CO2 hydrogenation to methanol via ZnO-SBA-15-supported Cu6 catalysts
Received
9th June 2025
, Accepted 22nd July 2025
First published on 22nd July 2025
Abstract
This study presents a mesoporous material-based catalyst for the CO2 hydrogenation to methanol reaction, utilizing copper nanoclusters (Cu6) immobilized on Zn-modified SBA-15. The Cu6–ZnO–SBA-15 catalysts are prepared by the wet impregnation of ZnO–SBA-15 with various metal loading contents. Multi-technique characterization shows uniform active phase dispersion within or on the mesoporous SBA-15 channels. The catalytic experiments reveal that the catalyst with the lowest copper content (2 wt%) exhibits superior activity with a methanol space–time yield of 232.5 (molMeOH kgCu−1 h−1) and a selectivity of 86%. Furthermore, the confinement of the active phase into the structure of SBA-15 prevents sintering to some extent, implying the excellent stabilization effect of the support. This study highlights the potential of using small nanoclusters prepared by wet chemistry in high-pressure CO2 hydrogenation to methanol.
1. Introduction
Global climate change caused by increasing atmospheric CO2 concentrations is one of our most serious environmental issues.1,2 As a great approach to sustainability, CO2 can be used as a promising carbon source for generating fossil fuel-derived hydrocarbons.3–6 Using such an approach, CO2 can be removed from the atmosphere and recycled and reused, resulting in a static CO2 loop.7,8
In recent decades, a significant amount of work has been dedicated to converting CO2 into value-added products to mitigate CO2 emissions. Among these studies, CO2 reduction to produce methanol has become a popular research topic due to its unique chemical and physical properties, making it an excellent alternative disposal process for CO2.9 Copper-based catalytic systems are the most promising for methanol synthesis, especially Cu/ZnO/support ones.10–12 The catalytic activity of these materials depends on the size and metal dispersion on the support, as well as support composition and metal–support interactions.13–16 In this context, CO2 hydrogenation to CH3OH on small Cu nanoclusters has gained attention due to their exceptional electronic and catalytic properties, making them different from bulk metals and larger nanoparticles.17–20 Vajda et al. demonstrated that methanol synthesis could be significantly improved using deposited ultra-small copper nanoclusters as the catalyst.21,22 However, their gas-phase-prepared nanoclusters are ideal for identifying the fundamental characteristics of immobilized nanoclusters, and their fabrication method is not realistic for large-scale production. As a result of the attractive features of small nanoclusters, solution-phase preparation methods are being applied to produce nanoclusters with narrow size distributions in significant quantities.23–26 The well-characterized Cu6 nanocluster stands out among the small Cu nanoclusters because it can be easily made at high yields from inexpensive materials and sold commercially.27,28
Although great effort has been made to obtain a homogeneously distributed and well-dispersed metal phase, poor stability has always been an obstacle regarding copper-based catalysts. Even the catalytic activity of the commercial Cu/ZnO/Al2O3 decreases to half after 15 h.29–33 One possible approach to increase the stability of the catalyst involves using appropriate supports that can increase the active site surface area and decrease sintering by stabilizing or confining the metal centers.34 This work reports the preparation of a new catalyst (Cu6–ZnO–SBA-15) containing highly dispersed ultra-small copper nanoclusters confined in SBA-15 mesoporous channels. Mesoporous SBA-15 with a high-surface area (up to 1000 m2 g−1) and 6–7 nm wide regular channels allowing easy diffusion is utilized for immobilizing Cu6 nanoclusters with sizes of 1.6 nm.33,35
Furthermore, the presence of ZnO prevents the sintering of copper species, increasing both the Cu dispersion and its promotion effect on the catalytic activity.36,37 This work is the first report on using a Cu-based catalyst derived from the deposition of chemically synthesized, atomically precise Cu nanoclusters in a mesoporous material and its application in CO2 hydrogenation to methanol. The as-made catalyst is tested for CO2 hydrogenation to methanol under different reaction conditions to study the correlation between its catalytic performance and physicochemical features. The catalyst stability over time was investigated by ex situ experiments.
2. Experimental
2.1. Chemicals
All reactants were reagent grade and purchased from Sigma-Aldrich, except for copper(I) chloride (M&B Laboratory Chemicals, May & Baker Ltd, Dagenham, England). All solvents used in this study were purchased from Merck and degassed and dried before use. Cu6 clusters were synthesized through wet impregnation under completely air-free conditions using the Schlenk-line technique.
2.2. Characterization of materials
Powder X-ray diffraction (PXRD) analysis of the samples was conducted using a Rigaku Smartlab diffractometer with Cu Kα radiation (1.5418 Å, operating at 40 kV and 30 mA) equipped with a crossbeam optics Bragg–Brentano (BB) selection slit. The diffracted X-rays were collected using a Rigaku D/tex Ultra 250 1D detector. All diffraction patterns were collected using capillary tubes in Bragg–Brentano focusing mode at room temperature. The Scherrer equation was used to determine the crystal sizes of the nanoparticles:| | 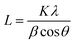 | (1) |
where L is the crystal dimension perpendicular to the reflection plane, λ is the wavelength of incident X-rays, β is the full-width half maximum of the diffraction peak, and K is a constant.
The surface area and pore characteristics of the materials were measured by the adsorption/desorption of N2 at 77 K using an Autosorb iQ-XR (Quantachrome Instruments, USA). Prior to the analysis, approximately 50 mg of the sample was degassed for 12 h at 120 °C. The specific surface area and pore volume were calculated using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) models. N2O titration was performed to determine the copper dispersion characteristics. An experiment involves typically weighing 30–50 mg of the sample into a quartz tube and attaching it to the instrument (BELCAT II (MicrotracBEL, Japan)). Before the test, the Cu nanoparticle and cluster-based catalyst were pretreated under a flow of 5% H2/Ar and He, respectively, at 220 °C for 30 min, after which 25–30 successive pulses of the titration gas mixture (1% N2O in He) were introduced using a calibrated injection valve (0.8650 mL N2O (STP) per pulse). To measure the amount of N2O consumed, a thermal conductivity detector (TCD) was used to monitor the ratio of N2O to N2 in the exhaust. The measurement was performed until the last three pulses demonstrated comparable areas (±1.2%). The quantities of surface metal sites were then determined by considering the following titration equations and calculations:
| | | 2Cu(s) + N2O → Cu2O(s) + N2 | (2) |
| | | Vm (cm3 g−1) = Vchem/m | (3) |
where
Vchem (cm
3) is the amount of N
2O adsorbed and
m is the sample weight, measured in grams (g).
| | 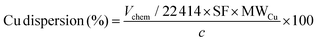 | (4) |
where MW
Cu (g mol
−1) is the atomic weight of Cu, the stoichiometric factor (SF) is assumed as 2, and
c is the copper weight in the sample, measured in grams (g) which were calculated using the following equation:
where
pCu is the weight percentage of Cu in the sample (wt%).
The surface areas of Cu per gram of the sample were calculated using the following equation:
| | 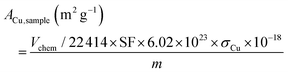 | (6) |
where
σCu (nm
2) is the cross-sectional area of one Cu atom.
While the surface area of Cu per gram of Cu in the sample was calculated using the following function:
| | 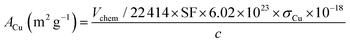 | (7) |
the particle size of copper, assuming a spherical particle, was calculated using the equation:
| | 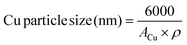 | (8) |
where
ρ (g cm
−3) is the density of Cu.
An Agilent 4210 microwave plasma-atomic emission spectrometer (MP-AES) equipped with an SPS 4 auto sampler was used for MP-AES. Before the test, the catalysts were treated with 2 mL of freshly prepared concentrated aqua regia solution, HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCl, at a ratio of 1:3 v/v at 80 °C for 2 d and then diluted to 250 mL with 2% v/v HNO3 solution in Milli-Q water. The aliquots of this solution were transferred into sample tubes for analysis by MP-AES. Transmission electron microscopy (TEM) images were obtained on a TECNAI F-20 transmission electron microscope operating at an acceleration voltage of 200 kV. Scanning transmission electron microscopy with energy-dispersive spectroscopy (STEM-EDS) elemental mapping of the catalysts was performed using the same spectrometer operated at 200 kV using a high-angle annular dark-field detector (HAADF) in STEM mode. The samples were analyzed using X-ray absorption spectroscopy (XAS) at the XAS beamline at the australian synchrotron, ANSTO Melbourne. XAS spectra were recorded at the Cu-K absorption edge in fluorescence mode in a helium atmosphere at room temperature using a 100-element solid-state HP-Ge detector (Canberra/Mirion, France). The excitation energy was selected using a Si(111) double crystal monochromator, which was calibrated at the Cu-K absorption edge using an inline Cu metal foil (first maximum of the first derivative at 8980.48 eV). Data were processed using Sakura (in-house program) and Athena for scan averaging, background subtraction and edge-height normalization.
:HCl, at a ratio of 1:3 v/v at 80 °C for 2 d and then diluted to 250 mL with 2% v/v HNO3 solution in Milli-Q water. The aliquots of this solution were transferred into sample tubes for analysis by MP-AES. Transmission electron microscopy (TEM) images were obtained on a TECNAI F-20 transmission electron microscope operating at an acceleration voltage of 200 kV. Scanning transmission electron microscopy with energy-dispersive spectroscopy (STEM-EDS) elemental mapping of the catalysts was performed using the same spectrometer operated at 200 kV using a high-angle annular dark-field detector (HAADF) in STEM mode. The samples were analyzed using X-ray absorption spectroscopy (XAS) at the XAS beamline at the australian synchrotron, ANSTO Melbourne. XAS spectra were recorded at the Cu-K absorption edge in fluorescence mode in a helium atmosphere at room temperature using a 100-element solid-state HP-Ge detector (Canberra/Mirion, France). The excitation energy was selected using a Si(111) double crystal monochromator, which was calibrated at the Cu-K absorption edge using an inline Cu metal foil (first maximum of the first derivative at 8980.48 eV). Data were processed using Sakura (in-house program) and Athena for scan averaging, background subtraction and edge-height normalization.
2.3. Synthesis of mesoporous SBA-15
In a typical preparation, 8.0 g of Pluronic P123 was dissolved in 240 mL of 2 M HCl solution with stirring at room temperature. Next, 60 mL of Milli-Q water and 17.00 g of tetraethyl orthosilicate were added to the mixture. After stirring at room temperature for 1 h, the mixture was aged for 24 h at 40 °C. The resulting gel was transferred into an autoclavable polypropylene bottle, tightly capped and kept at 100 °C for 48 h without stirring. Afterward, the capped bottle was cooled with running tap water to room temperature. The product was separated through filtration, followed by washing and overnight drying at 90 °C. The resulting as-made SBA-15 was calcined at 550 °C for 5 h.
2.4. Preparation of [(PPh3)CuH]6 denoted as Cu6
The preparation of Cu6 nanoclusters was done following the method reported by Albert et al.38 Typically, 1.340 g of triphenylphosphine and 0.525 g of copper(I) chloride were stirred in 10 mL of dry tetrahydrofuran (THF) under N2 for 10 min to obtain a white precipitate, which was then dissolved by adding K-selectride solution (5 mL of 1.0 M in THF, 5 mmol). After continuous stirring for 1 h, the mixture was filtered under vacuum, and the filtrate volume was reduced to half. Cu6 nanoclusters were crystallized after a few days in the presence of O2-free dry hexane and N2 gas at ambient temperature.
2.5. Preparation of ZnO–SBA-15
ZnO species were incorporated by the solid-state grinding of Zn(NO3)2·6H2O with as-made SBA-15 at room temperature for approximately 1 h. The resulting powder was calcined in the air at 500 °C for 5 h with a flow rate of 100 mL min−1. The calcined powder was denoted as nZnO–SBA-15, where n represents the wt% of ZnO.
2.6. Fabrication of nanocluster-based catalysts
To fabricate nanocluster-based catalysts, 0.2 g of Cu6 nanoclusters was rapidly transferred into a Schlenk flask under N2 to minimize decomposition, followed by adding 10 mL of dry and O2-free THF with a syringe. Based on a total catalyst weight of 1 g, 0.98 g of the support was dispersed in THF (10 mL) under a N2 atmosphere. An appropriate volume (5.3 mL) of the Cu6 solution containing 0.02 g of copper, equivalent to 2 wt% Cu, was added to the dispersed mixture, followed by stirring at room temperature for 5 h. After wet impregnation, the catalyst was dried under vacuum to evaporate the solvent and the sample was denoted as 2 wt% Cu6–support. The Cu content was varied to investigate the loading effect on the catalytic performance.
2.7. Fabrication of Cu nanoparticle-based catalysts
To prepare 1 g of the catalyst, 0.98 g of the support was dispersed and stirred in 10 mL deionized water. Then, 76 mg of Cu(NO3)3·3H2O was dissolved in 5 mL of deionized water to make an aqueous metal nitrate solution. Copper loading of 2 wt% was obtained by adding the prepared aqueous solution to the dispersed support, followed by stirring at room temperature for 5 h. The catalyst was dried at 80 °C and then calcined in an Ar flow of 100 mL min−1 at 400 °C for 3 h. The calcined catalyst was reduced in 10% H2/Ar for 1 h at 300 °C. The sample was denoted as 2%CuNP–support, with the NP referring to the fact that copper was present on the support in the nanoparticle form.
2.8. CO2 hydrogenation to methanol
The catalytic activity was investigated using a high-pressure fixed bed FlowCAT reactor. The column was packed with glass beads and quartz wool to support the catalytic bed (0.2 g, volume of ca. 0.5 cm3) inside the reactor. The activity measurements were conducted after a 30-min stabilisation period in which the inlet gas composition was monitored through a bypass line. The experiments were carried out at a pressure of 5.0 MPa and a gas hourly space velocity (GHSV) of 7500 mL g−1 h−1 at 220 °C. The catalytic performance under different GHSVs and temperatures was also investigated. The reaction gas mixture was CO2 and H2 at a ratio of 1:3. A heating tape was used to keep the exit gas temperature over 90 °C to be analysed using an online gas chromatograph. The gaseous products from the CO2 reduction experiment were detected using an SRI 8610C Gas Chromatograph (multi-gas configuration) with TCD and FID detectors and Alltech HayeSep D and molecular sieve 13 × 80/100 6′ × 1/8′′ × 0.085′′ stainless steel packed columns. The CO2 conversion, X(CO2), the carbon-based selectivity, S(MeOH), and the space–time yield of methanol, STY(MeOH), were calculated using the the following equations:| | 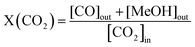 | (9) |
| | 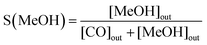 | (10) |
| | 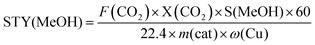 | (11) |
where m(cat) is the catalyst mass (g), F(CO2) is the inlet CO2 flow rate (mL min−1), and ω(Cu) is the copper mass percentage in the catalyst, and the STY unit would be molMeOH kgCu−1 h−1.
3. Results and discussion
3.1. Catalyst characterization
X-ray diffraction (XRD) revealed that the structure of the pure Cu6 nanocluster matched that which was previously reported (CCDC 1864974) (Fig. 1). It is worth mentioning that the slight change in the peak intensity might be indicative of a change in the amount of tetrahydrofuran (THF) between the two structures, i.e., solvent loss. In addition, the XRD patterns did not show any crystalline phases related to copper species, e.g., metallic Cu, Cu(I) oxide, and Cu(II) oxide, which would normally appear after 2-theta of 30°.39–41
 |
| | Fig. 1 XRD patterns of (a) the synthesized Cu6 nanocluster and (b) the simulated Cu6 nanocluster (CCDC 1864974). | |
Fig. 2a shows the low-angle XRD patterns of the catalyst series. As can be seen, all the patterns display three well-resolved peaks associated with the hexagonal symmetry of the support and can be identified as the (100), (110), and (200) reflections. This finding is consistent with a two-dimensional hexagonal (p6mm) structure, implying the intact structure of SBA-15 after nanocluster deposition.35 The wide-angle XRD patterns obtained for the catalysts are shown in Fig. 2b. Clearly, there are no ZnO crystalline phases in the XRD patterns of 10ZnO–SBA-15, confirming the homogeneous distribution of ZnO inside SBA-15 channels. This phenomenon could be due to the strong interactions between the silica matrix and zinc species, preventing ZnO crystallization, but instead forming zinc silicate layers with a two-dimensional disordered structure on the surface.42
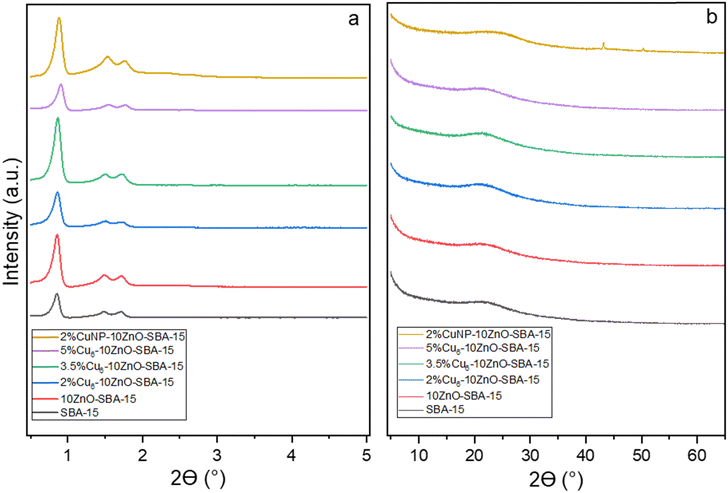 |
| | Fig. 2 (a) Low-angle and (b) wide-angle XRD patterns of the fresh catalysts. | |
Interestingly, no diffraction peaks of any form of copper are found in any of the samples. This can be ascribed to the ultra-small sizes of individual Cu6 nanoclusters and the uniform dispersion of CuNCs on the support. The broad peak at 2θ = 23.1° present in all the XRD patterns is attributed to the mesoporous SBA-15 support. The catalysts with supported copper nanoparticles show two diffraction peaks at 2θ = 43.2° and 50.3°, indexing to the (111) and (200) characteristic facets of metallic Cu, respectively. The average size of Cu NPs (Dcrystallite) from the Scherrer equation using the most intense peak (111) is about 26 nm.
Fig. 3 demonstrates the nitrogen adsorption–desorption isotherms of the as-made catalysts with various metal loadings. The support exhibits the characteristic features of mesoporous materials with uniform cylindrical pores, a type IV isotherm with an H1 hysteresis loop.43 It is evident from the type IV shape of the isotherms that the solid support retains its mesoporous nature after the impregnation of the clusters. Nonetheless, the adsorbed volume decreases as the pores are partially filled by increasing the active phase loading.44 As shown in Table 1, the pore volume and surface area of the SBA-15 are decreased after loading the active phase, indicating that the active phase is confined within the SBA-15 channels.45SBET and Vp for the primary support are higher than those of the as-made catalysts, implying that the mesopores are partially filled.37 As seen from Table 1, a high metal loading (3.5 and 5%Cu6–10ZnO–SBA-15) significantly decreases the specific surface area and mesopore volume compared with the primary support. The textural properties change dramatically for metal loadings higher than 2 wt% of Cu. It is worth noting that the values for the supported Cu nanoparticles are very close but slightly greater than that of the nanocluster (Table 1).
 |
| | Fig. 3 Nitrogen adsorption–desorption isotherms of the catalysts and SBA-15. | |
Table 1 Textural properties of the catalysts, support, and the prepared SBA-15
| Sample |
S
BET
(m2 g−1) |
S
meso
(m2 g−1) |
S
micro
(m2 g−1) |
D
p
(nm) |
V
p (cm3 g−1)d |
|
BET-specific surface area.
Determined by using the BJH model for the adsorption branch of the isotherm.
Determined as the difference between the total surface area and the mesopore surface area.
Total pore volume estimated at P/P0 = 0.98.
|
| SBA-15 |
773.2 |
529.7 |
243.5 |
6.89 |
1.05 |
| 10ZnO–SBA-15 |
679.0 |
513.0 |
163.0 |
6.57 |
0.95 |
| 2%Cu6–10ZnO–SBA-15 |
562.7 |
433.2 |
129.5 |
6.43 |
0.89 |
| 3.5%Cu6–10ZnO–SBA-15 |
359.3 |
282.9 |
76.4 |
6.17 |
0.61 |
| 5%Cu6–10ZnO–SBA-15 |
262.1 |
207.1 |
13.4 |
5.11 |
0.43 |
| 2%CuNP–10ZnO–SBA-15 |
595.4 |
486.9 |
108.5 |
6.44 |
0.92 |
N2O titration is used to determine the specific surface areas of copper in the catalysts, and the corresponding information is summarized in Table 2. As the active phase loading increases, metal dispersion decreases from 15.2% to 0.99%, implying the formation of larger copper nanoclusters. The metal surface area of 2%Cu6–10ZnO–SBA-15 is the best compared to the catalysts with higher metal loadings. Interestingly, the catalyst with deposited copper nanoparticles (2%CuNP–10ZnO–SBA-15) shows a significantly lower Cu dispersion than that of the same amount of metal loading with the copper nanoclusters, contributing to the improved performance of the supported copper nanocluster catalytic system, which is discussed in later parts.
Table 2 Copper dispersion and surface area characteristics of the supported nanoclusters
| Samplea |
Cub (wt%) |
Cu particle diameter (nm) |
Cu dispersion (%) |
Cu surface area per gram sample (m2 g−1) |
Cu surface area per gram Cu (m2 g−1) |
|
The support was 10ZnO–SBA-15 for all the samples.
Actual values from the atomic emission spectroscopy (AES) analysis.
|
| 2%Cu6 |
1.89 |
6.83 |
15.27 |
1.969 |
98.43 |
| 3.5%Cu6 |
3.38 |
12.86 |
8.11 |
1.830 |
52.28 |
| 5%Cu6 |
4.84 |
21.89 |
4.77 |
1.537 |
30.74 |
| 2%CuNP |
1.93 |
27.40 |
3.81 |
0.491 |
24.55 |
The transmission electron microscopy (TEM) image of the 2%Cu6–10ZnO–SBA-15 sample (Fig. 4a) does not exhibit isolated metal particles on the support. Meanwhile, for 2%CuNP–10ZnO–SBA-15 (Fig. 4b), randomly distributed Cu particles are observed on the external surface of the support. High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) (Fig. 5) and the energy-dispersive X-ray analysis (EDS) of 2%Cu6–10ZnO–SBA-15 confirm that Cu nanoclusters and Zn species are distributed homogenously throughout the architecture. These results are consistent with those derived from XRD, N2 adsorption–desorption, and N2O chemisorption analyses discussed earlier. It is worth noting that although the elemental mapping shows the presence of ligands, it is a well-established fact that the immobilized triphenylphosphine-ligated nanoclusters lose a significant amount of PPh3 ligands during deposition. The ligands remain on the support.24,26,46–49 However, the formation of ligand-free sites on phosphine-stabilized deposited nanoclusters strongly depends on the support properties, which require in-depth studies.
 |
| | Fig. 4 TEM images of (a) 2%Cu6–10ZnO–SBA-15 and (b) 2%CuNP–10ZnO–SBA-15. | |
 |
| | Fig. 5 HAADF-STEM image and the corresponding EDS elemental mappings of 2%Cu6–10ZnO–SBA-15. | |
3.2. Catalytic activity
The catalytic activity of the catalysts is evaluated at 220 °C under 5 MPa and a gas hourly space velocity (GHSV) of 7500 mL g−1 h−1. Below a 5 MPa reaction pressure, CO is the only product detected. Table 3 shows the activity results of various Cu6–10ZnO–SBA-15 catalysts for synthesizing methanol through CO2 hydrogenation. Under the reaction conditions (220 °C, 5 MPa, and a GHSV of 7500 mL g−1 h−1), the only produced hydrocarbons are methanol and CO. As can be seen, Cu loading significantly affects the methanol yield in Cu6–10ZnO–SBA-15. Among the investigated catalysts, 2%Cu6–10ZnO–SBA-15 shows the best STY (232.5 molMeOH kgCu−1 h−1) and the highest methanol selectivity (86%).
Table 3 Activity of the catalysts with various metal loadings. Reaction conditions: T = 220 °C; P = 5.0 MPa; H2/CO2 = 3; GHSV = 7500 mL g−1 h−1; mcat = 0.2 g
| Sample |
STY (molMeOH kgCu−1 h−1) |
CO2 conv. (%) |
MeOH select. (%) |
| 2%Cu6–10ZnO–SBA-15 |
232.5 |
6.7 |
86 |
| 3.5%Cu6–10ZnO–SBA-15 |
168.3 |
8.6 |
85 |
| 5%Cu6–10ZnO–SBA-15 |
126.4 |
9.5 |
83 |
| 2%CuNP–10ZnO–SBA-15 |
118.5 |
5.6 |
53 |
Contrary to expectations, the methanol yield does not improve with increasing the active phase loading from 2 to 5 wt%. According to the characterization results, this phenomenon can be explained by the fact that there are fewer active sites exposed as the metal loading increases. Another possible reason is the partial blocking of pores by copper nanoclusters, limiting access to the active sites inside SBA-15 channels. It is worth noting that with the same active phase loading, 2%Cu6–10ZnO–SBA-15 shows significantly better activity and selectivity than 2%CuNP–10ZnO–SBA-15.
On the other hand, according to previous studies, two active centers are involved during catalytic CO2 reduction to methanol. One center is the active support that adsorbs CO2 as the carbonate and bicarbonate species. Later, methanol is produced by stepwise hydrogenation of these intermediates. The other center is the Cu phase, which chemisorbs H2 and thus provides atomic hydrogen to the surface.50–53 Therefore, the catalytic performance depends not only on the Cu nanocluster properties but also on other parameters, such as the position of the active phase and the Cu/Zn molar ratio. To investigate the role of ZnO and Cu/Zn ratio effect, a series of 2%Cu6–XZnO–SBA-15 catalysts with the same amount of copper are prepared, the performances of which are shown in Fig. 6. It is evident that ZnO addition affects the catalytic performance remarkably. The methanol selectivity for 2%Cu6–0ZnO–SBA-15 (38.8%) increases to 60% with the addition of 2% ZnO for 2%Cu6–2ZnO–SBA-15 and reaches its optimum when the Cu/Zn molar ratio is 0.2. By increasing the Zn content to 20%, a slight reduction in methanol productivity to 226.5 (molMeOH kgCu−1 h−1) is observed for 2%Cu6–20ZnO–SBA-15. Increasing the zinc oxide content to >10% decreases catalyst activity and selectivity. As explained before, there is no ZnO crystalline phase in the XRD pattern of the catalyst, implying the full dispersion of ZnO. However, zinc is predominantly deposited in the micropores of SBA-15, which is not accessible for our nanoclusters due to their larger size.42 Therefore, although catalytic results demonstrate that the catalyst with a Cu/Zn molar ratio of 1 exhibits the best catalytic performance for Cu/ZnO–SBA-15,37 it requires a higher loading of ZnO for the catalytic system to ensure that a sufficient amount of ZnO is accessible to each copper center.
 |
| | Fig. 6 Performance comparison of the catalysts with different Cu/Zn ratios in terms of the space–time yield and selectivity to methanol. | |
Previous studies have demonstrated that the undercoordinated sites of nanoclusters can improve CO2 and hydrogen adsorption at low pressures, thereby promoting CO2 activation, which results in methanol production.20–22 One study revealed that the cluster size affects the catalytic activity of CO2 hydrogenation to methanol. An increased particle size significantly weakens the binding strength between intermediate adsorbates and the Cu cluster. As a result, the intermediate states become less stable, and therefore the overall energy of the reaction pathway.22 Moreover, it has been reported that the calculation of the reaction pathway for CO2 hydrogenation to methanol on Cu cluster resembles that of large Cu particles,20,21,54,55 where HCOO* species are formed first from the beginning of methanol synthesis, then hydrogenated to form HCOOH and subsequently H2COOH*. The next step involves the C–OH bond cleavage, where H2COOH* dissociates into H2CO* and OH*. OH* is then further hydrogenated to H2O*, while H2CO* yields H3CO*. Finally, the hydrogenation of H3CO* results in the formation of the methanol product (CH3OH). Prior research suggests that the energy barrier of the rate-limiting step for methanol production via hydrogenation of the HCOO* species on Cu nanoclusters is lower than the projected rate-limiting barriers for the Cu (111) surface. This implies that, unlike bulk and large Cu materials, Cu clusters may exhibit superior catalytic activity for CH3OH synthesis.21,22,56,57 On the other hand, the addition of ZnO can enhance Cu dispersion through strong interactions between Cu and ZnO. ZnO plays the role of regulating the electronic properties of the catalyst.58–61 The interaction between Cu and ZnO is crucial for methanol synthesis as the interface enhances the adsorption and activation of CO2 and H2, thereby promoting methanol synthesis.62 Recent density functional theory (DFT) calculations have also further demonstrated that Zn alloying into the Cu step can improve the adsorption strength and lower the barrier for HCO, H2CO, and H3CO, thus promoting the formation of CH3OH.54 The reaction mechanism of the ZnO–SBA-15 supported Cu6 catalyst can be deduced within the larger context of Cu–ZnO catalysis, in which the interfacial sites are mainly responsible for the catalytic activity. The cluster effect may provide an advantage by increasing the interfacial sites per Cu atom when compared to bulk and large Cu particles.
The catalyst is tested at various temperatures (200–280 °C) and gas hourly space velocities (7500–12000 mL g−1 h−1) to investigate the catalytic activity under different reaction conditions. In line with previous studies, increasing the reaction temperature increases CO2 conversion while it decreases the selectivity toward methanol production.63,64 It is widely accepted that reverse water–gas shift (RWGS) reactions and methanol synthesis coincide and compete, as shown below:
| | | CO2 + 3H2 ↔ CH3OH + H2O ΔH298K = −49.4 kJ mol−1 | (12) |
| | | CO2 + H2 ↔ CO + H2O ΔH298K = +41.0 kJ mol−1 | (13) |
Methanol formation is thermodynamically exothermic as opposed to the RWGS reaction, which is endothermic. Therefore, high temperature increases the RWGS reaction rate but inhibits methanol formation. Nonetheless, considering low reaction rates and the inert nature of CO2, increasing the reaction temperature to more than 200 °C boosts carbon dioxide activation and methanol formation.65 The catalyst activity under various GHSVs is also investigated, and the results are shown in Fig. 7. As the GHSV increases, the CO2 conversion decreases due to the unsaturated contact time between the reactants and the catalyst. However, the methanol selectivity remains at approximately 85% with minor fluctuations. According to the results, the catalyst contact time significantly affects the CO2 conversion but not the selectivity, which is in line with what was reported by other researchers previously.66
 |
| | Fig. 7 Catalytic performance of 2%Cu6–10ZnO–SBA-15 at different (a) GHSVs and (b) reaction temperatures. | |
Table 4 compares the catalyst prepared in this study with the most recent similar catalytic systems, from which the superior performance of the catalyst in the present study is evident. It should be noted that throughout the investigation, CO2/H2 = 1:3 is used as the reactant gas mixture for all the tested catalysts. This comparison shows the superior activity of the catalyst studied in this work compared to that of the previous catalytic systems.
Table 4 A comparison of the catalytic performance of the synthesized catalyst with that of the catalysts was reported in the literature
| Catalyst |
Temp. and pressure (°C bar−1) |
GHSV (mL g−1 h−1) |
WHSV (mL g−1 h−1) |
Methanol yield (mol kgCu−1 h−1) |
CO2 conv. (%) |
Select. (%) |
Ref. |
| Cu1La0.2/SBA-15 |
240/30 |
12000 |
— |
59.6 |
5.7 |
81.2 |
67
|
| Cu/ZnO–SBA-15 |
250/30 |
44000 |
— |
90.0 |
8.9 |
27.7 |
37
|
| Cu/ZnO/SBA-15 |
180/40 |
— |
60000 |
162.2 |
7.7 |
97.3 |
68
|
| Cu/ZnO@m-SiO2 |
250/50 |
— |
6000 |
36.3 |
9.8 |
66.6 |
69
|
| 2%Cu6–10ZnO–SBA-15 |
220/50 |
7500 |
— |
232.5 |
6.7 |
86.1 |
This work |
| 2%Cu6–10ZnO–SBA-15 |
220/50 |
12000 |
— |
173.0 |
5.2 |
83.0 |
This work |
3.3. Catalyst stability
The catalyst stability is investigated by performing a 9-h continuous test. The catalyst stability with regard to the space–time yield (STY) shows a decrease within the first 3 h for the supported nanoclusters, as shown in Fig. 8.
 |
| | Fig. 8 (a) Space–time yield and (b) selectivity of methanol versus time-on-stream of the catalysts. Reaction conditions: T = 220 °C; P = 5.0 MPa; H2/CO2 = 3 mol mol−1; GHSV = 7500 mL g−1 h−1; mcat = 0.2 g. | |
The 2%Cu6–10ZnO–SBA-15 catalyst shows the best activity for methanol production together with the latest STY reduction (3 h). A faster reduction is observed for the 3.5%Cu6–10ZnO–SBA-15 and 5%Cu6–10ZnO–SBA-15 catalysts (2 h and 1 h, respectively). This effect can be attributed to copper sintering, consistent with the previous work.70–72 The result can be further explained by XRD, TEM, and N2O titration data. As discussed, the confinement of a high copper content (3.5 and 5 wt%) leads to lower copper dispersion and faster sintering during the reaction, decreasing methanol activity. It is important to note that catalyst deactivation via sintering and agglomeration differ in the mechanism and impact. Sintering involves atomic migration and coalescence of active metal particles, reducing the surface area and catalytic activity irreversibly. In contrast, agglomeration is the physical clumping of catalyst particles due to mechanical forces or thermal instability, often leading to blocked active sites, but sometimes reversible. While sintering typically affects supported metal catalysts at the nanoscale, agglomeration is more common in powdered catalysts and occurs at a larger, physical scale. In this work, the Cu nanoclusters may undergo sintering, which reduces their activity. It is also worth noting that although 2%CuNP–ZnO–SBA-15 seems more stable compared to the supported nanocluster catalytic systems with the same copper loading, the activity of the latter remains significantly higher.
To gain further insight into the stability of the catalyst, the ex situ characterization of 2%Cu6–10ZnO–SBA-15 was conducted every 3 h, and the relevant results are explained below. The copper surface areas of the collected catalyst are determined by N2O titration (Table 5). As can be seen, metal dispersion and surface areas decrease over time, confirming the activity reduction due to sintering. Interestingly, the Cu surface area per Cu weight reduces by about 52% over time, reaching 47.23 m2 g−1. This value remains approximately twice as high as the Cu surface area for 2%CuNP–10ZnO–SBA-15, with a value of 24.55 m2 g−1.
Table 5 Copper dispersion for 2%Cu6–10ZnO–SBA-15 collected at different time-on-stream (TOS)
| TOS |
Cu particle diameter (nm) |
Cu dispersion (%) |
Cu surface area per gram sample (m2 g−1) |
Cu surface area per gram Cu (m2 g−1) |
| 0 h |
6.83 |
15.27 |
1.967 |
98.43 |
| 3 h |
9.47 |
11.02 |
1.420 |
71.02 |
| 6 h |
13.26 |
7.87 |
1.014 |
50.73 |
| 9 h |
14.24 |
7.33 |
0.944 |
47.23 |
The XANES, EXAFS and Fourier-transform EXAFS spectra of the supported nanoclusters and nanoparticles are presented in Fig. 9. Because of the element selectivity of XAS and its independence of crystalline order, the data can be highly informative about the structure and redox state. The edge position of deposited Cu6 clusters and nanoparticles lies between the edge position of metallic Cu and CuO. However, the phase appears to differ from any standard (i.e., not just a linear combination). It is important to note that all the samples were exposed to air before XAS measurements, so the results do not reflect the state of the catalyst in situ during the reaction. However, it can be seen from Fig. 9 that there is a systematic change that occurs over time. The edge energy of the supported clusters is shifted to a higher energy with an increase in the running time, which can be attributed to the loss of PPh3 ligands as time passes. The P/Cu ratio is essential because the coordination of electron-donating PPh3 to Cu affects the edge energy. Therefore, the edge position of the spent samples is at the higher energies; however, the difference is minor.
 |
| | Fig. 9 (a) X-ray absorption near-edge structure (XANES), (b) extended X-ray absorption fine structure (EXAFS) and (c) Fourier-transform (FT) EXAFS of the supported nanoclusters and nanoparticles. | |
Interestingly, HAADF-STEM characterization and EDS elemental mapping on 2%Cu6–10ZnO–SBA-15 after being on stream for 9 h (Fig. 10) shows the co-existence of larger nanoparticles and small nanoclusters. This phenomenon is because the confined nanoclusters inside the mesoporous channels survive sintering to some extent. In contrast, the nanoclusters positioned on the surface of the support undergo severe sintering, leading them to form larger copper nanoparticles.
 |
| | Fig. 10 HAADF-STEM image and EDS elemental mappings of the used 2%Cu6–10ZnO–SBA-15. | |
It is evident from the low-angle diffraction pattern that the used 2%Cu6–10ZnO–SBA-15 (9 h on the stream) catalyst maintains its hexagonal structure (Fig. 11a) compared to the fresh catalyst pattern. Only a slight shift toward higher angles indicates a slight shrinkage in mesoporous structures. Based on the wide-angle XRD patterns of the sample (Fig. 11b), a low-intensity diffraction peak associated with the metallic copper phase is seen at 2θ = 43.2° apart from the broad peak related to amorphous silica (PDF card 4-0836), confirming the presence of larger nanoparticles.
 |
| | Fig. 11 XRD patterns of the fresh and used 2%Cu6–10ZnO–SBA-15 catalysts at a (a) low-angle and (b) wide-angle. | |
4. Conclusions
Cu6 nanoclusters were synthesized and deposited on ZnO–SBA-15, a mesoporous silica framework with deposited ZnO, with different metal loadings (2, 3.5, and 5 wt%) and Cu/Zn molar ratios (0, 1, 2.5, 5 and 10). Characterization of the catalyst was evaluated by XRD, TEM, SEM with STEM-EDS, nitrogen adsorption–desorption analysis (BET), N2O titration, atomic emission spectroscopy (AES), and XAS. The characterization results demonstrated that the active phase was highly dispersed and properly confined inside the support channels. The activity of immobilized nanoclusters was tested under high pressure in the gas phase for CO2 hydrogenation and compared with supported nanoparticles. The methanol yield for the supported small nanoclusters (232.5 (molMeOH kgCu−1 h−1)) was approximately twice as high as that of the supported copper nanoparticles (118.5 (molMeOH kgCu−1 h−1)). Furthermore, the catalyst was tested at different reaction temperatures (200–280 °C) and gas hourly space velocities (7500–12000 mL g−1 h−1) to investigate its behavior under different reaction conditions. To study the stability of the catalyst, the catalyst samples were collected every 3 h from the reactor and were characterized by different methods. The activity and selectivity slightly decreased over the first 3 h of the reaction due to the sintering of small nanoclusters. However, the activity of the supported nanoclusters remained significantly higher than that of the supported nanoparticles even after sintering. The synthesis cost of the ZnO–SBA-15 supported Cu6 catalyst is highly associated with the synthesis cost of the Cu6 cluster, which requires the use of expensive ligands. Further research on a low-cost ligand suitable for making Cu6 clusters is key to realizing process scale-up.
Author contributions
Sara Mirzakhani: conceptualization, methodology, validation, formal analysis, investigation, and writing – original draft. Angie F. J. Tan: investigation and writing – review & editing. Deborah Crittenden: formal analysis, writing – original draft, and supervision. Majid Masteri-Farahani: formal analysis, investigation, and writing – original draft. Alex C. K. Yip: formal analysis, writing – original draft and review & editing, and supervision.
Conflicts of interest
There are no conflicts to declare.
Data availability
Readers may contact the corresponding author, A. C. K. Y., to inquire about the data supporting the findings in this study.
Acknowledgements
The authors thank the University of Canterbury Doctoral Scholarship for supporting Sara Mirzakhani. We also acknowledge the Australian Synchrotron facilities for conducting the X-ray absorption spectroscopy experiment in this work.
References
-
Global Climate Change-The Technology Challenge, ed. F. T. Princiotta, Springer, New York, 2011, pp. 1–50 Search PubMed.
-
Climate change: observed impacts on planet Earth, ed. T. M. Letcher, Elsevier, 2021 Search PubMed.
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS.
- M. Aresta, A. Dibenedetto and E. Quaranta, J. Catal., 2016, 343, 2–45 CrossRef CAS.
-
I. Landälv, Methanol as a renewable fuel–a knowledge synthesis, The Swedish Knowledge Centre for Renewable Transportation Fuels, Sweden, 2017, p. 6 Search PubMed.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- F. A. Rahman, M. M. A. Aziz, R. Saidur, W. A. W. A. Bakar, M. Hainin, R. Putrajaya and N. A. Hassan, Renewable Sustainable Energy Rev., 2017, 71, 112–126 CrossRef.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS.
- J. Yao, J. Yan, Y. Huang, Y. Li, S. Xiao and J. Xiao, Front. Chem., 2018, 6, 442 CrossRef CAS.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- T. Phongamwong, U. Chantaprasertporn, T. Witoon, T. Numpilai, Y. Poo-Arporn, W. Limphirat, W. Donphai, P. Dittanet, M. Chareonpanich and J. Limtrakul, Chem. Eng. J., 2017, 316, 692–703 CrossRef CAS.
- X. Cui and S. K. Kær, Chem. Eng. J., 2020, 393, 124632 CrossRef CAS.
- E. Lam, K. Larmier, P. Wolf, S. Tada, O. V. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 10530–10535 CrossRef CAS.
- J.-Y. Li, L. Yuan, S.-H. Li, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2019, 7, 8676–8689 RSC.
- L. Zheng, X. Li, W. Du, D. Shi, W. Ning, X. Lu and Z. Hou, Appl. Catal., B, 2017, 203, 146–153 CrossRef.
- Z. Yuan, L. Wang, J. Wang, S. Xia, P. Chen, Z. Hou and X. Zheng, Appl. Catal., B, 2011, 101, 431–440 CrossRef.
- S. Vajda and M. G. White, ACS Catal., 2015, 5, 7152–7176 CrossRef.
- C. Liu, H. He, P. Zapol and L. A. Curtiss, Phys. Chem. Chem. Phys., 2014, 16, 26584–26599 RSC.
- D. R. Kauffman, D. Alfonso, C. Matranga, H. Qian and R. Jin, J. Am. Chem. Soc., 2012, 134, 10237–10243 CrossRef PubMed.
- Y. Yang, J. Evans, J. A. Rodriguez, M. G. White and P. Liu, Phys. Chem. Chem. Phys., 2010, 12, 9909–9917 RSC.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef.
- B. Yang, C. Liu, A. Halder, E. C. Tyo, A. B. F. Martinson, S. Seifert, P. Zapol, L. A. Curtiss and S. Vajda, J. Phys. Chem. C, 2017, 121, 10406–10412 CrossRef.
- E. C. Tyo and S. Vajda, Nat. Nanotechnol., 2015, 10, 577–588 CrossRef PubMed.
- D. P. Anderson, J. F. Alvino, A. Gentleman, H. Al Qahtani, L. Thomsen, M. I. Polson, G. F. Metha, V. B. Golovko and G. G. Andersson, Phys. Chem. Chem. Phys., 2013, 15, 3917–3929 RSC.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef.
- H. S. Al Qahtani, K. Kimoto, T. Bennett, J. F. Alvino, G. G. Andersson, G. F. Metha, V. B. Golovko, T. Sasaki and T. Nakayama, J. Chem. Phys., 2016, 144, 114703 CrossRef PubMed.
- W. U. Khan, I. K. M. Yu, Y. Sun, M. I. Polson, V. Golovko, F. L. Y. Lam, I. Ogino, D. C. W. Tsang and A. C. K. Yip, Environ. Pollut., 2021, 279, 116899 CrossRef PubMed.
- L. Baharudin, A. C. K. Yip, V. B. Golovko, M. I. Polson, K.-F. Aguey-Zinsou and M. J. Watson, Appl. Catal., B, 2020, 262, 118265 CrossRef.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef.
- M. V. Twigg and M. S. Spencer, Top. Catal., 2003, 22, 191–203 CrossRef.
- H. H. Kung, Catal. Today, 1992, 11, 443–453 CrossRef.
- B. An, J. Zhang, K. Cheng, P. Ji, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 3834–3840 CrossRef.
- Z. Zhao, ChemCatChem, 2020, 12, 3960–3981 CrossRef.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- T. Fujitani and J. Nakamura, Catal. Lett., 1998, 56, 119–124 CrossRef CAS.
- M. Mureddu, F. Ferrara and A. Pettinau, Appl. Catal., B, 2019, 258, 117941 CrossRef CAS.
- C. F. Albert, P. C. Healy, J. D. Kildea, C. L. Raston, B. W. Skelton and A. H. White, Inorg. Chem., 1989, 28, 1300–1306 CrossRef CAS.
- L. Baharudin, A. C. K. Yip, V. B. Golovko, M. I. Polson and M. J. Watson, Chem. Eng. J., 2019, 377, 120278 CrossRef CAS.
- X. Liu, B. Geng, Q. Du, J. Ma and X. Liu, Mater. Sci. Eng., A, 2007, 448, 7–14 CrossRef.
-
C. F. Holder and R. E. Schaak, Tutorial on powder X-ray diffraction for characterizing nanoscale materials, ACS Publications, 2019, pp. 7359–7365 Search PubMed.
- O. Tkachenko, K. Klementiev, E. Löffler, I. Ritzkopf, F. Schüth, M. Bandyopadhyay, S. Grabowski, H. Gies, V. Hagen and M. Muhler, Phys. Chem. Chem. Phys., 2003, 5, 4325–4334 RSC.
- A. Gervasini and S. Bennici, Appl. Catal., A, 2005, 281, 199–205 CrossRef CAS.
- Q. Zhang, T. Gao, J. M. Andino and Y. Li, Appl. Catal., B, 2012, 123, 257–264 CrossRef.
- A. García-Trenco and A. Martínez, Catal. Today, 2013, 215, 152–161 CrossRef.
- D. P. Anderson, R. H. Adnan, J. F. Alvino, O. Shipper, B. Donoeva, J.-Y. Ruzicka, H. Al Qahtani, H. H. Harris, B. Cowie and J. B. Aitken, Phys. Chem. Chem. Phys., 2013, 15, 14806–14813 RSC.
- R. H. Adnan, G. G. Andersson, M. I. J. Polson, G. F. Metha and V. B. Golovko, Catal. Sci. Technol., 2015, 5, 1323–1333 RSC.
- A. Longo, E. J. de Boed, N. Mammen, M. van der Linden, K. Honkala, H. Häkkinen, P. E. de Jongh and B. Donoeva, Chem. – Eur. J., 2020, 26, 7051–7058 CrossRef PubMed.
- C. C. Chusuei, X. Lai, K. A. Davis, E. K. Bowers, J. P. Fackler and D. W. Goodman, Langmuir, 2001, 17, 4113–4117 CrossRef.
- J. Xiao, D. Mao, X. Guo and J. Yu, Appl. Surf. Sci., 2015, 338, 146–153 CrossRef.
- I. A. Fisher and A. T. Bell, J. Catal., 1997, 172, 222–237 CrossRef.
- X. Guo, D. Mao, G. Lu, S. Wang and G. Wu, J. Mol. Catal. A: Chem., 2011, 345, 60–68 CrossRef.
- F. Arena, G. Mezzatesta, G. Zafarana, G. Trunfio, F. Frusteri and L. Spadaro, Catal. Today, 2013, 210, 39–46 CrossRef.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef PubMed.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef.
- A. Mravak, S. Vajda and V. Bonačić-Koutecký, J. Phys. Chem. C, 2022, 126, 18306–18312 CrossRef CAS.
- J. Yao, B. Wang, H. Chen, Z. Han, Y. Wu, Z. Cai, G. W. Manggada, M. A. Elsayed and S. Zhou, Int. J. Hydrogen Energy, 2024, 78, 1089–1098 CrossRef CAS.
- J. Li, T. Du, Y. Li, H. Jia, Y. Wang, Y. Song and X. Fang, J. Catal., 2022, 409, 24–32 CrossRef CAS.
- F. Arena, G. Italiano, K. Barbera, S. Bordiga, G. Bonura, L. Spadaro and F. Frusteri, Appl. Catal., A, 2008, 350, 16–23 CrossRef CAS.
- A. Karelovic, G. Galdames, J. C. Medina, C. Yévenes, Y. Barra and R. Jiménez, J. Catal., 2019, 369, 415–426 CrossRef CAS.
- Y. Jiang, H. Yang, P. Gao, X. Li, J. Zhang, H. Liu, H. Wang, W. Wei and Y. Sun, J. CO2 Util., 2018, 26, 642–651 CrossRef CAS.
- X. Wei, W. Su, Y. Shi, J. Wang, P. Lv, X. Song, Y. Bai, G. Xu and G. Yu, Int. J. Hydrogen Energy, 2024, 58, 128–136 CrossRef CAS.
- C. Zhong, X. Guo, D. Mao, S. Wang, G. Wu and G. Lu, RSC Adv., 2015, 5, 52958–52965 RSC.
- D. Chen, D. Mao, G. Wang, X. Guo and J. Yu, J. Sol-Gel Sci. Technol., 2019, 89, 686–699 CrossRef CAS.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS.
- Q. Tan, Z. Shi and D. Wu, Ind. Eng. Chem. Res., 2018, 57, 10148–10158 CrossRef CAS.
- K. Chen, H. Fang, S. Wu, X. Liu, J. Zheng, S. Zhou, X. Duan, Y. Zhuang, S. C. E. Tsang and Y. Yuan, Appl. Catal., B, 2019, 251, 119–129 CrossRef CAS.
- M. K. Koh, Y. J. Wong, S. P. Chai and A. R. Mohamed, J. Ind. Eng. Chem., 2018, 62, 156–165 CrossRef CAS.
- H. Yang, P. Gao, C. Zhang, L. Zhong, X. Li, S. Wang, H. Wang, W. Wei and Y. Sun, Catal. Commun., 2016, 84, 56–60 CrossRef CAS.
- T. Witoon, J. Chalorngtham, P. Dumrongbunditkul, M. Chareonpanich and J. Limtrakul, Chem. Eng. J., 2016, 293, 327–336 CrossRef CAS.
- M. Kurtz, H. Wilmer, T. Genger, O. Hinrichsen and M. Muhler, Catal. Lett., 2003, 86, 77–80 CrossRef CAS.
- H. Lei, R. Nie, G. Wu and Z. Hou, Fuel, 2015, 154, 161–166 CrossRef CAS.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Majid
Masteri-Farahani
c and
Alex C. K.
Yip
b,
Majid
Masteri-Farahani
c and
Alex C. K.
Yip


















