Pt–Re dehydrogenation catalysts synthesized via solvent deficient precipitation
Received
28th December 2025
, Accepted 16th March 2026
First published on 30th March 2026
Abstract
This study demonstrates the feasibility of producing Pt–Re/Al2O3 catalysts via the scalable solvent deficient precipitation method achieving enhanced performance in the dehydrogenation of liquid organic hydrogen carriers. Effects similar to wet impregnated Pt–Re catalysts were observed, indicating the transferability of the Re promotion concept across catalyst synthesis approaches. In continuous gas-phase dehydrogenation of methyl cyclohexane, Re addition improved the Pt-based activity, selectivity, and stability, while reducing coking and effective activation energy. For perhydro benzyltoluene dehydrogenation, Re increased catalyst activity across various loadings with a molar ratio of Re/Pt = 0.5 yielding the highest hydrogen production rate. Higher Re loadings led to a rapid performance decline due to side reactions and catalyst deactivation, likely caused by deep dehydrogenation pathways and strong binding of the dehydrogenated product benzyltoluene to the active sites. Kinetic studies revealed that Re reduces the effective activation energy, although diffusional limitations were observed due to the small pore sizes of the produced catalyst materials. Successful 10-fold scale-up of the solvent-deficient precipitation was achieved without water, binders, or additives with performances comparable to small-scale syntheses. This highlights the potential of this simple, cost-effective, and scalable approach for the high-throughput production of bimetallic Pt-based catalysts.
1. Introduction
In the context of green hydrogen supply for the energy sector, liquid organic hydrogen carriers (LOHCs) offer a promising solution for hydrogen storage and long-range transportation by reversibly binding H2 to storage molecules such as aromatic hydrocarbons through hydrogenation/dehydrogenation reactions.1,2 This technology is scalable and compatible with existing fuel transportation infrastructure, as LOHCs possess similar physical properties to fossil fuels.3–6 Examples of LOHC pairs include methyl cyclohexane (MCH, H2-rich) and toluene (TOL, H2-lean), which are widely used solvents in the chemical industry, as well as perhydro benzyltoluene (H12-BT, H2-rich) and benzyltoluene (H0-BT, H2-lean), the latter being broadly utilised as heat transfer fluid.2,5 The hydrogen-loaded molecules enable safe and loss-free storage as liquids under atmospheric conditions of pressure and temperature, posing minimal safety risks, while hydrogen can be released on-site and on demand with high purity.5
The cyclic loading and unloading of the LOHC molecules occurs through catalytic hydrogenation and dehydrogenation processes, which can be repeated multiple times.5–8 These reactions are highly dependent on the efficiency of the catalysts, with platinum (Pt) being one of the most suitable active metals for hydrogen release from homocyclic LOHC molecules owing to its high selectivity for C–H bond cleavage.9 Furthermore, minimizing side product formation is crucial, as it facilitates the reloading of the resulting product mixture and ensures the circularity of the carrier molecule.5,9
Under the demanding conditions of power-dense dehydrogenation reactors, Pt-based catalysts are prone to deactivation through sintering and coking, as well as catalysing undesired side reactions. To mitigate this, adjusting the catalyst properties is essential, as improved Pt dispersion and electronic configuration can ensure a stable and robust active material.10 This can be achieved through the addition of promoters, which introduce geometric and electronic modifications that can enhance catalyst activity, selectivity, and stability.11 Such a beneficial ensemble effect has been reported for Pt–Re catalysts in MCH dehydrogenation, where the presence of Re reduces catalyst deactivation and increases the stability by altering the nature of the coke formed, making it less carboxylic.12–14 In H12-BT dehydrogenation, Pt–Re/Al2O3 catalysts outperformed the monometallic counterpart, achieving higher Pt-based H2 production rates, which was attributed to a modified reaction mechanism favouring fully dehydrogenated species (H0-BT) over partially hydrogenated (H6-BT) molecules.15
Nevertheless, successful modifications of Pt-based catalysts depend on the synthesis method. The latter is known to significantly influence key material properties, such as structure and surface chemistry.5 Solid-state syntheses, which involve nucleation and growth processes, have been employed to prepare catalyst supports and simplify aqueous-based synthesis methods, such as wet impregnation (WI). A notable example is the solvent-deficient precipitation (SDP) method developed by Woodfield et al.,16 which is a simple and versatile approach for preparing mesoporous mixed-metal oxide nanoparticles. This method uses only a metal salt (such as chloride or nitrate) and a base, such as ammonium (bi)carbonate or hydroxide (e.g., NH4HCO3).17 Initially, these solid precursors are mixed and ground for a few minutes, followed by calcination to form the metal oxide. By simply adding an active metal precursor to the mixture, metal oxide supported catalysts can be prepared. The solvent-free conditions not only reduce catalyst preparation times and significantly lower production costs, they also result in small, uniformly distributed nanoparticles, which are beneficial for improving the catalytic performance.18
Due to the versatility of this synthesis method, it has been successfully applied to prepare catalysts for the dehydrogenation of perhydro 2-(N-methylbenzyl)pyridine, perhydro dibenzyltoluene (H18-BT) and H12-BT, where the catalysts even outperformed their counterparts synthesised by WI.9,19 Additionally, the incorporation of secondary elements, such as sulphur and manganese, into Pt-based catalysts through the SDP method has been shown to produce effects similar to those achieved with WI catalysts.20,21
This study focuses on synthesising Pt-based catalysts modified with Re via the SDP method to enhance H2 release from MCH. The performance of Pt–Re/Al2O3 catalysts is evaluated using continuous MCH gas phase dehydrogenation in the temperature range between 240 and 320 °C. For comparison, the dehydrogenation of liquid H12-BT in semi-batch mode at 250 °C with the same catalysts is discussed. Catalytic properties are assessed through microscopic characterisation and CO-pulse chemisorption. Additionally, successful scaling up of the synthesis without the use of surfactants, additives, or binders is demonstrated.
2. Methodology
Materials
The compounds utilised to prepare the catalysts, namely aluminium nitrate nonahydrate (>98% purity), ammonium perrhenate(VII) ((NH4)ReO4, 99.999% purity), tetraamine platinum(II) nitrate (Pt(NH3)4(NO3)2, 99.99% purity), were supplied by Thermo Scientific and used as received. For dehydrogenation experiments, methyl cyclohexane (MCH, 99% purity) was purchased from Thermo Scientific and perhydro benzyltoluene (H12-BT, 98% purity) was supplied by Hydrogenious LOHC Technologies GmbH (https://www.hydrogenious.net).
