendritic 2D structures with primary and secondary branches. These corncob-like structures might actually represent the central trunk being formed with a high degree of roughness due to incomplete fusion of the Cu nanoparticles that are covered (partially) with PVP.
 | ||
| Fig. 5 Effect of surfactant (a–d) and temperature (e–f) on the morphology/structure of the Cu dendrites. Copper dendrites prepared at room temperature using CuSO4 (50 mM) and HCl (100 mM) at varying concentrations (mM) of PVP: (a) 100, (b) 10, (c) 1 and (d) 0.1. The inset shows higher magnification images of the structures formed. Cu dendrites prepared at 4 °C using CuSO4 (50 mM) with (e) NaCl (100 mM) and (f) HCl (100 mM). | ||
A further decrease in the PVP concentration to 1 mM starts to manifest tailoring of the structures of the Cu deposit. At this low concentration of PVP, the rate of nucleation and growth and the tendency to fuse and self-assemble is expected to be greater than with 10 mM PVP. As shown in Fig. 5c, the Cu deposit represents a ‘paddy-like’ networked structure with very short primary “branches” composed of tiny nanoparticles aggregated through self-assembly. This in effect shows that the experimental conditions now favor dendritic structures (although not fully grown). Interestingly, at 0.1 mM PVP, clear signs of size/morphology changes in the dendrites are observed, where dendritic like-structures with many short branches, stretching in random directions, “cemented” to the trunk can be observed (Fig. 5d). Some of the copper also forms smaller dendritic structures. These dendrites possess comparatively shorter side branching and are much smaller compared to dendrites prepared without any PVP. Even at such low concentrations of PVP, the smaller sizes of these dendritic-like structures clearly show the effect of the surfactant on the structure/morphology of the Cu dendrites. This effect of surfactant concentration on the morphology of the Cu dendrites is anticipated to yield surface-textured Cu structures with very high surface areas, which can be useful in a variety of applications, such as catalysis, wettability and SERS-based sensing.
