
A nanoengineered vanadium oxide composite as a high-performance anode for aqueous Li-ion hybrid batteries†
Ailun
Huang‡
 a,
Zhiyin
Yang‡
a,
Xueying
Chang
a,
Cheng-Wei
Lin
a and
Richard B.
Kaner
*ab
a,
Zhiyin
Yang‡
a,
Xueying
Chang
a,
Cheng-Wei
Lin
a and
Richard B.
Kaner
*ab
aDepartment of Chemistry and Biochemistry, University of California, Los Angeles (UCLA), Los Angeles, CA 90095, USA
bDepartment of Materials Science and Engineering, California NanoSystems Institute (CNSI), University of California, Los Angeles (UCLA), Los Angeles, CA 90095, USA. E-mail: kaner@chem.ucla.edu
First published on 3rd April 2024
Abstract
Aqueous lithium-ion batteries (LIBs) have received increasing attention as a promising solution for stationary energy storage systems due to their low environmental impact, non-flammability and low cost. Despite recent progress in electrolyte development and cathode manufacturing, the lack of anode materials with high specific capacity presents difficult challenges for a wide range of applications. In this study, we propose a novel synthetic strategy to fabricate a pseudocapacitive V2O5/graphene composite as a highly functional anode material for aqueous LIBs. The designed synthesis combines a fast laser-scribing step with controlled calcination to tune the morphology and oxidation state of the electrochemically active vanadium oxide species while obtaining a highly conductive graphene scaffold. The optimized V2O5/graphene anode shows an outstanding specific capacity of 158 mA h g−1 in three-electrode measurements. When the V2O5/graphene anode is paired with an LiMn2O4 cathode, the charge storage mechanism of the full cell is revealed to be dominantly surface-controlled, resulting in remarkable rate performance. Specifically, the full cell can reach a specific capacity of 151 and 107 mA h (g anode)−1 at C/6 and 3C, respectively. Moreover, this hybrid battery can achieve a high power density and an energy density of 650 W kg−1 at 15.6 W h kg−1 and 81.5 W h kg−1 at 13.6 W kg−1, respectively, outperforming most aqueous LIBs reported in the literature. This innovative strategy provides a pathway to incorporate pseudocapacitive electrodes for improving aqueous lithium-ion storage systems, enabling safe operation of large-scale energy storage without compromising their electrochemical performance.
New conceptsThis work reports an original synthetic approach combining laser-scribing and calcination to make a pseudocapacitive V2O5/laser-scribed graphene (LSG) composite made from VCl3/graphene oxide. The laser step generates the 3D graphene scaffold with nanosized vanadium oxide particles. The calcination step converts all remaining vanadium species to V2O5 nanoparticles in a controlled manner. In this way, the resulting V2O5 nanoparticles in the V2O5-LSG electrode are electrochemically active, small in grain size and easily accessible for charge transport via the LSG structure. The V2O5-LSG composite can be used as an anode in aqueous lithium-ion batteries. Based on previous literature reports, the choice of a suitable transition metal redox couple for an aqueous LIB anode is limited, restricted by low specific capacity. This new synthetic V2O5-LSG anode shows high capacity compared with other aqueous lithium-ion battery anodes. When paired with a LiMn2O4 cathode, the full cell displays a predominantly surface-controlled charge storage mechanism that is Faradaic in nature, leading to excellent fast charging properties, and high power and energy density that compare favorably to previously reported aqueous LIBs. Because of its high capacity, fast charging capability and safe operation, the V2O5-LSG||LiMn2O4 system provides a potential solution for future energy storage applications. |
Introduction
Aqueous rechargeable batteries have emerged as a promising technology for applications in large-scale energy storage systems due to their safe operation, non-toxicity, and cost-efficiency.1 Among the storage systems based on various cations such as Na+, K+, Zn2+, Mg2+, Al3+, etc., aqueous lithium-ion batteries (LIBs) are the most prominent candidates because of the well-developed manufacturing capability for LIB component production. The progress of aqueous LIB research has been significantly advanced by the development of numerous aqueous electrolytes, extending the electrochemical window beyond the conventional 1.23 V limit imposed by the electrolytic decomposition of water.2–5 A “water-in-salt” electrolyte was therefore obtained by dissolving lithium bis(trifluoromethane sulfonyl)imide (LiTFSI) at extremely high concentrations (molality >20 M) in water, which can widen the electrochemical window to >3 V. LiTFSI was chosen as the salt because of its high solubility in water (>20 M at 25 °C) and high stability against hydrolysis. In such a super-concentrated electrolyte, the decomposition of salt anion occurs preferentially on the anode before hydrogen evolution occurs, leading to the formation of a dense solid electrolyte interphase (SEI) primarily consisting of LiF to solve the hydrogen and oxygen evolution problem.5 While the Li/Li+ intercalation potential of commercially available LIB cathodes including LiCoO2, LiFePO4, and LiMn2O4 conveniently falls below the upper potential limit of the operational window of aqueous electrolytes, carbon-based anodes that are widely used in organic electrolytes are not compatible with the aqueous system due to their low Li/Li+ intercalation potential.6–11 Thus, unconventional transition metal-based anodes must be employed in aqueous LIBs.Restricted by the relatively high lower potential limit of aqueous lithium-ion electrolytes, the choice of suitable transition metal redox couples for an aqueous LIB anode is limited. Most reported aqueous LIBs use an intercalation-type Mo6S8 or TiO2 anode. Although high average full-cell voltage can be achieved, the energy density is restricted by their low specific capacity, since a high capacity ratio between the negative electrode (anode) and positive electrode (cathode), known as the n:p ratio, is required to compensate for the initial loss of anode capacity by reactions with the electrolyte.5 Therefore, there is an urgent need for anode materials that exhibit high specific capacity. Vanadium oxide anodes have been reported to show enhanced capacity in aqueous LIBs; however, the added capacity is not sufficient to account for the lower average voltage owing to their unfavorable redox potential, leading to disadvantageous energy density.12–15 Moreover, aqueous LIBs have great potential for fast charging applications due to the enhanced diffusion in an aqueous electrolyte, which can be further improved by integrating electrodes with multi-dimensional diffusion pathways or with redox reactions confined to the surface. The spinel LiMn2O4 cathode allows three-dimensional Li migration, and V2O5, as an intrinsically intercalation-type material, can show extrinsic pseudocapacitive behavior when nanosized.16–19 Therefore, the combination of these three components is expected to deliver a hybrid battery with excellent fast charging capability.
In this work, we report a novel V2O5/laser-scribed graphene (LSG) composite material in which V2O5 nanoparticles are uniformly anchored on a conductive graphene nanosheet scaffold. The V2O5–LSG composite is made from VCl3/graphite oxide via an originally designed two-step laser scribing/calcination approach where the former results in an expanded LSG network as well as nanosized vanadium particles and the latter further converts all vanadium species to V2O5 nanoparticles in a controlled manner. In this way, the resulting V2O5 nanoparticles in the V2O5–LSG electrode are electrochemically active, small in grain size and easily accessible for charge transport via the LSG structure. This V2O5–LSG electrode shows pseudocapacitive features and remarkable rate capability in three-electrode measurements. When the V2O5–LSG anode is paired with a LiMn2O4 cathode, the hybrid battery exhibits predominantly surface-controlled charge storage behavior, leading to excellent fast charging capability. The full-cell specific capacity can reach 151 mA h (g anode)−1 giving high power and energy density, 650 W kg−1 at 15.6 W h kg−1 and 81.5 W h kg−1 at 13.6 W kg−1, which outperform those of the majority of previously reported aqueous LIBs. Moreover, the effects of the calcination time were studied by transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS), revealing how the chemical position and structural morphology of the electrochemically active vanadium species can be effectively optimized.