Preparation of catalysts
Pt-based catalysts on a γ-Al2O3 mesoporous support were prepared following the SDP method. Single batches of 3 g were synthesised by mixing 0.02 g of Pt(NH3)4(NO3)2, 21 g of Al(NO3)3·9H2O and 14 g of NH4HCO3 in a mortar, followed by grinding by hand with a pestle. For Pt–Re/Al2O3 catalysts, (NH4)ReO4 was simultaneously added in desired proportions, maintaining a fixed molar ratio of 1 between total nitrate and ammonium hydrogen carbonate to ensure a stoichiometric reaction. During mixing, the reactants liquefied and bubbled vigorously due to hydration water and CO2 release. The end of this phase was marked by the absence of bubbles resulting in a slurry-like final mixture, indicating the precipitation of metal oxide precursors as depicted in Fig. 1. The resulting wet solid was calcined in a muffle oven from Nabertherm at 600 °C (temperature ramp: 2 K min−1) for 1 h with a synthetic air flow of 10 L min−1. The final solid was again ground for 30 seconds in a mortar to yield a fine powder. Finally, the catalyst was reduced in a quartz tube inside a tubular furnace from Carbolite at 400 °C (temperature ramp: 12.67 K min−1) under 10 vol% H2/N2 at a total flow rate of 500 mL min−1 for 1 h.
 |
| | Fig. 1 Summary of synthesis of a Pt–Re/Al2O3via the solvent deficient precipitation (SDP) method, showing the grinding, calcination and reduction steps. | |
Scaled batch sizes
For the production of larger batches of the catalyst in a semi-automatic process, the SDP synthesis was carried out in a 5 L three-necked round-bottom flask with a permanently attached KPG stirrer (Fig. 2). Stoichiometric amounts of NH4HCO3 were slowly added to the entire amount of Al(NO3)3·9H2O as a fine powder while stirring continuously. Pt(NH3)4(NO3)2 and (NH4)ReO4 were added, if appropriate, and the mixture was stirred for several hours. A transformation of the mixture in different stages could be observed via a thick slurry, a thin liquid, a sticky material, and finally a moist paste. During the reaction process, a significant cooling of the mixture below 0 °C was observed. The reaction is complete once a slurry-like consistency is achieved. With around four hours, the reaction time is significantly longer than for the small-scale synthesis described above. The moist slurry was freeze-dried in a beta 1–8 freeze dryer (Martin Christ GmbH). Drying was carried out in two steps at −30 °C and 0.3 mbar for 84 hours and then at −55 °C and 0.08 mbar for 12 hours. Calcination was carried out in evaporation dishes in a muffle furnace (Nabertherm L 9/13 B150) at a constant air flow of 10 L min−1 at 600 °C (heating rate 1.2 K min−1). Adequate ventilation was ensured to remove any nitrous gases produced. The reduction was carried out in small batches according to the procedure described above, followed by recombination and homogenisation. Example weigh-ins for the preparation of 40 g Pt/Al2O3 are 293.5 g (0.78 mol) Al(NO3)3·9H2O, 185.7 g (2.35 mol) NH4HCO3, 0.238 g (0.615 mmol) Pt(NH3)4(NO3)2.
 |
| | Fig. 2 Mixing stages during the solvent deficient precipitation (SDP) synthesis of 3 g of Pt/Al2O3 using a core drawn precision glassware (KPG) mixer. | |
Characterisation of catalysts
The metal loadings of the prepared catalysts were determined by means of inductively coupled plasma with optical emission spectroscopy (ICP-OES), using a Ciros CCD from Spectro Analytical Instruments GmbH. The samples were prepared by mixing approximately 200 mg with 10 mL of concentrated aqua regia (volumetric ratio HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 = 3:2) and subjected to microwave digestion at 200 °C for 30 min. Prior to measurements, the instrument was calibrated using standard solutions of the target elements, and the characteristic lines evaluated were 167.020 nm for Al, 214.423 nm for Pt, 221.426 nm for Re.
:HNO3 = 3:2) and subjected to microwave digestion at 200 °C for 30 min. Prior to measurements, the instrument was calibrated using standard solutions of the target elements, and the characteristic lines evaluated were 167.020 nm for Al, 214.423 nm for Pt, 221.426 nm for Re.
The textural properties of the supports and catalysts were evaluated by means of low temperature N2 physisorption using a TriStar II Plus from Micromeritics. The samples were degassed overnight at 250 °C in vacuum. The specific surface area was calculated via the Brunauer–Emmett–Teller (BET) method for a relative pressure range p/p0 of 0.05 to 0.35. The pore size distribution was obtained according to the Barrett–Joyner–Halenda (BJH) model for cylindrical pores from the desorption curve of the physisorption isotherm. Considering an uniform cylindrical shape for all pores, the average pore diameter of the catalytic systems, daverage, was estimated by eqn (1), where SBET represents the BET specific surface area and VBJH,cumulative, the cumulative pore volume calculated via the BHJ method.
| |  | (1) |
The particle size distribution (PSD) of Pt particles in the synthesised catalysts was analysed using conventional and high-resolution transmission electron microscopy (TEM), high-angle annular dark field scanning TEM (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDXS) for elemental mapping of Pt and Re. Samples were prepared by dispersing the catalyst in ethanol
via ultrasonication and depositing a few droplets onto a Cu-carbon grid. Imaging was performed with a Thermo Fisher Scientific Talos F200i microscope equipped with a Schottky emitter and two Bruker XFlash 6T-100 EDS detectors, operated at 200 kV. Data evaluation was performed using the Velox software and Pt particle diameters were determined from HAADF images using the open-source software Fiji ImageJ.
22 The average particle size was calculated using the volume–area diameter equation (
i.e. Sauter mean diameter),
23 represented in
eqn (2), where
di represents the particle diameter and
ni is the corresponding particle count (at least 75 particles).
| | 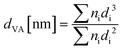 | (2) |
The metallic active centres in the synthesised catalysts were evaluated
via CO pulse chemisorption. First, all materials were reduced
in situ under 5 vol% of H
2 in Ar at 400 °C for 60 min. Following this, successive pulses of 0.5117 mL
N of CO/He were injected at 35 °C into the sample chamber until the catalyst surface was saturated (
i.e. no change in TCD signal). Subsequently, a temperature-programmed desorption (TPD) was conducted by heating the sample to 300 °C (temperature rate: 15 °C min
−1) under He, recording the desorbed amount of CO. It was considered that 1 mol of CO adsorbs onto 1 mol of available Pt.
The coke content of the spent SDP catalysts in the dehydrogenation of MCH was quantified via temperature-programmed oxidation (TPO). The weight change was monitored by means of thermogravimetric analysis (TGA) using a TGA 8000 from Perkin Elmer. A sample mass within the range of 5 to 20 mg was placed in a ceramic crucible, heated to 120 °C (heating rate: 10 K min−1), and held for 30 min under an inert atmosphere of N2 to remove physisorbed molecules. Subsequently, the sample was heated to 1000 °C (heating rate: 50 K min−1) and held for 10 min to guarantee the complete oxidation of all carbonaceous deposits in 21% O2/N2. The overall flow rate was 50 mL min−1 throughout the TPO. The mass loss in the final temperature ramp and at 1000 °C was compared to the sample mass after 2 h at 120 °C to assess to coke content.
Catalytic testing in the gas phase
Continuous gas-phase dehydrogenation of MCH was performed in a tubular inconel reactor (diameter: 9.26 mm, length: 60 cm) loaded with 1.0 g of catalyst, which was positioned between two plugs of glass wool in the middle of the reactor. The testing set-up is described in detail elsewhere.24 The reactor was heated to the desired reaction temperature and a programmed temperature variation between 240 and 320 °C was conducted for catalyst testing at 1 bara. The inlet gas mixture, consisting of 57 mL min−1 He and 0.05 g mL−1 liquid MCH, was regulated by Bronkhorst MFCs. MCH was vapourised at 200 °C using an evaporator, and all subsequent connecting lines were maintained at this temperature to prevent condensation. The mean residence time was approximately 0.6 s and the weight hourly space velocity (WHSV) was 8.31 Lgas gcat−1 h−1.
The outlet gas composition was analysed in a mini-GC 7820 A from Agilent Technologies equipped with an FID. Separation was achieved in a Restek Rtx-100-DHA column (diameter: 250 μm, length: 105 m). The molar fraction of each species was calculated based on the peak areas. The conversion of MCH XMCH was calculated from the total peak area Atotal and the peak area corresponding to unreacted MCH AMCH, as shown in eqn (3).
| | 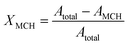 | (3) |
The selectivity of toluene
STOL was obtained based on the toluene peak area
ATOL in comparison to the total peak area excluding the one for MCH, as shown in
eqn (4).
| |  | (4) |
Toluene productivity Prod
TOL was calculated from the converted amount of MCH
ṅMCH,0, toluene selectivity and its molar mass
MTOL, as well as the Pt mass in the catalyst bed
mPt, as described in
eqn (5). Cumulative productivity Cum Prod
TOL,i was determined by integrating the productivity over a specific time interval
t as indicated in
eqn (6).
| | 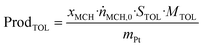 | (5) |
| |  | (6) |
Toluene selectivity exceeded 98% for all experiments. Assuming an ideal reaction where 3 mols of H
2 are released per mol of MCH, as shown in
Fig. 3, the amount of released hydrogen
ṅH2 was calculated based on the produced TOL, as described in
eqn (7). Thus, the productivity of H
2 was calculated according to
eqn (8).
| | 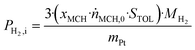 | (8) |
The dehydrogenation equilibrium of MCH was simulated
via Gibbs free energy minimisation using the software Aspen Plus
25 to compare with experimental results. The Predictive Redlich Kwong–Soave (PSRK) property method was chosen for gas processing. A sensitivity analysis was performed over a range of temperatures between 240 and 320 °C at 1 bara mirroring the lab experiments.
 |
| | Fig. 3 Reaction equation for the dehydrogenation of methylcyclohexane (MCH) to the desired product toluene (TOL). | |
Catalytic testing in the liquid phase
Semi-batch liquid-phase dehydrogenation experiments were conducted in a four-neck round-bottom flask with 0.1 mol of H12-BT (19.5 g) as feedstock. The system was supplied with an Ar flow of 300 mLN min−1, controlled by a MFC from Bronkhorst, for inertisation, and temperature was set to 250 °C with a heating mantle from Heraeus–Wittmann and a Fitron TP controller from Fiege. The catalyst was added through a metallic dispensing tool after reaching the target temperature. A condenser operating at −5 °C was used to condense and reflux Hx-BT species, with the outlet stream passing through a sulphuric acid bubbler and an active carbon filter to prevent interference or damage to the H2 analyser. Hydrogen concentration was continuously monitored using a calibrated FTC 300 thermal conductivity detector (TCD) from Messkonzept. Standard experiments used 0.3 wt% Pt catalysts with a molar ratio between Pt and LOHC of 0.0001 molPt molLOHC−1.
The volumetric flow rate of H2![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) H2 was obtained from the H2 concentration φH2 given by the TCD in ppm, according to eqn (9), while the cumulative volume of H2VH2,t was calculated viaeqn (10).
H2 was obtained from the H2 concentration φH2 given by the TCD in ppm, according to eqn (9), while the cumulative volume of H2VH2,t was calculated viaeqn (10).
| |  | (9) |
| |  | (10) |
The degree of dehydrogenation DoDh was obtained from the ratio of released H
2 volume and the maximum theoretically releasable amount of H
2Vmax, following
eqn (11).
| |  | (11) |
Hydrogen productivity Prod
H2 was calculated
viaeqn (12), taking into consideration the H
2 volumetric flow
H2,i,H
2 density
ρH2, Pt loading of the used catalyst
wPt and catalyst mass
mcatalyst.
| |  | (12) |
The liquid phase composition during dehydrogenation was analysed, clustering the LOHC species into five groups: H12-BT (fully hydrogenated reactant), H6-BT (intermediate product), H0-BT (fully dehydrogenated product), and methyl fluorene (MF), as shown in
Fig. 4. The formation of MF, which is a undesired side product, serves as marker to evaluate the catalyst selectivity. For this, liquid samples were analysed using a TRACE 1310 gas chromatograph (GC) from Thermo Scientific, equipped with a flame ionisation detector (FID). Separation was achieved with a Rxi17Sil column (diameter: 250 μm, length: 30 m). Samples were prepared by diluting 0.1 mL of taken sample with 0.9 mL of isopropanol. Approximately 2 μL of this mixture were injected for GC analysis.
 |
| | Fig. 4 Reaction paths for the dehydrogenation of perhydro benzyltoluene (H12-BT), including the intermediate H6-BT, desired product H0-BT and undesired side product methylfluorene (MF). | |
The molar fraction xi of each species was determined based on the GC peak areas. The degree of hydrogenation DoH was calculated disregarding dehydrogenated species (i.e. H0-BT, MF), as in eqn (13). The DoDh was then derived from the DoH, as shown in eqn (14).
| |  | (13) |
The hydrogen productivity based on the GC data was obtained from
eqn (15), where ΔDoDh represents the difference of the DoDh between two consecutive liquid samples,
nH12-BT,t=0 the initial amount of H12-BT,
nH2,per H12-BT the amount of H
2 mols per H12-BT molecule.
| |  | (15) |
3. Results and discussion
Catalyst characterisation
Structural and textural properties.
In the following, characterisation data of the Re-free Pt/Al2O3 catalyst obtained from the SDP synthesis method is exemplarily shown. XRD analysis of the calcined Pt/Al2O3 catalyst (see SI Fig. A1) confirmed the presence of an amorphous γ-Al2O3 phase and N2 physisorption indicated an average BET surface area of 259 m2 g−1 and an average pore diameter of 6.0 nm (see SI Fig. A2). The corresponding BHJ pore size distribution is shown in Fig. 5. The measured values are in good agreement with the ones reported by Smith et al.17 for γ-Al2O3 synthesised via the SDP method, namely a specific surface area of 267 m2 g−1 and an average pore diameter of 4.0 nm.
 |
| | Fig. 5 Pore volume (VBHJ) and pore size distribution (PSD) obtained for Pt/Al2O3 (0.3 wt% Pt) and Pt–Re/Al2O3 (0.3 wt% Pt, 0.27 wt% Re, Re/Pt = 0.88) via N2 physisorption obtained using the adsorption branch. | |
The structural and textural properties of the Re containing catalyst do not differ significantly from the one without Re. The data of a Pt–Re/Al2O3 catalyst with Re/Pt = 0.88 is also presented in Fig. 5 (BET surface area of 276 m2 g−1, average pore diameter of 5.8 nm).
Pt-dispersion.
TEM images of a Pt/Al2O3 catalyst (0.28 wt% Pt) reveal that the majority of Pt nanoparticles fall within the 1–2 nm diameter range (see Fig. 6), with an average size of 1.8 nm. In contrast, the addition of Re at a molar ratio of Re/Pt = 0.5 (0.31 wt% Pt, 0.14 wt% Re) resulted in smaller particles with an average size of 1.4 nm (see Fig. 7), indicating a higher dispersion. This observation is consistent with previous reports on Pt–Re catalysts synthesised via WI, where increasing Re loadings reduced Pt particle size.15 Nevertheless, further increasing the Re/Pt molar ratio to 3.19 (0.29 wt% Pt, 0.87 wt% Re) yielded an average particle size of 1.8 nm, comparable to that of the monometallic catalyst (see SI Fig. A3). TEM(-EDX) analyses of Pt–Re systems, shown in SI Fig. A4, were limited by low concentrations of Pt and Re, rapid charging of the Al2O3 support, and similar atomic numbers of Pt and Re.
 |
| | Fig. 6 Representative TEM image of a solvent deficient precipitation (SDP) Pt/Al2O3 catalyst (0.28 wt% Pt) showing Pt nanoparticles distributed over γ-Al2O3 (left) and corresponding particle size distribution (PSD) based the measurement of 128 particles (right). | |
 |
| | Fig. 7 TEM image of a solvent deficient precipitation (SDP) Pt–Re/Al2O3 with Re/Pt = 0.5 (0.31 wt% Pt, 0.14 wt% Re), showing metal nanoparticles distributed over γ-Al2O3 (left) and particle size distribution (PSD) based the measurement of 175 particles (right). Due to the low metal loadings, Pt and Re could not be distinguished. | |
Additionally, CO-pulse chemisorption was used to examine the accessibility of Pt surface sites and Pt–Re interactions. As shown in Fig. 8, the catalysts containing Re, especially the one with a Re/Pt ratio of 0.5, exhibited a decrease in chemisorbed CO compared to monometallic Pt. This observation was also reported by Pieck et al.,26 while the mean particle diameter measured by TEM remained nearly unchanged. They found that CO is only adsorbed on Pt surface atoms and concluded an interaction between Pt and Re altering the properties of Pt surface atoms and therefore the CO adsorption capacity.
 |
| | Fig. 8 Cumulative amount of CO adsorbed onto the Pt–Re catalysts with Re/Pt molar ratios of 0 (0.28 wt% Pt), 0.5 (0.31 wt% Pt, 0.14 wt% Re) and 1.6 (0.29 wt% Pt, 0.44 wt% Re). | |
For the SDP catalysts in this study, TEM pictures also indicate that the addition of low amounts of Re to Pt results in unchanged or slightly reduced average particle sizes, while, contrary to this observation, the CO uptake decreases. According to the literature on WI catalysts, Re might be present as ReOx as reduction processes conducted below 500 °C are not sufficient to fully reduce Re species. Moreover, previous studies have confirmed that the contribution of ReOx is negligible in CO-chemisorption at temperatures below 50 °C.26,27 However, these species could partially cover Pt sites and reduce adsorption relative to pure Pt.28 Besides this geometric effect, CO adsorption on Pt could also be altered via an electronic effect of Re species. In either way, the decreased CO uptake at similar particle sizes in Pt–Re catalysts points towards an interaction between the two compounds.
Dehydrogenation in the gas phase with Pt–Re catalysts
A series of Pt–Re/Al2O3 catalysts with Re/Pt molar ratios ranging from 0 to 1 was synthesised via SDP and tested in the continuous gas phase dehydrogenation of MCH. The contents of Pt and Re were measured by means of ICP-OES, which are shown in Table 1. The target values were reached with small deviations for all catalysts.
Table 1 Composition of Pt–Re/Al2O3 catalysts prepared via the solvent deficient precipitation (SDP) method, as confirmed by ICP-OES analysis, which were tested in the dehydrogenation of methylcyclohexane (MCH)
| Pttarget/wt% |
Ptmeasured/wt% |
Retarget/wt% |
Remeasured/wt% |
Measured Re/Pt molar ratio |
| 0.30 |
0.29 |
0 |
0 |
0 |
| 0.30 |
0.29 |
0.15 |
0.14 |
0.5 |
| 0.30 |
0.29 |
0.30 |
0.27 |
1.0 |
As shown in Fig. 9, Re addition enhanced both activity and selectivity. After 10 hours of time on stream (TOS), the cumulative toluene productivity was, compared to the Re-free Pt/Al2O3 system, by 42% and 27% higher for the Re-containing catalysts. Moreover, the Re/Pt = 0.5 catalyst showed an enhanced stability over 20 h TOS, which becomes evident when comparing the achieved conversion levels at 280 °C at different stages of the temperature program. TGA analysis (see SI Fig. A5) revealed a reduction in coke formation from (5.6 ± 0.2) wt% mass loss for the Pt catalysts to (4.9 ± 0.5) wt% and (5.2 ± 0.2) wt% for the systems with Re/Pt = 0.5 and 1.0. This equals to a reduction of 14 and 8%, respectively, and suggests the self-reactivation mechanism proposed by Sachtler.29 According to this mechanism, Re enhances the ability of reorganising the coke structure on Pt particles, preventing permanent deactivation of active sites. Our results indicate that Re addition increases the selectivity of Pt-based SDP catalysts and reduces coking during MCH dehydrogenation, which is fully aligned with findings from the literature for impregnated systems.14,30
 |
| | Fig. 9 Methylcyclohexane (MCH) conversion (left) and cumulative productivity from toluene (right) from different Pt–Re/Al2O3 catalysts (Re/Pt molar ratios of 0, 0.5 and 1.0) during the dehydrogenation of MCH in the gas phase. Experimental conditions: 1.0 g catalyst, 57 mL min−1 He and 0.05 g min−1 MCH, WHSV 8.31 Lgas h−1 gcat−1, 240–320 °C, 1 atm, ca. 20 h. Theoretical equilibrium data from Aspen Plus simulation is shown in the left graph. | |
Additionally, the kinetics of MCH dehydrogenation were evaluated for the three catalysts. The average hydrogen productivity was calculated for temperatures of 240, 260, 280 (final) and 300 °C and the resulting Arrhenius plot is shown in SI Fig. A6. Notably, Pt–Re catalysts demonstrated lower temperature sensitivity compared to monometallic Pt. From the Arrhenius plots, the effective activation energy was calculated for all systems. For the catalyst with Re/Pt = 0.5, EA,eff was reduced by 19% (59 kJ mol−1) and for Re/Pt = 1.0 by 14% (62 kJ mol−1) in comparison to monometallic Pt (72 kJ mol−1). This supports the assumption of an ensemble effect in Pt–Re systems as reported in the literature, where Re addition divides Pt into smaller ensembles, promoting a single-site mechanism and facilitating the release of H2.30
Dehydrogenation in the liquid phase with Pt–Re catalysts
A series of Pt–Re/Al2O3 catalysts with Re/Pt molar ratios ranging from 0 to 3.19 was synthesised and tested in the liquid phase dehydrogenation of H12-BT. The Pt and Re contents were determined by ICP-OES and are presented in Table 2. The measured values closely matched the target compositions, with only minor deviations. All subsequent analyses are based on these measured values.
Table 2 Composition of Pt–Re/Al2O3 catalysts prepared via the solvent deficient precipitation (SDP) method, as confirmed by ICP-OES analysis, which were tested in the dehydrogenation of perhydro benzyltoluene (H12-BT)
| Pttarget/wt% |
Ptmeasured/wt% |
Retarget/wt% |
Remeasured/wt% |
Measured Re/Pt molar ratio |
| 0.3 |
0.28 |
0 |
0 |
0 |
| 0.3 |
0.32 |
0.05 |
0.03 |
0.09 |
| 0.3 |
0.30 |
0.10 |
0.08 |
0.28 |
| 0.3 |
0.31 |
0.15 |
0.14 |
0.48 |
| 0.3 |
0.32 |
0.30 |
0.27 |
0.88 |
| 0.3 |
0.29 |
0.50 |
0.44 |
1.60 |
| 0.3 |
0.29 |
1.00 |
0.87 |
3.19 |
As presented in Fig. 10, the dehydrogenation activity is enhanced for Re containing catalysts with low loadings over the whole course of the reaction. Catalysts with Re/Pt molar ratios exceeding 0.88 demonstrated high initial dehydrogenation rates, which was less evident at higher DoDhs. The analysis of MF formation revealed that these systems led to higher by-product formation. For instance, the catalyst with Re/Pt = 1.60 achieved the highest maximal productivity of 2.9 gH2 gPt−1 min−1, which is 2.3 times higher than the catalyst without Re, but also exhibited significant MF formation (0.15 vs. 0.08 mol% at DoDh 27%), likely contributing to the rapid productivity loss observed over time.
 |
| | Fig. 10 Degree of dehydrogenation, H2 productivity and methylfluorene (MF) formation during semi-batch dehydrogenation of perhydro benzyltoluene (H12-BT) with solvent deficient precipitation (SDP) Pt–Re/Al2O3 catalysts with different Re/Pt molar ratios. Experimental conditions: T = 250 °C, p = 1 bara, nLOHC = 0.1 mol, wPt = 0.3 wt%, nPt/nLOHC = 0.0001, tR = 360 min, Ar = 300 mLN min−1. DoDh and productivity data was obtained from TCD measurements, whereas product composition data were determined via the GC method. Average values and standard deviation are shown. | |
To provide a broader overview over the tested range of Re loadings, the average hydrogen productivity was calculated for each catalyst within a DoDh range of 2–25% (see Fig. 11). An optimal Re/Pt molar ratio of 0.5 was identified, yielding an average productivity of 0.85 gH2 gPt−1 min−1. This ratio aligns with the results reported by Strauch et al.15 for the Re/Pt catalysts prepared according to the WI method under similar reaction conditions. Considering that the maximal productivity is reached within the first 5 min of dehydrogenation, this maximum value increased with the Re addition to the catalyst from 0.9 (Re/Pt = 0) to 2.0 gH2 gPt−1 min−1 (Re/Pt = 0.5) for our SDP systems. For WI systems, this value increased from 2.0 (Re/Pt = 0) to 4.1 gH2 gPt−1 min−1 (Re/Pt = 0.5). For the same Re/Pt molar ratio of 0.5, this represents a 2.3-fold and a 2.1-fold increase for SDP systems and WI systems, respectively. Despite WI systems achieving higher maximal DoDhs and productivity values, the same correlation regarding Re addition to Pt/Al2O3 catalysts was observed in SDP systems hinting at a similar activation mechanism. The difference in the absolute productivity values might stem from the small pore size of the SDP catalysts in comparison to the WI counterparts leading to diffusional limitations.31 A detailed comparison of the corresponding dehydrogenation data between SDP and WI systems can be found in the SI (Fig. A7).
 |
| | Fig. 11 Average H2 productivity (calculated from the productivity between a dehydrogenation degree of 2–25%) of different Pt–Re/Al2O3 catalysts (Re/Pt = 0–1.60) during dehydrogenation of perhydro benzyltoluene (H12-BT). For Pt–Re catalysts with Re/Pt = 3.19, the average productivity was calculated only up to a DoDh of 18% as no higher levels were reached. Experimental conditions: T = 250 °C, p = 1 bara, nLOHC = 0.1 mol, wPt = 0.3 wt%, nPt/nLOHC = 0.0001, tR = 360 min, Ar = 300 mLN min−1. Productivity data was obtained from TCD measurements. Average values and standard deviation are shown. | |
A detailed analysis of the product composition for Pt and Pt–Re systems, shown in SI Fig. A7, revealed that Re containing catalysts with higher Re loadings lead to reduced H6-BT levels. This suggests that Re addition favours the complete dehydrogenation of H12-BT to H0-BT by accelerating the conversion of particularly H6-BT, consistent with findings by Strauch et al.15 with WI Pt–Re catalysts. They attributed these changes in the individual reaction steps to an optimised dispersion of Pt and electronic modification in Pt–Re catalysts. Moreover, an increased formation of MF was observed, which suggests strong binding of the dehydrogenated reaction product H0-BT to the active sites and further dehydrogenation.15 MF is a consecutive reaction product formed by a deep dehydrogenation step.32 This can also be seen for the Re containing catalysts in this study and can explain the loss in productivity at higher DoDhs.
To further investigate the adsorption of aromatic species in Pt–Re systems, the dehydrogenation of a mixture containing 90% H12-BT and 10% H0-BT was conducted with Pt and Re/Pt = 0.5. The results, summarised in Fig. 12, show that when the initial mixture contains H0-BT, the activity of both catalysts is significantly decreased due to the stronger adsorption of H0-BT in comparison to H12-BT33 and thereby induced blockage of active sites. The average productivity of Pt/Al2O3 is reduced from 0.4 to 0.2 gH2 gPt−1 min−1; the catalyst with Re/Pt = 0.5 displays a reduction from 0.9 to 0.3 gH2 gPt−1 min−1. This indicates a strong deactivation effect of H0-BT on the Pt–Re system. Again, for the Re containing catalyst a lower H6-BT fraction can be found at the same DoDh. More interestingly, for the monometallic Pt catalyst the H0-BT fraction remains almost constant over the DoDh range considered, whereas Pt–Re shows a steady increase of H0-BT levels. This supports the finding that the addition of Re to Pt accelerates the dehydrogenation of H6-BT to H0-BT, while H6-BT builds up from the dehydrogenation of H12-BT when using the monometallic catalyst. In addition, an enhanced MF formation can be observed with the Re/Pt = 0.5 catalyst right from the start of the experiment. This points towards a further reaction of H0-BT through deep dehydrogenation on the Pt–Re system as discussed before. Overall, the experiments with added H0-BT support the observations made previously in a fully conclusive manner.
 |
| | Fig. 12 Degree of dehydrogenation over time (a) and H6-BT (b), H0-BT (c) and methylfluorene (MF) (d) formation as a function of DoDh during the semi-batch dehydrogenation of perhydro benzyltoluene (H12-BT) using Pt/Al2O3 and Pt–Re/Al2O3 (Re/Pt = 0.5) catalysts synthesised via the solvent deficient precipitation (SDP) method. Experimental conditions: T = 250 °C, p = 1 bara, nLOHC = 0.1 mol, wPt = 0.3 wt%, nPt/nLOHC = 0.0001, t = 360 min, Ar = 300 mL min−1. DoDh and product composition were determined via GC analysis. | |
The kinetics of H12-BT dehydrogenation for the SDP systems were evaluated for Pt and Pt–Re (molar ratio of 0.5) catalysts at temperatures of 230, 240 and 250 °C. The dehydrogenation data, shown in SI Fig. A9, revealed that the reaction rate of Pt/Al2O3 exhibited greater sensitivity to temperature changes compared to Pt–Re/Al2O3 due to the excellent low-temperature activity of Re-promoted catalysts.15 The average productivity within 2–25% DoDh was calculated for each temperature and the corresponding Arrhenius plot, shown in SI Fig. A10, enabled the determination of the effective activation energy (EA,eff), as presented in SI Table A1. The mean EA,eff for the Pt–Re system (113.0 ± 11.5 kJ mol−1) was found 11% lower than for Pt (126.6 ± 13.7 kJ mol−1). Comparatively, for WI catalysts, the addition of Re resulted in a reduction of 6% (−182.2 kJ mol−1 for Pt/Al2O3 and −170.5 kJ mol−1 for Pt–Re/Al2O3 with Re/Pt = 0.5),34 which also reveals a lower effective activation energy in SDP systems in general. This observation suggests that the dehydrogenation with SDP catalysts occurs in the transport-limited regime due to the small pore size of the alumina support in the present study (6 nm).35 Nevertheless, the general trend regarding reaction kinetics for the addition of Re to Pt/Al2O3 catalysts is consistent for WI and SDP catalysts.
Qualitative assessment of mobility of Pt and Re during calcination
To assess possible inhomogeneities during up-scaling and to better understand the Pt–Re and Re–Al interactions during SDP synthesis, the mobility of Pt and Re species was evaluated during calcination. Two experimental approaches were employed following the grinding phase of the synthesis. In the first, the wet solid was shaped into a spherical body and directly calcined. In the second, the solid was dried overnight before being crushed, spread into a crucible and calcined. The results, presented in Table 3, indicate that Pt and Re species are mobile during calcination, as evidenced by variations in their concentrations between the outer shell and inner region of the calcined sphere. However, the molar ratio of Re/Pt remained constant across both approaches and between the outer and inner regions of the spherical catalyst.
Table 3 Images of the resulting materials after the calcination experiment to test Re mobility and corresponding composition as confirmed by ICP-OES analysis
|
|
Sphere |
Spread |
| Before calcination |

|

|
| After calcination |

|

|
| Outside = 2.88 wt% Pt, 0.91 wt% Re |
2.70 wt% Pt, 0.91 wt% Re |
| Inside = 1.96 wt% Pt, 0.64 wt% Re |
| Molar ratio Re/Pt |
Outside = 0.3 |
0.3 |
| Inside = 0.3 |
Synthesis scale up
To scale up the synthesis, a glass flask and a precision agitator powered by an electrical motor (core-drawn precision glassware, KPG mixer) were used instead of a mortar and pestle. Details on the scale up process can be found in the SI. To assess its effectiveness, 3 g of Pt/Al2O3 catalyst were synthesised using the KPG setup. The yield was 97% and similar synthesis stages were observed like for the small-scale method with mortar and pestle, including liquefaction, bubbling and precipitation of a wet solid.
The synthesis was scaled up to prepare 40 g of Pt/Al2O3 and 30 g Pt–Re/Al2O3 catalysts. A freeze-drying step was included to remove excess moisture and prevent the extensive displacement of decomposition gases and inhomogeneous drying during calcination (see Table A2) due to the large material volume, which could lead to inhomogeneous products. Neither the longer reaction time of scaled batch sizes nor the freeze-drying step affected the achieved specific surface area and pore size distribution remarkably. The exact compositions of the obtained materials are shown in Table 4.
Table 4 Composition of Pt/Al2O3 and Pt–Re/Al2O3 catalysts prepared via the solvent deficient precipitation (SDP) method with a core drawn precision glassware (KPG) mixer (scaled up syntheses), as confirmed by ICP-OES analysis
| System |
Pt/wt% |
Re/wt% |
Re/Pt molar ratio |
Amount synthesised |
| Pt |
0.24 |
0 |
— |
40 g (increase by factor 13) |
| Pt–Re |
0.30 |
0.13 |
0.43 |
30 g (increase by factor 10) |
The catalysts were tested in the dehydrogenation of H12-BT and the results are summarised in Fig. 13. The Pt/Al2O3 system achieved an average H2 productivity of 0.51 gH2 gPt min−1 within the DoDh range of 2–25%, while the Pt–Re system reached 0.97 gH2 gPt min−1, nearly doubling the productivity with the presence of Re at a Re/Pt molar ratio of 0.43. A comparison with the Pt/Al2O3 and Pt–Re/Al2O3 catalysts from the small-scale synthesis via manual grinding is also shown indicating only minimal performance deviations and therefore successful up-scaling. The small deviations most probably stem from the slightly different Pt-loadings and Pt/Re molar ratios between the catalysts synthesized via mortar & pestle and KPG mixing.
 |
| | Fig. 13 Degree of dehydrogenation, H2 productivity and methylfluorene (MF) formation during dehydrogenation of perhydro benzyltoluene (H12-BT) with solvent deficient precipitation (SDP) Pt/Al2O3 and Pt–Re/Al2O3 catalysts synthesised either with a mortar & pestle or with a core drawn precision glassware (KPG) mixer. Experimental conditions: T = 250 °C, p = 1 bara, nLOHC = 0.1 mol, wPt = 0.3 wt%, nPt/nLOHC = 0.0001, tR = 360 min, Ar = 300 mL min−1. DoDh and productivity data was obtained from TCD measurements, whereas product composition data was determined via GC method. | |
4. Summary and outlook
This study demonstrates the feasibility of producing Pt–Re/Al2O3 catalysts via the SDP method that show attractive performance in the dehydrogenation of the LOHC compounds methylcyclohexane and perhydro benzyltoluene. Thereby, similar effects as for wet impregnated Pt–Re catalysts could be observed, which highlights the transferability of results between the two different synthesis approaches. Analytical characterization reveals similar textural properties than for pure SDP Al2O3 materials with specific surface areas of approx. 300 m2 g−1 and pore sizes in the range of 5 nm. Moreover, small metallic nanoparticles (in the range of 1.8 nm for pure Pt and 1.4 nm for Pt–Re) and indication for Pt–Re interactions influencing catalytic behavior were found.
In the continuous gas-phase dehydrogenation of MCH, the presence of Re enhances activity and selectivity, reduces coking, and promotes catalyst stability. After 20 hours of time on stream, the cumulative toluene productivity was higher by 42% for a catalyst with Re/Pt = 0.5 in comparison to monometallic Pt/Al2O3, whereas coke formation was reduced by 14%. Kinetic analyses reveal that Re lowers the effective activation energy of the reaction.
In the dehydrogenation of H12-BT, Re addition increased catalyst activity across most tested Re/Pt molar ratios loadings, with Re/Pt = 0.5 yielding the highest average hydrogen productivity (2–25% DoDh) of 0.85 gH2 gPt−1 min−1. This trend aligns with previous findings for WI catalysts. Catalysts containing Re demonstrated higher initial dehydrogenation rates and maintained better performance over the course of the semi-batch reactions compared to Re-free Pt catalysts. However, higher Re loadings (Re/Pt > 0.8) led to rapid productivity decline, likely due to increased formation of by-products, such as MF. The formation of MF and the accumulation of H0-BT suggest that Re facilitates the complete dehydrogenation pathway but also accelerates deep dehydrogenation to MF isomers that causes catalyst deactivation. Experiments with initial H0-BT in the feedstock confirmed that H0-BT inhibits catalyst activity due to its strong adsorption, especially on Re-promoted catalysts. Kinetic studies showed that Re reduces the effective activation energy with SDP catalysts showing diffusional limitations because of their small pore sizes.
Successful upscaling of the SDP synthesis to 10-fold batch size was demonstrated without the need for water, binders, or additives. The scale-up produced catalysts with performances comparable to small-scale samples, confirming the robustness and reproducibility of the synthesis approach. Overall, the results highlight the potential of the SDP method for high-throughput and large-scale experiments, effectively producing suitable bimetallic catalysts and mimicking trends also observed in WI catalysts. SDP proves to be a simple, cost-effective, and scalable ‘one-pot’ method for preparing both monometallic and bimetallic Pt-based catalysts by co-adding support and active phase precursors.
Conflicts of interest
Peter Wasserscheid is founder and minority shareholder of the company Hydrogenious LOHC technologies (https://www.hydrogenious.net) that offers commercially hydrogen storage systems based on the LOHC technology. There is no conflict of interest to declare about the specific scientific results reported in this paper.
Data availability
Data for this article are available at Zenodo under https://doi.org/10.5281/zenodo.18078485.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5cy01600b.
Acknowledgements
This work was funded by the Bavarian Ministry of Economic Affairs, Regional Development and Energy through the project “Erforschung und Entwicklung eines emissionsfreien und stark emissionsreduzierten Antriebssystems am Beispiel des Schienenverkehrs”. The authors would like to thank Dr. Andreas Hutzler, Dr. Birk Fritsch and M.Sc. Andreas Körner for obtaining the TEM images of the synthesized catalysts, Dr. Katharina Volz and Dr. Luca Lucarelli from Micromeritics for conducting the CO-pulse chemisorption analysis, M.Sc. Timo Schärfe for developing the Python method to analyse the GC results for H12-BT dehydrogenation, M.Sc. Oshin Sebastian for pre-calibrating the GC used during the off-gas analysis of the MCH dehydrogenation.
References
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74 CrossRef CAS.
- M. M. Distel, J. M. Margutti, J. Obermeier, A. Nuß, I. Baumeister, M. Hritsyshyna, A. Weiß and M. Neubert, Energy Technol., 2025, 13, 2301042 Search PubMed.
- T. Rüde, A. Bösmann, P. Preuster, P. Wasserscheid, W. Arlt and K. Müller, Energy Technol., 2018, 6, 529 Search PubMed.
- D. Teichmann, K. Stark, K. Müller, G. Zöttl, P. Wasserscheid and W. Arlt, Energy Environ. Sci., 2012, 5, 9044 RSC.
- F. Auer, D. Blaumeiser, T. Bauer, A. Bösmann, N. Szesni, J. Libuda and P. Wasserscheid, Catal. Sci. Technol., 2019, 9, 3537 RSC.
- P. Perreault, L. van Hoecke, H. Pourfallah, N. B. Kummamuru, C.-R. Boruntea and P. Preuster, Curr. Opin. Green Sustainable Chem., 2023, 41, 100836 CrossRef CAS.
- M. G. Rasul, M. Hazrat, M. A. Sattar, M. I. Jahirul and M. J. Shearer, Energy Convers. Manage., 2022, 272, 116326 CrossRef CAS.
- Q. N. Dao, E. On, S. Ramadhani, K. Lee, H. Sohn, S. H. Choi, S. Y. Lee, H. Jeong and Y. Kim, Int. J. Hydrogen Energy, 2024, 56, 1284 CrossRef CAS.
- J. Oh, Y. Jo, T. W. Kim, H. B. Bathula, S. Yang, J. H. Baik and Y.-W. Suh, Appl. Catal., B, 2022, 305, 121061 CrossRef CAS.
- J. Barbier, Appl. Catal., 1986, 23, 225 CrossRef CAS.
-
D. Y. Murzin, Engineering Catalysis, de Gruyter, Berlin, Boston, 2013 Search PubMed.
- P. Biloen, J. N. Helle, H. Verbeek, F. M. Dautzenberg and W. Sachtler, J. Catal., 1980, 63, 112 CrossRef CAS.
- K. Jothimurugesan, S. Bhatia and R. D. Srivastava, Ind. Eng. Chem. Fundam., 1985, 24, 433 CrossRef CAS.
- M. S. Akram, R. Aslam, F. S. Alhumaidan and M. R. Usman, Int. J. Chem. Kinet., 2020, 52, 415 CrossRef CAS.
- D. Strauch, P. Weiner, B. B. Sarma, A. Körner, E. Herzinger, P. Wolf, A. Zimina, A. Hutzler, D. E. Doronkin, J.-D. Grunwaldt, P. Wasserscheid and M. Wolf, Catal. Sci. Technol., 2024, 14, 1775 RSC.
-
B. F. Woodfield, S. Liu, J. Boerio-Goates and Q. Liu, WO Pat., WO/2007/098111, 2007 Search PubMed.
- S. J. Smith, B. Huang, S. Liu, Q. Liu, R. E. Olsen, J. Boerio-Goates and B. F. Woodfield, Nanoscale, 2015, 7, 144 RSC.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893 Search PubMed.
- J. Oh, H. B. Bathula, J. H. Park and Y.-W. Suh, Commun. Chem., 2019, 2, 1 Search PubMed.
- Y. Jo, T. Wan Kim, J. Oh, D. Kim and Y.-W. Suh, J. Catal., 2022, 413, 127 Search PubMed.
- Y. Jo, D. Kim, T. W. Kim, D. Yoon and Y.-W. Suh, Appl. Catal., B, 2023, 334, 122848 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld and B. Schmid,
et al.
, Nat. Methods, 2012, 9, 676 CrossRef CAS PubMed.
- M. Alderliesten, Part. Part. Syst. Charact., 1991, 8, 237 CrossRef CAS.
- O. Sebastian, S. Nair, N. Taccardi, M. Wolf, A. Søgaard, M. Haumann and P. Wasserscheid, ChemCatChem, 2020, 12, 4533 CrossRef CAS.
-
Aspen Technology Inc., Aspen Plus, aspenONE Engineering, 2020 Search PubMed.
- C. L. Pieck, C. R. Vera, J. M. Parera, G. N. Giménez, L. R. Serra, L. S. Carvalho and M. C. Rangel, Catal. Today, 2005, 107–108, 637 CrossRef CAS.
- K. G. Azzam, I. V. Babich, K. Seshan, B. L. Mojet and L. Lefferts, ChemCatChem, 2013, 5, 557 CrossRef CAS.
- C. G. Michel, W. E. Bambrick, R. H. Ebel, G. Larsen and G. L. Haller, J. Catal., 1995, 154, 222 Search PubMed.
- W. M. H. Sachtler, J. Mol. Catal., 1984, 1 CrossRef CAS.
- P. A. Van Trimpont, G. B. Marin and G. F. Froment, Ind. Eng. Chem. Fundam., 1986, 25, 544 Search PubMed.
- F. Auer, T. Solymosi, C. Erhardt, C. C. Collados, M. Thommes and P. Wasserscheid, Int. J. Hydrogen Energy, 2025, 100, 1282 Search PubMed.
- T. Rüde, S. Dürr, P. Preuster, M. Wolf and P. Wasserscheid, Sustainable Energy Fuels, 2022, 6, 1541 RSC.
- M. Geißelbrecht, S. Mrusek, K. Müller, P. Preuster, A. Bösmann and P. Wasserscheid, Energy Environ. Sci., 2020, 13, 3119 Search PubMed.
-
D. Strauch, PhD thesis, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2023 Search PubMed.
-
A. Jess and P. Wasserscheid, Chemical Technology: An Integral Textbook, Zweite, erweiterte Auflage, Wiley-VCH, Weinheim, Germany, 2013 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  cd,
Peter
Wasserscheid
cd,
Peter
Wasserscheid





