:[Cl−] = 1:2, 1 hour reaction time). Surprisingly, we find that the yield of the reaction with nitrate counter anions is very low when compared to the corresponding sulfate case, as depicted in Fig. 6a–d.
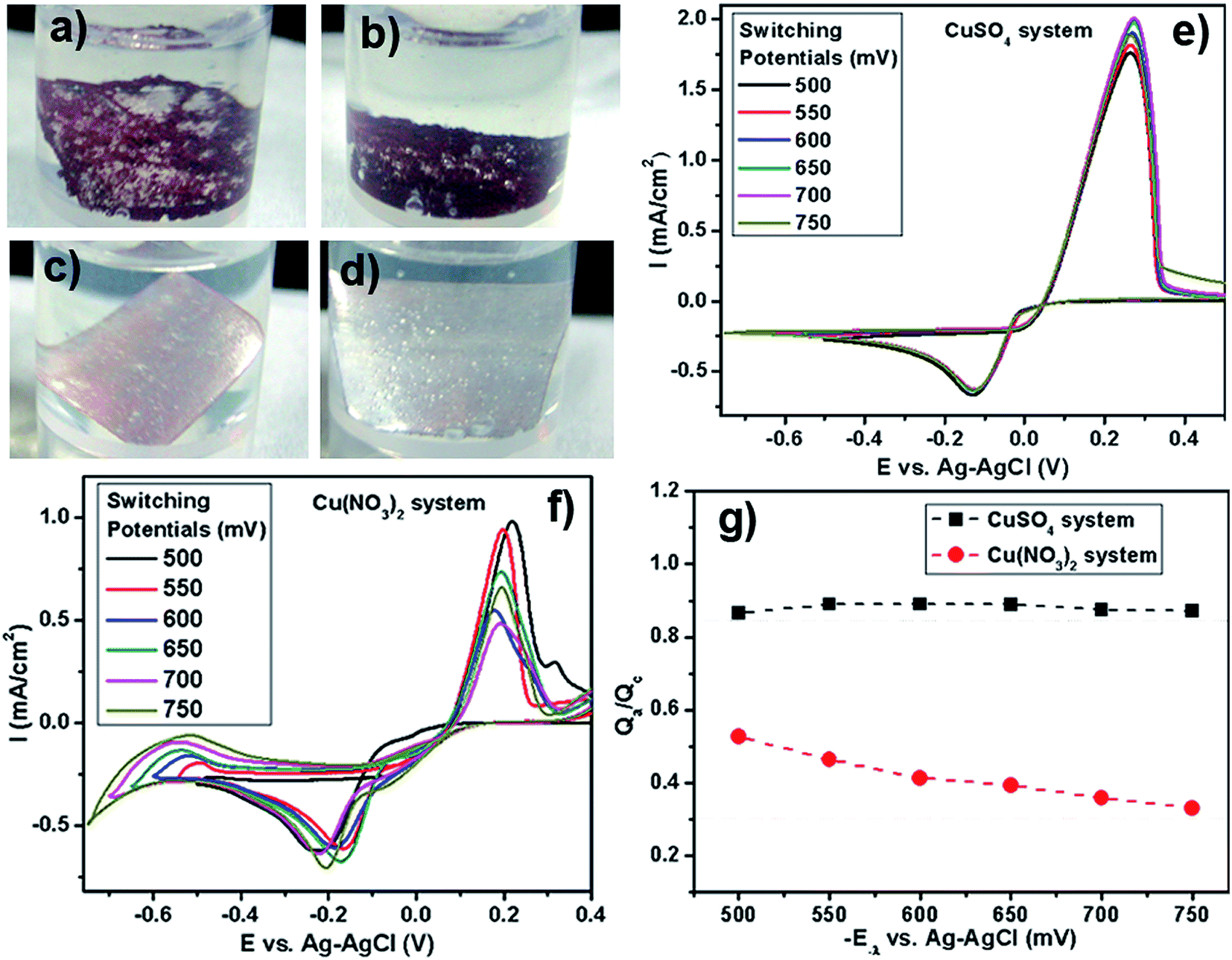 | ||
| Fig. 6 Images of the Al foil immersed for 1 hour in vials containing (a) CuSO4 and HCl, (b) CuSO4 and NaCl, (c) Cu(NO3)2 and HCl and (d) Cu(NO3)2 and NaCl. Voltammograms obtained for different switching potentials at a scan rate of 20 mV s−1 for copper electrodeposition on GCE from (e) CuSO4, (f) Cu(NO3)2 and (g) Cathodic efficiency of Cu deposition (Qa/Qc) at various switching potentials (−E−λ). | ||
This perplexing observation led us to further investigate the effect of the nitrate anions on dendrite formation. SEM images of the products formed with Cu(NO3)2 (50 mM) with NaCl/HCl (100 mM) after 2 hours of reaction time at room temperature are shown in Fig. S8a and b, ESI.† It is clearly observable that no dendrite formation takes place when nitrate anions are the counter ions. Even after 12 hours of reaction time, no dendrite formation was observed (Fig. S7c and d, ESI†), albeit the yield of the Cu deposit increased. In order to further probe this effect of counter anions, we ran control experiments where we added Na2SO4 or NaNO3 to the CuSO4 and Cu(NO3)2 reaction system. Specifically, when NaNO3 is added to the CuSO4 with HCl/NaCl system, we obtain a very low yield for the reaction, with no formation of dendritic morphology in the Cu deposit. The same holds true when Na2SO4 is added to the Cu(NO3)2 with HCl/NaCl system. Also, the reaction yield remains high when Na2SO4 is added to the CuSO4 with HCl/NaCl system. The corresponding photographs of the reaction vials are shown in Fig. S9, ESI.† This led us to believe that the nitrate ions do play a key role, acting as a “poison” and inhibiting the formation of Cu dendrites.
