
First report of the application of simple molecular complexes as organo-catalysts for Knoevenagel condensation†
Sumit Kumar Panja,
Nidhi Dwivedi and
Satyen Saha*
Department of Chemistry, Faculty of Science, Banaras Hindu University, Varanasi-221005, India. E-mail: satyen.saha@gmail.com; Fax: +91-542-2368127; Tel: +91-9935913366
First published on 20th July 2015
Abstract
A series of molecular complexes have been designed, synthesized and used as organo-catalysts for the first time for very efficient Knoevenagel condensation. Molecular complexes are thermally stable, easily recyclable, and have a low cost of preparation. The role of acidic protons in molecular complexes in Knoevenagel condensations has been identified as the key factor and helps us to provide useful information about the reaction pathway. The acidic proton of a catalyst enhances the electrophilicity of an aldehyde and accelerates the dehydration process of the reaction at room temperature (RT). An eco-friendly, green synthetic protocol for the Knoevenagel condensation is used to synthesize a series of important cyano group containing synthetic precursors for synthesis of biologically active molecules at RT using a minimum amount of catalyst (∼5 mol%) without the need for chromatographic separation techniques. Detailed mechanistic studies and substituent effects of aromatic aldehydes on the reaction have been investigated. In addition, biologically active 2-amino-4H-chromene derivatives have also been synthesized by the Knoevenagel condensation of salicylic aldehydes with active methylene compounds, followed by intramolecular cyclization (via Michael addition) delivering higher yields within shorter reaction times at RT without any need for chromatographic separation.
Introduction
Synthetic organic chemists have focused on converting difficult synthetic protocols to easier and greener ones with simple organic synthones to produce entirely new organic molecules.1 This useful synthetic technique is expected to contain a one-pot single step using environmentally benign non-hazardous catalysts and solvent-free conditions.2 Performing an organic reaction in an environmentally suitable mode is still a challenge to chemists today. One of the important objectives in organic synthesis, especially in the synthesis of fine chemical products such as pharmaceuticals, is the facile synthesis of new carbon–carbon bonds etc. The Knoevenagel reaction is a well-known carbon–carbon bond-forming reaction, discovered by E. Knoevenagel in 1896, it is also widely used in the chemical, pharmaceutical and perfume industries.3 The reaction is also well known for the synthesis of a large variety of intermediates useful in the manufacturing of top selling drugs (such as atorvastatin, pioglitazone, AMG 837, pregabalin, lumefantrine, entacapone) in the world.4Traditionally, Knoevenagel condensations are carried out under homogeneous conditions in the presence of catalysts such as organic bases5 solid bases,6 ionic liquids,7 amino acids,8 Lewis acids,9 organometallic catalysts,10 metal complexes immobilized on a solid support such as oxide and zeolite deteriorates,11 oxides modified by salts,12 and amine-functionalized materials affording variable yields with different reaction times.13 Recently, the synthesis of 2-amino-4H-chromene derivatives has attracted great interest due to their biological and pharmacological activities.14 2-Amino-4H-chromenes constitute a major class of naturally occurring compounds, and interest in their chemistry continues because of their utility as biologically active agents.15 They occur widely in plants, including edible vegetables and fruits.16 Synthetic chromene analogues have been developed over the years, and some of them have been employed in pharmaceuticals,17 which includes antifungal18 and antimicrobial agents.19 2-Amino-4H-chromenes are employed as pigments,20 cosmetics, agrochemicals21 and are major constituents of many biologically active natural products (such as HA 14-1, inhibitor of MK-2, IRSP inhibitor and MX58151) as shown in Fig. 1.22
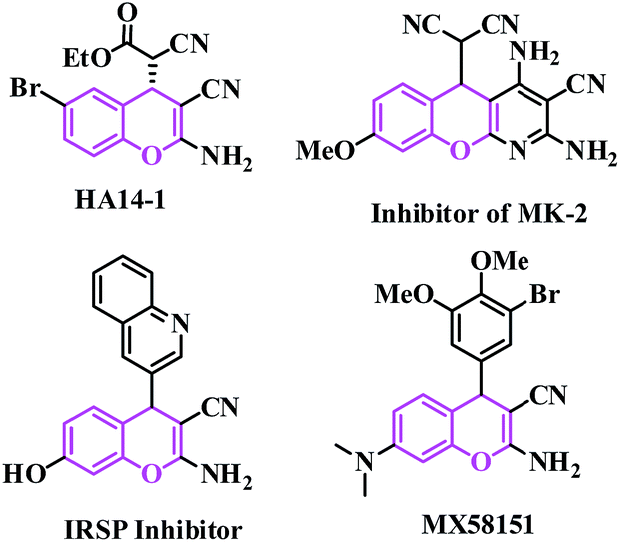 | ||
| Fig. 1 Example of biologically active 2-amino-4H-chromenes. | ||
Recently Li et al. have used task specific ionic liquids as catalysts for Knoevenagel condensations at room temperature (RT) with catalyst loadings as high as 15 mol%.23 Vakili et al. have reported L-histidine and L-arginine catalysed Knoevenagel condensation reactions though the protocol is not superior in terms of reaction conditions, reaction times and yields.24 Further Mase et al. have also reported eco-friendly organo-catalysed Knoevenagel condensation reactions but preparation, stability and recyclability of the organo-catalyst are still challenging factors.25 In our continuation of work with various kinds of molecular complexes, we are reporting for the first time the use of simple molecular complexes (Chart 1) based on weak acids and tertiary amines as organo-catalysts for the popular Knoevenagel condensation reaction. The mechanistic aspects of the Knoevenagel condensation using these molecular complexes have been investigated and are presented in our present work. Further, our synthetic protocol is used to synthesize a biologically active 2-amino-4H-chromene derivative.
