
Effective cerebral tuberculosis treatment via nose-to-brain transport of anti-TB drugs using mucoadhesive nano-aggregates†
Krishna
Jadhav
 a,
Agrim
Jhilta
a,
Raghuraj
Singh
a,
Eupa
Ray
a,
Vimal
Kumar
b,
Awadh Bihari
Yadav
c,
Amit Kumar
Singh
*b and
Rahul Kumar
Verma
*a
a,
Agrim
Jhilta
a,
Raghuraj
Singh
a,
Eupa
Ray
a,
Vimal
Kumar
b,
Awadh Bihari
Yadav
c,
Amit Kumar
Singh
*b and
Rahul Kumar
Verma
*a
aPharmaceutical Nanotechnology lab, Institute of Nano Science and Technology (INST), Sector-81, Mohali, Punjab 140306, India. E-mail: rahulverma@inst.ac.in; Fax: +91-172-2211074; Tel: +91-172-2210073/75 Ext-315 Tel: +91-8968439969
bExperimental Animal Facility, ICMR-National JALMA Institute for Leprosy and Other Mycobacterial Diseases, Tajganj, Agra, 282004, India
cCenter of Biotechnology, Nehru Science Centre, University of Allahabad, Prayagraj-211002, India
First published on 30th July 2024
Abstract
Central nervous system tuberculosis (CNS-TB) is a severe form of extra-pulmonary tuberculosis with high mortality and morbidity rates. The standard treatment regimen for CNS-TB parallels that of pulmonary TB, despite the challenge posed by the blood–brain barrier (BBB), which limits the efficacy of first-line anti-TB drugs (ATDs). Nose-to-brain (N2B) drug delivery offers a promising solution for achieving high ATD concentrations directly at infection sites in the brain while bypassing the BBB. This study aimed to develop chitosan nanoparticles encapsulating ATDs, specifically isoniazid (INH) and rifampicin (RIF). These nanoparticles were further processed into micro-sized chitosan nano-aggregates (NA) via spray drying. Both INH-NA and RIF-NA showed strong mucoadhesion and significantly higher permeation rates across RPMI 2650 cells compared to free ATDs. Intranasal administration of these NAs to TB-infected mice for four weeks resulted in a significant reduction of mycobacterial load by approximately ∼2.86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Log10 CFU compared to the untreated group. This preclinical data highlights the efficacy of intranasal chitosan nano-aggregates in treating CNS-TB, demonstrating high therapeutic potential, and addressing brain inflammation challenges. To our knowledge, this study is the first to show nasal delivery of ATD nano-formulations for CNS-TB management.
Log10 CFU compared to the untreated group. This preclinical data highlights the efficacy of intranasal chitosan nano-aggregates in treating CNS-TB, demonstrating high therapeutic potential, and addressing brain inflammation challenges. To our knowledge, this study is the first to show nasal delivery of ATD nano-formulations for CNS-TB management.
1. Introduction
Central nervous system tuberculosis (CNS-TB) is an extra-pulmonary form of tuberculosis (TB) that occurs when Mycobacterium tuberculosis (Mtb) spreads to the cerebrospinal fluid (CSF) and meninges. It is characterized by sub-acute or chronic meningitis, intracranial tuberculoma, and spinal tuberculous arachnoiditis.1 CNS-TB is a highly fatal and debilitating disease, affecting 1–2% of TB cases worldwide, with over 100000 cases per year.2,3 The current treatment regimen, which involves high oral doses of anti-tuberculosis drugs (ATDs) and chronic treatment durations of about six to nine months, presents significant challenges.4 Poor penetration and sub-therapeutic concentrations of key drugs in the CSF have led to studies on intensified treatment regimens and novel drug combinations to improve drug delivery to the CSF.5 Effective treatment of CNS-TB requires ATDs to cross significant barriers, such as the blood–brain barrier (BBB) and the blood-cerebrospinal fluid barrier (BCSFB), to reach therapeutic concentrations in the CSF. Therefore, there is a clear need for new approaches to enhance drug concentrations in the CSF of CNS-TB patients. Improved drug delivery could increase survival rates, reduce the development of drug resistance, and potentially shorten treatment durations.
The BBB presents a significant challenge for effectively delivering therapeutic agents in vivo. However, recent advancements have shown that the intranasal route of administration, known as the Nose-to-Brain (N2B) pathway, is highly effective in delivering drugs to the brain.6,7 It utilizes a large absorption surface area within the nasal cavity, facilitating rapid attainment of therapeutic drug levels and bypassing initial metabolic processes, thereby enhancing drug bioavailability in the CNS.8,9 Compared to traditional parenteral routes, N2B delivery enhances patient comfort and compliance, while also avoiding hepatic first-pass drug metabolism, improving patient adherence, and reducing systemic side effects.10,11 The N2B delivery route, despite its numerous benefits, faces significant challenges such as accurate drug dosing, rapid elimination by mucociliary clearance, and inter-patient variability.12 One of the primary challenges in utilizing nose-to-brain delivery in humans is the need for high pressure to effectively transport therapeutic agents across the nasal mucosa and into the brain, which raises safety and feasibility concerns.13 Nanocarrier-based formulations can address these challenges by incorporating mucoadhesive properties to improve nasal permeation and increase residence time in the nasal cavity, thereby avoiding rapid mucociliary clearance.14–16 Chitosan nanoparticles (CSNP) has been explored as nanocarriers for nasal delivery of several small molecules and biologicals in different formulations.17
In this study, we designed and evaluated chitosan nano-aggregates encapsulating a combination of ATD, specifically isoniazid (INH) and rifampicin (RIF), to target virulent Mtb both in vitro and in an experimental CNS-TB murine model. The nano-aggregates (NA) are designed to de-aggregate into nanoparticles upon reaching the nasal mucosa when they encounter nasal fluids after nasal deposition. This dual functionality combines the benefits of both nano- and micro-particles, offering optimal aerodynamic properties and enhanced penetration at the target site, thereby optimizing therapeutic outcomes. The formulation was evaluated for chemotherapeutic efficacy, toxicity, and brain inflammation in a murine model of virulent tuberculosis. Our findings provide compelling evidence for the superior efficacy of ATD-NA in targeting the brain in a CNS-TB murine model via the intranasal route. Compared to the oral route, intranasal administration resulted in enhanced efficacy and increased mycobacterial clearance from the brain.
2. Experimental section
2.1. Fabrication of chitosan nanoparticles (CSNP)
CSNP encapsulated with anti-TB drugs were fabricated employing a standard ionotropic gelation process reported by our group with some modifications.18,19 Briefly, chitosan (20–300 cP) is dissolved in glacial acetic acid (1% v/v) with a concentration of 1.75 mg ml−1 under constant overnight magnetic stirring at 300 rpm at room temperature and filtered through 0.22 μm filters to remove undissolved materials. Similarly, sodium tripolyphosphate (TPP) was solubilized in MilliQ water to produce a concentration of 1.5 mg ml−1. Then, 4 ml of TPP solution was added dropwise into 10 ml of chitosan solution under constant mechanical stirring (REMI 5ML-H, India) at 800 rpm. A zone of opalescence appeared on the optimized ratio of chitosan to TPP (3.3:1) and was upheld to produce blank CSNP. To prepare ATD-loaded CSNP, INH or RIF-loaded CSNP were prepared individually as above and with optimized chitosan to TPP ratio. INH (5 mg) was added to the TPP solution, and RIF (5 mg) was dissolved in chitosan solution under constant stirring before the addition of TPP solution. The resultant nanoparticles were centrifuged (Sorvall ST 8R, Thermo Fisher Scientific) at 14000 rpm at 4 °C for 30 minutes. The obtained sediment was dispersed in MilliQ water and supernatant was centrifuged again twice. The dispersed nanoparticles were used for further processing.
2.2. Processing of nanoparticles to nano-aggregates
The dry powder based on microencapsulated CSNP was developed using spray drying with some modifications.20 The prepared INH-CSNP or RIF-CSNP were centrifuged, and further formed pallets were resuspended in a mannitol solution (2 mg ml−1) with a solid content of 2%. INH-NA and RIF-NA dry powder were formulated using a lab-scale spray dryer (Techno Search Instruments, Mumbai, India) equipped with a 0.7 mm nozzle. The following parameters were adopted in the spray dryer during NA formation: the inlet air temperature was ∼110 °C, the aspirator flow rate was −60 mm of WC, the feed solution flow was 3 ml min−1, and atomization air at ∼1.25 kg cm−2 (fixed for both formulations). The nanoparticle suspension was fed into the feeding system and atomized through the nozzle. The resultant powder formulations were driven through the cyclone separator and deposited in the collecting bowl. Afterwards, the powders were removed from the collecting bowl and placed in a desiccator for further examination.21–23 Physicochemical characterization including detailed analysis of both physical and chemical properties of nanoparticles and resultant nanoaggregates are mentioned in ESI, S3.†2.3. Redispersion of nanoparticles from nano-aggregates
Achieving effective therapeutic outcomes depends on the ability of nano-aggregates to readily disperse into primary nanoparticles in an aqueous environment. This capability ensures the nanoparticles can function as intended in therapeutic applications. To assess re-dispersibility, 10 mg of ATD-NA powder was meticulously dispersed in 1 ml of Milli-Q water and vortexed for 30 seconds to achieve a stable and uniform colloidal dispersion. Subsequently, the particle size of the re-dispersed CSNP was measured. The effectiveness of re-dispersion was characterized by examining the change in particle size before and after redispersion. The Redispersibility Index (RDI) value, a metric that compares particle size before spray-drying to after redispersion, was utilized. An RDI value close to 1 indicates complete redispersion, whereas values exceeding 1.5 suggest poor redispersion.24–26 These measurements provide critical insights into the ability of the nanoparticles to maintain their functional properties after undergoing the spray drying process.2.4. Solid state characterization
2.5. Mucoadhesive strength of nano-aggregates
The mucoadhesive strength of NA was evaluated by investigating the interaction between nanoparticles and mucin using a previously reported protocol with minor modifications.27 In brief, porcine mucin type-III (0.5 mg ml−1) was suspended in SNF (pH 5.5) to mimic nasal conditions. Subsequently, an equal volume of mucin solution and reconstituted CSNP were mixed, vortexed, and incubated at 35 °C in a shaking incubator bath for 60 min. Subsequent to incubation, the resulting suspensions were subjected to centrifugation at 12000g for 40 minutes at 4 °C to pellet the CSNP. The residual free mucin present in the supernatant was quantified using a UV-vis spectrophotometer at 255 nm. The quantification of mucin-adsorbed nanoparticles was achieved by calculating the disparity between the initial mucin quantity added and the remaining mucin present in the supernatant. The pellet was resuspended in SNES and ς-potential of resuspended CSNP was recorded.
2.6. In vitro nasal deposition study
The aerodynamic properties and in vitro nasal deposition evaluation of ATD-NA were carried out using a Next Generation Cascade Impactor (NGI) (Copley, Nottingham, UK) with a well-equipped nasal extension chamber (1 L).28–30 The 25 mg of ATD-NA powder samples were loaded into a nasal expansion chamber via an entry port to mimic the nasal administration of powder. The apparatus was actuated at an airflow rate of 15 L min−1 using a calibrated pump and flow meter, considered the standard inspiration flow rate of the nose. Upon actuation of the nasal apparatus, the released powder was pulled through NGI at a constant airflow rate of 15 L min−1. The particles were segregated via their aerodynamic diameter with seven stages (1 to 7) with an effective cut-off diameter of 14.1, 8.61, 5.39, 3.30, 2.08, 1.36, and 0.98 μm, respectively. The collected powder sample in different stages was analyzed for aerodynamic properties such as mass median aerodynamic diameter (MMAD), geometric standard deviation (GSD), and fine particle fraction (FPF) to evaluate drug deposition in the nasal mucosa.2.7. Cell culture experiments
 | (1) |
2.8. In vitro antimycobacterial activity
The in vitro efficacy of ATD-NA was determined by using resazurin microplate assay (REMA). Virulent MTB C3 strain was cultured in Middlebrook 7H9 medium (Difco Laboratories, MI, USA) with 10% Albumin-dextrose-Catalase (ADC) supplement, 0.2% glycerol, and 0.05% Tween-80. The mycobacterial culture was procured at 2500g, and concentration was attuned to No. 1 McFarland standard and 1:20 dilution of the culture was prepared with 7H9 medium resulting in ∼1 × 105 colony-forming units (CFU) per ml. The ATD and ATD-NA ranging from 0.64 to 20 μg ml−1 were dissolved in 7H9 medium and introduced into respective wells. The culture media containing C3 strain was utilized as the negative control, whereas the culture medium alone served as the blank control. Following an incubation period of six days, a solution of resazurin dye (0.02% w/v) in a volume of 25 μL was added to each well of the plates and subsequently incubated for an additional day. The determination of the minimum inhibitory concentration (MIC) value was based on the extent to which the resazurin dye colour remained unchanged from blue to pink, indicating the absence of mycobacterial growth.23
2.9. Animal efficacy trials
| Animal group | Groups of mice (n = 6) | Infection and/or treatment |
|---|---|---|
| a ATD @ human equivalent doses i.e., INH-25 mg kg−1 and RIF-10 mg kg−1. | ||
| Group A | Uninfected | No infection, no treatment |
| Group B | +TB/untreated | Infection, no treatment, estimation of CFU after 10 weeks of infection (At the end of experiment) |
| Group C | +TB/oral ATD | ATDa (INH-625 μg, RIF-250 μg)/mice by oral route (5 days per week) |
| Group D | +TB/ATD-NA (insufflation) | ATD-NAa (INH-625 μg, RIF-250 μg)/mice insufflation by intranasal route (5 days per week) |
| Group E | +TB/ATD-NA (instillation) | ATD-NAa (INH-625 μg, RIF-250 μg)/mice instillation by intranasal route (5 days per week) |
In this study, a specialized engineered in-house insufflation device specifically designed for nasal administration entailed the release of a puff of powder from a small delivery tube (ESI S7 and Fig. S3A†).36,37 For the intranasal instillation, ATD-NA powder was suspended in PBS or normal saline, and a fine pipette tip was used to deliver 25 μL of the solution into each nare. The dosing regimen was administered five days per week for four weeks. For oral administration, a ball-tipped 20-gauge cannula was utilized. The antimycobacterial efficacy of ATD-NA powder was evaluated using histopathological analysis and bacterial load determination through the enumeration of CFUs in the brain, lungs, and spleen. Pharmacokinetic evaluation was conducted on naïve BALB/c mice to quantify the concentration reaching the brain following intranasal administration, as mentioned in ESI, S8† (Fig. 4).
2.10. Statistical analysis
All experimental results of different experiments were presented as the mean ± SD. All statistical analyses were executed using the GraphPad Prism (Version 8.4.2) California, USA statistical software. Student's t-test was employed to compare the two groups, whereas One-way Analysis of Variance (ANOVA) followed by the Bonferroni post hoc test was used to assess the significance among multiple groups. Statistical significance was denoted by p-values, with significance levels depicted as follows: * p < 0.05, ** P < 0.01, *** p < 0.001, **** p < 0.0001.3. Results