In order to comprehend the underlying chemistry behind this “poisoning” effect by nitrates, we have performed cyclic voltammetry (CV) experiments with CuSO4 and Cu(NO3)2 as the reactants in the presence of Na2SO4 as the background electrolyte under N2 purging. Typical cyclic voltammograms of copper(II) sulfate and copper(II) nitrate systems at varying negative switching potentials (−E−λ) are presented in Fig. 6e–f. The cathodic process for both the copper sulfate and the copper nitrate system is evidenced by the peak in the negative current density region, characterizing the reduction of Cu2+ to Cu0. The anodic process for both the copper sulfate and the copper nitrate system is evidenced by the peak in the positive current density region, which is due to the oxidation of Cu0 to Cu2+ by the applied scanning potential. These voltammograms display a crossover on the cathodic branches for both the sulfate and nitrate systems, which indicates that copper deposition proceeds via nucleation and growth phenomena.50–52 For the copper nitrate system in the cathodic and anodic region, the peaks are observed to shift slightly as the switching potential is varied. More interestingly, in the reduction region of the CV where E < −0.3 V, the nitrate systems present a notable difference in the cathodic current after the metallic copper has been deposited. With the potential varying towards more negative values, the current keeps growing as observed for the nitrate system. Such voltammetric behaviors for nitrate systems indicate an additional supply of electroactive species in the reduction process. Such behavior has been previously observed for Cu52 and Co51 reduction, wherein a secondary chemical reaction of the freshly deposited metal with the nitrates has been observed. We believe that a similar secondary chemical reaction involving the deposited Cu with the nitrates, that generates additional electroactive species, might be operative here, leading to an increasing cathodic current as the potential scans more negative values. This difference in the cathodic region for the sulfate vs. nitrate system indicates that the nitrate counter anions are far from being “spectator anions” and play an important chemical role during the deposition of metallic Cu.
In order to understand the effect of the nitrates on the yield of the Cu deposit, analysis of the cathodic efficiency of Cu deposition was performed by a voltammetric study with different cathodic switching potentials (−E−λ). The cathodic (Qc) and anodic (Qa) charges were calculated by integrating the cathodic and anodic branches of the I vs. −E−λ curves, respectively. This, in essence, reflects the amount of copper that undergoes oxidation from the electrode surface during the anodic scan by the applied potential. This charge-deposition ratio can very well be utilized as a relative metric for ascertaining the reaction yield of Cu deposits. Fig. 6g shows a plot of the ratio Qa/Qc against the increasing negative switching potential for both copper sulfate and copper nitrate systems. The charge recovery for copper sulfate is around 0.9 (90%), indicating that almost all of the deposited copper is stripped away by the applied potential during the anodic scan. However, for the copper nitrate system, the charge recovery at a switching potential of −550 mV is low, around 0.52 (∼52%). On further increasing the negative switching potential, the ratio goes down to 0.3 (30%). Therefore, low charge recoveries observed in the medium containing nitrate anions indicate an additional contribution that could be explained in terms of some process that can consume electrons or recently deposited Cu nuclei coupled to the cathodic reaction.
The only difference between the reactant compositions is the presence of nitrate counter ions, so it is likely that an interaction between newly deposited copper and nitrate ions in solution is occurring. This may include a direct redox reaction and/or nitrate reduction on the surface of the copper nuclei. The former reaction would consume the copper deposit and the latter would provide electrons to the external circuit. Jointly or separately, these reactions explain the low copper recovery efficiency for reactants containing nitrate counter-ions. In fact, there are reports of the electrodeposition of metals such as copper,52 cobalt51 and thallium,53 during which the nitrates have been found to interact with the freshly deposited metallic nuclei and undergo reduction, thereby oxidizing the deposited metal and decreasing the yield of the metal deposit. The striking similarity between these prior observations during an electrodeposition process and our electroless deposition process involving the GRR on Al foil leads us to believe that nitrate anions can act as an oxidant in a direct redox reaction with the freshly deposited Cu nuclei, thereby decreasing the reaction yield of Cu deposits during our GRR deposition process. This nitrate-augmented oxidative dissolution of the Cu nuclei will cause restrained nucleation and growth of the Cu deposit will be continuously interrupted. This reduced nucleation and growth rate hinders the kinetically-controlled formation of dendritic structures, thereby affecting the morphology of the obtained Cu deposits, as has been observed here in our case.
