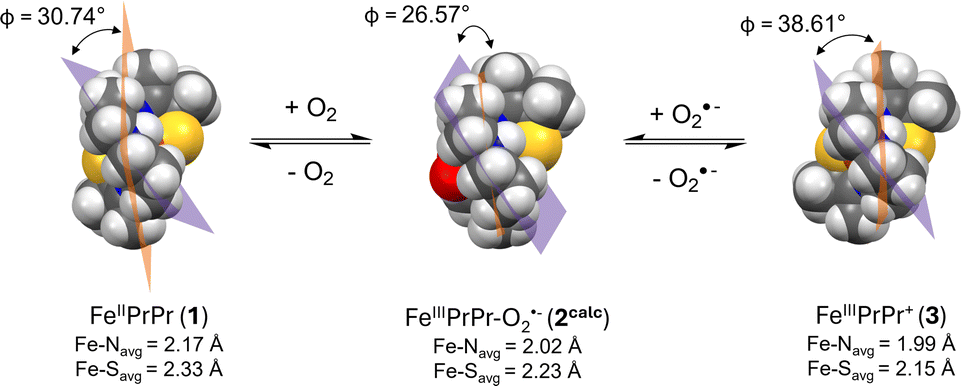DOI:
10.1039/D4SC02787F
(Edge Article)
Chem. Sci., 2024,
15, 12710-12720
Exploring the influence of H-bonding and ligand constraints on thiolate ligated non-heme iron mediated dioxygen activation†
Received
26th April 2024
, Accepted 5th July 2024
First published on 9th July 2024
Abstract
Converting triplet dioxygen into a powerful oxidant is fundamentally important to life. The study reported herein quantitatively examines the formation of a well-characterized, reactive, O2-derived thiolate ligated FeIII-superoxo using low-temperature stopped-flow kinetics. Comparison of the kinetic barriers to the formation of this species via two routes, involving either the addition of (a) O2 to [FeII(S2Me2N3(Pr,Pr))] (1) or (b) superoxide to [FeIII(S2Me2N3(Pr,Pr))]+ (3) is shown to provide insight into the mechanism of O2 activation. Route (b) was shown to be significantly slower, and the kinetic barrier 14.9 kJ mol−1 higher than route (a), implying that dioxygen activation involves inner-sphere, as opposed to outer sphere, electron transfer from Fe(II). H-bond donors and ligand constraints are shown to dramatically influence O2 binding kinetics and reversibility. Dioxygen binds irreversibly to [FeII(S2Me2N3(Pr,Pr))] (1) in tetrahydrofuran, but reversibly in methanol. Hydrogen bonding decreases the ability of the thiolate sulfur to stabilize the transition state and the FeIII-superoxo, as shown by the 10 kJ mol−1 increase in the kinetic barrier to O2 binding in methanol vs. tetrahydrofuran. Dioxygen release from [FeIII(S2Me2N3(Pr,Pr))O2] (2) is shown to be 24 kJ mol−1 higher relative to previously reported [FeIII(SMe2N4(tren))(O2)]+ (5), the latter of which contains a more flexible ligand. These kinetic results afford an experimentally determined reaction coordinate that illustrates the influence of H-bonding and ligand constraints on the kinetic barrier to dioxygen activation an essential step in biosynthetic pathways critical to life.
Introduction
Understanding dioxygen binding and activation by Fe centers in metalloenzymes and synthetic complexes can provide insights into how to perform challenging oxidative reactions with abundant and environmentally friendly components.1–4 Foundational studies evaluating nitric oxide and O2 binding to non-heme Fe in enzymes found that the charge donation by the thiolate (RS−), and a highly covalent Fe(III)–SR bond5 produces an energetically favorable FeIII-superoxo species, thereby driving the formation of an Fe(III)–O2˙−.6 These findings strongly suggest that the incorporation of a thiolate (RS−) in the coordination sphere would likely lower the activation barrier to O2 binding. Understanding how thiolates provide favorable conditions for O2 binding should provide important insight into enzyme reaction mechanisms and bio-inspired catalyst design especially since thiolates have been experimentally shown to increase the reactivity of iron-peroxo and -oxo intermediates.7–10
Studying O2 binding is challenging given the complex nature of the initial interaction between triplet O2 and the high-spin (S = 2) FeII center as well as the transient nature of the electronically ambiguous intermediate, which could either be described as an FeII–O2 adduct or an FeIII-superoxo species. The mechanism of its formation could involve either an outer or inner sphere electron transfer and requires changes to the spin multiplicity and geometry to accommodate the O2 molecule. Direct kinetic evidence for this reaction in thiolate-ligated non-heme iron metalloenzymes such as cysteine dioxygenase (CDO) and isopenicillin N synthase (IPNS), is lacking. In part, this is because the O2 binding event occurs too quickly for a precise determination of the activation barrier.11,12 Very few model systems have looked at the mechanistic details of O2 binding to non-heme Fe.13–15 One example comes from Lacy and coworkers who carried out the first kinetic study evaluating the inner vs. outer sphere reduction of O2 by a tunable model system lacking a thiolate ligand, finding that the system preferred an outer-sphere mechanism.13 We recently reported a five-coordinate thiolate-ligated ferrous complex, [FeII(SMe2N4(tren))]+ (4, (Scheme 1)), that binds O2 at diffusion-controlled rates (k(233.15 K) = 5.82(3) × 107 M−1 s−1) to afford a fleeting superoxo intermediate [FeIII(SMe2N4(tren))(O2)]+ (5) only observable (λmax = 490 nm) on the millisecond timescale with limiting [O2] at −40 °C.14,16 It's fleeting nature prevented detailed characterization of superoxo 5 (Scheme 1) or a more thorough analysis of the factors involved in the low barrier to its formation.
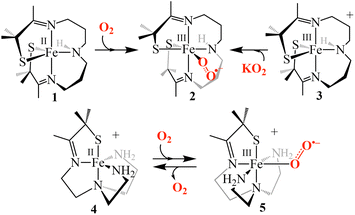 |
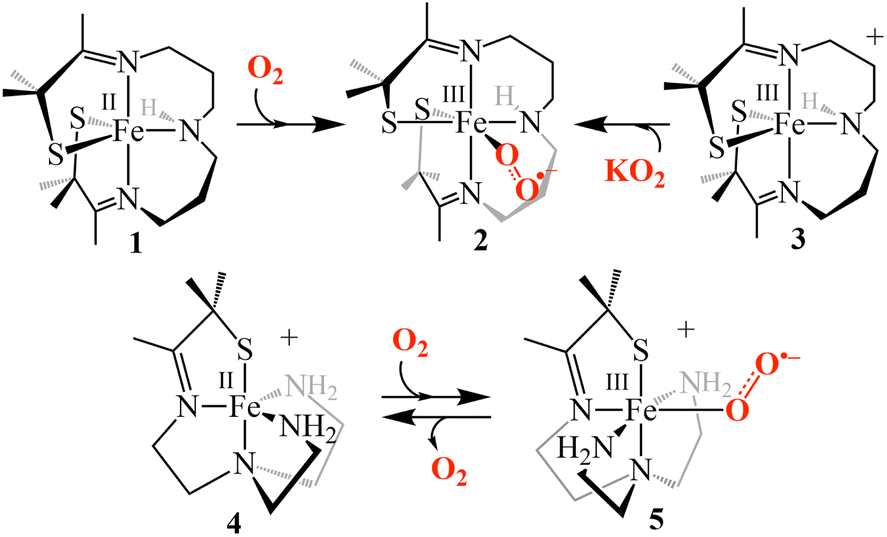
| | Scheme 1 Pathways for the formation of [FeIII(S2Me2N3(Pr,Pr))(O2)] (2) (top), and reversible dioxygen binding to [FeII(SMe2N4(tren))]+ (4) to form a putative superoxo intermediate [FeIII(SMe2N4(tren))(O2)]+ (5) (bottom). | |
Deciphering how the coordination environment, thiolate ligand, solvent and even counterions influence the reaction mechanism of dioxygen binding is best carried out with systems where the initial Fe(III)–O2˙− intermediate is well-characterized. However, isolated Fe(III)–O2˙− model compounds are rare17–21 and only two well-characterized thiolate-ligated RS–Fe(III)–O2˙− compounds have been reported.17,22 This includes our reported five-coordinate, alkyl thiolate-ligated Fe(II) complex, [FeII(S2Me2N3(Pr,Pr))] (1) that reacts with O2 (Scheme 1) to form an observable metastable ferric superoxo species, [FeIII(S2Me2N3(Pr,Pr))(O2)] (2).22 Vibrational (resonance Raman) data, computational studies, electron paramagnetic resonance, and 1H NMR spectroscopic data established that the electronic structure of 2 consists of a low-spin (S = ½) FeIII ion strongly coupled antiferromagnetically (Jcalc = −450 cm−1) to an S = ½ superoxo radical,22 indicating that electron transfer takes place during its formation (i.e., FeII (1) + O2 → FeIII–O2˙− (2)).22
The study herein explores the mechanism of formation of our well-characterized iron-superoxo compound, [FeIII(S2Me2N3(Pr,Pr))(O2)] (2),22via two distinct pathways, using low temperature stopped-flow kinetics. We will experimentally demonstrate that an inner-sphere, as opposed to outer-sphere, electron transfer mechanism is involved. In addition, hydrogen-bond donors are shown to modulate the thiolate ligand's influence,23–28 and ligand constraints are shown to dramatically influence the activation barrier to O2 binding.
