
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crosslinked polymeric bis(triphenylphosphine)iminium chloride gels as recyclable catalysts
Ziwei
Xu
ab and
Michael P.
Shaver
 *ab
*ab
aDepartment of Materials, School of Natural Sciences, University of Manchester, Manchester, M1 3BB, UK. E-mail: michael.shaver@manchester.ac.uk
bSustainable Materials Innovation Hub, Henry Royce Institute, University of Manchester, Manchester, M13 9BL, UK
First published on 9th December 2025
Abstract
Bis(triphenylphosphine)iminium chloride, PPNCl, is an increasingly important catalyst due to its broad reactivity in both small molecule and polymer synthesis. A recently reported polymeric variant, poly(PPNCl), offers limited recyclability in small molecule synthesis, but its soluble, thermoplastic nature complicates its recovery and reuse. To address this limitation, we developed a crosslinked poly(PPNCl) thermoset that dramatically improves performance and reaction scope. As a catalyst in CO2/epoxide coupling reactions the crosslinked derivative is faster and more readily recycled. In addition, the reaction scope can be extended to polymer synthesis, with soluble polyesters formed from coupling epoxides with maleic anhydrides isolated by filtration, with activity retained over multiple reaction cycles. Remarkably, the reaction scope can be further extended by anion exchange, with crosslinked poly(PPNCo(CO)4) used to catalyse the carbonylative ring expansion of epoxides to β-lactones. Together these show that crosslinked poly(PPNCl) is a promising platform for metal-free, robust, recyclable catalysis.
Introduction
Catalysts are essential enablers of sustainable chemical synthesis. Metal-free catalysts are of growing importance, with reduced cost, footprint and toxicity sparking significant academic and industrial interest.1–4 Bis(triphenyl-phosphine)iminium (PPN) salts are a flourishing sub-class of catalysts, particularly notable for their excellent solubility, stability, reaction scope and ease of use.5–9 The chloride derivative, PPNCl, has become a valuable catalyst, particularly for CO2 coupling to form small molecules10–12 and synthesis of degradable polyesters from the copolymerisation of cyclic anhydrides and epoxides.13–17However, the economic and environmental cost of the complex PPN cation limits sustainability assurances. Previous work in our group reported a polymeric bis(triphenylphosphine)iminium chloride (poly(PPNCl)) variant as a catalyst for multiple small molecule transformations; precipitation allowed for catalyst recovery and reuse (Fig. 1a).18 Our efforts to extend the use of poly(PPNCl) to polymer synthesis, such as phthalic anhydride and cyclohexene oxide copolymerisation, highlighted significant limitations, with their analogous solubilities preventing separation and recycling. The use of dissolution and precipitation consistently led to product loss, meaning that even with good activity retention the system lacked long term sustainability.
 | ||
| Fig. 1 Comparing previously reported linear poly(PPNCl) with crosslinked poly(PPNCl) systems enabling recyclable catalysis of epoxides reacting through CO2 insertions, copolymerisations with anhydrides, and CO insertions. | ||
Crosslinked polymer-supported catalysts, with the active catalytic species immobilized onto a crosslinked polymer matrix, are solid supports reacting with gases or liquids.19–21 Compared to soluble polymer-supported catalysts, they offer easy separation from the reaction mixture through filtration or in flow, offering future opportunities to manufacture in large-scale reactors and with continuous production.22–24 These catalysts can offer improved thermal and mechanical stability and anti-solvent free recyclability while maintaining good catalytic efficiency.25 Owing to these advantages, they have been explored widely, in diverse reactions, suggesting a crucial role in future sustainable manufacturing processes.26–28 Together, this suggested the need to shift topology, framing the new target of a crosslinked poly(PPNCl) that would swell but not dissolve to facilitate recovery.
In this work, we report the preparation of a crosslinked poly(PPNCl) (Fig. 1b). A comparison of the catalytic reactivity between soluble linear catalysts and crosslinked poly(PPNCl) gels showed increased catalytic activity and improved recyclability for both small molecule and, for the first time, polymeric syntheses. We further used the robustness of these gels to support anion exchange to synthesise a poly(PPNCo(CO)4) variant, inspired by a rare report of a small molecule derivative functioning in CO coupling reactions in conjunction with a Lewis acid.29 Indeed, we were able to show that crosslinked poly(PPNCo(CO)4) is an effective catalyst for the carbonylative ring expansion of epoxides to β-lactones.
Results and discussion
Synthesis of crosslinked poly(PPNCl)
A crosslinked precursor, P1, was formed via radical copolymerisation of styrene, St, p-(diphenylphosphino)styrene, StPPh2 and divinyl benzene (DVB), as shown in Scheme 1a. After optimisation, a feed ratio of 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:1 of St, StPPh2 and DVB afforded a stiff gel. 31P NMR spectroscopy of the swollen gel still afforded good resolution, with the crude product showing a narrow resonance at −5.8 ppm and a broad resonance at −6.1 ppm, associated with unpolymerised StPPh2 monomers and the incorporated PPh3 anchors, respectively (Fig. S1). Conversion of StPPh2 was 98% from integrating these resonances. Immersion of P1 in several aliquots of toluene for 4 h facilitated the extraction of residual monomers, offering pure P1 which was subsequently dried in vacuo and manually ground into a fine powder.
:5:1 of St, StPPh2 and DVB afforded a stiff gel. 31P NMR spectroscopy of the swollen gel still afforded good resolution, with the crude product showing a narrow resonance at −5.8 ppm and a broad resonance at −6.1 ppm, associated with unpolymerised StPPh2 monomers and the incorporated PPh3 anchors, respectively (Fig. S1). Conversion of StPPh2 was 98% from integrating these resonances. Immersion of P1 in several aliquots of toluene for 4 h facilitated the extraction of residual monomers, offering pure P1 which was subsequently dried in vacuo and manually ground into a fine powder.
 | ||
| Scheme 1 (a) Synthetic procedures of the crosslinked precursor P2 from St, StPPh2 and the crosslinker DVB with a feed ratio of 95:5:1; (i) AIBN, toluene, 70 °C, 16 h and (ii) C2Cl6, THF, r.t., 16 h. (b) Synthesis of Ph3PNLi from triphenylphosphine; (iii) hydroxylamine-O-sulfonic acid, methanol/DCM, r.t., 1 h; (iv) n-butyllithium, THF, initiated at −50 °C, −15 °C 2 h. (c) Synthesis of P3: (v) THF, r.t., 16 h. | ||
Post-polymerisation of this thermoset polymer was accomplished by modifying the methodology used to prepare poly(PPNCl).18 Dichlorination of P1 with C2Cl6 accessed the phosphorus(V) StPPh2Cl2 crosslinked gel P2 (Scheme 1a). A broad resonance at 63.5 ppm was observed in the 31P NMR spectrum (Fig. S2), comparable to that of its small molecule analogue (65.0 ppm). No signal associated with P1 was observed, suggesting full conversion. To remove any remaining C2Cl6, the crude product was immersed in aliquots of THF for 4 h, the solvent was decanted, and the product was dried in vacuo and manually ground into a fine powder.
Finally, the targeted crosslinked poly(PPNCl), P3, was synthesised by reaction between a swollen P2 gel and Ph3PNLi (Scheme 1c). A broad peak at 20.7 ppm was attributed to the crosslinked poly(PPNCl) functionalities (Fig. S3), with the disappearance of the P2 resonance again suggesting full conversion. Small signals at 29.3 ppm, suggesting phosphine oxidation, and 35.6 ppm, attributed to the residual Ph3PNH2Cl (35.6 ppm), were observed. Purification of P3 by dialysis removed the majority of small molecule impurities; however ca. 6% of partially oxidised phosphine remained in the product (Fig. S4). As this phosphine oxide was not expected to interfere with catalyst performance, P3 was used without further purification. Importantly, no progression in the extent of oxidation was observed through catalyst use or reuse.
CO2/GC coupling reaction
Crosslinked catalysts often depend on swelling to promote catalytic activity, improving the accessibility of reactants to active sites within the polymer network. While linear poly(PPNCl) showed good catalytic reactivity for the insertion of CO2 to epoxides,18 we wished to assess the comparative catalytic reactivity of the crosslinked variant, P3, in the coupling reaction between CO2 and glycidyl chloride (GC) to yield cyclic carbonates (Scheme 2).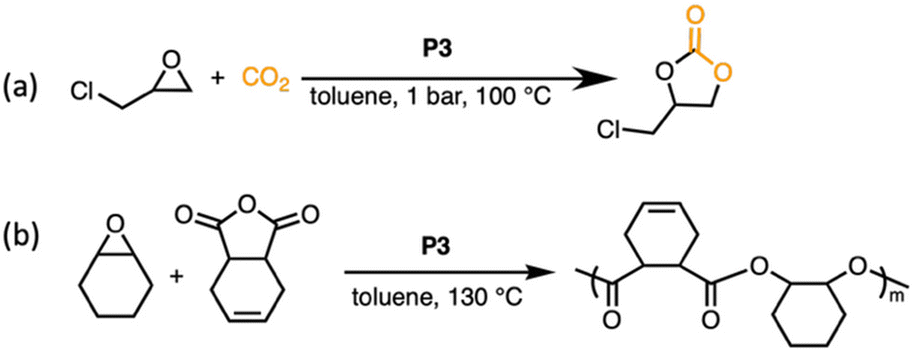 | ||
| Scheme 2 (a) CO2/GC coupling reaction catalysed by crosslinked poly(PPNCl). General reaction conditions: reactions were carried out in a sealed flask on a 12.8 mmol scale of GC and 0.4 mol% crosslinked poly(PPNCl) (mol% as calculated from the number of PPNCl moieties) in 1 mL toluene at 100 °C and 1 bar. (b) Ring-opening polymerisation catalysed by crosslinked poly(PPNCl). General reaction conditions: polymerisation was conducted in toluene at 130 °C. CHO:CDA:PPNCl group = 200:100:4. | ||
Under previously optimised conditions using linear poly(PPNCl), the conversion of GC to chloropropene carbonate reached up to 98% in neat reactions.18 With the same % functional loading (0.4%), P3 was employed for the CO2/GC coupling reaction. Given that the buried catalytic sites of crosslinked poly(PPNCl) become more accessible in the swollen state, toluene was introduced to assist in swelling the gel, despite decreasing effective catalyst concentration. The results are summarized in Table S1. As a control experiment, the reaction catalysed by linear poly(PPNCl) in toluene gave 97% conversion of GC (entry 2, Table S1). Notably, when switching to crosslinked poly(PPNCl), full conversion of GC to chloropropene carbonate was achieved (entry 3, Table S1), displaying comparable catalytic reactivity to that of its linear analogue poly(PPNCl). The porous structure of the swollen network allows both CO2 and GC to diffuse through the network and readily access catalytic sites embedded within the gel. No polyether formation was observed in the 1H NMR spectrum (Fig. S5), indicating high selectivity for cyclic carbonate formation.
Exploring the limits of this new crosslinked system for CO2/GC coupling, we reduced the reaction times (Table S2). Only at 4 h (95%) or 2 h (73%) did conversion not reach 100%. Compared to its linear counterpart, the surprising increase in activity for the crosslinked variant suggests more than simply a retention of the number of active sites in the cross-linked system. The access of reactants to catalytic sites is potentially improved due to compartmentalisation effects that minimise diffusion distances for substrates. Solvent choice can also improve CO2 solubility in active pockets. Indeed, reactions using the more polar anisole to swell the crosslinked gel gave lower conversions (66%), suggesting that CO2 solubility may be an important factor.