Fig. 7a–c shows a plot of the instantaneous absorbance as a function of reaction time for the various Cu structures utilized as catalysts here. As can be visually inferred, the apparent rate of the reduction reaction is fastest for Cu dendrites fabricated using HCl, followed by the Cu dendrites fabricated using NaCl and then followed by the commercial Cu powder (Fig. 7a–c). Since NaBH4 was present in excess during the reduction reaction, pseudo-first-order kinetic analysis was performed to calculate the apparent rate constants for the three systems as shown in Fig. 7d. The observed linear relationship allows us to calculate the apparent rate constant (kapp) from the slope of the plots shown in Fig. 7d. However, to compare the activities of the different solid catalysts, each kapp from the slope should be normalized to another comparable kinetic parameter, knor (mmol s−1 g−1), according to the following equation:29 knor = (10−3 × C0 × V × m) × kapp; where C0, V and m are the initial concentration of 4-NP (mM), the volume of the reactant (4-NP) solution (mL) and the mass of catalyst (g), respectively. The normalized rate constants knor (mmol s−1 g−1) for the Cu dendrites fabricated with HCl, NaCl and commercial copper powder are found to be 18.2 × 10−3, 6.1 × 10−3 and 1.9 × 10−4, respectively.
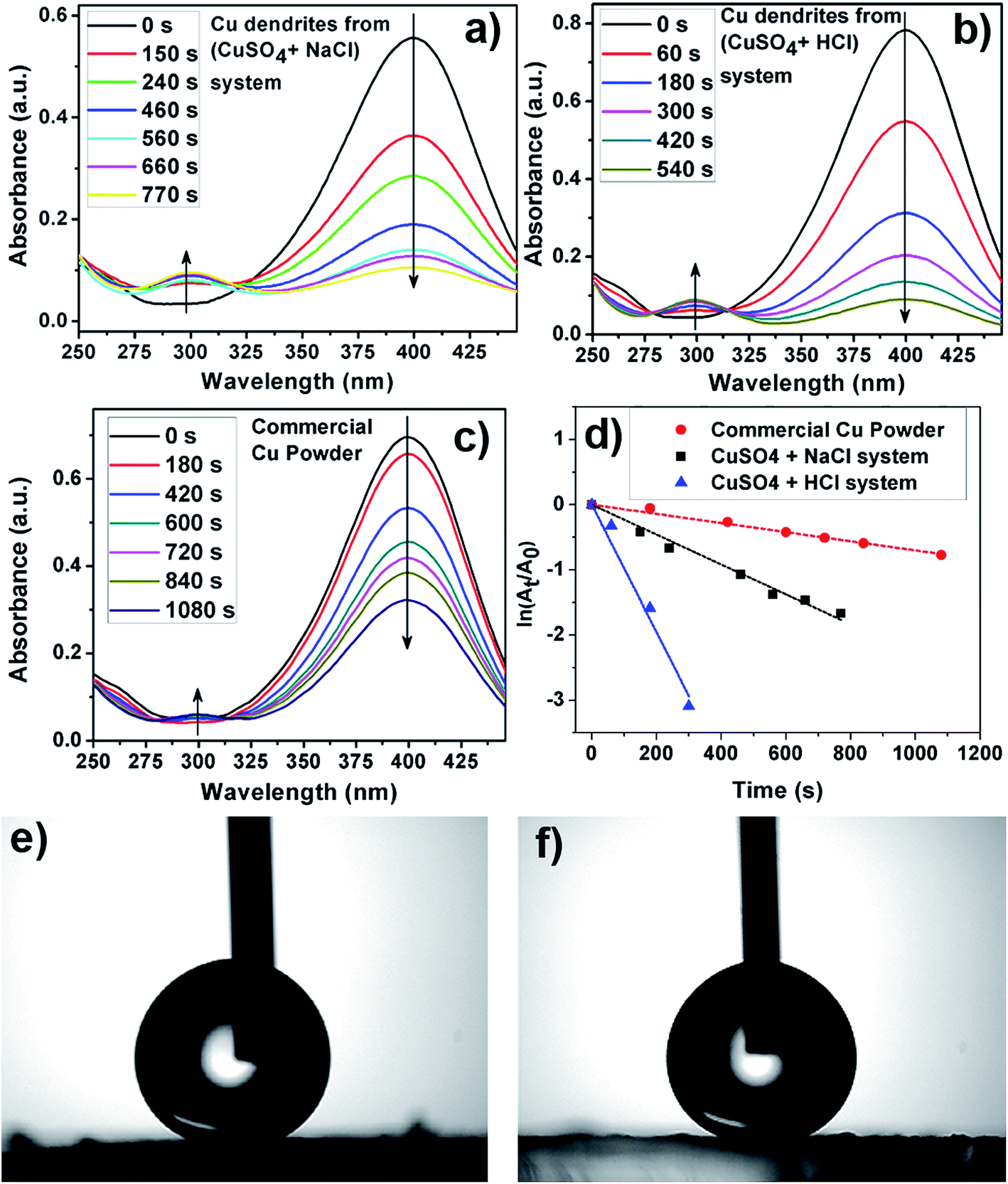 | ||
| Fig. 7 Reduction of 4-nitrophenol (NP) catalyzed by Cu dendritic structures prepared from 50 mM CuSO4 and (a) 100 mM NaCl, (b) 100 mM HCl, (c) commercial Cu powder; (d) plot of ln(At/A0) against time for the reduction of NP obtained by monitoring absorbance at 400 nm. Static water contact angle (SWCA) measurement of films of n-dodecanethiol-modified Cu dendrites prepared in the presence of 50 mM CuSO4 with (e) 100 mM NaCl and (f) 100 mM HCl, exhibiting superhydrophobicity. | ||
The observed normalized rate constants for Cu dendrites obtained using NaCl are similar to those published in an earlier report.29 However, it is interesting to find that the Cu dendrites fabricated using HCl have the highest normalized rate constant ever reported. This indicates that increased nano-structuring may play a vital role in the catalytic reduction reaction. The catalytic reduction of 4-NP with NaBH4 involves two steps: (1) diffusion and adsorption of 4-NP to metal surfaces and (2) electron transfer mediated by metal surfaces from NaBH4 to 4-NP.55,56 Moreover, the first step is the rate-determining step, which has a direct proportionality relationship with the available surface area. As discussed earlier in this report, the dendrites fabricated using HCl have more nano-structuring/a higher surface area than the ones prepared using NaCl (Fig. 2). Hence, the higher rate constant obtained for dendrites fabricated using HCl (vs. NaCl) is attributed to their higher surface texturing/surface area. We thus believe that the nano-structured Cu dendrites obtained by our facile method are promising candidates for the efficient catalytic reduction of nitrophenol compounds.
Superhydrophobicity
Wettability of a surface is a basic property that, when tailored, can be very useful for a variety of applications such as self-cleaning, anti-icing, anti-fogging and antifouling.57–59 Surface wettability for smooth surfaces is usually quantified in terms of the static contact angle (CA) as defined by the Young–Dupre equation (see Fig. S11a, ESI†). Contact angle hysteresis (CAH), which gives an indication of the stickiness of the surface, is the difference between the dynamic CAs measured during the growth (advancing; CAa) and shrinkage (receding; CAr) of a water droplet. Nature has plenty of examples of surfaces that are highly hydrophobic, which are characterized by a static water CA (SWCA) above 150°. Such surfaces are referred to as superhydrophobic surfaces and the classic example is a self-cleaning lotus leaf, characterized by a high SWCA (160°) and very low CAH (2°).59 Inspired by nature's art of preparing superhydrophobic surfaces, researchers now know that the following surface parameters and their interplay have key roles in designing superhydrophobic surfaces: intrinsic hydrophobicity, surface morphology and surface roughness.58,59 To obtain superhydrophobic surfaces with CAs above 150°, it is imperative to have surface roughness. After physical micro/nano structuring of surfaces, a chemical post-treatment with hydrophobic materials is performed routinely to lower the surface energy of the metallic structures.58,59 It is generally accepted that a low surface energy coating material needs to be present as the topmost layer on the surface of the material. Typically utilized low-energy coating materials are fluorinated or thiolated hydrocarbons. Such coatings turn the surface hydrophobic and help repel water from the surface. This hydrophobicity, coupled with the presence of surface roughness, increases the air fraction present at the interface of a liquid drop on the solid surface, thereby acting as a cushion for suspending the liquid droplet, resulting in an increased CA.58,59The utility of the highly nanostructured Cu dendrites as a potential material for fabricating superhydrophobic surfaces was tested. Thoroughly washed Cu dendrites deposited on a PDDA-PSS-functionalized glass slide are exposed to a solution of n-dodecanethiol, which acts as a low-energy coating material rendering the surface hydrophobic. Such films exhibit superhydrophobicity, as observed by measurement of static contact angles with a water droplet as probe liquid, as shown in Fig. 7e–f. The Cu dendrites prepared using NaCl were found to have a static CA (CAH) of 151° (7°), while the Cu dendrites prepared in the presence of HCl are found to have a static CA (CAH) of 159° (2°). Clearly, both the surfaces are superhydrophobic in nature. Moreover, it is found that the water droplet cannot rest on, and rolls off the n-dodecanethiol-modified copper dendrite surface immediately, showing great potential in self-cleaning applications. A corresponding real-time video showing the water droplet rolling off the surface is provided in the ESI.†
The observed differences in the CA for the Cu dendrites prepared using NaCl vs. HCl primarily arise due to inherent differences in the surface morphology and roughness of the dendrites as evidenced by SEM analysis. The higher degree of roughness of the Cu dendrites prepared with HCl compared to NaCl is thought to be the reason behind the differences in the measured static CA. The suspension of a water droplet on these nanostructured materials is described by a composite state, where the liquid–solid interface is now a composite of a liquid–solid and a liquid–vapor interface with an increased air fraction due to increased nano-structured asperities. In such a scenario, the Cassie–Baxter model (CB model) can be qualitatively implemented. As per the CB model, the apparent contact angle can be expressed solely in terms of the solid fraction for a given solid surface with CA θ (note that the apparent CA is directly proportional to the solid fraction, see ESI, Fig. S11b†). The increased nano-structuring with an increased air fraction, with a concomitant decrease in the solid fraction, then directly translates to an increased apparent CA as expected from the CB model. Clearly, the Cu dendrites prepared with HCl have higher nano-structuring than that of those prepared with NaCl and hence the former has a higher SWCA. We believe that the surface-textured Cu dendrites prepared by this simple method are an efficient superhydrophobic material with self-cleaning abilities that may attract many pragmatic applications.
Conclusions
Here, we present a simple, rapid, scalable, environment-friendly and uninterrupted galvanic replacement reaction (GRR)-based solution chemistry methodology to fabricate copper dendritic structures with tunable surface structure/morphology targeted towards desired applications. This facile methodology provides various reaction parameters that can be effectively utilized to control the deposition rate and surface morphology of the dendrites. The presence of acid in the reaction system is demonstrated to have a profound effect on the nano-structuring of the dendrites. Moreover, we also find that the choice of Cu2+ precursor salt has a dramatic effect on the structure of the obtained copper deposits. Careful control of the other reaction parameters, such as concentration of metal ions, concentration of surfactant and reaction temperature also provides a useful means of modulating the surface structure/morphology of the dendrites. The growth process of these dendrites has been probed by extensive temporal SEM investigations. The acid-induced nano-structured Cu dendrites show the highest ever reported normalized rate constant for the catalytic reduction of 4-nitrophenol (18.2 × 10−3 mmol s−1 g−1). Wettability studies utilizing these nano-structured dendrites demonstrate excellent self-cleaning abilities due to superhydrophobicity with high static water contact angle and low contact angle hysteresis.This report highlights how the underlying chemical reactions involved in the synthesis of these materials affect their material properties and hence their application efficiency. The merits of our solution chemistry methodology and the properties of the synthesized Cu dendrites can be beneficially exploited for the production of low-cost Cu nano/micro structures that can find practical applications as efficient catalysts and self-cleaning materials. Our ongoing efforts involve the utilization of dendritic structures with higher nanostructuring, obtainable by modulating the other reaction parameters demonstrated here, for applications in catalysis, wettability and surface-enhanced Raman scattering (SERS) and metal-enhanced fluorescence (MEF)-based chemical sensing.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to acknowledge Dr H. S. Panda, DIAT Pune for HRTEM images, Mr A. B. Gaikwad for preliminary SEM imaging and Mr S. S. Deo for acquiring XPS spectra. The authors would like to thank Prof. S. B. Ogale, Dr P. A. Joy and Dr A. Nag for insightful discussions. This work was supported by DST grant number (SB/S2/RJN-61/2013) and CSIR-NCL start-up grant number (MLP 030326).References
- Y. Xia and N. J. Halas, MRS Bull., 2005, 30, 338–348 CrossRef CAS.
- F. Kim, S. Connor, H. Song, T. Kuykendall and P. Yang, Angew. Chem., Int. Ed., 2004, 43, 3673–3677 CrossRef CAS PubMed.
- J. Rongchao, C. Y. Charles, H. Encai, G. S. Metraux, G. C. Schatz and C. A. Mirkin, Nature, 2003, 425, 487–490 CrossRef PubMed.