Results and discussion
The synthesis of V2O5-laser scribed graphene (LSG) was performed to adapt a pseudocapacitive material for battery chemistry. As shown in Fig. 1, the VCl3 solution was added to a graphite oxide (GO) dispersion to ensure uniform mixing of vanadium ions and GO sheets. The vanadium cations are believed to act as spacers to avoid aggregation of GO layers. As reported in our prior work, laser-scribing of a drop-cast VCl3/GO film can simultaneously lead to the formation of low-valent vanadium oxides (VOx, predominantly V2O3 and VO2) and an expanded reduced graphene oxide (rGO) network due to the rapid release of gases, denoted here as VOx–LSG.20,21 Although laser-scribing alone can fabricate thin-film electrodes for supercapacitor systems that only rely on surface charge storage processes, the electrodes usually suffer from low areal mass loading22,23 and incompatibility with battery-type electrochemistry, which require more uniformly formed electrodes to carry out ion transport deeper beneath the surface. Therefore, after the transient heat exposure induced by a CO2 laser, the VOx–LSG was exposed to a much longer thermal treatment in air, resulting in the oxidation of lower-valent VOx with an intrinsic tunnel-like structure and one-dimensional charge transfer pathways to a layered V2O5 structure where two-dimensional diffusion is enabled.24 By choosing a sufficiently low temperature, thermal decomposition of graphene to CO is prevented and the integrity of the LSG structure is preserved. In the end, the transformation from any unconverted VCl3/GO and VOx–LSG intermediate into a V2O5–LSG composite was completed.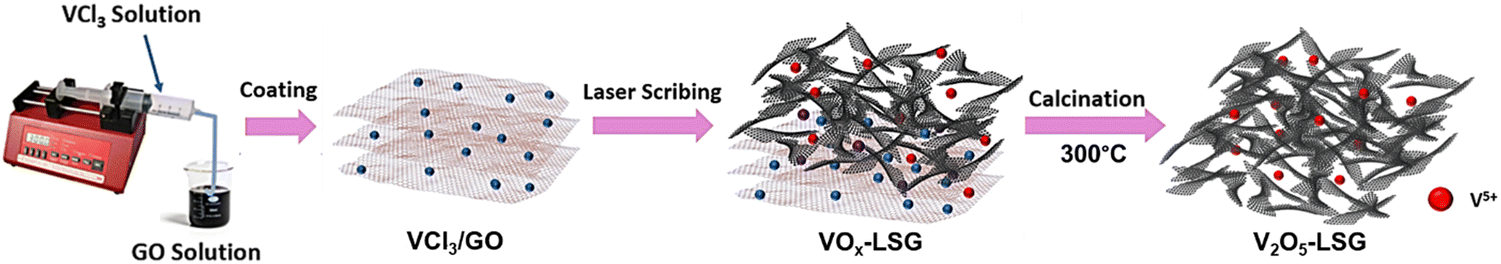 | ||
| Fig. 1 Schematic illustration of V2O5–LSG synthesis via laser scribing and calcination. | ||
The structural features of the as-synthesized V2O5–LSG composite were investigated by electron microscopy (SEM) and transmission electron microscopy (TEM). In Fig. 2a, randomly stacked rGO flakes with visible wrinkles are observed in the SEM image, suggesting the successful formation of the conductive LSG network. Under high magnification, Fig. 2b shows that the LSG sheets are evenly and fully coated with V2O5 nanoparticles. From the TEM images (Fig. 2c), it can be observed that nanoparticles are uniformly distributed on the graphene sheets and are ∼40–50 nm in size. Fig. S15 (ESI†) shows the size distribution of VOx particles based on Fig. 2c and the vast majority of the sizes are ∼40–50 nm. The SEM images of the precursor VCl3/GO and the intermediate VOx–LSG are displayed in Fig. S2 (ESI†). Fig. S2a and b (ESI†) show flat GO sheets closely stacked together mixed with an aggregated network of VCl3 while the VOx–LSG in Fig. S2c and d (ESI†) has expanded spacing between the rGO sheets and no sign of agglomerates of VOx, demonstrating that the laser step simultaneously achieves reduction of GO and nanosizing of the vanadium species. The morphological evidence indicates that the interconnected and porous LSG structures are preserved and the V2O5 particle size remained within the same order of magnitude after calcination.
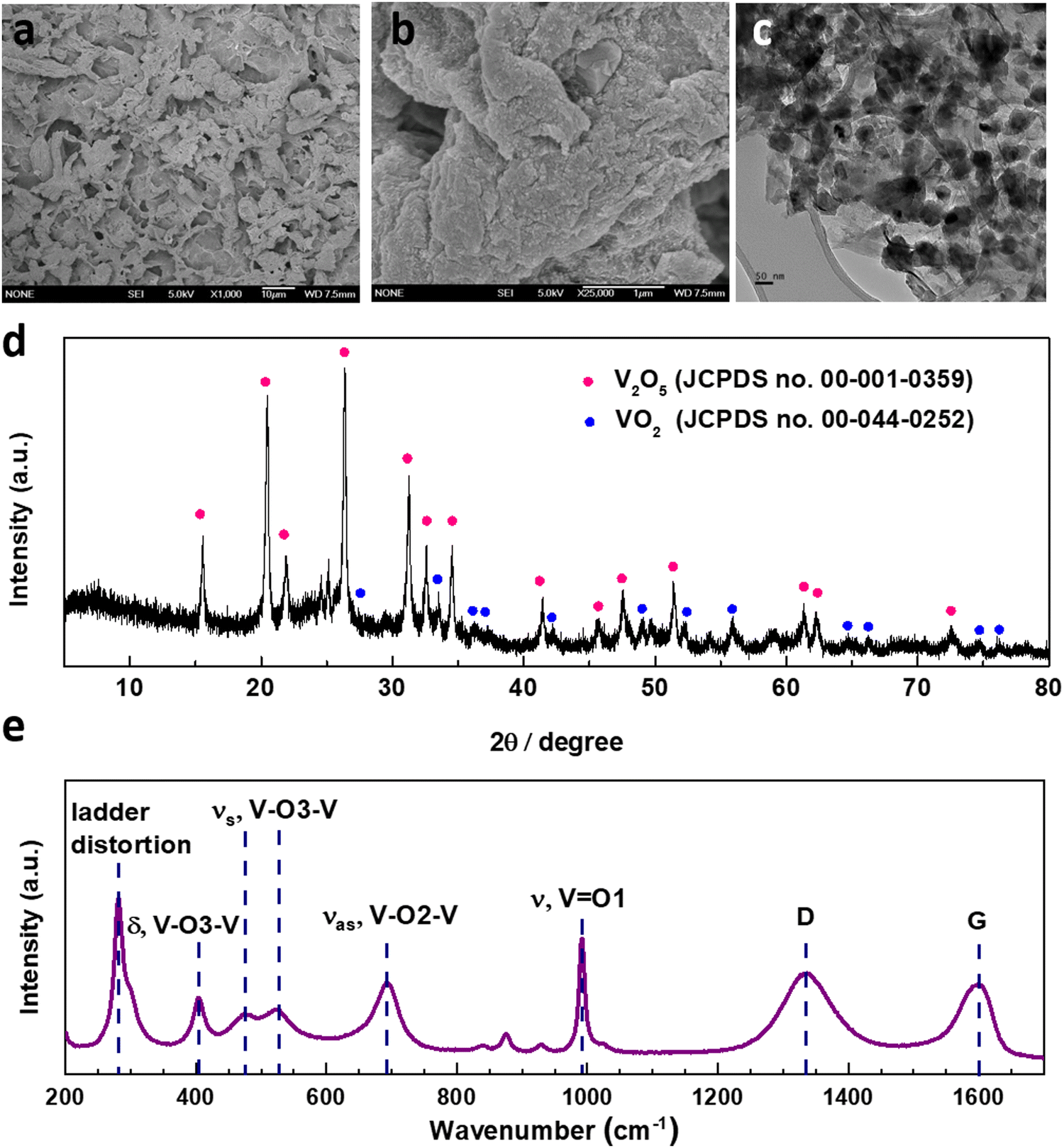 | ||
| Fig. 2 Characterization of the as-synthesized V2O5–LSG composite (calcination time = 1 h). (a) Low- and (b) High-magnification SEM images of the V2O5–LSG composite. (c) A TEM image showing the V2O5 particles on the rGO sheets. (d) XRD pattern of V2O5–LSG matching V2O5 (JCPDS no. 00-001-0359) and VO2 (JCPDS no. 00-044-0252). (e) Raman spectra of the composite showing V2O5 and graphene features. | ||
The compositions of the synthesized V2O5–LSG composites were studied by X-ray powder diffraction (XRD) and Raman spectroscopy. Fig. 2d presents the diffraction pattern of the V2O5–LSG composite and it matches that of orthorhombic V2O5, confirming the formation of V2O5 from the lower-valent VCl3 precursor and the VOx intermediate. The most prominent peaks at 20.4°, 26.3° and 31.2° correspond to the (001), (110) and (310) planes, respectively. The rest of the peaks are VO2 and match with JCPDS no. 00-044-0252. Fig. S3a and b (ESI†) show the XRD patterns of VCl3/GO and VOx–LSG, respectively. The former resembles the patterns of graphite and graphite oxide with no signs of any crystalline vanadium oxide species. The latter shows peaks attributed to V2O3, VO2 and mixed-valent vanadium oxides, indicating that the laser-scribing step involves the oxidation from VCl3 to the multivalent VOx and the following calcination completes further oxidation to V2O5. The successful synthesis of V2O5–LSG was confirmed using Raman spectra, which show all the V2O5 vibrational features as well as the D and G bands of graphene (Fig. 2e). O1, O2 and O3 denote the out-of-plane terminal, in-plane terminal and in-plane bridging oxygen atoms in V2O5, and all the peaks are labeled with the corresponding vibrational modes.25 Furthermore, the complete oxidation of VCl3 to V2O5 is confirmed by the absence of both Cl 2s and Cl 2p peaks in the XPS survey spectrum of V2O5–LSG (Fig. S1, ESI†). Furthermore, since the calcination temperature was chosen to be below 400 °C, above which LSG is vulnerable to thermal oxidation to CO, ∼10% of the final V2O5 composite is LSG by weight, as signified by the sharp decrease around 400 °C in the thermogravimetric analysis (TGA), as shown in Fig. S4 (ESI†). Therefore, we choose a temperature lower than 400 °C to prevent the thermal decomposition of graphene to CO and preserve the integrity of the LSG structure. We made the assumption that at 300 °C the conversion to V2O5 can be completed without risking the loss of the rGO scaffold. Overall, all characterization results indicate that the post-laser-scribing calcination step ensures full reaction converting VCl3 to V2O5 without compromising the conductive LSG scaffold, as illustrated in Fig. 1.