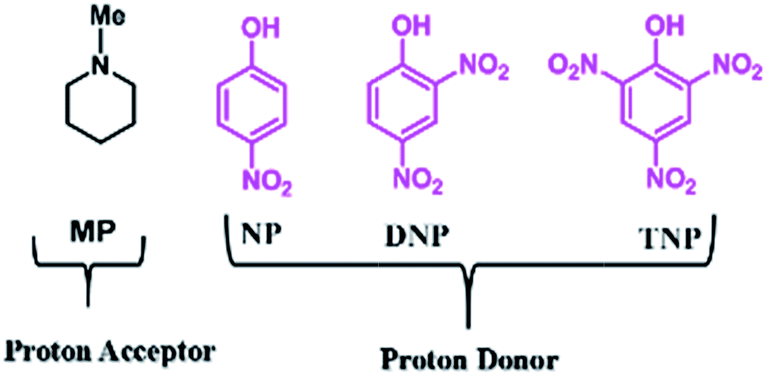 | ||
| Chart 1 Structural formulas and abbreviations for the components of molecular complexes. | ||
Results and discussion
As a well-known simple condensation reaction, the Knoevenagel condensation is preferred as the model reaction for studies of our newly synthesised molecular complexes as organo-catalysts. As depicted in Scheme 1, 1 mmol of benzaldehyde (1a) and 1.01 mmol of malononitrile (2a) are taken in different solvent systems and with different amounts of catalyst loading (range 0.02 to 0.1 mmol) at RT. Several sets of reactions are performed to optimize the reaction in terms of yield, reaction time, solvent conditions, and the amount of catalyst used. It was observed that the reaction does not proceed in the absence of the catalyst (molecular complex) either in ethanol or under neat conditions at room temperature (Table 1, entry 1). But when the reaction is carried out using 0.02 mmol of the catalyst (MP(DNP)) in ethanol, the yield of the desired product (3aa) is found to be 85% at RT (Table 1, entry 2). Further, when the catalyst loading is increased to 0.05 mmol under the same conditions, the yield of 3aa (desired product) is increased to 98% (Table 1, entry 3). However, no change in the yield is observed when a higher mole% of catalyst is used (Table 1, entry 8).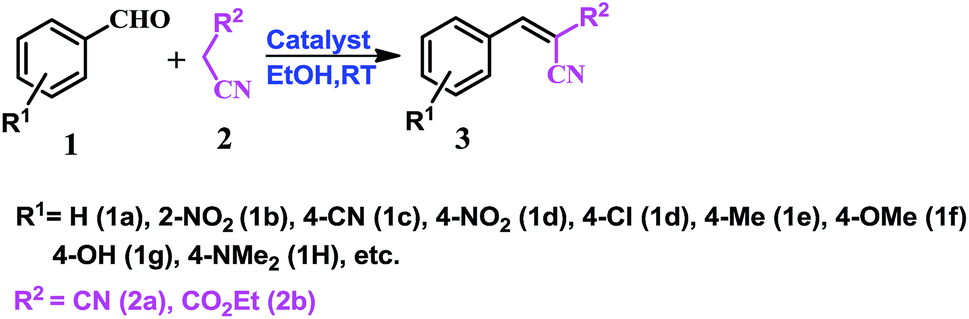 | ||
| Scheme 1 Knoevenagal condensation. | ||
| Sl. | Catalyst (mmol) | Solvent | Time (min) | Yieldb |
|---|---|---|---|---|
| a Reaction conditions: benzaldehyde (1 mmol) and malononitrile (1 mmol) were stirred with different amounts of catalyst under ethanol or neat (solvent-free) conditions.b Isolated pure yield.c No reaction. | ||||
| 1 | — | EtOH | 90 | —c |
| 2 | MP(DNP) (0.02) | EtOH | 3 | 85 |
| 3 | MP(DNP) (0.05) | EtOH | 3 | 98 |
| 4 | MP(DNP) (0.05) | Neat | 3 | 98 |
| 5 | MP(DNP) (0.05) | H2O | 5 | 92 |
| 6 | MP(DNP) (0.05) | DCM | 120 | 88 |
| 7 | MP(DNP) (0.05) | ACN | 45 | 90 |
| 8 | MP(DNP) (0.1) | EtOH | 3 | 98 |
| 9 | MP(DNP) (0.05) | Neat | 3 | 98 |
| 10 | MP (0.05) | EtOH | 180 | 80 |
| 11 | DNP (0.05) | EtOH | 360 | —c |
| 12 | DNP (0.05) + MP (0.05) | EtOH | 5 | 98 |
| 13 | MP(NP)2 (0.05) | EtOH | 20 | 85 |
| 14 | MP(TNP) (0.05) | EtOH | 15 | 84 |
Therefore, the optimized reaction conditions are found to be 0.05 mmol (5 mol%) of catalyst in ethanol with the reaction time of 3 min at RT. We also carried out the reaction under solvent-free conditions using 0.05 mmol (5 mol%) of catalyst at RT. We found that 98% of the desired product (3aa) is obtained within 3 min in neat conditions making the reaction even more environmentally friendly. We have also examined the yield of the reaction in different solvents. When the reaction is carried out in H2O, 92% of the desired product is obtained using 0.05 mmol (5 mol%) of catalyst, for 3 min at RT (Table 1, entry 5). A slight decrease in the yield is observed in DCM (88%) using 0.05 mmol (5 mol%) of catalyst, and a reaction time of 120 min at RT (Table 1, entry 6). Similarly, a decrease in the yield is also observed when the reaction is carried out in an aprotic polar solvent like acetonitrile (ACN) at RT (Table 1, entry 7).