Results and discussion
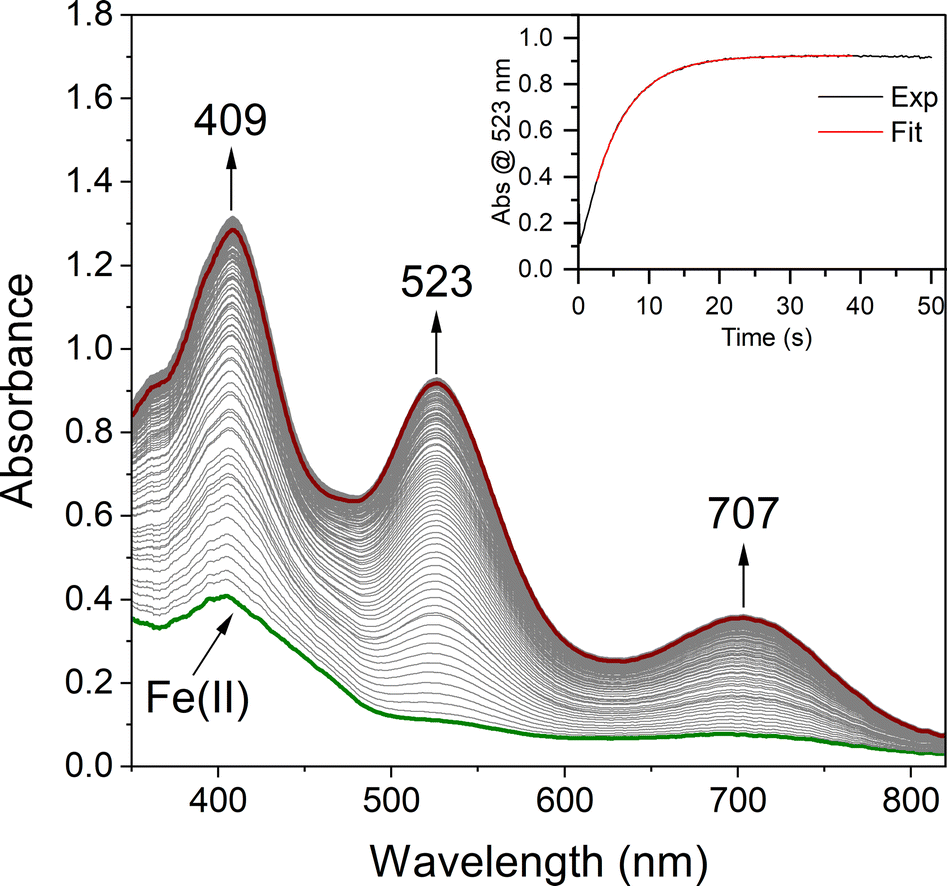
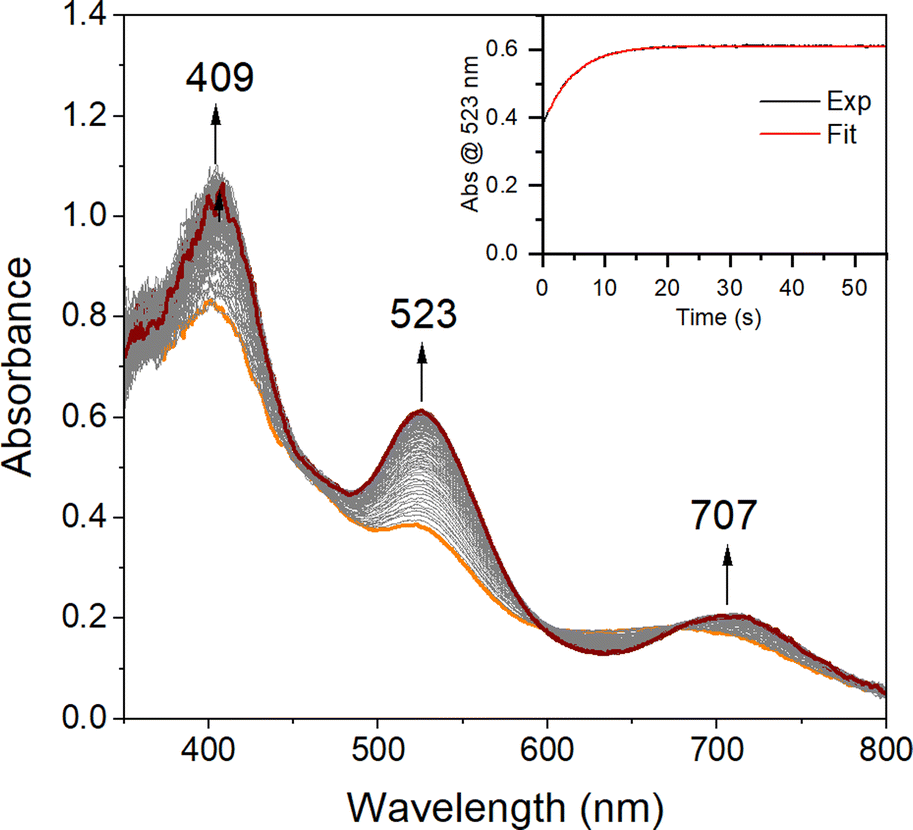
In order to quantitatively determine the barrier to, and reversibility of, O2 binding to five-coordinate [FeII(S2Me2N3(Pr,Pr))] (1, Scheme 1) and the likely mechanism of formation of the corresponding superoxo compound, [FeIII(S2Me2N3(Pr,Pr))(O2)] (2),22 we investigate herein variable temperature stopped-flow kinetics. Time-resolved electronic absorption spectroscopy was used to monitor the reaction between 1 and O2 using a TgK cryogenic stopped-flow instrument. The growth of spectral features (Fig. 1) associated with 2 were monitored in two different solvents, THF and MeOH. Kinetics were monitored at λ = 523 nm under pseudo first order conditions with excess O2. As illustrated in the time-resolved absorption spectrum and kinetic trace of Fig. 1, superoxo 2 forms in less than 20 s at −40 °C in THF. The kinetic trace (Fig. 1, insert) can be fit to the single exponential of eqn (1) with very small residuals (Fig. S1†).| | | At = A∞ − (A∞ − A0)e−kobst | (1) |
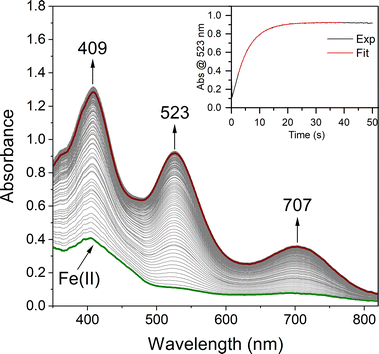 |
| | Fig. 1 Time-resolved spectral changes obtained upon mixing THF solutions of five – coordinate [FeII(S2Me2N3(Pr,Pr))] (1, 0.25 mM) and O2 (3.95 mM) at −40 °C. Inset: kinetic trace (λ = 523 nm) showing the formation of the Fe–O2 intermediate 2. All reported concentrations are after mixing in the stopped-flow cell. | |
Low temperature kinetics for the reaction between O2 and 1
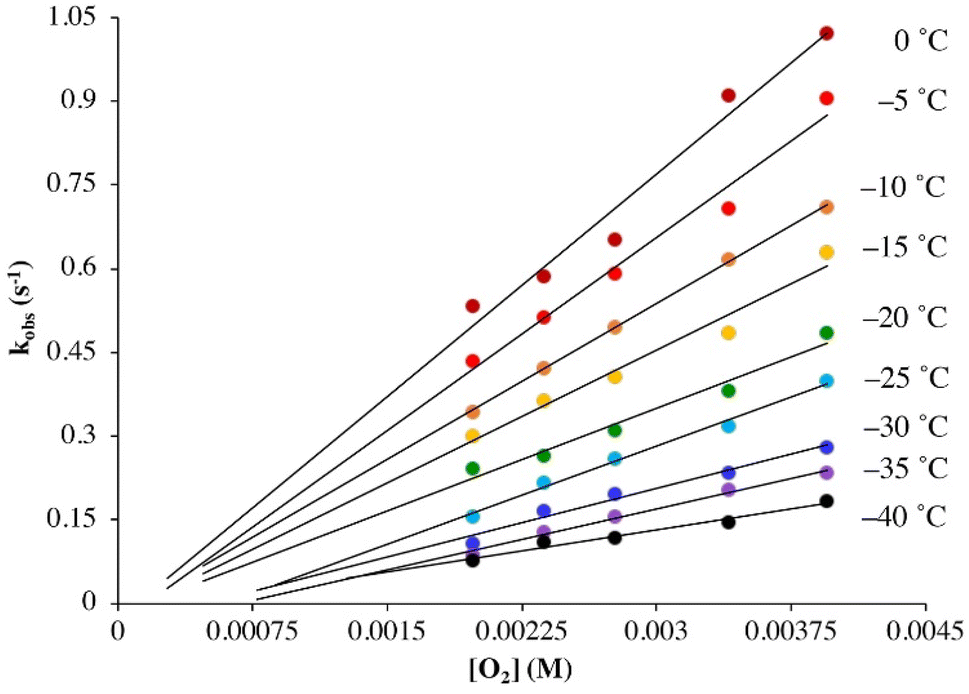
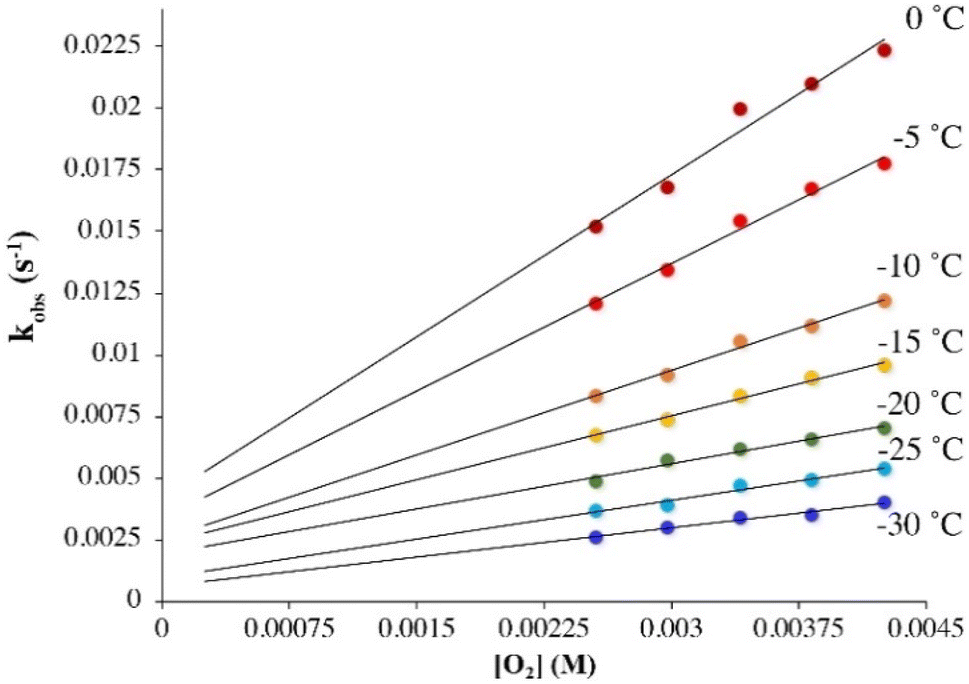
Under pseudo first-order conditions with excess O2, kobs values obtained from fits to eqn (1) (Tables S1 and S2†) were found to be independent of the concentration of 1 in both THF (Fig. S2†) and MeOH (Fig. S3†), confirming that the reaction is first-order overall with respect to 1. This would be consistent with the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dioxygen adduct. Observed rate constants, kobs, were found to increase linearly with increasing O2 concentration in both THF (Fig. 2) and MeOH (Fig. 3) consistent with first-order dependence on O2 (kobs = kon[O2] + koff, eqn (S10)†). Second order rate constants (Table 1), kon, were obtained from the slope of the kobsversus [O2] plot (Fig. 2 and 3) over the temperature range −40 °C to 0 °C in THF, and −30 °C to 0 °C in MeOH, in 5 °C increments. In MeOH, O2 binds to 1, and is released from 2, too slowly to collect data at temperatures below −30 °C using a stopped-flow instrument. In THF, the intercept of these plots was found to be approximately zero over the entire temperature range examined (Fig. 2), indicating that O2 binds irreversibly to 1 (eqn (S10)†) in this solvent. This is supported by the fact that no trend in koffvs. temperature is observed in THF (Fig. S4†). In MeOH, the non-zero intercept (Fig. 3) was found to increase with increasing temperature (Fig. S5†) consistent with reversible O2 binding in this solvent. Dioxygen dissociation rate constants in MeOH, koff, obtained from the intercept of the kobsversus [O2] plot of Fig. 3, are assembled in Table 1.
:1 dioxygen adduct. Observed rate constants, kobs, were found to increase linearly with increasing O2 concentration in both THF (Fig. 2) and MeOH (Fig. 3) consistent with first-order dependence on O2 (kobs = kon[O2] + koff, eqn (S10)†). Second order rate constants (Table 1), kon, were obtained from the slope of the kobsversus [O2] plot (Fig. 2 and 3) over the temperature range −40 °C to 0 °C in THF, and −30 °C to 0 °C in MeOH, in 5 °C increments. In MeOH, O2 binds to 1, and is released from 2, too slowly to collect data at temperatures below −30 °C using a stopped-flow instrument. In THF, the intercept of these plots was found to be approximately zero over the entire temperature range examined (Fig. 2), indicating that O2 binds irreversibly to 1 (eqn (S10)†) in this solvent. This is supported by the fact that no trend in koffvs. temperature is observed in THF (Fig. S4†). In MeOH, the non-zero intercept (Fig. 3) was found to increase with increasing temperature (Fig. S5†) consistent with reversible O2 binding in this solvent. Dioxygen dissociation rate constants in MeOH, koff, obtained from the intercept of the kobsversus [O2] plot of Fig. 3, are assembled in Table 1.
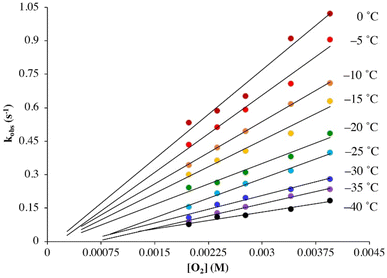 |
| | Fig. 2 Temperature-dependent rate constants kobs for the formation of superoxo 2 in the reaction between 1 (0.25 mM) and O2 in THF plotted against [O2]. The intercept of approximately 0.0 would be consistent with irreversible O2 binding. | |
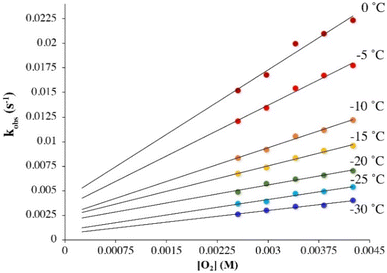 |
| | Fig. 3 Temperature-dependent rate constants kobs for the formation of superoxo 2 in the reaction between 1 (0.1 mM) and O2 in MeOH, plotted against [O2]. The non-zero intercepts would be consistent with reversible O2 binding in MeOH. | |
Table 1 Temperature-dependent rate constants for irreversible O2 binding to [FeII(S2Me2N3(Pr,Pr))] (1) in THF, reversible O2 binding to 1 in MeOH and KO2 binding to [FeIII(S2Me2N3(Pr,Pr))]+ (3) under constant ionic strength (0.01 M) maintained via the addition of Bu4NPF6
| Temperature (°C) |
k
on (M−1 s−1) in THF |
k
on (M−1 s−1) in MeOH |
k
off (s−1) in MeOH |
k
superoxide (M−1 s−1) in THF |
| 0 |
2.66(28) × 102 |
4.4(5) |
4(2) × 10−3 |
N/A |
| −5 |
2.31(19) × 102 |
3.4(2) |
3.4(8) × 10−3 |
N/A |
| −10 |
1.86(3) × 102 |
2.3(1) |
2.5 (4) × 10−3 |
N/A |
| −15 |
1.59(16) × 102 |
1.7(1) |
2.4(3) × 10−3 |
N/A |
| −20 |
1.23(13) × 102 |
1.2(1) |
1.9(4) × 10−3 |
2.98(11) × 102 |
| −25 |
1.17(6) × 102 |
1.0(1) |
1.0(4) × 10−3 |
2.35(24) × 102 |
| −30 |
8.2(8) × 101 |
0.79(6) |
6(2) × 10−4 |
1.42(17) × 102 |
| −35 |
7.28(4) × 101 |
N/A |
N/A |
9.5(1.2) × 101 |
| −40 |
5.02(5) × 101 |
N/A |
N/A |
5.8(3) × 101 |
| −45 |
N/A |
N/A |
N/A |
4.6(7) × 101 |
| −50 |
N/A |
N/A |
N/A |
2.9(2) × 101 |
| −55 |
N/A |
N/A |
N/A |
2.0(2) × 101 |
| −60 |
N/A |
N/A |
N/A |
1.16(5) × 101 |
| ΔH‡ (kJ mol−1) |
19.3(8) |
30(2) |
32(4) |
34.3(9) |
| ΔS‡ (J mol−1 K−1) |
−127(4) |
−123(7) |
−170(20) |
−60(4) |
|
E
a (kJ mol−1) |
21.4(8) |
32(2) |
34(4) |
36.3(9) |
| ΔG‡ (243 K, kJ mol−1) |
50(1) |
60(3) |
73(4) |
N/A |
Kinetic barrier to O2 binding
Comparison of the second order rate constants, kon, in THF versus MeOH (Table 1) indicates that O2 binds roughly two orders of magnitude more slowly to 1 in MeOH. Activation parameters for O2 binding were obtained from Eyring (Fig. 4) and Arrhenius plots (Fig. S6 and S7†) and are assembled in Table 1. The negative entropy of activation (ΔS‡ = −127(4) J mol−1 K−1 in THF; ΔS‡ = −123(7) J mol−1 K−1 in MeOH) is consistent with an associative mechanism, as opposed to one that involves initial solvent dissociation, consistent with a five-coordinate structure in both solvents. If, however, the entropy change associated with solvent release is less than that of O2 binding, then it would be possible that solvent release is involved. However, we have no evidence for the formation of six-coordinate ferrous derivatives 1-solv (solv = coordinated solvent molecule) in any solvent, despite crystallization from coordinating solvents MeCN or MeOH, which reproducibly affords a five-coordinate complex.
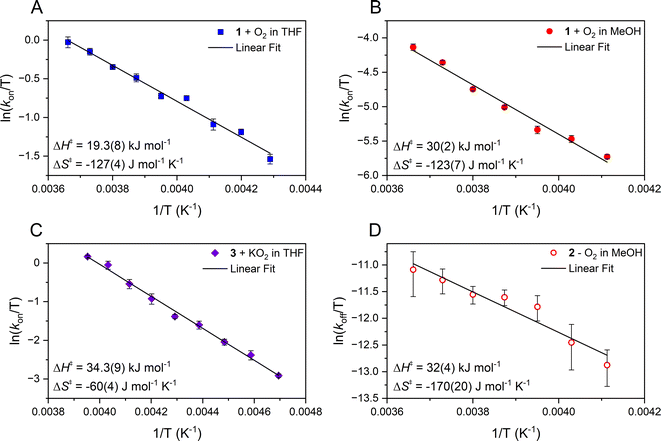 |
| | Fig. 4 Eyring plots from which the activation parameters reported were obtained for the following reactions. (A) O2 binding to 1 in THF and (B) O2 binding to 1 in MeOH, which use the second order rate constants, kon, obtained from the slope of kobsvs. [O2] plots (Fig. 2 and 3) with [FeII] = 0.25 mM and 0.1 mM respectively. (C) The reaction of 3 and KO2 (solubilized with KryptoFix) in THF plotted with second order rate constants, kon, obtained from the slope of kobsvs. [KO2] plots. [FeIII] = 0.1 mM, after mixing. (D) The release of O2 from 2 in MeOH which uses first order rate constants, koff, obtained from the intercept of kobsversus [O2] plots (Fig. 3). | |
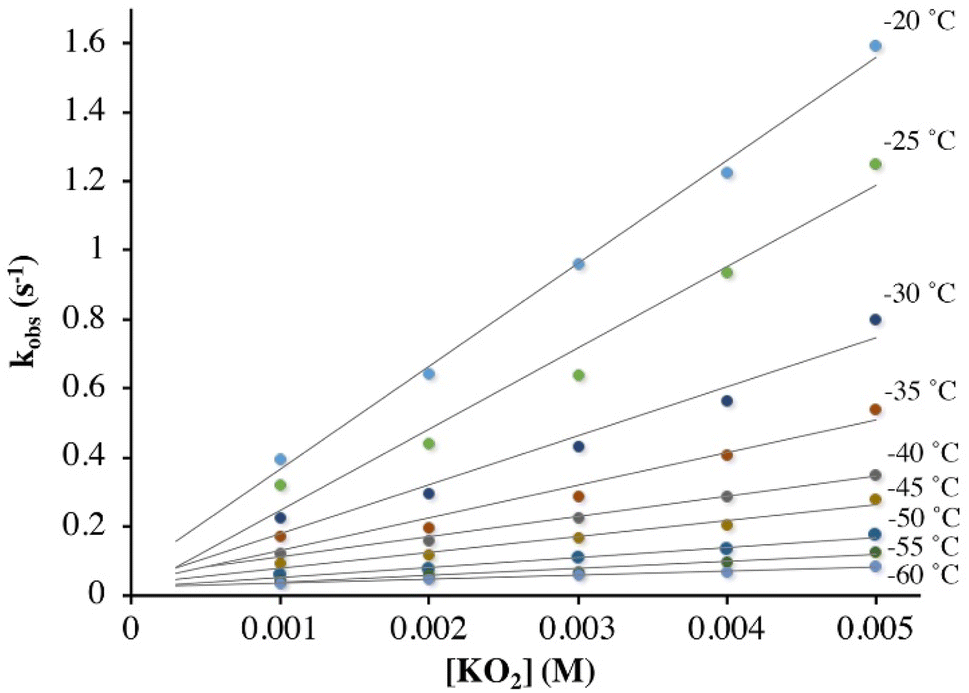
Low temperature kinetics for the reaction between KO2 and 3
As shown in Scheme 1, superoxo intermediate 2 forms both via the addition of O2 to Fe(II)-1 as well as via the addition of KO2 to [FeIII(S2Me2N3(Pr,Pr))]PF6 (3).22 Evidence for this was described in a recent publication,22 where we showed that an electronic absorption spectrum identical to that of Fig. 1 is observed in the reaction between 3 and KO2 (Fig. 5). Here we examine the variable temperature kinetics for the reaction between KO2 and Fe(III)-3, in order to determine whether the activation barrier is lower or higher than that for Fe(II) + O2, and thus whether an inner- or outer-sphere electron transfer mechanism is involved. Observed pseudo first order rate constants (kobs), obtained from fits to eqn (1) (Fig. S8†), assembled in Table S3,† increase linearly as [KO2] is increased (Fig. 6) under pseudo first-order conditions with excess KO2, and are independent of the concentration of 3 (Fig. S9†). The latter indicates a 1st-order dependence on Fe(III) overall. The reaction order with respect to superoxide (O2˙−) was determined by varying the concentration of KO2 (Fig. 6) over the range 1.0 mM to 5.0 mM. Second-order rate constants, ksuperoxide, were obtained from the slope of the kobsvs. [KO2] plot of Fig. 6 and are assembled in Table 1. Activation parameters for O2˙− binding to 3 in THF (Ea = 36.3(9) kJ mol−1; Table 1) under constant ionic strength conditions (0.01 M; maintained with Bu4NPF6) were obtained from Eyring (Fig. 4) and Arrhenius (Fig. S10†) plots. The Arrhenius parameter, Ea, for KO2 binding to 3 (Table 1) is 14.9 kJ mol−1 greater than the barrier (Ea = 21.4(8) kJ mol−1) to O2 binding to 1 (Scheme 1 and Table 1) in the same solvent. This has implications about the mechanism of the reaction between 1 and O2 (vide infra). The negative entropy of activation, ΔS‡, (Table 1) is consistent with an associative process involving KO2 binding to 3. The smaller magnitude of ΔS‡ for the KO2 reaction relative to the O2 reaction (Table 1) likely reflects the smaller ligand reorganization required to convert Fe(III)-3 to 2 relative to that required to convert Fe(II)-1 to 2 (vide infra).
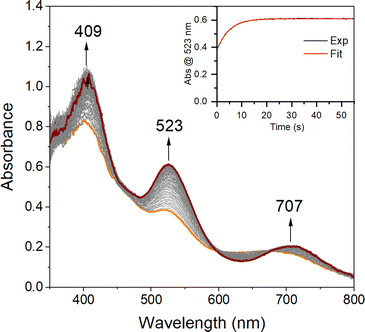 |
| | Fig. 5 Time-resolved spectral changes observed in the reaction between 3 (0.1 mM) and KO2 (5.0 mM; solubilized with 222-Kryptofix) in THF at −40 °C. Inset: kinetic trace (λ = 523 nm) showing the formation of 2. All reported concentrations are after mixing in the stopped-flow cell. | |
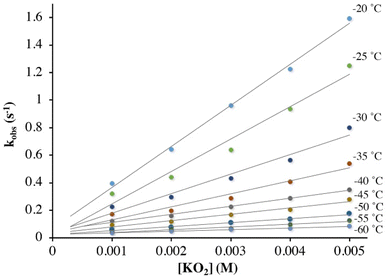 |
| | Fig. 6 Temperature-dependent rate constants, kobs, for the formation of superoxo 2 in the reaction between 3 and KO2 (solubilized with KryptoFix) in THF, plotted against [KO2]. The ∼ zero intercepts would be consistent with irreversible O2˙− binding. [Fe(III)] = 0.1 mM. Concentrations listed correspond to after mixing in the stopped-flow cell. | |
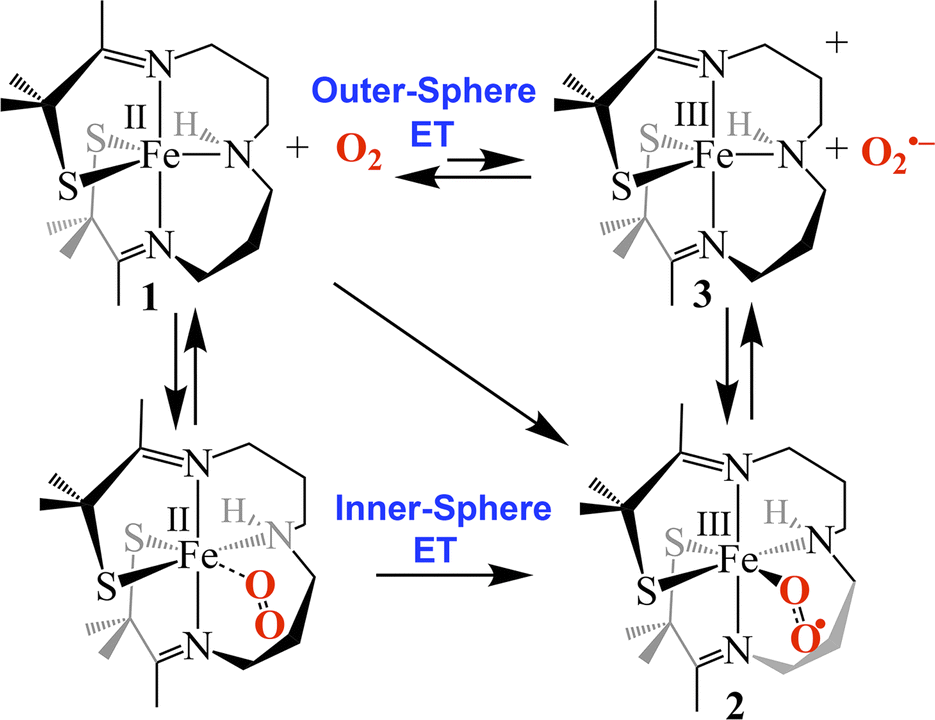
Possible mechanisms (Fig. 7) for the formation of 2 include (a) outer-sphere electron transfer (ET) from Fe(II) to O2 to afford oxidized Fe(III)-3 and superoxide (O2˙−), followed by O2˙− binding to 3 to afford superoxo 2, (b) dioxygen binding to 1 to afford an Fe(II)–O2 dioxygen intermediate followed by inner-sphere electron transfer (ET) to afford superoxo 2, or, (c) a concerted mechanism across the diagonal. The latter two are indistinguishable. The fact that the activation barrier (Ea) to O2˙− binding to 3 was experimentally determined to be 14.9 kJ mol−1 higher than Ea for dioxygen binding to 1 (Table 1) implies that mechanism (b) or (c), involving inner sphere ET, are the most likely mechanisms. The unfavorable thermodynamics of electron transfer from 1 to O2 (Keq = 4.66 × 10−12 M−1 calculated using E1/2(Fe(III)-3/Fe(II)-1) = −0.40 V and E1/2(O2/O2˙−) = −1.07 V vs. SCE in THF),29 provides additional support for this conclusion.
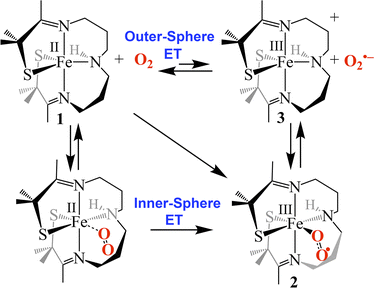 |
| | Fig. 7 Possible mechanisms for the reaction between 1 and O2 involving either outer-sphere electron transfer (ET) followed by superoxide (O2˙−) binding to oxidized 3, O2 binding followed by inner-sphere ET, or a concerted mechanism (diagonal). | |
Kinetic barrier for O2 release from 2
In MeOH, O2 binds reversibly to [FeII(S2Me2N3(Pr,Pr))] (1), in contrast to the irreversible binding in THF. Activation parameters for O2 release from [FeIII(S2Me2N3(Pr,Pr))(O2)] (2) in MeOH (ΔH‡ = 32(4) kJ mol−1, ΔS‡ = −170(20) J mol−1 K−1, Ea = 34(4) kJ mol−1) were obtained from Eyring (Fig. 4) and Arrhenius plots (Fig. S11†) and are assembled in Table 1. The negative entropy of activation for O2 release is unexpected given its dissociative nature. A negative entropy has also been observed for O2 release from thiolate-ligated [FeIII(SMe2N4(tren))(O2)]+ (4) in MeOH as well as [MnIII(LQuino)(O2)]+.14,30 These complexes both contain flexible ligands for which bond rearrangement during the oxidation state change (vide infra) is proposed to contribute to a negative entropy of activation large enough to offset O2 release.14,30 To understand the negative entropy of activation for O2 release from 2, we evaluated the requisite spin-state and oxidation state changes that require changes to the Fe–L bond lengths. Because 1 is high-spin S = 2 (μeff = 4.69 BM), whereas 2 contains a low-spin S = ½ Fe(III) ion, a spin-state change must take place during O2 release. To prepare for this electron transfer step a more ordered state involving adjusted Fe–L bond lengths is therefore required. In addition to the spin-state change, oxidation state-dependent H-bonding between MeOH and the thiolate sulfurs of 1 relative to 2 (vide infra) influences the entropy changes involved in O2 release. Both factors are examined in greater detail in the context of crystallographically determined structures, electronic absorption spectra and DFT calculations, below.
Influence of H-bonds on kinetic barriers
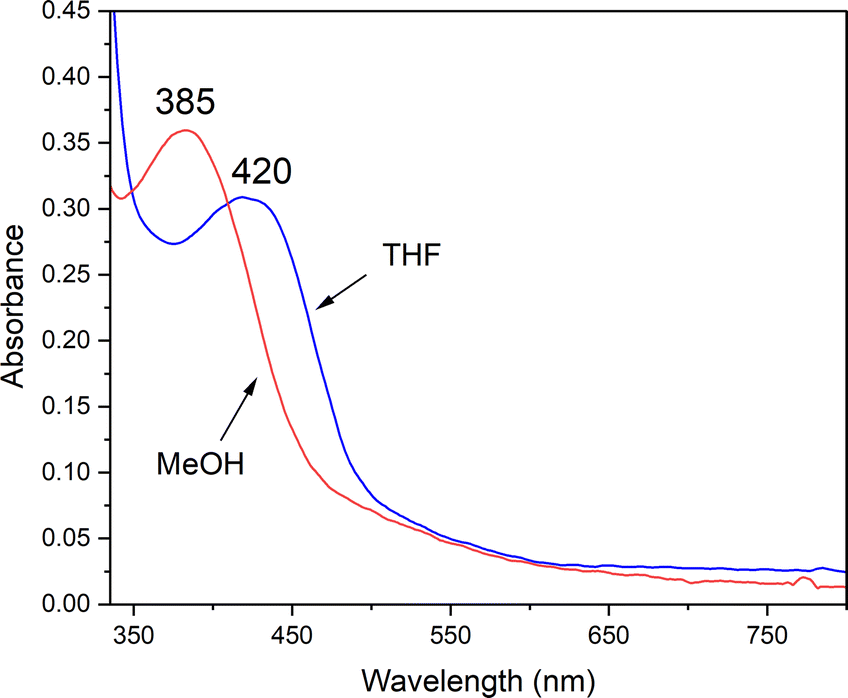
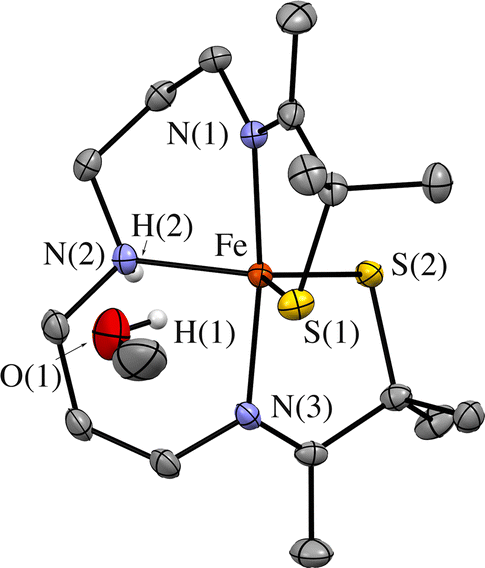
An observed blue-shift (by 2160 cm−1) of the dominant feature in the electronic absorption spectrum from 420 nm (23800 cm−1) in THF to 385 nm (26000 cm−1) in MeOH provides experimental evidence for the influence of either the dielectric constant ((THF) = 7 vs. ε(MeOH) = 33), or H-bonding on the orbitals involved in the transition (Fig. 8). The crystallographically determined structure of 1 (Fig. 9 and Tables S5–S9†), contains a MeOH nestled in the O2 binding pocket and the MeOH proton H(1), points towards S(1) with an H(1)⋯S(1) distance of 2.318 Å, providing evidence for H-bonding in MeOH. The O2 binding site was deduced from previous structures that show exogenous ligands such as N3− and NO bind to Fe in the pocket that lies opposite to the secondary amine proton,31,32 H(2), in the more open N(2)–Fe–S(1) angle. Therefore at a minimum there would be release of the MeOH that is in proximity to the binding site upon O2 binding which would contribute a favorable positive ΔS‡(solvent release) term that slightly offsets the unfavorable negative ΔS‡(O2 binding) term. Conversely, O2 release from 2 would involve the addition of at least one H-bonded MeOH to 1 (Fig. 9) offsetting the favorable entropy of activation associated with O2 release.
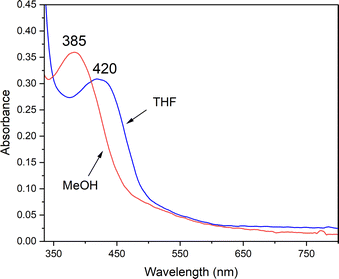 |
| | Fig. 8 H-bonding to MeOH causes the RS → Fe band of reduced 1 (0.238 mM) to blue-shift relative to its energy in THF. | |
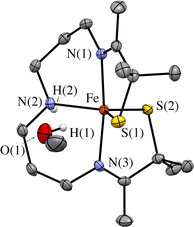 |
| | Fig. 9 ORTEP diagram of 1 crystallized from MeOH showing the MeOH that is H-bonded to one of the thiolate sulfurs, S(1). Shown with 50% probability ellipsoids and numbering scheme. All hydrogen atoms excluding H(2) and H(1) have been deleted for clarity. | |
To evaluate the properties likely to affect H-bonding in 1 relative to 2 we pursued density functional theory (DFT) calculations. The DFT calculated structure of 1 (Fig. S12†), shows that the thiolate sulfurs of reduced 1-Fe(II) have an average Mulliken charge of −0.670 (Table S4†), whereas the thiolate sulfurs of oxidized superoxo 2 have Mulliken charges of −0.519 and −0.482, for the trans RS− and cis RS−, respectively (Table S4†). Hydrogen-bonding to the thiolates would therefore be less likely, and/or involve fewer MeOH molecules, with oxidized 2, relative to 1. The electronic absorption spectra of 2 in THF vs. MeOH shows a distinct, but less amplified blue-shift relative to 1 (ΔE = 721 cm−1 for 2 (Fig. S13†) vs. ΔE = 2114 cm−1 for 1).
In the absence of a more polar H-bonding solvent, the barrier to O2 binding was experimentally determined to be lower (Fig. 10). The blue-shift in the electronic absorption spectrum (Fig. 8) and insights from DFT calculations indicate this is in part caused by a stabilization of the Fe(II)–SR molecular orbitals involved in the RS → Fe charge transfer transition in protic MeOH solvent (Fig. S14 and S15†).33 This has a noticeable effect on the kinetic barrier, ΔG‡, to O2 binding and release. If solvent polarity alone were responsible, then one would expect the kinetic barrier to be lower in MeOH, as opposed to higher as is experimentally observed. This is because charge separation increases as neutral 1 and O2 convert to 2-O2˙−.
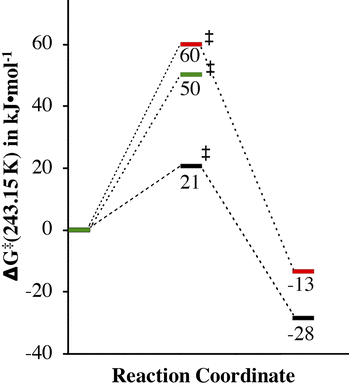 |
| | Fig. 10 Comparison of the experimentally determined barrier to O2 binding to 1 in THF (green) and MeOH (red) and 4 in MeOH (black), as well as the release of O2 from 2 and 5 in MeOH. | |
An explanation as to why H-bonding to thiolate sulfurs would influence the barrier to O2 binding is that it would tie up lone pairs that would otherwise be involved in π-bonding to the metal ion. Thiolates have been shown by our group to form highly covalent Fe(III)–SR bonds5 with partial Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond character. This stabilizes oxidized products such as 2,14,22 and any transition states with developing Fe(III)–SR character. If, on the other hand, a lone pair on the thiolate sulfur is tied up in H-bonding to a protic solvent (e.g., MeOH),33 then the incipient Fe(III)–SR bond of the transition-state would have less double bond character, and would thus be less stable. This, in addition, to the stabilization of the Fe(II) precursor provided by H-bonding would result in slower O2 binding to 1 in MeOH, as is experimentally observed (Table 1). The experimentally measured free energy of activation, ΔG‡, for dioxygen binding to 1 in MeOH (60(3) kJ mol−1) is 10 kJ mol−1 higher than in THF (50(1) kJ mol−1) at 243.15 K (Fig. 10 and Table 1), consistent with this.
S double bond character. This stabilizes oxidized products such as 2,14,22 and any transition states with developing Fe(III)–SR character. If, on the other hand, a lone pair on the thiolate sulfur is tied up in H-bonding to a protic solvent (e.g., MeOH),33 then the incipient Fe(III)–SR bond of the transition-state would have less double bond character, and would thus be less stable. This, in addition, to the stabilization of the Fe(II) precursor provided by H-bonding would result in slower O2 binding to 1 in MeOH, as is experimentally observed (Table 1). The experimentally measured free energy of activation, ΔG‡, for dioxygen binding to 1 in MeOH (60(3) kJ mol−1) is 10 kJ mol−1 higher than in THF (50(1) kJ mol−1) at 243.15 K (Fig. 10 and Table 1), consistent with this.
Influence of ligand constraints on kinetic barriers
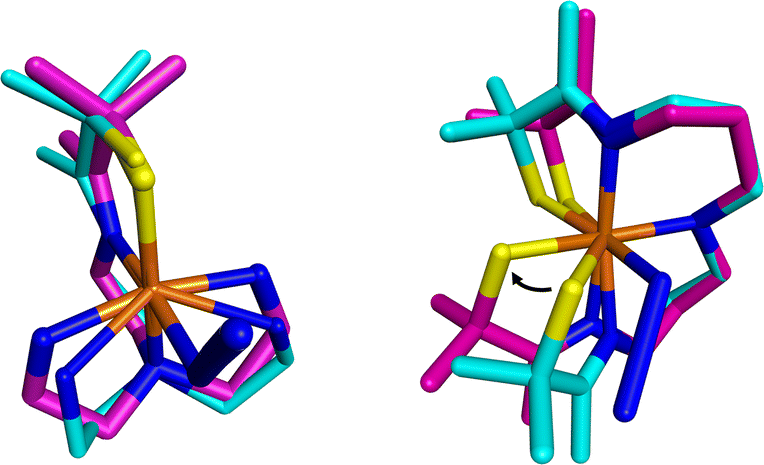
Comparison of the free energy of activation, ΔG‡ (243.15 K), for O2 release from 2 (73(4) kJ mol−1) in MeOH at 243.15 K (Table 1) relative to that for previously reported [FeIII(SMe2N4(tren))(O2)]+ (5, Scheme 1, 49(7) kJ mol−1)14 under the same conditions, shows that the barrier to O2 release is 24 kJ mol−1 higher for 2. This likely reflects ligand constraints31,34,35 and the ligand rearrangement required to form the transition-state necessary for O2 release. The multidentate ligand of 1 and 2 consists of a single chain that wraps around the metal ion in a helical fashion (Fig. 11).35 Even minor angle changes at the metal ion in preparation for O2 release translate into significant movement of the thiolate containing arms that terminate the single chain ligand backbone (Fig. 11). An experimental demonstration of this can been seen when comparing the crystallographically determined structures of Fe(II) structures 4versus1, relative to the corresponding Fe(III)–N3 structures (Fig. 12).22,31,36,37 The geometry of each 5-coordinate Fe(II) structure, 4 and 1 (Scheme 1) is best described as distorted trigonal-bipyramidal τ = 0.63 and 0.79, respectively. Structure 4 (Scheme 1) is closer to square pyramidal, and thus requires less geometric rearrangement upon N3− binding (Fig. 12, left). In contrast, as the entire S(R) arm of 1 swings away from the binding site (Fig. 12, right), the thiolate is pushed 1.8 Å opening the binding site by 46°. This reorganizational barrier would be mostly entropic in nature. The entropy of activation (Table 1) for O2 binding to 1 in MeOH (ΔS‡ = −123(7) J mol−1 K−1), is less favorable (by 178 J mol−1 K−1) than for O2 binding to 4 (+55(68) J mol−1 K−1).14 This factor dominates and is partially responsible for the large differences in ΔG‡ (Fig. 10). The lower barrier for O2 release from 5 relative to 2 (Scheme 1), likely reflects its less constraining tripodal ligand with its three independent arms (Fig. 12). The less constraining ligand of 4 also results in a lower free energy of activation for dioxygen binding to 4 (ΔG‡ (243.15 K) = 21(2) kJ mol−1) relative to 1 (ΔG‡ (243.15 K) = 60(3) kJ mol−1) in the same solvent (MeOH).14
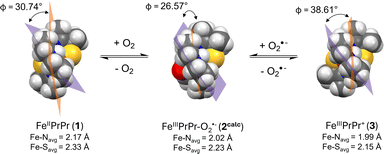 |
| | Fig. 11 Space-filling models generated from the crystallographically-determined structure of 1 (left) and 3 (left), and the DFT-optimized geometry of 2calc (middle) displaying differences in the helical wrapping angle, Φ, and the larger amount of structural rearrangement required for O2˙− binding to 3 than O2 binding to 1. The least squared planes of N(1)FeN(2)N(3) and C(6)N(2)C(7)Fe shown in orange and purple respectively. | |
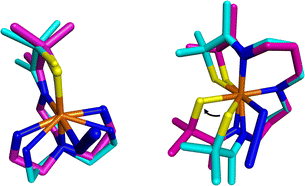 |
| | Fig. 12 Visualization of experimentally obtained structural changes that occur upon oxidation and binding of a sixth ligand, azide. Left: [FeII(SMe2N4(tren))]+ (4, cyan)36 to [FeIII(SMe2N4(tren))N3] (magenta).37 Right: [FeII(S2Me2N3(Pr,Pr))] (1, cyan)22 and [FeIII(S2Me2N3(Pr,Pr))(N3)] (magenta).31 Depiction utilized previously reported crystallographically determined structures. | |
Understanding why the kinetic barrier to O2˙− binding is higher than the kinetic barrier to O2 binding
Given that oxidized [FeIII(S2Me2N3(Pr,Pr))]+ (3) is cationic, one might expect electrostatic attraction to lower the barrier to O2˙− binding to 3, relative to O2 binding to neutral 1. This likely would be the case, if it weren't for the oxidation state-dependent ligand constraints imposed by the single chain helical ligand.31,35 With five-coordinate 3, the helical twist is “tighter” relative to five-coordinate 1 (Fig. 11) due to the shorter Fe(III)–L bond lengths. The bond lengths of 3 expand upon binding a sixth ligand, e.g. O2˙−.31 This difference can be quantified by comparing the helical twist angle, Φ, of crystalographically characterized 1, 3 and the DFT geometry optimized structure of 2 (Fig. 11).35 The angle Φ is defined as the angle separating the least squared plane containing the Fe center, the two carbons flanking the equatorial nitrogen, N(2), and the equatorial nitrogen (C(6)N(2)C(7)Fe) from the mean plane containing N(1)FeN(2)N(3). One can see in Fig. 11 that the twist angle decreases (from 30.74° to 26.57°) when O2 binds to Fe(II)-1 to form to Fe(III)–O2˙−2, whereas a larger decrease (from 38.61° to 26.57°) is required when O2˙− binds to Fe(III)-3. This helps to explain the larger barrier to superoxide binding to 3, relative to O2 binding to 1.
Relevance to metalloenzymes and transition-metal O2 chemistry
The contribution of ligand constraints and H-bond donors on the reactivity reported herein is reminiscent of the influence of protein constraints and H-bond donors on metalloenzymes. Protein constraints have been shown to have a dramatic effect on electronic structure and reactivity.38–42 For example, the redox potential and electron transfer kinetics blue copper proteins is controlled by protein constraints.43 Hydrogen-bond donors can also influence the stability of metastable enzyme intermediates and influence reaction pathways.44–47
If we compare the activation barriers and rate constants associated with O2 binding to the Fe(II) complex, 1, reported herein, with those reported in the literature for Fe(II), both heme and non-heme, Co(II), Mn(II), and Cu(I) complexes,30,48–58 (Table 2), we see that rates are faster for porphyrin-ligated systems,59,60 both synthetic and biological.61–65 This can be attributed to the smaller rearrangement required for ligands to bind to the more readily accessible open face of a porphyrin-ligated metal ion— a property that would be important to facilitate O2 release in O2 transport proteins.
Table 2 Comparison of kinetic parameters for dioxygen binding to mononuclear Fe(II), Mn(II), Co(II), and Cu(I) model complexesg and biological active sites
|
|
Solvent (T) |
k
on (M−1 s−1) |
ΔH‡ (kJ mol−1) |
ΔS‡ (J mol−1 K−1) |
Ref. |
|
NMe2L = DMAE of Fig. S16.
ImL = impy1 of Fig. S16.
L1 = β-diketiminate (R = Me).
These values were obtained using flash photolysis methods.
IPr = 1,3-bis(diisopropyl)phenylimidazol-2-ylidene which is an N-heterocyclic carbene.
H-NOX = Heme nitric oxide/oxygen binding.
See ESI, Fig. S16 for drawings of the ligands.
|
| 3-, 4-, and 5-Coordinate complexes |
| [FeII(S2Me2N3(Pr,Pr))] (1) |
THF (−40 °C) |
5.02(5) × 101 |
19.3(8) |
−127(4) |
This work |
| THF (0 °C) |
2.66(28) × 102 |
|
|
This work |
| MeOH (−30 °C) |
0.79(6) |
30(2) |
−123(7) |
This work |
| MeOH (0 °C) |
4.4(5) |
|
|
This work |
| [FeII(SMe2N4(tren))]+ (4) |
MeOH (−40 °C) |
5.82(3) × 107 |
34(15) |
55(68) |
14
|
| [MnII(SMe2(6-MeDPEN)]+ |
MeCN (−40 °C) |
7.17(45) × 102 |
26(2) |
−76(7) |
30 and 48 |
| [CuI(TPA)(THF)]+ |
THF (25 °C) |
1.3 × 109 |
7.62 |
−45.1 |
49
|
| [NMe2LCuI]a |
THF (−80 °C) |
6.90(2) × 107 |
32.1 |
80.1 |
50
|
|
ImLCuI(MeCN)b |
THF (−80 °C) |
1.80(3) × 108 |
23.4 |
35.1 |
50
|
| L1CuI(MeCN)c |
THF (−50 °C)d |
1.56(2) × 103 |
18(2) |
−100(10) |
51
|
| (Me6tren)CuI(EtCN) |
EtCN (−50 °C) |
8.7(4) × 105 |
17.1(6) |
−52(3) |
52
|
| (TMPA)CuI(EtCN) |
EtCN (−50 °C) |
5.0(3) × 105 |
31.6(5) |
10(3) |
53
|
| CoII(acacMeDPT) |
Acetone (−50 °C) |
1.0(1) × 106 |
5.0 |
−109 |
54
|
| CoII(SalMeDPT) |
Acetone (−50 °C) |
2.10(4) × 105 |
12.6 |
−80 |
54 and 56 |
| CoII(pXyacacMeDPT) |
Acetone (−50 °C) |
1.74(5) × 106 |
28 |
−13 |
54
|
| V(N[tBu]Ar)3 |
Toluene (−53 °C) |
40(3) × 103 |
13.8(8) |
−92(4) |
55
|
| [Pd(IPr)2]e |
Toluene (−80 °C) |
6.1(1) × 103 |
18(3) |
−54(4) |
57
|
| [Pd(IPr)(P(p-tolyl)3)]e |
THF (−80 °C) |
1.2 × 103 |
9.2 |
−117 |
58
|
|
| Five coordinate porphyrin complexes |
| [Fe(Piv35CIm)] |
Toluene (25 °C) |
4.3 × 108 |
0.75(33) |
−117(3) |
59
|
| [FeII(Piv2C9)(RS)]1− |
Toluene (20 °C) |
3.5 × 106 |
|
|
60
|
|
| Biological systems |
| Cyt bo3 ubiquinol oxidase |
H2O (25 °C) |
3.8 × 107 |
|
|
62
|
| H-NOXf |
H2O (25 °C) |
2.5(3) × 107 |
|
|
63
|
| Hemoglobin (α-human) |
H2O (25 °C) |
2.8 × 107 |
|
|
64
|
| Fe-Mb (horse) |
H2O (22 °C) |
9.8 × 107 |
23 |
−30 |
65
|
Irreversible O2 binding, such as that observed with 1 in THF, is important if the enzyme function involves the metal-superoxo species, as is the case with isopenicillin N-synthase (IPNS)11 and cysteine dioxygenase (CDO).12 With thiolate-ligated systems like 1, and IPNS, one can see from this study (Table 2) that the reversibility of O2 binding can be controlled via the removal of H-bond donors in the vicinity of the thiolate sulfur. The influence of steric effects on activation enthalpy, ΔH‡, has been previously observed for O2 binding to a series of five-coordinate CoII complexes containing pentadentate ligands, in which the bulkiest ligand was shown to have a considerably larger (by 28 kJ mol−1) ΔH‡ value.54 Complexes containing more flexible ligands with independent arms (such as TPA, TMPA, tren, Tp; Fig. S16†), or those that favor a planar geometry (such as salen, porphyrins,59 or acac; Fig. S16†) bind O2 more readily than 1 (Table 2).
Conclusions
Low-temperature stopped-flow kinetics have been used to determine the most likely mechanism, and reversibility of O2 binding to [FeII(S2Me2N3(Pr,Pr))] (1) to afford a well-characterized, reactive, thiolate-ligated ferric superoxo compound, [FeIII(S2Me2N3(Pr,Pr))(O2)] (2).22 Comparison of the kinetics of formation of 2via the addition of KO2 to oxidized [FeIII(S2Me2N3(Pr,Pr))]+ (3) versus the addition of O2 to 1 provided evidence for a mechanism involving inner-sphere electron transfer from Fe(II) to O2 upon binding, as opposed to an outer-sphere electron transfer process to afford O2˙− and Fe(III) followed by O2˙− binding to the oxidized metal ion.
H-bonding and structural constraints were shown to have a noticeable effect on the O2 binding and release. The kinetic barrier to O2 binding was shown to be 10 kJ mol−1 higher in MeOH relative to THF, indicating that the transition-state is destabilized in the presence of H-bond donors. Thiolates have been shown by our group to form highly covalent Fe(III)–SR bonds with partial FeS double bond character. An explanation as to why H-bonding would influence the barrier to O2 binding is that any transition states with developing Fe(III)–SR character would have less FeS double bond character if the thiolate sulfur lone pairs were engaged in RS⋯H–OMe H-bonding. Hydrogen bonding would also, for the same reasons, destabilize the Fe(III)–superoxo intermediate in MeOH relative to THF thereby decreasing the barrier to O2 release in MeOH. We cannot experimentally verify this, since irreversible binding in THF prevents us from determining the barrier to O2 release in THF from an Fe(III)–superoxo that is presumably more stable.
In addition, the constraints of the single chain ligand system of 1 was shown to significantly increase the barrier to O2 binding and release relative to the less constraining tripodal ligand system of 4. From this study, H-bonding as well as ligand flexibility are shown to have substantial impact on not only O2 binding but also its release.
The experimentally determined kinetic parameters provided herein for O2 binding to 1 and O2 release from 2, both of which require spin-state changes and electron transfer steps, will provide a point of reference for future in-depth computational studies of transition states for thiolate containing compounds. Further knowledge of this critical first step of O2 activation has relevance to understanding enzymes important for our health as well as for the design of new environmentally friendly bio-inspired catalysts.
Data availability
Crystallographic data for 1·MeOH has been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number 2212311.†
Author contributions
JAK raised the funding and conceptualized the project with contributions from MNL. Experimental work which included collecting and analysing the stopped flow electronic absorption spectra was carried out by MNL, MBG, and MCP. MNL and MBG performed the DFT calculations. PMG and WK collected and refined the crystallographic data. JAK and MNL wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the NIH (GM123062). Additional funds (NIH GM123062-S1) for the purchase of a cryogenic stopped-flow UV-vis spectrometer is also gratefully acknowledged. We thank Dylan Rogers, Bennet Karel, Chris Lowe, and Doug Baumgardner for helpful discussion.
Notes and references
- E. I. Solomon, D. E. DeWeese and J. T. Babicz Jr, Mechanisms of O2 activation by mononuclear non-heme iron enzymes, Biochemistry, 2021, 60, 3497–3506 CrossRef CAS PubMed.
- M. Gennari and C. Duboc, Bio-inspired, multifunctional metal–thiolate motif: from electron transfer to sulfur reactivity and small-molecule activation, Acc. Chem. Res., 2020, 53, 2753–2761 CrossRef CAS PubMed.
- D. Jeong, J. Selverstone Valentine and J. Cho, Bio-inspired mononuclear nonheme metal peroxo complexes: synthesis, structures and mechanistic studies toward understanding enzymatic reactions, Coordin. Chem. Rev., 2023, 480, 215021 CrossRef CAS.
- F. Nastri, M. Chino, O. Maglio, A. Bhagi-Damodaran, Y. Lu and A. Lombardi, Design and engineering of artificial oxygen-activating metalloenzymes, Chem. Soc. Rev., 2016, 45, 5020–5054 RSC.
- P. Kennepohl, F. Neese, D. Schweitzer, H. L. Jackson, J. A. Kovacs and E. I. Solomon, Spectroscopy of non-heme iron thiolate complexes: insight into the electronic structure of the low-spin active site of nitrile hydratase, Inorg. Chem., 2005, 44, 1826–1836 CrossRef CAS PubMed.
- C. D. Brown, M. L. Neidig, M. B. Neibergall, J. D. Lipscomb and E. I. Solomon, VTVH-MCD and DFT studies of thiolate bonding to {FeNO}7/{FeO2}8 complexes of isopenicillin N synthase: substrate determination of oxidase versus oxygenase activity in nonheme Fe enzymes, J. Am. Chem. Soc., 2007, 129, 7427–7438 CrossRef CAS PubMed.
- T. H. Yosca, J. Rittle, C. M. Krest, E. L. Onderko, A. Silakov, J. C. Calixto, R. K. Behan and M. T. Green, Iron(IV)hydroxide pKa and the role of thiolate ligation in C–H bond activation by cytochrome P450, Science, 2013, 342, 825–829 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Cytochrome P450 compound I: capture, characterization, and C–H bond activation kinetics, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- M. T. Green, C–H bond activation in heme proteins: the role of thiolate ligation in cytochrome P450, Curr. Opin. Chem. Biol., 2009, 13, 84–88 CrossRef CAS PubMed.
- C. M. Krest, E. L. Onderko, T. H. Yosco, J. C. Calixto, R. F. Karp, J. Livada, J. Rittle and M. T. Green, Reactive intermediates in cytochrome P450 catalysis, J. Biol. Chem., 2013, 288, 17074–17081 CrossRef CAS PubMed.
- E. Y. Tamanaha, B. Zhang, Y. Guo, W.-C. Chang, J. M. Bollinger Jr and C. Krebs, Spectroscopic evidence for the two C–H-cleaving intermediates of Aspergillus nidulans isopenicillin N synthase, J. Am. Chem. Soc., 2016, 138, 8862–8874 CrossRef CAS.
- E. P. F. Tchesnokov, A. S. Faponle, C. G. Davies, M. G. Quesne, R. Turner, M. Fellner, R. J. Souness, S. M. Wilbanks, S. P. deVisser and G. N. L. Jameson, An iron–oxygen intermediate formed during the catalytic cycle of cysteine dioxygenase, Chem. Comm., 2016, 52, 8814–8817 RSC.
- R. Surendhran, A. A. D'Arpino, B. Y. Sciscent, A. F. Cannella, A. E. Friedman, S. N. MacMillan, R. Gupta and D. C. Lacy, Deciphering the mechanism of O2 reduction with electronically tunable non-heme iron enzyme model complexes, Chem. Sci., 2018, 9, 5773–5780 RSC.
- M. A. Dedushko, M. B. Greiner, A. N. Downing, M. K. Coggins and J. A. Kovacs, Electronic structure and reactivity of dioxygen-derived thiolate-ligated Fe-peroxo and Fe(IV) oxo compounds, J. Am. Chem. Soc., 2022, 144, 8515–8528 CrossRef CAS PubMed.
- S. V. Kryatov, E. V. Rybak-Akimova and S. Schindler, Kinetics and mechanisms of formation and reactivity of non-heme iron oxygen intermediates, Chem. Rev., 2005, 105, 2175–2226 CrossRef CAS PubMed.
-
M. A. Dedushko, PhD thesis, University of Washington, 2020.
- A. A. Fischer, S. V. Lindeman and A. T. Fiedler, A synthetic model of the nonheme iron–superoxo intermediate of cysteine dioxygenase, Chem. Comm., 2018, 54, 11344–11347 RSC.
- S. Hong, K. D. Sutherlin, J. Park, E. Kwon, M. A. Siegler, E. I. Solomon and W. Nam, Crystallographic and spectroscopic characterization and reactivities of a mononuclear non-haem iron(III)-superoxo complex, Nat. Commun., 2014, 5, 5440–5446 CrossRef CAS PubMed.
- C.-W. Chiang, S. T. Kleepsies, H. D. Stout, K. K. Meier, P.-Y. Li, E. L. Bominaar, L. Que Jr, E. Munck and W.-Z. Lee, Characterization of a paramagnetic mononuclear nonheme iron–superoxo complex, J. Am. Chem. Soc., 2014, 136, 10846–10849 CrossRef CAS PubMed.
- F. O’ddon, Y. Chiba, J. Nakazawa, T. Ohta, T. Ogura and S. Hikichi, Characterization of mononuclear non-heme iron(III)–superoxo complex with a five-azole ligand set, Angew. Chem., Int. Ed., 2015, 54, 7336–7339 CrossRef PubMed.
- C. Winslow, H. B. Lee, M. J. Field, S. J. Teat and J. Rittle, Structure and reactivity of a high-spin, nonheme iron(III)–superoxo complex supported by phosphinimide ligands, J. Am. Chem. Soc., 2021, 143, 13686–13693 CrossRef CAS PubMed.
- M. N. Blakely, M. A. Dedushko, P. C. Y. Poon, G. Villar-Acevedo and J. A. Kovacs, Formation of a reactive, alkyl thiolate-ligated Fe(III)–superoxo intermediate derived from dioxygen, J. Am. Chem. Soc., 2019, 141, 1867–1870 CrossRef CAS PubMed.
- M. Couture, S. Adak, D. J. Stuehr and D. L. Rousseau, Regulation of the properties of the Heme–NO complexes in nitric-oxide synthase by hydrogen bonding to the proximal cysteine*, J. Biol. Chem., 2001, 276, 38280–38288 CrossRef CAS PubMed.
- C. M. Krest, A. Silakov, J. Rittle, T. H. Yosca, E. L. Onderko, J. C. Calixto and M. T. Green, Significantly shorter Fe–S bond in cytochrome P450-I is consistent with greater reactivity relative to chloroperoxidase, Nat. Chem., 2015, 7, 696–702 CrossRef CAS PubMed.
- P. K. Das, S. Chatterjee, S. Samanta and A. Dey, EPR, resonance raman, and DFT calculations on thiolate- and imidazole-bound iron(III) porphyrin complexes: role of the axial ligand in tuning the electronic structure, Inorg. Chem., 2012, 51, 10704–10714 CrossRef CAS.
- N. Ueyama, N. Nishikawa, Y. Yamada, T.-a. Okamura, S. Oka, H. Sakurai and A. Nakamura, Synthesis and properties of octaethylporphinato(arenethiolato)iron(III) complexes with intramolecular NH⋯S hydrogen bond: chemical function of the hydrogen bond, Inorg. Chem., 1998, 37, 2415–2421 CrossRef CAS.
- R. Davydov, S. Im, M. Shanmugam, W. A. Gunderson, N. M. Pearl, B. M. Hoffman and L. Waskell, Role of the proximal cysteine hydrogen bonding interaction in cytochrome P450 2B4 studied by cryoreduction, electron paramagnetic resonance, and electron–nuclear double resonance spectroscopy, Biochemistry, 2016, 55, 869–883 CrossRef CAS PubMed.
- E. I. Solomon, S. I. Gorelsky and A. Dey, Metal–thiolate bonds in bioinorganic chemistry, J. Comput. Chem., 2006, 27, 1415–1428 CrossRef CAS PubMed.
- M. A. Dedushko, J. Pikul and J. A. Kovacs, Superoxide oxidation by a thiolate-ligated iron complex and anion inhibition, Inorg. Chem., 2021, 60, 7250–7261 CrossRef CAS PubMed.
- P. C. Y. Poon, M. A. Dedushko, X. Sun, G. Yang, S. Toledo, E. C. Hayes, A. Johansen, M. C. Piquette, J. A. Rees, S. Stoll, E. Rybek-Akimova and J. A. Kovacs, How metal ion lewis acidity and steric properties influence the barrier to dioxygen binding, peroxo O–O bond cleavage, and reactivity, J. Am. Chem. Soc., 2019, 141, 15046–15057 CrossRef PubMed.
- J. J. Ellison, A. Nienstedt, S. C. Shoner, D. Barnhart, J. A. Cowen and J. A. Kovacs, Reactivity of five-coordinate models for the thiolate-ligated fe site of nitrile hydratase, J. Am. Chem. Soc., 1998, 120, 5691–5700 CrossRef CAS.
- D. Schweitzer, J. J. Ellison, S. C. Shoner, S. Lovell and J. A. Kovacs, A synthetic model for the NO-inactivated form of nitrile hydratase, J. Am. Chem. Soc., 1998, 120, 10996–10997 CrossRef CAS.
- H. L. Jackson, S. C. Shoner, D. Rittenberg, J. A. Cowen, S. Lovell, D. Barnhart and J. A. Kovacs, Probing the influence of local coordination environment on the properties of Fe-type nitrile hydratase model complexes, Inorg. Chem., 2001, 40, 1646–1653 CrossRef CAS PubMed.
- D. Schweitzer, J. Shearer, D. K. Rittenberg, S. C. Shoner, J. J. Ellison, R. Loloee, S. Lovell, D. Barnhart and J. A. Kovacs, Enhancing reactivity via structural distortion, Inorg. Chem., 2002, 41, 3128–3136 CrossRef CAS PubMed.
- S. C. N. Shoner, A. Nienstedt, J. J. Ellison, I. Kung, D. Barnhart and J. A. Kovacs, Structural comparison of thiolate–ligated M(II)= Fe(II), Co(II), Ni(II), and Zn(II) ions wrapped in a chiral helical ligand, Inorg. Chem., 1998, 37, 5721–5725 CrossRef CAS.
- J. Shearer, J. Nehring, S. Lovell, W. Kaminsky and J. A. Kovacs, Modeling the reactivity of superoxide reducing metalloenzymes with a nitrogen and sulfur coordinated iron complex, Inorg. Chem., 2001, 40, 5483–5484 CrossRef CAS PubMed.
- J. Shearer, S. B. Fitch, W. Kaminsky, J. Benedict, R. C. Scarrow and J. A. Kovacs, How does cyanide inhibit superoxide reductase? Insight from synthetic FeIIIN4S model complexes, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3671–3676 CrossRef CAS PubMed.
- C. Guo, J. Yu, J. R. Horsley, M. Sheves, D. Cahen and A. D. Abell, Backbone-constrained peptides: temperature and secondary structure affect solid-state electron transport, J. Phys. Chem. B, 2019, 123, 10951–10958 CrossRef CAS PubMed.
- J. S. Kavanaugh, P. H. Rogers and A. Arnone, Crystallographic evidence for a new ensemble of ligand-induced allosteric transitions in hemoglobin: the T-to-THigh quaternary transitions, Biochemistry, 2005, 44, 6101–6121 CrossRef CAS PubMed.
- K. M. Clark, Y. Yu, N. M. Marshall, N. A. Sieracki, M. J. Nilges, N. J. Blackburn, W. A. van der Donk and Y. Lu, Transforming a blue copper into a red copper protein: engineering cysteine and homocysteine into the axial position of azurin using site-directed mutagenesis and expressed protein ligation, J. Am. Chem. Soc., 2010, 132, 10093–10101 CrossRef CAS PubMed.
- E. L. Hegg, S. H. Mortimore, C. L. Cheung, J. E. Huyett, D. R. Powell and J. N. Burstyn, Structure-reactivity studies in copper(II)-catalyzed phosphodiester hydrolysis, Inorg. Chem., 1999, 38, 2961–2968 CrossRef CAS PubMed.
- J. S. Plegaria, M. Duca, C. Tard, T. J. Friedlander, A. Deb, J. E. Penner-Hahn and V. L. Pecoraro,
De novo design and characterization of copper metallopeptides inspired by native cupredoxins, Inorg. Chem., 2015, 54, 9470–9482 CrossRef CAS.
- P. M. Colman, H. C. Freeman, J. M. Guss, M. Murata, V. A. Norris, J. A. M. Ramshaw and M. P. Venkatappa, Blue copper structure & entatic state, Nature, 1978, 272, 319 CrossRef CAS.
- S. I. Mann, T. Heinisch, T. R. Ward and A. S. Borovik, Peroxide activation regulated by hydrogen bonds within artificial cu proteins, J. Am. Chem. Soc., 2017, 139, 17289–17292 CrossRef CAS PubMed.
- A. S. Borovik, Bioinspired hydrogen bond motifs in ligand design: the role of noncovalent interactions in metal ion mediated activation of dioxygen, Acc. Chem. Res., 2005, 38, 54–61 CrossRef CAS PubMed.
- S. A. B. Cook and A. S. Borovik, Molecular designs for controlling the local environments around metal ions, Acc. Chem. Res., 2015, 48, 2407–2414 CrossRef CAS PubMed.
- S. A. H. Cook, E. A. Hill and A. S. Borovik, Lessons from Nature: a bio-inspired approach to molecular design, Biochemistry, 2015, 54, 4167–4180 CrossRef CAS PubMed.
- M. K. Coggins, X. Sun, Y. Kwak, E. I. Solomon, E. Rybak-Akimova and J. A. Kovacs, Characterization of metastable intermediates formed in the reaction between a Mn(II) complex and dioxygen, including a crystallographic structure of a binuclear Mn(III)–peroxo species, J. Am. Chem. Soc., 2013, 135, 5631–5640 CrossRef CAS PubMed.
- H. C. S. Fry, D. V. Scaltrito, K. D. Karlin and G. J. Meyer, The Rate of O2 and CO Binding to a Copper Complex, Determined by a “Flash-and-Trap” Technique, Exceeds that for Hemes, J. Am. Chem. Soc., 2003, 125, 11866–11871 CrossRef CAS PubMed.
- H. R. Lucas, G. J. Meyer and K. D. Karlin, CO and O2 Binding to Pseudo-tetradentate Ligand−Copper(I) Complexes with a Variable N-Donor Moiety: Kinetic/Thermodynamic Investigation Reveals Ligand-Induced Changes in Reaction Mechanism, J. Am. Chem. Soc., 2010, 132, 12927–12940 CrossRef CAS PubMed.
- N. W. Aboelella, S. V. Kryatov, B. F. Gherman, W. W. Brennessel, V. G. Young, R. Sarangi, E. V. Rybak-Akimova, K. O. Hodgson, B. Hedman, E. I. Solomon, C. J. Cramer and W. B. Tolman, Dioxygen activation at a single copper site: structure, bonding, and mechanism of formation of 1:1 Cu-O2 adducts, J. Am. Chem. Soc., 2004, 126, 16896–16911 CrossRef CAS PubMed.
- M. Weitzer, S. Schindler, G. Brehm, E. Hörmann, B. Jung, S. Kaderli and A. Zuberbühler, Reversible Binding of Dioxygen by the Copper(I) Complex with Tris(2-dimethylaminoethyl)amine (Me6tren) Ligand, Inorg. Chem., 2003, 42, 1800–1806 CrossRef CAS PubMed.
- C. X. Zhang, S. Kaderil, M. Costas, E. Kim, Y.-M. Neuhold, K. D. Karlin and A. D. Zuberbuhler, Copper(I)–dioxygen reactivity of [(L)CuI]+ (L = tris(2-pyridylmethyl)amine): kinetic/thermodynamic and spectroscopic studies concerning the formation of Cu–O2 and Cu2–O2 adducts as a function of solvent medium and 4-pyridyl ligand substituent variations, Inorg. Chem., 2003, 42, 1807–1824 CrossRef CAS PubMed.
- E. V. Rybak-Akimova, W. Otto, P. Deardorf, R. Roesner and D. H. Busch, Kinetics and equilibrium of dioxygen binding to a vacant site in cobalt(II) complexes with pentadentate ligands, Inorg. Chem., 1997, 36, 2746–2753 CrossRef CAS PubMed.
- A. F. Cozzolino, D. Tofan, C. C. Cummins, M. Temprado, T. D. Palluccio, E. V. Rybak-Akimova, S. Majumdar, X. Cai, B. Captain and C. D. Hoff, Two-step binding of O2 to a vanadium(III) trisanilide complex to form a non-vanadyl vanadium(V) peroxo complex, J. Am. Chem. Soc., 2012, 134, 18249–18252 CrossRef CAS PubMed.
- R. Cini and P. Orioli, Crystal and molecular structure of two oxygen-inactive forms of [N,N′-(3,3′-dipropylmethylamine)bis(salicylideneiminato) cobalt(II)], Inorg. Chim. Acta, 1982, 63, 243 CrossRef CAS.
- T. D. Palluccio, X. Cai, S. Majumdar, L. F. Sarafim, N. C. Tomson, K. Wieghardt, C. S. J. Cazin, S. P. Nolan, A. F. Cozzolino, E. V. Rybak-Akimova, M. A. Fernandez-Gonzalez, M. Temprado, B. Captain and C. D. Hoff, Ligand-directed reactivity in dioxygen and water binding to cis-[Pd(NHC)2(η2–O2)], J. Am. Chem. Soc., 2018, 140, 264–276 CrossRef CAS PubMed.
- X. Cai, S. Majumdar, G. C. Fortman, C. S. J. Cazin, A. M. Z. Slawin, C. Lhermitte, R. Prabhakar, M. E. Germain, T. D. Palluccio, S. P. Nolan, E. V. Rybak-Akimova, M. Temprado, B. Captain and C. D. Hoff, Oxygen binding to [Pd(L)(L0)] (L= NHC, L0 = NHC or PR3, NHC = N-heterocyclic carbene) synthesis and structure of a paramagnetic trans-[Pd(NHC)2(η1-O2)2] complex, J. Am. Chem. Soc., 2011, 133, 1290–1293 CrossRef CAS PubMed.
- J. P. Collman, J. I. Brauman, B. L. Iverson, J. L. Sessler, R. M. Morris and Q. H. Gibson, O2 and CO binding to iron(II) porphyrins: a comparison of the “picket fence” and “pocket” porphyrins, J. Am. Chem. Soc., 1983, 105, 3052–3064 CrossRef CAS.
- M. R. Momenteau and C. A. Reed, Synthetic heme dioxygen complexes, Chem. Rev., 1994, 94, 659–698 CrossRef CAS.
- M. M. Purdy, L. S. Koo, P. R. Ortiz de Montellano and J. P. Klinman, Mechanism of O2 Activation by cytochrome P450cam Studied by isotope effects and transient state kinetics, Biochemistry, 2006, 45, 15793–15806 CrossRef CAS PubMed.
- I. Szundi, C. Kittredge, S. K. Choi, W. McDonald, J. Ray, R. B. Gennis and O. Einarsdottir, Kinetics and intermediates of the reaction of fully reduced escherichia coli bo3 ubiquinol oxidase with O2, Biochemistry, 2014, 53, 5393–5404 CrossRef CAS PubMed.
- E. E. Weinert, C. M. Phillips-Piro, R. Tran, R. A. Mathies and M. A. Marletta, Controlling conformational flexibility of an O2-binding H-NOX domain, Biochemistry, 2011, 50, 6832–6840 CrossRef CAS PubMed.
- B. A. Springer, S. G. Sligar, J. S. Olson and G. N. Phillips Jr, Mechanisms of ligand recognition in myoglobin, Chem. Rev., 1994, 94, 699–714 CrossRef CAS.
-
E. Antonini and M. Brunori, Hemoglobin and Myoglobin in Their Reactions with Ligands, North-Holland, Amsterdam, 1971 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full derivation of the rate expression and explanation. CCDC 2212311. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02787f |
| ‡ University of Washington staff crystallographer |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Maria B.
Greiner
a,
Marc C.
Piquette
b,
Paige M.
Gannon
a,
Werner
Kaminsky‡
a and
Julie A.
Kovacs
a,
Maria B.
Greiner
a,
Marc C.
Piquette
b,
Paige M.
Gannon
a,
Werner
Kaminsky‡
a and
Julie A.
Kovacs