3.1. Characterization of mucoadhesive chitosan nanoparticles
In the present work, we individually developed CSNP encapsulated with ATDs (INH and RIF) using the ionic gelation method. This method is simple and easily applied as it includes adding two aqueous phases, i.e., TPP and chitosan solution, under magnetic stirring at room temperature. The resulting blank CSNP had a mean hydrodynamic diameter of 180.3 ± 1.65 nm, a polydispersity index (PDI) of 0.229 ± 0.008, and a zeta potential of 16.4 ± 0.681 mV (Fig. 1E and F). The narrow PDI value for all formulations indicates a relatively narrow size distribution and homogeneity of the particles. The positive zeta potential values indicate a stable suspension with a low likelihood of aggregation. In the first formulation, INH-CSNP had a median hydrodynamic diameter of 188.2 ± 2.75 nm, a PDI of 0.174 ± 0.033, and a zeta potential of 14.2 ± 1.106 mV. In the second formulation, RIF-CSNP had a mean particle size of 190.5 ± 2.21 nm, a PDI of 0.194 ± 0.024, and a zeta potential of 15.3 ± 0.666 mV. The EE of INH-CSNP and RIF-CSNP was determined to be 66.86% and 49.70%, respectively, indicating a high capacity for efficient encapsulation of ATDs within the CSNP. The CSNPs were observed to be smooth and spherical according to TEM and FE-SEM images and were uniformly dispersed, as confirmed by AFM images. In addition, both SEM and TEM analyses unveiled that the particle size of CSNPs was below 200 nm (Fig. 1). These results were in agreement with the data obtained from DLS.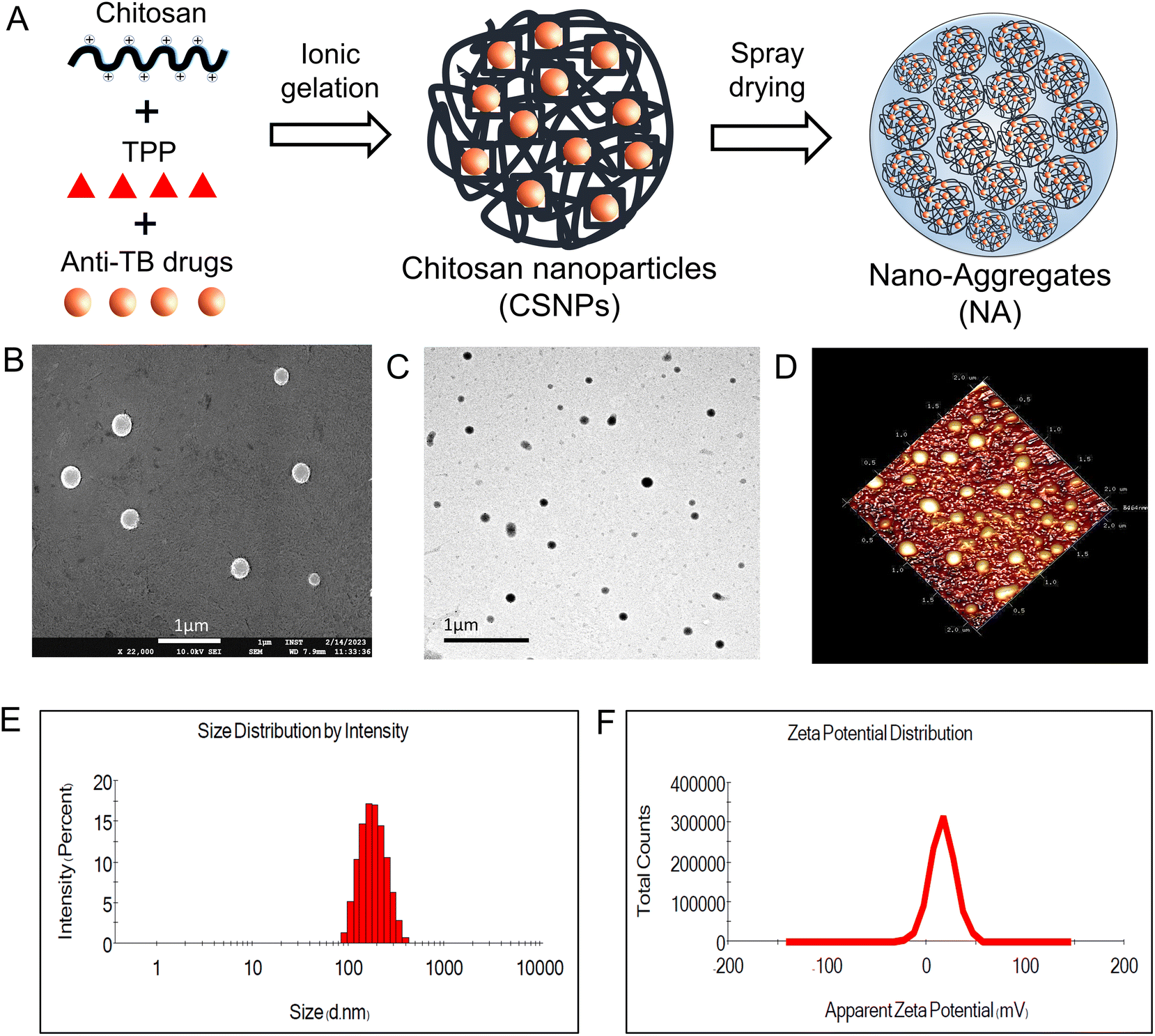 | ||
| Fig. 1 Fabrication and characterization of chitosan nanoparticles (CSNPs). (A) Schematic for the preparation of CSNP. (B) Scanning electron microscopy (SEM) showing spherical and smooth morphology of CSNP, (scale bar = 1 μm). (C) Transmission electron microscopy (TEM) of CSNP. (D) Atomic force microscope (AFM) image of CSNP showing 3D topographic representation of CSNP. (E) Particle size and (F) zeta potential of CSNP by dynamic light scattering (DLS). Data represents mean ± S.D (n = 3). | ||
3.2. Characterization of nano-aggregates
In this study, nano-aggregates encased with ATD-CSNP were created using spray drying to address drawbacks associated with liquid forms and impart suitable aerodynamic properties for effective nasal deposition of particles. The spray drying setting was optimized to get maximum process yield of powder formulation ranging from 48 to 52%. As observed in SEM micrographs, depicted in Fig. 2B, the produced NA powder shows a mulberry shape with a non-smooth topology revealing visible aggregated nanoparticles. The INH-NA and RIF-NA have a mean diameter of 5.83 ± 1.97 μm and 6.45 ± 1.38 μm, respectively, with particle size distribution of ∼5 to 10 μm Table 2. RDI is a key metric used to evaluate the ability of spray-dried NA powder to reconstitute into its native form. After redispersion of INH-NA and RIF-NA, the RDI value was found to be 1.12 and 1.08, close to 1, indicating that the powder can be effectively reconstituted into INH-CSNP and RIF-CSNP, respectively. The slight increase in particle size distribution of redispersed CSNP compared to the one observed with the freshly prepared CSNP, may be due to the presence of cryoprotectants, i.e., mannitol. Dynamics in particle size were evaluated for NA after three months of storage condition (ESI Fig. S2E†), indicating stability parameters relevant to the NA, including particle size of redispersed CSNP. The particle size and PDI of the blank-CSNP and INH-CSNP appeared relatively stable over the three months, with slight changes in the average particle size and PDI values. However, the RIF-NA showed a significant increase in particle size and PDI over the three months study period. The zeta potential values for all three formulations decreased slightly over the three months, indicating a possible change in the surface charge of the particles.| Formulations | Particle size (nm) | PDI | Zeta potential (mV) | EE (%) | DL% |
|---|---|---|---|---|---|
| Blank CSNP | 174.2 ± 1.65 | 0.136 ± 0.08 | 16.4 ± 0.681 | — | — |
| INH-CSNP | 188.2 ± 2.75 | 0.174 ± 0.03 | 14.2 ± 1.106 | 66.86 ± 3.1 | 9.8 ± 0.9 |
| RIF-CSNP | 193.0 ± 2.21 | 0.210 ± 0.02 | 15.3 ± 0.666 | 49.70 ± 1.9 | 8.47 ± 1.2 |
 | ||
| Fig. 2 Formulation and characterization of nano-aggregates (NA). Scanning electron microscopy (SEM) images of (A) CSNP (scale bar = 1 μm), (B) group of NA and single NA showed a spherical and non-smooth surface with aggregation of nanoparticles. (C) FT-IR spectra (D) PXRD patterns and (E) DSC thermogram of anti-TB drugs and ATD-NA formulations. (F) In vitro nasal deposition analysis of ATD-NA dry powder as a percentage of total emitted dose on each stage of the NGI. (G) Adsorption kinetic of formulations with 1 mg ml−1 of mucin solution at 35 °C. Mucin adsorption (%) of formulations represents the excellent interaction of CSNP with mucin (<70%), which renders it effective mucoadhesive characteristics. (H) Zeta potential measurement performed for mucin solution and mucin + ATD-NA before and after incubation at 35 °C with mucin (mean ± standard deviation, n = 3, ***P < 0.001). | ||
3.3. Solid state characterization
The FT-IR analysis of ATD and ATD-NA exhibits characteristic peaks and represents no interaction between drugs and polymers. The spectra revealed no chemical changes or interactions were observed between anti-TB drugs and excipients used for nanoparticle preparation (Fig. 2C). The characteristic peaks of INH [3300 cm−1 (N–H stretching, Amine), 3108 cm−1 (C–H asymmetric stretching), 3008 cm−1 (C–H symmetric stretching), 1664 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1552 cm−1 (N–H bending), 1327 cm−1 (NH2 wagging)], RIP [3483.85 cm−1 (v(–NH)), 2975 cm−1 (v(–OH)), 1648 cm−1 (v(–CO)), 1567 cm−1 (amide CO), 1726 cm−1 (furanone CO), 2883 cm−1 (N–CH3)] observed in ATR-FTIR spectra. The characteristic peaks for drugs were also reported in the physical mixture of polymers with drugs. We could conclude that no chemical modification was observed as polymers mixed with drugs. The diffractogram of ATD, such as INH and RIF exhibited several sharp crystalline peaks (Fig. 2D). Briefly, PXRD spectra of INH showed crystalline peaks at 2θ = 11.70, 14.10, 15.6, 16.50, 19.6, and 25.2 with some extra peaks in the between 26–30, and RIF exhibited major crystalline peaks at 2θ = 11.9, 13.8, 14.4, 16.3, 18.5, and 20, respectively. In the case of ATD, all samples exhibited reciprocal PXRD patterns, demonstrating the different XRD patterns from raw materials. Fig. 2E illustrates the DSC thermograms of chitosan, INH, RIF, INH-NA, and RIF-NA. The DSC thermogram of INH exhibited a distinct peak at 175.97 °C, indicating the presence of a crystalline region and representing its melting point. Pure RIF displayed an intense endothermic peak at 197.1 °C, along with two exothermic signals observed at approximately 212.2 and 259.1 °C, which corresponded to the melting point of its crystalline form. Chitosan, on the other hand, demonstrated an endothermic peak at 88.8 °C, signifying the loss of bound and absorbed water. Notably, similar endothermic peaks were observed in the DSC thermograms of INH-NA and RIF-NA, appearing at 91.1 °C and 89.7 °C, respectively. Although there were slight variations in the position and peak area of these endothermic peaks compared to pure chitosan, these differences could be attributed to the varying water-polymer interactions and different holding capacities. Importantly, the disappearance of the endothermic peaks corresponding to INH and RIF in the INH-NA and RIF-NA, respectively, confirmed the successful encapsulation and stabilization of the drugs into CSNP matrix and their presence in an amorphous state.
O stretching), 1552 cm−1 (N–H bending), 1327 cm−1 (NH2 wagging)], RIP [3483.85 cm−1 (v(–NH)), 2975 cm−1 (v(–OH)), 1648 cm−1 (v(–CO)), 1567 cm−1 (amide CO), 1726 cm−1 (furanone CO), 2883 cm−1 (N–CH3)] observed in ATR-FTIR spectra. The characteristic peaks for drugs were also reported in the physical mixture of polymers with drugs. We could conclude that no chemical modification was observed as polymers mixed with drugs. The diffractogram of ATD, such as INH and RIF exhibited several sharp crystalline peaks (Fig. 2D). Briefly, PXRD spectra of INH showed crystalline peaks at 2θ = 11.70, 14.10, 15.6, 16.50, 19.6, and 25.2 with some extra peaks in the between 26–30, and RIF exhibited major crystalline peaks at 2θ = 11.9, 13.8, 14.4, 16.3, 18.5, and 20, respectively. In the case of ATD, all samples exhibited reciprocal PXRD patterns, demonstrating the different XRD patterns from raw materials. Fig. 2E illustrates the DSC thermograms of chitosan, INH, RIF, INH-NA, and RIF-NA. The DSC thermogram of INH exhibited a distinct peak at 175.97 °C, indicating the presence of a crystalline region and representing its melting point. Pure RIF displayed an intense endothermic peak at 197.1 °C, along with two exothermic signals observed at approximately 212.2 and 259.1 °C, which corresponded to the melting point of its crystalline form. Chitosan, on the other hand, demonstrated an endothermic peak at 88.8 °C, signifying the loss of bound and absorbed water. Notably, similar endothermic peaks were observed in the DSC thermograms of INH-NA and RIF-NA, appearing at 91.1 °C and 89.7 °C, respectively. Although there were slight variations in the position and peak area of these endothermic peaks compared to pure chitosan, these differences could be attributed to the varying water-polymer interactions and different holding capacities. Importantly, the disappearance of the endothermic peaks corresponding to INH and RIF in the INH-NA and RIF-NA, respectively, confirmed the successful encapsulation and stabilization of the drugs into CSNP matrix and their presence in an amorphous state.
3.4. In vitro nasal deposition analysis
The aerodynamic properties were evaluated using NGI to confirm the suitability of the particle size range for nasal deposition. The aerodynamic particle diameter is a crucial factor for nasal delivery, as the particles needed to deposit in the nasal cavity with minimal lung deposition. Table S1† represents the aerodynamic properties, including ED, MMAD, and FPF of INH-NA and RIF-NA. At an airflow rate of Q = 15 L min−1, both formulations exhibited an excellent total emitted dose (ED) of ≥80%, signifying good powder dispersion from the Rotahaler for passive animal inhalation. The formulation distribution across various stages of the NGI, as demonstrated in Fig. 2F, revealed that a significant portion of the particles was deposited in the pre-separator and stage-1 (cut-off: 8.06 μm). The FPF value for both formulations was observed in the range of 34.4 ± 4.22% to 31.46 ± 3.16%, which represents the minimal proportion of particles that exist in the respirable size range. These results suggest that the tested formulations exhibit a particle size distribution suitable for nasal delivery, with a substantial proportion of the emitted dose deposited in the nasal cavity. Furthermore, the relatively low levels of fine particles that may penetrate the lower respiratory tract indicate that the formulations with particle size distributions conducive to nasal delivery may offer enhanced therapeutic benefits.3.5. Nano-aggregates powder shows mucoadhesion with the mucin
The in vitro mucoadhesion potential of NA was assessed by suspending them in diluted aqueous mucin solutions at room temperature, and mucin adsorption studies were conducted for unloaded and ATD-NA. The study found that the prepared unloaded NA had strong mucoadhesion potential after 30 min of incubation with a range of ∼71.57 to 78.62%, as inherent nature of CSNP (Fig. 2G). This outcome demonstrates that there is a strong electrostatic interaction between CSNP and mucin. In addition, INH-NA and RIF-NA exhibit strong mucoadhesion tendency with 65.17 and 72.10% percent mucin adsorbed, which was slightly less when compared to unloaded particles. This lesser depiction may be caused due to several factors which contribute to mucoadhesion potential, including the number of free amino groups, molecular weight, and degree of cross-linking of chitosan. Fig. 2H depicts the alterations in surface charge of the unloaded CSNP or formulated with anti-TB drugs upon interaction with mucin. The incubation of nanoparticles with mucin resulted in a decreased surface charge across all groups. This decrease can be ascribed to the ionic interaction taking place between the negatively charged sialic moieties inherent in mucin and the positively charged outer layer of the CSNP. The reduction in zeta potential observed prior to and subsequent to the mixing process signifies the nanoparticle's substantial mucoadhesive strength towards the nasal mucosa, suggesting their potential for prolonged residence and enhanced adhesion at the targeted site.3.6. Nano-aggregates promote the transepithelial permeability of anti-TB drugs
RPMI-2650 cell line is well-suited for evaluating the permeability of substances across epithelial barriers. These cell lines exhibit physiologically relevant TEER values that suggest they form similar barriers to inhaled xenobiotics as those found in living organisms. RPMI-2650 cells cultured for 21 days on transwell inserts, demonstrated a TEER value of 81.6 ± 7.6 Ω cm2, which falls within the range of measurements obtained from excised human/animal nasal mucosa (40–120 Ω cm2). Fig. 3D illustrates the gradual development of the tight junction barrier by RPMI 2650 cells cultured on SPLInsertTM over time. The TEER values steadily increased from around 21.67 Ω cm2 until reaching a plateau between 101.67 Ω cm2, which persisted up to day 17 before the TEER values started to decline. The findings imply that a minimum of 14 days is required for the cells to establish a cohesive and impermeable layer with the maximum TEER barrier when cultured under air–liquid interface (ALI) conditions. However, beyond day 17, a decrement in TEER values is observed, suggesting a potential deterioration in the integrity of the tight junctions among the cells. The study concludes that values above 75 Ω cm2 are adequate for conducting experiments, where the TEER values ranged from 90 to 150 Ω cm2. Transepithelial permeation of anti-TB drugs on the RPMI 2650 nasal septum cell monolayer was performed in the direction of the apical to basolateral compartment. Fig. 3C, D shows the flux of anti-TB drugs and equivalent formulations calculated from eqn (1). The Papp value of INH from INH-NA formulations was found to be 2.12 × 10−6 cm s−1, which was higher permeation compared to the free INH (0.79 × 10−6 cm s−1) on RPMI 2650 cell monolayer. The Papp of RIF from RIF-NA was found to be 2.02 × 10−6 cm s−1, comparatively higher than that of free RIF (1.12 × 10−6 cm s−1). Intriguingly, INH showed higher permeation than RIF-based formulations, which may be explained by the difference in solubility of compounds and efflux mechanisms. The findings of the study suggest that the observed effects of chitosan on the nasal absorption of INH can be attributed to the enhance the paracellular pathway by widening the tight junctions.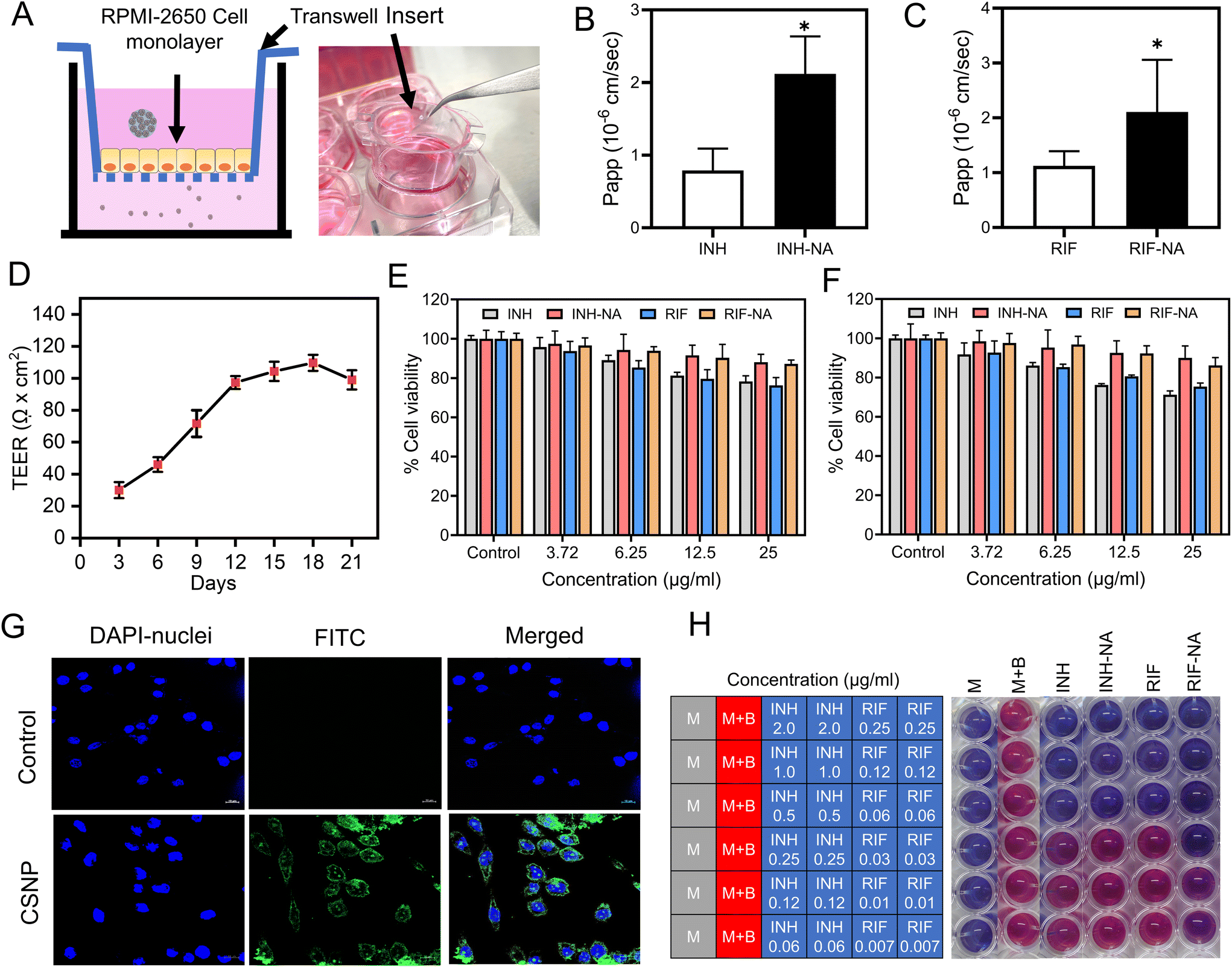 | ||
| Fig. 3 In-vitro permeability, biocompatibility and efficacy studies of nano-aggregates (A) Schematic and set up of transwell system (SPLinsert™) utilized for in vitro permeability experiment. Apparent permeability coefficient (Papp) of (B) INH and INH-NA (C) RIF and RIF-NA from RPMI 2650 monolayers under ALI conditions at 37 °C. (D) TEER measurement throughout cultivation and growth RPMI 2650 cell onto SPLinsert™ (n = 6, mean ± SD) for 21 days until constant TEER values are obtained. The viability of RPMI 2650 cells (E) and glioma cells (F) after treatment with blank-NA, INH, RIF, and their equivalent formulations (INH-NA and RIF-NA) at different concentrations (3.725, 6.25, and 12.5 μg ml−1) analyzed by MTT assay. (G) Cellular uptake of FITC-loaded CSNP by glioma cells for 12 h, measured by confocal microscopy (FITC-loaded CSNP, DAPI stained nuclei, and merged image, scale bar ∼10 μm). (H) The in vitro anti-TB activity of INH, RIF, and their formulations (INH-NA and RIF-NA) was evaluated using REMA assay (blue colour indicated anti-Mtb activity, while a pink colour signified bacterial growth). The blank medium was used as sterility control. (M – blank medium controls without inoculation, M + B – growth control without treatment. Values are expressed as mean ± SD (n = 3). | ||
3.7. Nanoparticles exhibited enhanced internalization in glioma cells
The cellular uptake of nanoparticles in the glioma cell line was evaluated using CSLM. The intracellular fate of nanoparticles notably depends on the size, shape, and surface composition of particles, which CSNP reportedly favoured clathrin-mediated endocytosis. FITC-tagged CSNP (FITC-CSNP) were fabricated to enumerate the potential uptake into brain cells. Confocal microscopy images from the FITC channel showed green fluorescence of FITC-CSNP, and pictures obtained from the DAPI channel showed distinct blue fluorescence of cell nuclei. FITC-CSNP has superior uptake and internalization into brain glioma cells compared to untreated cells depicted in Fig. 3G. The merged channel of FITC and DAPI revealed that cytoplasmic and nuclear localization of CSNP internalized by brain glioma cells. The results inferred that CSNPs have excellent brain targeting and membrane permeability. The collective findings presented herein provide strong evidence for the improved cellular uptake of CSNP, establishing a robust basis for their potential in vivo applications.3.8. Nano-aggregates show cytocompatibility with nasal epithelial cells and glioma cells
After nasal administration, nanoparticles must permeate the frontline barrier, i.e., nasal epithelial cells, to reach the brain. A cell viability assay was performed to address the concerns regarding the toxicity of the developed formulation. A cell viability assay was conducted using NA containing varying concentrations of 3.25 μg ml−1 to 12.5 μg ml−1 on RPMI-2650 and C6 cells for all samples to assess the safety of powder formulation for potential use in future animal studies and compared with the control group. Fig. 3E demonstrates that the cell viability of cells after exposure to INH and RIF for 24 h was more than 80%, indicating that both drugs are biocompatible with RPMI-2650 cells. Both INH-NA and RIF-NA were compatible with RPMI-2650 cells, with all samples revealing more than 90% cell viability.3.9. In vitro anti-mycobacterial activity
To assess the antimicrobial efficacy in vitro, the MIC values of the pure anti-TB drug and its respective formulations were determined using the REMA plate method. This assay relies on an oxidation–reduction reaction that changes colour from blue to pink in the presence of live bacteria. In our study, the MIC value of pure INH was observed at ∼0.25 μg ml−1, consistent with previously reported studies on INH (Fig. 3H).39 Interestingly, the MIC values of the prepared INH-NA exhibited similar efficacy to pure INH. Furthermore, the MIC value of RIF was observed to be 0.06 μg ml−1, consistent with the MIC value obtained from RIF-NA. These outcomes unveil that encapsulating anti-TB molecules into nanoparticles and subsequently into microparticles did not compromise their potential antimycobacterial effect.3.10. Animal trials
log10 CFU on 45 days in Mtb infected mice. The histopathology analysis revealed inflammatory cell infiltration in mice brains at 45 days, which was more aggravated at 75 days post-infection in infected but untreated mice (Fig. 4B). Infected mice were randomly allocated into six groups to receive oral and nasal administration of ATD and ATD-NA at human equivalent doses for four weeks (Table 1). At the end of the treatment period, the reduction of mycobacterial load and changes in histopathology in the brain, lungs, and spleens were evaluated. The infected mice tolerated the nasal administration of ATD-NA dry powder at human equivalent doses with no apparent adverse effects. Pharmacokinetic analysis revealed that intranasal delivery of the formulation attained therapeutic concentrations in the brain necessary for effective anti-mycobacterial activity (ESI S8, Fig. S4†).
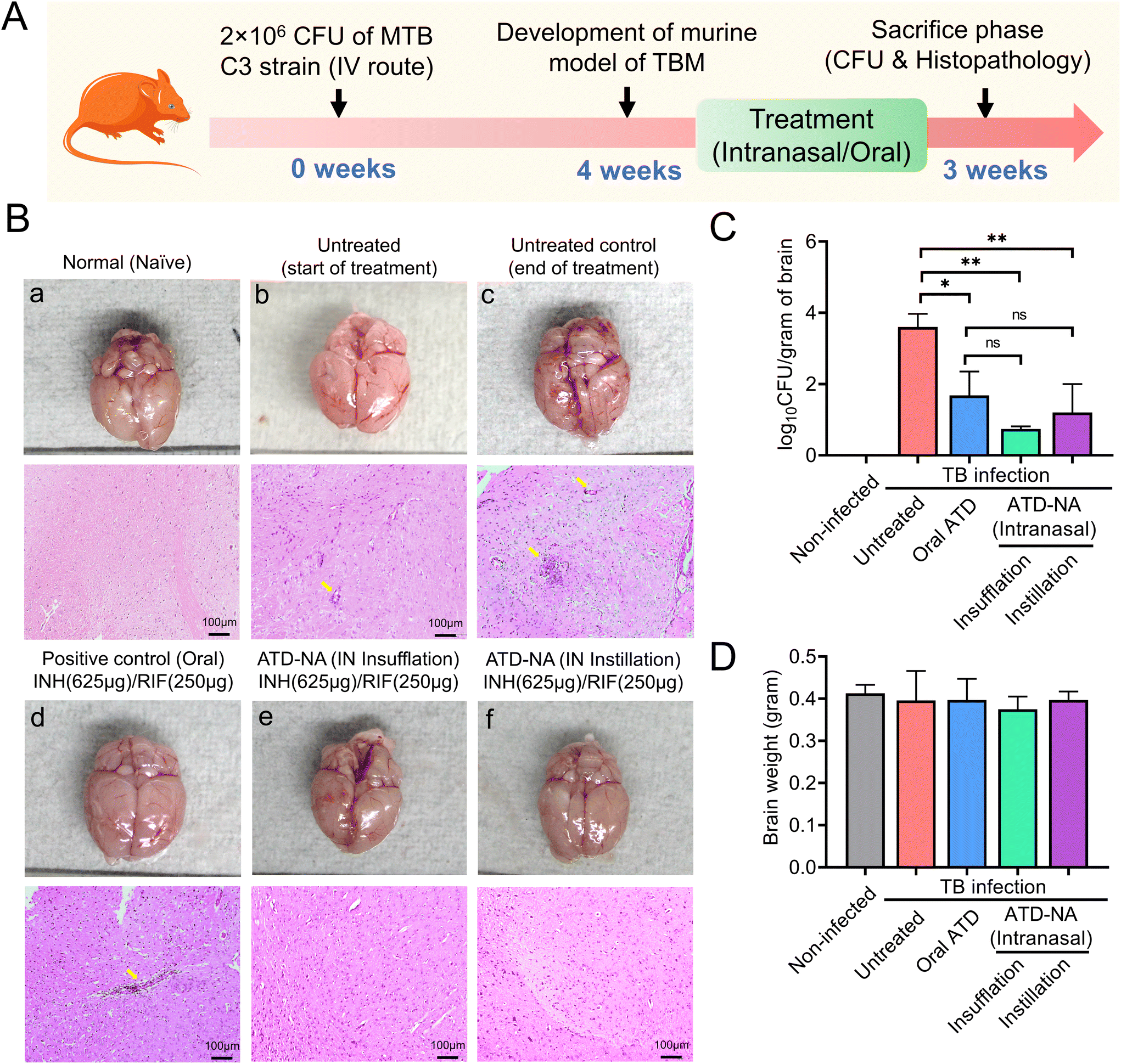 | ||
| Fig. 4 Efficacy evaluation. Brain morphology, histopathological analysis and mycobacterial load in mice were subjected to Mtb-exposed and diverse treatment groups. (A) Schematic for in vivo antimicrobial efficacy on CNS-TB murine model. (B) Illustrations of the whole brain of mice demonstrating brain healing, upper panel: representative morphological images from each group of animals exhibiting early and untreated control, oral ATD, and ATD-NA via intranasal route. Lower panel: H &E-stained sections of the brain of naïve, untreated and treated mice. (a, b, c, d, e and f). (C) The mean count of CFU in the brain of both untreated and treated mice serve as indicators of the bacterial load in the brain tissues. (D) Graph depict the average weight of the wet brain (n = 5). | ||
Brain. Morphological analysis of the organs harvested from Mtb-infected mice revealed noticeable swelling/oedema of the brain in the left hemisphere compared to control mice (Fig. 4B). Intranasal delivery of ATD-NA via dry powder insufflation or instillation exhibited notably higher bactericidal activity in the brain tissues four weeks after initiation of treatment (P < 0.001) (Fig. 4C) (Table 3). At the end of the 4-week treatment, oral INH and RIF reduced the brain CFU count to 1.90
log10 CFU per gram (CFU diminution compared to the untreated group). Mice treated with the ATD-NA delivered by insufflation had a significant reduction of mycobacterial burden after four weeks of treatment (CFU diminution compared to untreated group ∼2.86log10 CFU). Compared with the ATD-treated group, the ATD-NA insufflated group showed an additional reduction of approximately 1.0log10 CFU in the lungs of BALB/c mice (P < 0.05). The mice group treated with ATD-NA via the instillation route exhibited significant inhibition of mycobacterial growth compared to the untreated group (P < 0.05). Additionally, although the difference was not statistically significant, the ATD-NA treatment offered superior protection compared to oral dosing.
| Group | Experimentation animal group | Brain | Lung | Spleen |
|---|---|---|---|---|
| Group-3 | Infection + at end of treatment | 3.60 ± 0.37 | 5.86 ± 0.16 | 4.84 ± 0.46 |
| Group-4 | Infection + ATD (oral) (INH-625 μg, RIF-250 μg) | 1.68 ± 0.67 | 3.65 ± 0.77 | 2.34 ± 0.46 |
| Group-5 | Infection + ATD-NA (INH-625 μg/RIF-250 μg) insufflation | 0.74 ± 0.07 | 2.89 ± 1.98 | 2.37 ± 0.43 |
| Group-6 | Infection + ATD-NA (INH-625 μg, RIF-250 μg) instillation | 1.20 ± 0.80 | 4.06 ± 0.23 | 2.46 ± 0.23 |
Lung. Mice lung morphological analysis revealed the presence of greyish nodule-like lesions suggestive of active mycobacterial infection. Treatment with intranasal ATD resulted in a significant decrease in the number and density of lesions. Reductions in bacterial burden were also noted in the lungs of these mice, with a statistically significant increase in mycobacterial clearance compared to oral administration (P < 0.05). The mean bacterial burden in the ATD-treated mice lungs decreased to 3.65 ± 0.77
log10 CFU in the lungs. The mean CFU count reduced significantly to 2.89 ± 1.98log10 CFU in the mice group treated with ATD-NA delivered intranasally by insufflation (p < 0.001) (Fig. 5B).
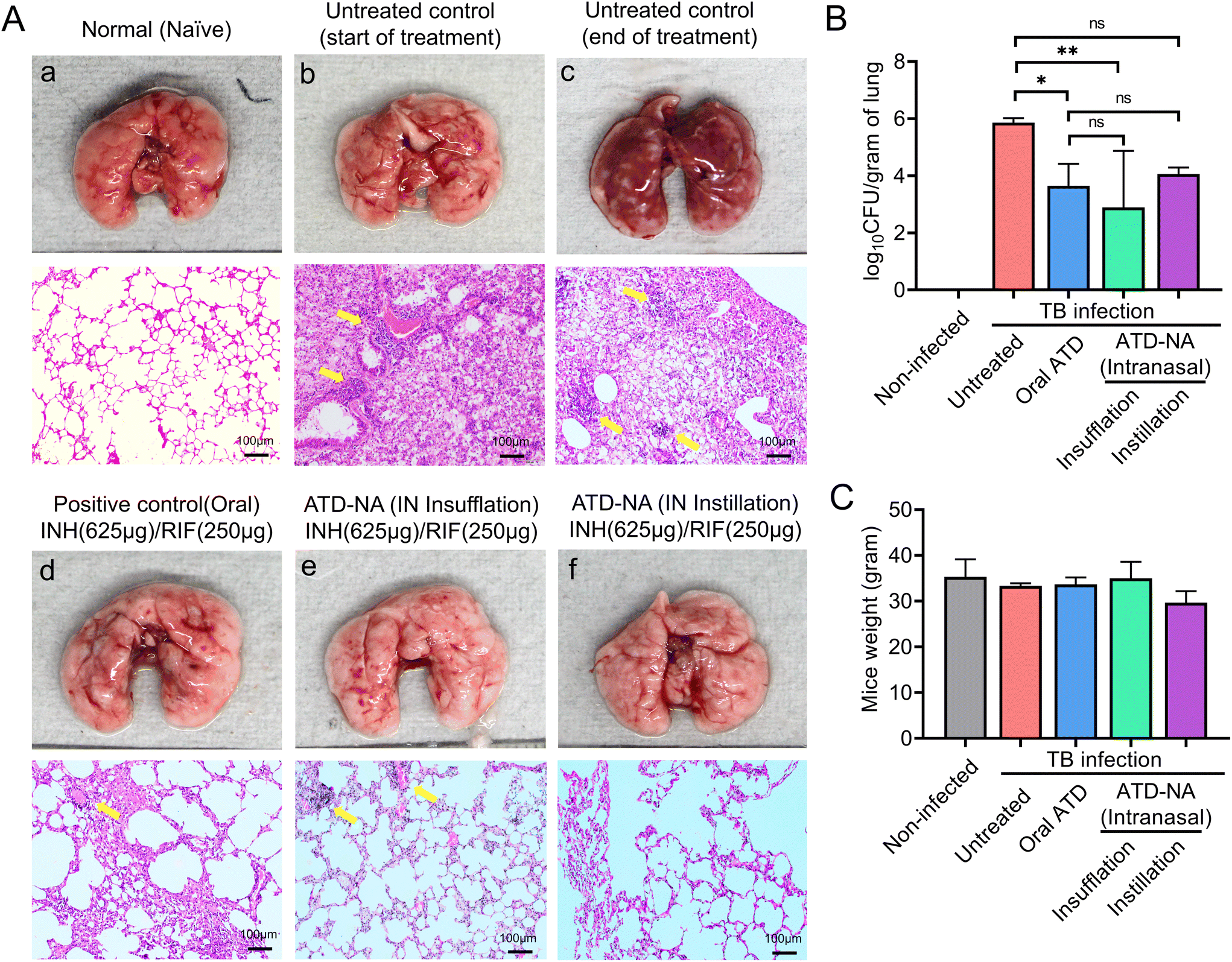 | ||
| Fig. 5 Lung morphology and mycobacterial burden in mice subjected to Mtb and diverse therapeutic interventions. (A) Depictions of the entire murine lungs were employed to elucidate pulmonary recuperation, as evidenced by distinct stages (a, b, c, d, e and f) characterized by greyish nodular lesions, denoted by yellow arrowhead markings. (B) The mean CFU in the lungs of untreated and treated mice were assessed, providing insights into bacteria load within the lung tissues. (C) The weight dynamics of mice during the therapeutic phase are depicted as an average plot. Values are expressed from five mice per animal group (n = 5). | ||
Spleen. A notable reduction in bacterial load was observed within the spleen of these mice. The analysis revealed a significant reduction in mycobacterial survival in the spleen of treated mice in all the groups compared to infected and untreated mice groups (p < 0.05). However, differences among the treated mice group were not statistically significant (P > 0.05) (Fig. 6B).
 | ||
| Fig. 6 (A) Morphological and histopathological assessments of murine splenic tissue of mice subsequent to Mtb infection and therapeutic interventions. Upper panel: representative images illustrating gross anatomic alterations of dissected spleen of BALB/c mice infected with Mtb (C3 strain) and following treatment with ATD and their respective formulations (INH-NA and RIF-NA) (a, b, c, d, e and f), lower panel: histopathological cross-sections of excised splenic tissue. (B) The mean CFU in the spleen tissue of untreated control and treated mice. (C) Graphical representations delineate the weight of wet spleen presented as an average plot. Values are expressed from five mice per animal group (n = 5). | ||
The lung pathology further deteriorated at 10 weeks post-infection in untreated animals, with notable lymphocytic infiltration (Fig. 5A). The treatment of ATD-NA with insufflation and instillation via the intranasal route shows a reduction in the severity of the infection and improved histopathology of the brain in murine mice. The ATD-NA treatment resulted in diminished swelling of neurons and lymphocytic infiltration, coupled with a decrease in the oedema of brain parenchyma.
In case of lung tissues, the decrease in severity is evidenced by the decrease in consolidation and fibrinous degeneration and the presence of only a small number of foam-like cells and small granulomatous nodular lesions. The mice group treated via the oral route did not exhibit as much restoration of lung tissue architecture, as evidenced by the presence of emphysematous-like changes with fibrinous degeneration and infiltration of mononuclear cells in their lungs.
The histopathological analysis of spleen tissues revealed distinct signs of TB, including granuloma and inflammation. The untreated control group exhibited the highest relative lesion burden, indicating significant damage caused by the infection. In contrast, treatment with ATD-NA (insufflation or instillation) effectively reduced the relative lesion burden in mice infected with Mtb, though findings were inferior to orally treated group (Fig. 6A). These findings were consistent with the CFU values of each group, indicating that treatment with ATD-NA may effectively reduce the relative lesion burden in TB-infected mice.
4. Discussion
This study investigates the effectiveness of CSNP encapsulated with anti-TB drugs, which are then converted to NA for N2B delivery. The objective is to enhance the treatment of CNS-TB by improving drug delivery directly to the brain. The study was performed on mice with experimentally induced CNS-TB and compared the efficacy of intranasal administration to the traditional oral route. Findings indicated that intranasal delivery leads to faster bacterial reduction compared to oral administration, likely due to increased drug exposure in the brain.40,41 The CSNP were optimized to be below 200 nm in size, enhancing their ability to reach the brain via the nasal route. These nanoparticles exhibited a positive zeta potential (∼+15 mV), which prevents aggregation and ensures stability. Chitosan's positive charge (∼+15 mV) facilitated strong binding with negatively charged cell membranes, prolonging nasal mucosa residence time and improving adhesion and transport properties.42 The olfactory epithelium in the upper nasal cavity was identified as the primary target for effective N2B delivery.43,44 Research indicates that particles with a size of approximately >5 μm may exhibit preferential deposition within the olfactory region when administered via intranasal route, under typical inhalation rates.45 The study also emphasized the non-toxicity and high biocompatibility of chitosan and mannitol, showing no toxicity towards RPMI-2650 cells.We compared the transport rates of INH and RIF formulations. Enhanced nasal permeability was observed, with INH demonstrating higher transport rates than RIF due to its hydrophilic nature. It is attributed to INH's hydrophilic nature and chitosan's inhibition of CYP450 enzymes responsible for INH metabolism in nasal mucosa. Notably, the study found no significant changes in the TEER values of RPMI-2650 cells, indicating that chitosan did not compromise the integrity of the nasal epithelium during the experiment. These insights suggest that chitosan-enhanced permeability of anti-TB drugs across nasal mucosa may facilitate their concentration in the brain, potentially aiding in the treatment of mycobacterial infections and reducing brain inflammation. Chitosan and mannitol, used in the formulation, are of natural origin, ensuring non-toxicity and high biocompatibility. These findings suggest the safety and efficacy of CSNPs for future animal studies.
In vivo studies in mice demonstrated that intranasal CSNPs significantly improved bacterial clearance in the brain and reduced bacteria in the lungs and spleen compared to oral administration. This effect is likely due to increased drug exposure in the brain. Daily intranasal doses for four weeks showed excellent tolerability without adverse effects. The mice model, mimicking human CNS-TB, confirmed the potential of N2B delivery to enhance treatment outcomes. Further investigation is warranted to corroborate these findings and ensure safety and efficacy before clinical application. This approach holds promise for better treatment outcomes in CNS-TB patients, potentially improving drug bioavailability and efficacy compared to oral administration. The integrative approach of using CSNP encapsulated with anti-TB drugs for nose-to-brain delivery shows great promise in enhancing the treatment of CNS-TB. This method could revolutionize TB therapeutics, especially for CNS infections, by offering a more effective and targeted treatment option.
5. Conclusion
This study underscores the potential of N2B delivery of anti-TB drugs encapsulated in CSNPs for treating CNS-TB. The findings suggest that intranasal administration can significantly enhance drug delivery to the brain, leading to improved treatment outcomes. The enhanced bacterial clearance in the brain, along with the safety and tolerability of the formulation, highlights the promise of this approach for future therapeutic strategies in managing CNS-TB.Abbreviations
| CNS-TB | Central nervous system tuberculosis |
| Mtb | Mycobacterium tuberculosis |
| MIC | Minimum inhibitory concentration |
| CSNPs | chitosan nanoparticles |
| ATD | Anti-tubercular drugs |
| NA | Nano-aggregates |
| PDI | Polydispersity index |
| FE-SEM | Field-emission scanning electron microscopy |
| FTIR | Fourier transform infrared spectroscopy |
| ANOVA | Analysis of variance |
| PBS | phosphate buffered saline |
| MTT | 3(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| DMSO | dimethyl sulfoxide |
Author contributions
Krishna Jadhav: formulation development, permeability studies, animal studies, data curation, original draft preparation: Agrim Jhilta: in vitro efficacy studies; Raghuraj Singh: solid state characterization studies; Eupa Ray: aerodynamic characterization; Vimal Kumar: animal infection studies; Awadh Bihari Yadav: histopathological studies; Amit Kumar Singh: conceptualization, funding acquisition, methodology, animal infection study, supervision, writing-review & editing; Rahul Kumar Verma: conceptualization, funding acquisition, methodology, supervision, writing-review & editing.Data availability
The data used to support the finding of this study are included within the article and ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests.Acknowledgements
The work was supported by the extramural grant of Indian Council of Medical Research, New Delhi (F. No. 5/8/5/68/Adhoc/2020/ECD-I) and Science and Engineering Research Board, New Delhi (CRG/2019/001483). We are grateful for the support and helpful assistance from Technical staff of ABSL-3 lab, ICMR-NJIL & OMD, Agra specially Nilesh Pal, Amit Kumar and Hemant Singh Thakur for their helpful assistance. KJ acknowledge Institute of Nano Science and Technology (INST), India, for funding his fellowship. Some segments of the figures were drawn by utilizing images sourced from Servier Medical Art. Servier Medical Art, provided by Servier, is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).References
- J. M. Leonard, Microbiol. Spectrum, 2017, 5(2) DOI:10.1128/microbiolspec.TNMI7-0044-2017.
- S. M. Katrak, J. Neurol. Sci., 2021, 421, 117278 CrossRef PubMed.
- R. J. Wilkinson, U. Rohlwink, U. K. Misra, R. Van Crevel, N. T. H. Mai, K. E. Dooley, M. Caws, A. Figaji, R. Savic and R. Solomons, Nat. Rev. Neurol., 2017, 13, 581–598 CrossRef PubMed.
- G. A. Ellard, M. J. Humphries and B. W. Allen, Am. Rev. Respir. Dis., 1993, 148, 650–655 CrossRef PubMed.
- R. Ruslami, A. R. Ganiem, S. Dian, L. Apriani, T. H. Achmad, A. J. van der Ven, G. Borm, R. E. Aarnoutse and R. van Crevel, Lancet Infect. Dis., 2013, 13, 27–35 CrossRef PubMed.
- C. Velasco-Aguirre, F. Morales, E. Gallardo-Toledo, S. Guerrero, E. Giralt, E. Araya and M. J. Kogan, Int. J. Nanomed., 2015, 10, 4919 Search PubMed.
- S. Sood, K. Jain and K. Gowthamarajan, J. Drug Targeting, 2014, 22, 279–294 CrossRef PubMed.
- R. M. Singh, A. Kumar and K. Pathak, Expert Opin. Drug Delivery, 2013, 10, 115–130 CrossRef PubMed.
- F. Sonvico, A. Clementino, F. Buttini, G. Colombo, S. Pescina, S. Stanisçuaski Guterres, A. Raffin Pohlmann and S. Nicoli, Pharmaceutics, 2018, 10, 34 CrossRef PubMed.
- P. Kaur, T. Garg, G. Rath and A. K. Goyal, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 1167–1176 Search PubMed.
- J. Rohrer, N. Lupo and A. Bernkop-Schnürch, Int. J. Pharm., 2018, 553, 8–20 CrossRef PubMed.
- P. G. Djupesland, J. C. Messina and R. A. Mahmoud, Ther. Delivery, 2014, 5, 709–733 CrossRef.
- M. M. Miyake and B. S. Bleier, Am. J. Rhinol. Allergy, 2015, 29, 124–127 CrossRef.
- A. Kumar, A. N. Pandey and S. K. Jain, Drug Delivery, 2016, 23, 671–683 CrossRef.
- E. Kole, K. Jadhav, N. Sirsath, P. Dudhe, R. K. Verma, A. Chatterjee and J. Naik, J. Drug Delivery Sci. Technol., 2023, 81, 104261 CrossRef.
- E. Kole, K. Jadhav, R. Singh, S. Mandpe, A. Abhang, R. K. Verma and J. Naik, Curr. Drug Delivery, 2024, 21, 1–12 CrossRef.
- M. Török, Br. Med. Bull., 2015, 113, 117–131 CrossRef.
- N. M. Elbaz, I. A. Khalil, A. A. Abd-Rabou and I. M. El-Sherbiny, Int. J. Biol. Macromol., 2016, 92, 254–269 CrossRef CAS PubMed.
- D. K. Jakhar, V. K. Vishwakarma, R. Singh, K. Jadhav, S. Shah, T. Arora, R. K. Verma and H. N. Yadav, Int. J. Pharm., 2023, 122585 CrossRef CAS.
- P. Calvo, C. Remunan-Lopez, J. L. Vila-Jato and M. Alonso, J. Appl. Polym. Sci., 1997, 63, 125–132 CrossRef CAS.
- A. Sharma, K. Vaghasiya, E. Ray, P. Gupta, U. D. Gupta, A. K. Singh and R. K. Verma, ACS Biomater. Sci. Eng., 2020, 6, 4126–4140 CrossRef CAS.
- S. Al-Qadi, A. Grenha, D. Carrión-Recio, B. Seijo and C. Remuñán-López, J. Controlled Release, 2012, 157, 383–390 CrossRef CAS.
- K. Jadhav, A. Jhilta, R. Singh, E. Ray, N. Sharma, R. Shukla, A. K. Singh and R. K. Verma, Biomater. Adv., 2023, 154, 213594 CrossRef CAS.
- Y. Chen, Y. Gui, Y. Luo, Y. Liu, L. Tu, Y. Ma, P. Yue and M. Yang, Powder Technol., 2021, 377, 128–138 CrossRef CAS.
- R. Singh, K. Jadhav, R. Kamboj, H. Malhotra, E. Ray, A. Jhilta, V. Dhir and R. K. Verma, Biomater. Adv., 2024, 160, 213853 CrossRef CAS.
- K. Jadhav, E. Kole, R. Singh, S. K. Rout, R. K. Verma, A. Chatterjee, A. Mujumdar and J. Naik, Drying Technol., 2024, 42, 1415–1441 CrossRef.
- L. Godfrey, A. Iannitelli, N. L. Garrett, J. Moger, I. Imbert, T. King, F. Porreca, R. Soundararajan, A. Lalatsa and A. G. Schätzlein, J. Controlled Release, 2018, 270, 135–144 CrossRef CAS PubMed.
- Q. Meng, A. Wang, H. Hua, Y. Jiang, Y. Wang, H. Mu, Z. Wu and K. Sun, Int. J. Nanomed., 2018, 13, 705 CrossRef.
- S. Md, R. A. Khan, G. Mustafa, K. Chuttani, S. Baboota, J. K. Sahni and J. Ali, Eur. J. Pharm. Sci., 2013, 48, 393–405 CrossRef.
- J. W. Suh, J.-S. Lee, S. Ko and H. G. Lee, J. Agric. Food Chem., 2016, 64, 5384–5388 CrossRef PubMed.
- I. Y. Wu, T. E. Nikolaisen, N. Škalko-Basnet and M. P. Di Cagno, J. Pharm. Sci., 2019, 108, 2570–2579 CrossRef PubMed.
- T. Jullaphant, T. Nakpeng and T. Srichana, Pharm. Dev. Technol., 2019, 24, 494–503 CrossRef PubMed.
- K. Vaghasiya, E. Ray, R. Singh, K. Jadhav, A. Sharma, R. Khan, O. P. Katare and R. K. Verma, Mater. Sci. Eng., C, 2021, 123, 112027 CrossRef PubMed.
- K. Vaghasiya, E. Ray, A. Sharma, R. Singh, K. Jadhav, R. Khan, O. P. Katare and R. K. Verma, J. Mater. Sci., 2021, 56, 14700–14716 CrossRef.
- A. A. Husain, U. D. Gupta, P. Gupta, A. R. Nayak, N. H. Chandak, H. F. Daginawla, L. Singh and R. S. Kashyap, Indian J. Med. Res., 2017, 145, 833 CrossRef PubMed.
- C. Mercier, S. Hodin, Z. He, N. Perek and X. Delavenne, Mol. Pharmaceutics, 2018, 15, 2246–2256 CrossRef CAS PubMed.
- S. G. Thakkar, Z. N. Warnken, R. F. Alzhrani, S. A. Valdes, A. M. Aldayel, H. Xu, R. O. Williams III and Z. Cui, J. Controlled Release, 2018, 292, 111–118 CrossRef CAS.
- A. Sharma, K. Vaghasiya, P. Gupta, A. K. Singh, U. D. Gupta and R. K. Verma, J. Controlled Release, 2020, 324, 17–33 CrossRef CAS PubMed.
- N. I. N. Jamil, W. N. A. W. A. Wahab, I. A. Ali and M. L. Yahaya, Malays. J. Med. Sci., 2018, 25, 59 Search PubMed.
- L. T. Fasiolo, M. D. Manniello, E. Tratta, F. Buttini, A. Rossi, F. Sonvico, F. Bortolotti, P. Russo and G. Colombo, Eur. J. Pharm. Sci., 2018, 113, 2–17 CrossRef PubMed.
- P. R. Donald, Tuberculosis, 2010, 90, 279–292 CrossRef CAS PubMed.
- D. Lee and T. Minko, Pharmaceutics, 2021, 13, 2049 CrossRef CAS.
- M. L. Bruschi, S. B. de Souza Ferreira and J. B. da Silva, in Nanotechnology for oral drug delivery, ed. J. P. Martins and H. A. Santos, Elsevier, 2020, pp. 77–141 Search PubMed.
- L. Kozlovskaya, M. Abou-Kaoud and D. Stepensky, J. Controlled Release, 2014, 189, 133–140 CrossRef CAS.
- G. Drin, S. Cottin, E. Blanc, A. R. Rees and J. Temsamani, J. Biol. Chem., 2003, 278, 31192–31201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02621g |
| This journal is © The Royal Society of Chemistry 2024 |