Catalyst recyclability
Catalyst recyclability was evaluated at both 4 h and 24 h to both compare with those from linear polymer recycling studies and to assess performance below full conversion limits. Recycling studies were conducted in air. After each cycle, gels were washed with three aliquots of diethyl ether to extract cyclic carbonate products. The gels were dried in vacuo between catalytic runs. As illustrated in Fig. 2, the catalytic reactivity of P3 decreases minimally over three reaction cycles (1st cycle: 95%; 2nd: 90%; 3rd: 88%) with a fixed reaction time of 4 h. No phosphine oxidation occurred after three reaction cycles as demonstrated by 31P NMR spectroscopy (Fig. S6). In contrast, when the reaction time was extended to 24 h, a well-maintained conversion of GC was observed over three cycles, achieving complete transformation into chloropropene carbonate in each run. This suggests no decomposition of the catalytic sites, but reduced access to these sites in the gels after repeated drying and swelling cycles; indeed swelling appeared to be qualitatively slower in repeatedly dried systems. The recycling process was much more straightforward than those with previous systems, suggesting future application in flow may be possible. The consistency of the catalytic reactivity of crosslinked poly(PPNCl) over prolonged reaction times (24 h) supports robust stability for longer term manufacturing. | ||
| Fig. 2 Conversions of GC in the CO2/GC coupling reaction catalysed by cross-linked poly(PPNCl) with reaction times of 4 h and 24 h over three reaction cycles. Conversion was calculated by 1H NMR spectroscopy. | ||
Epoxide anhydride copolymerisation
Poly(PPNCl) was unable to facilitate productive polymerisation or copolymerisation reactions due to separation challenges (Table S3). While P3 slightly improves catalyst performance in small molecule coupling reactions, we believe that this new topology would offer an extended reaction scope. Indeed, the system exhibits high reactivity in the ring-opening copolymerisation of cyclohexene oxide (CHO) and cis-4-cyclohexene-1,2-dicarboxylic anhydride (CDA) (Scheme 2b and Table S3).A near-quantitative conversion of CHO:CDA into the desired polymer was observed after an initial catalyst screen (24 h, entry 2, Table S3), matching previous reports of the small molecule PPNCl serving as a standalone catalyst in the ring-opening polymerisation of CHO and anhydrides.17,30 Reducing reaction times to assess catalyst performance showed that full conversion of CDA was achieved in as little as 2 h (Table S3). The polymerisation was monitored over the course of 2 h by 1H NMR spectroscopy (Fig. S7). The reaction showed first order kinetics over the limited time range, with k = 0.26 h−1 (Fig. S8). Notably, at the latter stage of the polymerisation, when nearly all CDA was consumed, new resonances in the region of 3.5 ppm were observed, with these ether linkages suggesting homopolymerisation in the absence of a comonomer.
To assess the recyclability of P3 in this copolymerisation, reaction times of 1 h and 24 h were selected (Fig. 3), with the former assessing activity below full conversion and the latter assessing long term robustness. As with small molecule transformations, catalysts were washed, dried and reswollen between catalytic runs. CDA conversion decreased slightly over three reaction cycles (1st cycle: 93%; 2nd: 85%; and 3rd: 80%) while consistent conversions (1st cycle: 99%; 2nd: 99%; and 3rd: 99%) were observed in longer reactions, suggesting excellent recyclability and robustness.
 | ||
| Fig. 3 Conversion of CDA in the ring-opening polymerisation of CHO and CDA catalysed by crosslinked poly(PPNCl) with reaction times of 4 h and 24 h over three reaction cycles. Conversion was calculated by 1H NMR spectroscopy. | ||
Carbonylative ring expansion of epoxides to β-lactones
Finally, we exploited the robustness of these gels to facilitate anion exchange to further increase reaction scope. The carbonylative ring expansion of epoxides forms valuable β-lactone intermediates, and has recently attracted significant attention,31–33 often relying on complex bimetallic catalytic systems incorporating a Lewis acid and Co(CO)4 salt.34–36 We hypothesized that P3 could be developed into a unique carbonylation catalyst. Thus, crosslinked poly(PPNCo(CO)4), P4 (Fig. 4), was prepared through an anion exchange reaction between P3 and NaCo(CO)4 with conditions based on unpublished optimisation of the linear system.29 | ||
| Fig. 4 A two-step synthetic method to synthesise crosslinked poly(PPNCo(CO)4) and its structure. | ||
Building from our previous work on polymeric frustrated Lewis pairs,29,37–41 we explored the catalytic activity of P4 in conjunction with our poly(BPh3) Lewis acid, with the carbonylation of propylene oxide (PO) as a substrate. As observed in other systems,36,42–44 the solvent significantly influenced both selectivity and reaction rates. Highly coordinating solvents competed with binding sites and slowed ring-opening, while non-coordinating solvents favoured polyether formation. The weakly coordinating solvent diphenyl ether, DPE, gave optimal results (Fig. 5). Importantly, the reaction was productive at much lower pressures (12 bar) than those previously reported for many systems, including a report of immobilized Co(CO)x onto reduced carbon nanotubes (60 bar).45
 | ||
| Fig. 5 Carbonylation reaction of PO catalysed by P4/poly(BPh3). General reaction conditions: reactions were performed in a high-pressure reactor with 1 mol% crosslinked poly(PPNCo(CO)4) and 1 mol% poly(BPh3) (mol% as calculated from the number of functional groups) in 0.9 mL diphenyl ether (DPE) at 50 °C with a CO pressure of 12 bar. | ||
PO conversion to β-butyrolactone, BBL, was 77% as determined by 1H NMR spectroscopy (Fig. S9 and Table S4), suggesting that crosslinked poly(PPNCo(CO)4)/poly(BPh3) is an unusually active catalyst combination. The catalytic system had high selectivity for β-lactone over polymer formation, as no polyether is observed. The formation of the off-cycle by-product acetone was observed. Varying reaction times changed selectivity for the lactone over acetone (Table S4) resulting in lower BBL yields at extended reaction times (Fig. S10 and S11). Preliminary ICP-OES measurements suggest no leaching of cobalt from catalytic reactions from the analysis of the supernatant, although more work is needed to ensure sample preparation does not lead to false negative values. Based on the above results, the crosslinked poly(PPNCo(CO)4)/poly(BPh3) catalytic system is a promising candidate for more sustainable carbonylation reactions and remains an active area of future research efforts.