The role of the individual counter parts of the catalyst (MP(DNP)) towards product formation has been investigated with an aim to understand the detailed reaction mechanism. Therefore, MP and DNP, two components of this organo-catalyst have been used separately as a probable catalyst at otherwise optimized reaction conditions. Interestingly, when MP is used as a catalyst at RT, the reaction takes place but with very long times (∼3 h) and drastically low yields (Table 1, entry 10). On the other hand, no reaction is observed even after 6 h when DNP is used alone (Table 1, entry 11). For finding out the combining effect of MP and DNP in the reaction, the same molar ratio of MP is added to the reaction mixture containing DNP. Interestingly, the reaction is completed within 5 minutes at RT. This clearly indicates that the DNP component actively participates in enhancing the reaction rate and completing the reaction in a short time (Table 1, entry 4 and 12). The experimental results indicate that the in situ generated molecular complex shows very similar catalytic activity to that of the externally added molecular complex (MP(DNP)). This fact is very important considering the reduction of effort in preparing the molecular complex separately for use as a catalyst. Other molecular complexes, (such as MP(NP)2 and MP(TNP)) have also been studied. It is observed that MP(NP)2 and MP(TNP) require longer reaction times and give lower percentage of yields compared to that of MP(DNP) (Table 1, entry 13 and 14). The higher efficiency of DNP containing molecular complexes compared with the other two is due to the relative stability of the nitrophenolate ion in the molecular complex and can be explained considering the pKa values of nitrophenols. The pKa values of NP, DNP and TNP are 7.16, 4.11 and 0.38 respectively.26 The intermediate value for DNP suggests that it efficiently acts as both a proton donor and acceptor depending on the situation. Since Schemes 2 and 3 indicate that the catalyst should preferably have both proton donor and accepting efficiencies depending on the reaction steps, a DNP containing catalyst therefore works best. Further, it is evident that a very weak acid additive such as nitrophenol derivatives and/or its salts (molecular complex: tertiary amine) enhances the electrophilicity of the aldehyde accelerating the dehydration process shown in Scheme 2. As a result, the Knoevenagel condensation reported here is found to take place at much lower temperatures with higher yields than those reported to date. Substituent effects on the yield are also investigated. It is found that excellent yields in all most all cases are observed except for the aldehyde with an electron donating group at the para position (Table 2, entry 6 and 18).
