
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeminal-dithiol-based precursors for reactive sulfur species†
Shi
Xu
 ,
Geat
Ramush
,
Iris J.
Yang
,
Eshani
Das
,
Meg
Shieh
and
Ming
Xian
*
,
Geat
Ramush
,
Iris J.
Yang
,
Eshani
Das
,
Meg
Shieh
and
Ming
Xian
*
Department of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: ming_xian@brown.edu
First published on 30th April 2024
Abstract
Caged gem-dithiols have been developed as the donors of reactive sulfur species (RSS), but the chemistry of free gem-dithiols as RSS donors has not been well understood. Herein, we report the study of a free gem-dithiol, 1,3-diphenylpropane-2,2-dithiol, as the precursor for several RSS. It releases H2S under physiological conditions and can be converted to a mono-S-nitrosothiol, which serves as a NO donor. Furthermore, it can be converted to 3,3-dibenzyldithiirane, which is an active sulfur transfer reagent and can induce S-persulfidation.
Reactive sulfur species (RSS) are important regulating molecules in biological systems. These include thiols (RSH), hydrogen sulfide (H2S), persulfides (RSSH), polysulfides (RSSnSR), and post-translational modification products on protein thiols such as S-nitrosothiol (RSNO) and sulfenic acid (RSOH).1–3 Many RSS are fleeting species that exist in low concentrations, while exhibiting a variety of biological functions such as antioxidation and vasodilation. It is therefore important to develop chemicals (e.g. donors) that can controllably deliver RSS under physiological conditions. These compounds are useful for understanding the properties and functions of RSS, and have therapeutic potentials. In the past several years, many such donors have been developed, including molecules releasing H2S, H2Sn, and RSSH.4–6 These compounds have greatly advanced the understanding of RSS.
Among the reported H2S donors, gem-dithiol-based compounds are particularly interesting. Free gem-dithiols are normally unstable under physiological conditions and quickly decompose to release H2S. Therefore, bio-compatible protecting groups have been introduced to cage the –SH groups of gem-dithiols to create the desired H2S donors (Scheme 1).7–9 Interestingly, some free gem-dithiol compounds were synthesized and characterized back in the 1950's.10 We wondered if these relatively stable gem-dithiols could be directly used as RSS donors. Herein, we report the study of a representative free gem-dithiol, 1,3-diphenylpropane-2,2-dithiol (1), and its derivatives. Their preparation, stability, reactivity, and ability to serve as RSS donors were studied.
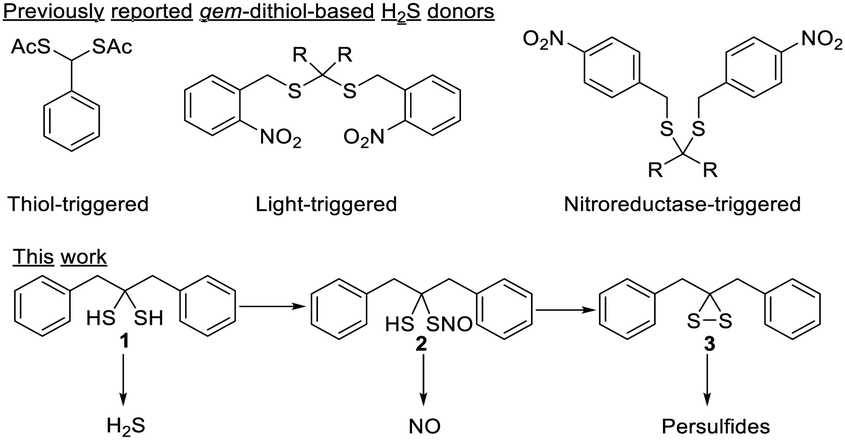 | ||
| Scheme 1 gem-dithiol-based RSS donors. | ||
While several gem-dithiols (such as methanedithiol, 2,2-propanetithiol, cyclohexanedithiol, phenylmethanedithiol) have been reported before, they are unstable and volatile liquids with pungent smells, making them unsuitable for our studies. 1,3-Diphenylpropane-2,2-dithiol (1), first reported by Carmack in 1959,11 was prepared via treating ketone 4 with H2S (g) and HCl, with thioketone 5 as an intermediate (Scheme 2). This method allowed us to obtain 1 in the gram scale with 65% yield. Compound 1 is a colorless solid with a faint smell of mercaptan. It was found to be stable at room temperature in its neat form or in organic solvents, with no NMR-observable decomposition after storage for days. In water-containing solutions (for example 20% D2O/CD3CN), 1 also exhibited good stability, with no decomposition over 12 hours (Fig. S1, ESI†). However, the decomposition of 1 was noted in buffer- containing solutions (such as acetonitrile with 20% pH 7.4 PBS buffer). NMR studies demonstrated that thioketone 5 (in its enethiol form 6) was the main decomposition product (Fig. S1, ESI†).
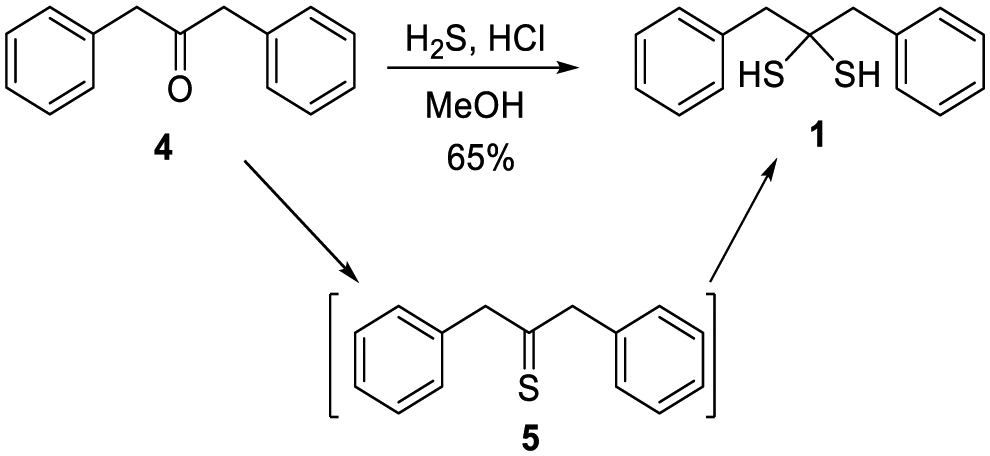 | ||
| Scheme 2 The synthesis of free gem-dithiol 1. | ||
The decomposition of compound 1 in buffer solutions prompted us to examine its ability to release H2S. A Unisense H2S microsensor was used to measure H2S production from the solutions of 1 (50 μM) in different buffers (pH 5, 6, 7, 8). Fig. 1a shows that 1 released H2S in a pH-dependent manner. At pHs 6 and 7, ∼50 μM H2S was produced after 150 min, while only ∼30 μM H2S was produced at pH 5. These results suggest that under these pHs, roughly one equivalent of H2S could be produced from 1, while slightly more than one equivalent of H2S (∼68 μM) was produced at pH 8.
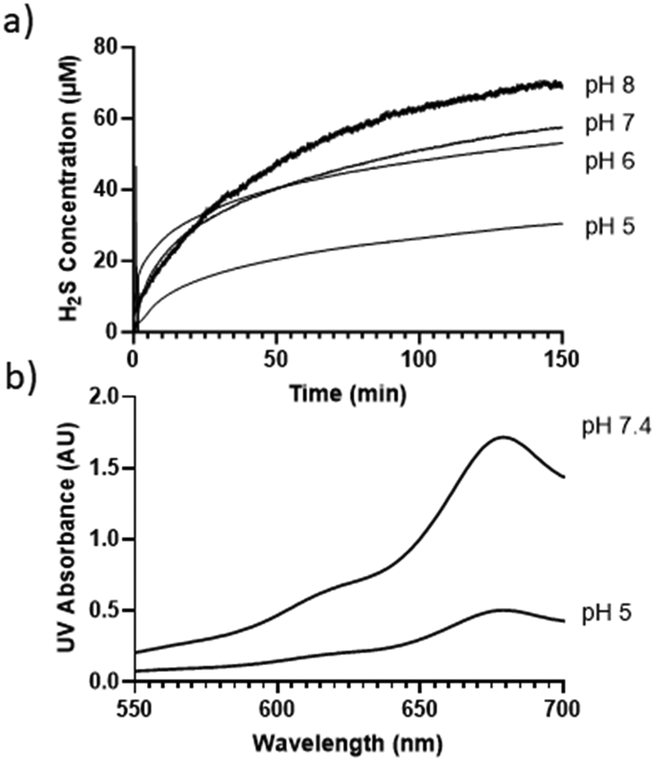 | ||
| Fig. 1 (a) H2S release profiles of 1 (50 μM) in different buffers. (b) Methylene blue UV-vis absorbance spectra of H2S released from 1 as measured by the gas trapping method (1.5 mM 1 was incubated in buffers for 3 hours. H2S gas was trapped by zinc acetate and measured by the methylene blue method). | ||
To further validate H2S release from 1, a gas trapping experiment was performed. Briefly, in sealed vials, the solutions of 1 (1.5 mM) in different buffers (pH 5 and 7.4) were placed with a trapping vial containing 10% zinc acetate solution under room temperature for 3 hours. H2S generated from 1 would diffuse into the trapping vial to form zinc sulfide, which was then treated with the methylene blue cocktail. The concentration of H2S was analyzed by UV-vis absorbance at 670 nm. As shown in Fig. 1b, a strong absorbance peak at 670 nm was observed, demonstrating the generation of H2S from 1. These results suggested the following mechanism: in aqueous solutions, 1 undergoes elimination to produce H2S (likely in the form of HS−) and thioketone 5 (the major form is enethiol 6) (Scheme 3). This occurs both in acidic and basic pHs but basic conditions might be more efficient. Aliphatic thioketones are known to be unstable and can form complex decomposition products including oligomers and ketones.12 Nonetheless, it is possible for 5 to undergo hydrolysis and release H2S, forming ketone 4 as a product. Indeed, 4 was obtained when 1 or 6 was incubated in buffers, with 19% and 18% yields, respectively. This finding suggests that thioketones like 5 could release H2S in buffers, but the efficiency is low.
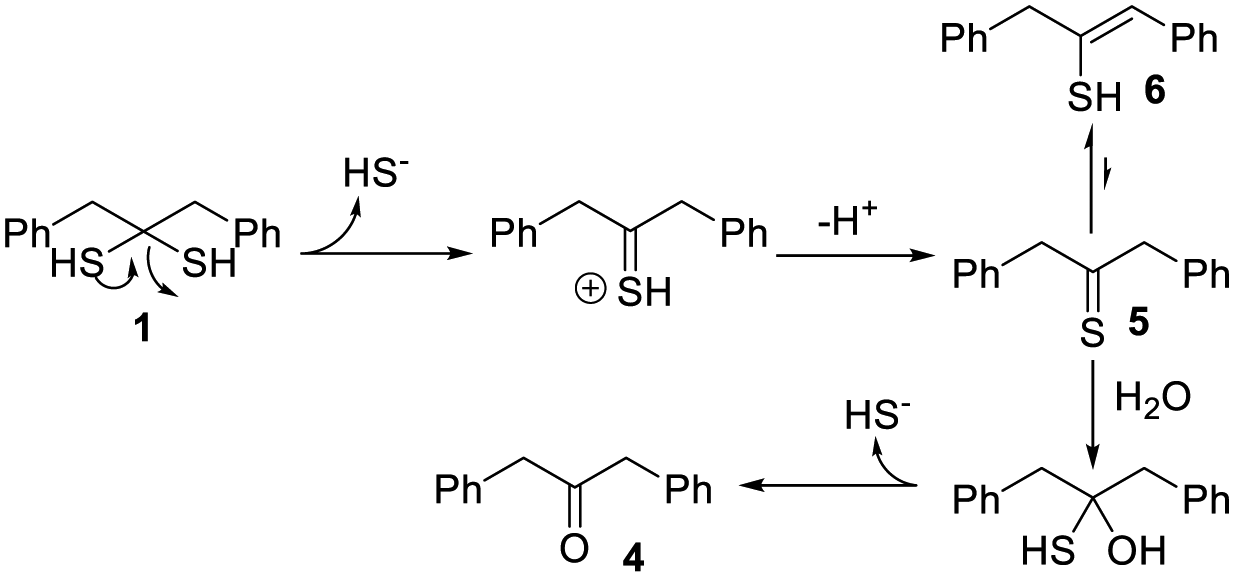 | ||
| Scheme 3 The mechanism of H2S release from 1. | ||
Some thiocarbonyl compounds like arylthioamides are known H2S donors, but their H2S releasing mechanisms are still unclear.13 The interactions between thiocarbonyls with certain biomolecules (such as cysteine and GSH) may be responsible for their H2S release. We next wondered if 5/6 could release more H2S under biomimicking environments. Freshly prepared solutions of 6 (10 mM) were incubated with cysteine (20 mM), HeLa cell lysate, or human plasma for 12 hours. The H2S released was measured by the H2S trapping method. As shown in Fig. 2, a notable increase of UV-vis absorbance was detected at 670 nm under these conditions. In contrast, only very weak H2S production was detected when 6 was incubated in buffer alone. These results indicated biologically-relevant conditions could promote H2S release from 6. However, it should be noted that even under these biologically-relevant conditions, the H2S released from 6 was still significantly less than that of the positive control (5 mM Na2S), suggesting that it may not be an effective H2S donor. This adds to the speculation that the bioactivities of thiocarbonyl-based H2S donors may not come from H2S, but rather from the thiocarbonyl group itself.
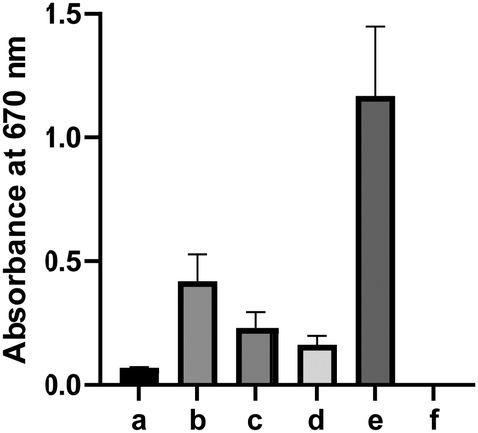 | ||
| Fig. 2 H2S release from 6 measured by the gas trapping method. 6 (10 mM) or Na2S (5 mM) was incubated in PBS buffer (pH 7.4) under various conditions. H2S gas was trapped by zinc acetate and measured by the methylene blue method. (a) 6 only; (b) 6 with 20% HeLa cell lysate; (c) 6 with 10% plasma; (d) 6 with 20 mM cysteine; (e) Na2S only; (f) blank. | ||
S-Nitrosothiols (SNOs) are one of the key molecules involved in nitric oxide (NO) signal transduction and possess unique chemical and biological properties.14 Since we had gem-dithiol 1 in hand, we tested its nitrosation in the hopes of preparing the gem-bis-SNO compound. Interestingly, we were only able to access the mono-SNO compound 2 (in 90% purity, via nitrosation of 1 with organic nitrites at low temperature, −20 °C). SNO 2 showed the characteristic green color of tertiary SNO compounds with an obvious UV-vis absorbance peak at 618 nm (Fig. S2 (ESI†), ε = 26 M−1cm−1 in CHCl3). It also exhibited decent stability in solutions as determined by clean NMR spectra (Fig. 3). Attempts to further nitrosate 2 were unsuccessful, and this may be explained by the hydrogen bonding between the –SH and NO group of 2. SH-based hydrogen bonding is known to stabilize certain structures.15 DFT calculations were carried out to estimate the H-bond energy (see ESI†). Nevertheless, the solution of 2 could degrade in a few hours to form dithiirane 3.
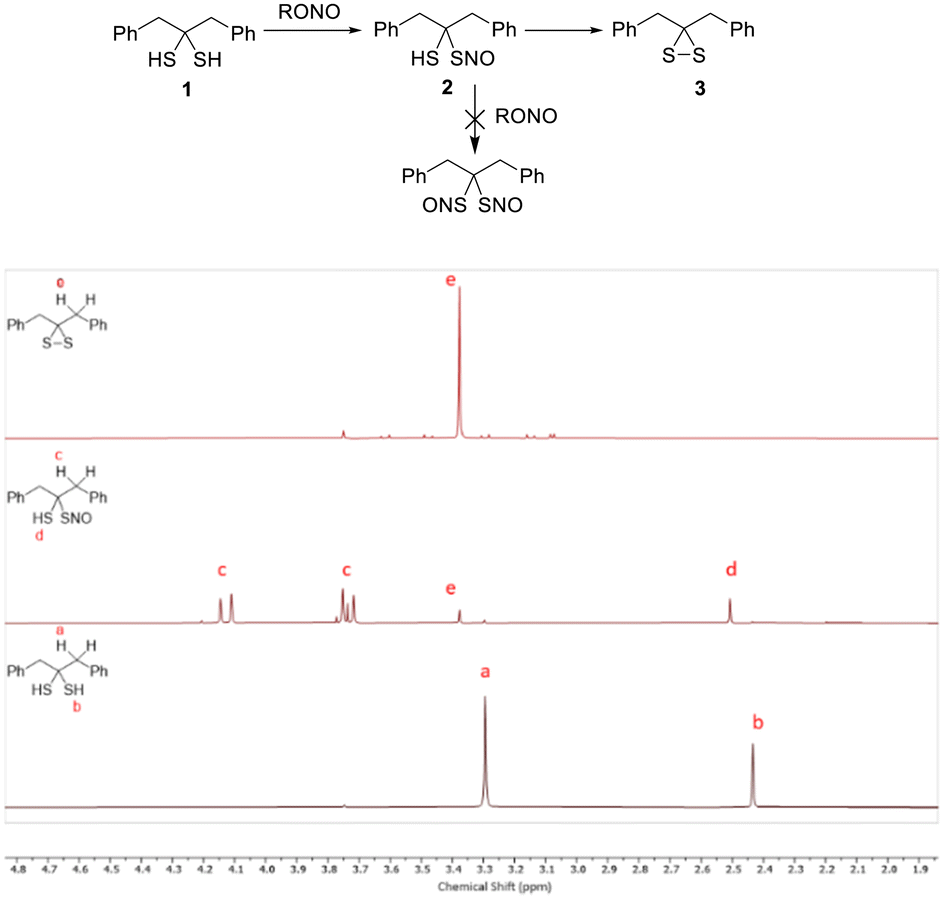 | ||
| Fig. 3 Nitrosation of 1 and 1H-NMR spectra of compounds 1, 2, and 3. | ||
With the relatively stable SNO 2 in hand, we wondered if its conversion to 3 could release NO. To this end, an indirect NO trapping and detection experiment was performed. The in situ generated 2 was incubated in buffers under different pHs in a sealed 20 mL vial. NO generated in the solution was diffused into an Eppendorf vial containing NO red, an NO-specific fluorescent probe.16 As shown in Fig. 4, the formation of NO was noted in a pH-dependent manner in all conditions. In CHCl3 and in more acidic buffers (pH 3 and 5), NO released from 2 was comparable to the positive control, a pyrrolidine-based NONOate. Under pH 7.4, 9, and 10, less NO production from 2 was observed. The formation of NO from 2 suggested a homolytic S–N bond cleavage process with a thiyl radical intermediate. This was confirmed by TEMPO-9-Ac, a thiyl radical fluorescent probe.17 When 2 was incubated with TEMPO-9-AC in 10% THF/PBS, significant fluorescence turn on at 432 nm was observed (Fig. S3, ESI†) while other more stable SNOs (trityl SNO and GSNO) gave much weaker fluorescence. It is interesting to note that NO formation from 2 at higher pH (10) decreased. The results suggested that the conversion of 2 to 3 could also proceed via an anionic pathway to form nitroxyl (HNO). We attempted to trap HNO using GSH,18 but were unable to obtain the corresponding sulfinamide product. Nevertheless, HNO generation was still possible. These results suggest 2 could be considered as an interesting NO (or HNO) donor.
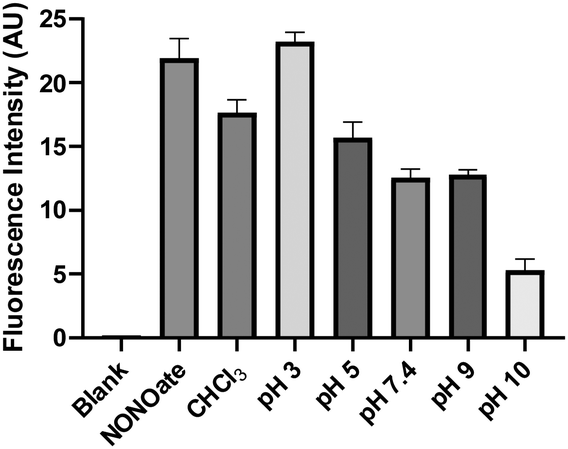 | ||
| Fig. 4 NO release from 2 measured by NO red. 6.4 mM 2 was incubated with CHCl3 or PBS buffers (containing 10% THF) for 1.5 h. NO was then trapped by NO red (40 μM), and its fluorescence (λem 593 nm, λex 540 nm) was measured. | ||
Dithiiranes are the smallest cyclic disulfides.19 The S–S bond of dithiiranes is forced to adopt a “cisoid” geometry, resulting in maximum lone pair interactions and a weak S–S bond.20 Due to this property, most dithiiranes are unstable and can only exist as reaction intermediates.21,22 A few isolable dithiiranes23–25 have been reported (Scheme 4), and they are derived from highly sterically hindered substrates. To verify the compound obtained in our reactions was indeed a dithiirane, we studied its properties and reactions. 3 could be generated from the direct reaction between 1 and an excess of t-butyl nitrite. It was isolated as a yellow liquid (∼90% purity) once the solvent was removed. 3 showed a UV-vis absorbance at 441 nm (Fig. S4, ESI†), which corresponds to the nπ → σ* transition of the dithiirane ring and is similar to the dithiirane reported before (at 452 nm).24 In its neat form, 3 was found to be unstable at room temperature and could degrade completely in 6 hours. When it was dissolved in organic solvents, it appeared to be more stable, but still decomposed in ∼4 days. The degradation products were complex, with thioketone 5 being one of the identified compounds, and this matched the properties of reported dithiiranes.26 The treatment of 3 with PPh3 or Et3N resulted in more rapid formation of 5. In the case of PPh3, triphenyl phosphine sulfide (Ph3P = S) was isolated in 75% yield (Scheme 4). The reaction between 3 and PPh3 indicated that dithiiranes could be effective sulfur transfer reagents while the corresponding thioketones appeared to lack this reactivity.
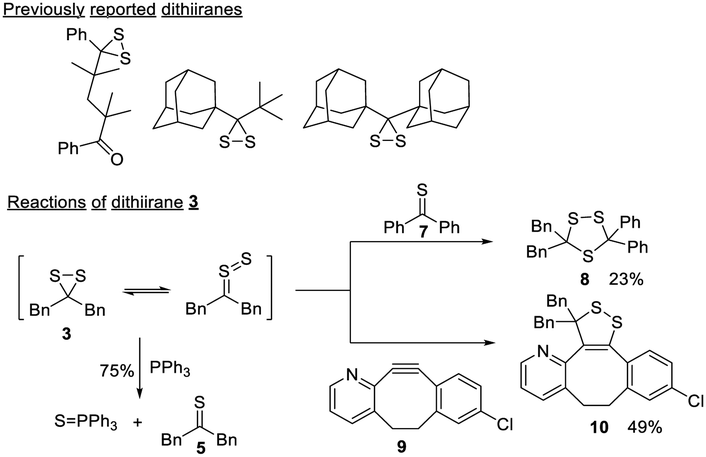 | ||
| Scheme 4 The structures of reported dithiiranes and dithiirane 3's reactions. | ||
Dithiiranes can be considered as the isomers of thiocarbonyl S-sulfides,19 which are usually in situ generated by the prolonged heating of trithiolanes. These thiocarbonyl S-sulfides are known to react with alkynes or thioketones to form cycloaddition products.27 We thus treated 3 with thiobenzophenone 7 and a strained cyclooctyne 9 (Scheme 4). The corresponding products (trithiolane 8 and the cyclic disulfide 10) were obtained in modest yields. These results demonstrated that isolated dithiiranes could perform thiocarbonyl S-sulfide-mediated 1,3-dipolar cycloadditions under mild conditions.
Given the possibility of 3 being a unique sulfur transfer reagent, we wondered if it could transfer its sulfur to thiols (RSH), which would form persulfides (RSSH). This type of sulfur species plays important roles in redox biology,1 but currently reliable methods to induce persulfidation in biological systems are still needed. We treated 3 with a model RSH compound 13 in both organic and water-containing solvents (Scheme 5). The desired sulfur transfer product 14 was obtained in good yields. Although 3 was found to be unstable and would degrade to complex products (including 6) in aqueous solutions, we tested its ability in inducing protein persulfidation. Briefly, papain (a cysteine protease) was treated with 3 in tris buffer for 30 min. The protein was then purified by a PD-10 column to remove excess small molecules and their byproducts. The level of papain persulfidation was analyzed by an established protocol using the fluorescent probe SSP4.28 As shown in Fig. 5, papain treated with 3 gave significant fluorescence, albeit not as strong as the signal from papain treated with DTNB and Na2S, an authentic method for protein persulfide formation. The combination of compound 1 with DTNB also resulted in considerable fluorescence. Other controls, including papain treated with 6, did not give detectable fluorescence. The control experiment with only compound 3 was to demonstrate that the PD-10 column could effectively remove small molecules. As such, the signal from papain treated with 3 was not due to the incomplete removal of 3 from the sample. These results suggest that dithiirane compounds have the potential to be developed as useful persulfidation reagents. However, structural modifications to enhance their stability in aqueous solutions will be needed.
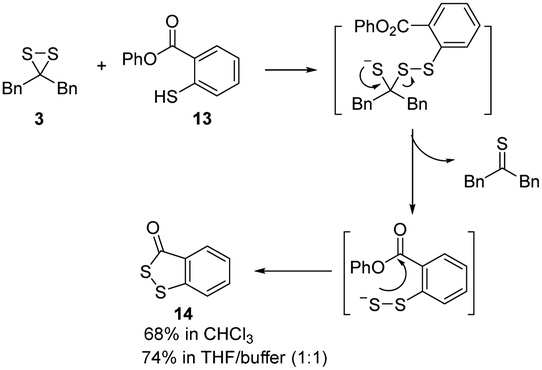 | ||
| Scheme 5 Assessing the sulfur transferring capability of 3 using 13. | ||
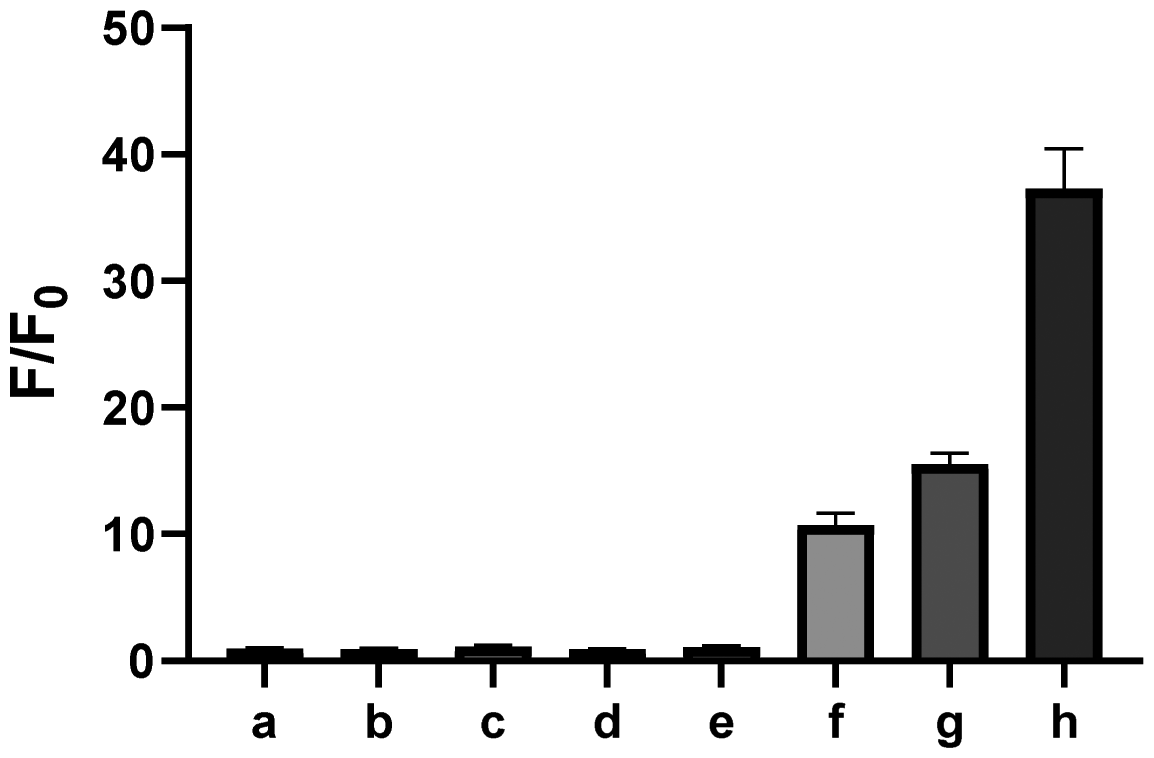 | ||
| Fig. 5 Fluorescence enhancements of SSP4 (5 μM) with persulfidated papain samples and controls. (a) blank; (b) 3 only; (c) 6 only; (d) papain only; (e) papain + 6; (f) papain + 3; (g) papain + DTNB + 1; (h) papain + DTNB + Na2S; results are represented as the mean ± SEM (n = 3). | ||
In conclusion, we studied the reactions and properties of a stable gem-dithiol 1 and its derivatives. 1 can serve as a H2S donor. Its SNO derivative 2 shows good stability. To the best of our knowledge, compound 2 is the first compound with both free thiol and nitrosothiol moieties in the structure, making it a unique precursor for both NO (or HNO) and reactive sulfur species. Dithiirane 3, which is generated from 2 after NO release, is a sulfur transfer reagent and can be used to induce protein persulfidation. Further structural optimization on these molecules may provide reagents with better stability and specificity (as H2S donors, NO donors, or protein persulfidation reagents), and that is ongoing research in our lab.
This work was supported by NIH (R35GM149170) to M. X. M. S. is supported by a NIH F31 Predoctoral Fellowship (F31HL170516).
Conflicts of interest
There are no conflicts to declare.References
- M. R. Filipovic, et al. , Chem. Rev., 2018, 118, 1253–1337 CrossRef CAS PubMed.
- K. Ono, et al. , Free Radical Biol. Med., 2014, 77, 82–94 CrossRef CAS PubMed.
- C. E. Paulsen and K. S. Carroll, Chem. Rev., 2013, 113, 4633–4679 CrossRef CAS PubMed.
- X. Ni, et al. , Acc. Chem. Res., 2021, 54, 3968–3978 CrossRef CAS PubMed.
- P. Bora, et al. , RSC Adv., 2018, 8, 27359–27374 RSC.
- S. Xu, et al. , Chem. – Eur. J., 2019, 25, 4005–4016 CrossRef CAS PubMed.
- Y. Zhao, et al. , Org. Lett., 2014, 16, 4536–4539 CrossRef CAS PubMed.
- N. O. Devarie-Baez, et al. , Org. Lett., 2013, 15, 2786–2789 CrossRef CAS PubMed.
- P. Shukla, et al. , Chem. Sci., 2017, 8, 4967–4972 RSC.
- T. L. Cairns, et al. , J. Am. Chem. Soc., 1952, 74, 3982–3989 CrossRef CAS.
- G. A. Berchtold, et al. , J. Am. Chem. Soc., 1959, 81, 3148 CrossRef CAS.
- P. Metzner, Synthesis, 1992, 1185–1199 CrossRef CAS.
- Z.-L. Song, et al. , Med. Res. Rev., 2022, 42, 1930–1977 CrossRef CAS PubMed.
- C. Zhang, et al. , Chem. Commun., 2017, 53, 11266–11277 RSC.
- K. Mazmanian, et al. , J. Phys. Chem. B, 2016, 120, 10288–10296 CrossRef CAS PubMed.
- H. Zheng, et al. , Org. Lett., 2008, 10, 2357–2360 CrossRef CAS PubMed.
- G. G. Borisenko, et al. , J. Biol. Chem., 2004, 279, 23453–23462 CrossRef CAS PubMed.
- S. Donzelli, et al. , Free Radical Biol. Med., 2006, 40, 1056–1066 CrossRef CAS PubMed.
- A. Senning, Angew. Chem., Int. Ed. Engl., 1979, 18, 941–942 CrossRef.
- J. Nakayama and A. Ishii, Adv. Heterocycl. Chem., 2000, 77, 221–284 CrossRef CAS.
- F. A. G. El-Essawy, et al. , J. Org. Chem., 1998, 63, 9840–9845 CrossRef CAS.
- R. W. Saalfrank and W. Rost, Angew. Chem., Int. Ed. Engl., 1985, 24, 855–856 CrossRef.
- A. Ishii, et al. , J. Am. Chem. Soc., 1993, 115, 4914–4915 CrossRef CAS.
- A. Ishii, et al. , Angew. Chem., Int. Ed. Engl., 1994, 33, 777–779 CrossRef.
- A. Ishii, et al. , J. Org. Chem., 2003, 68, 1555–1558 CrossRef CAS PubMed.
- K. Shimada, et al. , Chem. Lett., 1999, 695–696 CrossRef CAS.
- R. Huisgen and J. Rapp, Tetrahedron, 1997, 53, 939–960 CrossRef CAS.
- M. Shieh, et al. , Redox Biol., 2022, 56, 102433 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01003e |
| This journal is © The Royal Society of Chemistry 2024 |