
An efficient polymer acceptor with fluorinated linkers enables all polymer solar cells with an efficiency of 15.7%†
Haiqin
Xiao
a,
Junfang
Lv
a,
Miao
Liu
a,
Xia
Guo
b,
Xinxin
Xia
c,
Xinhui
Lu
c and
Maojie
Zhang
 *ab
*ab
aLaboratory of Advanced Optoelectronic Materials, Suzhou Key Laboratory of Novel Semiconductor-optoelectronics Materials and Devices, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: mjzhang@sdu.edu.cn
bNational Engineering Research Center for Colloidal Materials, School of Chemistry & Chemical Engineering, Shandong University, Jinan, Shandong 250100, China
cDepartment of Physics, Chinese University of Hong Kong, New Territories, Hong Kong 999077, P. R. China
First published on 4th February 2023
Abstract
Despite the significant progress in all-polymer solar cells (all-PSCs) in recent years, obtaining both high open-circuit voltage (VOC) and short-circuit current density (JSC) simultaneously has been a challenging issue. Herein, a novel polymer acceptor PY-DF was developed by polymerizing small molecule acceptor (SMA) monomers with difluorothiophene linkers. Compared to non-fluorinated PYT, PY-DF exhibits a more coplanar and rigid molecular conformation, which leads to better intra-molecular conjugation and enhanced interchain packing, resulting in improved electron mobility and reduced energetic disorder. Furthermore, PY-DF exhibits a relatively up-shifted lowest unoccupied molecular orbital (LUMO) energy level (−3.76 eV) than PYT (−3.80 eV), which is favorable for improving VOC. In addition, the polymer acceptor demonstrates good miscibility with polymer donor, thus leading to optimized phase segregation for superior exciton dissociation and charge transport. As a result, the PY-DF-based all-PSCs achieved a higher PCE of 15.7% with simultaneously enhanced JSC (23.1 mA cm−2) and VOC (0.97 V) in comparison with PYT-based all-PSCs (PCE = 13.2%, JSC = 21.7 mA cm−2, and VOC = 0.93 V). This work provides a promising polymer acceptor for all-PSCs and shows that fluorination of linkers is a potential strategy to build high-performance polymer acceptors.
1. Introduction
Polymer solar cells (PSCs) are considered one of the most promising solar harvesting technologies due to the potential of combining low manufacturing cost, light weight, flexibility, and transparency.1,2 PSCs based on polymer donors and small molecule acceptors (SMAs) have achieved power conversion efficiencies (PCEs) surpassing 19% to date because of the extensive investigations on efficient photovoltaic materials and device optimization.3,4 Unlike SMA-based PSCs, all-polymer solar cells (all-PSCs), which comprise conjugated polymers as both donors and acceptors, provide the additional merits of excellent mechanical flexibility, outstanding morphological stability, and great light/thermal stability for large-scale commercialization.5–7 However, the PCEs of all-PSCs lag behind those of SMA-based PSCs mainly due to the lack of high-performance polymer acceptors.Before 2017, many classical polymer acceptors had been developed based on naphthalene diimide (NDI),8 perylene diimide (PDI),9 bithiophene imide (BTI),10 B←N-bridged bipyridine (BN-Py),11 and their derivatives.12 However, these polymer acceptors usually suffer from some intrinsic flaws such as the weak absorption intensity for NDI-, PDI-, and BTI-based polymer acceptors and the low electron mobility for BN-Py-based polymers, which limit the performance of all-PSCs.5 Regarding these issues, Li et al. creatively proposed a successful strategy by copolymerizing SMAs with π-linkers to construct polymer SMAs (PSMAs).13 The resulting PSMAs share the same skeleton structure as SMAs and show the advantages of high absorption coefficients, extended photon response, and good crystallinity, which significantly improve the PCEs of all-PSCs.14,15 Encouraged by the rapid progress of Y-series SMAs,16,17 researchers in this field have been attracted to using Y-series SMAs to construct high-performance PSMAs.6,14 Afterwards, many efforts were devoted to further developing PSMAs by modulating π-conjugated fused-ring cores,18–20 electron-deficient end groups,21–25 and π-linkers.26–31 Recently, the PCEs of all-PSCs have reached 18%, narrowing the efficiency gap with SMA-based OSCs.24,32
Recent studies have confirmed that the molecular conformations of PSMAs play a crucial role in device performance.23,33,34 For example, the commonly used thiophene linkers contribute to twisted molecular conformations and “randomness” to the polymer backbone, which negatively impact the properties of polymers.27,34 Yu et al. reported a vinyl linker-based polymer PY-V-γ, which exhibits a more coplanar and rigid molecular conformation leading to tighter interchain packing and higher mobility.27 Therefore, the PY-V-γ-based device shows simultaneously enhanced JSC (24.75 mA cm−2) and fill factor (FF = 75.8%) than those of PM6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PY-T-γ (JSC = 24.1 mA cm−2 and FF = 71.9%). However, the PY-V-γ-based device exhibits a lower VOC (0.91 V) than that based on PYT (0.93 V) due to the downshifted lowest unoccupied molecular orbital (LUMO) level of PY-V-γ.27 Furthermore, severe phase separation morphology is observed in all-PSCs due to reduced entropic contribution of PSMAs relative to SMAs and significantly suppressing the miscibility of donors and acceptors, which is a long-term challenge.6,14 Therefore, it is essential to take a comprehensive consideration of molecular conformations, energy levels, and microscopic morphology to achieve high VOC and JSC simultaneously in all-PSCs.
:PY-T-γ (JSC = 24.1 mA cm−2 and FF = 71.9%). However, the PY-V-γ-based device exhibits a lower VOC (0.91 V) than that based on PYT (0.93 V) due to the downshifted lowest unoccupied molecular orbital (LUMO) level of PY-V-γ.27 Furthermore, severe phase separation morphology is observed in all-PSCs due to reduced entropic contribution of PSMAs relative to SMAs and significantly suppressing the miscibility of donors and acceptors, which is a long-term challenge.6,14 Therefore, it is essential to take a comprehensive consideration of molecular conformations, energy levels, and microscopic morphology to achieve high VOC and JSC simultaneously in all-PSCs.
Introducing fluorine atoms into polymers is an effective strategy to enhance photovoltaic performance.35–38 The fluorine atom can effectively enhance the electron affinity of a polymer and facilitate electron transport.35,39–41 It has been verified that fluorinated end groups can enhance the intramolecular charge transfer (ICT) effect to improve absorption and molecular packing, which ultimately enhance JSC for the corresponding devices.21,22 Unfortunately, the fluorinated end group strategy results in lower VOC due to the downshifted LUMO levels of acceptors.21,22,42 Modifying linker units can fine-tune the energy levels and electron transport properties of polymers.26–28 Previously reported work verified that a difluorothiophene-substituted polymer (BN-2fT) exhibits a slightly up-shifted LUMO level relative to that of nonfluorinated thiophene analogs (BN-T), which contributes to high VOC.43 Numerous noncovalent interactions have been found in fluorinated polymers, which can increase the planarity of the backbone.21,35,44 The fluorination strategy can also change the miscibility of donors and acceptors and further fine-tune microscopic morphology.22,28,45 Therefore, difluoro-substituted thiophenes as linking units of polymer acceptors may provide another effective approach to design high-performance polymer acceptors.
Here we developed a novel polymer acceptor PY-DF by introducing difluorothiophenes as linkers to optimize molecular conformations and optoelectronic properties. PY-DF exhibits more planar molecular conformation and tighter inter-chain stacking, resulting in higher electron mobility (7.56 × 10−4 cm2 V−1 S−1) than PYT (1.68 × 10−4 cm2 V−1 s−1). Furthermore, the up-shifted LUMO level (−3.76 eV) for PY-DF in comparison with that of PYT (−3.80 eV) can contribute to increasing the VOC of the resulting devices. In addition, the fluorination effect improves the miscibility of the donor and acceptor and enables more suitable phase segregation, thus promoting exciton dissociation and charge transport. When blended with PM6, the PY-DF-based device showed a higher PCE of 15.7% with simultaneously enhanced VOC (0.97 V), JSC (23.1 mA cm−2), and FF (70.2%) in comparison with the PM6:PYT-based device (PCE = 13.2%, VOC = 0.93 V, JSC = 21.7 mA cm−2, and FF = 65.6%).
2. Results and discussion
2.1. Materials synthesis, theoretical calculations and crystalline properties
The synthetic routes of polymer acceptors are shown in Fig. 1a. Both PYT and PY-DF were all synthesized via Stille cross-coupling polymerization of Y5–C20–Br (M1) and 2,5-bis(trimethylstannyl)thiophene (M2) or (3,4-difluorothiophene-2,5-diyl)bis(trimethylstannane) (M3). The synthetic routes are detailed in the ESI.† The two polymers can be readily dissolved in common solvents including chlorobenzene (CB), toluene (Tol), and chloroform (CF). As measured by gel permeation chromatography (GPC), the PYT and PY-DF acceptors exhibit comparable number average molecular weights (Mn)/polydispersity index (PDI) of 10.1 kDa/1.7 and 10.0 kDa/1.8, respectively. As shown in Fig. S1,† both polymers show good thermal stability with decomposition temperature (Td, 5% weight loss) above 300 °C under a nitrogen atmosphere. | ||
| Fig. 1 (a) The synthetic routes and molecular structures of monomers and polymer acceptors. Simulated chemical geometry from DFT calculations: (b) top-view and (c) side-view of PYT and PY-DF. (d) The 2D GIWAXS patterns and (e) the corresponding in-plane (IP) and out-of-plane (OOP) line cuts of neat films. | ||
Density functional theory (DFT) calculations of Gaussian simulation at the B3LYP/631G(d,p) level were carried out to study the impact of linkers on molecular geometry. All alkyl chains were replaced by methyl groups to simplify DFT calculations. For PY-DF, there is an obvious noncovalent C–F⋯H interaction effect between H atoms on end groups and F atoms on difluorothiophene linkers (Fig. 1b), which facilitates obtaining excellent backbone coplanarity.35,44 Compared to PYT with dihedral angles of 17.51° and 18.51° between the end group and adjacent thiophene linker and 2.04° between the end group and adjacent BTTP unit, PY-DF shows smaller dihedral angles between the end group and adjacent difluorothiophene linker (11.97° and 12.65°) and between the end group and adjacent BTTP unit (1.03°). The enhanced molecular planarity of PY-DF compared to PYT is beneficial for interchain packing and charge transport.21,27,46
The molecular orientations and packing of neat films were investigated by grazing incident wide-angle X-ray diffraction (GIWAXS) measurements and the corresponding results are exhibited in Fig. 1d and e, and Table S1.† Both PY-DF and PYT neat films display quite similar stacking behaviors with preferred “face-on” packing. The PY-DF film displays a smaller π–π stacking spacing (d-spacing) of 3.81 Å with a higher crystal coherence length (CCL) of 18.85 Å compared to the PYT film (d-spacing = 3.85 Å and CCL = 16.15 Å) in the out-of-plane (OOP) direction. In addition, the CCL value in the in-plane (IP) direction for PY-DF (CCL = 28.27 Å) is higher than that of PYT (20.19 Å). The higher CCLs for PY-DF than PYT in both π–π stacking in the OOP direction and lamellar stacking in the IP direction indicate that PY-DF exhibits more ordered intermolecular packing and strong crystallization propensity, which is favorable for charge transport.46 The charge carrier mobilities of PY-DF and PYT films were measured by the space-charge-limited current (SCLC) method. The electron mobility (μe) for the PY-DF neat film (7.56 × 10−4 cm2 V−1 s−1) is higher than that of PYT (1.68 × 10−4 cm2 V−1 s−1), probably mainly due to the stronger crystallization propensity and intermolecular interaction of PY-DF (Fig. S2†).47
2.2. Optical and electrochemical properties
The ultraviolet-visible-near-infrared (UV-vis-NIR) absorption spectra of PYT and PY-DF in chloroform solution and thin films are shown in Fig. 2a and b, respectively. In solution, PY-DF shows a blue-shifted maximum absorption peaks (λmax,sol = 740 nm) relative to that of PYT (779 nm), which can be ascribed to the weaker ICT induced by difluorothiophene linkers.43 In thin films, PY-DF and PYT display similar maximum absorption peaks (λmax,film) at 792 and 794 nm, respectively, while PY-DF exhibits a broader absorption range. PY-DF shows an obvious red-shift (52 nm) from solutions to films while PYT exhibits a smaller red-shift (15 nm), implying the enhanced molecular packing feature of PY-DF.42 The optical bandgaps (Eoptg) of PY-DF and PYT are 1.43 and 1.40 eV, respectively, calculated from absorption onsets in films. As shown in Fig. 2b, the maximum extinction coefficient (αmax,film) increases from 1.11 × 105 cm−1 of the PYT film to 1.21 × 105 cm−1 of the PY-DF film, which might favor photon utilization and facilitate higher JSC in the corresponding all-PSCs. As shown in Fig. 2c and Fig. S3,† PY-DF exhibits a comparable highest occupied molecular orbital (HOMO) level (−5.68 eV) to PYT (−5.67 eV). However, the LUMO level of PY-DF (−3.76 eV) is slightly higher than that of PYT (−3.80 eV) by the cyclic voltammetry (CV) method, which facilitates higher VOC for the corresponding devices.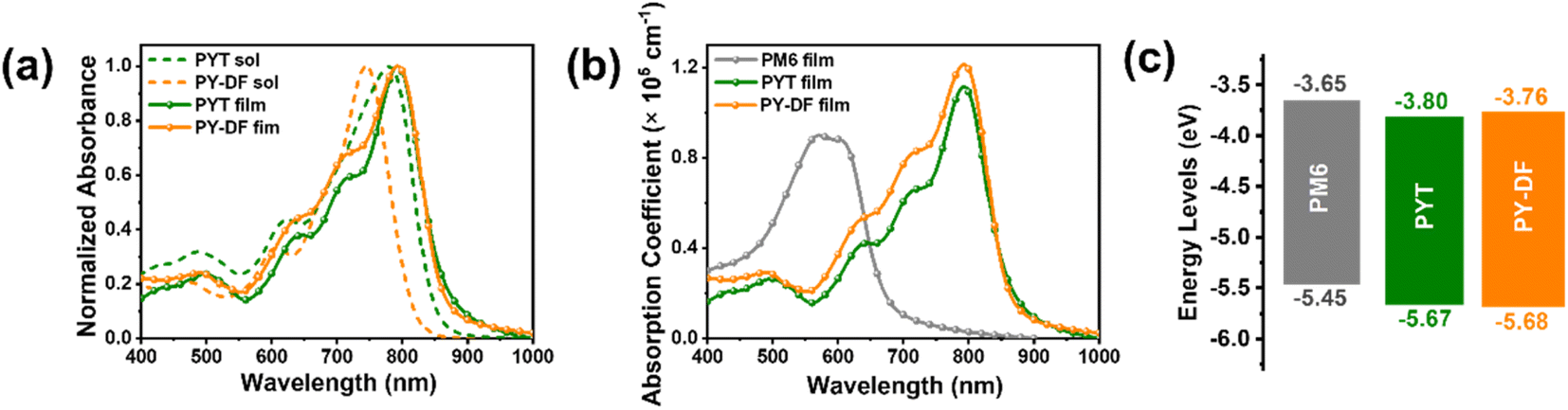 | ||
| Fig. 2 (a) Normalized UV-vis-NIR absorption spectra of PYT and PY-DF in solution and as thin films. (b) Absorption coefficient of PM6, PYT and PY-DF films. (c) Energy level diagram of PM6, PYT, and PY-DF. | ||
2.3. Photovoltaic performance of all-PSCs
All-PSCs were fabricated with a conventional device structure of ITO/PEDOT:PSS/active layer/PFN-Br/Ag. The photovoltaic properties were optimized by changing D/A weight ratios, adjusting thermal annealing (TA) temperature, and adding solvent additives (Fig. S4; Tables S2–S4†). The optimal current density–voltage (J–V) curves are plotted in Fig. 3a, and the detailed photovoltaic parameters are collected in Table 1. The PM6:PYT-based device yields a VOC of 0.93 V, a JSC of 21.7 mA cm−2, and a FF of 65.6%, resulting in a PCE of 13.2%. As expected, the PM6:PY-DF-based device shows a higher VOC of 0.97 V (∼40 mV higher than that of the PYT-based device), which may benefit from the high-lying LUMO level of PY-DF and reduced Eloss (discussed below). Compared to the PM6:PYT-based device, the PM6:PY-DF-based device exhibits higher JSC (23.1 mA cm−2) and FF (70.2%), which may result from optimized blend morphology. Therefore, a high PCE of 15.7% can be achieved in PM6:PY-DF-based devices. These two devices both have over 70% external quantum efficiency (EQE) in the absorption region of 450–850 nm, while the overall EQE values (the maximum is close to 80%) of the PY-DF-based device are much higher than those of the PYT-based device (Fig. 3b). The calculated integrated JSC value of the PM6:PY-DF device (22.1 mA cm−2) is higher than that of PM6:PYT (20.9 mA cm−2). Fig. 3c shows a histogram of PCEs for PM6:PYT and PM6:PY-DF devices of 30 independent cells and shows Gaussian distribution, indicating good reproducibility of photovoltaic performance in the corresponding all-PSCs. Fig. 3d and Table S5† summarize the key device parameters for representative high-efficiency binary all-PSCs. To the best of our knowledge, the VOC value in this work is one of the highest among those of binary all-PSCs with a JSC exceeding 18 mA cm−2. The simultaneous enhancements in VOC, JSC and FF from PY-DF-based devices indicate that difluorothiophenes as linkers are very meaningful for molecular design of PSMAs.
 | ||
| Fig. 3 (a) J–V curves of optimal devices based on PM6:PYT and PM6:PY-DF. (b) EQE curves of the corresponding devices. (c) Histograms of the PCE measurements for over 30 individual PM6:PYT, and PM6:PY-DF-based devices. (d) Plots of JSC against VOC for binary all-PSCs reported previously with PCEs of over 9% and this work. | ||
:acceptors (1:1, w/w) under AM 1.5 G illumination (100 mW cm−2)
| Active layers | V OC (V) | J SC (mA cm−2) | Cal. JSCb (mA cm−2) | FFa (%) | PCEa (%) |
|---|---|---|---|---|---|
| a The mean values and standard deviations of device parameters based on 30 devices are shown in parentheses. b The integral JSC from EQE curves. | |||||
| PM6:PYT |
0.93 (0.92 ± 0.01) | 21.7 (21.5 ± 0.2) | 20.9 | 65.6 (64.4 ± 1.2) | 13.2 (12.9 ± 0.3) |
| PM6:PY-DF |
0.97 (0.96 ± 0.01) | 23.1 (22.9 ± 0.2) | 22.1 | 70.2 (69.1 ± 1.1) | 15.7 (15.4 ± 0.3) |
2.4. Exciton dissociation, and charge recombination and transport
As shown in Fig. 4a and S5,† the PM6:PY-DF-based blends show higher and more balanced charge carrier mobilities (hole mobilities, μh = 7.86 × 10−4 cm2 V−1 s−1, μe = 7.09 × 10−4 cm2 V−1 s−1, and μh/μe = 1.11) than PM6:PYT-based blends (μh = 6.10 × 10−4 cm2 V−1 s−1, μe = 4.56 × 10−4 cm2 V−1 s−1, and μh/μe = 1.34). More balanced charge transport for PM6:PY-DF can minimize the impact of space-charge formation and high mobility charge carriers can be transported to the electrode more quickly, contributing to higher JSC and FF.48 To understand the exciton dissociation and charge collection process, the photocurrent density (Jph) versus effective voltage (Veff) of PYT and PY-DF-based devices were investigated and are shown in Fig. 4b. The Veff is obtained from Veff = VOC − Vapp, where Vapp is applied voltage.47,49 The values of exciton dissociation probability (Pdiss) and charge collection probability (Pcoll) for PM6:PY-DF and PM6:PYT were calculated to be 98.2%/84.5% and 96.5%/79.5% under the short-circuit and maximum power output conditions, respectively. Obviously, the PM6:PY-DF devices show the more efficient processes of exciton dissociation and charge collection. Photoluminescence (PL) was recorded to further study exciton dissociation and charge transfer behavior of PY-DF and PYT neat films and their blends. As shown in Fig. S6,† compared to PL spectra of PM6 and PYT neat films, more than 99.3% and 83.8% of PL quenching for the donor and acceptor, respectively, can be achieved for PM6:PYT blends. Notably, the PM6:PY-DF blends show higher PL quenching of over 99.8% and 85.9% for the donor and acceptor, respectively, meaning more effective charge transfer between PM6 and PY-DF.50
 | ||
| Fig. 4 (a) Hole and electron mobilities. (b) The plots of Jphversus Veff curves of PM6:PYT and PM6:PY-DF all-PSCs. (c) TPC and (d) TPV measurements. (e) and (f) The dependence of VOC and JSC on Plight of the corresponding all-PSCs. | ||
Transient photocurrent (TPC) measurement was also performed to evaluate the charge carrier generation and transportation properties. As shown in Fig. 4c, the photocurrent decay times (τ1) of PM6:PYT, and PM6:PY-DF were determined to be 0.32 and 0.24 μs, respectively. The shorter extraction lifetime suggests that the PM6:PY-DF device has a faster charge sweep-out and a superior charge extraction capacity than PM6:PYT devices. In addition, transient photovoltage (TPV) measurement was performed to evaluate the recombination rate of charge carriers in all-PSCs (Fig. 4d). The PY-DF-based device shows a higher charge carrier lifetime (τ2) than the PYT-based device, indicating less carrier recombination, mainly attributed to the more balanced carrier mobility and reduced charge trap states. The dependence of JSC and VOC on light intensity (Plight) was investigated to analyze charge recombination behavior. As shown in Fig. 4e, the VOC − Plight measurements reveal that the slope of the PM6:PYT device (1.26 kT/q) is higher than that of the PM6:PY-DF device (1.20 kT/q), suggesting that trap-assisted recombination of charge carriers in the PM6:PYT device is more significant than in the PY-DF-based device.51 The Plight dependence of JSC demonstrated the weaker bimolecular recombination in the PM6:PY-DF device (S = 0.996) than the PM6:PYT device (S = 0.990), which can be calculated using the power-law equation JSC ∝ PlightS (where S is an exponential constant) (Fig. 4f).52 All results are well consistent with the higher JSC and FF in PY-DF-based all-PSCs.
2.5. Energy loss
To gain insight into the higher VOC delivered by PM6:PY-DF devices, the Eloss characteristic was explored via Fourier transform photocurrent spectroscopy (FTPS–EQE) and electroluminescence external quantum efficiency (EQEEL) spectra (Fig. S7† and Table 2).27 According to detailed balance theory, the energy loss can be described by the following equation:53| Eloss = (EPVg − qVOC,SQ) + (qVOC,SQ − qVOC,Rad) + (qVOC,Rad − qVOC) = ΔE1 + ΔE2 + ΔE3 |
:PYT and PM6:PY-DF blends were calculated to be ∼1.45 eV. Both devices show the same ΔE1 value of 0.26 eV. ΔE2 and ΔE3 are caused by radiative recombination below the gap and non-radiative recombination, respectively.25 Compared with the PM6:PYT device (ΔE2 = 0.04 eV), the PM6:PY-DF device shows a slight reduction of ΔE2 (0.02 eV). ΔE3 can be directly estimated from EQEEL spectra using the equation: ΔE3 = −KTlnEQEEL.50 As shown in Fig. S7d,† the PM6:PY-DF device exhibits higher EL emission and produces lower EQEEL, and finally shows a lower ΔE3 value (0.20 eV) than the PM6:PYT device (ΔE3 = 0.22 eV). The total Eloss values are determined to be 0.48 and 0.52 eV for PM6:PY-DF and PM6:PYT, respectively, and the reduced Eloss in PY-DF-based all-PSCs is mainly attributable to the lower ΔE2 and ΔE3 values (Fig. 5a).
:PYT and PM6:PY-DF based devices
| Active layer | V OC (V) | E PVg (eV) | E loss (eV) | EQEELd (%) | ΔE1 (eV) | ΔE2 (eV) | ΔE3 (eV) |
|---|---|---|---|---|---|---|---|
| a The VOC values were calculated from J–V curves. b The EPVg values were determined from derivatives of EQE spectra. c E loss is equal to the difference between EPVg and VOC. d EQEEL is EL quantum efficiency. | |||||||
| PM6:PYT |
0.93 | 1.45 | 0.52 | 1.67 × 10−2 | 0.26 | 0.04 | 0.22 |
| PM6:PY-DF |
0.97 | 1.45 | 0.48 | 3.87 × 10−2 | 0.26 | 0.02 | 0.20 |
 | ||
| Fig. 5 (a) Eloss and its detailed three part values (ΔE1, ΔE2, and ΔE3) of PM6:PYT and PM6:PY-DF-based devices. (b) Plots of eVOC against EPVg and (c) PCE against Eg − eVOC in all-PSCs with PCEs of over 9% reported in the literature. | ||
The urbach energy (EU) values were further calculated from the exponential tail near the band edge of the corresponding FTPS–EQE spectra (Fig. S7c†) using the following equation:55
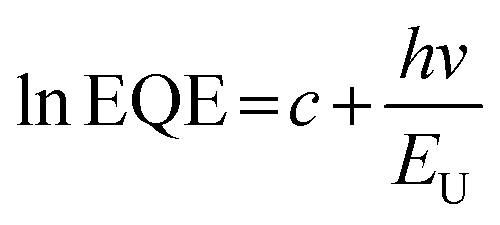 :PY-DF-based devices are both high PCE and low Eg − eVOC simultaneously. The results indicate that introducing the difluorothiophenes as linkers in PSMAs is one of the most effective ways to achieve a high VOC with low Eloss values.
:PY-DF-based devices are both high PCE and low Eg − eVOC simultaneously. The results indicate that introducing the difluorothiophenes as linkers in PSMAs is one of the most effective ways to achieve a high VOC with low Eloss values.
2.6. Film morphology
To investigate the fluorination effect on blend film morphology, atomic force microscopy (AFM) and transmission electron microscopy (TEM) were carried out. As shown in Fig. 6a and b, both blends display distinct uniform and fibrillar bicontinuous interpenetrating networks. The PM6:PYT films show a relatively coarse surface with a root-mean-square surface roughness (Rq) value of 1.58 nm. By contrast, the PM6:PY-DF films demonstrate a uniform and relatively smooth surface with a Rq of 1.31 nm, facilitating efficient exciton separation, charge transfer and extraction.28 Furthermore, the TEM results demonstrate that delicate bright and dark regimes could be clearly observed in PM6:PY-DF films (Fig. S8b†), while large size bright regimes are shown in PM6:PYT films (Fig. S8a†). Unlike the larger phase separation of PM6:PYT, the suitable phase separation of PM6:PY-DF provides more donor–acceptor interfaces for efficient charge dissociation.40 Contact angle measurements were further performed to explore the fundamental origin of the morphological difference. As shown in Fig. S9 and Table S6,† the surface energy (γ) of PM6 was calculated to be 31.25 mN m−1 using the Wu method.58,59 For PSMAs, PY-DF (38.73 mN m−1) shows a lower γ value than PYT (40.82 mN m−1). Furthermore, the miscibility of PM6 with the two polymer acceptors was evaluated using Flory–Huggins interaction parameters (χ).50 The χ between PM6 and PY-DF was calculated to be 0.40, which is lower than 0.64 for PM6/PYT. The weaker interaction between PM6 and PY-DF indicates better miscibility, and thus a relatively small and suitable domain size in blends, which is consistent with the TEM results.22
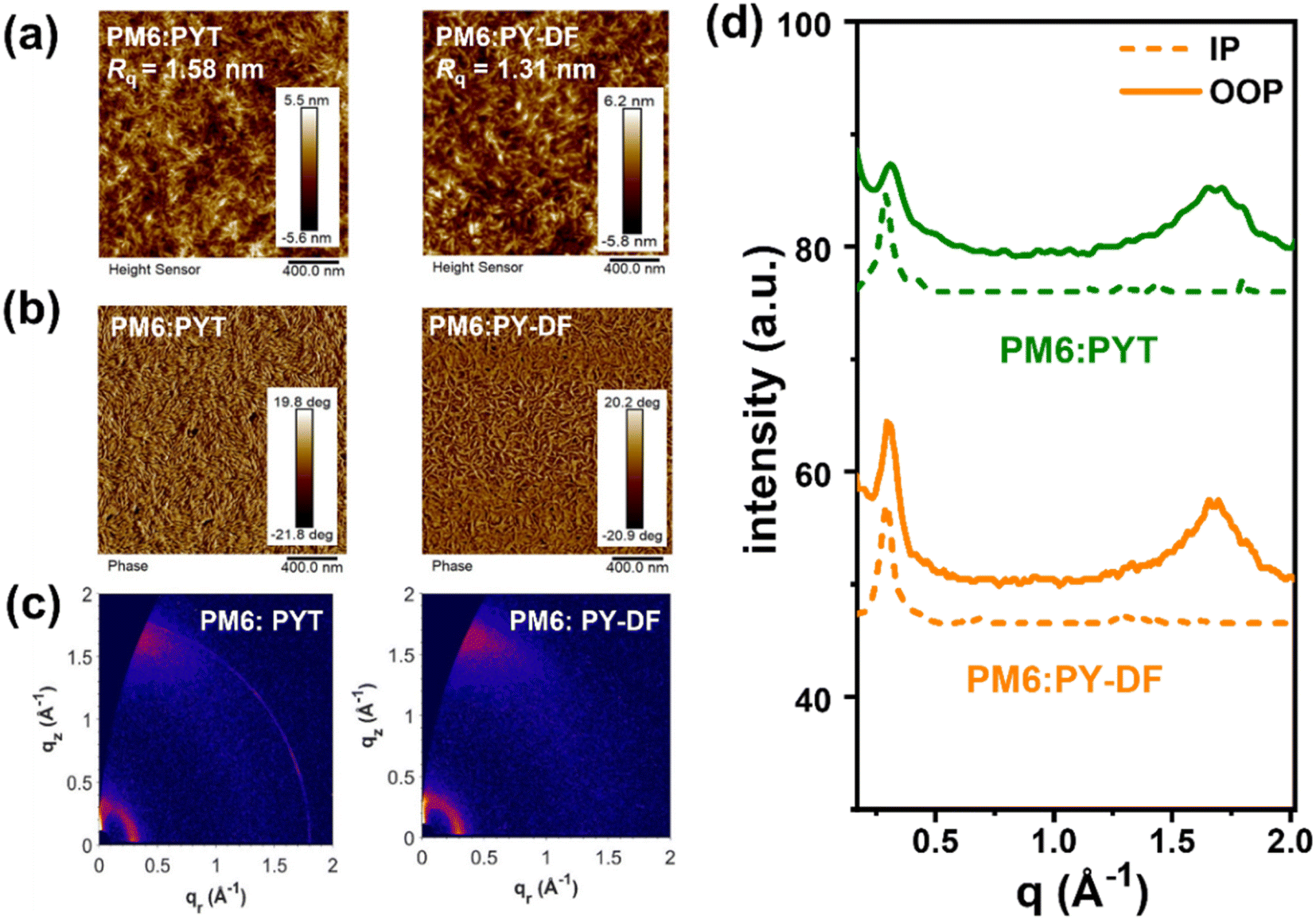 | ||
| Fig. 6 The AFM (a) height images and (b) phase images. (c) The 2D GIWAXS profiles and (d) the corresponding IP and OOP line-cuts of blend films. | ||
The crystalline nature and molecular packing of blend films were investigated by GIWAXS measurements. Fig. 6c and d show the 2D patterns and the relevant crystallographic parameters are shown in Table S7.† Both blends show sharp (010) π–π stacking diffraction peaks in the OOP direction, which exhibits a clear ‘‘face-on’’ dominant orientation. For the (010) peaks in the OOP direction, the PM6:PY-DF blend exhibits smaller d-spacing and higher coherence lengths (3.74 Å and 23.56 Å at 1.67 Å−1) compared to the PM6:PYT blend (3.78 Å and 21.74 Å at 1.66 Å−1). The CCL in the IP direction also increases from 23.56 Å for the PM6:PYT blend to 25.70 Å for the PM6:PY-DF blend, indicating the improved crystallite size in PM6:PY-DF blends. These above results explain the reason why the charge mobility is higher for PY-DF than for PYT, which eventually results in higher JSC and FF for PM6:PY-DF-based all-PSCs.
3. Conclusion
In summary, we designed and synthesized a novel polymer acceptor PY-DF with difluorothiophenes as linkers for all-PSC fabrications. The C–F⋯H noncovalent interaction between the difluorothiophene linkers and end groups results in more planar and rigid molecular conformation than that of PYT based on thiophene linkers. Therefore, PY-DF displays enhanced electron mobility, molecular crystallinity, and reduced energy disorder. In addition, PY-DF exhibits a lower surface energy than nonfluorinated PYT, leading to improved miscibility with PM6. The increased crystallinity and miscibility enable PM6:PY-DF to achieve an optimized active layer morphology, facilitating charge separation and transport. PY-DF exhibits up-shifted LUMO levels than PYT, leading to higher VOC in all-PSCs. As a result, PM6:PY-DF-based all-PSCs achieved an optimal PCE of 15.7% with both high VOC (0.97 V) and JSC (23.1 mA cm−2), corresponding to a ∼19% improvement in the PCE of the PM6:PYT-based device. To the best of our knowledge, this value of VOC is one of the highest among all-PSCs. This study indicates that fluorination of linker units is a potential strategy to build high-performance polymer acceptors for all-PSC application.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 51973146), the Shandong Provincial Natural Science Foundation for Distinguished Young Scholars (ZR2022JQ09), and the Collaborative Innovation Center of Suzhou Nano Science & Technology. Grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements were carried out with a Xeuss 2.0 SAXS/WAXS laboratory beamline using a Cu X-ray source, which is supported by the Department of Physics, Chinese University of Hong Kong.References
- J. Wang, P. Xue, Y. Jiang, Y. Huo and X. Zhan, Nat. Rev. Chem., 2022, 6, 614–634 CrossRef.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- W. Gao, F. Qi, Z. Peng, F. R. Lin, K. Jiang, C. Zhong, W. Kaminsky, Z. Guan, C. S. Lee, T. J. Marks, H. Ade and A. K. Jen, Adv. Mater., 2022, 34, 2202089 CrossRef CAS PubMed.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C. C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- C. Lee, S. Lee, G. U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- G. Wang, A. F. Ferdinand, S. Melkonyan and T. J. Marks, Angew. Chem., Int. Ed., 2019, 58, 4129–4142 CrossRef CAS PubMed.
- Z. Genene, W. Mammo, E. Wang and M. R. Andersson, Adv. Mater., 2019, 31, 1807275 CrossRef PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- Q. Shi, J. Wu, X. Wu, A. Peng and H. Huang, Chem.–Eur. J., 2020, 26, 12510–12522 CrossRef CAS PubMed.
- K. Feng, H. Guo, H. Sun and X. Guo, Acc. Chem. Res., 2021, 54, 3804–3817 CrossRef CAS PubMed.
- R. Zhao, J. Liu and L. Wang, Acc. Chem. Res., 2020, 53, 1557–1567 CrossRef CAS PubMed.
- J. Yang, B. Xiao, A. Tang, J. Li, X. Wang and E. Zhou, Adv. Mater., 2019, 31, 1804699 CrossRef CAS PubMed.
- Z. G. Zhang, Y. Yang, J. Yao, L. Xue, S. Chen, X. Li, W. Morrison, C. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 13503–13507 CrossRef CAS PubMed.
- B. Wu, B. Yin, C. Duan and L. Ding, J. Semicond., 2021, 42, 080301 CrossRef CAS.
- S. Ma, H. Zhang, K. Feng and X. Guo, Chem.–Eur. J., 2022, 28, 202200222 Search PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- X. Gu, Y. Wei, X. Liu, N. Yu, L. Li, Z. Han, J. Gao, C. Li, Z. Wei, Z. Tang, X. Zhang and H. Huang, Sci. China: Chem., 2022, 65, 926–933 CrossRef CAS.
- H. Fu, Y. Li, J. Yu, Z. Wu, Q. Fan, F. Lin, H. Y. Woo, F. Gao, Z. Zhu and A. K. Jen, J. Am. Chem. Soc., 2021, 143, 2665–2670 CrossRef CAS PubMed.
- W. Wang, Q. Wu, R. Sun, J. Guo, Y. Wu, M. Shi, W. Yang, H. Li and J. Min, Joule, 2020, 4, 1070–1086 CrossRef CAS.
- F. Peng, K. An, W. Zhong, Z. Li, L. Ying, N. Li, Z. Huang, C. Zhu, B. Fan, F. Huang and Y. Cao, ACS Energy Lett., 2020, 5, 3702–3707 CrossRef CAS.
- H. Yu, Z. Qi, J. Yu, Y. Xiao, R. Sun, Z. Luo, A. M. H. Cheung, J. Zhang, H. Sun, W. Zhou, S. Chen, X. Guo, X. Lu, F. Gao, J. Min and H. Yan, Adv. Energy Mater., 2020, 11, 2003171 CrossRef.
- Z. Luo, T. Liu, R. Ma, Y. Xiao, L. Zhan, G. Zhang, H. Sun, F. Ni, G. Chai, J. Wang, C. Zhong, Y. Zou, X. Guo, X. Lu, H. Chen, H. Yan and C. Yang, Adv. Mater., 2020, 32, 2005942 CrossRef CAS PubMed.
- R. Sun, T. Wang, Q. Fan, M. Wu, X. Yang, X. Wu, Y. Yu, X. Xia, F. Cui, J. Wan, X. Lu, X. Hao, A. K. Y. Jen, E. Spiecker and J. Min, Joule, 2023, 7, 221–237 CrossRef CAS.
- Y. Li, J. Song, Y. Dong, H. Jin, J. Xin, S. Wang, Y. Cai, L. Jiang, W. Ma, Z. Tang and Y. Sun, Adv. Mater., 2022, 34, 2110155 CrossRef CAS PubMed.
- H. Sun, B. Liu, Y. Ma, J. W. Lee, J. Yang, J. Wang, Y. Li, B. Li, K. Feng, Y. Shi, B. Zhang, D. Han, H. Meng, L. Niu, B. J. Kim, Q. Zheng and X. Guo, Adv. Mater., 2021, 33, 2102635 CrossRef CAS PubMed.
- H. Yu, Y. Wang, H. K. Kim, X. Wu, Y. Li, Z. Yao, M. Pan, X. Zou, J. Zhang, S. Chen, D. Zhao, F. Huang, X. Lu, Z. Zhu and H. Yan, Adv. Mater., 2022, 34, 2200361 CrossRef CAS PubMed.
- D. Zhou, C. Liao, S. Peng, X. Xu, Y. Guo, J. Xia, H. Meng, L. Yu, R. Li and Q. Peng, Adv. Sci., 2022, 9, 2202022 CrossRef CAS PubMed.
- Q. Fan, Q. An, Y. Lin, Y. Xia, Q. Li, M. Zhang, W. Su, W. Peng, C. Zhang, F. Liu, L. Hou, W. Zhu, D. Yu, M. Xiao, E. Moons, F. Zhang, T. D. Anthopoulos, O. Inganäs and E. Wang, Energy Environ. Sci., 2020, 13, 5017–5027 RSC.
- Z. Yin, Y. Wang, Q. Guo, L. Zhu, H. Liu, J. Fang, X. Guo, F. Liu, Z. Tang, M. Zhang and Y. Li, J. Mater. Chem. C, 2020, 8, 16180–16187 RSC.
- Y. Wang, N. Wang, Q. Yang, J. Zhang, J. Liu and L. Wang, J. Mater. Chem. A, 2021, 9, 21071–21077 RSC.
- J. Wang, Y. Cui, Y. Xu, K. Xian, P. Bi, Z. Chen, K. Zhou, L. Ma, T. Zhang, Y. Yang, Y. Zu, H. Yao, X. Hao, L. Ye and J. Hou, Adv. Mater., 2022, 34, 2205009 CrossRef CAS PubMed.
- H. Wang, H. Chen, W. Xie, H. Lai, T. Zhao, Y. Zhu, L. Chen, C. Ke, N. Zheng and F. He, Adv. Funct. Mater., 2021, 31, 2100877 CrossRef CAS.
- S. Seo, C. Sun, J. W. Lee, S. Lee, D. Lee, C. Wang, T. N. L. Phan, G. U. Kim, S. Cho, Y. H. Kim and B. J. Kim, Adv. Funct. Mater., 2021, 32, 2108508 CrossRef.
- Q. Zhang, M. A. Kelly, N. Bauer and W. You, Acc. Chem. Res., 2017, 50, 2401–2409 CrossRef CAS PubMed.
- A. Tang, B. Xiao, F. Chen, J. Zhang, Z. Wei and E. Zhou, Adv. Energy Mater., 2018, 8, 1801582 CrossRef.
- J. Yang, P. Cong, L. Chen, X. Wang, J. Li, A. Tang, B. Zhang, Y. Geng and E. Zhou, ACS Macro Lett., 2019, 8, 743–748 CrossRef CAS PubMed.
- T. Dai, X. Li, P. Lei, A. Tang, Y. Geng, Q. Zeng and E. Zhou, Nano Energy, 2022, 99, 107413 CrossRef CAS.
- K. Kawashima, T. Fukuhara, Y. Suda, Y. Suzuki, T. Koganezawa, H. Yoshida, H. Ohkita, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 10265–10275 CrossRef CAS PubMed.
- G. Sun, X. Jiang, X. Li, L. Meng, J. Zhang, S. Qin, X. Kong, J. Li, J. Xin, W. Ma and Y. Li, Nat. Commun., 2022, 13, 5267 CrossRef CAS PubMed.
- J. W. Jung, J. W. Jo, C. C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef CAS PubMed.
- H. Yu, S. Luo, R. Sun, I. Angunawela, Z. Qi, Z. Peng, W. Zhou, H. Han, R. Wei, M. Pan, A. M. H. Cheung, D. Zhao, J. Zhang, H. Ade, J. Min and H. Yan, Adv. Funct. Mater., 2021, 31, 2100791 CrossRef CAS.
- Y. Li, H. Meng, T. Liu, Y. Xiao, Z. Tang, B. Pang, Y. Li, Y. Xiang, G. Zhang, X. Lu, G. Yu, H. Yan, C. Zhan, J. Huang and J. Yao, Adv. Mater., 2019, 31, 1904585 CrossRef CAS PubMed.
- S. Yu, A. Peng, S. Zhang and H. Huang, Sci. China: Chem., 2018, 61, 1359–1367 CrossRef CAS.
- J. Wu, C. Y. Liao, Y. Chen, R. M. Jacobberger, W. Huang, D. Zheng, K. W. Tsai, W. L. Li, Z. Lu, Y. Huang, M. R. Wasielewski, Y. M. Chang, T. J. Marks and A. Facchetti, Adv. Energy Mater., 2021, 11, 2102648 CrossRef CAS.
- J. Lin, Q. Guo, Q. Liu, J. Lv, H. Liang, Y. Wang, L. Zhu, F. Liu, X. Guo and M. Zhang, Chin. J. Chem., 2021, 39, 2685–2691 CrossRef CAS.
- Y. Wang, Y. Wang, L. Zhu, H. Liu, J. Fang, X. Guo, F. Liu, Z. Tang, M. Zhang and Y. Li, Energy Environ. Sci., 2020, 13, 1309–1317 RSC.
- J. Wu, Q. Fan, M. Xiong, Q. Wang, K. Chen, H. Liu, M. Gao, L. Ye, X. Guo, J. Fang, Q. Guo, W. Su, Z. Ma, Z. Tang, E. Wang, H. Ade and M. Zhang, Nano Energy, 2021, 82, 105679 CrossRef CAS.
- Y. Ren, X. Liu, H. Li, J. Qin, S. Du, X. Lu, J. Tong, C. Yang and J. Li, Opt. Mater., 2022, 129, 112520 CrossRef CAS.
- Q. Guo, J. Lin, H. Liu, X. Dong, X. Guo, L. Ye, Z. Ma, Z. Tang, H. Ade, M. Zhang and Y. Li, Nano Energy, 2020, 74, 104861 CrossRef CAS.
- L. J. A. Koster, V. D. Mihailetchi, R. Ramaker and P. W. M. Blom, Appl. Phys. Lett., 2005, 86, 123509 CrossRef.
- P. Schilinsky, C. Waldauf and C. J. Brabec, Appl. Phys. Lett., 2002, 81, 3885–3887 CrossRef CAS.
- J. Yao, T. Kirchartz, M. S. Vezie, M. A. Faist, W. Gong, Z. He, H. Wu, J. Troughton, T. Watson, D. Bryant and J. Nelson, Phys. Rev. Appl., 2015, 4, 014020 CrossRef.
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS.
- F. Urbach, Phys. Rev., 1953, 92, 1324 CrossRef CAS.
- K. Vandewal, K. Tvingstedt, A. Gadisa, O. Inganäs and J. V. Manca, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 125204 CrossRef.
- C. Zhang, S. Mahadevan, J. Yuan, J. K. W. Ho, Y. Gao, W. Liu, H. Zhong, H. Yan, Y. Zou, S.-W. Tsang and S. K. So, ACS Energy Lett., 2022, 7, 1971–1979 CrossRef CAS.
- S. Nilsson, A. Bernasik, A. Budkowski and E. Moons, Macromolecules, 2007, 40, 8291–8301 CrossRef CAS.
- L. Xue, X. Liu, Q. Wang, M. Yang, S. Du, C. Yang, J. Tong, Y. Xia and J. Li, Energy Technol., 2022, 10, 2200504 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09364b |
| This journal is © The Royal Society of Chemistry 2023 |