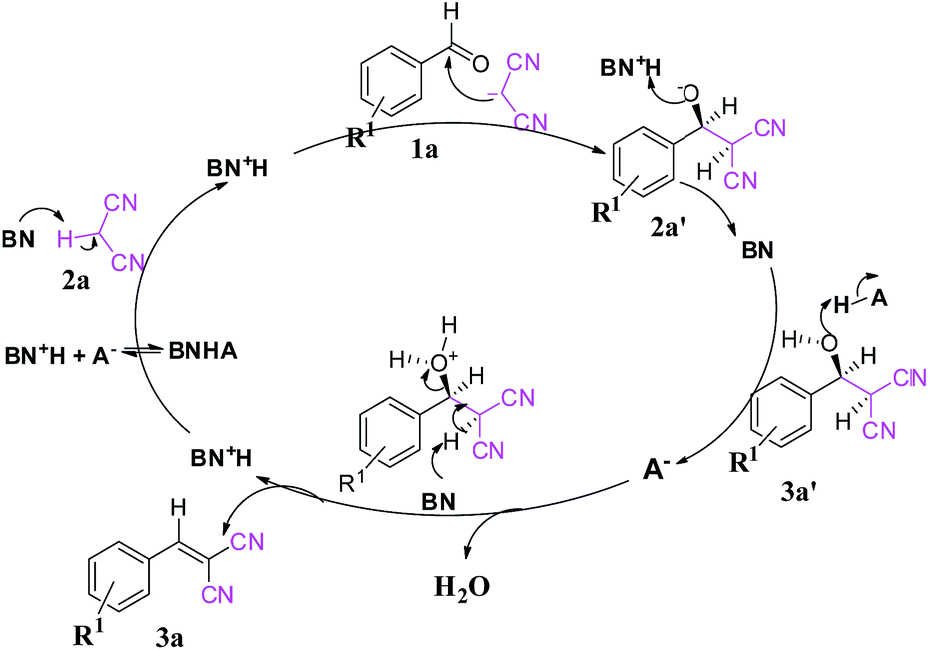 | ||
| Scheme 2 Plausible reaction mechanism of the Knoevenagel condensation. Here, BN represents a proton acceptor (i.e., MP) and HA represents a proton donor (i.e., NP, DNP and TNP). | ||
 | ||
| Scheme 3 Synthesis of 2-amino-4H-chromene derivatives. | ||
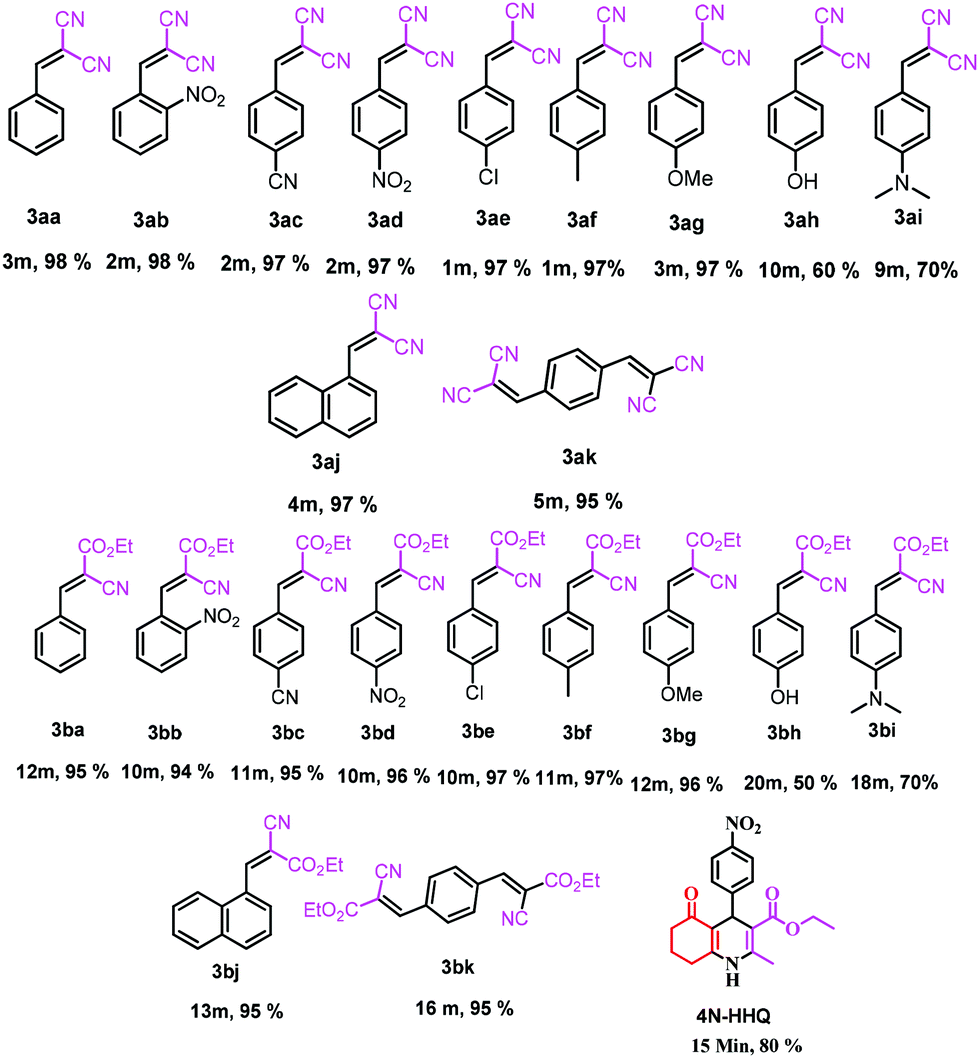 |
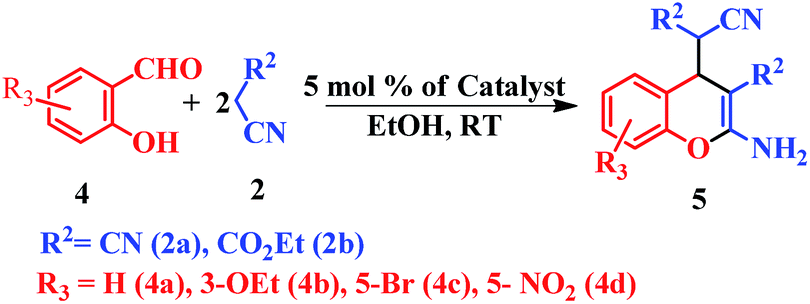
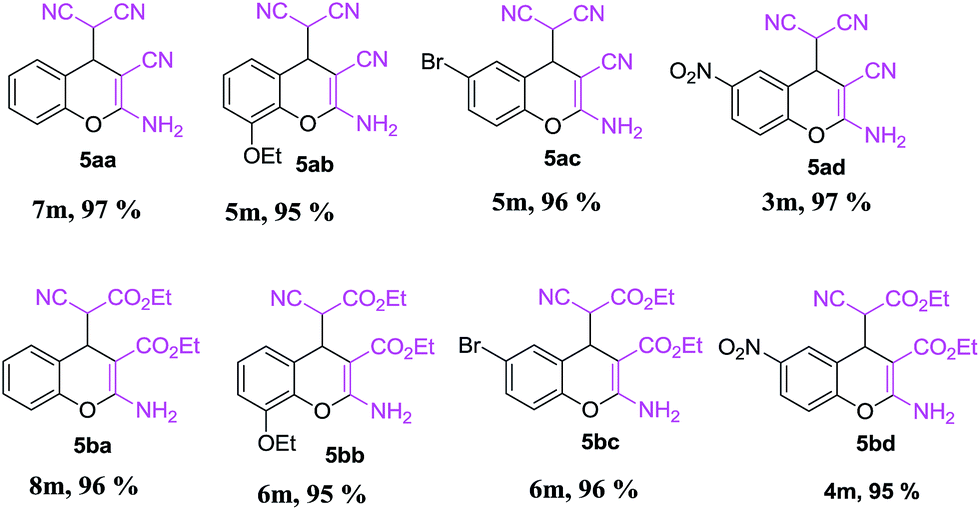
This is due to the increase of electron density at the carbonyl centre and reduction in the formation of the stable intermediate as depicted in 3a′ (Scheme 2). Using the method and optimized reaction conditions that are described in this report, a large number of derivatives have been synthesized and are depicted in Table 2. As a further application of the catalyst (MP(DNP)) and synthone from the Knoevenagel condensation, 2-amino-4H-chromene derivatives were synthesised via the Knoevenagel condensation of salicylic aldehydes with active methylene compounds, followed by intramolecular cyclization (via Michael addition). Scheme 3 shows the reaction in ethanol at RT. The yield of the desired reaction product, 2-amino-4H-chromene (5aa) is quite high (∼97%) using 0.05 mmol (5 mol%) of MP(DNP) as the catalyst in ethanol with the reaction completing within 7 minutes at RT (Table 3, entry 5aa). The Michael addition becomes faster and thereby favours the formation of 2-amino-4H-chromenes at room temperature (Scheme 4).
 | ||
| Scheme 4 Probable reaction mechanism of 2-amino-4H-chromene.Here, BN represents a proton acceptor (i.e., MP) and HA represents a proton donor (i.e., NP, DNP and TNP). | ||
 |
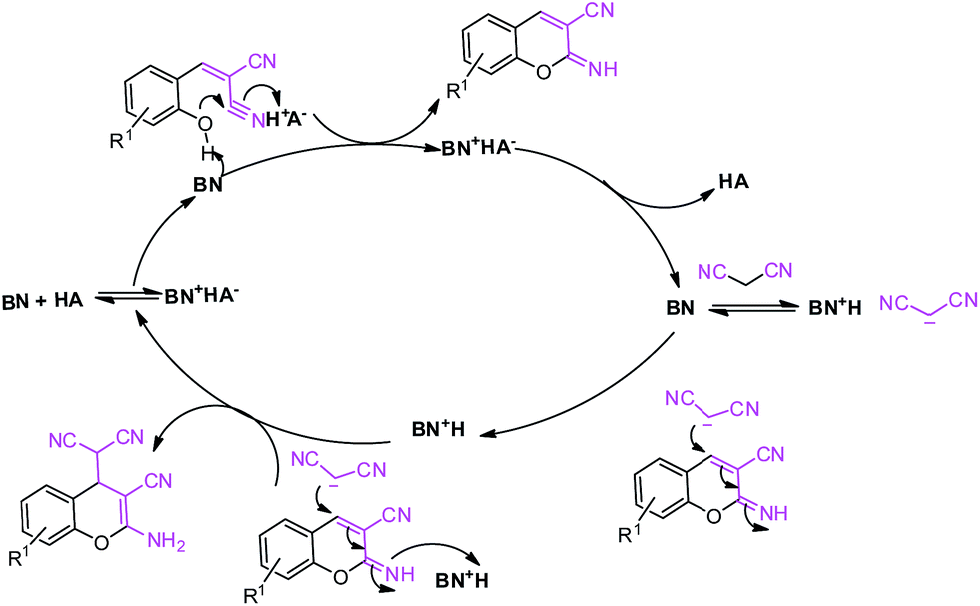
Based on the experimental results, a conceivable mechanism for the Knoevenagel condensation is proposed in Scheme 2. Initially the tertiary amine component (MP, BN) of the catalyst acts as a Lewis base and makes 2a′ the nucleophile form of substrate 2a (pKa ∼ 11.1). Then 2a′ further takes part in the condensation reaction with substrate 1a to provide intermediate 3a′. The intermediate 3a′ is then dehydrated in the presence of the acidic part (HA, DNP) of the catalyst to give us the desired product (3a).