- P. Lignier, R. Bellabarba and R. P. Tooze, Chem. Soc. Rev., 2012, 41, 1708–1720 RSC.
- S. Habas, H. Lee, V. Radmilovic, G. Somorjai and P. Yang, Nat. Mater., 2007, 6, 692–697 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2008, 48, 60–103 CrossRef PubMed.
- S. Lal, N. K. Grady, J. Kundu, C. S. Levin, B. Lassiter and N. J. Halas, Chem. Soc. Rev., 2008, 37, 898–911 RSC.
- B. Wiley, Y. Sun, J. Chen, H. Cang, Z. Y. Li, X. Li and Y. Xia, MRS Bull., 2005, 30, 356–361 CrossRef CAS.
- B. Wiley, Y. Sun and Y. Xia, Acc. Chem. Res., 2007, 40, 1067–1076 CrossRef CAS PubMed.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS PubMed.
- A. Gutés, C. Carraro and R. Maboudian, J. Am. Chem. Soc., 2010, 132, 1476–1477 CrossRef PubMed.
- M. H. Rashid and T. K. Mandal, J. Phys. Chem. C, 2007, 111, 16750–16760 CAS.
- W. Ren, S. J. Guo, S. J. Dong and E. K. Wang, Nanoscale, 2011, 3, 2241–2246 RSC.
- M. Matsushita, M. Sano, Y. Hayakawa, H. Honjo and Y. Sawada, Phys. Rev. Lett., 1984, 53, 286–290 CrossRef CAS.
- D. Barkey, F. Oberholtzer and Q. Wu, Phys. Rev. Lett., 1995, 75, 2980–2984 CrossRef CAS PubMed.
- T. A. Witten and L. M. Sander, Phys. Rev. Lett., 1981, 47, 1400–1403 CrossRef CAS.
- S. R. Forrest and T. A. Witten, J. Phys. A, 1979, 12, L109 CrossRef CAS.
- J. X. Fang, H. J. You, P. Kong, Y. Yi, X. P. Song and B. J. Ding, Cryst. Growth Des., 2007, 7, 864–867 CAS.
- Y. C. Han, S. H. Liu, M. Han, J. C. Bao and Z. H. Dai, Cryst. Growth Des., 2009, 9, 3941–3947 CAS.
- P. Meakin, Phys. Rev. Lett., 1983, 51, 1119 CrossRef.
- M. Kolb, R. Botet and R. Jul-lien, Phys. Rev. Lett., 1983, 51, 1123 CrossRef.
- G. X. Zhang, S. H. Sun, M. N. Banis, R. Y. Li, M. Cai and X. Sun, Cryst. Growth Des., 2011, 11, 2493–2499 CAS.
- H. P. Ding, G. Q. Xin, K. C. Chen, M. L. Zhang, Q. Y. Liu, J. C. Hao and H. G. Liu, Colloids Surf., A, 2010, 353, 166–171 CrossRef CAS.
- R. Qiu, H. G. Cha, H. B. Noh, Y. B. Shim, X. L. Zhang, R. Qiao, D. Zhang, Y. I. Kim, U. Pal and Y. S. Kang, J. Phys. Chem. C, 2009, 113, 15891–15896 CAS.
- W. Shao and G. Zangari, J. Phys. Chem. C, 2009, 113, 10097–10102 CAS.
- B. K. Barman and K. K. Nanda, Dalton Trans., 2015, 44, 4215–4222 RSC.
- S. Sun, C. Kong, L. Wang, S. Yang, X. Song, B. Ding and Z. Yang, CrystEngComm, 2011, 13, 1916–1921 RSC.
- C. Yan and D. Xue, Cryst. Growth Des., 2008, 8, 1849–1854 CAS.
- J. Liu, Q. Wu, F. Huang, H. Zhang, S. Xu, W. Huang and Z. Li, RSC Adv., 2013, 3, 14312–14321 RSC.
- W. Ye, Y. Chen, F. Zhou, C. Wang and Y. Li, J. Mater. Chem., 2012, 22, 18327–18334 RSC.
- J. Y. Zheng, A. P. Jadhav, G. Song, C. W. Kim and Y. S. Kang, Thin Solid Films, 2012, 524, 50–56 CrossRef CAS.
- K. H. Kim, J. Y. Zheng, W. Shin and Y. S. Kang, RSC Adv., 2012, 2, 4759–4767 RSC.
- J. Y. Zheng, M. J. Kang, G. Song, S. I. Son, S. P. Suh, C. W. Kim and Y. S. Kang, CrystEngComm, 2012, 14, 6957–6961 RSC.
- R. Qiu, J. Y. Zheng, H. G. Cha, M. H. Jung, K. J. Lee and Y. S. Kang, Nanoscale, 2012, 4, 1565–1567 RSC.
- A. Sinha and B. P. Sharma, Mater. Res. Bull., 2002, 37, 407–416 CrossRef CAS.
- M. D. Susman, Y. Feldman, A. Vaskevich and I. Rubinstein, Chem. Mater., 2012, 24, 2501–2508 CrossRef CAS.
- Y. Wang, A. V. Biradar, G. Wang, K. K. Sharma, C. T. Duncan, S. Rangan and T. Asefa, Chem.–Eur. J., 2010, 16, 10735–10743 CrossRef CAS PubMed.
- A. Sarkar, T. Mukherjee and S. Kapoor, J. Phys. Chem. C, 2008, 112, 3334–3340 CAS.
- M. Jin, G. He, H. Zhang, J. Zeng, Z. Xie and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 10560–10564 CrossRef CAS PubMed.
- X. Zhang, G. Wang, X. Liu, H. Wu and B. Fang, Cryst. Growth Des., 2008, 8, 1430–1434 CAS.
- J. Fu, W. Ye and C. Wang, Mater. Chem. Phys., 2013, 141, 107–113 CrossRef CAS.
- Q. Wu and D. Barkey, J. Electrochem. Soc., 2000, 147, 1038–1045 CrossRef CAS.
- Y. Zhang, S. Sun, X. Zhang, L. Tang, X. Song and Z. Yang, Phys. Chem. Chem. Phys., 2014, 16, 18918–18925 RSC.
- R. M. Brady and R. C. Ball, Nature, 1984, 309, 225–229 CrossRef CAS.
- J. Xu and D. Xue, J. Phys. Chem. B, 2006, 110, 11232–11236 CrossRef CAS PubMed.
- J. Fang, B. Ding and X. Song, Cryst. Growth Des., 2008, 8, 3616–3622 CAS.
- J. Fang, X. Ma, H. Cai, X. Song and B. Ding, Nanotechnology, 2006, 17, 5841–5845 CrossRef CAS.
- J. Fang, B. Ding, X. Song and Y. Han, Appl. Phys. Lett., 2008, 92, 173120 CrossRef.
- I. Haas, S. Shanmugam and A. Gedanken, J. Phys. Chem. B, 2006, 110, 16947–16952 CrossRef CAS PubMed.
- S. Fletcher, Electrochim. Acta, 1983, 28, 917–923 CrossRef CAS.
- E. Barrera, M. P. Pardavé, N. Batina and I. González, J Electrochem. Soc., 2000, 147, 1787–1796 CrossRef CAS.
- A. Ramos, M. Miranda-Hernández and I. González, J Electrochem. Soc., 2001, 148, C315–C321 CrossRef CAS.
- A. Serruya and B. R. Scharifker, in Extended abstracts of the Tenth Congreso de la Sociedad Venezolana de Electroquímica, 1997, p. 57 Search PubMed.
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2009, 10, 30–35 CrossRef PubMed.
- K. Esumi, K. Miyamoto and T. Yoshimura, J. Colloid Interface Sci., 2002, 254, 402–405 CrossRef CAS PubMed.
- K. Hayakawa, T. Yoshimura and K. Esumi, Langmuir, 2003, 19, 5517–5521 CrossRef CAS.
- T. C. Rangel, A. F. Michels, F. Horowitz and D. E. Weibel, Langmuir, 2015, 31, 3465–3472 CrossRef CAS PubMed.
- T. Darmanin, E. T. Givenchy, S. Amigoni and F. Guittard, Adv. Mater., 2013, 25, 1378–1394 CrossRef CAS PubMed.
- X.-M. Li, D. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 1350–1368 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra22683j |
| This journal is © The Royal Society of Chemistry 2016 |