The electrochemical features of the V2O5–LSG composite electrodes were first studied using a three-electrode setup in the potential window 0 to −1.4 V vs. Ag/AgCl with 21 M lithium bis(trifluoromethane)sulfonimide (LiTFSI) as the electrolyte. As shown in Fig. 3a, the CV curves possess broad peaks with small peak-to-peak separations, which is a feature of fast Faradaic charge storage processes and is typical for a redox-active pseudocapacitive material. The voltage profile in Fig. 3b shows two sloping regions with drastically different gradients. From −1.1 V to −0.5 V vs. Ag/AgCl, there is a sloping plateau and the capacity contribution gradually decreases with increasing current density, indicting the rate-dependent battery-type behavior. This is consistent with CV measurements since the most pronounced redox peaks are also observed in the same region. On the other hand, a sloping voltage is always observed between −0.5 V and 0 V vs. Ag/AgCl and the gradient remains constant with changing current density, signifying capacitive behavior. As described in Fig. 3c, the V2O5–LSG composite electrode demonstrated an excellent rate capability in 21 M LiTSFI. At 0.1, 0.2, 0.5 and 1 A g−1, the average discharge capacity can reach 158, 145, 131 and 119 mA h g−1, respectively, based on the total mass of the V2O5–LSG composite. The LSG contribution is limited to ∼11 mA h g−1 based on the three-electrode test shown in Fig. S14 (ESI†). The specific capacity per VOx is calculated as follows:
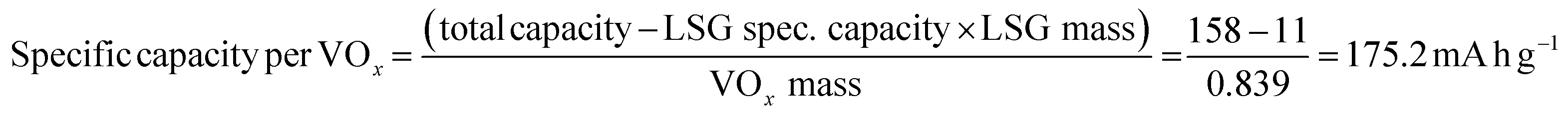 | (1) |
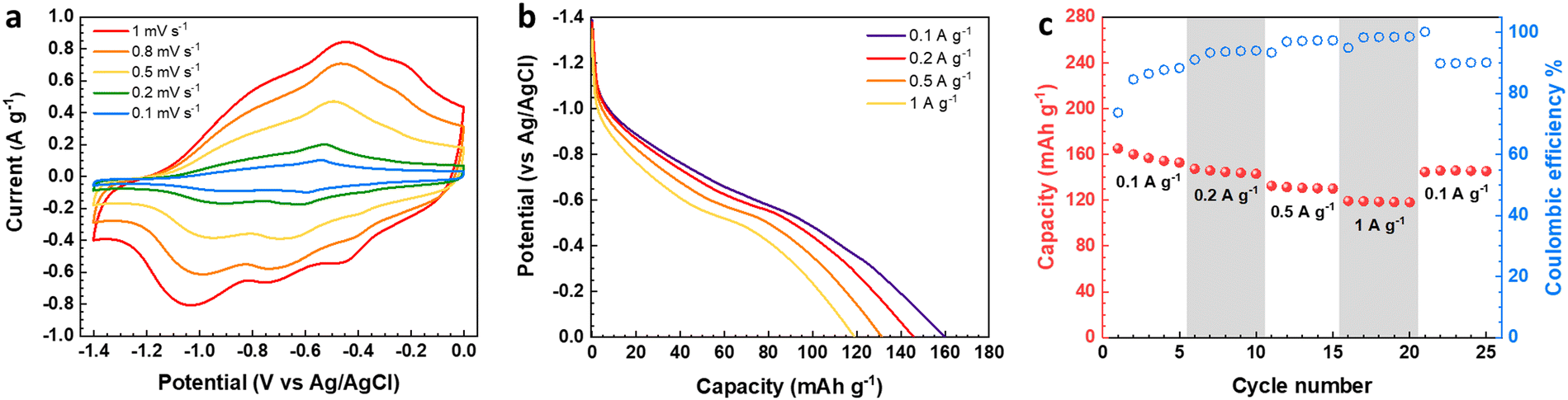 | ||
| Fig. 3 Electrochemical measurements of the V2O5–LSG electrode in a three-electrode setup. (a) Cyclic voltammetry curves for V2O5–LSG at 0.1, 0.2, 0.5, 0.8, and 1 mV s−1. (b) Capacity–voltage profiles at 0.1, 0.2, 0.5, and 1 A g−1. (c) Rate performance and corresponding Coulombic efficiency of V2O5–LSG cycled at different current densities. | ||
Impressively, 75% of the maximum capacity was retained upon a 10-fold rate increase from 0.1 to 1 A g−1. Additionally, ∼7% capacity loss was observed in the first cycle, which is speculated to be caused by irreversible formation of LiF from the electrode reaction with LiTSFI.5 When the rate is returned to 0.1 A g−1 in the end, ∼95% of the initial capacity was recovered after accounting for the loss from LiF formation. The outstanding rate performance of the V2O5–LSG electrode provides a promising foundation for its fast-charging applications in full cells.
To evaluate the electrochemical performance of the V2O5–LSG composite in a more practical setting, it was tested as the anode in full cells and paired with an intercalation-type cathode LiMn2O4, as illustrated in Fig. 4a. Fig. 4b shows the CV curves of the V2O5–LSG anode and the LiMn2O4 cathode in 21 M LiTFSI electrolyte. The 21 M LiTFSI electrolyte has a wider electrochemical window with its hydrogen and oxygen evolution potentials extended to −0.9 V and 2 V, respectively, compared with 5 M LiTFSI which only has an electrochemical window of 1.7 V (Fig. S16a, ESI†), indicating a sufficiently wider electrochemical window of ∼3 V that ensures stable operation of both the V2O5–LSG anode and the LMO cathode. The anode is electrochemically active between 1.8 V and 3.2 V vs. Li/Li+, which covers the potential range of common aqueous lithium-ion battery anodes such as Mo6S8 and TiO2.4,26 The LMO cathode is electrochemically active between 3.8 V and 4.6 V vs. Li/Li+, which enables a wider voltage window. Another cathode, LiFePO4, was tried to pair with the anode. However, LFP has a lower potential range compared to LMO (Fig. S17a, ESI†). Therefore, LMO with a higher potential was used enabling a wider voltage window and a higher capacity (Fig. S17b, ESI†).