The formation of 2a′enhances the electrophilicity of the aldehyde and the dehydration process, leading to a faster reaction. Further, the creation of 2a′ is controlled by the base (MP), enhancing the electrophilicity of the aldehyde whereas the dehydration process is controlled by acidic protons of the catalyst. The synergetic effect is found to be the key factor for the faster reaction rate.
Separation of the desired product and recovery of the catalyst from the reaction mixture are also important part ofs any catalysed reaction. In this case, the crude reaction mixture is filtered and washed several times with ethanol (3 × 10 mL each time) until the catalyst is completely removed. The solid residue is then collected and dried under vacuum. This is found to provide the pure desired product. Therefore, column chromatographic separation is not necessary. The collected ethanol is evaporated to obtain the pure catalyst. Finally, the product is crystallized from ethanol. The reusability of the catalyst is tested by carrying out repeated runs of the reaction under standard reaction conditions. After each cycle, the catalyst is collected in ethanol and the solvent is evaporated. It is then washed with diethyl ether several times (at least three times) to obtain the pure catalyst for another set of reactions. We tested the recyclability five times and observed that there is no loss of activity and selectivity (Fig. 2).
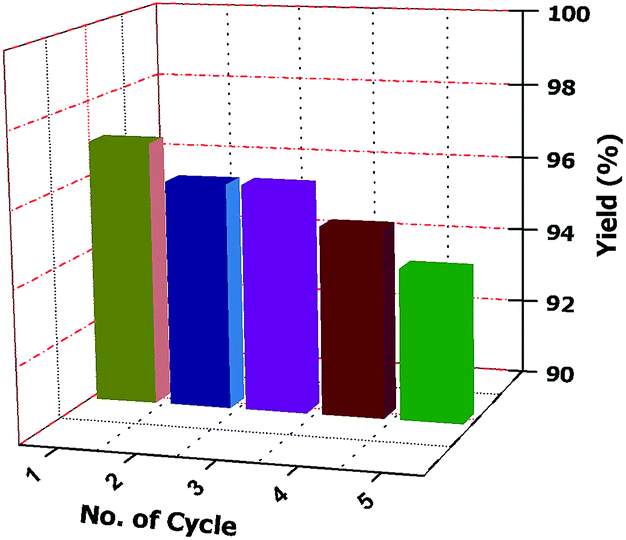 | ||
| Fig. 2 Recyclability chart (recyclability of the catalyst was tested during the reaction under standard reaction conditions). | ||
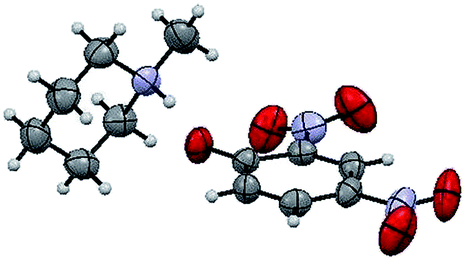
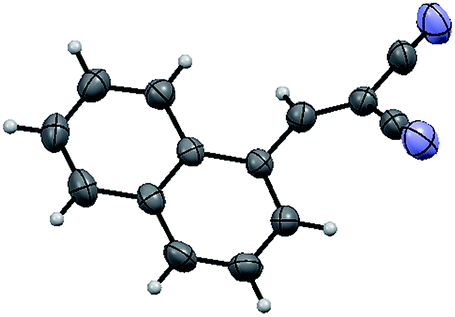
The structure of the catalyst was confirmed beyond doubt by measuring the single crystal X-ray diffraction (SCXRD) as depicted in Fig. 3. In the case of condensation products, the confirmations are established by the detailed analysis of 1H,13C-NMR, FT-IR and ESI-MS spectra. In some cases, the desired products (as in 3aj or 5ba) are further confirmed by SCXRD measurements. The ORTEP diagram obtained from these SCXRD measurements are presented in Fig. 4 and 5. Further hexahydroquinoline synthetic protocols were chosen to test the wider applicability of our molecular complex as an efficient catalyst.27 We successfully obtained the desired product (Table 2, entry 4N-HHQ) with this molecular complex at high yield (80%) within 15 min at room temperature (ESI†).
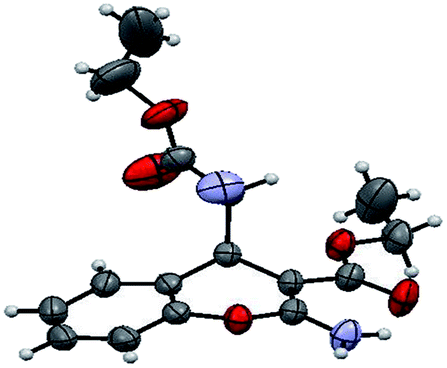 | ||
| Fig. 5 ORTEP diagram of ethyl 2-amino-4-(1-cyano-2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (5ba) (CCDC no. 946632†). | ||
Conclusions
In conclusion, we have reported here some simple molecular complexes which can be used as remarkable organo-catalysts for Knoevenagel condensation and Michael addition reactions. The in situ generation of these molecular complexes works as efficiently as when separately preparing and adding in the complex during the condensation step. Among all of the catalysts, MP(DNP) is found to be the most efficient catalyst. Easily accessible acidic proton of the molecular complexes is the key for the high efficiency and remarkable catalytic activity. Further, detailed mechanistic studies reveal that the electrophilicity of the aldehyde and dehydration process are enhanced in the presence of the acidic proton of the catalyst to make the reaction feasible at RT. Therefore, an eco-friendly, green (in addition to low cost) synthetic protocol has been developed for synthesizing important organic precursors via the Knoevenagal condensation using a highly stable catalyst. This protocol has higher yields and shorter reaction times at room temperature, and uses a minimum amount of catalyst (∼5 mol%) without the need for chromatographic separation. The catalytic activity is not significantly lost after several uses. In a further application of the above procedure, 2-amino-4H-chromene derivatives were synthesized by Knoevenagel condensation of salicylic aldehydes with active methylene compounds, followed by intramolecular cyclization (via Michael addition) delivering higher yields within shorter reaction times at RT without any need for chromatographic separation.Synthesis and characterization of catalyst
N-Methylpiperidine (1.01 mmol) was taken in a 50 mL round bottom flask containing 5 mL CHCl3 and a nitrophenol derivative (1 mmol) was added. Immediately a yellowish coloured liquid was obtained. The resulting yellowish solution was stirred for 1–3 h at room temperature and was kept in a refrigerator. Finally yellow colour crystals were separated out from the solvent (ESI†).General procedure for synthesis of Knoevenagal products
Substituted aromatic aldehyde 1 (1 mmol), active methylene 2 (1.01 mmol), and 0.05 mmol of catalyst (MP(DNP)) were taken in a round bottom flask and stirred at room temperature for a stipulated period of time (Table 2) until the completion of the reaction (monitored using TLC). After completion of the reaction, a solid precipitate was separated out, filtered and washed with water (3 × 10 mL) followed by ethanol (3 × 10 mL), and dried under high vacuum to afford an NMR pure product.General procedure for synthesis of 2-amino-4H-chromene derivatives
Substituted salicylic aldehyde (1 mmol, 4), malononitrile (2.01 mmol, 2) and 0.05 mmol of catalyst (MP(DNP)) were taken in ethanol solvent at room temperature for a stipulated period of time (Table 3) until the completion of the reaction (monitored using TLC). After completion of the reaction, a solid precipitate was separated out which was filtered and washed with water (3 × 10 mL) followed by ethanol (3 × 10 mL) and dried under high vacuum to afford an NMR pure product.Note: in case the final product does not separate from the reaction mixture, cold water needs to be added after the completion of the reaction with vigorous shaking. The solid precipitate is then filtered and washed with cold water several times (3 × 10 mL) followed by ethanol (3 × 10 mL) and dried under high vacuum to afford an NMR pure product.
General procedure for synthesis of hexahydroquinolines
A mixture of aldehyde (1 mmol), dimedone (1 mmol), ethyl acetoacetate (1 mmol), ammonium acetate (2.5 mmol) and MP(DNP) containing (5 mol%) as the catalyst were stirred at room temperature for 15 min in ethanol. After completion of the reaction (monitored using TLC), ethanol wass evaporated and 3 mL of water was added to the mixture. A solid product was obtained and washed several times with water (at least four times). For recycling of the catalyst, after washing the solid product with water, the water containing the molecular complex (MP(DNP)) was evaporated under reduced pressure and the molecular complex (catalyst, MP(DNP)) was recovered and reused after washing with diethylether (∼three times). The solid product was purified by recrystallization from ethanol.Acknowledgements
We thank the CSIR (01(2792)/14/EMR-11), New Delhi and HEMRL-DRDO (Pune), Govt. of India, India for the research grants and fellowship awarded to SKP. The department of Chemistry, BHU is also gratefully acknowledged for providing instruments (including SCXRD) and infrastructure facilities.Notes and references
-
(a) S. L. Schreiber, Science, 2000, 287, 1964 CrossRef CAS
; (b) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS PubMed
-
(a) I. Ugi, Pure Appl. Chem., 2001, 73, 187 CrossRef CAS
-
(a) E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1898, 31, 2585 CrossRef CAS PubMed
-
(a) A. Graul and J. Castaner, Drugs Future, 1997, 22, 956 CrossRef CAS
-
(a) P. Formentin, H. Garcia and A. Leyva, J. Mol. Catal. A: Chem., 2004, 214, 137 CrossRef CAS PubMed
- Y. Goa, P. Wu and T. Tatsumi, J. Catal., 2004, 224, 107 CrossRef CAS PubMed
-
(a) B. C. Ranu and R. Jana, Eur. J. Org. Chem., 2006, 3767 CrossRef CAS PubMed
-
(a) A. Lee, A. Michrowska, S. Sulzer-Mosse and B. List, Angew. Chem., Int. Ed., 2011, 50, 1707 CrossRef CAS PubMed
-
(a) A. Dandia, V. Parewa, A. K. Jain and K. S. Rathore, Green Chem., 2011, 13, 2135 RSC
- D. Fildes, V. Caignaert, D. Villemin and P.-A. Jaffres, Green Chem., 2001, 3, 52 RSC
- T. I. Reddy and R. S. Varma, Tetrahedron Lett., 1997, 38, 1721 CrossRef CAS
-
(a) V. S. R. R. Pullabhotla, A. Rahman and S. B. Jonnalagadda, Catal. Commun., 2009, 10, 365 CrossRef CAS PubMed
-
(a) J. Bennazha, M. Zohouily, S. Sebti, A. Boukhari and E. M. Holt, Catal. Commun., 2001, 2, 101 CrossRef CAS
-
(a) M. M. Khafagy, A. H. F. A. El-Wahas, F. A. Eid and A. M. El-Agrody, Farmaco, 2002, 57, 715 CrossRef CAS
-
(a) H. Miao and Z. Yang, Org. Lett., 2000, 2, 1765 CrossRef CAS PubMed
- M. Curini, G. Cravotto, F. Epifano and G. Giannone, Curr. Med. Chem., 2006, 13, 199 CrossRef CAS
- F. Borges, F. Roleira, N. Milhazes, L. Santana and E. Uriarte, Curr. Med. Chem., 2005, 12, 887 CrossRef CAS
- J. G. Tangmouo, A. L. Meli, J. Komguem, V. Kuete, F. N. Ngounou, D. Lontsi, V. P. Beng, M. I. Choudhary and B. L. Sondengam, Tetrahedron Lett., 2006, 47, 3067 CrossRef CAS PubMed
- G. A. Kraus and I. Kim, J. Org. Chem., 2003, 68, 4517 CrossRef CAS PubMed
- G. P. Ellis, The Chemistry of Heterocyclic Compounds, Chromenes, Chromanes, and Chromones, ed. A. Weissberger and E. C. Taylor, John Wiley, New York, 1977, ch. II, pp. 11–141 Search PubMed
- E. A. Hafez, M. H. Elnagdi, A. G. A. Elagamey and F. M. A. A. El-Taweel, Heterocycles, 1987, 26, 903 CrossRef CAS
-
(a) J. Adams and S. Cory, Science, 1998, 281, 1322 CrossRef CAS
- A. Ying, Y. Ni, S. Xu, S. Liu, J. Yang and R. Li, Ind. Eng. Chem. Res., 2014, 53(14), 5678 CrossRef CAS
- A. Rahmati and K. Vakili, Amino Acids, 2010, 39, 911 CrossRef CAS PubMed
- N. Mase and T. Horibe, Org. Lett., 2013, 15, 1854 CrossRef CAS PubMed
-
(a) M. D. Liptak, K. C. Gross, P. G. Seybold, S. Feldgus and G. C. Shields, J. Am. Chem. Soc., 2002, 124, 6421 CrossRef CAS PubMed
- M. Ghorbani, S. Noura, M. Oftadeh, E. gholamia and M. A. Zolfigol, RSC Adv., 2015, 5, 55303–55312 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedure, separation techniques, electronic spectra, NMR spectra of all compounds. CCDC: 873550, 937875 and 946632. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra09036a |
| This journal is © The Royal Society of Chemistry 2015 |