
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlternatives to water oxidation in the photocatalytic water splitting reaction for solar hydrogen production
Yaqiang
Wu
 a,
Takuya
Sakurai
a,
Takumi
Adachi
a and
Qian
Wang
*ab
a,
Takuya
Sakurai
a,
Takumi
Adachi
a and
Qian
Wang
*ab
aGraduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: wang.qian@material.nagoya-u.ac.jp; Tel: +81-527893250
bInstitute for Advanced Research, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8601, Japan
First published on 7th March 2023
Abstract
The photocatalytic water splitting process to produce H2 is an attractive approach to meet energy demands while achieving carbon emission reduction targets. However, none of the current photocatalytic devices meets the criteria for practical sustainable H2 production due to their insufficient efficiency and the resulting high H2 cost. Economic viability may be achieved by simultaneously producing more valuable products than O2 or integrating with reforming processes of real waste streams, such as plastic and food waste. Research over the past decade has begun to investigate the possibility of replacing water oxidation with more kinetically and thermodynamically facile oxidation reactions. We summarize how various alternative photo-oxidation reactions can be combined with proton reduction in photocatalysis to achieve chemical valorization with concurrent H2 production. By examining the current advantages and challenges of these oxidation reactions, we intend to demonstrate that these technologies would contribute to providing H2 energy, while also producing high-value chemicals for a sustainable chemical industry and eliminating waste.
 Qian Wang | Qian Wang is currently an Associate Professor at Nagoya University, Japan. She received her Ph.D. in 2014 from the University of Tokyo, Japan and then worked as a postdoctoral researcher for the Japan Technological Research Association of Artificial Photosynthetic Chemical Processes (ARPChem) project at the University of Tokyo. In 2018, she became a Marie Sklodowska-Curie Research Fellow at the University of Cambridge. She joined Nagoya University as an Associate Professor in May 2021 and established her research group, which is currently developing new materials, approaches, and technologies for solar energy storage in the form of renewable fuels via artificial photosynthesis. |
As humanity moves forward into the 21st century, the supplementation of clean and sustainable energy is one of our greatest scientific and technical challenges. The photocatalytic water splitting process using nanoparticulate photocatalysts and solar energy is a leading candidate to achieve low-carbon production of H2 as a clean and sustainable energy carrier. In this process, water is split into H2 and O2via the following redox reactions (eqn (1) and (2); pH 0 in aqueous solution versus (normal hydrogen electrode) NHE, 25 °C, 1 atm gas pressure, and 1 M for other solutes):
| 2H+ + 2e− → H2 E° = 0 V | (1) |
| 2H2O → O2 + 4H+ + 4e− E° = +1.23 V | (2) |
Water splitting is a non-spontaneous chemical reaction involving water oxidation to O2, which requires four electrons to simultaneously transfer from at least two water molecules. Photocatalysis is a light-driven chemical process that involves oxidation and reduction reactions by the photogenerated electrons and holes. To achieve the photocatalytic water splitting reaction, the conduction band minimum must be more negative than the H+ to H2 reduction potential, while the valence band maximum must be more positive than the H2O to O2 oxidation potential. Consequently, the minimum theoretical energy required to initiate the water-splitting reaction is 1.23 eV. However, O2 evolution from water leads to sluggish kinetics and high additional kinetic overpotentials,1,2 making photocatalytic H2 evolution need the use of sacrificial reagents to provide an electron source.3–5 Specifically, the overpotential losses of the oxygen evolution reaction (OER) can reach up to 30%, resulting in a significant amount of wasted energy.6 In addition, the lack of effective methods to separate the produced O2 and H2 gases is a key problem for the practical application of photocatalytic water splitting.7 As a result of these challenges, increasing attention is being focused on emerging replacements for the OER that may support an expanding H2 economy globally.
In this minireview, we highlight a variety of key reactions that are well suited as potential replacements for the OER, focusing on the recent developments in the study of photoreforming and photochemical oxidation reactions to produce value-added organic and inorganic products that have received considerable attention in the literature, such as formic acid (∼1.1 $ per kg) production from waste PET (≤0 $ per kg) and H2O2 (∼0.7 $ per kg) formation by water (∼2.9 × 10−4 $ per kg) oxidation.8,9 These reactions are appealing because they would enable (1) the production of more valuable chemicals than O2 (∼0.1 $ per kg) alongside the production of H2; (2) the elimination of the need for a H2 and O2 mixture separation unit, thereby lowering the cost, improving the safety, and simplifying the design of photocatalytic processes; and (3) reduced overpotential due to the simper 1 or 2 electron net oxidation mechanisms, which would result in an increased conversion efficiency and reaction rate.6 Under light irradiation, an inorganic or organic substrate is oxidized by the photoexcited holes at the valence band, which subsequently generated valuable chemicals or CO2 or intermediate products and protons (Fig. 1a). The protons are reduced by electrons at the conduction band to produce H2. While the direct oxidation of alternative substrates combined with the production of clean H2 can be feasibly carried out using various technologies including electrolysis, photoelectrochemical and photocatalytic systems,6,7,10 we have targeted cases that have been demonstrated using nanoparticulate photocatalysts due to their potential for large-scale operations.
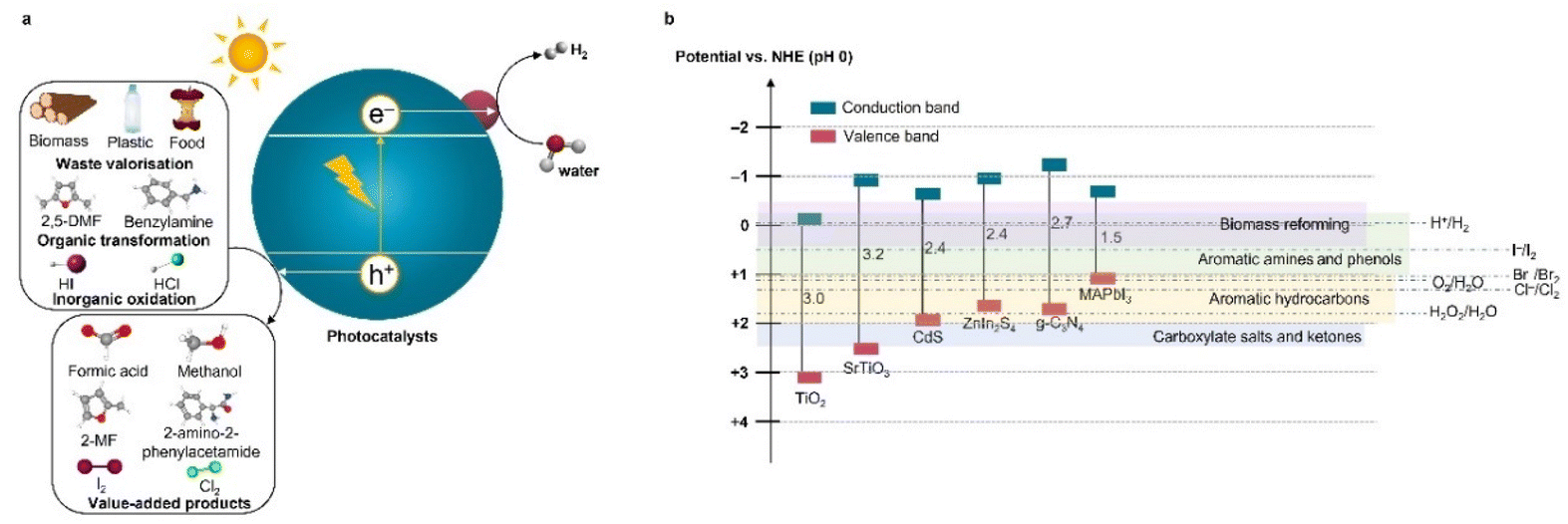 | ||
| Fig. 1 (a) Diagram of the photooxidation reactions rather than water-to-O2 oxidation combing with solar H2 production. (b) Band structures of representative photocatalysts and standard redox potentials for some common oxidation reactions. Data were obtained from ref. 6. | ||
Photoreforming
Photocatalytic reforming that harnesses solar energy to convert sustainable or waste feedstocks for H2 generation has become one of the most rapidly evolving applications in the field due to its ability to valorize a range of feedstocks especially biomass, food and plastics (≤0 $ per kg) to both energy and value-added products (such as formic acid (∼1.1 $ per kg), methanol (∼0.6 $ per kg), acetaldehyde (∼2 $ per kg) and lactic acid (∼3 $ per kg)), as listed in Table 1.9 Research trends highlight the shift from the early work which focused on simple model substrates such as ethanol, glycerol, and cellulose to more recent work that utilizes real waste, making the process more complex but more economically and environmentally sustainable.11 The energetics of coupling the oxidation of a variety of common organic waste substrates with H2 evolution are nearly neutral (Fig. 1b), for example, glucose oxidation (eqn (3)), making it more favorable than water splitting, although these oxidation reactions involve multiple electron transfer steps.12 Theoretically, coupling photoreforming with H2 production would no longer restrict the use of semiconductors with deep valence bands, allowing the application of those with a smaller bandgap. The photocatalytic reforming could therefore be driven by a large portion of incident sunlight including visible and IR parts.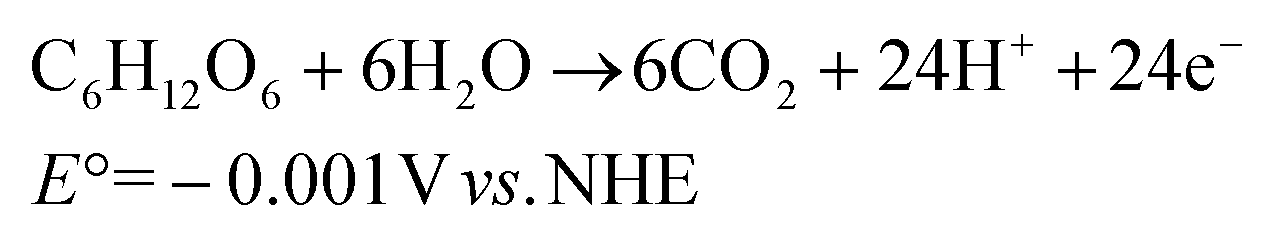 | (3) |
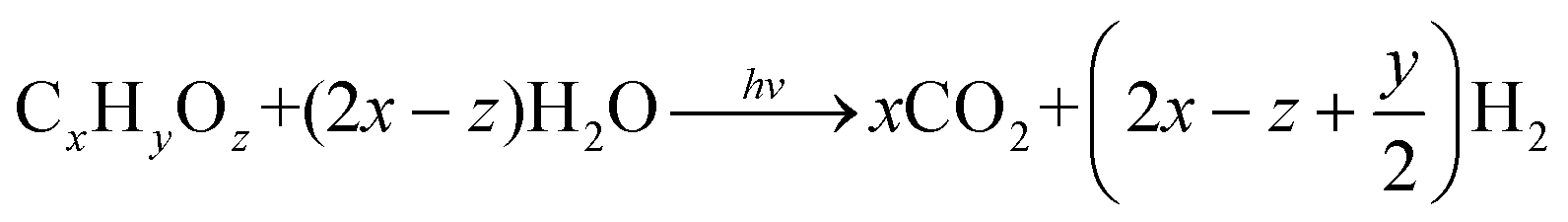 | (4) |
| Substrates | Photocatalysts | Reaction conditions | Products | Efficiency | Reaction time | Ref. |
|---|---|---|---|---|---|---|
| Glucose | Pt/C3N4 (∼ 1 μm) | 40 W blue LED (λ = 427 nm) | H2: 3.39 mmol g−1 h−1 | Glucose conversion: 100% | 16 hours | 28 |
| 10 M KOH | Lactic acid | Selectivity (lactic acid): 86% | ||||
| Microcrystalline cellulose | Pt/TiO2 | 16 W UV-A lamp (λ = 365 nm) | H2: 13.3 μmol h−1 | QE: 42.8% at 365 nm | 5 hours | 25 |
| Cellulose | Pt/S- and N-doped graphene oxide dots (2–7 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: 100 μmol h−1 | QE: 23.3% at 420 nm | 6 days | 108 |
| pH 12.5 | Acetaldehyde, methanol, formate, etc. | |||||
| Glucose | TiO2/Co(terpyridine)2 (∼20 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼0.43 mmol per gcatalyst per 24 h | Glucose conversion efficiency: 20% | 24 hours | 48 |
| Aqueous sodium acetate and MeCN purged with CO2 | CO: ∼0.43 mmol per gcatalyst per 24 h | QE: 0.22–0.41% at 360 nm for CO; 1.05–1.45% at 360 nm for formate | ||||
| Formate: ∼1.18 mmol per gcatalyst per 24 h | ||||||
| Cellobiose | TiO2/Co(terpyridine)2 (∼20 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼0.72 mmol per gcatalyst per 24 h | Cellobiose conversion efficiency: 8.6% | 24 hours | 48 |
| Aqueous sodium acetate and MeCN purged with CO2 | CO: ∼0.92 mmol per gcatalyst per 24 h | |||||
| Formate: ∼1.03 mmol per gcatalyst per 24 h | ||||||
| Glycerol | Cu/TiO2 nanorods | LED irradiation (λ = 365 nm) | H2: 22 mmol per gcatalyst per 12 h | QE: 3.4% at 365 nm | 12 hours | 49 |
| MeCN–water solution | Methanol, CO, and CO2 | |||||
| Cellulose | Pt/NCNCNx | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼190 mmol gcatalyst−1 h−1 | 24 hours | 50 | |
| LiBr and LiOH in 0.1 M H2SO4 solution | Formic acid, lactic acid, arabinose, erythrose, etc. | |||||
| Cellulose | NiP/carbon dots (<10 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼5 μmol per 24 h | 24 hours | 26 | |
| C6H12O6 and C6H10O5 compounds | ||||||
| Glucose | NiP/carbon dots (<10 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼6 μmol per 24 h | 24 hours | 26 | |
| C6H12O6 and C6H10O5 compounds | ||||||
| Cellulose | Pt/P25 TiO2 (20–30 nm) | UV–vis light (250 W iron doped halide lamp) | H2: ∼170 μmol per 10 h | 10 hours | 35 | |
| 0.6 M H2SO4 solution 403 K; 3.0 bar | HMF, erythrose, and arabinose | |||||
| α-Cellulose | CdS/CdOx quantum dots (diameter ∼5 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼2.57 mmol gcatalyst−1 h−1 | 6 days | 24 | |
| 10 M KOH | Formate, carboxylic acid, CO2, etc. | |||||
| Fructose | CdS/CdOx quantum dots | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼1070 mmol per gsubstrate per 20 h | QE: 2.73% at 430 nm | 20 hours | 45 |
| 10 M KOH | Formate, etc. | |||||
| Starch | CdS/CdOx quantum dots | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼462 mmol per gsubstrate per 20 h | 20 hours | 45 | |
| 10 M KOH | Formate, etc. | |||||
| Apple | CdS/CdOx quantum dots | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼6070 mmol gcatalyst−1 h−1 | 20 hours | 45 | |
| 10 M KOH | ||||||
| Cheese | CdS/CdOx quantum dots | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼9350 mmol gcatalyst−1 h−1 | 20 hours | 45 | |
| 10 M KOH | ||||||
| Artificial mixed waste | CdS/CdOx quantum dots | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼9900 mmol gcatalyst−1 h−1 | 20 hours | 45 | |
| 10 M KOH | ||||||
| Municipal waste | CdS/CdOx quantum dots | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼5920 mmol gcatalyst−1 h−1 | 20 hours | 45 | |
| 10 M KOH | ||||||
| Wooden branch | CdS/CdOx quantum dots (diameter ∼5 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼5.59 mmol gcatalyst−1 h−1 | 24 hours | 24 | |
| 10 M KOH | ||||||
| Municipal solid waste | CNx/Ni2P | Simulated sunlight (100 mW cm−2) | Formate, carbonate, etc. H2: 130 mmol h−1 m−2 | 20 hours | 39 | |
| 0.5 M KOH | ||||||
| PLA | CdS/CdOx quantum dots (diameter ∼5 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼64.3 mmol gcatalyst−1 h−1 | QE: 15.0% at 430 nm | 4 hours | 37 |
| 10 M NaOH | Pyruvate, etc. | |||||
| PET | CdS/CdOx quantum dots (diameter ∼5 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼3.42 mmol gcatalyst−1 h−1 | QE: 3.74% at 430 nm | 4 hours | 37 |
| 10 M NaOH | Formate, glycolate, ethanol, acetate, and lactate | |||||
| PUR | CdS/CdOx quantum dots (diameter ∼5 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼0.85 mmol gcatalyst−1 h−1 | QE: 0.14% at 430 nm | 4 hours | 37 |
| 10 M NaOH | Formate, acetate, pyruvate, and lactate | |||||
| PET water bottle | CdS/CdOx quantum dots(diameter ∼5 nm) | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼4.13 mmol gcatalyst−1 h−1 | QE: 2.17% at 430 nm | 24 hours | 37 |
| 10 M NaOH | Formate, glycolate, ethanol, acetate, lactate, glyoxylate, methylglyoxal, and methanol | Conversion: 5.15% | ||||
| Methanol | CoP/Zn2In2S5 nanosheets | Simulated sunlight (AM 1.5G) | H2: ∼21 mmol gcatalyst−1 h−1 | 12 hours | 67 | |
| Ethylene glycol: 18.9 mmol gcatalyst−1 h−1 | ||||||
| HCHO: ∼4 mmol gcatalyst−1 h−1 | ||||||
| Ethanol | CoP/Zn2In2S5 nanosheets | 300 W Xe lamp (λ = 400–780 nm) | H2: ∼8.5 mmol gcatalyst−1 h−1 | 12 hours | 67 | |
| 2,3-Butanediol: 3.2 mmol gcatalyst−1 h−1 | ||||||
| CH3CHO: ∼5 mmol gcatalyst−1 h−1 | ||||||
| Benzyl alcohol | Zn0.2In2S3.2 | 6 W LED (λ = 365 ± 5 nm) | H2 yield: 172% | 12 hours | 58 | |
| Benzoin yield: 61% | ||||||
| Deoxybenzoin yield: 30% | ||||||
| Benzaldehyde yield: 9% | ||||||
| Furfural alcohol | Ti3C2Tx/CdS nanosheets (∼60 nm) | 300 W Xe lamp (λ ≥ 420 nm) | H2: 773 μmol per gcatalyst per 4 h | 4 hours | 54 | |
| Furfural: 777 μmol per gcatalyst per 4 h | ||||||
| Cetophenone | Pd/TiO2 | 6 W LED (λ = 365 ± 5 nm) | H2: ∼0.25 mmol per 3 h | 3 hours | 109 | |
| Ethanol | Phenethanol: ∼0.55 mmol per 3 h | |||||
| Ethylbenzene: ∼0.05 mmol pre 3 h | ||||||
| Acetaldehyde: ∼0.4 mmol per 3 h | ||||||
| DMF | Ni/CdS nanosheets | 300 W Xe lamp (λ ≥ 420 nm) | H2: ∼1 mmol per 3 h | QE: ∼44% at 420 nm | 3 hours | 69 |
| N-Benzylidenebenzylamine: ∼2 mmol per 5 h | ||||||
| Benzyl alcohol | Zn3In2S6 (1–3 μm) | 300 W Xe lamp (λ > 380 nm) | H2: 708.8 μmol per 4 h | QE: 6.48% at 380 nm | 4 hours | 64 |
| Benzaldehyde: 732 μmol per 4 h | 5.46% at 400 nm | |||||
| 4.02% at 420 nm | ||||||
| 1.03% at 450 nm | ||||||
| DMA | Pt/CNx | 14.4 W blue LED (λmax = 470 nm) | H2: ∼12 μmol per 10 h | 10 hours | 59 | |
| Minisci oxidation products: ∼15 μmol per 10 h | ||||||
| THF | Pt/CNx | 14.4 W blue LED (λmax = 470 nm) | H2: ∼9 μmol per 10 h | 10 hours | 59 | |
| Minisci oxidation products: ∼9.5 μmol per 10 h | ||||||
| 2,5-DMF | Ru–ZnIn2S4 (0.6–1.6 μm) | 9 W blue LED (λ = 455 nm) | H2: ∼6.0 mmol gcatalyst−1 h−1 DFPs: ∼1.04 g gcatalyst−1 h−1 | QE: 15.2% at 452 nm | 6 hours | 68 |
| 2-MF | Ru–ZnIn2S4 (0.6–1.6 μm) | 9 W blue LED (λ = 455 nm) | H2: ∼1.2 mmol gcatalyst−1 h−1 DFPs: ∼0.1 g gcatalyst−1 h−1 | QE: 1.9% at 452 nm | 12 hours | 68 |
| HMF | Pt/porous carbon nitride | Simulated sunlight (λ > 400 nm, 100 mW cm−2) | H2: 12 μmol m−2 h−1 | DFF yield: 13.8% after 6 h | 6 hours | 53 |
| DFF: 1.3μmol m−2 h−1 | Selectivity >99% | |||||
| Benzylamine | Ni/CdS nanoparticles | 1.85 W Xe lamp (λ > 420 nm) | H2: 21.4 mmol gcatalyst−1 h−1 | Conversion: 99% | 45 minutes | 60 |
| Imine | Selectivity for imine: 97% | |||||
| QE: 11.2% at 450 nm | ||||||
| Benzyl mercaptane | PtS/ZnIn2S4 nanocomposites | 5 W LED (λ = 450 nm) | H2 yield: 100% | Conversion: ∼100% after 6 h | 6 hours | 61 |
| Dibenzyl disulfide | Selectivity of dibenzyl disulfide: ∼100% | |||||
| QE: 24.96% at 420 nm | ||||||
| Methanol | MoS2 foam/CdS nanorods | 300 W Xe lamp (λ = 420–780 nm) | H2: 12 mmol gcatalyst−1 h−1 | 12 hours | 66 | |
| Ethylene glycol, etc. | ||||||
| MBA | RuCat/Pt–g-C3N4 | 300 W Xe lamp (λ > 400 nm) | H2: 56 μmol per 10 h | QE: 0.28% at 400 nm | 10 hours | 65 |
| MBAd | ||||||
| Furfural alcohol | Ni/CdS nanosheets (thickness ∼1 nm) | 8 W blue LED (λ = 450 nm) | H2: ∼5 mL per 8 h | 8 hours | 52 | |
| Furfural | ||||||
| HMF | Ni/CdS nanosheets (thickness ∼1 nm) | 8 W blue LED (λ = 450 nm) | H2: ∼0.5 mL per 8 h | 8 hours | 52 | |
| DFF | ||||||
| 4-MBA | NiP/NCNCNx | Simulated sunlight (AM 1.5G, 100 mW cm−2) | H2: ∼21.3 μmol per 24 h | QE: 15% at 360 nm | 24 hours | 57 |
| 4-MBAd: ∼19.8 μmol per 24 h | ||||||
| H2O | CdS-ZnIn2S4 nanosheets | 300 W Xe lamp (λ > 400 nm) | H2: ∼0.5 mmol gcatalyst−1 h−1 | QE: 1.63% at 400 nm | 12 hours | 86 |
| H2O2: ∼0.6 mmol gcatalyst−1 h−1 | ||||||
| HI | MoS2/MAPbI3 | 280 W Xe lamp (λ ≥ 420 nm) | H2: ∼29.4 mmol gcatalyst−1 h−1 | QE: 13–22% in the range of 420–850 nm | 90 hours | 102 |
| I3− | STH: 7.35% | |||||
Initially, photocatalytic reforming research centered on the production of H2 from a variety of lignocellulose biomass-derived feedstocks, the most abundant and non-edible biomass resource, including monosaccharides, such as pentoses (ribose and arabinose) and hexoses (glucose, fructose, galactose, and mannose), alcohols (methanol, propanol, ethanol, and butanol) and organic acids (formic acid and acetic acid).12–19 Theoretically, in the proposed biomass photoreforming process, photoexcited holes lead to the oxidation of biomass components and derivatives (denoted as CxHyOz), accompanied by hydrogen generation through the reduction of protons by electrons (eqn (4)). The irreversible reaction of CxHyOz with photogenerated holes results in the concomitant suppression of electron–hole recombination and back reactions involving the produced H2, thereby increasing the quantum efficiency and the rate of H2 evolution.13
Nevertheless, lignocellulose refining is costly and inefficient, typically necessitating acid hydrolysis, enzymatic hydrolysis, or pyrolysis to produce more manageable substrates.20 Therefore, viable H2 production systems should directly reform lignocellulose. The main components of lignocellulosic biomass are cellulose (35–50%), hemicellulose (25–30%), and lignin (15–30%).21,22 Cellulose,23–28 hemicellulose24,29,30 and lignin24,26,31,32 have been reported to produce H2 using various photocatalysts such as TiO2, CdS/CdOx quantum dots, carbon dots, carbon nitride, and CdS (Table 1). Metallic nanoparticulate cocatalysts (e.g., Pt, Au, and Pd) and non-precious cocatalysts (e.g., NiP and NiS) are frequently loaded on photosensitizers by wet impregnation and photodeposition methods, with Pt the most commonly studied.33 As an example, CdS/CdOx quantum dots loaded Co species as cocatalysts were reported to perform photoreforming of cellulose, hemicellulose and lignin to H2 in alkaline solution under simulated sunlight irradiation (H2 evolution rates for cellulose, hemicellulose, and lignin were 2.57, 2.32, and 0.53 mmol gcatalyst−1 h−1, respectively).24
Reports on using raw biomass as the substrate for photoreforming are limited. Although direct H2 production from unprocessed biomass has the advantage of lower production costs, it is limited by low substrate solubility. Additionally, light is scattered and absorbed by insoluble biomass and colored components, lowing the light absorption efficiency. The recalcitrance of raw biomass may lead to a large overpotential for biomass oxidation.12 TiO2 is frequently used as a photocatalyst for raw biomass reforming, despite its large bandgap (around 3.2 eV) and limited sunlight utilization ability. The H2 evolution rates of TiO2 for the photoreforming of raw biomass, such as poplar wood chips,27 rice husk,34 paper pulp,35 chlorella algae,36 and turf,36 are in the range of 0.01–0.1 mmol gcatalyst−1 h−1, which are typically lower than those obtained using carbon nitride and CdS with narrower bandgaps (carbon nitride: ∼2.7 eV; CdS: ∼2.4 eV) (Table 1).24,29 For instance, a rate of over 5 mmol H2 gcatalyst−1 h−1 was reported through photoreforming a wooden branch on Co/CdS/CdOx, as shown in Fig. 2. The system was stable for more than 6 days and was capable of reforming unprocessed lignocellulose, demonstrating a cost-effective approach to produce H2 from waste biomass. The high H2 evolution rate may also be attributed to the alkaline conditions that not only provide an in situ pretreatment of lignocellulose by dissolving hemicellulose, lignin, cellulose, and other saccharides but also facilitate CdOx deposition on CdS, thereby producing a photocatalyst that is active and resistant to corrosion.24
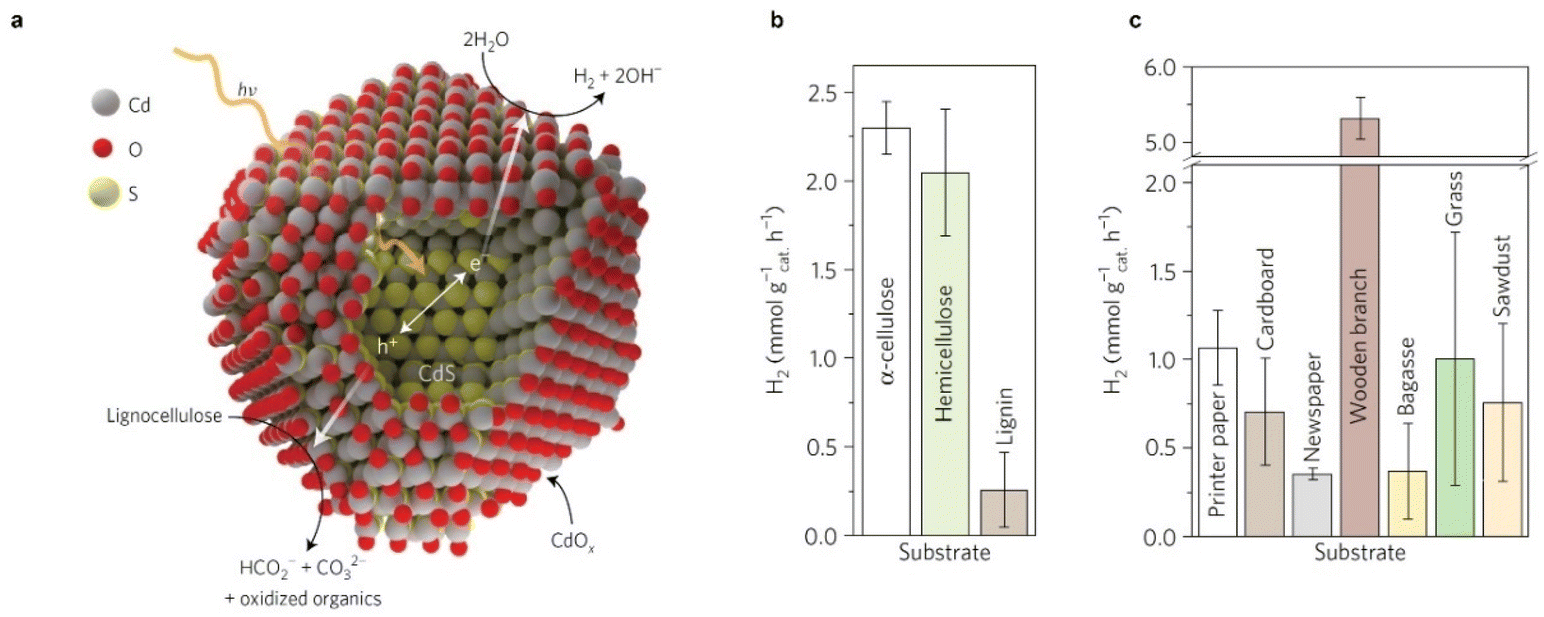 | ||
| Fig. 2 (a) Diagram of the photoreforming of lignocellulose to H2 on CdS/CdOx. (b and c) Photocatalytic production rates of H2 from α-cellulose, hemicellulose and lignin (b), and raw and waste biomass substrates (c) using Co/CdS/CdOx quantum dots in 10 M KOH aqueous solution under simulated solar light (AM 1.5G, 100 mW cm−2). Reproduced with permission from ref. 24. Copyright 2017 Macmillan Publishers Limited, part of Springer Nature. | ||
The ideal feedstock for photoreforming comes from waste streams that cannot be recycled or reused in any other way.11 To this end, the photooxidation of food waste and nonrecycled plastics has recently become an emerging topic in the field.11,37–44 The chemical composition of food waste varies greatly. Due to their hydrophobicity and typically chemically inert hydrocarbon chain, photoreforming fats in an aqueous solution is more difficult than photoreforming proteins composed of long chains of amino acid residues (Table 1).11 Under simulated solar irradiation, simultaneous production of H2 and the oxidation of a variety of carbohydrates, proteins and fats as well as real-world waste (apples, bread, and cheese) to generate formate and CO2 or carbonate was observed over CdS/CdOx quantum dots and Ni2P/CNx.45 The carbon footprints of these processes were estimated to be 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600–68800 gCO2 per kWh H2 with a conversion efficiency of 1.9–22% after 3 days, which were still high due to the need for alkaline solutions as well as energy for stirring, and pre-treatment (accounts for more than 90% of the values).45 The carbon footprint can be reduced to a negative value of −3200 gCO2 per kWh H2 if the waste in the water is 100% converted to H2 and formate. Therefore, improvements in the conversion efficiency and photooxidation process selectivity are required.
600–68800 gCO2 per kWh H2 with a conversion efficiency of 1.9–22% after 3 days, which were still high due to the need for alkaline solutions as well as energy for stirring, and pre-treatment (accounts for more than 90% of the values).45 The carbon footprint can be reduced to a negative value of −3200 gCO2 per kWh H2 if the waste in the water is 100% converted to H2 and formate. Therefore, improvements in the conversion efficiency and photooxidation process selectivity are required.
In comparison with the photodegradation of plastics that converts plastics into CO2 and H2O, photorefoming can not only reduce plastic pollution but also produce fuels and products with added value, including pyruvate (∼190 $ per kg), glycolate (∼450 $ per kg), formic acid (∼1.1 $ per kg), ethanol (∼1.2 $ per kg) and acetic acid (∼1.2 $ per kg) (Table 1),9 allowing for a substantial economic advantage and a higher atom economy. While hydrocarbon chains such as polyethylene (PE), polystyrene (PS), and polypropylene (PP) in plastics are currently difficult to reform due to their highly stable C–C bonds, the oxygen-containing esters including polyethylene terephthalate (PET) (eqn (5) and (6)), polylactic acid (PLA) (eqn (7) and (8)) and polyurethane (PUR) (eqn (9) and (10)) can facilitate photoreforming after proper hydrolysis treatment.37
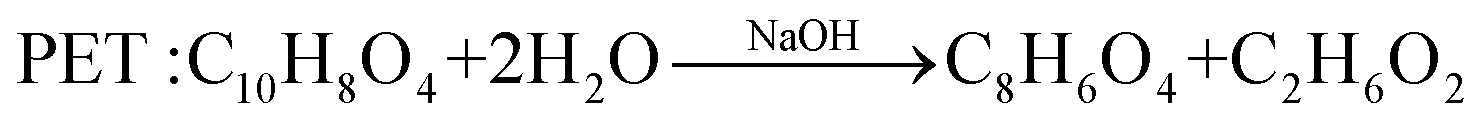 | (5) |
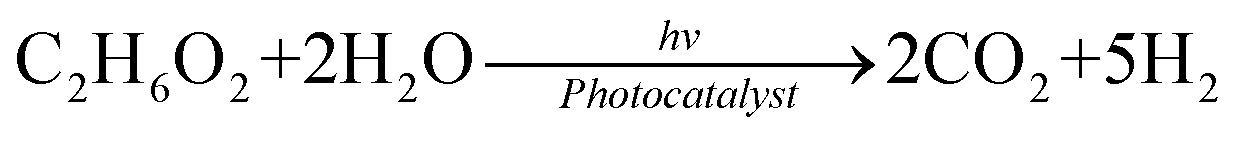 | (6) |
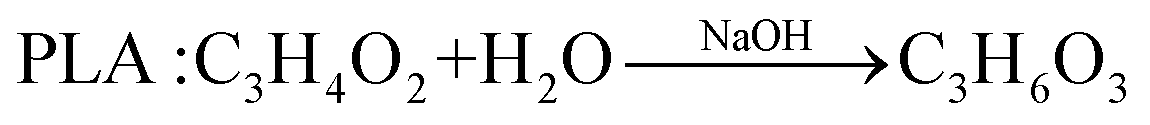 | (7) |
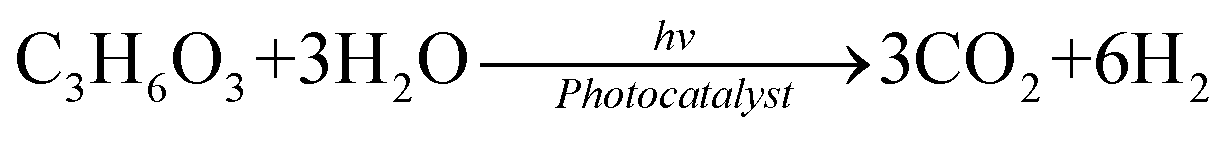 | (8) |
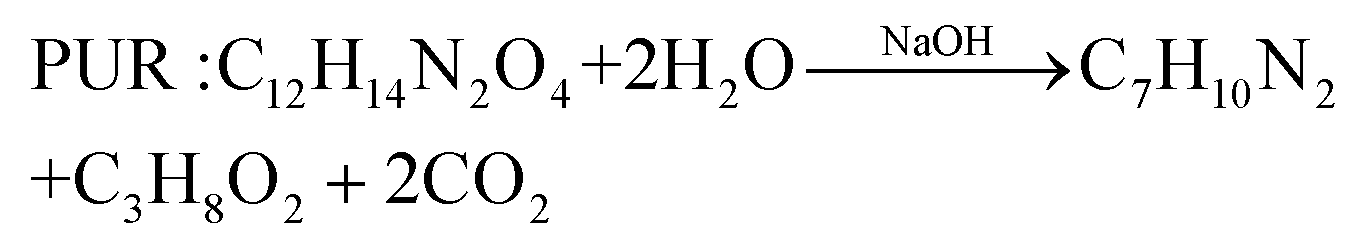 | (9) |
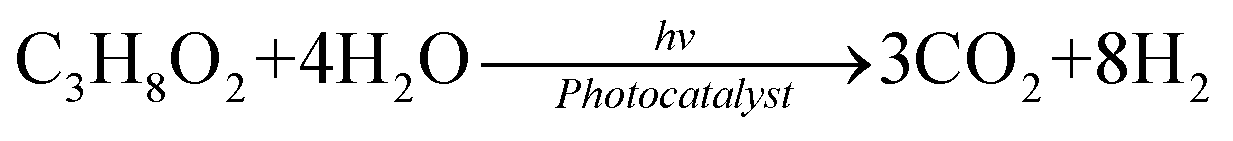 | (10) |
The reforming of plastics conjugating with proton reduction to H2 is also nearly energy neutral.40 For instance, the Gibbs free energy changes for the reforming of ethylene glycol (a monomer of PET) and lactic acid (a monomer of PLA) are +9.2 kJ mol−1 and +27 kJ mol−1, respectively. A variety of plastics including PLA, PET, PUR, and a PET water bottle were oxidized by photogenerated holes in CdS/CdOx quantum dots into a range of substrate-dependent organic products (e.g., formate, glycolate, ethanol, acetate, and pyruvate) in 10 M NaOH aqueous solution, while the photogenerated electrons reduced protons to produce H2 with a rate of 3–65 mmol gcatalyst−1 h−1 (Table 1, and Fig. 3).37 A Ni2P/CNx photocatalyst which is inexpensive and nontoxic has also shown promising activities for visible-light-driven PET and PLA reforming to produce H2 fuel and a variety of organic chemicals under alkaline aqueous conditions.46 The challenge in plastic photoreforming is the competition with plastic dehydrogenation to H2 and selective oxidation to a single product rather than CO2.47 The reported conversion of plastics was up to 30%–40%, which requires further improvement. Furthermore, these studies imply that photoreactions can cleave C–C, C–N, and C–O bonds in the aliphatic chains of plastics as effectively as hydrolysis.
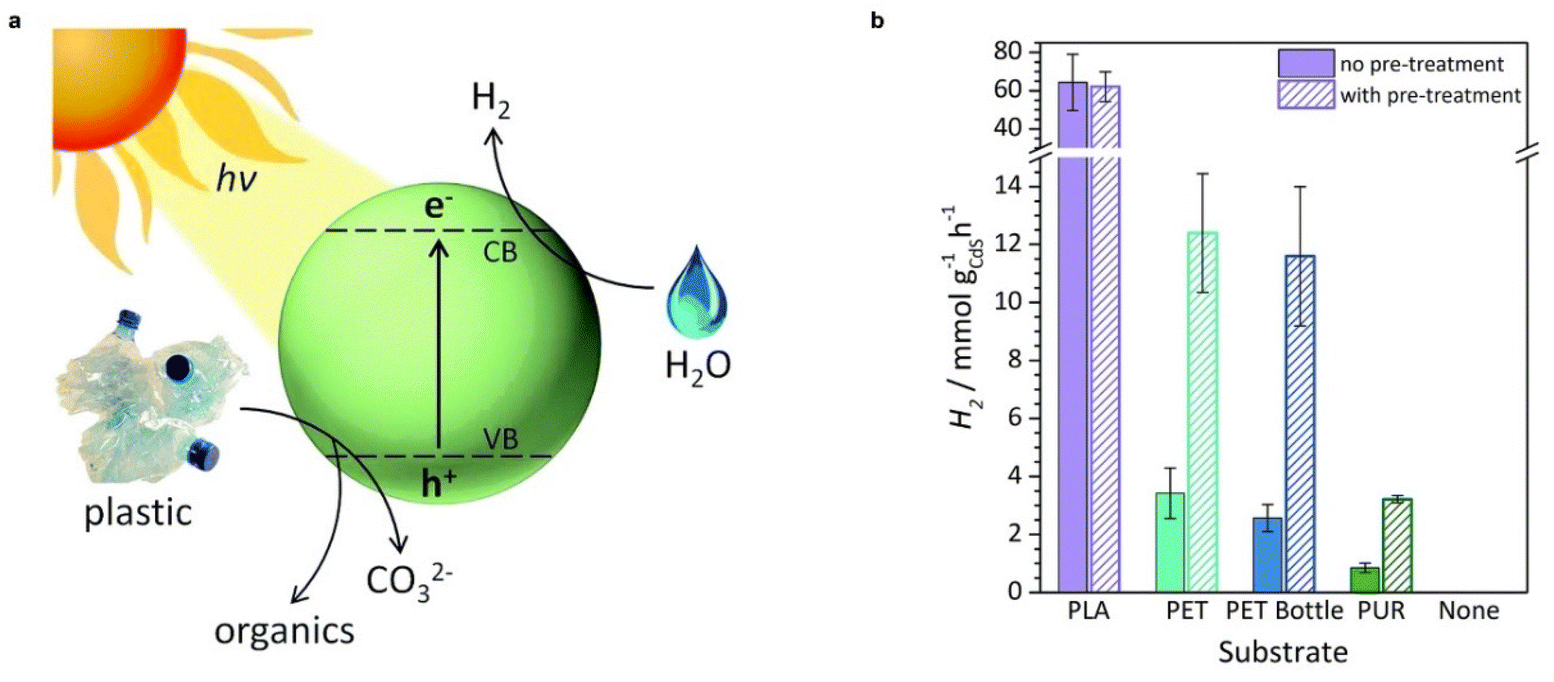 | ||
| Fig. 3 (a) Diagram of the photoreforming of plastic waste using a CdS/CdOx quantum dot photocatalyst. (b) Photoreforming of polymers to H2 using CdS/CdOx quantum dots under simulated solar light (AM 1.5G, 100 mW cm−2). Reproduced with permission from ref. 37. Copyright 2018 The Royal Society of Chemistry. | ||
It is suggested that a theoretical maximum of 310–650 Mt of H2 could be produced each year (the equivalent of ∼6–13% of annual global energy consumption) if the above biomass, food and plastic components were all utilized for photoreforming.11 The preliminary techno-economic and life cycle assessment of photoreforming has shown that it has a lower carbon footprint than or is comparable to existing methods for producing H2, converting waste to fuel, and managing waste, but the improvement in production costs and energy balance is still required before industrial applications can be envisaged. Although the product of the oxidation reaction in photoreforming is typically CO2, it is preferred that valuable organic chemicals can be produced instead of CO2 to improve the sustainability and overall process value of the system.26,28,37,46,48–50 For example, Cu dispersed on titanium oxide nanorods was reported to be effective for the conversion of polyols and sugars into syngas and methanol under UV light irradiation and ambient conditions.49 Decreased water content inhibited the formation of CO2, whereas the copper loading amount controlled the decomposition way of the formic acid intermediate. Controlling the oxidation half-reaction is essential to produce high-value organic products instead of CO2.21,41 In addition, pre-treatment, including physical pre-treatment (such as crushing, shredding, and grinding) and chemical pre-treatment (such as hydrolysis and solubilization under highly alkaline or acidic conditions), is one of the most critical barriers to the practical application of the current photocatalytic reforming systems, adding significant cost and time to the overall process. Recently, it was demonstrated that metal salt hydrate solutions allowed for the complete solubilization of biomass and could be used as a reaction medium for the photocatalytic reforming of lignocellulose to produce H2 and organic products under more benign conditions than the typically required extremely alkaline aqueous solutions, though the photocatalysts suffered from partial deactivation due to metal salt hydrate adsorption, necessitating future development.50
Organic transformation
Since the pioneering work of Kolbe that reported the electrochemical oxidation of organic molecules with the goal of developing new methods for synthesizing organic small molecules,51 a larger body of research has grown around this topic in the ensuing decades. Over the past decade, photocatalytic processes that use light as the energy input have emerged as a new alternative approach in the repertoire of the organic synthesis toolbox. In particular, interest has been aroused in waste biomass valorization in order to produce high-value commodity chemicals instead of CO2. Researchers have succeeded in converting biomass-derived small molecules such as 5-hydroxymethylfurfural (HMF) and furfural alcohol into higher-value materials (e.g., aldehydes and acids) (Fig. 4a).52–54 For example, photocatalytic H2 evolution coupled with HMF (7110 $ per kg) oxidation to 2,5-diformylfuran (DFF) (198000 $ per kg)55 using porous carbon nitride was reported to reach a DFF yield of 13.8% with >99% selectivity after 6 h under visible light.53
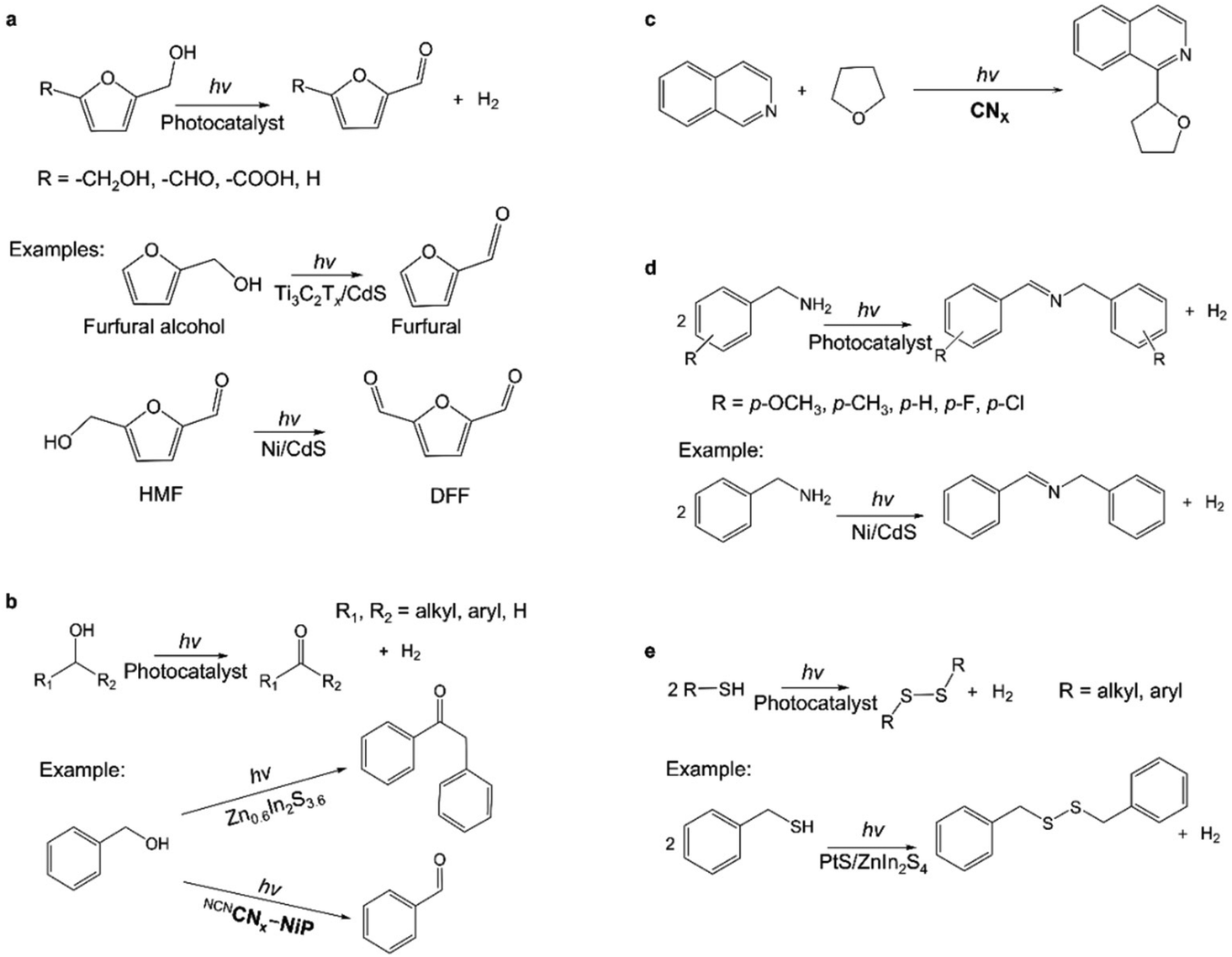 | ||
| Fig. 4 (a) Photocatalytic oxidation of biomass-derived intermediate compounds.52,54,56 HMF: 5-hydroxymethylfurfural; DFF: 2,5-diformylfuran. (b) Photocatalytic oxidation of alcohols.57,58 (c) Photocatalytic Minisci coupling.59 (d) Photocatalytic C–N coupling reactions.56,60 (e) Photocatalytic S–S coupling reactions.56,61 | ||
Recent research has revealed a wide range of photocatalysts for oxidizing alcohols in conjunction with H2 production.57,58,62–65 Alcohols such as methanol have been widely used as sacrificial reagents for photocatalytic H2 generation from water due to the lower thermodynamic requirements, and alcohols are oxidized to CO2.3–5 We will not discuss these examples because the focus of this work is on the co-production of high-value chemicals with H2. For instance, under visible light irradiation, the coupling of H2 generation with the oxidation of benzyl alcohol (273 $ per kg) yielded deoxybenzoin (57200 $ per kg) over ZnIn sulfides,55,58 and 4-methylbenzyl alcohol (2110 $ per kg) was converted into an aldehyde (4-methylbenzaldehyde; 3440 $ per kg) over a cyanamide surface functionalized with melon-type carbon nitride.55,57 Multi-carbon alcohols and polyalcohols have been reported to be photocatalytically produced by selectively activating the inert sp3 α-C–H bonds of alcohols and directly forming C–C bonds with the coupling partners (Fig. 4b).62 For instance, Zn2In2S5 and CdS were reported as visible-light-responsive photocatalysts for the selective activation of the α-C–H bond of methanol to generate ethylene glycol with a selectivity of as high as 90%.66,67 The CdS-based photocatalytic system was beneficial for the activation of the C−H bond in methanol without affecting the O–H group, forming ethylene glycol via a ˙CH2OH radical intermediate with a yield of 16% and a quantum efficiency (QE) of above 5.0% at wavelengths ≤450 nm.66 The high ethylene glycol selectivity (90%) was maintained throughout the 100-hour reaction, with a yield of 16% after 100 hours, demonstrating an alternative nonpetroleum strategy for ethylene glycol synthesis. The CoP/Zn2In2S5 photocatalyst also was demonstrated to be the first example of the visible-light-driven dehydrogenative coupling of ethanol to 2,3-butanediol with a selectivity of 53%.67 The α-C–H bond of ethanol was selectively activated to form ˙CH(OH)CH3 radicals for subsequent coupling to 2,3-butanediol, presenting that the α-C–H bond can be preferentially activated in the presence of the O–H group under mild conditions.
Aside from alcohols, the C–H bonds in furanics can also be activated for C–C coupling reactions through photocatalysis. The production of diesel fuel precursors from 2,5-dimethylfuran (2,5-DMF) and 2-methylfuran (2-MF), with concurrent H2 production, over a Ru–ZnIn2S4 photocatalyst was demonstrated under visible light irradiation (Fig. 5).68 Dehydrocoupling of 2,5-DMF and 2-MF yielded mainly H2, dimers and trimers as products with a small number of tetramers produced in 12 h. Photogenerated holes oxidized the furfuryl C–H bond of 2,5-DMF/2-MF, delivering protons and furfuryl radicals that, through C–C coupling, form the desired DFPs. Over Ru–ZnIn2S4, the QE for the dehydrocoupling of 2,5-DMF was 15.2% at 452 nm, which was approximately double that of ZnIn2S4 because Ru dopants substituted for indium ions in the ZnIn2S4 matrix facilitated charge separation efficiency and thus accelerated C–H activation. Diesel fuel precursors were produced with selectivities of more than 96% in conjunction with H2 generation and were subsequently transformed into diesel fuels composed of straight- and branched-chain alkanes that were comparable to petroleum diesel in terms of alkane constituents. Future work should focus on improving the yields of C–C coupling products and the QE by the use of more efficient catalysts.
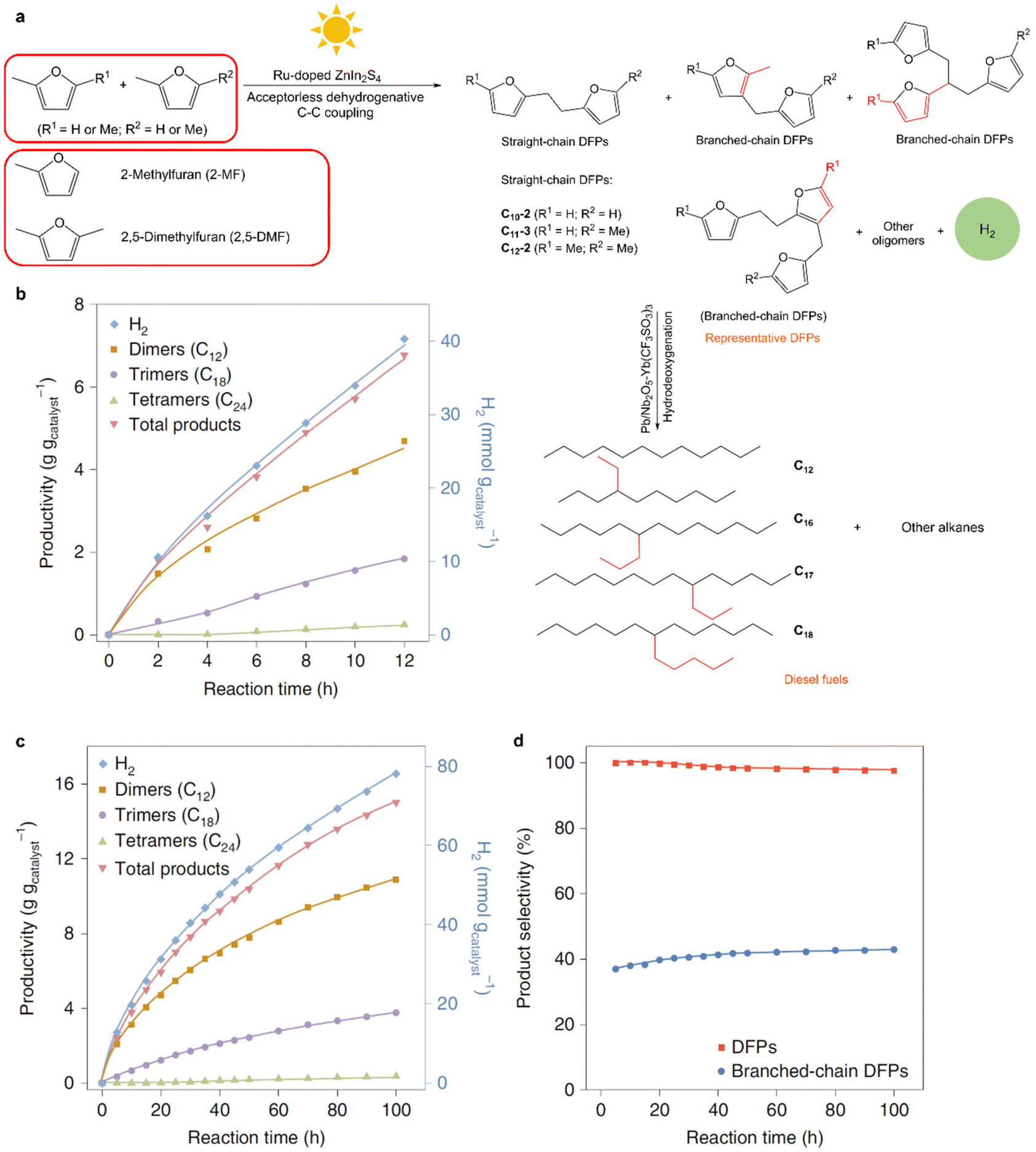 | ||
| Fig. 5 (a) Schematic representation of the photocatalytic transformation of 2,5-DMF/2-MF into diesel fuel. (b–d) Photocatalytic dehydrocoupling of 2,5-DMF as a substrate: standard experiment (b), catalyst lifetime evaluation (c), and selectivity to oxygenated DFPs and branched-chain DFPs (d). Reproduced with permission from ref. 68. Copyright 2019 Springer Nature Limited. | ||
Lately, it has been demonstrated that H2 production could be coupled with more complex organic synthesis. The Minisci reaction is a known strategy for the direct functionalization of C–H bonds in heteroarenes, involving the radical coupling of electron-deficient heteroarenes with oxidatively-generated nucleophilic radicals.59 A visible light-mediated Minisci coupling of N-heteroarenes with ethers, alcohols, and amides in the presence of aerobic oxygen as an oxidant was reported to conjugate with proton reduction to H2 using a cyanamide functionalized carbon nitride photocatalyst (Fig. 4c).59
Photocatalytic C–N coupling of amines for the efficient synthesis of imines may have important applications in pharmaceutical and agricultural chemistry research (Fig. 4d).56 Ni/CdS was reported to photocatalyze the non-oxygen coupling of amines to yield their corresponding imines under visible light irradiation, concurrent with H2 generation.60,69 In particular, the photocatalyst showed a high conversion of benzylamine (99%) with 97% selectivity for imines, associated with a QE of 11.2% at 450 nm for simultaneous H2 evolution.60 By constructing in situ photoimmobilized Ni clusters on two-dimensional ultrathin CdS nanosheets, the generation of imines was dramatically improved with a QE of ∼44% at 420 nm for the conversion of 4-methoxybenzylamine.69 It was proposed that photogenerated holes initiated the oxidation of amines via the deprotonation process with the creation of C-centered α-amine radicals to form aldimine intermediates and release protons. The protons were reduced by the photogenerated electrons and produced H2 with the assistance of a Ni cocatalyst. Additionally, the photocatalytic system converted a wide range of primary and secondary amines (i.e., heterocyclic, aliphatic, and N-heterocycles) to form their corresponding imines with high yields (≥84%) and selectivity (≥95%), thereby offering a promising and versatile pathway for organic chemical synthesis.
The photocatalytic synthesis of disulfides through the coupling of thiols is also of interest because disulfides function as protecting groups in synthetic applications and as vulcanizing agents for rubber (Fig. 4e). One of the examples is the visible-light irradiation of CdSe quantum dots to result in virtually quantitative coupling of a variety of thiols, exemplified by 3-mercaptopropionic acid (MPA), to produce disulfides and H2 in aqueous solution.70 The conversion of MPA reached 99% in 1.5 hours and the yield of 3,3′-dithiodipropanoic acid was near 100%. Likewise, PtS/ZnIn2S4 completely transformed benzyl mercaptan to produce dibenzyl disulfide in 6 h with a selectivity of ∼100% under optimal conditions.61 These examples demonstrated an attractive alternative to existing procedures for synthesizing disulfides from thiols.
Controlling the selectivity of products is the most important issue of organic synthesis, as well as the existence of various potential reactive oxygen species in photocatalysis, including superoxide radicals (˙O2−), hydrogen peroxide (H2O2), hydroxyl radicals (˙OH) and singlet oxygen (1O2).71 For selective oxidation, the reactive oxygen species and adsorption–desorption behavior must be carefully controlled.
Inorganic oxidation
Photocatalytic H2 production along with more valuable inorganic products rather than O2via a two-electron oxidation pathway represents an alternatively appealing way. For instance, hydrogen peroxide (H2O2) production from water via the two-electron oxidation process is kinetically more favorable than the water-splitting reaction, which requires four electrons to produce O2, despite the O2 generation (1.23 eV vs. NHE) showing a thermodynamic advantage over the H2O2 production (1.78 eV vs. NHE). The photocatalytic reaction to simultaneously produce H2 and H2O2 from water (eqn (11)) has attracted increasing attention in recent years because it obtains clean H2 energy by utilizing abundant water resources and inexhaustible sunlight,72,73 as well as being environmentally friendly for producing the value-added H2O2 (∼0.7 $ per kg),9 which was widely utilized in the chemical industry, and disinfection, energy, and environmental fields.74–77 In addition, this reaction not only realizes the automatic separation of the oxidation product with gaseous H2 but also effectively avoids the reverse reaction of generated H2 and O2 existing in the thoroughly overall water splitting reaction.78,79| 2H2O → H2 + H2O2 | (11) |
Recent efforts, including optimizing reaction conditions, doping, heterojunctions, cocatalyst loading, and using biological hybrids, have been made to enhance the carrier utilization efficiency of the photocatalyst and increase the selectivity for H2O2 production, thereby advancing this technology. Graphitic carbon nitride (g-C3N4), for example, as a notable photocatalyst with advantageous features of low cost and tunable optical and electronic properties was widely employed in this field. A sulfur doping treatment was performed on thin g-C3N4 nanosheets to compensate for the light absorption loss caused by the exfoliation process while improving the charge transfer efficiency.80 Moreover, CoxNiyP nanoclusters were loaded onto a P-doped g-C3N4 photocatalyst, and the doping and cocatalyst effects enhanced the photocatalytic water reduction and oxidation with a H2 evolution rate of 239.3 μmol gcatalyst−1 h−1 and stoichiometric H2O2 production.81 It is also reported that the living Chlorella vulgaris could accelerate the photocatalytic activity of g-C3N4 to produce H2O2 and H2 from water, giving a QE of 0.86% at 420 nm.82 The defect control is also a proposed way applicable to increase H2O2 production. Although defects are typically regarded as charge recombination sites, it has been demonstrated that if properly controlled, they can also contribute to photocatalytic reactions.83 Defects can introduce mid-gap states in the forbidden band of semiconductor photocatalysts to harness more visible light, serve as active sites for reactants, and offer more trapping sites to hamper the recombination of carriers.84 For example, the introduced carbon vacancies in C3N4 can not only extend the light absorption range and improve carrier separation efficiency but also alter the H2O2 production pathway from a two-step indirect reaction to a one-step direct reaction, thereby enhancing the H2O2 production.85 Construction of heterojunctions appears to be an effective strategy for inorganic semiconductor photocatalysts to generate H2 and H2O2 simultaneously. For instance, a system consisting of CdS and ZnIn2S4 hollow cubes was employed for the stoichiometric generation of H2 and H2O2 from pure water, achieving a QE of 1.63% at 400 nm.86 Noble metal deposition remains a common but effective way to promote photocatalytic reactions. Production of H2 and H2O2 with rates of 9.8 μmol mgcatalyst−1 h−1 and 8.2 μmol mgcatalyst−1 h−1, respectively, from pure water (pH ∼ 7) was realized using Pt/porous brookite TiO2 nanoflutes as a photocatalyst, and the QE reached 43.4% at 365 nm.87 The simulation and experimental results revealed that a two-electron reaction is kinetically favorable on the surface of brookite TiO2 due to the unique surficial chemistry micro-circumstance, promoting the adsorption of activated hydroxyl groups and the subsequent step of hydrogen peroxide generation.
Though some promising progress has been made, photocatalytic H2O2 production is still far from the market requirement for practical applications, and the industrial production of H2O2 still mainly relies on the anthraquinone oxidation process (accounting for over 95% of production).88 Facing the same problems with other semiconductor photocatalysis, photocatalytic H2O2 production also suffers from the drawbacks of low light utilization ability and the high recombination of photogenerated carriers. Another tricky problem is the rapid decomposition of the generated H2O2 occurs at the surface of the photocatalyst/cocatalyst, which can be caused by UV irradiation. More efforts should be devoted to the surface modification and fine control of the photocatalyst/cocatalyst and new cocatalyst development to promote H2O2 evolution and inhibit its decomposition process. For example, the combination of common inorganic semiconductor photocatalysts with bacteria, biomaterials, and organic semiconductors may have some positive effects. Additionally, a deeper understanding of the photocatalytic mechanism and H2O2 evolution process is significant and will contribute to the optimization and design of highly active photocatalysts.
| 2HX → H2 + X2 | (12) |
| 2Cl− → Cl2 + 2e− E° = +1.36 V | (13) |
| 2Br− → Br2 + 2e− E° = +1.09 V | (14) |
| 2I− → I2 + 2e− E° = +0.54 V | (15) |
Photocatalytic hydrohalic acid (HX, X = Cl−, Br−, I−) splitting (eqn (12)) is another attractive approach for producing H2 with a valuable byproduct X2, which has numerous applications in the energy and hygiene industries.89–91 Halide ion (Br−, I−) oxidation (eqn (13)–(15)) is thermodynamically much easier than H2O oxidation, and more importantly, these photocatalytic reactions are conducted under the conditions of a relatively high H+/X− concentration, allowing for a faster reaction rate in dynamics. In addition, the oxidation of halide ions does not involve a net proton transfer, distinct from the H–O breakage and proton loss during O2 formation in H2O splitting, and thus are kinetically favorable. As an example, chloride oxidation catalyzed by silver ions in concentrated Cl− solution has been reported, and the chloride complex-assisted catalysis mechanism has been proposed.92 The research on chloride oxidation could promote the development of photocatalytic seawater splitting, which is still in its infancy due to its low product yield and a plausible catalytic mechanism involving the effect of inorganic ions, yet having been studied for years.93,94
Photocatalytic HBr and HI splitting to produce H2 based on halide perovskites has aroused a new research upsurge in recent years since the first report about the hydrogen generation from hydriodic acid using MAPbI3 as a photocatalyst in 2016 (Fig. 6a–c).95 Subsequently, material compositing and heterojunction construction were extensively employed to boost hydrohalic acid splitting. Reduced graphene oxide (rGO) was first used to composite with MAPbI3 perovskites for the photocatalytic hydrogen evolution reaction, resulting in a hydrogen evolution rate (93.9 μmol h−1 under visible-light irradiation) 67 times higher than that of pure MAPbI3.96 Afterwards, various materials such as TiO2, Ni3C, carbon dots, black phosphorus, MoS2, and CoP were used as cocatalysts or heterojunction counterparts to promote carrier separation.97–103 In particular, a MoS2/MAPbI3 composite exhibited a remarkable photocatalytic hydrogen evolution activity of 29.4 μmol gcatalyst−1 h−1 under visible light and an H2 production of 103 mL day−1 under outdoor natural sunlight irradiation.103 In addition to the heterostructures, composition regulation is also a feasible way. For instance, a graded and gradient halide ion distribution was realized in both organometal and all-inorganic mixed halide perovskites,104,105 and a resulting bandgap funneling was constructed for efficient carrier transfer, causing an enhanced photocatalytic activity and a solar-to-H2 conversion efficiency (STH) of 1.05% for MAPbBr3−xIx/Pt.104 In addition, single-atom decoration is also an effective strategy to improve the perovskite photocatalytic HI splitting efficiency,106,107 an excellent photocatalytic H2 evolution rate of 682.6 mmol h−1 under simulated sunlight irradiation (area of π cm) and a corresponding STH of 4.5% were recorded using Pt/FAPbBr3−xIx (Fig. 6d and e).107
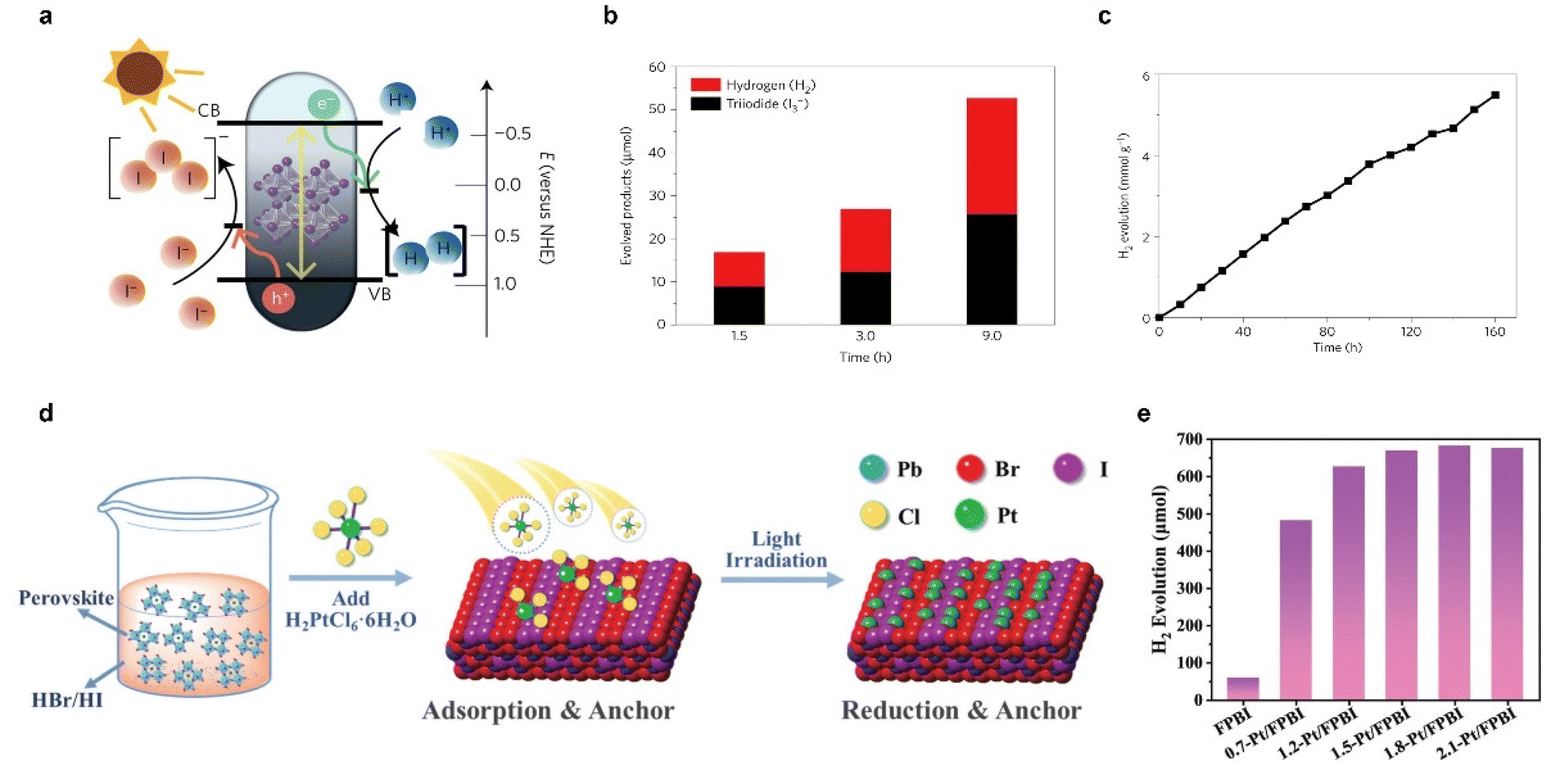 | ||
| Fig. 6 (a) Schematic band diagram of MAPbI3 powder for the HI splitting photocatalytic reaction. (b) Quantitative comparison between the evolved H2 and I3−. (c) Stable photocatalytic H2 evolution produced by MAPbI3 powder in the saturated solution for 160 h (H3PO2 was added to the HI solution). Reproduced with permission from ref. 95. Copyright 2016 Springer Nature Limited. (d) Mechanism illustration of the synthesis process of Pt/FAPbBr3−xIx. (e) Photocatalytic H2 evolution activities of ω-FPBI (FAPbBr3−xIx loaded ω% Pt in mass percentage). Reproduced with permission from ref. 107. Copyright 2022 The Royal Society of Chemistry. | ||
Halide perovskites possess several excellent material properties, such as a wide light absorption range, a high absorption coefficient, and a long carrier diffusion length, allowing them to be ideal photocatalysts. However, satisfactory performance of this kind of material for photocatalytic H2 production has not been achieved. More efforts should be devoted to the investigation of the mechanism and fundamental principles of the physical/chemical process during photocatalysis for this special system, where exists a dynamic equilibrium of dissolution-crystallization between the catalyst and the corresponding saturated solution.
Conclusions and outlook
Although the research in this field is at too early a stage to be towards industry-level operation and deployment, exciting work in combining H2 fuel generation with value-added chemical production or waste valorization is already showing to have a wide impact on large-scale challenges in global energy (Table 1). When looking at both inorganic and organic chemistries, the water-to-O2 conversion can be replaced by a variety of photoreactions that can occur in an aqueous solution.In any proton-to-H2 generation, the availability of substrates at the scale required for the process is a key consideration. Biomass, food, and plastic wastes are abundant, inexpensive, and carbon- and hydrogen-rich feedstocks. Photocatalytic methods offer upcycling routes that yield higher-value products, as opposed to conventional mechanical strategies that typically downcycle to lower-quality products.11,40 In recent years, much effort has been devoted to the study of the simultaneous production of H2 and chemicals with added value through reduction and oxidation, respectively. A preliminary techno-economic assessment demonstrates that photoreforming of waste can become economically competitive with existing technologies if the efficiency and lifetime can be substantially improved.11 For large-scale applications to be economically and environmentally viable, H2 evolution rates must increase by a factor of at least 50 (rates of >0.004 mol H2 gsubstrate−1 h−1, conversions of >50% per day, and QEs of >50%). It will be imperative to develop more effective and durable photocatalytic systems. In addition, the current pre-treatments commonly reported for photoreforming to improve the solubilization of the feedstock are unlikely to be economically viable on a large scale.
The photocatalytic oxidation of organic compounds, including alcohols, hydrocarbons, aromatics, and amines, differs from conventional methods conducted at high temperatures and pressures in that it is environmentally friendly, operates under mild conditions, and is sustainable. However, selective photocatalytic oxidations present huge challenges. Ideally, the oxidation process would selectively produce a single high-value product. Controlling reactive oxidation species and adsorption–desorption behavior is the primary challenge in selective oxidation. Therefore, it may be necessary to observe the reaction mechanism in situ for a better understanding of the reaction kinetics.71 To yield a single higher-value product in both organic transformation and waste valorization processes, selective oxidation cocatalysts may be required.
For inorganic reactions, the H2O-to-H2O2 conversion and halide oxidation present themselves as an attractive alternative to O2 evolution due to the higher value of the products and the simpler mechanism resulting from the lower electron demand. Nonetheless, the reaction efficiency of these reactions must be enhanced.
The H2 production cost for a waste photoreforming pilot plant (400 m2, 200 kgwaste per day, 14.4 kgH2 per day) was estimated to be £11.80 per kg, which is more than H2 sale targets of £3–5 per kg and predictions for H2 produced by photocatalytic water splitting (£1–3 per kg).1,11 It can, however, be reduced by increasing the plant scale and photoreforming efficiency. If the plant scale is enlarged 30 times, the cost of producing H2 can be decreased to £3 per kg. Furthermore, economic gains can be realized by producing high-value compounds such as pharmaceutical components at the same time. As an example, oxidizing biomass to 3-hydroxybutyrolactone (3-HBL), a precursor for chiral drugs, could cut H2 production costs to £3 per kg even if the photoreforming conversion efficiency is as low as 0.2% per day. However, although 3-HBL and its hydrolyzed form 3,4-dihydroxybutyric acid (3,4-DHBA) have been reported to be produced from biomass (glucose, glycolic acid, and xylose) via biological synthesis using bacteria (Escherichia coli), enzymes and yeast, its production via a photocatalytic process has not yet been realized.110–112 Future efforts would combine photocatalysts with microbes to create abiotic–biotic hybrids capable of producing photocatalytic H2 while oxidizing biomass to 3-HBL. Likewise, the H2 production costs can be reduced by photocatalytic transformations to produce value-added organic and inorganic chemicals. For instance, a recent techno-economic analysis suggested that by oxidizing water to produce H2O2 instead of O2 using the photoelectrochemical technique, a levelized cost of hydrogen of $6.19 per kg can be obtained with an STH of 10.1% and an H2O2 price of $0.85 per kg, which is a significant improvement over the values found in an analogy of the photoelectrochemical water splitting system.113 Aside from producing cost-effective H2, the procedures would also reduce the cost of organic and inorganic products. A minimal selling price of glucaric acid was predicted to be $6.94 per kg under a base case scenario employing a potential photo-biorefinery approach for sunlight-driven electro-oxidation of glucose, which was 70% lower than the current market price.114
In spite of the obstacles listed in Table 2, significant progress has been made in the study of coupling H2 production with diverse oxidation chemistry and will continue. With continued material and technological advancements, these technologies have the potential to produce fuels and chemicals with added value, while also reducing waste.
| Oxidation reaction | Substrates | Main products | Challenges |
|---|---|---|---|
| Reforming | Lignocellulose biomass-derived feedstocks (pentoses, hexoses, alcohols, and organic acids) | CO2 | • Costly and inefficient lignocellulose refining processes required |
| • Low-value products | |||
| Cellulose, hemicellulose, and lignin | CO2 | • Low substrate solubility | |
| • Low-value products | |||
| Raw biomass | CO2 | • Low substrate solubility | |
| • Costly and environmentally unfriendly pre-treatment processes required | |||
| • Low-value products | |||
| Waste streams (biomass, food, and plastic) | CO2, formate | • Low substrate solubility | |
| • Costly and environmentally unfriendly pre-treatment processes required | |||
| • Competition with water reduction and plastic dehydrogenation to H2 | |||
| Organics | Alcohols | • Low selectivity | |
| Furans | Dimers and trimers | • Low selectivity | |
| Amines | Imine | ||
| Thiols | Disulfides | ||
| Inorganics | Water | H2O2 | |
| HX (X = Cl−, Br−, I−) | X2 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS Leading Initiative for Excellent Young Researchers Program (to Q. W.), the JST Fusion Oriented REsearch for Disruptive Science and Technology Program (no. 21470281, to Q. W.), and the JSPS Grant-in-Aid for Young Scientists (Start-up) (no. 21K20485; to Q. W.).References
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173–179 CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- K. Zhang and L. Guo, Catal. Sci. Technol., 2013, 3, 1672–1690 RSC.
- Y. Xu, Y. Huang and B. Zhang, Inorg. Chem. Front., 2016, 3, 591–615 RSC.
- C. R. Lhermitte and K. Sivula, ACS Catal., 2019, 9, 2007–2017 CrossRef CAS.
- A. Y. Ru Ng, B. Boruah, K. F. Chin, J. M. Modak and H. S. Soo, ChemNanoMat, 2020, 6, 185–203 CrossRef.
- V. Andrei, Q. Wang, T. Uekert, S. Bhattacharjee and E. Reisner, Acc. Chem. Res., 2022, 55, 3376–3386 CrossRef CAS PubMed.
- . ChemAnalyst, https://www.chemanalyst.com, (accessed February 10, 2023).
- L. M. Reid, T. Li, Y. Cao and C. P. Berlinguette, Sustainable Energy Fuels, 2018, 2, 1905–1927 RSC.
- T. Uekert, C. M. Pichler, T. Schubert and E. Reisner, Nat. Sustain., 2021, 4, 383–391 CrossRef.
- M. F. Kuehnel and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 3290–3296 CrossRef CAS PubMed.
- D. I. Kondarides, V. M. Daskalaki, A. Patsoura and X. E. Verykios, Catal. Lett., 2008, 122, 26–32 CrossRef CAS.
- M. R. St. John, A. J. Furgala and A. F. Sammells, J. Phys. Chem., 1983, 87, 801–805 CrossRef.
- H. Bahruji, M. Bowker, P. R. Davies, L. S. Al-Mazroai, A. Dickinson, J. Greaves, D. James, L. Millard and F. Pedrono, J. Photochem. Photobiol., A, 2010, 216, 115–118 CrossRef CAS.
- T. Shiragami, T. Tomo, H. Tsumagari, R. Yuki, T. Yamashita and M. Yasuda, Chem. Lett., 2012, 41, 29–31 CrossRef CAS.
- Q. Xu, Y. Ma, J. Zhang, X. Wang, Z. Feng and C. Li, J. Catal., 2011, 278, 329–335 CrossRef CAS.
- Y. Li, D. Gao, S. Peng, G. Lu and S. Li, Int. J. Hydrogen Energy, 2011, 36, 4291–4297 CrossRef CAS.
- R. Su, R. Tiruvalam, A. J. Logsdail, Q. He, C. A. Downing, M. T. Jensen, N. Dimitratos, L. Kesavan, P. P. Wells, R. Bechstein, H. H. Jensen, S. Wendt, C. R. A. Catlow, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2014, 8, 3490–3497 CrossRef CAS PubMed.
- A. V. Puga, Coord. Chem. Rev., 2016, 315, 1–66 CrossRef CAS.
- W. Deng, H. Zhang, L. Xue, Q. Zhang and Y. Wang, Chin. J. Catal., 2015, 36, 1440–1460 CrossRef CAS.
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- T. Kawai and T. Sakata, Nature, 1980, 286, 474–476 CrossRef CAS.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- L. Lan, H. Chen, D. Lee, S. Xu, N. Skillen, A. Tedstone, P. Robertson, A. Garforth, H. Daly, C. Hardacre and X. Fan, ACS Sustainable Chem. Eng., 2022, 10, 4862–4871 CrossRef CAS PubMed.
- D. S. Achilleos, W. Yang, H. Kasap, A. Savateev, Y. Markushyna, J. R. Durrant and E. Reisner, Angew. Chem., Int. Ed., 2020, 59, 18184–18188 CrossRef CAS PubMed.
- Q. Cheng, Y.-J. Yuan, R. Tang, Q.-Y. Liu, L. Bao, P. Wang, J. Zhong, Z. Zhao, Z.-T. Yu and Z. Zou, ACS Catal., 2022, 12, 2118–2125 CrossRef CAS.
- E. Wang, A. Mahmood, S.-G. Chen, W. Sun, T. Muhmood, X. Yang and Z. Chen, ACS Catal., 2022, 12, 11206–11215 CrossRef CAS.
- H. Kasap, D. S. Achilleos, A. Huang and E. Reisner, J. Am. Chem. Soc., 2018, 140, 11604–11607 CrossRef CAS PubMed.
- C. Rao, M. Xie, S. Liu, R. Chen, H. Su, L. Zhou, Y. Pang, H. Lou and X. Qiu, ACS Appl. Mater. Interfaces, 2021, 13, 44243–44253 CrossRef CAS PubMed.
- S. R. Kadam, V. R. Mate, R. P. Panmand, L. K. Nikam, M. V. Kulkarni, R. S. Sonawane and B. B. Kale, RSC Adv., 2014, 4, 60626–60635 RSC.
- C. Li, H. Wang, S. B. Naghadeh, J. Z. Zhang and P. Fang, Appl. Catal., 2018, 227, 229–239 CrossRef CAS.
- N. Skillen, H. Daly, L. Lan, M. Aljohani, C. W. J. Murnaghan, X. Fan, C. Hardacre, G. N. Sheldrake and P. K. J. Robertson, Top. Curr. Chem., 2022, 380, 33 CrossRef CAS PubMed.
- A. Speltini, M. Sturini, D. Dondi, E. Annovazzi, F. Maraschi, V. Caratto, A. Profumo and A. Buttafava, Photochem. Photobiol. Sci., 2014, 13, 1410–1419 CrossRef CAS PubMed.
- J. Zou, G. Zhang and X. Xu, Appl. Catal., A, 2018, 563, 73–79 CrossRef CAS.
- K. Tomoji and S. Tadayoshi, Chem. Lett., 1981, 10, 81–84 CrossRef.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- C. M. Pichler, S. Bhattacharjee, M. Rahaman, T. Uekert and E. Reisner, ACS Catal., 2021, 11, 9159–9167 CrossRef CAS PubMed.
- T. Uekert, M. A. Bajada, T. Schubert, C. M. Pichler and E. Reisner, ChemSusChem, 2021, 14, 4190–4197 CrossRef CAS PubMed.
- S. Chu, B. Zhang, X. Zhao, H. S. Soo, F. Wang, R. Xiao and H. Zhang, Adv. Energy Mater., 2022, 12, 2200435 CrossRef CAS.
- C. M. Pichler, S. Bhattacharjee, E. Lam, L. Su, A. Collauto, M. M. Roessler, S. J. Cobb, V. M. Badiani, M. Rahaman and E. Reisner, ACS Catal., 2022, 12, 13360–13371 CrossRef CAS PubMed.
- M. Han, S. Zhu, C. Xia and B. Yang, Appl. Catal., B, 2022, 316, 121662 CrossRef CAS.
- Y. Li, S. Wan, C. Lin, Y. Gao, Y. Lu, L. Wang and K. Zhang, Sol. RRL, 2021, 5, 2000427 CrossRef CAS.
- B. Cao, S. Wan, Y. Wang, H. Guo, M. Ou and Q. Zhong, J. Colloid Interface Sci., 2022, 605, 311–319 CrossRef CAS PubMed.
- T. Uekert, F. Dorchies, C. M. Pichler and E. Reisner, Green Chem., 2020, 22, 3262–3271 RSC.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- K. Su, H. Liu, C. Zhang and F. Wang, Chin. J. Catal., 2022, 43, 589–594 CrossRef CAS.
- E. Lam and E. Reisner, Angew. Chem., Int. Ed., 2021, 60, 23306 CrossRef CAS PubMed.
- M. Wang, M. Liu, J. Lu and F. Wang, Nat. Commun., 2020, 11, 1083 CrossRef CAS PubMed.
- C. M. Pichler, T. Uekert and E. Reisner, Chem. Commun., 2020, 56, 5743–5746 RSC.
- H. Kolbe, Justus Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef.
- G. Han, Y.-H. Jin, R. A. Burgess, N. E. Dickenson, X.-M. Cao and Y. Sun, J. Am. Chem. Soc., 2017, 139, 15584–15587 CrossRef CAS PubMed.
- V. R. Battula, A. Jaryal and K. Kailasam, J. Mater. Chem. A, 2019, 7, 5643–5649 RSC.
- Y.-H. Li, F. Zhang, Y. Chen, J.-Y. Li and Y.-J. Xu, Green Chem., 2020, 22, 163–169 RSC.
- MilliporeSigma, https://www.sigmaaldrich.com/US/en, (accessed February 10, 2023).
- J.-H. Tang and Y. Sun, Mater. Adv., 2020, 1, 2155–2162 RSC.
- H. Kasap, C. A. Caputo, B. C. M. Martindale, R. Godin, V. W.-h. Lau, B. V. Lotsch, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2016, 138, 9183–9192 CrossRef CAS PubMed.
- N. Luo, T. Hou, S. Liu, B. Zeng, J. Lu, J. Zhang, H. Li and F. Wang, ACS Catal., 2020, 10, 762–769 CrossRef CAS.
- A. Vijeta and E. Reisner, Chem. Commun., 2019, 55, 14007–14010 RSC.
- W. Yu, D. Zhang, X. Guo, C. Song and Z. Zhao, Catal. Sci. Technol., 2018, 8, 5148–5154 RSC.
- L. Xu, X. Deng and Z. Li, Appl. Catal., B, 2018, 234, 50–55 CrossRef CAS.
- Z. Gao, N. Luo, Z. Huang, S. H. Taylor and F. Wang, ACS Sustainable Chem. Eng., 2021, 9, 6188–6202 CrossRef CAS.
- D. Antón-García, E. Edwardes Moore, M. A. Bajada, A. Eisenschmidt, A. R. Oliveira, I. A. C. Pereira, J. Warnan and E. Reisner, Nat. Synth., 2022, 1, 77–86 CrossRef.
- X. Ye, Y. Chen, Y. Wu, X. Zhang, X. Wang and S. Chen, Appl. Catal., B, 2019, 242, 302–311 CrossRef CAS.
- F. Li, Y. Wang, J. Du, Y. Zhu, C. Xu and L. Sun, Appl. Catal., B, 2018, 225, 258–263 CrossRef CAS.
- S. Xie, Z. Shen, J. Deng, P. Guo, Q. Zhang, H. Zhang, C. Ma, Z. Jiang, J. Cheng, D. Deng and Y. Wang, Nat. Commun., 2018, 9, 1181 CrossRef PubMed.
- H. Zhang, S. Xie, J. Hu, X. Wu, Q. Zhang, J. Cheng and Y. Wang, Chem. Commun., 2020, 56, 1776–1779 RSC.
- N. Luo, T. Montini, J. Zhang, P. Fornasiero, E. Fonda, T. Hou, W. Nie, J. Lu, J. Liu, M. Heggen, L. Lin, C. Ma, M. Wang, F. Fan, S. Jin and F. Wang, Nat. Energy, 2019, 4, 575–584 CrossRef CAS.
- Y. Huang, C. Liu, M. Li, H. Li, Y. Li, R. Su and B. J. A. C. Zhang, ACS Catal., 2020, 10, 3904–3910 CrossRef CAS.
- X. B. Li, Z. J. Li, Y. J. Gao, Q. Y. Meng, S. Yu, R. G. Weiss, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2014, 126, 2117–2121 CrossRef.
- L. Xiong and J. Tang, Adv. Energy Mater., 2021, 11, 2003216 CrossRef CAS.
- Y. F. Zhang and S. J. Park, J. Mater. Chem. A, 2018, 6, 20304–20312 RSC.
- J. Chen, N. Kang, J. Fan, C. Lu and K. Lv, Mater. Today Chem., 2022, 26, 101028 CrossRef CAS.
- K. P. Bryliakov, Chem. Rev., 2017, 117, 11406–11459 CrossRef CAS PubMed.
- Z. Tang, P. Zhao, H. Wang, Y. Liu and W. Bu, Chem. Rev., 2021, 121, 1981–2019 CrossRef CAS PubMed.
- J. Xu, X. Zheng, Z. Feng, Z. Lu, Z. Zhang, W. Huang, Y. Li, D. Vuckovic, Y. Li, S. Dai, G. Chen, K. Wang, H. Wang, J. K. Chen, W. Mitch and Y. Cui, Nat. Sustain., 2021, 4, 233–241 CrossRef PubMed.
- J. Y. Tang, T. S. Zhao, D. Solanki, X. B. Miao, W. G. Zhou and S. Hu, Joule, 2021, 5, 1432–1461 CrossRef CAS.
- K. Li, M. Han, R. Chen, S. L. Li, S. L. Xie, C. Mao, X. Bu, X. L. Cao, L. Z. Dong, P. Feng and Y. Q. Lan, Adv. Mater., 2016, 28, 8906–8911 CrossRef CAS PubMed.
- R. Shi, H.-F. Ye, F. Liang, Z. Wang, K. Li, Y. Weng, Z. Lin, W.-F. Fu, C.-M. Che and Y. Chen, Adv. Mater., 2018, 30, 1705941 CrossRef PubMed.
- C. Feng, L. Tang, Y. Deng, J. Wang, Y. Liu, X. Ouyang, H. Yang, J. Yu and J. Wang, Appl. Catal., B, 2021, 281, 119539 CrossRef CAS.
- F. Xue, Y. Si, M. Wang, M. Liu and L. Guo, Nano Energy, 2019, 62, 823–831 CrossRef CAS.
- Y. Fu, C. a. Liu, M. Zhang, C. Zhu, H. Li, H. Wang, Y. Song, H. Huang, Y. Liu and Z. Kang, Adv. Energy Mater., 2018, 8, 1802525 CrossRef.
- Z. Pei, L. Ding, J. Hu, S. Weng, Z. Zheng, M. Huang and P. Liu, Appl. Catal., B, 2013, 142–143, 736–743 CrossRef CAS.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- S. Li, G. Dong, R. Hailili, L. Yang, Y. Li, F. Wang, Y. Zeng and C. Wang, Appl. Catal., B, 2016, 190, 26–35 CrossRef CAS.
- E. Zhang, Q. Zhu, J. Huang, J. Liu, G. Tan, C. Sun, T. Li, S. Liu, Y. Li, H. Wang, X. Wan, Z. Wen, F. Fan, J. Zhang and K. Ariga, Appl. Catal., B, 2021, 293, 120213 CrossRef CAS.
- S. Cao, T.-S. Chan, Y.-R. Lu, X. Shi, B. Fu, Z. Wu, H. Li, K. Liu, S. Alzuabi, P. Cheng, M. Liu, T. Li, X. Chen and L. Piao, Nano Energy, 2020, 67, 104287 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- B. Huskinson, J. Rugolo, S. K. Mondal and M. J. Aziz, Energy Environ. Sci., 2012, 5, 8690–8698 RSC.
- G. R. Taylor and M. Butler, Epidemiol. Infect., 1982, 89, 321–328 CAS.
- R. Yeo and D. T. Chin, J. Electrochem. Soc., 1980, 127, 549 CrossRef CAS.
- J. Du, Z. Chen, C. Chen and T. J. Meyer, J. Am. Chem. Soc., 2015, 137, 3193–3196 CrossRef CAS PubMed.
- Y. Wang, L. Shi and H. Hu, et al., Binary trinuclear metal-oxo sub-nanomaterials for photocatalytic hydrogen and chlorine production from seawater, SusMat, 2022, 2, 708–719 CrossRef CAS.
- J. Zhang, Y. Lei, S. Cao, W. Hu, L. Piao and X. Chen, Nano Res., 2022, 15, 2013–2022 CrossRef CAS.
- S. Park, W. J. Chang, C. W. Lee, S. Park, H.-Y. Ahn and K. T. Nam, Nat. Energy, 2016, 2, 16185 CrossRef.
- Y. Wu, P. Wang, X. Zhu, Q. Zhang, Z. Wang, Y. Liu, G. Zou, Y. Dai, M.-H. Whangbo and B. Huang, Adv. Mater., 2018, 30, 1704342 CrossRef PubMed.
- C. Cai, Y. Teng, J.-H. Wu, J.-Y. Li, H.-Y. Chen, J.-H. Chen and D.-B. Kuang, Adv. Funct. Mater., 2020, 30, 2001478 CrossRef CAS.
- Z. Zhao, J. Wu, Y.-Z. Zheng, N. Li, X. Li and X. Tao, ACS Catal., 2019, 9, 8144–8152 CrossRef CAS.
- X. Wang, H. Wang, H. Zhang, W. Yu, X. Wang, Y. Zhao, X. Zong and C. Li, ACS Energy Lett., 2018, 3, 1159–1164 CrossRef CAS.
- R. Li, X. Li, J. Wu, X. Lv, Y.-Z. Zheng, Z. Zhao, X. Ding, X. Tao and J.-F. Chen, Appl. Catal., B, 2019, 259, 118075 CrossRef CAS.
- Y. Zhao, Q. Zeng, Y. Yu, T. Feng, Y. Zhao, Z. Wang, Y. Li, C. Liu, J. Liu, H. Wei, S. Zhu, Z. Kang, H. Zhang and B. Yang, Mater. Horiz., 2020, 7, 2719–2725 RSC.
- W. Guan, Y. Li, Q. Zhong, H. Liu, J. Chen, H. Hu, K. Lv, J. Gong, Y. Xu, Z. Kang, M. Cao and Q. Zhang, Nano Lett., 2021, 21, 597–604 CrossRef CAS PubMed.
- X. L. Zhao, S. Chen, H. J. Yin, S. Y. Jiang, K. Zhao, J. Kang, P. F. Liu, L. X. Jiang, Z. J. Zhu, D. D. Cui, P. R. Liu, X. J. Han, H. G. Yang and H. J. Zhao, Matter, 2020, 3, 935–949 CrossRef.
- Y. Wu, P. Wang, Z. Guan, J. Liu, Z. Wang, Z. Zheng, S. Jin, Y. Dai, M.-H. Whangbo and B. Huang, ACS Catal., 2018, 8, 10349–10357 CrossRef CAS.
- Z. Guan, Y. Wu, P. Wang, Q. Zhang, Z. Wang, Z. Zheng, Y. Liu, Y. Dai, M.-H. Whangbo and B. Huang, Appl. Catal., B, 2019, 245, 522–527 CrossRef CAS.
- P. Zhou, H. Chen, Y. Chao, Q. Zhang, W. Zhang, F. Lv, L. Gu, Q. Zhao, N. Wang, J. Wang and S. Guo, Nat. Commun., 2021, 12, 4412 CrossRef CAS PubMed.
- Y. Wu, Q. Wu, Q. Zhang, Z. Lou, K. Liu, Y. Ma, Z. Wang, Z. Zheng, H. Cheng, Y. Liu, Y. Dai, B. Huang and P. Wang, Energy Environ. Sci., 2022, 15, 1271–1281 RSC.
- V.-C. Nguyen, D. B. Nimbalkar, L. D. Nam, Y.-L. Lee and H. Teng, ACS Catal., 2021, 11, 4955–4967 CrossRef CAS.
- H. Li, Z. Gao, L. Lei, H. Liu, J. Han, F. Hong, N. Luo and F. Wang, Green Chem., 2020, 22, 3802–3808 RSC.
- T. Yukawa, T. Bamba, M. Matsuda, T. Yoshida, K. Inokuma, J. Kim, J. Won Lee, Y.-S. Jin, A. Kondo and T. Hasunuma, Biotechnol. Bioeng., 2023, 120, 511–523 CrossRef CAS PubMed.
- C. H. Martin, H. Dhamankar, H.-C. Tseng, M. J. Sheppard, C. R. Reisch and K. L. J. Prather, Nat. Commun., 2013, 4, 1414 CrossRef PubMed.
- H. Dhamankar, Y. Tarasova, C. H. Martin and K. L. J. Prather, Metab. Eng., 2014, 25, 72–81 CrossRef CAS PubMed.
- K. Wenderich, W. Kwak, A. Grimm, G. J. Kramer, G. Mul and B. Mei, Sustainable Energy Fuels, 2020, 4, 3143–3156 RSC.
- M. A. Khan, T. A. Al-Attas, N. G. Yasri, H. Zhao, S. Larter, J. Hu and M. G. Kibria, Sustainable Energy Fuels, 2020, 4, 5568–5577 RSC.
| This journal is © The Royal Society of Chemistry 2023 |