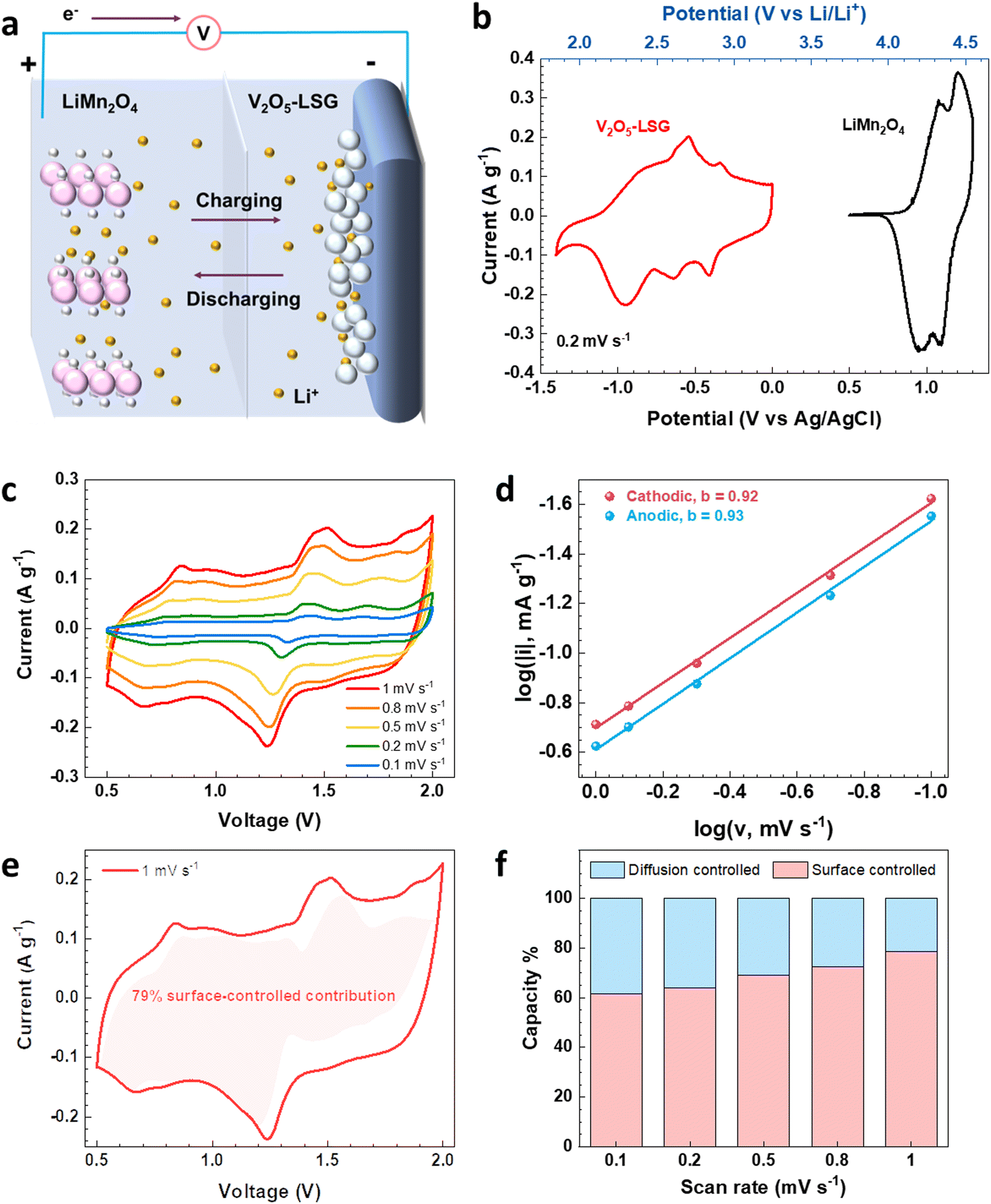 | ||
| Fig. 4 Electrochemical analysis of the V2O5–LSG||LiMn2O4 cells. (a) Illustration of Li-ion storage in the hybrid system based on a pseudocapacitive anode and an intercalation-type cathode. (b) Potential windows of V2O5–LSG and LiMn2O4 in a 21 M LiTFSI electrolyte. (c) Cyclic voltammetry curves of a V2O5–LSG||LiMn2O4 coin cell at 0.1, 0.2, 0.5, 0.8, and 1 mV s−1. (d) b-values of the most pronounced pair of peaks in the CV. (e) Current contribution by surface-controlled processes at 1 mV s−1. (f) Capacity contribution by surface-controlled and diffusion-controlled processes at a range of scan rates. | ||
The discharge capacity of the LiMn2O4 cathode was measured to be 112 mA h g−1 (Fig. S5, ESI†), which is 29% lower than that of the V2O5–LSG anode. Conventional LIB anodes like TiO24 or Mo6S8,5 which have similar specific capacities of 125 mA h g−1 and 118 mA h g−1, respectively, like LiMn2O4, often require doubling the mass of the anode to obtain a suitable n:p ratio in order to compensate for the anode capacity loss due to solid–electrolyte interphase formation;6 however, since the V2O5–LSG anode capacity is 41% higher than that of LiMn2O4, an anode![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cathode mass ratio of 1 was used in all full cell testing. In addition, the cell capacity is based on the total mass of the anode and cathode. We have carried out experiments with higher and lower cathode masses (Fig. S13, ESI†) and found that when the mass ratio is 1:1, the full cell has the highest capacity (mA h gtotal mass−1). When the mass ratio V2O5–LSG: LiMn2O4 is 1:2, the capacity decreases. The reason is that when increasing the cathode mass, while the capacity increases, the total mass also increases, but the ratio of capacity/total mass decreases because the total mass increases more. When the mass ratio V2O5–LSG:LiMn2O4 is 2:1, the capacity also decreases. The reason is the same. When increasing the anode mass, the capacity increases; however, the total mass also increases, but the ratio of capacity/total mass decreases because the total mass increases more. As shown in Fig. 4c, between 0.5 and 2 V, the CV curves of the V2O5–LSG||LiMn2O4 cell adopt a distorted rectangular shape with prominent redox peaks, which is typical for a hybrid energy storage system.
:cathode mass ratio of 1 was used in all full cell testing. In addition, the cell capacity is based on the total mass of the anode and cathode. We have carried out experiments with higher and lower cathode masses (Fig. S13, ESI†) and found that when the mass ratio is 1:1, the full cell has the highest capacity (mA h gtotal mass−1). When the mass ratio V2O5–LSG: LiMn2O4 is 1:2, the capacity decreases. The reason is that when increasing the cathode mass, while the capacity increases, the total mass also increases, but the ratio of capacity/total mass decreases because the total mass increases more. When the mass ratio V2O5–LSG:LiMn2O4 is 2:1, the capacity also decreases. The reason is the same. When increasing the anode mass, the capacity increases; however, the total mass also increases, but the ratio of capacity/total mass decreases because the total mass increases more. As shown in Fig. 4c, between 0.5 and 2 V, the CV curves of the V2O5–LSG||LiMn2O4 cell adopt a distorted rectangular shape with prominent redox peaks, which is typical for a hybrid energy storage system.
In order to investigate the kinetics of the charge storage processes in the V2O5–LSG||LiMn2O4 cell, the sharpest pair of peaks that appear between 1.2 and 1.6 V were analyzed. The peak current densities (i) and the scan rates (v) generally obey the following power law:27,28
| i = avb | (2) |
By plotting log(i) of each peak against log(v), the b-value of the peak can be determined, and the common consensus is that b-values of 0.5 and 1 indicate battery-type and capacitor-type behavior, respectively. Fig. 4d illustrates that the anodic and cathodic peaks, respectively, have b-values of 0.93 and 0.92 (with R2 = 0.998), suggesting that the charge storage mechanism of the V2O5–LSG||LiMn2O4 system consists of both battery-type and capacitor-type processes that is dominated by the latter. Furthermore, to gain more quantitative insights into the charge storage behavior and decouple the capacity contribution from the diffusion-controlled battery-type and the surface-controlled capacitor-type processes, a second kinetic analysis was carried out based on the following relationship:
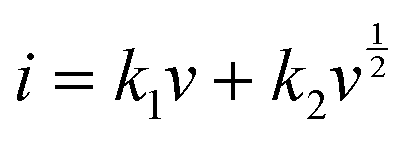 | (3) |
The surface-controlled component (k1v) and the diffusion-controlled component (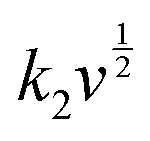 ) are proportional to the scan rate and the square root of the scan rate, respectively. Fig. 4e shows an example where the current contributions from the two types of processes were quantified based on the CV data of the system at 1 mV s−1. It is clear that the lower-voltage region (0.5–1.2 V) is more predominantly governed by surface-controlled processes, whereas the diffusion-controlled current starts to significantly grow going into the higher-voltage region (1.2–2 V). This is consistent with the observation in Fig. 3b, in which the V2O5–LSG anode also presents more battery-type behavior toward higher state-of-charge. This analysis was also carried out using CV data at 0.1, 0.2, 0.5 and 0.8 mV s−1 and the corresponding capacity quantification is summarized in Fig. 4f. The capacity contributed by the diffusion-controlled processes decreased from 79% at 1 mV s−1 to 61% at 0.1 mV s−1. This indicates that the charge storage mechanism of the V2O5–LSG||LiMn2O4 system is mainly governed by capacitive processes even at very slow scan rates although the contribution gap between the processes gradually lessens from 1 to 0.1 mV s−1. This capacity dominated by diffusion-controlled contributions of V2O5–LSG||LiMn2O4 gives an excellent foundation for its great electrochemical performance at high rates.
) are proportional to the scan rate and the square root of the scan rate, respectively. Fig. 4e shows an example where the current contributions from the two types of processes were quantified based on the CV data of the system at 1 mV s−1. It is clear that the lower-voltage region (0.5–1.2 V) is more predominantly governed by surface-controlled processes, whereas the diffusion-controlled current starts to significantly grow going into the higher-voltage region (1.2–2 V). This is consistent with the observation in Fig. 3b, in which the V2O5–LSG anode also presents more battery-type behavior toward higher state-of-charge. This analysis was also carried out using CV data at 0.1, 0.2, 0.5 and 0.8 mV s−1 and the corresponding capacity quantification is summarized in Fig. 4f. The capacity contributed by the diffusion-controlled processes decreased from 79% at 1 mV s−1 to 61% at 0.1 mV s−1. This indicates that the charge storage mechanism of the V2O5–LSG||LiMn2O4 system is mainly governed by capacitive processes even at very slow scan rates although the contribution gap between the processes gradually lessens from 1 to 0.1 mV s−1. This capacity dominated by diffusion-controlled contributions of V2O5–LSG||LiMn2O4 gives an excellent foundation for its great electrochemical performance at high rates.
The electrochemical performance of the V2O5–LSG||LiMn2O4 system was assessed in coin cell formats. First, a control experiment was carried out to eliminate any capacity contribution from the graphite paper substrate, and Fig. S6 (ESI†) shows that the substrate has a negligible capacity when paired with a LiMn2O4 cathode. The V2O5–LSG||LiMn2O4 cell was cycled at C/6, C/2, 1C, 3C and C/6 rates as shown in Fig. 5a. The average discharge capacity reached 151, 137, 123 and 107 mA h (g anode)−1, or 75, 69, 61 and 53 mA h g−1 based on the total electrode mass, at C/6, C/2, 1C, and 3C, respectively. With a 18-fold rate increase from C/6 to 3C, 71% capacity was retained, signifying outstanding fast charging capability. Again, the same initial decrease in capacity is observed, leading to 91% capacity retention when the rate is reversed to C/6, as seen in Fig. 3c. The voltage profiles of the V2O5–LSG||LiMn2O4 cell consist of a small plateau around 1.5 V and a voltage slope in the 0.5–1.4 V range, indicating that the capacity is predominantly from surface-controlled processes, as illustrated in Fig. 5b. The plateau is a feature of diffusion-controlled Faradaic processes, and its gradient increases with increasing rate, suggesting that the capacity contribution from battery-type behavior also falls with the increasing rate. This trend corroborates with the kinetic analysis results presented in Fig. 4f, and the plateau voltage matches the peak positions in the CV curves (Fig. 4c). Faster conditions have been tested with 6C (Fig. S18, ESI†). With a 6-fold rate increase from 1C to 6C, 65% of the capacity was retained, signifying outstanding fast charging capability. When the rate is returned to 1C, ∼95% of the initial capacity was recovered, confirming the fast charging capacity.
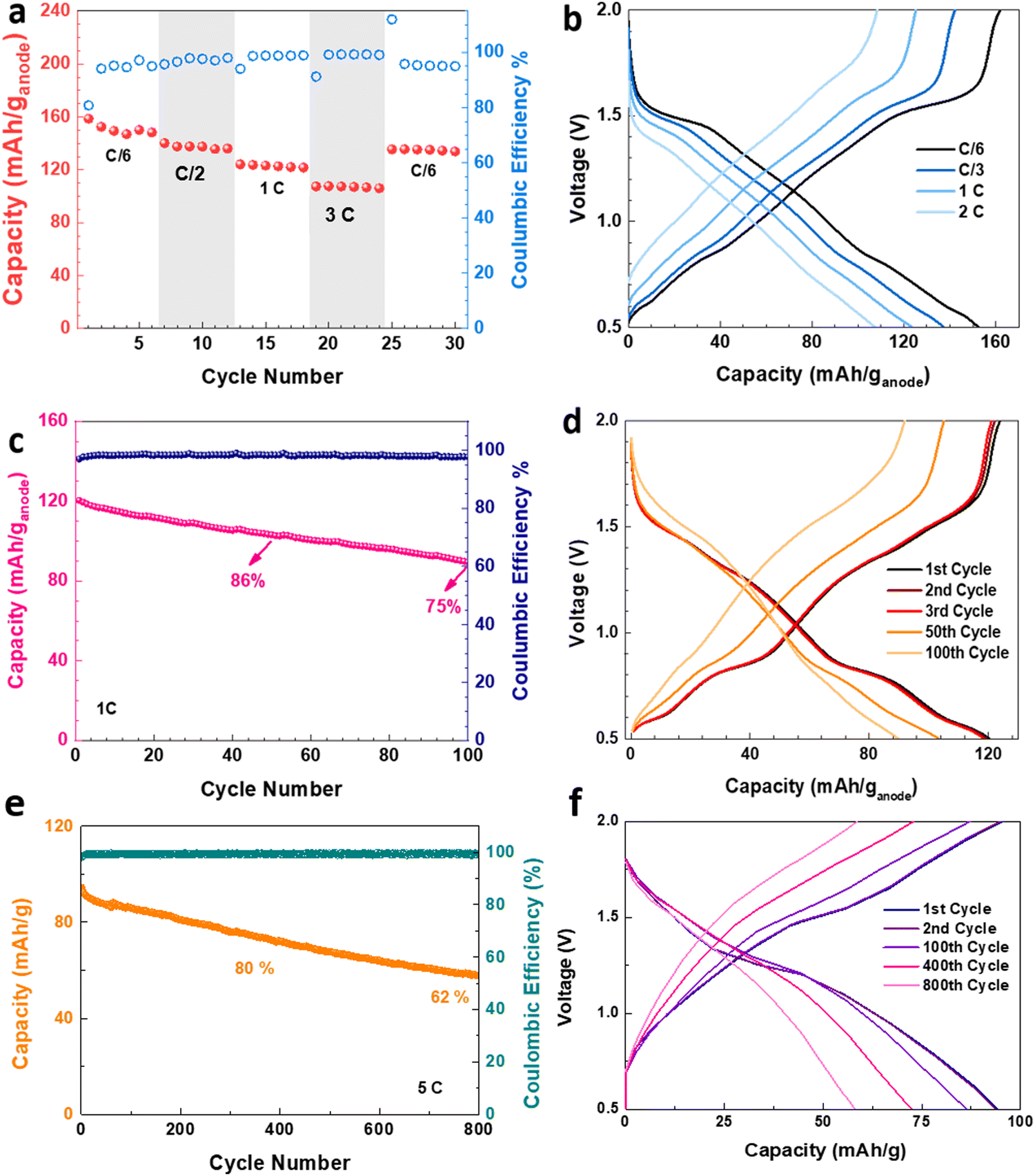 | ||
| Fig. 5 Electrochemical performance of the V2O5–LSG||LiMn2O4 coin cells. (a) Rate performance and corresponding Coulombic efficiency of a V2O5–LSG||LiMn2O4 cell cycled at C/6, C/2, 1C, 3C and C/6. (b) Capacity–voltage profiles at different rates. (c) Capacity and Coulombic efficiency of a V2O5–LSG||LiMn2O4 cell over 1C cycling. (d) Capacity–voltage profiles of the 1st, 2nd, 3rd, 50th, and 100th cycles during 1C cycling. (e) Capacity and Coulombic efficiency of a V2O5–LSG||LiMn2O4 cell over 5C cycling. (f) Capacity–voltage profiles of the 1st, 2nd, 100th, 400th and 800th cycles during 5C cycling. | ||
The V2O5–LSG||LiMn2O4 system was tested for long-term cycling at a 1C rate (Fig. 5c). 86% and 75% of the initial capacity were retained after 50 and 100 cycles, respectively. The SEM images of the V2O5–LSG electrodes before and after 100 cycles demonstrated that there are no significant morphological changes and the LSG network remains intact after 100 cycles (Fig. S11, ESI†). As illustrated in Fig. 5d, the capacity fading is associated with the disappearance of the voltage plateaus around 0.8 and 1.5 V, indicating reduced contribution from diffusion-controlled Faradaic processes. Moreover, the LSG||LiMn2O4 cell showed excellent long-term cycling stability at a high rate of 5C (Fig. 5e), reaching 80% and 62% capacity retention after 330 and 800 cycles, respectively. As shown in Fig. 5f, the individual plateaus are less observable in the charge and discharge curves during 5C cycling, displaying predominantly pseudocapacitive behavior. Furthermore, when a pair of V2O5–LSG||LiMn2O4 cells were connected in series, the battery pack can light up not only red, but also blue LED lights; impressively, the fully charged battery pack can power the LED for more than 24 h (Fig. S7, ESI†). Additionally, several control experiments were carried out to confirm the source of the great electrochemical performance and to evaluate the designed synthetic strategy. Fig. S8 (ESI†) compares the electrochemical properties of a physical mixture V2O5 + rGO to those of the nanoengineered V2O5–LSG, and the former shows poor rate capability and cyclability due to the lack of a conductive scaffold and V2O5 nanosizing. As illustrated in the electrochemical impedance spectroscopy (EIS) spectra, the system with V2O5–LSG possesses dramatically reduced charge transfer resistance compared to the physical mixture (Fig. S8c, ESI†). The V2O5–LSG composite electrode fabricated following the synthesis in Fig. 1 is also compared to a sample that only underwent one of either the laser-scribing or calcination steps (Fig. S9, ESI†). Both the calcination only and laser only samples experienced a sharp capacity decrease at the start of cycling and showed inferior rate capability, confirming the necessity of both steps. The poor performance of the laser only sample can be attributed to the incomplete oxidation and a large amount of the remaining VCl3 precursor (Fig. S1, ESI†), which is susceptible to dissolution into the electrolyte. As shown in Fig. S9c (ESI†), the Nyquist plots of the systems with V2O5–LSG and VOx–LSG are similar with significantly lower resistance than the VCl3/GO precursor mixture, confirming that a highly conductive network is formed upon laser scribing. As shown in the SEM images of the calcination only electrode (Fig. S10a, ESI†), without the laser-scribing step, the graphene sheets are less expanded in comparison to the porous 3D LSG structure (Fig. 2a). In addition, large V2O5 clusters of ∼2 μm are observed, which are much larger than the nanoparticles in the V2O5–LSG composite, leading to a reduced active surface area and poor rate capability (Fig. S10b and Fig. 2c, ESI†). Overall, the meticulously designed V2O5–LSG electrode exhibits a high specific capacity and excellent fast-charging properties due to its predominantly surface-controlled charge storage mechanism that is triggered by the V2O5-nanoparticle-on-LSG-scaffold structure.
Furthermore, the calcination time in the synthesis to convert VOx–LSG to V2O5–LSG (Fig. 1) was varied to investigate its effects on the resulting composite. The TEM images of V2O5–LSG that underwent post-laser calcination for 0.5 h, 1 h and 2 h are shown in Fig. 6a–c. In all three images, VOx/V2O5 is distributed on graphene sheets and its morphology changes significantly with increasing calcination time. With only 0.5 h post-laser calcination, the VOx/V2O5 clusters adopt undefined shapes and are uneven in size, resembling the morphology of spinodal decomposition products (Fig. 6a).29 As the calcination time increased to 1 h, defined V2O5 nanoparticles formed with an average size of ∼50 nm (Fig. 6b and Fig. 2c). The nucleation process continued from 1 h to 2 h, inducing much larger agglomeration of V2O5 particles by Ostwald ripening (Fig. 6c).30 X-ray photoelectron spectroscopy (XPS) was used to elucidate the changes in the chemical composition of the VOx/V2O5–LSG composites and the deconvoluted V 2p regions are shown in Fig. 6d–f and Fig. S12 (ESI†). In Fig. 6d, the peak at 516.6 eV represents V4+ and accounts for 58% of all V present, indicating that the major oxidation state of VOx is 4+ after 0.5 h post-laser calcination and the oxidation process is incomplete. As depicted in Fig. 6e and f, after 1 h and 2 h of calcination, the V5+ peak at 517.8 eV becomes the dominant peak in the spectrum and makes up ∼80% of all V present, suggesting that V5+ has overtaken V4+ as the main vanadium species present. Fig. S12 (ESI†) shows the V 2p sub-spectrum of the VOx–LSG intermediate that has never undergone any calcination, and it shows the presence of vanadium oxidation states +2 to +5, indicating incomplete oxidation of the VCl3 precursor without the calcination step. Thus, TEM images and XPS V 2p spectra suggest that it takes more than 0.5 h for the oxidation from VOx to V2O5 to reach completion and the optimal particle size is obtained after 1 h of calcination.
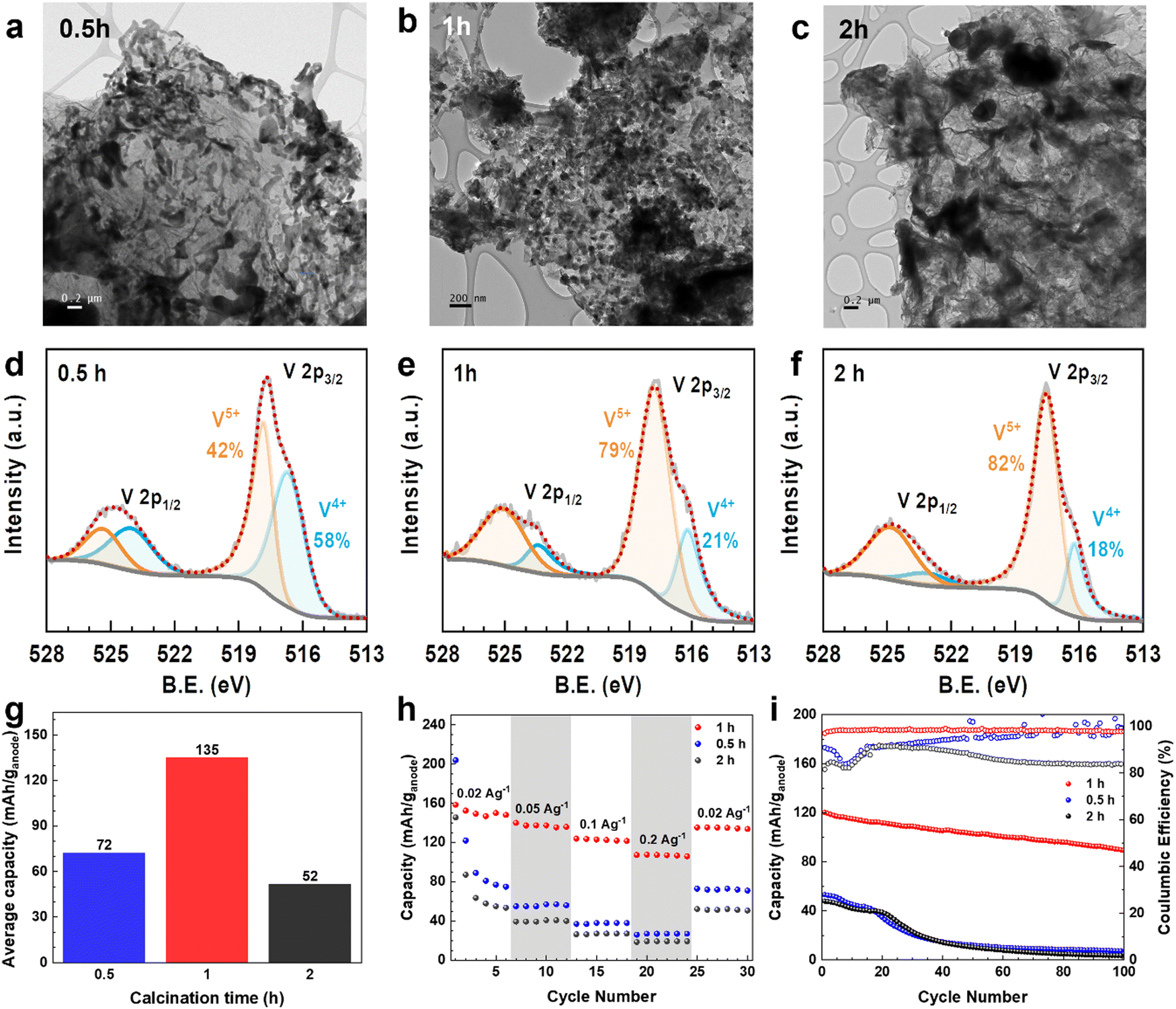 | ||
| Fig. 6 Effects of calcination time on the V2O5–LSG composite. TEM images of the eV2O5–LSG composite calcined for (a) 0.5 h, (b) 1 h, and (c) 2 h. XPS V2p spectra of the V2O5–LSG composite calcined for (d) 0.5 h, (e) 1 h, and (f) 2 h. (g) Specific capacity comparison between samples synthesized with 0.5, 1, and 2 h of calcination. (h) Rate performance of V2O5–LSG||LiMn2O4 cells with different calcination times cycled at C/6, C/2, 1C, 3C and C/6. (i) Capacity and Coulombic efficiency of V2O5–LSG||LiMn2O4 cells with different calcination times over cycling at 0.1 A g−1. | ||
The electrochemical behavior of the composites with various calcination times was also studied in full cells when they are paired with LiMn2O4 cathodes. Their average discharge capacities of cycle 25–30 (where the low-current capacities were stabilized) are compared in Fig. 6g. The cells with 0.5 h, 1 h and 2 h have specific capacity of 72, 135 and 52 mA h g−1, respectively. As shown by TEM and XPS data, on the one hand, with shorter calcination time, the conversion to V2O5–LSG is incomplete; on the other hand, the V2O5 nanoparticles become aggregated clusters with 2 h of calcination making a large proportion inaccessible near the core. Either way, this leads to a low proportion of the electrochemically active V2O5 in the resulting composites, reducing the specific capacity, suggesting 1 h as the optimal calcination duration. Fig. 6h shows the rate performance of full cells with these VOx/V2O5–LSG anodes. Both cells with anodes that underwent calcination for 0.5 h and 2 h show sharp capacity fading during the first 5 cycles at a low current density of 0.02 A g−1, suggesting possible structural instability caused by incomplete formation of V2O5 structures or pulverization accompanied by a large cluster size. When the current density was increased 10-fold, the cells with 0.5 h and 2 h calcination anodes can only reach a capacity retention of 35% and 37%, respectively, whereas 71% of capacity was retained for the anode with 1 h calcination time.31
The cycling stabilities of cells with anodes of different calcination times were also investigated (Fig. 6i). The capacity of cells with anodes of 0.5 h and 2 h calcination fell below 26% after only 40 cycles, while the cell with the 1 h calcination anode can retain 86% of its capacity after the same number of cycles. In addition, the Coulombic efficiencies of cells with anodes that underwent 0.5 h and 2 h calcination are low (∼80%) during the first 20 cycles. For the 0.5 h calcination anode, this phenomenon can be attributed to the dissolution of incompletely reacted VOx in the electrolyte. For the 2 h calcination anode, the Coulombic efficiency remained low throughout the cycling, which is likely caused by Li+ trapped deep into the bulk of the agglomerated V2O5 particles.
Finally, the electrochemical performance of the V2O5–LSG||LiMn2O4 system is compared to those of other lithium-ion batteries (LIBs) previously reported in the literature. Fig. 7a compares the full-cell specific capacity based on the total mass of both anode and cathode compositions when other aqueous LIB anodes are paired with the LiMn2O4 cathode.5,13,32–35 The V2O5–LSG||LiMn2O4 system gives the highest specific capacity of 67 mA h g−1 (after an initial capacity loss is accounted for) compared to other Ti, Mo and V-based anodes. This competitive advantage is attributed to the high capacity of the designed binder-free V2O5–LSG anode. The Ragone plot (Fig. 7b) compares the energy and power density of the V2O5–LSG||LiMn2O4 cell to those of the aqueous LIBs reported in the literature.5,13,15,32,33,35,36 The V2O5–LSG||LiMn2O4 system can reach a high power density of 650 W kg−1 at 15.6 W h kg−1 and a high energy density of 81.5 W h kg−1 at 13.6 W kg−1, outperforming the majority of prior aqueous LIBs. The detailed calculation is shown in Table S1 (ESI†). The outstanding power density can be attributed to the predominantly surface-controlled charge storage as illustrated in Fig. 4f. Notably, although V-based anodes have a relatively less negative redox potential that leads to lower average full-cell voltages in comparison to Ti and Mo-based anodes, the energy density of the V2O5–LSG||LiMn2O4 cell is actually comparable to if not higher than those of other aqueous LIBs due to its remarkably high anode capacity. Thus, the V2O5–LSG||LiMn2O4 system exhibits tunable power and energy density and is a promising candidate for fast-charging and safe energy storage applications.
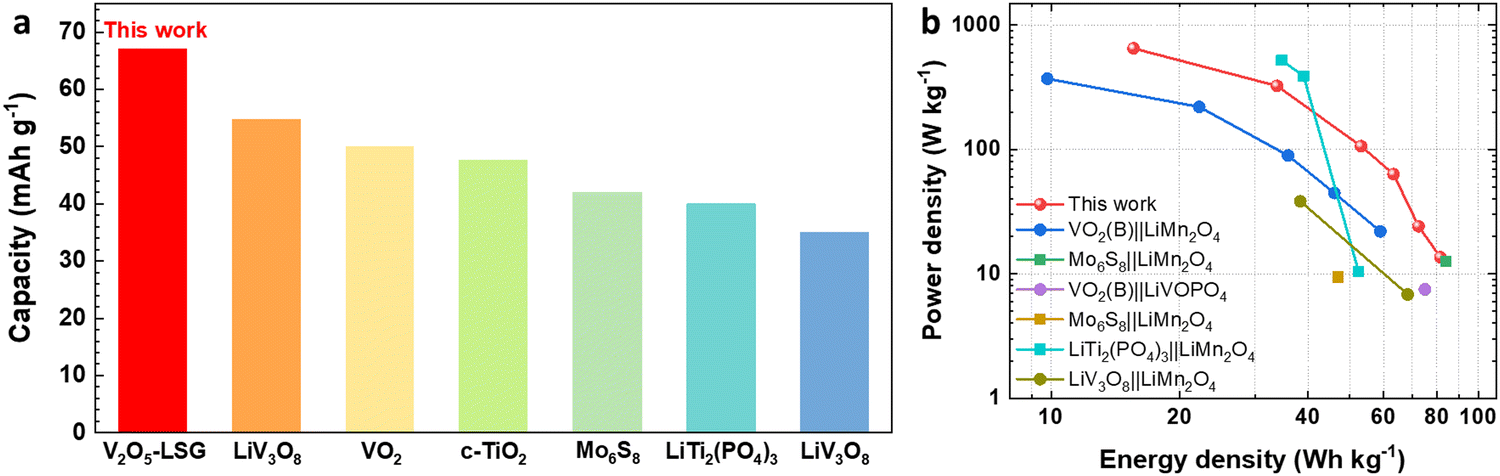 | ||
| Fig. 7 Performance comparison of V2O5–LSG||LiMn2O4 to previously reported systems in the literature. (a) A capacity comparison based on the total electrode mass of V2O5–LSG||LiMn2O4 to those of other aqueous Li-ion batteries reported in the literature.5,11,30–33 (b) A Ragone plot comparing power and energy densities based on the total electrode mass of V2O5–LSG||LiMn2O4 to those of other aqueous Li-ion batteries reported in the literature. | ||
Conclusions
In summary, a pseudocapacitive composite anode for aqueous lithium-ion storage in which V2O5 nanoparticles are distributed on a conductive graphene scaffold is reported for application in aqueous LIBs. The V2O5–LSG composite was synthesized from VCl3/GO precursors by a two-step laser-scribing/calcination approach, resulting in both an expanded LSG structure and fully oxidized V2O5 particles that are ∼50 nm in size. The V2O5–LSG anode can achieve an outstanding average specific capacity of 158 mA h g−1 and excellent rate capability as shown in three-electrode measurements. When paired with the intercalation-type cathode LiMn2O4 cathode, the full cell displays a predominantly surface-controlled charge storage mechanism that is faradaic in nature, leading to excellent fast charging properties. The V2O5–LSG||LiMn2O4 system shows an average specific capacity of 151 and 107 mA h (g anode)−1 at C/6 and 3C. Furthermore, the full-cell specific capacity as well as the power and energy density of this hybrid battery can reach 67 mA h g−1, 650 W kg−1 (at 15.6 W h kg−1) and 81.5 W h kg−1 at (13.6 W kg−1) respectively, all of which are favorable compared to those of previously reported aqueous LIBs. Thanks to its high capacity, fast charging capability and safe operation, the V2O5–LSG||LiMn2O4 system provides a potential solution for future energy storage applications.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank the University of California Climate Action Seed Grant R02CP7008 (R. B. K.), and the Dr. Myung Ki Hong Endowed Chair in Materials Innovation at UCLA, for financial support (R. B. K.).References
- Z. Ju, Q. Zhao, D. Chao, Y. Hou, H. Pan, W. Sun, Z. Yuan, H. Li, T. Ma, D. Su and B. Jia, Energetic Aqueous Batteries, Adv. Energy Mater., 2022, 12(27), 2201074, DOI:10.1002/aenm.202201074.
- S. Chen, M. Zhang, P. Zou, B. Sun and S. Tao, Historical Development and Novel Concepts on Electrolytes for Aqueous Rechargeable Batteries, Energy Environ. Sci., 2022, 15(5), 1805–1839, 10.1039/D2EE00004K.
- Q. Li, J. Chen, L. Fan, X. Kong and Y. Lu, Progress in Electrolytes for Rechargeable Li-Based Batteries and Beyond, Green Energy Environ., 2016, 1(1), 18–42, DOI:10.1016/J.GEE.2016.04.006.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Concentrated Mixed Cation Acetate “Water-in-Salt” Solutions as Green and Low-Cost High Voltage Electrolytes for Aqueous Batteries, Energy Environ. Sci., 2018, 11(10), 2876–2883, 10.1039/C8EE00833G.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, “Water-in-Salt” Electrolyte Enables High-Voltage Aqueous Lithium-Ion Chemistries, Science, 1979, 350(6263), 938–943, DOI:10.1126/science.aab1595.
- H. Ao, Y. Zhao, J. Zhou, W. Cai, X. Zhang, Y. Zhu and Y. Qian, Rechargeable Aqueous Hybrid Ion Batteries: Developments and Prospects, J. Mater. Chem. A, 2019, 7(32), 18708–18734, 10.1039/C9TA06433H.
- J. Liu, C. Xu, Z. Chen, S. Ni and Z. X. Shen, Progress in Aqueous Rechargeable Batteries, Green Energy Environ., 2018, 3(1), 20–41, DOI:10.1016/J.GEE.2017.10.001.
- Y. Wang, J. Yi and Y. Xia, Recent Progress in Aqueous Lithium-Ion Batteries, Adv. Energy Mater., 2012, 2(7), 830–840, DOI:10.1002/aenm.201200065.
- W. Zuo, R. Li, C. Zhou, Y. Li, J. Xia and J. Liu, Battery-Supercapacitor Hybrid Devices: Recent Progress and Future Prospects, Adv. Sci., 2017, 4(7), 1600539, DOI:10.1002/advs.201600539.
- Y. Fu, X. Guo, Z. Xu, G. Zhao, C. Xu, Y. Zhu and L. Zhou, Nanostructure-Mediated Phase Evolution in Lithiation/Delithiation of Co3O4, ACS Appl. Mater. Interfaces, 2021, 13(24), 28171–28180, DOI:10.1021/acsami.1c05591.
- S. Abouali, M. Akbari Garakani, Z.-L. Xu and J.-K. Kim, NiCo2O4/CNT Nanocomposites as Bi-Functional Electrodes for Li Ion Batteries and Supercapacitors, Carbon, 2016, 102, 262–272, DOI:10.1016/j.carbon.2016.02.055.
- Y. Sun, S. Jiang, W. Bi, C. Wu and Y. Xie, Highly Ordered Lamellar V2O3-Based Hybrid Nanorods towards Superior Aqueous Lithium-Ion Battery Performance, J. Power Sources, 2011, 196(20), 8644–8650, DOI:10.1016/J.JPOWSOUR.2011.06.050.
- W. Li, J. R. Dahn and D. S. Wainwright, Rechargeable Lithium Batteries with Aqueous Electrolytes, Science, 1979, 264(5162), 1115–1118, DOI:10.1126/science.264.5162.1115.
- M. Shao, J. Deng, F. Zhong, Y. Cao, X. Ai, J. Qian and H. Yang, An All-Vanadium Aqueous Lithium Ion Battery with High Energy Density and Long Lifespan, Energy Storage Mater., 2019, 18, 92–99, DOI:10.1016/J.ENSM.2018.09.029.
- G. J. Wang, H. P. Zhang, L. J. Fu, B. Wang and Y. P. Wu, Aqueous Rechargeable Lithium Battery (ARLB) Based on LiV3O8 and LiMn2O4 with Good Cycling Performance, Electrochem. Commun., 2007, 9(8), 1873–1876, DOI:10.1016/j.elecom.2007.04.017.
- V. Augustyn, P. Simon and B. Dunn, Pseudocapacitive Oxide Materials for High-Rate Electrochemical Energy Storage, Energy Environ. Sci., 2014, 7(5), 1597–1614, 10.1039/C3EE44164D.
- P. Simon, Y. Gogotsi and B. Dunn, Where Do Batteries End and Supercapacitors Begin, Science, 2014, 1210–1211, DOI:10.1126/science.1249625.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Achieving High Energy Density and High Power Density with Pseudocapacitive Materials, Nat. Rev. Mater., 2019, 5, 5–19, DOI:10.1038/s41578-019-0142-z.
- A. Manthiram, A Reflection on Lithium-Ion Battery Cathode Chemistry, Nat. Commun., 2020, 1–9, DOI:10.1038/s41467-020-15355-0.
- L. J. Wang, M. F. El-Kady, S. Dubin, J. Y. Hwang, Y. Shao, K. Marsh, B. McVerry, M. D. Kowal, M. F. Mousavi and R. B. Kaner, Flash Converted Graphene for Ultra-High Power Supercapacitors, Adv. Energy Mater., 2015, 5(18), 1500786, DOI:10.1002/aenm.201500786.
- A. Huang, M. F. El-Kady, X. Chang, M. Anderson, C. Lin, C. L. Turner and R. B. Kaner, Facile Fabrication of Multivalent VO x/Graphene Nanocomposite Electrodes for High-Energy-Density Symmetric Supercapacitors, Adv. Energy Mater., 2021, 11(26), 2100768, DOI:10.1002/aenm.202100768.
- J. Y. Hwang, M. F. El-Kady, Y. Wang, L. Wang, Y. Shao, K. Marsh, J. M. Ko and R. B. Kaner, Direct Preparation and Processing of Graphene/RuO2 Nanocomposite Electrodes for High-Performance Capacitive Energy Storage, Nano Energy, 2015, 18, 57–70, DOI:10.1016/j.nanoen.2015.09.009.
- J. Y. Hwang, M. F. El-Kady, M. Li, C. W. Lin, M. Kowal, X. Han and R. B. Kaner, Boosting the Capacitance and Voltage of Aqueous Supercapacitors via Redox Charge Contribution from Both Electrode and Electrolyte, Nano Today, 2017, 15, 15–25, DOI:10.1016/j.nantod.2017.06.009.
- P. Hu, P. Hu, T. D. Vu, M. Li, S. Wang, Y. Ke, X. Zeng, L. Mai and Y. Long, Vanadium Oxide: Phase Diagrams, Structures, Synthesis, and Applications, Chem. Rev., 2023, 123(8), 4353–4415, DOI:10.1021/acs.chemrev.2c00546.
- M. B. Smirnov, E. M. Roginskii, K. S. Smirnov, R. Baddour-Hadjean and J.-P. Pereira-Ramos, Unraveling the Structure–Raman Spectra Relationships in V 2 O 5 Polymorphs via a Comprehensive Experimental and DFT Study, Inorg. Chem., 2018, 57(15), 9190–9204, DOI:10.1021/acs.inorgchem.8b01212.
- Z. Hou, M. Dong, Y. Xiong, X. Zhang, Y. Zhu and Y. Qian, Formation of Solid–Electrolyte Interfaces in Aqueous Electrolytes by Altering Cation-Solvation Shell Structure, Adv. Energy Mater., 2020, 10(15), 1903665, DOI:10.1002/aenm.201903665.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, High-Rate Electrochemical Energy Storage through Li+ Intercalation Pseudocapacitance, Nat. Mater., 2013, 12(6), 518–522, DOI:10.1038/nmat3601.
- H. Lindström, S. Södergren, A. Solbrand, H. Rensmo, J. Hjelm, A. Hagfeldt and S.-E. Lindquist, Li + Ion Insertion in TiO 2 (Anatase). 1. Chronoamperometry on CVD Films and Nanoporous Films, J. Phys. Chem. B, 1997, 101(39), 7710–7716, DOI:10.1021/jp970489r.
- E. Seknazi, S. Kozachkevich, I. Polishchuk, N. Bianco Stein, J. Villanova, J.-P. Suuronen, C. Dejoie, P. Zaslansky, A. Katsman and B. Pokroy, From Spinodal Decomposition to Alternating Layered Structure within Single Crystals of Biogenic Magnesium Calcite, Nat. Commun., 2019, 10(1), 4559, DOI:10.1038/s41467-019-12168-8.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Mechanisms of Nucleation and Growth of Nanoparticles in Solution, Chem. Rev., 2014, 114(15), 7610–7630, DOI:10.1021/cr400544s.
- X. Tang, P. Wang, M. Bai, Z. Wang, H. Wang, M. Zhang and Y. Ma, Unveiling the Reversibility and Stability Origin of the Aqueous V 2 O 5 –Zn Batteries with a ZnCl 2 “Water-in-Salt” Electrolyte, Adv. Sci., 2021, 8(23), 2102053, DOI:10.1002/advs.202102053.
- C.-H. Lin, K. Sun, M. Ge, L. M. Housel, A. H. McCarthy, M. N. Vila, C. Zhao, X. Xiao, W.-K. Lee, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and Y. K. Chen-Wiegart, Systems-Level Investigation of Aqueous Batteries for Understanding the Benefit of Water-in-Salt Electrolyte by Synchrotron Nanoimaging, Sci. Adv., 2020, 6(10), 1–12, DOI:10.1126/sciadv.aay7129.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Advanced High-Voltage Aqueous Lithium-Ion Battery Enabled by “Water-in-Bisalt” Electrolyte, Angew. Chem., Int. Ed., 2016, 55(25), 7136–7141, DOI:10.1002/anie.201602397.
- J.-Y. Luo and Y.-Y. Xia, Aqueous Lithium-Ion Battery LiTi2(PO4)3/LiMn2O4 with High Power and Energy Densities as Well as Superior Cycling Stability, Adv. Funct. Mater., 2007, 17(18), 3877–3884, DOI:10.1002/adfm.200700638.
- L. Suo, F. Han, X. Fan, H. Liu, K. Xu and C. Wang, “Water-in-Salt” Electrolytes Enable Green and Safe Li-Ion Batteries for Large Scale Electric Energy Storage Applications, J. Mater. Chem. A, 2016, 4(17), 6639–6644, 10.1039/C6TA00451B.
- D. Sun, Y. Tang, K. He, Y. Ren, S. Liu and H. Wang, Long-Lived Aqueous Rechargeable Lithium Batteries Using Mesoporous LiTi2(PO4)3@C Anode, Sci. Rep., 2015, 5(1), 17452, DOI:10.1038/srep17452.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00579h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |