An ultra-high mass-loading transition metal phosphide electrocatalyst for efficient water splitting and ultra-durable zinc–air batteries†
Received
4th January 2024
, Accepted 7th June 2024
First published on 20th June 2024
Abstract
The development of sustainable energy conversion and storage technologies is an effective approach to relieve the increasingly severe global energy crisis. Herein, a facile reductive electrosynthesis approach, using Pluronic P123 as a structure-directing agent, is reported to prepare an electrically conductive, electrochemically stable, and porous Ni–Co–Mn phosphide (NCMP) electrocatalyst with a super-high mass loading of 22.6 mg cm−2, feasible for industrial-level applications. The NCMP electrocatalyst exhibits superior trifunctional electrocatalytic activities toward the hydrogen evolution reaction (ηj=10 = 100 mV), oxygen evolution reaction (ηj=50 = 218 mV), and oxygen reduction reaction (half-wave potential = 0.74 V vs. reversible hydrogen electrode) in alkaline electrolytes. The NCMP-based cell delivers an overall water-splitting voltage of 1.53 V at a rate of 10 mA cm−2, which is lower than that of the benchmark Pt/C(−)–RuO2/C(+) system. The NCMP-based zinc–air battery exhibits a high power density of 148 mW cm−2, a high specific energy of ∼932 W h kgZn−1, and excellent cycling stability of over 6000 cycles at 5 mA cm−2. Mechanistic studies through theoretical calculations revealed that a trimetallic species formed by Ni, Co, and Mn is the most catalytically active site. It is anticipated that this novel reductive electrosynthesis approach may extend to other electrodeposition processes and pave the way to better meet the existing and expected energy demands.
Broader context
Hydrogen (H2), is an indispensable clean energy carrier, and the electrocatalytic splitting of water is a sustainable and eco-friendly approach for hydrogen production. However, hydrogen/oxygen evolution reactions (HER/OER) are kinetically sluggish processes. Moreover, for hydrogen to play a significant role in a sustainable energy future, water electrolyzers should have a good mass loading of electrocatalyst, to be practically feasible. Most electrocatalysts suffer from drastic activity loss with an increase in mass loading and/or electrode thickness because of the blockade of the ion penetration and electron transport pathways. Similarly, despite the potential advantages of a century-old zinc–air battery (ZAB) technology, the practical implementation of ZABs has been impeded by sluggish OER and oxygen reduction reactions (ORR). Herein, we report a facile templated electrosynthesis method to prepare Ni–Co–Mn phosphide nanostructures. We demonstrate the practical applicability of Ni–Co–Mn–P as a highly efficient and cost-effective replacement for precious metals as electrocatalysts active in all the HER, OER, and ORR processes without being detached or pulverized during operation, even under an ultra-high mass loading of over 22 mg cm−2. This study shines light on the practical implementation of earth-abundant electrocatalysts to make hydrogen and metal–air batteries an attractive reality.
|
1 Introduction
In the post-oil era, hydrogen (H2), produced via electrocatalytic water splitting, is regarded as the cleanest (zero-carbon emission), most secure, and affordable energy carrier.1 Meanwhile, zinc–air batteries are fast becoming an indispensable technology for next-generation sustainable energy utilization owing to their ultrahigh theoretical specific energy (1086 W h kgZn−1), the abundance of the zinc anode, and safety merits.2 However, in both of these technologies, the sluggish kinetics with large overpotentials (η) of the involved redox processes—namely, the hydrogen evolution reaction (HER), the oxygen evolution reaction (OER), and the oxygen reduction reaction (ORR)—to deliver appreciable currents, are the major challenges hindering their extensive commercialization. Although transition metal-based electrocatalysts have experienced great breakthroughs over the past decade,3–8 tailoring advanced electrocatalysts with a commercial-level mass loading (≥10 mg cm−2), which is required for practical viability, represents one of the most promising contenders for advancement in the beyond noble-metal catalysts energy conversion and storage arena.
Among transition metal-based catalysts, transition metal phosphides (TMPs) have spurred extensive research interests in the pursuit of next-generation high-performance electrocatalysts.9–14 The higher electronegativity of phosphorus than transition metals triggers electron delocalization and leads to a partially positively charged metal and negatively charged phosphorus (Mδ+–Pδ−), which are structural mimics of the hydrogenase active site.15,16 The slightly positively charged metal atoms mimic that of a hydride (H−) acceptor and the slightly negatively charged phosphorus atoms serve as a proton (H+) acceptor, thereby enhancing the HER performance of TMPs. In addition, the incorporation of the phosphorus atom makes it possible to tailor the band gap and increase the density-of-states near the Fermi level, which signifies abundant charge carriers similar to noble metals for the electrocatalytic processes. Phosphorus is also important for the durability of the electrocatalysts since the covalent nature of the M–P bond guarantees the long-term stability of the catalyst. Note that in TMPs, the M–M metallic bond co-exists with P–P and M–P bonds, which not only confer TMPs with high chemical stability and mechanical robustness, but also enables facile compositional tailoring of TMPs over a wide range including metal-rich phosphides (MxPy, x > y), monophosphides (MxPy, x = y = 1), and P-rich phosphides (MxPy, y > x).15,17
Among TMPs, Ni- and Co-based phosphides can be considered as potent and economically viable electrocatalysts for HER and OER in alkaline electrolytes.18–22 However, since the work medium for HER activity of a mono-metallic phosphide is incompatible with that for the OER activity in a specific pH electrolyte (either acidic or alkaline), achieving remarkable electrochemical performance for catalyzing the overall water splitting reaction in a mono-metallic phosphide-based symmetrical two-electrode cell setup is still challenging.23 As a remedy to the above-mentioned difficulty, two approaches have been adopted to enhance the catalytic efficiency and durability of TMPs:24 (i) tailoring TMPs with unique nanostructures and morphology (extrinsic catalytic activity), and more importantly, (ii) modulating the electronic configuration of TMPs through vacancy engineering or heteroatom doping (intrinsic catalytic activity).25 Mixed metal phosphides exhibit higher catalytic activities than their single counterparts, revealing the positive synergy between the components. In the mixed-metal TMPs, the difference in electron-donating abilities of the metallic species results in the regulation of the electronic structure, offering more exposed active sites, and boosting the adsorption energy of the catalytic reaction intermediate, thus, enhancing the catalytic activities. Taking CoP as a model alkaline HER catalyst, Men et al. established a correlation between catalytic activity and the proportion of the unoccupied 3d orbitals (Pun) of the doped elements.26 Their theoretical calculations indicate that the transition metals with higher Pun boost the kinetics of the CoP catalytic performance, by providing more unoccupied d-orbitals and regulating the water (ΔG*H2O) and hydrogen adsorption/desorption strengths (ΔG*H). Yet, despite the intriguing merits of TMPs, including high electrical conductivity, chemical stability, mechanical durability, and their environmentally friendly nature, progress towards the widespread implementation of TMPs is hampered by a series of challenges. One of the most important ones is that scaling up the catalyst to commercial-level mass-loadings (≥10 mg cm−2) has remained unexplored because the mass transport kinetics deteriorates rapidly with increasing electrode thickness (this problem, of course, is not unique to TMPs, but pervasive in essentially all kinds of active materials across the whole research community). The mass loading of the electrocatalyst in commercial water-splitting cells and air cathodes of metal–air batteries can vary depending on the specific design and configuration of the cell, as well as the type of electrocatalyst being used. While plentiful research efforts have been devoted to down-sizing the loading of the precious metal electrocatalysts to nanoclusters or single atom level to reach the trade-off of the low cost and high catalytic performance,27 developing a high loading of non-noble metal-based electrocatalysts along with optimal percolation networks for the ions and electrons is also a long-standing scientific challenge towards lab-scale-to-realistic prototype performance development. For a catalyst, the lower the required mass, the better the catalytic system will be. However, in this regard, air cathode mass loadings of ≥10 and ≥15 mg cm−2 have been recommended for practical Zn–air pouch cell-level specific energies of 300 and 500 W h kg−1, respectively.28,29
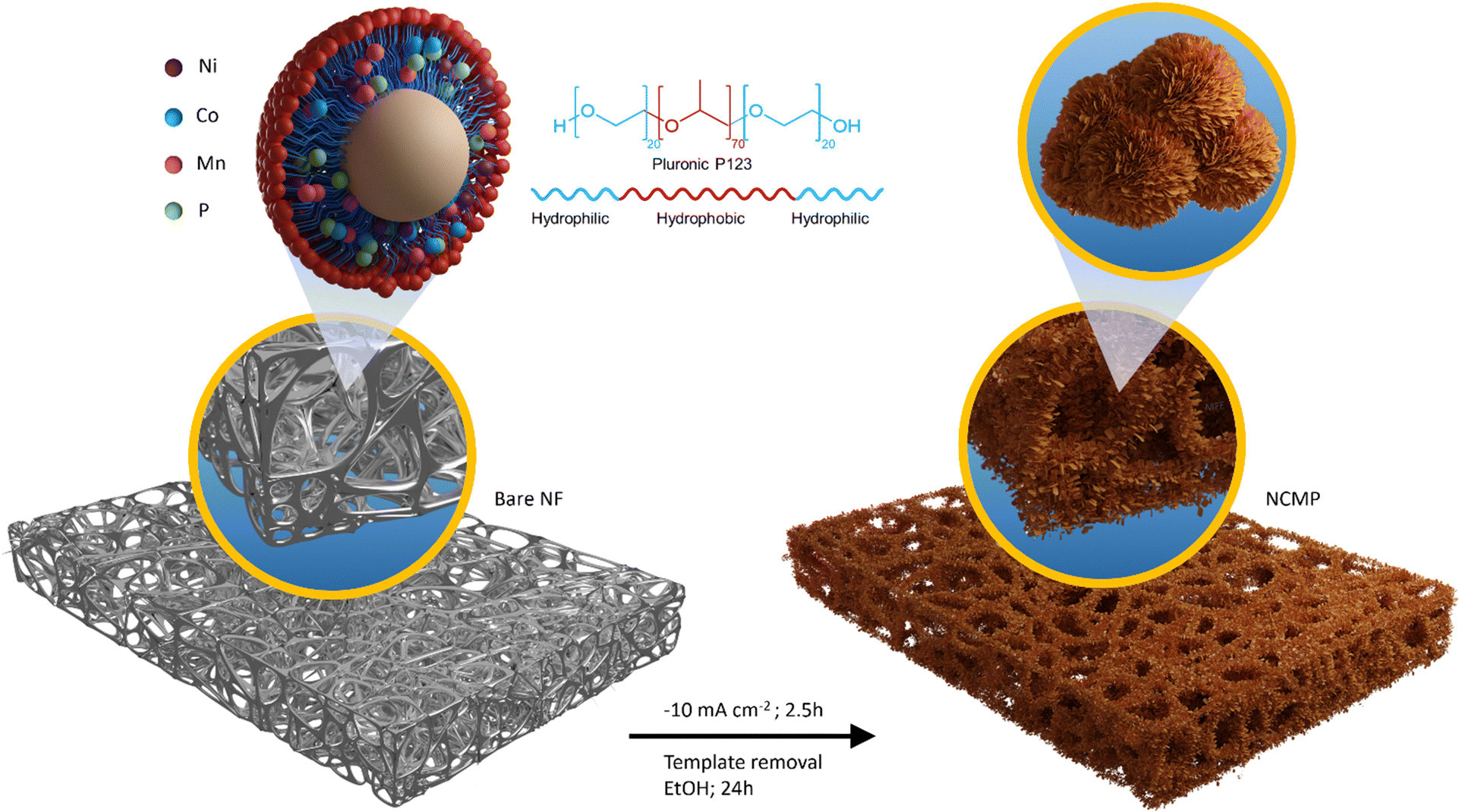
In this study, an innovative approach for a one-pot electrosynthesis of nanoporous Ni–Co–Mn–P (NCMP) with a superhigh mass-loading of 22.6 mg cm−2 on nickel foam is proposed, in which Pluronic P123 (as a non-ionic surfactant) is used as a structure-directing agent (Fig. 1). Benefiting from the tailored trimetallic electronic structure and engineered morphology, the NCMP exhibits excellent trifunctional catalytic activity toward HER, OER, and ORR. In addition, we employed density functional theory (DFT) calculations to elucidate the catalytic activity of the NCMP catalyst, revealing that the active site of the catalyst is a trimetallic species consisting of Mn, Ni, and Co elements. The outstanding trifunctional catalytic activity of the catalyst makes it a promising candidate for practical applications as a bifunctional HER/OER catalyst in water splitting cells and an OER/ORR catalyst in zinc–air batteries. In spite of a super-high mass loading with no need for any binders or conductive additives, the catalytic performance of the NCMP catalyst outperforms most reported metal catalysts, the improved performances of which are harvested at laboratory-level mass loadings (typically ∼1 mg cm−2) and even surpasses the benchmark RuO2 catalyst in the anode of the water electrolyzer as well as Pt/C + RuO2 in the cathode of the Zn–air battery (see comparison tables, Tables S1 and S2, in the ESI†). By leveraging the unique advantages of the P123-templated synthesis approach and controlled current electrosynthesis method a hierarchically porous electrocatalyst was prepared that demonstrates practical viability. Benefiting from these advantageous characteristics, this work holds particular promise by paving the way towards the practical implementation of advanced materials across a wide spectrum of energy fields.
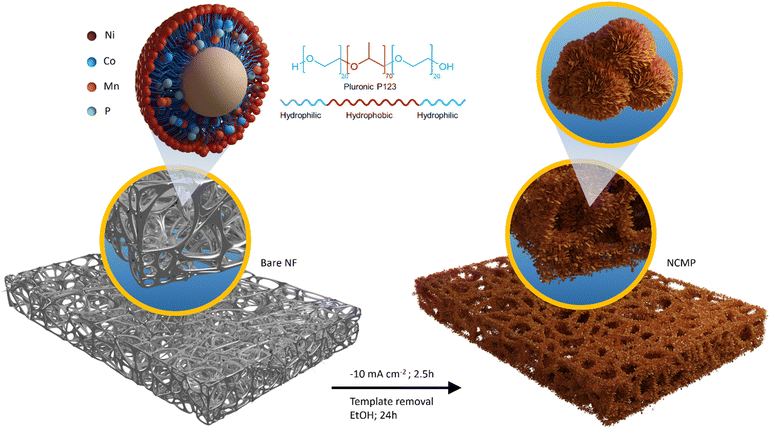 |
| | Fig. 1 Schematic representation of the fabrication process for the NCMP electrode. Schematic illustration of the one-pot and constant current (−10 mA cm−2, 2.5 h) electrosynthesis of the NCMP film on a nickel foam substrate. The chemical structure, amphiphilic character, and the possible morphological transition of Pluronic P123 in aqueous electrolyte, which results in a morphology-controlled electrocatalyst film, is also presented. | |
2 Experimental section
2.1 Chemicals
NiCl2·6H2O (≥98%), CoCl2·6H2O (≥98.0%), MnCl2·6H2O (≥98%), NaH2PO2·H2O (≥99%), Pluronic P123 (≥99.0%, 5750 g mol−1), NH4Cl (≥98%), and the Pt/C catalyst (20 wt%, loaded on graphitized carbon with a mean particle size of ≤5.0 nm) were purchased from Sigma-Aldrich (USA). RuO2 and zinc coarse powder with a particle size in the range from 0.3–1.5 mm (14–50 mesh ASTM) were obtained from Merck (Germany). All the other chemicals were of analytical reagent grade and used without further purification. Doubly distilled water was used for the preparation of all aqueous solutions.
2.2 Apparatus
The crystal structures of the powdered samples were characterized using a Philips X’pert X-ray diffractometer (XRD) and those of the thin films were recorded using a PANalytical X’Pert Pro-MPD diffractometer, both equipped with a Cu-Kα radiation source (λ = 1.5406 Å) generated at 40 kV and 30 mA with a step size of 0.04° s−1. Xpert software was used to fit the peaks of the XRD spectra. X-ray photoelectron spectroscopy (XPS) measurements of the samples were performed using a VG Microtech instrument consisting of an XR3E2 X-ray source and a Twin anode (AlKα = 1486.6 eV) without Ar+ sputtering. All XPS spectra were calibrated based on the C 1s photoemission peak at 284.8 eV as a reference. The surface morphology of the samples was characterized using a field-emission scanning electron microscope (FE-SEM, Philips) and a high-resolution transmission electron microscope (HR-TEM, FEI Tecnai F20), and elemental analysis was conducted using an energy dispersive X-ray spectrometer (EDX, as an attachment to SEM instrument). Specific surface area, pore volume and pore width of the electrocatalyst were measured using Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) procedures by an ASAP 2020 (Micromeritics, USA) instrument. All electrochemical measurements were carried out on a BioLogic SP-300 potentiostat/galvanostat (BioLogic, France) instrument controlled via EC-Lab v11.01 software. In all three-electrode studies, the reference electrode was a plastic-bodied double junction Ag/AgCl (Sat. KCl) and the counter electrode was a platinum plate. A Solartron 1470A multichannel battery test unit (Solartron Analytical, UK) equipped with Cell Test software (v. 3.5.0) was used for cycle stability tests. All electrochemical experiments were conducted at room temperature.
2.3 Electrosynthesis of the NCMP film
Electrosynthesis of the mesoporous NCMP film was carried out in a typical three-electrode cell configuration with a piece of pretreated Ni foam (1 × 1 cm2) as a working electrode, Pt plate as a counter electrode, and Ag/AgCl (Sat. KCl) as a reference electrode. Before electrodeposition, Ni foam was rinsed in 1.0 M HCl in an ultrasound bath to remove the surface oxide layer. Then, the electrode was washed thoroughly with deionized water and absolute ethanol to ensure the surface of the Ni foam was well-cleaned. Under optimized conditions, the electrosynthesis solution comprised of NiCl2·6H2O (0.2 M), CoCl2·6H2O (0.2 M), MnCl2·6H2O, NaH2PO2 H2O (0.2 M), Pluronic P123 (0.174 g cm−3, 0.03 M), and NH4Cl (0.25 M) dissolved in 10.0 mL of water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH (60:40 v/v%) solvent. A constant current of −10 mA cm−2 was applied for 2.5 h to electrosynthesize a porous NCMP film on the Ni foam substrate. Then, the as-deposited NCMP film was rinsed with doubly distilled water and dried and aged overnight at room temperature. Prior to use, the NCMP film electrode was dipped into absolute EtOH and maintained under moderate magnetic stirring for 24 h to extract the P123 surfactant.
:EtOH (60:40 v/v%) solvent. A constant current of −10 mA cm−2 was applied for 2.5 h to electrosynthesize a porous NCMP film on the Ni foam substrate. Then, the as-deposited NCMP film was rinsed with doubly distilled water and dried and aged overnight at room temperature. Prior to use, the NCMP film electrode was dipped into absolute EtOH and maintained under moderate magnetic stirring for 24 h to extract the P123 surfactant.
2.4 The HER/OER/ORR measurements
The electrocatalytic HER/OER performances of the materials were evaluated by recording the linear sweep voltammetry (LSV) curves in a 1.0 M KOH electrolyte at a scan rate of 5 mV s−1. The data collected with respect to the Ag/AgCl (Sat. KCl) reference electrode were converted to a reversible hydrogen electrode (RHE) scale where ERHE = EAg/AgCl + 0.197 V + 0.059pH. The overpotential data represent the mean of at least three independent measurements. The overall water splitting process was performed in a two-electrode cell comprising the NCMP electrocatalyst as both the anode and the cathode (NCMP‖NCMP) in a 1.0 M KOH electrolyte. The ORR studies were conducted in a standard three-electrode cell setup with a glassy carbon (GC, 0.159 cm2) rotating disk electrode (RDE) in a 0.1 M KOH electrolyte. To prepare the RDE, 1.0 mg of the electrosynthesized catalyst, carefully scratched off from the Ni foam, was mixed with 4 mg of Vulcan carbon and 30 μL of Nafion (5 wt%), and dispersed in 455 μL of ethanol. Then, the mixture was sonicated in an ultrasonic bath to obtain a homogeneous catalyst ink. The as-prepared catalyst ink (5 μL) was finally drop-cast onto the surface of a pre-cleaned GC RDE. For comparison, a mixture of commercial 20 wt% Pt/C, 20 wt% RuO2, and 80 wt% of Vulcan carbon were prepared in the same way.
2.5 Fabrication and test of Zn–air battery
The prototype Zn–air battery was assembled using Zn powder (pressed on a Cu mesh current collector) as an anode, a piece of Ni foam, the inner surface of which was coated with the NCMP catalyst and its outer surface was coated with a gas diffusion layer (GDL), as a cathode, a ZnO saturated 6.0 M KOH solution as an electrolyte, and cellulosic paper or Celgard M824 as a separator. Note that during the electrosynthesis process, NCMP catalyst is also slightly deposited on the second side of the Ni foam. The GDL was coated on the second side without scratching off the NCMP film. For the GDL, carbon nanotube (CNT), activated carbon (AC), and carbon black (CB) with CNT:AC:CB mass ratios of 10:2.5:1 were well-dispersed in 100 μL of ethanol containing 10 μL of PTFE (40 wt%). For wet-proofing the GDL layer, an O-ring was used around the GDL to prevent electrolyte creeping into it. The mass loading of NCMP was ∼22.6 mg cm−2 for all the catalyst electrodes. Similarly, an air cathode using a commercial Pt/C + RuO2 catalyst with a mass-loading of ∼1 mg cm−2 was also fabricated as a basis for comparison. Note that increasing the mass loading of the Pt/C + RuO2 catalyst did not translate into better catalytic performance.
2.6 Theoretical method
Density functional theory (DFT) calculations were carried out using the generalized gradient approximation (GGA) and ultrasoft pseudopotentials with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional included in the Quantum ESPRESSO package.30 Additionally, to account for long-range van der Waals (vdW) interactions, the Grimme vdW correction method was incorporated into the calculations. The self-consistent field (SCF) convergence threshold was set to 10−6 atomic units, with an energy cutoff of 600 eV. The maximum change in energy and force throughout the relaxation was considered to be 0.01 meV and 0.01 eV Å−1, respectively. A vacuum space of 20.0 Å was introduced along the z-axis to minimize the interaction between neighboring cells under periodic boundary conditions. The GGA+U method was taken for the correction of the strong correlation between electrons. The effective U parameter (Ueff) values were chosen from previous studies, which agree well with other theoretical predictions based on diverse criteria.31,32 For structural optimization, a k-point mesh of 6 × 6 × 1 was employed across all the investigated systems. A 12 × 12 × 1 Monkhorst–Pack grid was also used for density-of-states (DOS) calculations. The additional details of the DFT convergence tests, performed on several key parameters including k-point mesh, energy cutoff, and structural relaxations, is provided in the ESI.†
3 Results and discussion
3.1 Controlled-current electrosynthesis
In this designation, a controlled-current (galvanostatic) electrodeposition method was purposefully chosen for the electrosynthesis of the NCMP electrocatalyst. In contrast to the controlled-potential (potentiostatic) techniques that enable highly selective material deposition, controlled-current methods, that impose a constant electrochemical reaction rate regardless of the reactions that occur, are better suited for exquisite co-deposition of multiple species and alloys (see ESI†). The morphology, composition, and thickness of the electrodeposited catalyst can be tailored by varying the current rate and time. Herein, sodium hypophosphite, NaH2PO2, was used as a phosphorus source, which under negative potentials is reduced to elemental P. For the electrosynthesis of the NCMP electrocatalyst, the following reactions are likely to occur:33,34| | | Ni2+ + 2e− → Ni (E0 = −0.257 V) | (1) |
| | | Co2+ + 2e− → Co (E0 = −0.280 V) | (2) |
| | | Mn2+ + 2e− → Mn (E0 = −1.185 V) | (3) |
| | | H2PO2− + 2H+ + e− → P + 2H2O (E0 = −0.508 V) | (4) |
It is known that the reduction of hypophosphite in the presence of metal species leads to the formation of metal phosphides due to the diffusion of the reduced P into the metallic phase of the Ni, Co, and Mn. Moreover, the reduction of metal cations induces the reduction of hypophosphite at affordable potentials, thus, spontaneous M–P bond formation occurs. Therefore, the net reaction can be written as:| | | xNi2+ + yCo2+ + zMn2+ + H2PO2− + 2H+ + (2x + 2y + 2z + 1)e− → NixCoyMnzP + 2H2O | (5) |
Since the radius and electronegativity of Ni (149 pm, 1.91) and Co (152 pm, 1.88) are close to each other they can substitute for each other in the TMP structure.35 However, the radius of Mn (161 pm) is higher than those of Ni and Co, and its electronegativity (1.55) also differs significantly from that of P (2.19), this is why the Mn content of the NCMP electrocatalyst plays a critical role in its catalytic performance. Since Mn is less electronegative compared to Ni and Co, Mn contributes more electrons in the NCMP electrocatalyst. Because of this, by modulating the charge distribution and tailoring the d-band center of the Mn, more electrocatalytic active sites will be created. On the other hand, owing to the stronger electronegativity of P than metals, the incorporated P (Lewis base) withdraws electrons and induces more positive charges on the neighboring metal centers, therefore, facilitating the chemisorption of the water and tuning the electronic structure of metals to accelerate the HER process.6,36 The decreased electron-occupied states of metallic sites, M(2+δ)+, are in favor of the gradual switch from the M2+ to the M3+ state, thus accelerating the adsorption of reaction intermediates (*OH and *OOH) responsible for the OER/ORR activity enhancement.4 Since the standard reduction potential of Mn is remarkably more negative than those of Ni and Co, the deliberately chosen controlled-current reductive approach, which provides a sequence-controlled reduction of metal species, afforded a highly exquisite control over the Mn content of the catalyst. The plausible redox reactions of the composition-tailored NCMP electrocatalyst during the catalysis process or charge–discharge of the Zn–air battery in an alkaline electrolyte can be written as:17,24| | | NixCoyMnzP + 3OH− ⇄ NixPAOH + CoyPBOH + MnzP(1−A−B)OH + 3e− | (6) |
| | | NixPAOH + OH− ⇄ NixPAO + H2O + e− | (7) |
| | | CoyPBOH + OH− ⇄ CoyPBO + H2O + e− | (8) |
3.2 Structural and morphological characterizations
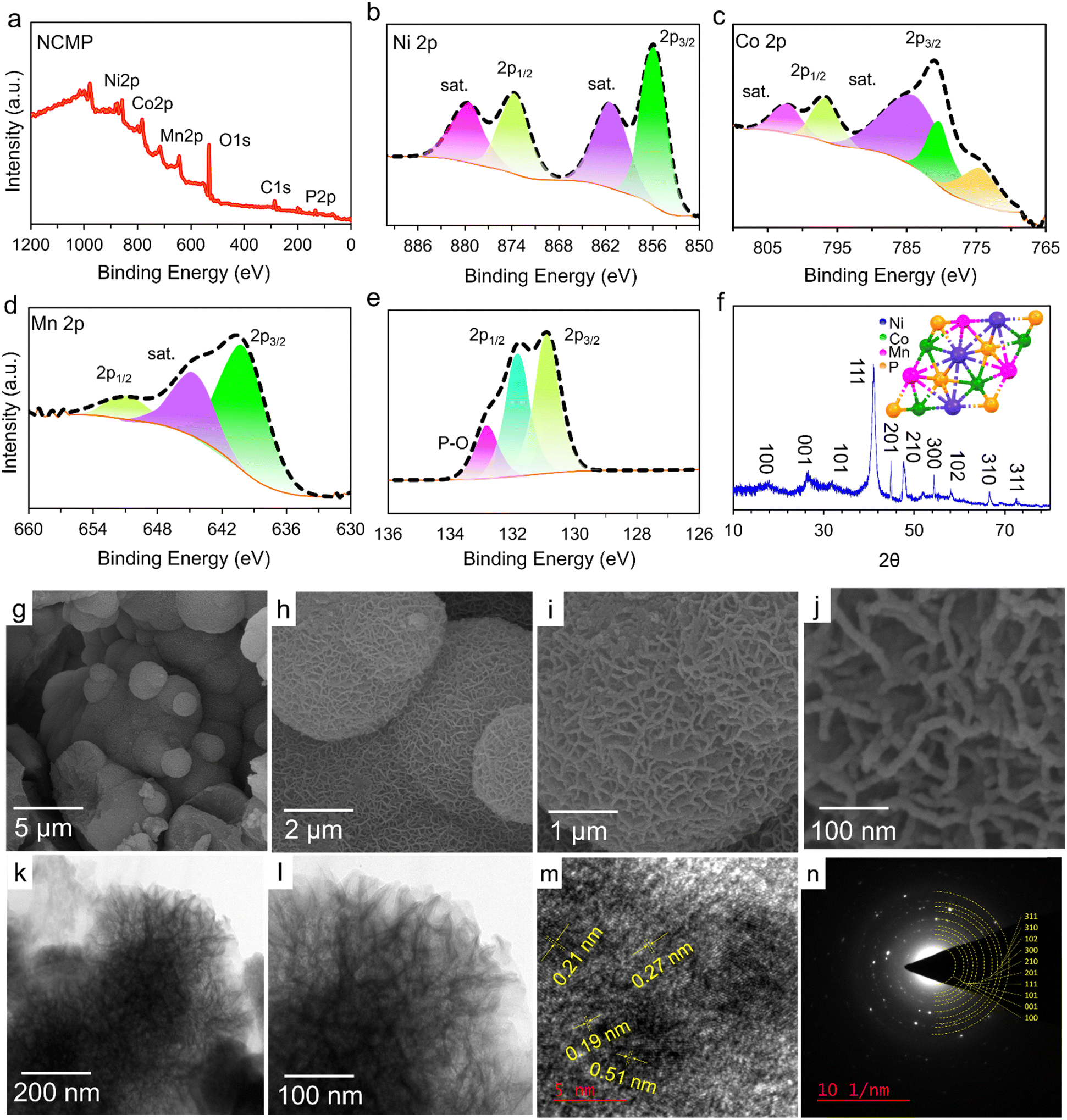
X-ray photoelectron spectroscopy (XPS) measurements were performed to analyze the surface chemistry of the as-prepared NCMP electrocatalyst (Fig. 2a–e). The survey XPS spectrum shows the presence of Ni, Co, Mn, P, and O elements (Fig. 2a). Furthermore, a comprehensive analysis of the XPS survey spectrum reveals that the atomic percentages of nickel, cobalt, manganese, and phosphorus in the sample are 20.3%, 26.4%, 18.8%, and 34.4% respectively. While diverse stoichiometric compositions of NixCoyMnzPW are possible, based on the XPS analysis, the formulation of the as-synthesized NCMP electrocatalyst is essentially Ni1.1Co1.4MnP1.8 (∼M2P). Note that the true TMP-based electrocatalyst for the OER contains the in situ formed (oxy)hydroxide or phosphate species on the surface. However, for simplicity of the representations, the abbreviated form of the catalyst (NCMP), regardless of the oxidation state, is used throughout the text. The core-level XPS spectrum of Ni 2p consists of two broad spin–orbit doublets at 855.8 eV (Ni 2p3/2) and 874.1 eV (Ni 2p1/2), along with two shake-up satellites (marked as Sat.), confirming the dominance of Ni–P and Ni2+ oxidation state (Fig. 2b).37,38 The high-resolution XPS spectrum of Co 2p displays two broad peaks at binding energies of 780.4 eV (Co 2p3/2) and 796.2 eV (Co 2p1/2) that accord well with the Co–P and Co2+ oxidation state (Fig. 2c).39–41 The core level Mn 2p spectrum shows two peaks at binding energies of 640.7 (Mn 2p3/2) and 652.5 eV (Mn 2p1/2) that cover those of metallic Mn (Mn–P) and Mn2+ species (Fig. 2d).42,43 The core-level XPS spectrum of P 2p shows two main peaks at binding energies of 130.8 and 131.9 eV corresponding to the P 2p3/2 and P 2p1/2, respectively, which can be well fit with the M–P bonds (Fig. 2e).44,45 The peak fitting analysis of P 2p also displays a weak band for P–O bonding (132.8 eV) indicating that the metal phosphide is indeed the predominant species. The presence of oxygen in the sample can thus be mainly ascribed to the presence of metal oxides and partially to the surface-oxidized P–O species.
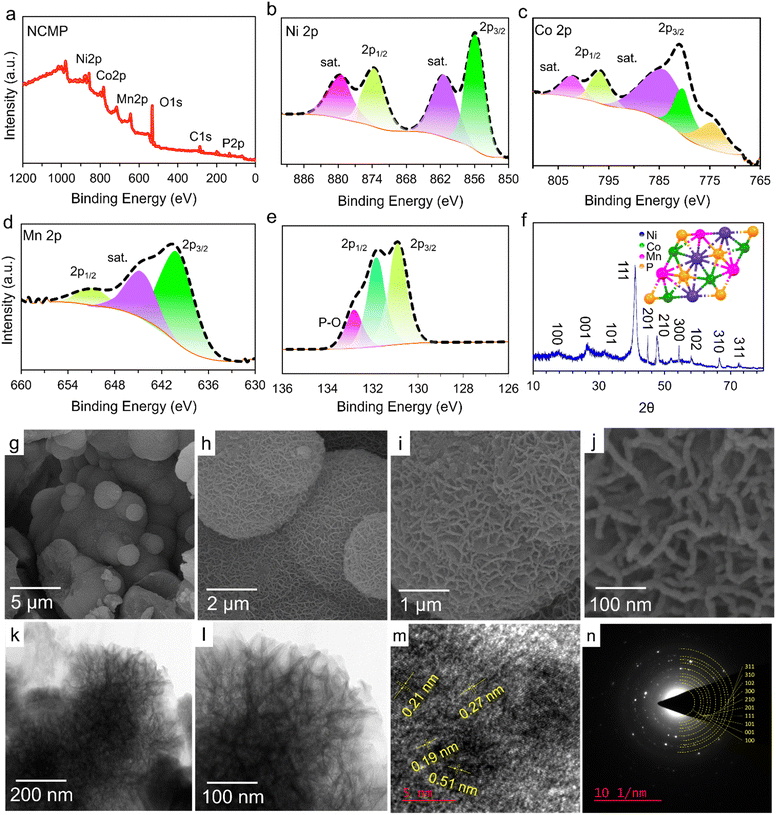 |
| | Fig. 2 Structural and morphological characterization of the NCMP catalyst. (a) The survey XPS spectrum of the NCMP catalyst. High resolution core-level XPS spectra of (b) Ni 2p, (c) Co 2p, (d) Mn 2p, and (e) P 2p. (f) XRD pattern of the NCMP catalyst that matches the XRD pattern of NiCoP (JCPDS no. 01-071-2336). The inset shows the possible crystalline structure of the NCMP catalyst, provided with modification of the NiCoP crystalline structure. (g)–(j) The FE-SEM and (k) and (l) TEM images of the NCMP at different magnifications. (m) The HR-TEM image, and (n) SAED pattern of the trimetallic NCMP catalyst. | |
Fig. 2f shows the X-ray diffraction (XRD) spectrum of the as-synthesized NCMP electrocatalyst. The spectrum displays distinct diffraction peaks at 2θ values of 16.6°, 26.4°, 32.1°, 41.1°, 44.8°, 47.6°, 54.3°, 58.1°, 66.5°, and 72.4°, which is in very good agreement with the hexagonal crystal structure of NiCoP (JCPDS no. 01-071-2336). Field emission-scanning electron microscopy (FE-SEM) images were recorded to investigate the morphology of the NCMP nanostructure (Fig. 2g–j). As can be seen, the NCMP particles display a micro-spherical morphology. The microspheres themselves are built up from worm-like nanoparticles with a mean diameter of ∼10 nm, resulting in a hierarchically porous texture. A catalyst with a hierarchical porosity benefits from the fast transport of the species to a large surface area, which significantly enhances the reactivity of heterogeneous catalysis. In addition, the FE-SEM-energy dispersive X-ray spectroscopy (FE-SEM-EDX) spectrum shows explicit elemental peaks related to Ni, Co, Mn, P, and O (Fig. S1a, ESI†). Also, the FE-SEM-EDX elemental mapping of the NCMP shows a homogeneous distribution of the constituent elements throughout the catalyst (Fig. S1b–g, ESI†).
To reveal that P123, as a structure-directing agent, dictates a specific shape with the desired morphology, we electrosynthesized NCMP on Ni foam without using P123. Fig. S2 (ESI†) shows the FE-SEM images and EDX elemental mapping of the control NCMP sample. As can be seen, in the absence of P123, a dense film with a compact morphology is formed, although the constituent elements are uniformly distributed throughout the film. P123 is an amphiphilic triblock copolymer, composed of a central hydrophobic poly(propylene oxide) chain (70 units) flanked by two hydrophilic poly(ethylene oxide) chains (20 units each). P123 behaves similar to surfactants and forms micelles in water when its concentration exceeds the critical micelle concentration (CMC = 0.052 mM at 25 °C).46 P123 (0.03 M, ∼580 × CMC) interacts non-covalently with the water-coordinated metal ion precursors and provides a scaffold around which the material can form. By using P123 as a structure-directing agent, the NCMP electrocatalyst can be electrosynthesized with controlled size, shape, and structure without being agglomerated, because the adsorption of P123 molecules on certain crystal facets reduces the surface energy and tension, and regulates the growth of nanoparticles as well. The tailored texture and controlled morphology of the porous NCMP catalyst are essential factors that determine the electrocatalytically active surface area, facilitated mass transport to and away from the catalytic centers inside the hierarchical pores, and accessibility of active catalytic sites. Typically, the nanostructured materials synthesized using templates exhibit lower crystallinity and benefit from more abundant defect-rich facets, edges, and corners with plentiful unsaturated active sites.47
The HR-TEM images of the pristine NCMP nanostructure also confirm that the catalyst is porous, which could help to increase the accessible active sites that participate in electrocatalytic reactions (Fig. 2k and l).48 The HR-TEM image of NCMP displays lattice fringes with d-spacings of 0.19, 0.21, 0.27, and 0.51 nm that correspond to the (210), (201), (101), and (100) lattice facets of the trimetallic Ni–Co–Mn–P (Fig. 2m). The selected area electron diffraction (SAED) pattern displays concentric electron diffraction rings with varying diameters, indicating the polycrystalline nature of the NCMP electrocatalyst (Fig. 2n). The SAED pattern also reveals the (100), (001), (101), (111), (201), (210), (300), (102), (310), and (311) planes, which all align well with the XRD pattern associated with the trimetallic Ni–Co–Mn–P (Fig. 2f).
After template extraction by solvent washing, the NCMP catalyst exhibited a Brunauer–Emmett–Teller (BET) surface area of 101 m2 g−1 (Fig. S3a, ESI†). The pore size distribution obtained using Barrett–Joyner–Halenda (BJH) method (Fig. S3b, ESI†) demonstrated the presence of hierarchical porosity (<3 nm, 5–8 nm, 25 nm, >40 nm) in the NCMP electrocatalyst. The 3D hierarchical porous structure of the catalyst benefits from obvious advantages in which the macropores (>50 nm) serve as ion-buffering reservoirs, the mesopores (2–50 nm) shorten the ion-transport pathways, and the micropores (<2 nm) provide ample sites for catalytic activities.49,50 Note that while micropores provide ample sites for catalytic activities, a sole-microporous electrocatalyst suffers from obstacles on the diffusional transport of species with poor efficiency and deteriorated catalytic rates. This challenge can be circumvented via the introduction of meso and macropores. For example, poor diffusivity of species, especially tightly fitting ones, into the single-sized micropores of zeolites is often the rate-determining step of their catalytic activity and causes their rapid deactivation, whereas, a hierarchically porous zeolite is an efficient catalyst because of its excellent diffusivity.51
3.3 Electrochemical studies
While the ultra-low loading of precious metal electrocatalysts36,52 as well as the relatively high loading of single atom catalysts anchored on a substrate53,54 are hot research topics, the high loading of non-noble metal catalysts without losing active sites is also a long-standing scientific challenge, especially for industrial-level applications. As evidence, the performance of electrocatalysts in practical Zn–air batteries and water electrolyzers is still unsatisfactory due to the deterioration of the overall catalytic performance under high mass loadings. The high loading affects the transport of electrons in the electrode active material as well as the transport of ions in the electrolyte, thus, leading to the deviation of the apparent catalytic activity from the real intrinsic catalytic performance. To investigate the effect of mass loading on the electrocatalytic activity of the NCMP catalyst, a series of mesoporous NCMPs with different mass loadings were synthesized and studied for the best-performance electrocatalyst. Surprisingly, the results demonstrate that the NCMP electrode with a high mass loading of 22.6 mg cm−2 shows the best electrocatalytic activities towards the HER and the OER (Fig. S4a and b, ESI†). The outstanding catalytic activity at such a high mass loading can be ascribed to the specific composition and morphology of the electrocatalyst (morphological changes, FE-SEM images, of the NCMP electrocatalyst at different time intervals of the electrosynthesis process are presented in Fig. S5, ESI†). Note, however, that due to the controlled-current electrosynthesis approach adopted in this study, the composition of the electrosynthesized NCMP electrode can be different in different stages of the electrosynthesis process. Tailoring the electrocatalyst in a hierarchical structure is also a nature-learned winning approach that can realize desirable electrochemical performance even under high mass loading.
OER activity.
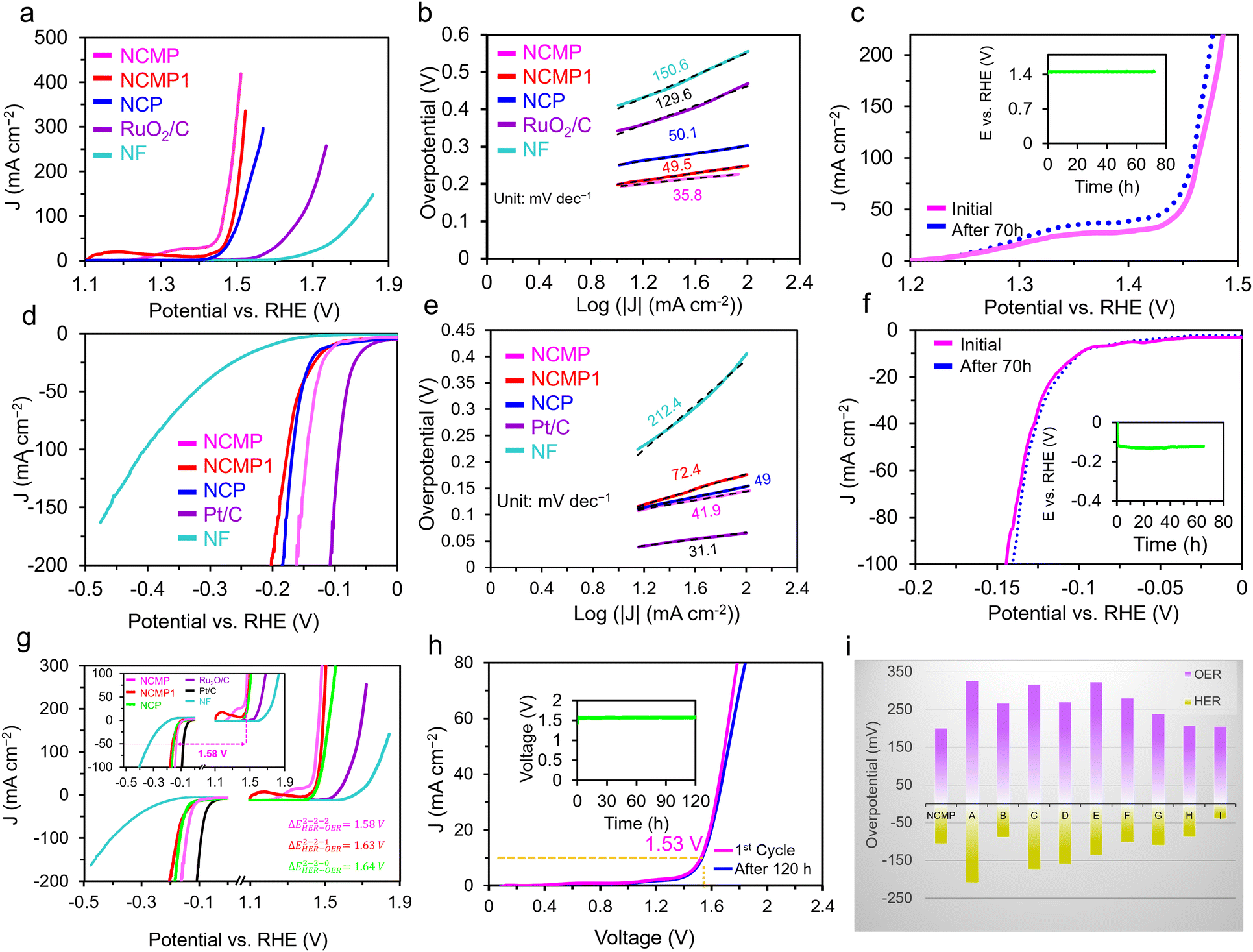
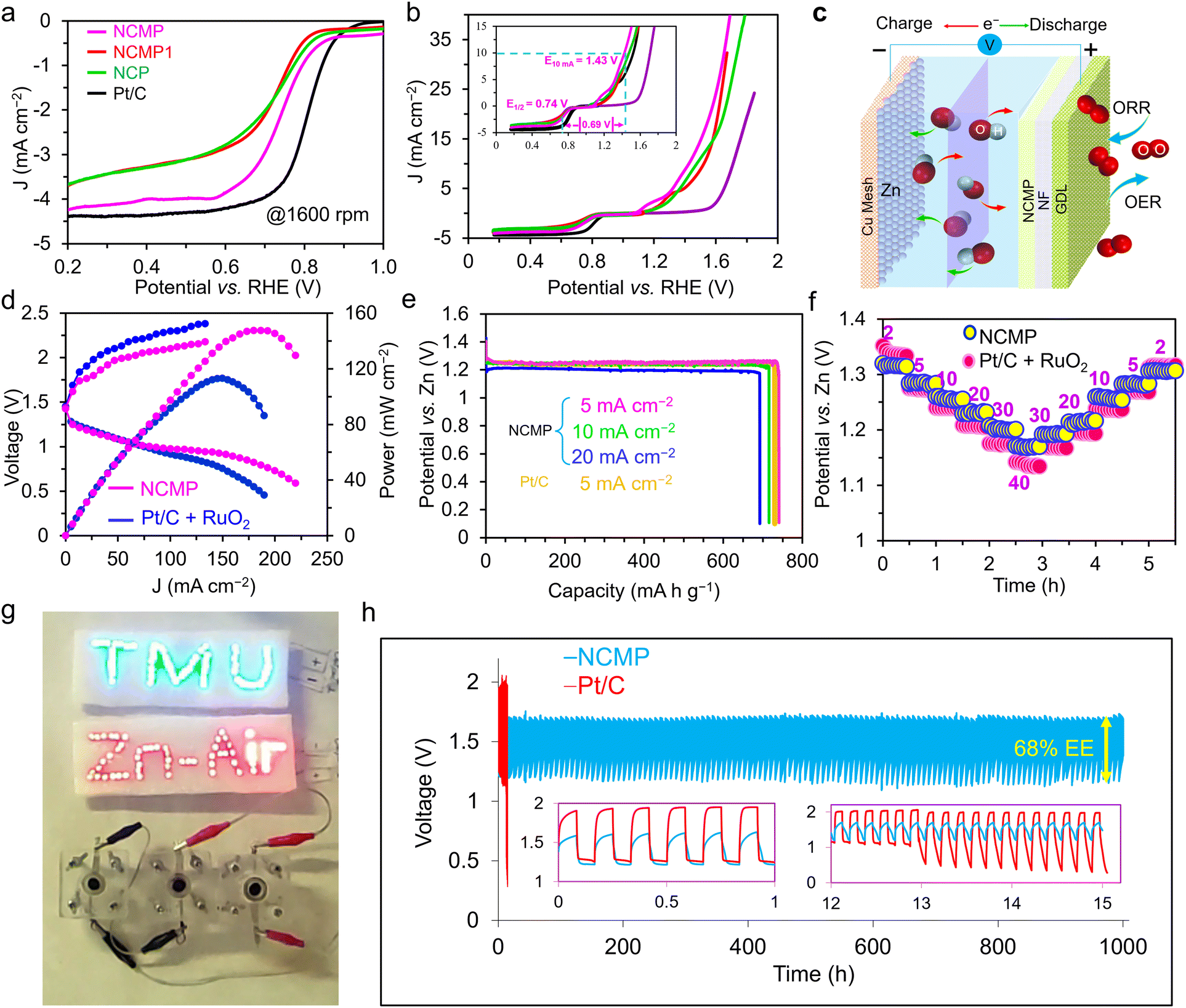
The OER activity of the NCMP catalyst was evaluated in a 1.0 M KOH solution in a standard three-electrode cell setup, comprising the NCMP working electrode, a double-junction Ag/AgCl reference electrode, and a Pt plate counter electrode. In addition to Ni–Co–Mn–P samples prepared with different Ni:Co:Mn molar ratios of 2:2:0 (NCP), 2:2:1 (NCMP1), and 2:2:2 (NCMP) in the synthesis solution, catalytic activities of the bare Ni foam and a RuO2/C electrode were also measured as control experiments. Fig. 3a displays the linear sweep voltammetry (LSV) curves of the samples at a scan rate of 5 mV s−1. The OER activity trend is NCMP > NCMP1 > NCP > RuO2/C > Ni foam. The bare Ni foam displays the least OER activity with an overpotential of 533 mV at a rate of 50 mA cm−2 (η50 = 533 mV). Note that the LSV curves of the Ni–Co–Mn–P catalysts exhibit a plateau, the current range of which exceeds 10 mA cm−2 at some potentials. Thus, to avoid overestimation of the catalytic activities, a rate of 50 mA cm−2 was chosen for comparison. With increasing Mn content in the starting material, the OER activity improves (NCP (zero Mn content), η50 = 254 mV and η200 = 311 mV; NCMP1, η50 = 244 mV and η200 = 277 mV). However, the NCMP sample displays the best OER activity (η50 = 218 mV and η200 = 253 mV) that is even far greater than that of the RuO2/C catalyst (η50 = 395 mV and η200 = 488 mV) and superior to most non-noble-metal based electrocatalysts reported in the literature (Table S1, ESI†). The importance of the excellent catalytic performance of NCMP becomes more apparent when considering the point that the reported values in Table S1 (ESI†) are mostly harvested at laboratory-level mass loadings (typically 0.2 to 2.0 mg cm−2). Most importantly, the industrially relevant current regime is essential for profitable oxygen/hydrogen production. Yet, increasing the operational gas evolution rate is problematic because the evolving gas bubbles contribute substantially to enhancing the electrolyte ohmic resistance. This is due to the fact that adherent bubbles reduce the amount of active surface available for the electrocatalysis and the dispersed bubbles impede the transport of species in the electrolyte and increase the electrolyte resistance by diminishing the electrolyte conductivity.55,56 Interestingly, as the current rate increases, the catalytic performance of NCMP becomes better and better compared to the RuO2 benchmark catalyst. The excellent OER activity of the NCMP catalyst can be attributed to its 3D architecture with well-defined hierarchical pores that allow rapid escape of gas bubbles. While the precise mechanisms underlying the relationship between the 3D hierarchical pore structure and the release of gas bubbles are not yet fully understood, studies have shown that strategies that promote efficient removal of gas bubbles from the electrode interface result in significant enhancements in device performance.56,57 It is expected that the rate of gas evolution from a hierarchically porous material, with shorter diffusion path lengths, is much higher than that of a single-sized microporous electrocatalyst. It is worth noting that the construction of such an efficient electrode using conventional manufacturing methods is challenging. This critically important goal was accomplished thanks to the advantageous characteristics of the templated electrosynthesized NCMP film, with interconnected pores, in which the blocking effect of the polymer binders and conductive additives, as well as the trapped bubbles, can be simply avoided.
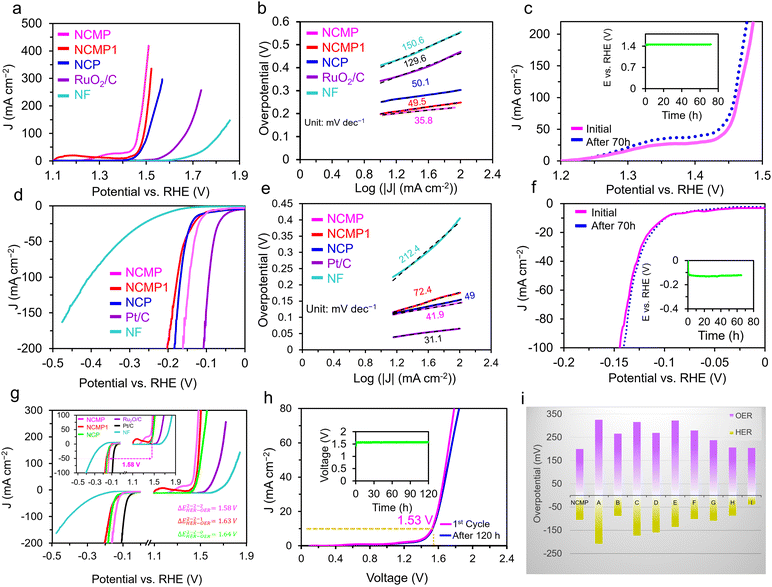 |
| | Fig. 3 OER and HER activities of NCMP and overall water splitting studies. (a) The OER polarization curves and (b) Tafel plots of the Ni–Co–Mn–P catalysts as well as the RuO2/C and the bare Ni foam (NF) in a 1.0 M KOH electrolyte at a scan rate of 5 mV s−1. (c) Durability study: the OER polarization curves of the as-synthesized NCMP catalyst as well as the one after 70 h of continuous operation. The inset shows a V–t plot for the NCMP catalyst at a constant current density of 10 mA cm−2 for 70 h. (d) The HER polarization curves and (e) Tafel plots of the Ni–Co–Mn–P catalysts as well as the Pt/C and the bare NF electrodes in a 1.0 M KOH electrolyte at a scan rate of 5 mV s−1. (f) Durability study of the NCMP catalyst for the HER over the course of 70 h of continuous operation at a rate of −10 mA cm−2. (g) Overall OER and HER polarization curves of the NCMP catalysts along with Pt/C, RuO2/C, and the bare Ni foam electrodes in three-electrode cell setups. (h) Durability study of the NCMP catalyst in 1.0 M KOH for overall water splitting at a rate of 10 mA cm−2 for 120 h. (i) Comparison of the overpotentials to drive a current density of 10 mA cm−2 for the NCMP catalyst with some other transition metal phosphide-based electrocatalysts namely, (A) Co2P/Co-, N-, and P-doped carbons;58 (B) CoFeO@black phosphorus;59 (C) CoP@P, N co-doped carbon;60 (D) CoP N-doped carbon@CNT;61 (E) CoP NFs;62 (F) CoP/Ti3C2;63 (G) Mo–Ni3S2/NixPy/NF;64 (H) (Fe0.1Ni0.9)2P(O)/NF;65 and (I) NF@Fe2–Ni2P/C.66 | |
Fig. 3b displays the Tafel plot of the investigated catalysts. As can be seen, the Tafel slope of NCMP (35.8 mV dec−1) is lower than that of the NCMP1 (49.5 mV dec−1) and NCP (50.1 mV dec−1), and far lower than that of RuO2/C (129.6 mV dec−1), and Ni foam (150.6 mV dec−1) control catalysts, confirming faster kinetics and more favorable OER activity for the NCMP catalyst. In addition, the NCMP catalyst shows excellent durability over 70 h of continuous operation with a negligible activity loss (Fig. 3c).
HER activity.
We also compared the HER performance of the three Ni–Co–Mn–P samples along with Pt/C and Ni foam as controls in a 1.0 M KOH electrolyte. As shown in Fig. 3d, the HER activity trends of the catalysts are as follows: Pt/C (η10 = 44.2 mV) > NCMP (η10 = 100 mV) > NCP (η10 = 110 mV) > NCMP1 (η10 = 112 mV) > Ni foam (η10 = 205 mV). The Tafel slope of the Pt/C for the HER is the lowest (31.1 mV dec−1). Among the Ni–Co–Mn-P samples, the Tafel slope of the NCMP catalyst for the HER (41.9 mV dec−1) is also lower than those of the NCP (49 mV dec−1) and NCMP1 (72.4 mV dec−1) catalysts, demonstrating the favorable reaction rates in the HER process (Fig. 3e). As shown in Fig. 3f (inset), the electrochemical stability test shows that NCMP is highly durable, with positive changes in LSV and chronoamperometric response curves after continuous operation for 70 hours. Although none can compete with Pt in intrinsic HER activity, the HER activity of the NCMP catalyst is outstanding among the other reported non-precious electrocatalysts (Table S1, ESI†). The key advantages mentioned for the catalytic OER performance of NCMP are also held for the HER activity, demonstrating the worthy place of the NCMP catalyst for commercial applications.
OER and HER simultaneously.
Due to the excellent OER and HER performances of the NCMP electrocatalyst, we studied the combination of OER and HER for overall water splitting. The LSV profiles for OER and HER (individually investigated in 3E cells) show that a potential difference (ΔEOER–HER) of 1.58 V is required to attain a current density of 50 mA cm−2 for the NCMP-based system, which is much lower than those obtained for the systems based on NCMP1 (ΔEOER–HER = 1.63 V), NCP (ΔEOER–HER = 1.64 V), and Ni foam (ΔEOER–HER = 2.10 V), and even surpassing the one based on the benchmark Pt/C(−)–RuO2/C(+) electrocatalyst (ΔEOER–HER = 1.70 V) (Fig. 3g). This indicates that the NCMP‖NCMP water electrolyzer could be powered by a single AA battery with a nominal voltage of ≈1.5 V (Video S1, ESI†). The long-term stability of the two-electrode cell was evaluated by means of LSV as well as chronopotentiometry (at a current density of 10 mA cm−2) over 120 h (Fig. 3h, inset). According to the LSV profiles and chronopotentiometry response, the potential change after 120 h is negligible, indicating the excellent long-term stability of the NCMP catalyst, thanks to its porous architecture and a rather firm connection to the Ni foam substrate. Note, however, that under a high current regime where bubbles vigorously evolve, some parts of the electrocatalyst may detach due to the interfacial adhesion force between the catalyst and bubbles. This can compromise the mechanical stability of the electrocatalyst during high-rate water splitting and warrants further investigation. The cell voltages to drive a current density of 10 mA cm−2 for the NCMP electrocatalyst is smaller than that of some other transition metal phosphide-based electrocatalysts reported in the literature (Fig. 3i).
To further investigate the reasons behind the long-term stability of the NCMP catalyst, the morphological changes of the positive and negative electrodes of the water splitting cell were investigated before and after long-term operation using FE-SEM (Fig. S6 and S7, ESI†). Note that the durability of a catalyst does not necessarily mean that the starting material is still the active form of the catalyst. Post-operation SEM analysis of the negative electrode after 120 h of HER reveals that the morphology of the negative electrode remains almost unchanged, and the surface composition is also preserved (Fig. S6, ESI†). However, the morphology and composition of the positive electrode changes significantly after 120 h of OER. Many studies have shown that TMPs are susceptible to in situ phase transformation and structural reorganization during the OER process.67–69 It has been well demonstrated that TMPs are thermodynamically less stable than metal oxides when subjected to oxidizing potentials, typical of the alkaline OER environment.70 Thus, transition metal oxides or (oxy)hydroxides (TMOs) as the thermodynamically stable end-point, are the true active form of most transition metal-based OER catalysts.71 To evaluate the crystalline structure of the true TMP-derived OER electrocatalyst, the NCMP electrocatalyst was characterized using a surface XRD technique, performed on an NCMP thin film, after 21 h. Note, the electrocatalyst electro-synthesized on a porous Ni foam substrate cannot be easily scratched off after the OER operation and surface XRD analysis of the rough Ni foam is challenging because of the diffuse scattering. Thus, we electrosynthesized a thin film of the electrocatalyst on a Ni-plated fluorine-doped tin oxide (FTO) smooth substrate that partially resembles the real environment of the NCMP catalyst. The XRD pattern of the thin film indicates that the true electrocatalyst is mostly NiMnO3 and CoMnO3, whereas POx species are less likely to play a significant role (Fig. S8, ESI†).
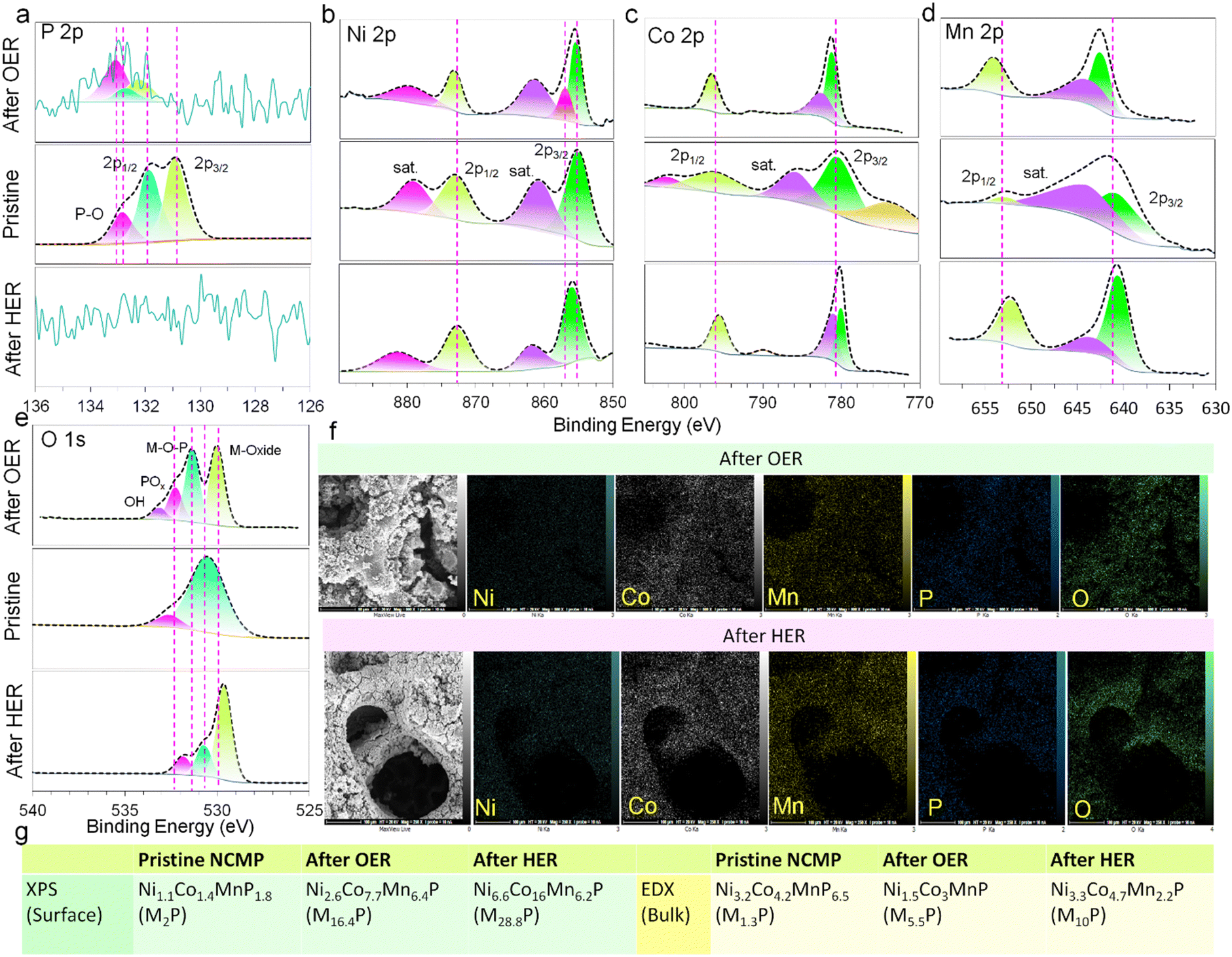
The chemical states and elemental compositions of the NCMP electrocatalyst before and after HER and OER processes were further explored using the XPS (Fig. 4a–e) and FE-SEM-EDX methods. The bulk composition of the pristine NCMP electrocatalyst according to EDX elemental analysis is Ni3.2Co4.2MnP6.5 (generally, M1.3P, M = Ni + Co + Mn), whereas the XPS analysis shows that the surface of pristine electrocatalyst is slightly phosphorus-deficient than the bulk, Ni1.1Co1.4MnP1.8 (M2P). After OER catalysis, the overall bulk composition by EDX is Ni1.5Co3MnP (M5.5P) but the surface XPS analysis demonstrates a significant depletion in phosphorus (Ni2.6Co7.7Mn6.4P, M16.4P). This can be ascribed to the leaching of phosphorus as phosphate under an oxidizing alkaline OER condition. Based on the EDX data, the post-HER electrocatalyst undergoes a significant change in bulk composition (Ni3.3Co4.7Mn2.2P, ∼M10P), though elemental mapping analysis confirmed the even distribution of Ni, Co, Mn, and P in the post-HER catalyst (Fig. 4f). According to the XPS data, the surface composition is, however, quite different from the bulk, and phosphorus is almost completely depleted (Ni6.6Co16Mn6.2P, M28.8P), which is consistent with displacement of the P-containing species by metal hydroxides during operation under an alkaline electrolyte.72,73 Thus, during both OER and HER processes, the pristine metal phosphide plays a sacrificial role and undergoes severe in situ chemical modification in alkaline media.74 We also probed the oxidation state of Ni, Co, and Mn. Generally, the binding energies of Ni 2p, Co 2p, and Mn 2p for the post-OER electrocatalyst shift to higher binding energies and those of the post-HER electrocatalyst shift to slightly lower values (these shifts for Ni are less pronounced). A shift to a higher binding energy after OER indicates that the original M2+ species are partially oxidized to M3+ during OER, whereas a shift to a lower binding energy after HER implies that the M0/M2+ ratio of the electrocatalyst increases during HER.75 After both OER and HER, the oxygen content increases significantly. The O 1s spectra confirm the presence of metal oxides and hydroxide species. Note that the pristine electrocatalyst also contains oxygen, however, it was omitted from the empirical formula for simplicity of representation and due to the fact that parts of the oxygen content originate from the remaining P123 surfactants (makes its proper estimation challenging). Fig. 4g summarizes the XPS and EDX analysis of the empirical formula of the electrocatalyst before and after electrocatalytic activities, providing information about the surface (since the penetration depth of XPS technique is generally limited to a few nm) and bulk of the material, respectively. Depending on the extent of oxidation, a fully oxidized species or a TMP-core@TMO-shell structure can be formed. Generally, the OER performance of the in situ generated metal oxides/(oxy)hydroxides is much better than that of their original counterparts synthesized directly. TMP can act as a conductive scaffold for the true catalytically active TMO on the surface, and the TMO shell serves as an impermeable barrier to protect the underlying TMP core from corrosion as well. The phase transformation from TMP precatalyst to TMO true catalyst leads to the formation of metastable metal oxide/hydroxide phases rich in vacancies, edges, corners, kinks, and facets that are more catalytically active and substantially beneficial to boost the intrinsic catalytic activity.
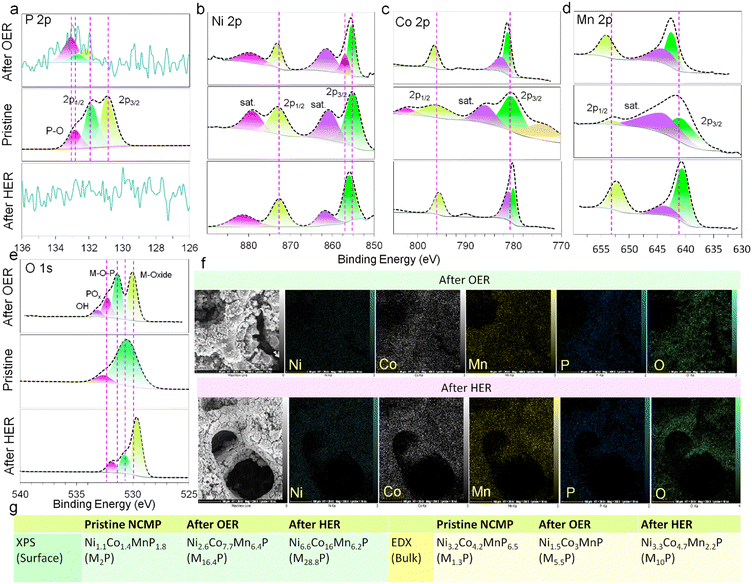 |
| | Fig. 4 XPS and EDX characterization after OER and HER processes. Deconvoluted core-level XPS spectra of (a) P 2p, (b) Ni 2p, (c) Co 2p, (d) Mn 2p, and (e) O 1s after OER (top) and HER (bottom) compared to the pristine one (middle). (f) FE-SEM-EDX elemental mappings of Ni, Co, Mn, P, and O of the NCMP electrocatalyst after OER and HER. (g) Deduction of the elemental composition and empirical formula of the pristine, post-OER, and post-HER electrocatalysts using XPS and EDX techniques. | |
The outstanding water splitting activity of the NCMP catalyst is well ahead of most other transition metal-based electrocatalysts reported in the literature (Table S1, ESI†). In addition, for an in-depth understanding of the reasons behind the superior electrocatalytic performance of the NCMP catalyst, we measured the electrochemically active surface area (ECSA) as an important parameter reflecting the intrinsic catalytic activity of the electrocatalysts.76–78 Fig. S9a–c (ESI†) represents the CV curves of the Ni–Co–Mn–P electrodes in a potential window from 0.05 to 0.1 V vs. Ag/AgCl, which is the non-faradaic region of the profiles. The extracted ECSA values from the slope of the j–ν plots (Fig. S9d, ESI†) are presented in Fig. S9e (ESI†). As can be seen, the ECSA of the NCMP electrode (48 cm2) is far higher than those of the NCMP1 (24 cm2), and NCP (11.5 cm2) electrodes. Considering the point that the mass loadings of the three Ni–Co–Mn–P electrodes are almost the same, the huge difference in their ECSA cannot be explained simply by the film thickness, instead, it is because of the high number of the electrocatalytically active sites of the NCMP. The ECSA-normalized current density of the NCMP electrocatalyst at a fixed overpotential of 240 mV was 2.81 mA cm−2 and those of the control samples were obtained as JNCMP1ECSA = 1.71 mA cm−2 and JNCPECSA = 2.56 mA cm−2. This finding provides further evidence for the high intrinsic catalytic activity of the NCMP electrocatalyst. Note, however, that while considering practical applicability of an electrocatalyst, the influence of ECSA-normalized current (JECSA) can be neglected since the overall outcome depends solely on the geometrically-normalized (Jgeo) activity. Thus, the trade-off between practical application and intrinsic activity is important to keep in mind when comparing different electrocatalysts. Combining the experimental results obtained from the LSV curves and the mechanisms frequently discussed in the literature, namely, Volmer–Heyrovsky and Volmer–Tafel, we tried to unravel the involved HER mechanism. The HER reactions under alkaline conditions can be expressed as:
Volmer reaction (bond formation step):
| | | M + H2O + e− → M–H* + OH− | (9) |
Heyrovsky reaction (bond cleavage step):
| | | M–H* + H2O + e− → M + H2 + OH− | (10) |
or Tafel reaction (bond cleavage step):
where, M is the active site of the catalyst. If the Volmer, Heyrovsky, or Tafel step is the rate-determining step (RDS), the Tafel slope will be 120, 40, or 30 mV dec
−1. A Tafel slope of 41.9 mV dec
−1 demonstrates that the Heyrovsky and Tafel reactions are likely equally effective. The lower the Tafel slope, the faster the reaction kinetics, and the more efficient the electrocatalyst will be. All the results above clearly support that NCMP is a high-performance and stable electrocatalyst for overall water splitting. The outstanding OER/HER performances of the NCMP catalyst can be attributed to the following characteristics: (1) tailoring the morphology of the catalyst so that mass transport into and out of the electroactive materials takes place readily. (2) Interfacial engineering that ensures exposure of abundant catalytically active sites toward the OER/HER processes. (3) A boosted catalytic activity arising from a strong synergistic effect. (4) The hierarchically porous architecture of the catalyst that facilitates mass transport, and thus enables full utilization of the active sites for catalysis. (5) Direct electrodeposition of the electrically conductive TMP on the substrate surface that makes and maintains intimate contact between them with no need for any binders or conductive additives. In a nutshell, benefiting from these advantageous characteristics makes the NCMP one of the most efficient water splitting electrocatalysts.
ORR activity.
Development of advanced ORR catalysts is crucial for the implementation of different electrochemical energy conversion and storage technologies including fuel cells, metal–air batteries, as well as a variety of chemical syntheses in which the electrogenerated superoxide ion, O2−, serves as a base, nucleophile, electron transfer shuttle, free radical, and so forth. We used CV to investigate the ORR activities of the NCMP catalysts as well as the Pt/C control sample in N2- and O2-saturated 0.1 M KOH electrolytes. As shown in Fig. S10 (ESI†), the Ni–Co–Mn–P series catalysts display a semi-rectangular CV curve in an N2-saturated electrolyte. However, after purging the solution with O2 for about 20 min, a significantly high intensity reduction peak appears at the potential of ∼0.72 V vs. RHE, demonstrating the relatively high and efficient ORR catalytic activities of the Ni–Co–Mn–P catalysts. Furthermore, the smaller the potential gap between the ORR and OER processes, the higher the efficiency of a bifunctional OER/ORR catalyst will be. In order to investigate the ORR activity of the NCMP catalyst as well as the NCMP1, NCP, and Pt/C control samples, the polarization curves of the catalysts were evaluated on a glassy carbon (GC) rotating disc electrode (RDE) in an O2-saturated 0.1 M KOH electrolyte. The LSV curves of all the catalysts at a rotation rate of 1600 rpm are shown in Fig. 5a. The extracted thermodynamic parameters, including the onset potential (Eonset) and the half-wave potential (E1/2) for the investigated catalysts, are summarized in Table S3 (ESI†). As can be seen, the NCMP catalyst displays excellent ORR activity with a more positive Eonset of 0.85 V and E1/2 of 0.76 V (vs. RHE), the characteristics of which approach that of Pt/C benchmark (Eonset = 0.91 V, E1/2 = 0.82 V). To obtain kinetic information about the ORR activity of the catalysts, LSV curves of the individual catalysts were recorded at various rotation speeds of 400, 625, 900, 1225, 1600, and 2025 rpm in an O2-saturated 0.1 M KOH solution at a scan rate of 5 mV s−1 (Fig. S11, ESI†). From the slope of the Koutecky–Levich plot, the number of electrons per oxygen molecule (n) was calculated to be 3.97, manifesting a nearly 4e−-dominated electron transfer pathway for the ORR process (the n values of the control catalysts are presented in Table S4, ESI†).
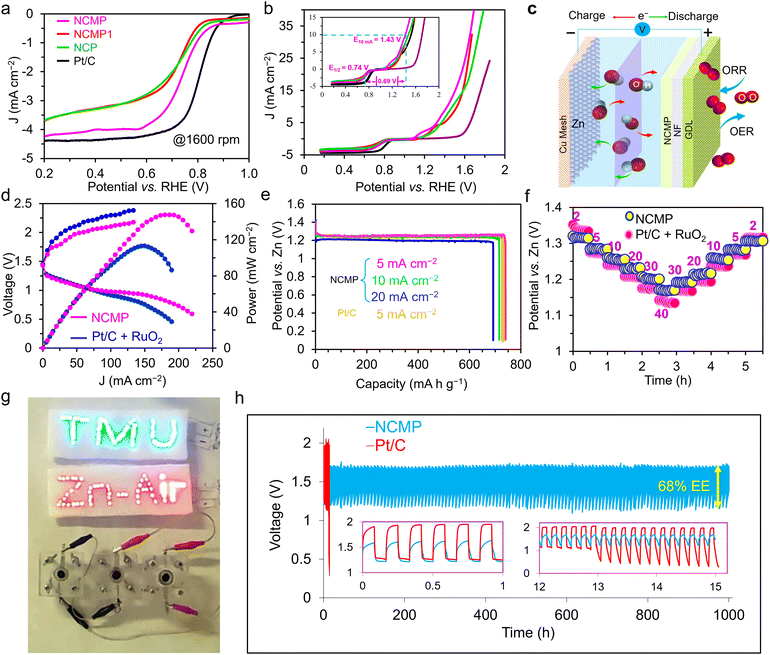 |
| | Fig. 5 Electrochemical performance of the rechargeable ZAB based on the NCMP air cathodes. (a) ORR polarization curves of the NCMP, NCMP1, and NCP catalysts as well as the Pt/C at a rotation speed of 1600 rpm. (b) ORR and OER polarization curves of the catalysts as well as the bare GC electrode. The inset shows the magnified view of the potential gap between the OER and ORR (ΔEOER–ORR). (c) Schematic illustration of a ZAB based on the Zn powder anode and the NCMP air cathode. (d) Charging and discharging profiles and power density curves of the NCMP and Pt/C air cathodes of a ZAB. (e) Discharge profiles of the NCMP and Pt/C air cathodes at different current densities of 5, 10, and 20 mA cm−2. (f) Rate capability study of a ZAB based on the NCMP and Pt/C air cathodes at different rates from 2 to 40 mA cm−2 and back to 2 mA cm−2. (g) Practical application of the three prototype ZABs connected in series in lighting up 60 red (1.7 V) and 60 blue (3.3 V) LEDs. (h) Long-term cycling performances at the charging/discharging rate of 5 mA cm−2 (5 min charge and 5 min discharge). The left inset shows the GCD cycles during the 1st hour, and the right inset displays the cycles at the end of life of the Pt/C-based cell. The 3E ORR studies are performed in a 0.1 M KOH solution and the electrolyte of the Zn–air battery is a ZnO-saturated 6.0 M KOH solution. | |
Fig. 5b shows that the potential gap between the EOER at a current density of 10 mA cm−2 and the half-wave potential of ORR at a rotation speed of 1600 rpm (ΔEOER–ORR) for the NCMP catalyst is ∼0.69 V which is lower than or at least comparable to the other TMP-based catalysts, and superior to those of the benchmark Pt/C–RuO2 based catalysts, used in metal–air batteries (Table S2, ESI†).
The outstanding catalytic performance of the NCMP catalyst can be ascribed to (1) the hierarchical porous morphology with sufficient diffusion channels that facilitate mass transport of the species into and out of the catalyst, (2) easy escape of the generated gaseous species from the pores with a negligible blocking effect, (3) excellent distribution of active sites throughout the catalyst, (4) utilization of a high fraction of the catalytic active sites, in spite of an ultra-high mass loading, because of the direct template-assisted electrodeposition of the catalyst with no need for any polymer binders or additives, and (5) the synergistic effects between the constituent metal elements.
3.4 Zn–air battery assembly
The bottleneck for the development of high-performance Zn–air batteries (ZABs) lies in that OER and ORR processes have different requirements for catalytically active sites. In virtue of the superior bifunctional catalytic activity of the NCMP catalyst towards both the OER/ORR, it can be an ideal catalyst in an integrated air electrode structure. A rechargeable ZAB was assembled using pure zinc powder as an anode and NCMP as a cathode (Fig. 5c). The redox reactions that occur in a ZAB are as follows:| | | Positive electrode: O2 + 2H2O + 4e− ⇄ 4OH− | (12) |
| | | Negative electrode: Zn + 4OH− ⇄ Zn(OH)42− + 2e− | (13) |
| | | Zn(OH)42− ⇄ ZnO + H2O + 2OH− | (14) |
| | | Overall: 2Zn + O2 ⇄ 2ZnO | (15) |
In these equations, the forward arrows denote the discharging steps, and the backward arrows signify the charging steps. We also assembled a control device using Pt/C + RuO2 as an air cathode. Fig. 5d shows charge–discharge profiles and power density plots of the NCMP and Pt/C + RuO2 cathodes. As can be seen, the NCMP-based ZAB delivers a maximum power density of 148 mW cm−2, which is higher than that of the Pt/C + RuO2-based ZAB (113 mW cm−2), and also exhibits a lower charge–discharge voltage gap (≈0.5 V at 5 mA cm−2) than that of the Pt/Ru-based device (≈0.65 V), demonstrating the improved efficiency and excellent rate capability of the NCMP-based battery.
Fig. 5e displays the specific capacities obtained at various discharge rates from 5 to 20 mA cm−2 for the NCMP-based ZAB. The ZAB exhibits a high voltage plateau of ≈1.25 V. The device also delivers an outstanding specific capacity of 740.3 mA h gZn−1 (based on the consumed Zn) at a rate of 5 mA cm−2 (∼90% of the theoretical capacity of Zn, 824 mA h g−1) with a corresponding specific energy of ∼932 W h kgZn−1, whereas the Pt/C + RuO2-based ZAB provides a specific capacity of 729.6 mA h gZn−1 and a specific energy of 912 W h kgZn−1 at the same rate of 5 mA cm−2 (Fig. 5e). The NCMP-based device also demonstrates a remarkable rate capability and exhibits an output voltage of 1.28 V at a rate of 5 mA cm−2 that diminishes to 1.17 V at 40 mA cm−2 and returns to 1.26 V (∼98.4% of the initial output voltage) upon switching to the rate of 5 mA cm−2 over the course of about 6 h operation (Fig. 5f). To demonstrate the practical applicability and outlooks of the NCMP as the air cathode catalyst of a ZAB, three devices were connected in series, which were capable of turning on 60 red (1.8 V) and 60 blue (3.2 V) light-emitting diodes (LEDs) and running a rotor robustly (Fig. 5g).
As an important criterion for practical application, the durability of the NCMP-based ZAB along with the Pt/C + RuO2-based control device was investigated by performing successive GCD measurements at a fixed rate of 5 mA cm−2 with a 10 min cycle time (Fig. 5h). Outstandingly, the NCMP-based ZAB displays long-term stability of over 6000 cycles that is about two orders of magnitude more durable than the Pt/C + RuO2 benchmark-based battery, thanks to the excellent chemical stability of the NCMP catalyst or the true catalysts thereof. As a result, the NCMP-based ZAB preserved ∼96% of the initial energy even after 1000 h of cycling, whereas the Pt/C + RuO2-based ZAB quickly lost its catalytic performance, thus, its energy efficiency deteriorated severely upon cycling. Note, however, that the electrolyte was occasionally injected into the cell through a hole on top of the cell to compensate for the evaporation of the electrolyte. Given that the O-ring positioned on the GDL side of the electrode is sufficiently large to prevent a watertight seal, it may require cleaning after prolonged operation.
3.5 Theoretical studies
The OER/ORR mechanism begins with the adsorption of H2O molecules on the active site of the catalyst and its dissociation into OH− and H+. OER and ORR involve four proton-coupled electron transfer steps (see ESI† for the equations). In an alkaline medium, the 4e− reaction pathway typically begins with the adsorption of OH− at the active site to form M–OH* species. In the second step, by giving up a proton and an electron, M–OH* is transformed into M–O*. Third, the M–O* reacts with another OH− molecule to produce M–OOH*. Finally, via another proton-coupled electron transfer step, M–OOH* dissociates into O2 and H+. Note that, the ORR process can be considered as the reverse of the OER.
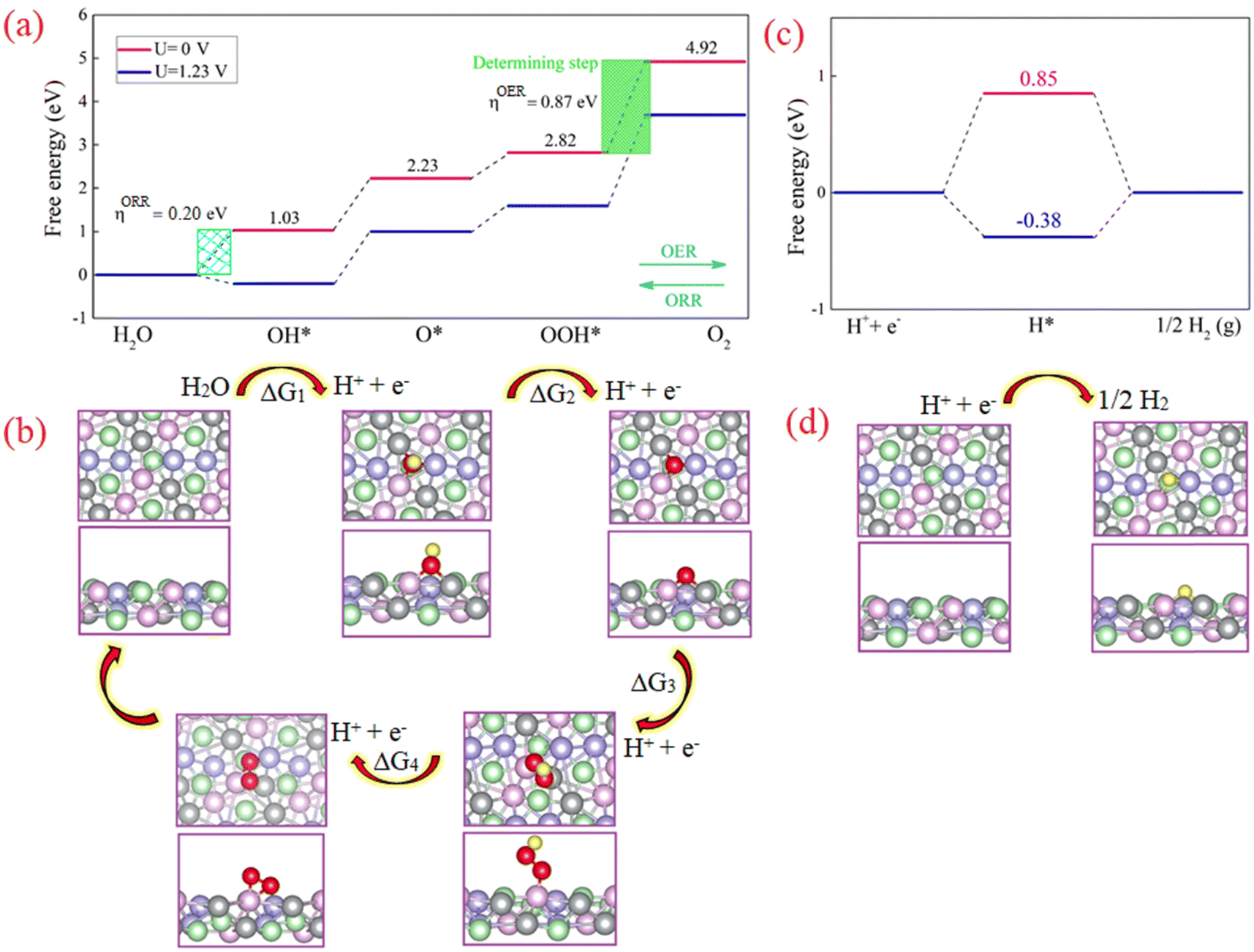
Density functional theory (DFT) calculations were performed to study the active sites in the NCMP electrocatalyst. The results indicate that the active site of the catalyst is likely a trimetallic species formed by Mn, Ni, and Co (see ESI,† Fig. S12). The reaction pathway and the corresponding energetically favorable structures for OER/ORR reactions are shown in Fig. 6a and b. The formation of O2 molecules is the most difficult step and has the largest free energy difference, thus, this is the rate-determining step. Under U = 1.23 V the adsorption free energy of OH is negative (ΔG1 = −0.20 eV) due to the strong interaction between OH− and the active sites. However, the adsorption free energies of the other steps are positive, indicating that the catalyst's surface remains clean during the ORR/OER. The calculated overpotential for the OER (ORR) is 0.87 V (0.20 V). The obtained ηOER/ηORR is relatively small, which means a small amount of energy is required (released) during the OER (ORR) process. Also, all ORR steps are downhill in the free energy diagram (at U = 0 V). The latter illustrates that the four elementary reactions in the ORR proceed spontaneously.
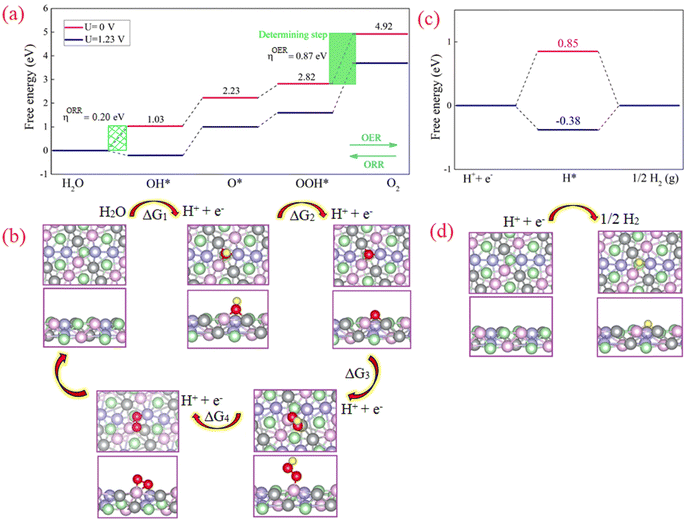 |
| | Fig. 6 DFT Studies of the NCMP catalyst. (a) and (c) The calculated free energy diagrams along with (b) and (d) the optimized structure and the microkinetic modeling of the electrocatalytic reaction processes for the (a) and (b) OER, and (c) and (d) HER on the surface of the NCMP catalyst. Color code: Mn (pink), Co (violet), Ni (gray), P (green), O (red), H (yellow). | |
Subsequently, the four produced electron–hole pairs (H+ + e−) enter the HER process. Simplifying the HER process, the reaction begins with the adsorption of H+ on the active site (M–H*). Then M–H* interacts with another electron–hole pair to generate a molecule of H2. The optimized structure and the free energy diagram of the HER are illustrated in Fig. 6c. The calculated ΔG values for proceeding with the HER process for the external potential values (ϕ) of 0.00 and 1.23 V are 0.85 and −0.38 eV, respectively. The latter indicates that without applying a negative external potential, the adsorption of H on the catalyst surface is weak. However, by applying a negative potential, hydrogen adsorption on the catalyst surface proceeds favorably.
4 Conclusions
In summary, we have designed and successfully synthesized a transition metal based trimetallic Ni–Co–Mn phosphide on nickel foam via a facile and fast reductive electrosynthesis approach, in which Pluronic P123 served as a structure directing agent. The resulting transition metal phosphide displays excellent trifunctional electrocatalytic activity for HER, OER, and ORR with superior practical applicability for overall water splitting and fabricating high performance and durable Zn–air batteries. The NCMP catalyst exhibits low overpotentials of 100 and 218 mV for HER and OER in 1.0 M KOH to reach a current density of 10 and 50 mA cm−2, respectively, along with a very low half-wave potential of 0.74 V for ORR. The fabricated water electrolyzer using NCMP as both the negative and positive electrodes, displays a cell voltage of 1.53 V to achieve a current density of 10 mA cm−2. Moreover, a zinc–air battery based on an NCMP air cathode displays a high open circuit voltage of 1.42 V, an excellent power density of 148 mW cm−2, an outstanding specific energy of ∼932 W h kgZn−1, along with a super-long life of over 1000 h (about two orders of magnitude more durable than the device based on the Pt/C + RuO2 air cathode). All these exceptional characteristics are obtained from the electrode with an ultra-high mass loading of 22.6 mg cm−2. The superior energy conversion and storage performances of the NCMP catalyst can be attributed to the following characteristics: (1) a firm connection of the NCMP catalyst to the Ni foam substrate owing to the direct electrodeposition of the catalyst, (2) the hierarchically porous architecture of the electrocatalyst that not only enables the full utilization of active sites towards electrocatalysis, but also facilitates mass transport into and out of the pores along with the escape of the generated gaseous species (O2 and H2), (3) synergistic effects between the constituent elements of the TMP that individually are among the highest performing non-precious metal catalysts residing near the top of the so-called HER/OER/ORR volcano plots, (4) fast electron transport through the TMP scaffold, and (5) uniform distribution of the catalytically active sites that guarantees the long-term stability and durability of the catalyst for practical applications. This study paves the way to unleash the full potential of multi-metallic TMPs for use in different realms of energy conversion and storage where non-precious transition metals are likely to make a difference.
Author contributions
Conceptualization and designing the experiments, N. K., A. N., and M. R.; data curation and formal analysis, N. K., M. M., and A. N.; writing – original draft, N. K. and A. N.; software, theoretical studies, formal analysis, N. H. and M. N.-A.; visualization and investigation, M. F. E. and N. B. M.; writing – review & editing, resources, and supervision, X. X., Y. Z., R. B. K. and M. F. M.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was made possible through financial support from the Tarbiat Modares University Research Council as well as the Research Core of Tarbiat Modares University, grant number IG-39802 (M. F. M.), the University of California Climate Action Seed Grant R02CP7008 (R. B. K.), and the Dr Myung Ki Hong Endowed Chair in Materials Innovation at UCLA (R. B. K.).
Notes and references
- S. Anantharaj, S. Noda, V. R. Jothi, S. Yi, M. Driess and P. W. Menezes, Angew. Chem., Int. Ed., 2021, 60, 18981–19006 CrossRef CAS PubMed.
- Q. Wang, Q. Feng, Y. Lei, S. Tang, L. Xu, Y. Xiong, G. Fang, Y. Wang, P. Yang, J. Liu, W. Liu and X. Xiong, Nat. Commun., 2022, 13, 3689 CrossRef CAS PubMed.
- F. Shahbazi Farahani, M. S. Rahmanifar, A. Noori, M. F. El-Kady, N. Hassani, M. Neek-Amal, R. B. Kaner and M. F. Mousavi, J. Am. Chem. Soc., 2022, 144, 3411–3428 CrossRef CAS PubMed.
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CrossRef CAS PubMed.
- A. Hameed, M. Batool, Z. Liu, M. A. Nadeem and R. Jin, ACS Energy Lett., 2022, 7, 3311–3328 CrossRef CAS.
- M. Moloudi, A. Noori, M. S. Rahmanifar, Y. Shabangoli, M. F. El-Kady, N. B. Mohamed, R. B. Kaner and M. F. Mousavi, Adv. Energy Mater., 2023, 13, 2203002 CrossRef CAS.
- Y. Wang, Y. Nian, A. N. Biswas, W. Li, Y. Han and J. G. Chen, Adv. Energy Mater., 2021, 11, 2002967 CrossRef CAS.
- A. B. Jorge, R. Jervis, A. P. Periasamy, M. Qiao, J. Feng, L. N. Tran and M. M. Titirici, Adv. Energy Mater., 2020, 10, 1902494 CrossRef CAS.
- Y. Li, Z. Dong and L. Jiao, Adv. Energy Mater., 2020, 10, 1902104 CrossRef CAS.
- P. M. Bodhankar, P. B. Sarawade, P. Kumar, A. Vinu, A. P. Kulkarni, C. D. Lokhande and D. S. Dhawale, Small, 2022, 18, 2107572 CrossRef CAS PubMed.
- X. Li and J. Wang, Adv. Mater. Interfaces, 2020, 7, 2000676 CrossRef CAS.
- J.-T. Ren, L. Chen, L. Wang, X.-L. Song, Q.-H. Kong and Z.-Y. Yuan, J. Mater. Chem. A, 2023, 11, 2899–2909 RSC.
- B. Liu, X. Lan, Q. Zhong and T. Wang, ACS Catal., 2024, 14, 757–775 CrossRef CAS.
- R. Anne Acedera, A. Theresse Dumlao, D. J. Donn Matienzo, M. Divinagracia, J. Anne Paraggua, P.-Y. Abel Chuang and J. Ocon, J. Energy Chem., 2024, 89, 646–669 CrossRef CAS.
- L. K. Putri, B.-J. Ng, R. Y. Z. Yeo, W.-J. Ong, A. R. Mohamed and S.-P. Chai, Chem. Eng. J., 2023, 461, 141845 CrossRef CAS.
- A. Parra-Puerto, K. L. Ng, K. Fahy, A. E. Goode, M. P. Ryan and A. Kucernak, ACS Catal., 2019, 9, 11515–11529 CrossRef CAS.
- N. Zhang, I. Amorim and L. Liu, Nanotechnology, 2022, 33, 432004 CrossRef CAS PubMed.
- R. Sun, X. Huang, J. Jiang, W. Xu, S. Zhou, Y. Wei, M. Li, Y. Chen and S. Han, Dalton Trans., 2022, 51, 15205–15226 RSC.
- K. Xu, H. Cheng, H. Lv, J. Wang, L. Liu, S. Liu, X. Wu, W. Chu, C. Wu and Y. Xie, Adv. Mater., 2018, 30, 1703322 CrossRef PubMed.
- M. F. Delley, Z. Wu, M. E. Mundy, D. Ung, B. M. Cossairt, H. Wang and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 15390–15402 CrossRef CAS PubMed.
- B. Ma, Z. Yang, Y. Chen and Z. Yuan, Nano Res., 2019, 12, 375–380 CrossRef CAS.
- S. Geng, F. Tian, M. Li, X. Guo, Y. Yu, W. Yang and Y. Hou, J. Mater. Chem. A, 2021, 9, 8561–8567 RSC.
- Y. Liu, W. Gong, S. Yao, Y. Liang, Y. Yang, T. Yu, C. Yuan and Y. Yang, Inorg. Chem., 2022, 61, 14201–14210 CrossRef CAS PubMed.
- M. Xie, M. Zhou, Y. Zhang, C. Du, J. Chen and L. Wan, J. Colloid Interface Sci., 2022, 608, 79–89 CrossRef CAS PubMed.
- R. Deng, X. Li, M. Wang and Q. Zhang, Appl. Surf. Sci., 2023, 616, 156456 CrossRef CAS.
- Y. Men, P. Li, J. Zhou, S. Chen and W. Luo, Cell Rep. Phys. Sci., 2020, 1, 100136 CrossRef CAS.
- F. Guo, T. J. Macdonald, A. J. Sobrido, L. Liu, J. Feng and G. He, Adv. Sci., 2023, 10, e2301098 CrossRef PubMed.
- S. S. Shinde, J. Y. Jung, N. K. Wagh, C. H. Lee, D.-H. Kim, S.-H. Kim, S. U. Lee and J.-H. Lee, Nat. Energy, 2021, 6, 592–604 CrossRef CAS.
- S. S. Shinde, N. K. Wagh, C. H. Lee, D. H. Kim, S. H. Kim, H. D. Um, S. U. Lee and J. H. Lee, Adv. Mater., 2023, 35, e2303509 CrossRef PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. De Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- P. A. Ignatiev, N. N. Negulyaev, D. I. Bazhanov and V. S. Stepanyuk, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 235123 CrossRef.
- M. Capdevila-Cortada, Z. Łodziana and N. López, ACS Catal., 2016, 6, 8370–8379 CrossRef CAS.
- F. H. Saadi, A. I. Carim, E. Verlage, J. C. Hemminger, N. S. Lewis and M. P. Soriaga, J. Phys. Chem. C, 2014, 118, 29294–29300 CrossRef CAS.
- C. Wang, Y. Wu, Z. Zhou, J. Wang, S. Pei and S. Liu, Int. J. Hydrogen Energy, 2022, 47, 40849–40859 CrossRef CAS.
- L. Chai, S. Liu, S. Pei and C. Wang, Chem. Eng. J., 2021, 420, 129686 CrossRef CAS.
- F. Guo, T. J. Macdonald, A. J. Sobrido, L. Liu, J. Feng and G. He, Adv. Sci., 2023, 10, 2301098 CrossRef CAS PubMed.
- M. Nazari, A. Noori, M. S. Rahmanifar, M. F. El-Kady, N. Hassani, M. Neek-Amal, R. B. Kaner and M. F. Mousavi, ACS Appl. Mater. Interfaces, 2022, 14, 50900–50912 CrossRef CAS PubMed.
- S. Li, P. Xu, M. K. Aslam, C. Chen, A. Rashid, G. Wang, L. Zhang and B. Mao, Energy Storage Mater., 2020, 27, 51–60 CrossRef.
- H. Yousefzadeh, A. Noori, M. S. Rahmanifar, N. Hassani, M. Neek-Amal, M. F. El-Kady, A. Vinu, R. B. Kaner and M. F. Mousavi, Adv. Energy Mater., 2023, 13, 2302137 CrossRef CAS.
- S. Cobo, J. Heidkamp, P.-A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS PubMed.
- Y. Shabangoli, M. S. Rahmanifar, M. F. El-Kady, A. Noori, M. F. Mousavi and R. B. Kaner, Energy Storage Mater., 2018, 11, 282–293 CrossRef.
- G. Zhu, L. Wang, H. Lin, L. Ma, P. Zhao, Y. Hu, T. Chen, R. Chen, Y. Wang and Z. Tie, Adv. Funct. Mater., 2018, 28, 1800003 CrossRef.
- N. Khodayar, A. Noori, M. S. Rahmanifar, Y. Shabangoli, A. Baghervand, M. F. El-Kady, N. Hassani, X. Chang, M. Neek-Amal, R. B. Kaner and M. F. Mousavi, Adv. Energy Mater., 2022, 12, 2202784 CrossRef CAS.
- Q. Yao, X. Zhou, S. Xiao, J. Chen, I. A. Abdelhafeez, Z. Yu, H. Chu and Y. Zhang, Water Res., 2019, 165, 114930 CrossRef CAS PubMed.
- A. Adam, M. H. Suliman, M. N. Siddiqui, Z. H. Yamani, B. Merzougui and M. Qamar, ACS Appl. Mater. Interfaces, 2018, 10, 29407–29416 CrossRef CAS PubMed.
- P. Alexandridis, J. F. Holzwarth and T. A. Hatton, Macromolecules, 1994, 27, 2414–2425 CrossRef CAS.
- C. Li, W. Yang, W. He, X. Zhang and J. Zhu, Energy Storage Mater., 2021, 43, 1–19 CrossRef.
- H. Chen, X. Liang, Y. Liu, X. Ai, T. Asefa and X. Zou, Adv. Mater., 2020, 32, 2002435 CrossRef PubMed.
- M. A. Isaacs, N. Robinson, B. Barbero, L. J. Durndell, J. C. Manayil, C. M. A. Parlett, C. D'Agostino, K. Wilson and A. F. Lee, J. Mater. Chem. A, 2019, 7, 11814–11825 RSC.
- Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC.
- L.-H. Chen, M.-H. Sun, Z. Wang, W. Yang, Z. Xie and B.-L. Su, Chem. Rev., 2020, 120, 11194–11294 CrossRef CAS PubMed.
- H. Li, H. J. Kim, T. Garcia, G. Park, Y. Ding, M. Liu, J. An and M. H. Lee, ACS Catal., 2023, 13, 11172–11181 CrossRef CAS PubMed.
- N. Wang, R. Mei, X. Lin, L. Chen, T. Yang, Q. Liu and Z. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 29195–29203 CrossRef CAS PubMed.
- B. Wang, X. Zhu, X. Pei, W. Liu, Y. Leng, X. Yu, C. Wang, L. Hu, Q. Su, C. Wu, Y. Yao, Z. Lin and Z. Zou, J. Am. Chem. Soc., 2023, 145, 13788–13795 CrossRef CAS PubMed.
- D. Kiuchi, H. Matsushima, Y. Fukunaka and K. Kuribayashi, J. Electrochem. Soc., 2006, 153, E138 CrossRef CAS.
- S. Park, D. Lohse, D. Krug and M. T. M. Koper, Electrochim. Acta, 2024, 485, 144084 CrossRef CAS.
- S. Yuan, C. Zhao, X. Cai, L. An, S. Shen, X. Yan and J. Zhang, Prog. Energy Combust. Sci., 2023, 96, 101075 CrossRef.
- H. Liu, J. Guan, S. Yang, Y. Yu, R. Shao, Z. Zhang, M. Dou, F. Wang and Q. Xu, Adv. Mater., 2020, 32, e2003649 CrossRef PubMed.
- X. Li, L. Xiao, L. Zhou, Q. Xu, J. Weng, J. Xu and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 21106–21113 CrossRef CAS PubMed.
- Y. Li, Y. Liu, Q. Qian, G. Wang and G. Zhang, Energy Storage Mater., 2020, 28, 27–36 CrossRef.
- L. Chai, Z. Hu, X. Wang, Y. Xu, L. Zhang, T. T. Li, Y. Hu, J. Qian and S. Huang, Adv. Sci., 2020, 7, 1903195 CrossRef CAS PubMed.
- L. Ji, J. Wang, X. Teng, T. J. Meyer and Z. Chen, ACS Catal., 2020, 10, 412–419 CrossRef CAS.
- L. Yan, B. Zhang, S. Wu and J. Yu, J. Mater. Chem. A, 2020, 8, 14234–14242 RSC.
- X. Luo, P. Ji, P. Wang, R. Cheng, D. Chen, C. Lin, J. Zhang, J. He, Z. Shi, N. Li, S. Xiao and S. Mu, Adv. Energy Mater., 2020, 10, 1903891 CrossRef CAS.
- C. Lin, D. Wang, H. Jin, P. Wang, D. Chen, B. Liu and S. Mu, J. Mater. Chem. A, 2020, 8, 4570–4578 RSC.
- H. Sun, Y. Min, W. Yang, Y. Lian, L. Lin, K. Feng, Z. Deng, M. Chen, J. Zhong, L. Xu and Y. Peng, ACS Catal., 2019, 9, 8882–8892 CrossRef CAS.
- B. R. Wygant, K. Kawashima and C. B. Mullins, ACS Energy Lett., 2018, 3, 2956–2966 CrossRef CAS.
- X. Hu, R. Wang, W. Feng, C. Xu and Z. Wei, J. Energy Chem., 2023, 81, 167–191 CrossRef CAS.
- L.-M. Cao, J. Zhang, L.-W. Ding, Z.-Y. Du and C.-T. He, J. Energy Chem., 2022, 68, 494–520 CrossRef CAS.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS.
- A. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2017, 8, 144–152 CrossRef CAS PubMed.
- Y. Zhang, L. Gao, E. J. M. Hensen and J. P. Hofmann, ACS Energy Lett., 2018, 3, 1360–1365 CrossRef CAS PubMed.
- Q. Fu, X. Wang, J. Han, J. Zhong, T. Zhang, T. Yao, C. Xu, T. Gao, S. Xi, C. Liang, L. Xu, P. Xu and B. Song, Angew. Chem., Int. Ed., 2021, 60, 259–267 CrossRef CAS PubMed.
- T. N. Batugedara and S. L. Brock, Chem. Mater., 2022, 34, 4414 CrossRef CAS.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251–6254 CrossRef CAS PubMed.
- Y. Yang, H. Yao, Z. Yu, S. M. Islam, H. He, M. Yuan, Y. Yue, K. Xu, W. Hao and G. Sun, J. Am. Chem. Soc., 2019, 141, 10417–10430 CrossRef CAS PubMed.
- X. Zhao, P. Pachfule, S. Li, J. R. J. Simke, J. Schmidt and A. Thomas, Angew. Chem., 2018, 130, 9059–9064 CrossRef.
- Z. Xue, Y. Li, Y. Zhang, W. Geng, B. Jia, J. Tang, S. Bao, H. P. Wang, Y. Fan and Z. W. Wei, Adv. Energy Mater., 2018, 8, 1801564 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; characterization of the samples; electrochemical studies in 3E and 2E cell setups; comparison tables, calculation formula, optimization steps, supplementary videos. See DOI: https://doi.org/10.1039/d4ee00042k |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Mohammad S.
Rahmanifar
b,
Masumeh
Moloudi
a,
Nasim
Hassani
a,
Mohammad S.
Rahmanifar
b,
Masumeh
Moloudi
a,
Nasim
Hassani