Conclusions
In summary, crosslinked poly(PPNCl) was prepared for the first time through post-polymerisation modification. Its utility was first demonstrated in the CO2/GC coupling reaction, where it surprisingly exhibited superior catalytic efficiency compared to its linear analogue poly(PPNCl), producing chloropropene carbonate. Furthermore, the crosslinked polymer demonstrated excellent recyclability across multiple reaction cycles. Leveraging this robustness, crosslinked poly(PPNCl) was subsequently employed as a sole catalyst for the ring-opening polymerisation of CHO and CDA. It achieved quantitative incorporation of CDA into the polyester backbone within 2 hours at 130 °C using only 4 mol% catalyst. Crucially, the crosslinked catalyst could be easily separated via centrifugation and maintained high reactivity over three consecutive cycles. Finally, to showcase its synthetic versatility, crosslinked poly(PPNCl) was transformed into crosslinked poly(PPNCo(CO)4). This derived catalyst successfully facilitated the formation of β-butyrolactone from PO and CO under mild conditions (50 °C, 12 bar CO), achieving 73.7% conversion in the presence of linear poly(BPh3). These results confirm crosslinked poly(PPNCl) as a versatile and robust metal-free catalytic platform with excellent recyclability.Author contributions
Ziwei Xu: investigation, methodology and manuscript writing. Michael P. Shaver: conceptualisation, manuscript writing – review and editing, supervision and funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details, additional characterisation data, and catalyst optimisation. See DOI: https://doi.org/10.1039/d5py01143d.Acknowledgements
We kindly thank the University of Manchester (President's Doctoral Scholar Award for Z. X.), the Leverhulme Trust (81420), and equipment funded through the Sustainable Materials Innovation Hub (European Regional Development Fund OC15R19P) and Henry Royce Institute (EP/X527257/1) for financial support.References
- Y.-C. Yang and D. E. Bergbreiter, Soluble polymer-supported organocatalysts, Pure Appl. Chem., 2012, 85, 493–509 CrossRef.
- X. Liu and L. Dai, Carbon-based metal-free catalysts, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed.
- E. Poursaitidis, P. L. Gkizis, I. Triantafillidi and C. G. Kokotos, Organocatalytic activation of hydrogen peroxide: towards green and sustainable oxidations, Chem. Sci., 2024, 15, 1177–1203 RSC.
- K. A. Ahrendt, C. J. Borths and D. W. MacMillan, New strategies for organic catalysis: the first highly enantioselective organocatalytic Diels− Alder reaction, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- T. W. Rave, Preparation and properties of bis (phosphoranylidene) ammonium chlorides, J. Org. Chem., 1967, 32, 3461–3466 CrossRef CAS.
- L. Mann, E. Hornberger, S. Steinhauer and S. Riedel, Further development of weakly coordinating cations: fluorinated bis (triarylphosphoranylidene) iminium salts, Chem. – Eur. J., 2018, 24, 3902–3908 CrossRef CAS PubMed.
- F. Lalor and S. Chaona, An economical small-scale synthesis of the μ-nitridobis (triphenylphosphonium) cation,[PPN]+, J. Organomet. Chem., 1988, 344, 163–165 CrossRef CAS.
- J. Ruff and W.J Schlientz, Inorg. Synth., 1974, 16, 84 CrossRef.
- M. A. Lacour, M. Zablocka, C. Duhayon, J. P. Majoral and M. Taillefer, Efficient Phosphorus Catalysts for the Halogen–Exchange (Halex) Reaction, Adv. Synth. Catal., 2008, 350, 2677–2682 CrossRef CAS.
- C. Knapp and R. Uzun, Solvate-free bis (triphenylphosphine) iminium chloride, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o3185 CrossRef CAS PubMed.
- D. Xie, Z. Yang, L. Wu, C. Zhang and M. H. Chisholm, One–pot regioselective and stereoselective terpolymerization of rac–lactide, CO2 and rac–propylene oxide with TPPMCl (M = Cr, Co, Al)/PPNCl binary catalyst, Polym. Int., 2018, 67, 883–893 CrossRef CAS.
- J. Ruff, W. Schlientz, R. Dessy, J. Malm, G. Dobson and M. Memering, µ-Nitridobis (triphenylphosphorus)(l+)(“PPN”) Salts with Metal Carbonyl Anions, Inorg. Synth., 1974, 15, 84–90 CrossRef CAS.
- O. Nuyken and S. D. Pask, Ring-opening polymerization—an introductory review, Polymers, 2013, 5, 361–403 CrossRef.
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Kinetic studies of the alternating copolymerization of cyclic acid anhydrides and epoxides, and the terpolymerization of cyclic acid anhydrides, epoxides, and CO2 catalyzed by (salen) CrIIICl, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- S. Penczek, J. Pretula and S. Slomkowski, Ring-opening polymerization, Chem. Teach. Int., 2021, 3, 33–57 CrossRef.
- R. Mundil, Z. Host'alek, I. Sedenkova and J. Merna, Alternating ring-opening copolymerization of cyclohexene oxide with phthalic anhydride catalyzed by iron(III) salen complexes, Macromol. Res., 2015, 23, 161–166 CrossRef CAS.
- B. Han, L. Zhang, B. Liu, X. Dong, I. Kim, Z. Duan and P. Theato, Controllable Synthesis of Stereoregular Polyesters by Organocatalytic Alternating Copolymerizations of Cyclohexene Oxide and Norbornene Anhydrides, Macromolecules, 2015, 48, 3431–3437 CrossRef CAS.
- Z. Xu, M. Wang and M. P. Shaver, Polymeric bis(triphenylphosphine)iminium chloride as a recyclable catalyst, Chem. Sci., 2024, 15, 15745–15750 RSC.
- Z. Xie, C. Wang, K. E. deKrafft and W. Lin, Highly stable and porous cross-linked polymers for efficient photocatalysis, J. Am. Chem. Soc., 2011, 133, 2056–2059 CrossRef CAS PubMed.
- M. Sultan, A. Javeed, M. Uroos, M. Imran, F. Jubeen, S. Nouren, N. Saleem, I. Bibi, R. Masood and W. Ahmed, Linear and crosslinked Polyurethanes based catalysts for reduction of methylene blue, J. Hazard. Mater., 2018, 344, 210–219 CrossRef CAS PubMed.
- B. Sharma and S. Striegler, Crosslinked microgels as platform for hydrolytic catalysts, Biomacromolecules, 2018, 19, 1164–1174 Search PubMed.
- M. Campanati, G. Fornasari and A. Vaccari, Fundamentals in the preparation of heterogeneous catalysts, Catal. Today, 2003, 77, 299–314 CrossRef CAS.
- G. J. Hutchings, Heterogeneous catalysts—discovery and design, J. Mater. Chem., 2009, 19, 1222–1235 RSC.
- I. Fechete, Y. Wang and J. C. Védrine, The past, present and future of heterogeneous catalysis, Catal. Today, 2012, 189, 2–27 CrossRef CAS.
- G. K. Boreskov, Heterogeneous catalysis, Nova Publishers, 2003 Search PubMed.
- C. E. Hobbs, Recoverable Polymer-supported DMAP Derivatives, 2017 Search PubMed.
- M. Benaglia, A. Puglisi and F. Cozzi, Polymer-supported organic catalysts, Chem. Rev., 2003, 103, 3401–3430 Search PubMed.
- B. Clapham, T. S. Reger and K. D. Janda, Polymer-supported catalysis in synthetic organic chemistry, Tetrahedron, 2001, 57, 4637–4662 CrossRef CAS.
- T. A. Horton, Polymeric Frustrated Lewis Pairs as Catalytic Systems and Stimuli-Responsive Materials, The University of Manchester, United Kingdom, 2022 Search PubMed.
- Z. Hošťálek, O. Trhlikova, Z. Walterová, T. Martinez, F. Peruch, H. Cramail and J. Merna, Alternating copolymerization of epoxides with anhydrides initiated by organic bases, Eur. Polym. J., 2017, 88, 433–447 CrossRef.
- F. Molnar, G. A. Luinstra, M. Allmendinger and B. Rieger, Multisite Catalysis: A Mechanistic Study of β–Lactone Synthesis from Epoxides and CO—Insights into a Difficult Case of Homogeneous Catalysis, Chem. – Eur. J., 2003, 9, 1273–1280 CrossRef CAS PubMed.
- J. W. Kramer, D. Y. Joh and G. W. Coates, Carbonylation of Epoxides to Substituted 3-Hydroxy-δ-Lactones, Org. Lett., 2007, 9, 5581–5583 CrossRef CAS.
- S. Rajendiran, V. Ganesan and S. Yoon, Balancing between Heterogeneity and Reactivity in Porphyrin Chromium-Cobaltate Catalyzed Ring Expansion Carbonylation of Epoxide into β-Lactone, Inorg. Chem., 2019, 58, 3283–3289 CrossRef CAS PubMed.
- Y. D. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, Synthesis of β-lactones: a highly active and selective catalyst for epoxide carbonylation, J. Am. Chem. Soc., 2002, 124, 1174–1175 CrossRef CAS.
- J. W. Kramer, E. B. Lobkovsky and G. W. Coates, Practical β-lactone synthesis: epoxide carbonylation at 1 atm, Org. Lett., 2006, 8, 3709–3712 CrossRef CAS PubMed.
- T. L. Church, Y. D. Getzler and G. W. Coates, The mechanism of epoxide carbonylation by [Lewis Acid] + [Co (CO) 4]-catalysts, J. Am. Chem. Soc., 2006, 128, 10125–10133 CrossRef CAS.
- M. Wang, F. Nudelman, R. R. Matthes and M. P. Shaver, Frustrated Lewis pair polymers as responsive self-healing gels, J. Am. Chem. Soc., 2017, 139, 14232–14236 CrossRef CAS.
- U. Yolsal, T. A. Horton, M. Wang and M. P. Shaver, Cyclic ether triggers for polymeric frustrated Lewis pair gels, J. Am. Chem. Soc., 2021, 143, 12980–12984 CrossRef CAS.
- T. A. Horton, M. Wang and M. P. Shaver, Polymeric frustrated Lewis pairs in CO 2/cyclic ether coupling catalysis, Chem. Sci., 2022, 13, 3845–3850 RSC.
- J. Holland and M. P. Shaver, Polymeric N-heterocyclic carbenes in frustrated Lewis pair chemistry, Can. J. Chem., 2025, 103, 822–831 Search PubMed.
- J. Holland, T. Horton and M. P. Shaver, Amine-Derived Polymeric Frustrated Lewis Pair Networks and Catalysts, ACS Appl. Polym. Mater., 2024, 6, 12321–12328 CrossRef CAS.
- S. Rajendiran, P. Natarajan and S. Yoon, A covalent triazine framework-based heterogenized Al–Co bimetallic catalyst for the ring-expansion carbonylation of epoxide to β-lactone, RSC Adv., 2017, 7, 4635–4638 RSC.
- A. Stirling, M. Iannuzzi, M. Parrinello, F. Molnar, V. Bernhart and G. A. Luinstra, β-lactone synthesis from epoxide and CO: Reaction mechanism revisited, Organometallics, 2005, 24, 2533–2537 CrossRef CAS.
- T. L. Church, Y. D. Getzler, C. M. Byrne and G. W. Coates, Carbonylation of heterocycles by homogeneous catalysts, Chem. Commun., 2007, 657–674 RSC.
- J. Luo, P. Liu, W. Yang, H. Niu, S. Li and C. Liang, Chemical kinetics and promoted Co-immobilization for efficient catalytic carbonylation of ethylene oxide into methyl 3-hydroxypropionate, Front. Chem., 2022, 945028 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |