
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards control of excitonic coupling in DNA-templated Cy5 aggregates: the principal role of chemical substituent hydrophobicity and steric interactions†
Sebastián A.
Díaz
 *a,
Gissela
Pascual
b,
Lance K.
Patten
b,
Simon K.
Roy
b,
Adam
Meares
a,
Matthew
Chiriboga
ac,
Kimihiro
Susumu
de,
William B.
Knowlton
bf,
Paul D.
Cunningham
g,
Divita
Mathur
h,
Bernard
Yurke
bf,
Igor L.
Medintz
a,
Jeunghoon
Lee
*bi and
Joseph S.
Melinger
*g
*a,
Gissela
Pascual
b,
Lance K.
Patten
b,
Simon K.
Roy
b,
Adam
Meares
a,
Matthew
Chiriboga
ac,
Kimihiro
Susumu
de,
William B.
Knowlton
bf,
Paul D.
Cunningham
g,
Divita
Mathur
h,
Bernard
Yurke
bf,
Igor L.
Medintz
a,
Jeunghoon
Lee
*bi and
Joseph S.
Melinger
*g
aCenter for Bio/Molecular Science and Engineering Code 6900, U.S. Naval Research Laboratory, Washington, D.C. 20375, United States. E-mail: sebastian.diaz@nrl.navy.mil
bMicron School of Materials Science & Engineering, Boise State University, Boise, Idaho 83725, USA. E-mail: jeunghoonlee@boisestate.edu
cVolgenau School of Engineering, George Mason University, Fairfax, Virginia 22030, USA
dOptical Sciences Division, Code 5600, U.S. Naval Research Laboratory, Washington, DC, USA
eJacobs Corporation, Hanover, MD, USA
fDepartment of Electrical & Computer Engineering, Boise State University, Boise, Idaho 83725, USA
gElectronics Science and Technology Division Code 6800, U.S. Naval Research Laboratory, Washington, D.C. 20375, USA. E-mail: joseph.melinger@nrl.navy.mil
hDepartment of Chemistry, Case Western Reserve University, Cleveland, OH 44106, USA
iDepartment of Chemistry & Biochemistry, Boise State University, Boise, Idaho 83725, USA
First published on 27th January 2023
Abstract
Understanding and controlling exciton coupling in dye aggregates has become a greater focus as potential applications such as coherent exciton devices, nanophotonics, and biosensing have been proposed. DNA nanostructure templates allow for a powerful modular approach. Using DNA Holliday junction (HJ) templates variations of dye combinations and precision dye positions can be rapidly assayed, as well as creating aggregates of dyes that could not be prepared (either due to excess or lack of solubility) through alternative means. Indodicarbocyanines (Cy5) have been studied in coupled systems due to their large transition dipole moment, which contributes to strong coupling. Cy5-R dyes were recently prepared by chemically modifying the 5,5′-substituents of indole rings, resulting in varying dye hydrophobicity/hydrophilicity, steric considerations, and electron-donating/withdrawing character. We utilized Cy5-R dyes to examine the formation and properties of 30 unique DNA templated homodimers. We find that in our system the sterics of Cy5-R dyes play the determining factor in orientation and coupling strength of dimers, with coupling strengths ranging from 50–138 meV. The hydrophobic properties of the Cy5-R modify the percentage of dimers formed, and have a secondary role in determining the packing characteristics of the dimers when sterics are equivalent. Similar to other reports, we find that positioning of the Cy5-R within the HJ template can favor particular dimer interactions, specifically oblique or H-type dimers.
Introduction
Considerable effort has been expended to understand and control exciton coupling in dye aggregates going as far back as the 1930s.1,2 The first successful transition, starting in the 1970s, was the spectral sensitization of the silver halide crystals embedded in photographic films.3,4 Since then potential applications have increased to include coherent exciton devices, nanophotonics, light harvesting/solar energy conversion, and biosensing.5–12 Dye-aggregates have been studied in solution, within crystals, and at surfaces and interfaces, demonstrating unique properties.13,14 A recent focus is on the use of DNA templates, which provide a unique handle for controlled aggregation both through covalent and non-covalent binding of dyes.15,16 DNA templates provide modular templates where designer 1-, 2-, and 3-D structures can be prepared at high fidelity with nm scale features.17 This allows for hundreds of variations of, for example, dye combinations and dye positions, to be rapidly assayed which could not be done with chemical approaches.18,19 Importantly, due to the programmable nature of DNA nanotechnology and the nm scale of the DNA double helix, the positioning of dyes can be controlled down to the single nm level.20,21 By placing multiple dyes within a small area of the DNA scaffold, precise aggregates can be formed at low concentrations, which is something particularly challenging when dealing with both highly soluble or highly insoluble dyes. While computational chemistry offers great insight,22 the varying factors that regulate coupling such as energy levels, transition dipole moments (TDM), electrostatics, hydrophobicity, etc., are hard to capture without experimental data to support the models. This holds particularly true for the complex landscape of DNA templates. Therefore, experimental confirmation of aggregates and their structure are required.Cyanine dyes are fluorescent molecules, composed of heterocyclic units conjugated through a polymethine bridge. Cyanines have become workhorses of many fields, including cellular microscopy, flow cytometry, and bioassays. Importantly, they are also readily available as labels of oligonucleotides from commercial vendors.20 The red-shifted indodicarbocyanines (Cy5, having a five methine bridge) have been a recent focus because of the large TDM (the absolute square of the TDM being proportional to the measured molar absorptivity, ∼250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at peak), which contributes to producing strong coupling.23–25 Additionally, their absorption and fluorescence spectra are in the visible light range, allowing for detailed characterization of monomers and aggregates on common spectroscopic systems. A final consideration is the capability of chemically synthesizing variants of the Cy5 dye.26 Specifically, position 5 and 5′ on the indole rings can be modified without disrupting the conjugation, while still leaving a biorthogonal handle available for DNA conjugation. Work by the Armitage and Takahashi labs demonstrated that 5 and 5′ substitutions did result in changed cyanine properties (e.g. sterics and hydrophobicity) and that this subsequently resulted in changes in the propensity to form aggregates from freely diffusing dyes, yet in these studies, the final form of the aggregates was not changed.26,27
000 M−1 cm−1 at peak), which contributes to producing strong coupling.23–25 Additionally, their absorption and fluorescence spectra are in the visible light range, allowing for detailed characterization of monomers and aggregates on common spectroscopic systems. A final consideration is the capability of chemically synthesizing variants of the Cy5 dye.26 Specifically, position 5 and 5′ on the indole rings can be modified without disrupting the conjugation, while still leaving a biorthogonal handle available for DNA conjugation. Work by the Armitage and Takahashi labs demonstrated that 5 and 5′ substitutions did result in changed cyanine properties (e.g. sterics and hydrophobicity) and that this subsequently resulted in changes in the propensity to form aggregates from freely diffusing dyes, yet in these studies, the final form of the aggregates was not changed.26,27
The study of the unmodified Cy5, which we will also denominate Cy5H, and its scaffold directed aggregation has been approached from a few angles in recent years. These include investigation into dimers formed by labeling two complementary ssDNA to initially form a Cy5H dimer on dsDNA.24,25,28,29 As the concentration of Mg2+ was modified the dsDNA dimer coupled with a second dsDNA dimer via branch migration to form a four-armed junction that resulted in a Cy5H tetramer.28 It was suggested that the dye properties were favoring these dimer-tetramer transitions. Subsequent work focused on using static four-armed junctions formed from four non-homologous ssDNA, i.e. Holliday junctions (HJ), to look at the Cy5H aggregates.30 They observed that the aggregate characteristics depended largely on the position of the dyes within the HJ structure. This key discovery motivated our work to determine what other variables can tune the aggregate properties of Cy5 dye, or Cy5-R, where R is the 5,5′-substituent of the indole rings.
Other work focused on modifying the DNA template, for example, the use of modified nucleotides (e.g. locked nucleic acids and bridged nucleic acids) replacing natural nucleotides resulted in an increase in Cy5H coupling strength.31 It was also reported that the properties of heterogeneous aggregates of Cy5H and Cy5.5 also depended on the position of the dyes within the HJ structure.32 This foundation of work focused on modifying the scaffold, either the design of the DNA itself or even chemically modifying the nucleotides of the oligos. The work of Mass and co-workers refocused the question of dye aggregate formation on the properties of the dyes themselves, even within DNA scaffolds.33 Specifically, their work harkened back to the classic work in the field where hydrophobicity of the dye moiety and solvent polarity were the principal levers driving aggregations.4,13,34 Their work looked at the effect of dye hydrophobicity of modified squaraines and the aggregates templated by DNA HJ structures in aqueous solutions.35 They concluded that the greater the hydrophobicity of the substituted squaraine dyes the greater the coupling strength of the exciton within the aggregates.33
We reiterate the interest in Cy5 dyes, due to the above-mentioned properties, and the observation that positioning within DNA scaffolds, along with the dye attributes themselves, strongly modify aggregate properties. Recently, we have tailored the properties of Cy5-R by chemically modifying the 5,5′-substituents, resulting in varying dye hydrophobicity/hydrophilicity, steric considerations, as well as the electron-donating/withdrawing character which tunes absorption and emission properties (i.e. energy levels).36 Here we utilize these chemically modified dyes to examine the formation and properties of 30 unique homodimers of Cy5-R. We find that positioning of the Cy5-R within the HJ template can favor either oblique dimers (adjacent HJ dimers) or H-type dimers (transverse HJ dimers), where HJ dimers refer to Cy5-R dimers assembled using DNA HJ as the template. More importantly, we show that in our system the sterics of Cy5-R play the determining factor, in orientation and coupling strength of dimers within DNA HJs. However, the hydrophobic properties of the Cy5-R do play a role in determining the percentage of dimers formed, and when steric considerations are equal, can determine the packing characteristics of the dimers.
Results
Assembling Cy5-R HJ structures
As noted above the intent was to utilize the Cy5-R we prepared previously through symmetric 5,5′-substituents to determine which physicochemical properties (e.g. hydrophobicity and electrostatics) affected dimer coupling and packing. As the Cy5-R shown in Fig. 1 (the substituents being: n-hexyloxy (hex), triethylene glycol monomethylether (Peg), tert-butyl (tBu), and chloro (Cl)) are not commercially available, nor can the dye-labeled DNA be purchased without expensive special orders, extensive work was done to synthesize and characterize the dyes, integrate them into DNA oligos through phosphoramidite incorporation, and purify the final product. The details of this and the characterization of the dyes and the dye-labeled oligos as single stranded (ss)DNA is available in our previous publication by Meares et al.36 The sequences of the specific DNA oligos and a structure showing the double attachment of the Cy5-R to the oligos are available in the ESI Table S1 and Fig. S1.† We note that our approach to dye integration into the DNA utilizes a double conjugation approach with relatively short three carbon linkers. This approach was undertaken to minimize dye mobility and sample heterogeneity.37,38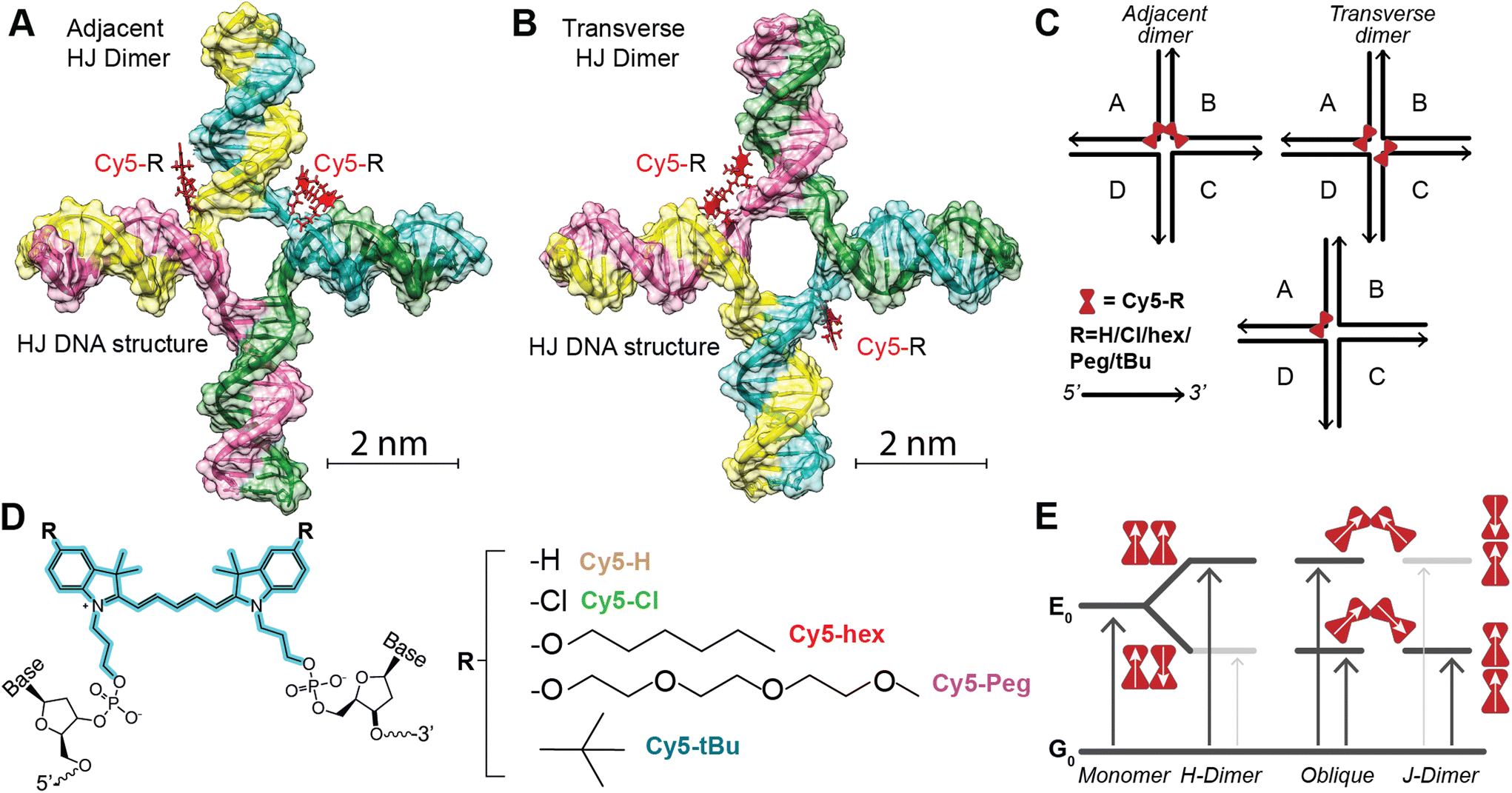 | ||
| Fig. 1 (A) Schematic of AB adjacent homodimer. Dye position represents initial localization based on base pair site and not the position adapted upon dimer formation. (B) Schematic of AC transverse homodimer. Dye position represents initial localization based on base pair site and not the position adapted upon dimer formation. (C) Simple line schematic of the representative HJ structures. Black lines in this case represent the DNA, with the arrow denoting the 5′ to 3′ direction, and red bowtie the Cy5-R dye. We note that adjacent dimers can be represented: [AB, BC, CD, or AD] while transverse dimers: [AC and BD]. Monomers include the [A, B, C, and D] conformations. (D) Chemical structure of the Cy5-R showing the two-point attachment to DNA. The R groups are shown to the right. (E) Schematic of the TDM alignments and coupling of the energy levels in the dimers. Red bowties as in C, while the white arrows represent the TDM alignment. The upward arrows indicate absorptive transitions from the ground state, where a dark grey arrow indicates an allowed transition and a light gray arrow indicates a forbidden transition. | ||
The work of Mass et al.35 confirmed the hypothesis that increasing dye hydrophobicity increases dye packing on DNA scaffolds when other factors, such as dye steric effects, are not in play. In this work we take the subsequent step of further testing this hypothesis by using Cy5-R variants where dye steric effects are varied through the properties of the added substituent. The HJ structures39 were prepared by combining equimolar amounts (10 μM) of each unlabeled or dye-labeled DNA strand [A, B, C, and D] in a 1× TAE (40 mM tris(hydroxymethyl)aminomethane, 20 mM acetic acid, 1 mM ethylenediaminetetraacetic acid, pH 8.0) + 15 mM MgCl2 aqueous buffer.32 Proper structural formation was driven by an overnight annealing program which heated the solution to 95 °C then very slowly lowered the temperature to 4 °C, at which point the samples were stored in the dark at 4 °C until measured. Within the HJ structures there are two conformations of homogenous dimers (see Fig. 1), the first being the adjacent dimers (i.e. dye modifications are on DNA strands that are partially dimerized to each other within the HJ: [AB, BC, CD, AD]), and the second the transverse dimers (i.e. dye modifications are on DNA strands that do not dimerize to each other within the HJ: [AC, BD]). As demonstrated in other works, when properly positioned within HJ DNA structures Cy5 dyes form dimers that support delocalized excitons.23,24,30 The position of the dimer, adjacent or transverse, can have considerable effect on the relative dye orientation and therefore the type of aggregate the dimer forms.
The three types of dimers, as seen in Fig. 1E, are: (1) H-dimer, a stacked form with parallel transition dipoles, (2) J-dimer, a collinear (i.e. head-to-tail) alignment of TDMs, (3) Oblique, an intermediate angled configuration.4,6,13 The coupled dimers have photophysical properties that depend on dye orientation.40 H-type aggregates have blue-shifted absorbance peaks relative to the monomer, strongly quenched fluorescence, and large Stokes shifts. J-type aggregates, have a red-shifted absorbance, and a relatively sharp emission peak with a small Stokes shift. The Oblique dimers have a mix of H- and J-aggregate properties, e.g. they display Davydov splitting of the absorbance spectrum. The degree to which these properties are observed is dependent on the coupling or delocalization strength, which in turn is dependent on the positioning (distances and angles) of the dyes within the dimers, as well as the inherent energetic and dipole properties of the particular dyes. To uncover the photophysical properties, a suite of biophysical characterization and steady-state spectroscopic measurements were performed on the varying structures. This data set was then analyzed with a Kühn–Renger–May (KRM)41 interdye vibronic coupling model to determine packing characteristics as detailed in later sections.
Comparing Cy5-R properties as ssDNA and within HJ
For a more in-depth look at the dyes and the dye labeled ssDNA we direct the reader to the previously published report by Meares et al.36 To summarize, the dye properties can be described in terms of increasing hydrophobicity (measured by log(P) determined through the use of Percepta software from Advanced Chemistry Development, Inc. (Toronto, Canada)): Peg ≈ H (1.7) < Cl (2.3) ≪ tBu (3.4) ≪ hex (5.7).36 The electron donating capability of the substituent group (measured by the para-position Hammett parameter, σp) is tBu (−0.20) < Peg (−0.26) ≈ hex(−0.27), with Cl in fact being a weak acceptor (0.22) and the unmodified Cy5H defined as null (H = 0).42 The greater the electron-donating character, the greater the bathochromic shift, which was observed in the dye-labeled ssDNA. Finally, we utilize A-values as a representation of bulkiness or steric hindrance of the substituent group.43 The A-value is defined as the difference in energy between the equatorial (favored) and axial cyclohexane conformation, i.e. the bulkier the substituent the larger the A-value. The traditional A-value is therefore a representation of gauche interactions. In this case for increasing A-values, Cl (0.48 kcal mol−1) < Peg ≈ hex (∼0.9 kcal mol−1) ≪ tBu (∼4.9 kcal mol−1), with again the unmodified Cy5H defined as null (H = 0).43 Deeper analysis led us to recognize that these values may not be the optimal representation for our system, Boiadjiev and Lightner developed an alternative approach that actually utilizes exciton coupling measured through CD on a bilirubin template to assess head-to-head steric compression, which better aligns with our system.44 The key result of that work is to diminish the relative bulkiness of the tBu as compared to other substituents; as such we consider the tBu value to still be the bulkiest but with a lower value of 1.85 kcal mol−1.44 The authors did not study our other substituents but their analysis appears to support the pre-existing values and relative steric considerations of each dye modification. Our analysis focused on these dyes within the more rigid structure of the DNA HJs, as such we refer to fully formed HJ structures where only one strand (A, B, C, or D) is dye modified as our monomer dyes.We find that the relative properties of the free dyes and the respective dye-labeled ssDNA generally transfer to the monomers. The electron donating or withdrawing character of the substituent determines the absorbance and emission maximum, with the stronger the electron donating or withdrawing character the more red-shifted the peak maximum (see Fig. 2, and Data in Table 1). The Stokes shift is smallest for Cy5H, 11 nm, and slightly larger the more electron donating/withdrawing the substituent group is going to 17 nm for Cy5Cl and 22 nm for Cy5hex. Consistent with observations in the ssDNA, the particular nanoenvironment of the dye has considerable effect on its photophysical properties, i.e. the HJ monomers are distinct if they are A, B, C, or D.45 This applies to the peak position, where a 5 nm range was observed for the absorbance maximum, and a 3–7 nm range was observed for the emission maximum just within different HJ. Interestingly, the HJ A monomer had the reddest absorbance and emission for all the measured samples, while the HJ C tended to be more blue-shifted. If we consider the nearest neighbor base pairs in each monomer we find that the A monomer is flanked by two –CG– pairs, monomers B and D, are flanked by a –CG– and a –AT– pair, while monomer C is flanked by two –AT– pairs.45 The Stokes shift value does not appear to correlate to any particular monomer.
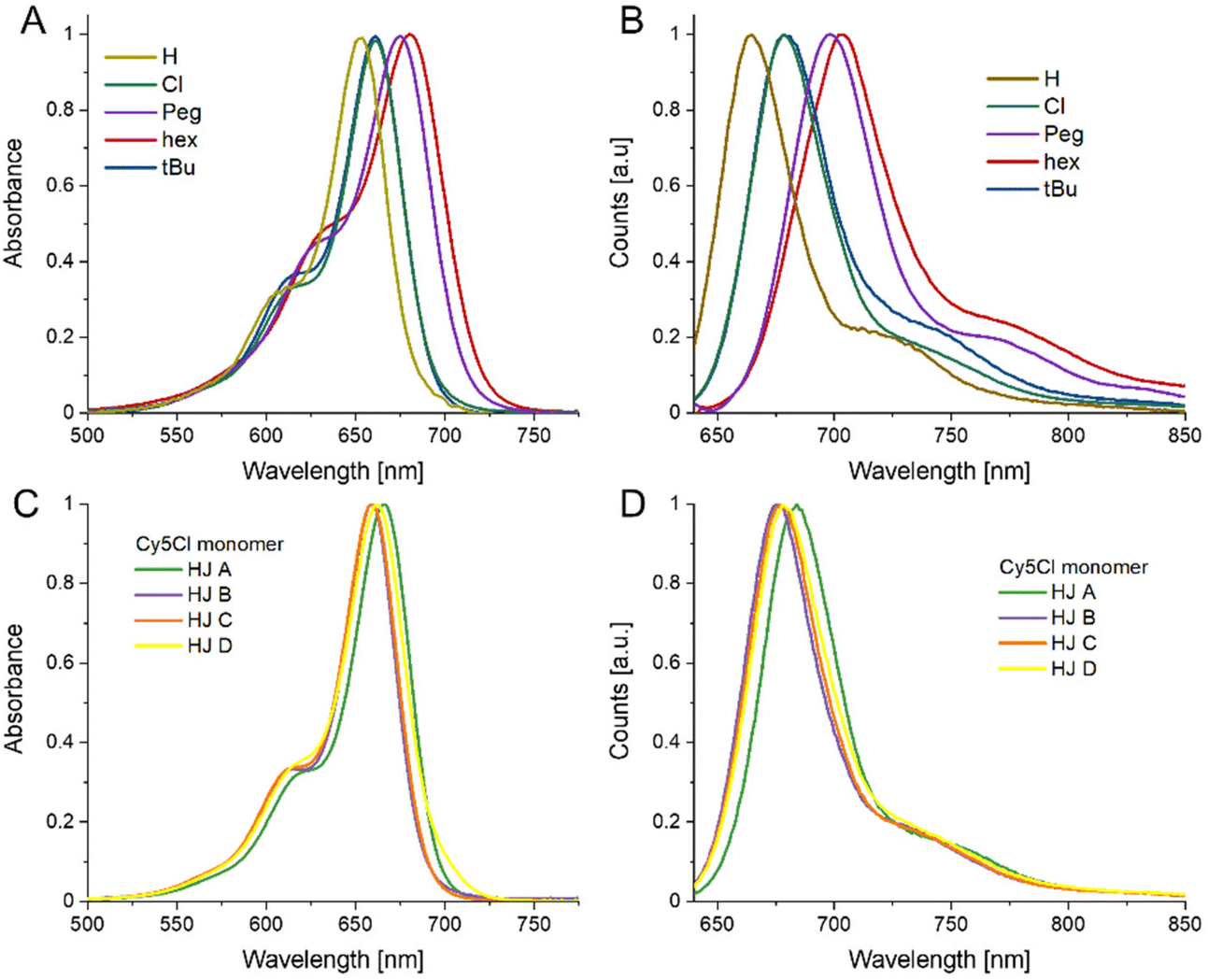 | ||
| Fig. 2 Monomer Spectra. (A) Normalized absorbance spectra of Cy5-R HJ monomers. (B) Normalized fluorescence emission spectra of Cy5-R HJ monomers excited at 615 nm. (C) Absorbance spectra of Cy5-Cl monomer. (D) Fluorescence emission spectra of Cy5-Cl monomer excited at 615 nm. The spectra in (C) and (D) are representative of the variation observed in the different DNA scaffolds. The spectra for the remaining dyes are available in the ESI Fig. S2.† All measurements were performed at 20 °C. | ||
| Dye | Abs max λ (range) [nm] | Fluor. Em. max λ (range) [nm] | Fluor. QY a |
|---|---|---|---|
| a The QY reported are the average of the four monomer positions, with uncertainty arising from the difference in the four positions as well as other experimental propagations. | |||
| Cy5H | 654 (650–656) | 665 (662–667) | 0.29 ± 0.07 |
| Cy5hex | 680 (678–683) | 702 (700–706) | 0.10 ± 0.02 |
| Cy5Peg | 675 (673–677) | 695 (694–697) | 0.09 ± 0.02 |
| Cy5tBu | 661 (658–663) | 679 (675–682) | 0.27 ± 0.03 |
| Cy5Cl | 661 (659–664) | 678 (675–682) | 0.26 ± 0.04 |
Fluorescence quantum yields (QY) were also determined for each sample. Here again, the QY generally correlated with the electron donating character of the substituent, the stronger electron donating substituents (hex and Peg) had QYs near 10% within the HJs while the others had QY near 26% (see Table 1). There did not appear to be a clear trend with the positional monomer QYs, except that HJ D monomer was generally on the low end of the QY range. As seen with most cyanine dyes the QY increased within the more rigid HJ DNA structures as compared to ssDNA.46 In this case the increase in QY appeared to correlate with dye hydrophobicity, the Cy5Peg and Cy5Cl increased about 1.5-fold while the Cy5hex and Cy5tBu increased around 2-fold over the ssDNA.
Steady-state spectral characterization of dimer HJ structures
The use of DNA scaffolding allows for the creation of dye aggregates that would otherwise be inaccessible due to their solubility. The DNA scaffold provides structural variance by comparing either adjacent or transverse dimers (see Fig. 1). It has been shown that the positional variations of dimers within HJ structures can lead to completely different energetic properties.32 As such we undertook a full steady-state spectroscopic characterization of the homodimers of the four Cy5 variants. This involved absorbance, circular dichroism (CD), and emission/excitation fluorescence spectroscopy for the six dimer variations (four adjacent and two transverse HJ dimers) for each dye. Fig. 3 shows the average spectra (the ESI contains all spectra in Fig. S3†) for each dye in the adjacent and transverse conformation. It is very important to note that though the transverse spectra are well represented by their average, in the case of the adjacent spectra there is greater variability and, for example, some adjacent dimers composed by the AB strands differ from the BC version. This will be discussed in greater detail in the modeling and discussion sections.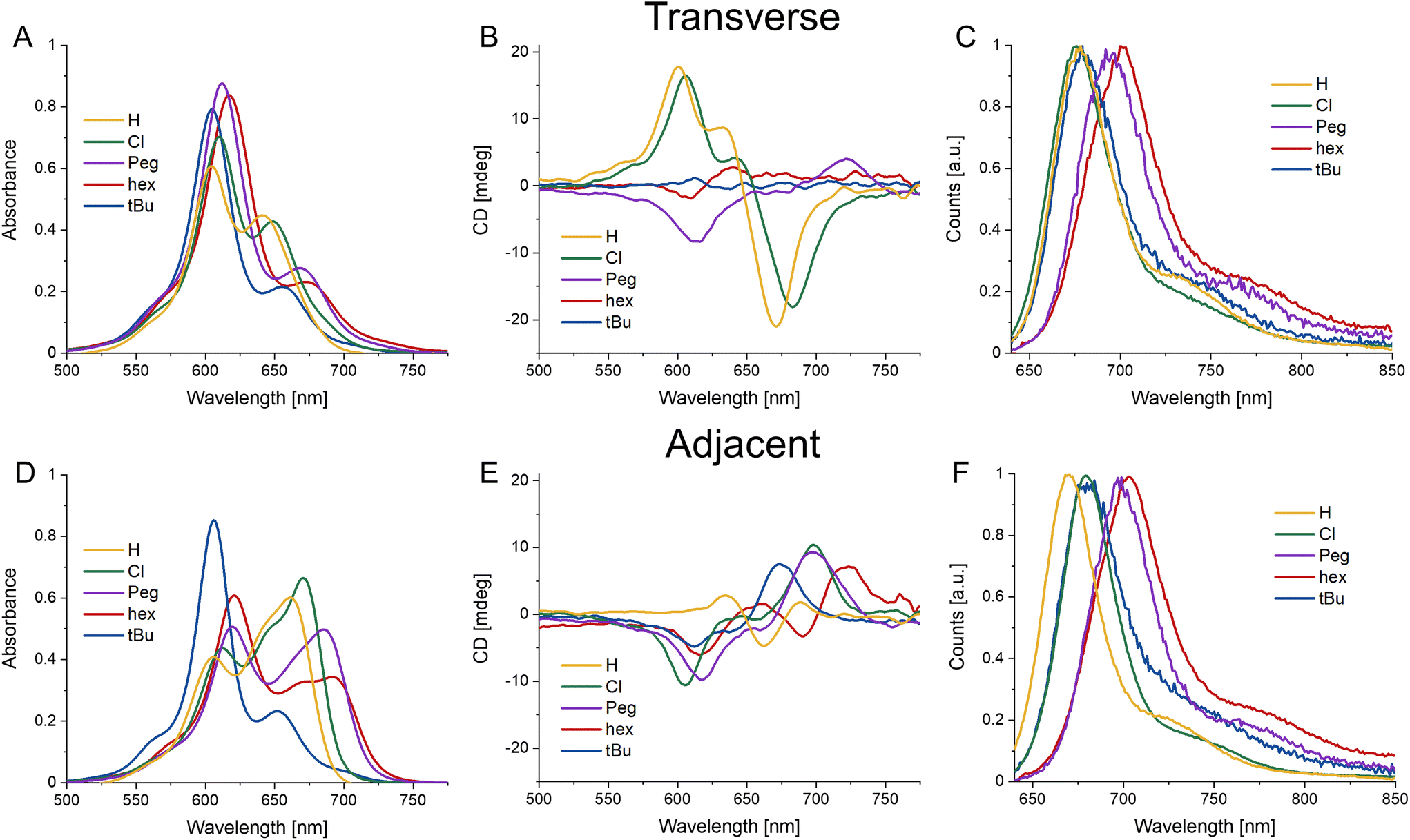 | ||
| Fig. 3 Steady state spectra of average HJ homodimers. (A–C) Absorbance, CD, and normalized emission spectra (excitation 615 nm) of HJ transverse homodimers of Cy5-R. (D–F) Absorbance, CD, and normalized emission spectra (excitation 615 nm) of HJ adjacent homodimers of Cy5-R. All measurements were performed at 20 °C. | ||
The absorbance spectral shapes of the transverse dimers are all generally similar, exhibiting a strongly coupled H-like blue shifted absorbance peak (with respect to the monomer absorption, see Fig. 2) with a less prominent red-shifted shoulder, with the particular positioning of the peak corresponding to the electronic properties of the substituent, in line with the monomer trend. Comparatively, the Cy5H and Cy5Cl have a much greater intensity of the red-shifted peak and the splitting is smaller (∼950 cm−1) than the other dyes (∼1350 cm−1). The red-shifted shoulder absorbance aligns well with the monomer peak absorbance of the same dye. Note that the ionic strength experiments detailed in the next section demonstrate that the absorbance peak is composed of a small monomer component in addition to its being principally part of the H-type aggregate spectra. The CD signals for the transverse conformations are not as similar, the more hydrophilic substituents (H, Peg, and Cl) have larger signals, for both the positive and negative Cotton effects; the hydrophobic substituents have minimal signals in the CD. When the absorbance and CD spectra are observed in conjunction, the far red edge of the absorption spectra (centered near 700–730 nm) aligns with the strong CD bands on the red edge (in cases where the CD signal is strong enough to see them). The Cy5-Peg transverse dimer is a good example of alignment of weak absorbance and a relatively strong CD peak near 725 nm. These correspond to the lowest energy weakly allowed exciton states for H-like aggregates.
The adjacent conformation has greater variability not only within each dye set, but from dye to dye as well. Initially focusing on the absorbance spectra, it can be seen that the Cy5tBu has a similar H-type spectral shape as the transverse conformation, while the other three present oblique type spectra with clear Davydov type splitting. Conversely, the CD spectra are similar, though shifted to lower or higher wavelengths according to the dyes respective energy profile. They all have an initial positive Cotton effect as they approach from the red-end of the spectra, with a subsequent negative signal as the spectra scans toward lower wavelengths.
Fig. 3C and F shows the fluorescence emission spectra. Consistent with previous reports, the fluorescence spectra are quite similar to the monomer version of the respective dyes.23,24,30 This is due to the strong quenching observed upon formation of H-type and oblique dimers of the Cy5 family of dyes.23 We suspect that a majority of the emission is arising from monomer type structures (i.e. small minority of HJ structures that have positioned the dyes in such a way that they are unable to come into close contact, e.g. increased dye separation from DNA breathing, or in perfectly crossed or X-aggregates).47 The work of Huff et al. went into detailed analysis on this mechanism for unmodified Cy5 and the evidence suggests it translates directly to our study as well.23,24 Additional evidence that the emission arises from monomers was provided by the relative quenching (Q) of each dimer. The fluorescence of the six dimers was compared to the fluorescence intensity of the respective monomers taking into account the extinction coefficient at the 615 nm excitation wavelength, represented by the following equation 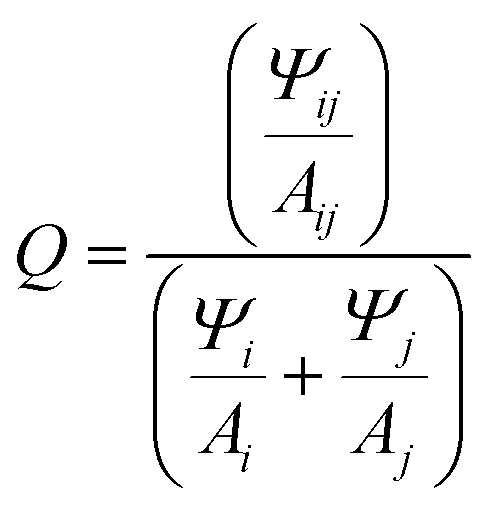 , where Ψ is the integrated fluorescence and A the absorbance at 615 nm of dimer ij, composed of monomer i and monomer j. The Ψ and A values for both monomers and dimers were obtained experimentally from the spectra in Fig. 2 and 3. For the more hydrophobic dyes the relative quenching was indistinguishable between the adjacent and transverse conformations, the values being Cy5tBu (0.990 ± 0.005) and Cy5hex (0.991 ± 0.003). For the more hydrophilic dyes there was a statistically significant difference between the adjacent and transverse conformations. For Cy5Peg the adjacent quenching was 0.96 ± 0.01 and the transverse was 0.982 ± 0.003, while for the Cy5Cl the adjacent quenching was 0.956 ± 0.005 and the transverse was 0.975 ± 0.003. In both cases the transverse conformation had greater quenching than the adjacent conformation.
, where Ψ is the integrated fluorescence and A the absorbance at 615 nm of dimer ij, composed of monomer i and monomer j. The Ψ and A values for both monomers and dimers were obtained experimentally from the spectra in Fig. 2 and 3. For the more hydrophobic dyes the relative quenching was indistinguishable between the adjacent and transverse conformations, the values being Cy5tBu (0.990 ± 0.005) and Cy5hex (0.991 ± 0.003). For the more hydrophilic dyes there was a statistically significant difference between the adjacent and transverse conformations. For Cy5Peg the adjacent quenching was 0.96 ± 0.01 and the transverse was 0.982 ± 0.003, while for the Cy5Cl the adjacent quenching was 0.956 ± 0.005 and the transverse was 0.975 ± 0.003. In both cases the transverse conformation had greater quenching than the adjacent conformation.
The steady-state absorbance and CD spectra of each particular dye and position homodimer were utilized in conjunction with theoretical models to determine coupling parameters, these results and analysis are presented in the KRM section.
Biophysical characterization of dimer HJ structures
The HJ DNA nanostructure has been very successful at templating dimers of dyes that could not be obtained in other ways due to their solubility. Yet, there are conformational distributions and limits on the formation efficiency of the DNA HJ that previous studies have shown can hamper the interpretation of optical characterization measurements.48 To address these issues biophysical characterization was undertaken using PAGE electrophoresis, the effect of divalent ions on HJ conformation, and determination of the HJ melting temperatures. The results demonstrated that our chosen structures were well formed, that the dyes were not disruptive of the HJ formation, and that 15 mM Mg2+ is sufficient to obtain the desired conformation.DNA hybridization must overcome the electrostatics of two very negatively charged biopolymers coming into close proximity, to this end, divalent ions, such as Mg2+, are required to obtain proper structure formation. Even beyond initial hybridization, Mg2+ may be utilized to direct the DNA towards the desired conformation and to tighten DNA hybridization. We prepared the HJ AB and HJ AC dimers of each Cy5 homodimer at 1× TAE with increasing amounts of Mg2+: 0, 3.75, 7.5, 15, and 30 mM. The absorbance, CD, and fluorescence spectra of each sample were recorded; example spectra are presented in Fig. 4A and B with the additional data available in the ESI.† The spectra show that for the HJ dimers the general spectral shape of all Cy5 dyes, including the unmodified Cy5, are obtained already at 3.75 mM Mg2+. At 0 mM Mg2+ we see greater intensity in the spectral region where the monomer has peak absorbance for all the samples, which correlates with the fact that it is always the sample with the greatest fluorescence intensity. In the case of the transverse HJ AC structures, they still have the H-type aggregate shape, even at 0 mM Mg2+, with the resonant strength becoming more distributed towards the principal blue-shifted peak as the ion concentration increases. The adjacent HJ AB structure has a clearer monomer absorbance peak (generally between the two split peaks) at 0 mM Mg2+ that decreases strongly at 3.75 mM Mg2+ with subtle changes as the Mg concentration continues to increase.
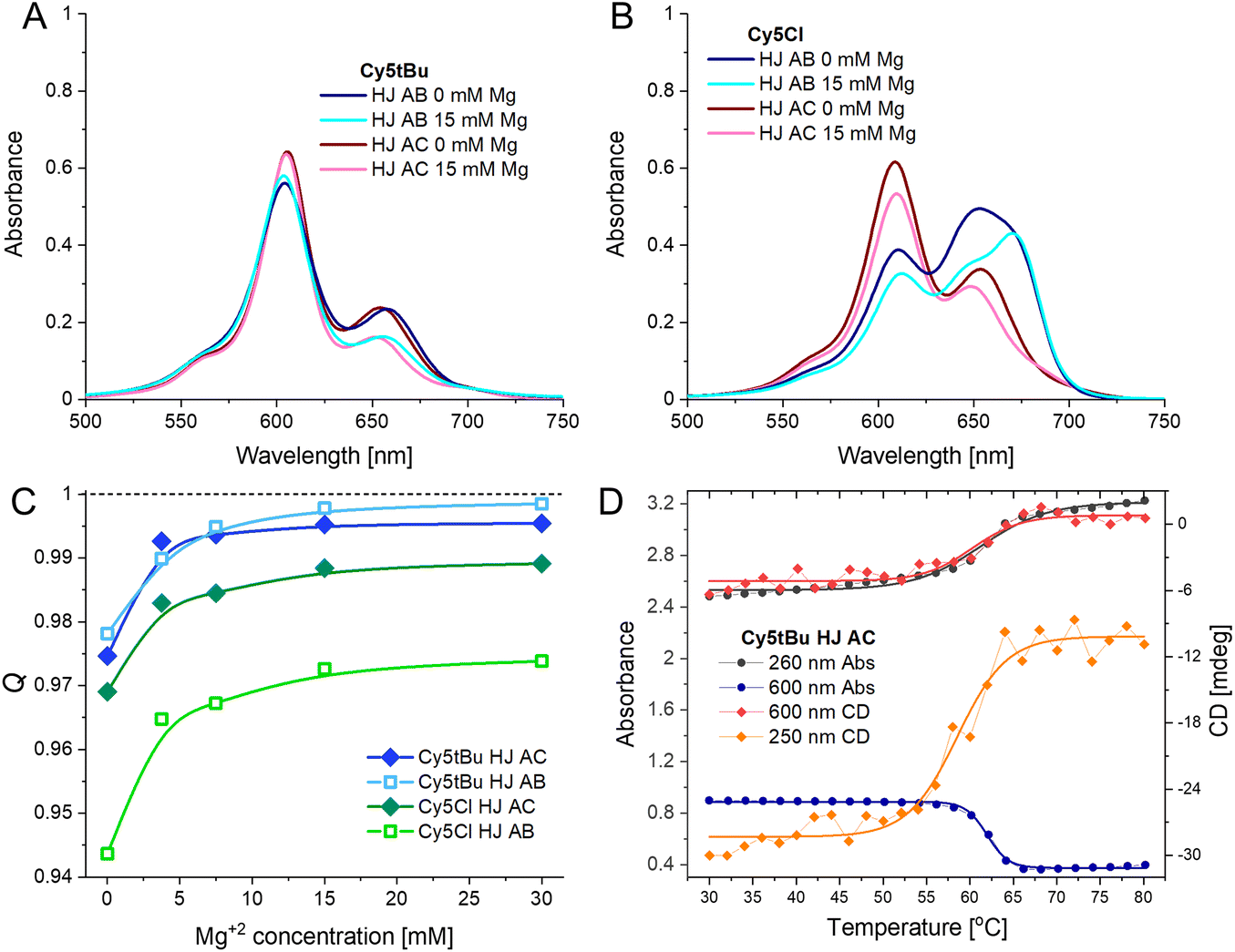 | ||
| Fig. 4 (A) Absorbance spectra of Cy5tBu AB and AC dimer at 0 or 15 mM Mg2+ concentration in buffer. (B) Absorbance spectra of Cy5Cl AB and AC dimer at 0 or 15 mM Mg2+ concentration in buffer. The spectra in panels A and B were measured at 20 °C. (C) Q value (for 615 nm excitation) of a representative selection of Cy5-R dimers in different conformations at varying Mg2+ concentration in 1× TAE buffer. (D) Melting curve of Cy5tBu AC dimer showing absorbance and CD changes. | ||
The fluorescence intensity has an asymptotic relation with the ionic concentration (see Fig. 4C), since the fluorescence intensity correlates to the monomer-like conformation, it is clear that as the Mg2+ concentration increases the amount of monomer decreases.23 Interestingly, when observing the HJ dimers the relative quenching at 0 mM Mg2+ appears to depend on both the conformation and the hydrophobicity of the dye. The hydrophobic dyes (tBu, hex) already have Q values of 0.978 ± 0.002 at 0 mM Mg2+ in both conformations. The Peg and Cl only have 0.947 ± 0.003 and 0.971 ± 0.002 for the AB and AC conformation, respectively. It would appear that at lower Mg2+ the hydrophobicity of the dyes still partially drives their interaction and that the transverse conformation positions the dyes closer together in the case of the hydrophilic dyes. Yet, as noted above, at higher Mg2+ concentrations the Q values continue to increase, as DNA further drives the interactions. These experiments confirmed that the choice of buffer with 15 mM Mg2+ provided an adequate experimental condition to study the dye coupling within HJ structures. A result confirmed by running a 10% PAGE gel showing both the decreased mobility upon complementary strand addition (i.e. A + B + C + D) as well that the structural integrity of the HJ was not adversely affected by inclusion of any of the dyes in this buffer condition (see Fig. S4†). Importantly, it also confirmed that the red-shifted peak in the H-type aggregate co-incidentally overlaps with the monomer peak. If this peak is entirely due to monomer-like species then they would account for 25% of the ensemble, which would result in a much stronger fluorescence signal than what was observed. As the samples are quenched down to more than 99% it cannot be the case that it is a true monomer peak. This same logic confirms that the structures are forming very efficiently, with Q values approaching 100%, almost all HJ structures must have been formed with both dyes integrated into the structure and almost every labeled ssDNA integrated into HJ structures. This, however, still does not eliminate the possibility of heterogeneity within the HJ dimers as discussed below.
The melting temperatures of the HJ structures were determined by following changes in the absorbance and CD spectra as a function of solution temperature (Fig. 4D and S9A–K†). The intent was to determine the effect of the particular dyes on structural stability. An example set of curves is present in Fig. 4D with the data presented in Table 2. We looked at both the DNA only part of the spectra (CD signal at 250 nm and absorbance at 260 nm) as well as the dye only part of the spectra (CD and absorbance signal at 600 or 620 nm depending on the dye). We observed no statistically significant difference upon determining the melting temperature by utilizing the DNA or dye portion of the spectra, signifying that local stabilizations within the structure are unlikely.
| Melting temp [°C] | |||||
|---|---|---|---|---|---|
| Cy5H | Cy5tBu | Cy5hex | Cy5Peg | Cy5Cl | |
| Unlabeled HJ structure: 59.6 ± 0.3 °C. Melting temperatures determined by fitting curves to Boltzmann sigmoidal equation. Reported values are the average of the four curves (CD and absorbance of DNA and dye spectral components). Uncertainty determined from curve fittings and value deviation of average. | |||||
| HJ AB | 57.0 ± 1.3 | 60.8 ± 0.9 | 60.1 ± 1.2 | 57.3 ± 0.9 | 58.6 ± 0.2 |
| HJ AC | 59.8 ± 0.7 | 61.1 ± 0.8 | 62.0 ± 1.6 | 58.2 ± 1.7 | 60.5 ± 0.7 |
Two observations can be made from the melting temperatures of the HJ structures. The first is that the hydrophobic dyes (tBu and hex) result in increases of 2–3 °C in the melting temperature over the unmodified Cy5; the PEG and Cl modifications do not alter the melting temperature. This is in-line with the Mg2+-dependent spectra, where the hydrophobic modifications of the Cy5 led to stronger dye-dye interactions, and as such had greater quenching at 0 mM Mg2+. The second observation is that the HJ AC had a higher melting temperature for all five dyes, though many of the differences were not statistically significant. This is noteworthy due to the overall stronger dye–dye coupling strengths observed for the transverse HJ conformation, discussed in detail in the next section.
Kühn–Renger–May theoretical modeling to determine dye coupling and packing
We employed the theoretical work of Kühn, Renger, and May (KRM)41 through our KRM Model Simulation Tool to understand how the Cy5 modifications influence dye coupling and aggregate packing.41 The KRM Model Simulation Tool utilizes experimental absorbance and CD spectra to extract relative orientation information for two or more coupled dyes. The excitonic coupling strength (given by Jm,n) was determined using an extended dipole approximation which gives more accurate results than the point-dipole approximation when dye separations are on the same order as the size of the dyes themselves.49–51 This approach has been applied successfully to cyanine dyes as well as a multiplicity of other dyes.29,31–33,35,51–53 The unmodified Cy5 and the four Cy5 modifications were modeled in each of the six conformations using this approach. Within the KRM Model Simulation Tool, dyes are modeled as TDM vectors that are assumed to be oriented along the long axis of the Cy5 molecule. The key parameters determined by the Tool (seen in Fig. 5) are: the excitonic hopping parameter (Jm,n), center-to-center dye–dye separation (R), oblique angle (the deviation from parallel of the TDMs, α), and relative slip and twist angles (θs and θt, respectively).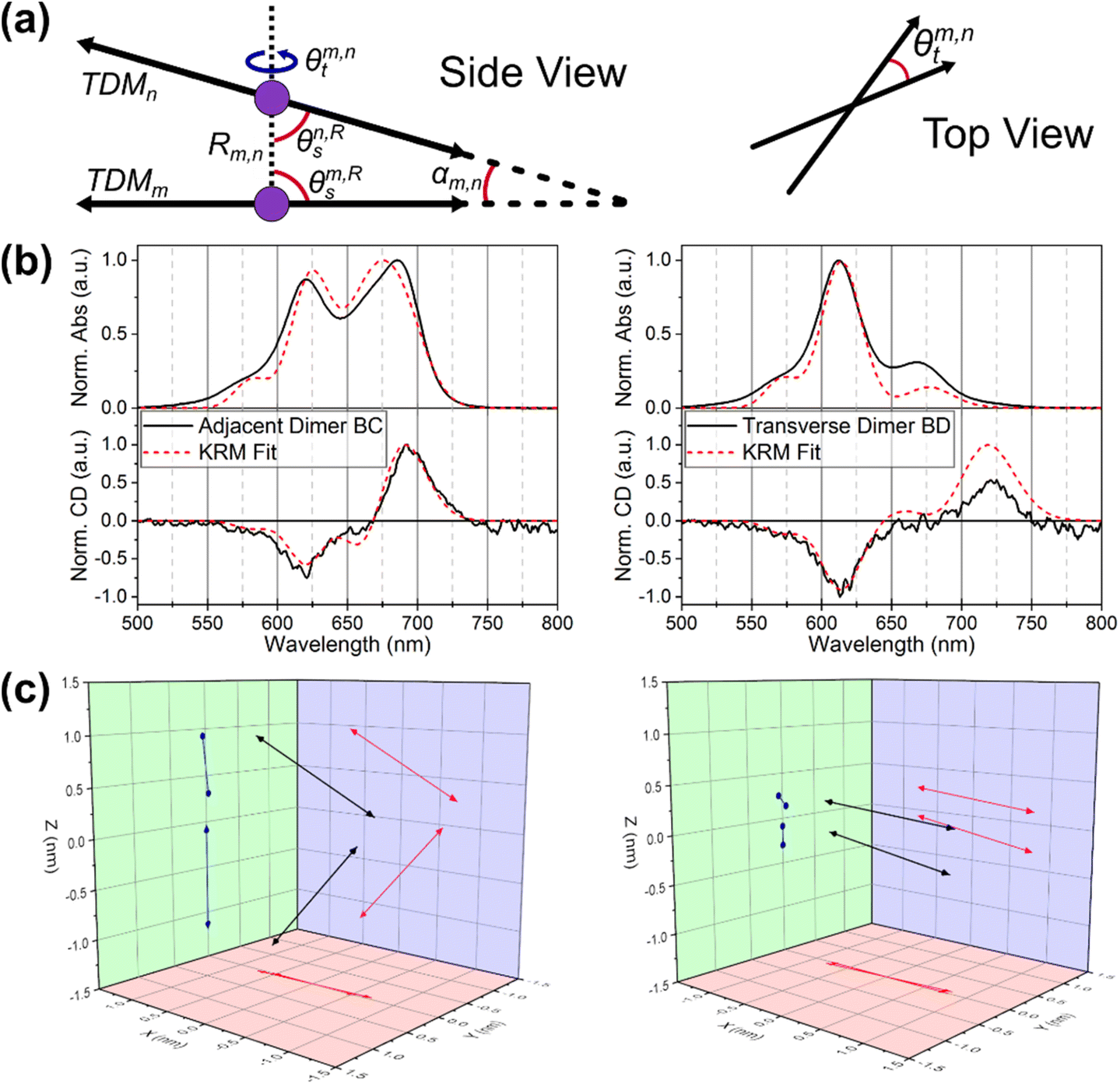 | ||
| Fig. 5 (a) Schematics describing KRM output geometric parameters (b) KRM fit results and (c) transition dipole vector diagrams of Cy5Peg BC adjacent dimer and BD transverse dimer. | ||
The first result presented by the model is the confirmation that the HJ dimers are composed of either H-type or oblique aggregates. Utilizing the results of the model (seen in Fig. 5 and Table 3, with the full data set available in the ESI†) we begin to discern the variability between the packing of the modified dyes. Depending on the dye, there can be differences between the adjacent and transverse conformations, and even between the specific structures of the adjacent conformation in some cases. We also note that in a few cases the data were best fit by assuming two distinct populations of aggregates. This approach was motivated by recent photo-selective transient absorption (TA) experiments, which indicated that structural heterogeneity in Cy5 HJ dimer can lead to co-existing sub-populations of dimer conformations.24 In most cases this resulted in dimer pairs with opposite chirality and nearly identical packing parameters. As such, the interpretation is not modified by representing these cases as a single population of dimers. Further insight and detail are provided in the ESI (Table S3 and Fig. S11† and complete fitting data for each system available in pages S12–S47†). We note that experiments which provide information on potential structural heterogeneity in Cy5-R HJ dimers would sharpen KRM fitting by justifying single-conformer or multi-conformer fitting.
| Dye | Type | J m,n [meV] | R m,n [nm] | α m,n [°] | θ s1/θs2 [°] | θ t |
|---|---|---|---|---|---|---|
| The mean ± standard deviation of each geometrical parameter (see Fig. 5) of four adjacent or two transverse dimers are reported.a The HJ BC structure is oblique.b The HJ AB structure is H-like. | ||||||
| Adjacent | ||||||
| Cy5tBu | H-like | 115.0 ± 3.0 | 0.4 ± 0.0 | 7.2 ± 1.7 | 85.0 ± 2.9/87.2 ± 2.5 | 4.4 ± 1.8 |
| Cy5hex | H-likea | 92.6 ± 24.1 | 0.7 ± 0.3 | 27.2 ± 25.6 | 69.4 ± 15.2/84.0 ± 10.8 | 4.5 ± 1.4 |
| Cy5Peg | Obliqueb | 64.9 ± 20.8 | 1.0 ± 0.3 | 57.9 ± 29.1 | 59.9 ± 18.6/62.4 ± 18.4 | 3.3 ± 1.3 |
| Cy5Cl | Oblique | 49.4 ± 2.3 | 1.3 ± 0.1 | 80.9 ± 9.1 | 36.4 ± 5.4/61.0 ± 5.6 | 1.9 ± 0.6 |
| Cy5-H | Oblique | 52.5 ± 3.3 | 1.2 ± 0.1 | 75.4 ± 11.6 | 57.3 ± 15.8/47.3 ± 9.7 | 1.3 ± 0.5 |
| Transverse | ||||||
| Cy5tBu | H-like | 131.2 ± 9.1 | 0.4 ± 0.0 | 3.2 ± 0.6 | 87.7 ± 1.2/85.7 ± 2.2 | 2.5 ± 0.1 |
| Cy5hex | H-like | 120.4 ± 22.8 | 0.5 ± 0.1 | 9.1 ± 6.1 | 88.4 ± 1.4/83.2 ± 8.3 | 2.6 ± 0.8 |
| Cy5Peg | H-like | 127.5 ± 4.9 | 0.4 ± 0.0 | 5.8 ± 0.1 | 82.9 ± 2.2 /82.1 ± 4.1 | 3.6 ± 1.3 |
| Cy5Cl | oblique | 82.3 ± 3.9 | 0.6 ± 0.0 | 20.2 ± 0.2 | 73.5 ± 1.2/86.9 ± 0.6 | 5.1 ± 1.3 |
| Cy5-H | oblique | 69.7 ± 4.0 | 0.8 ± 0.0 | 35.9 ± 0.6 | 80.8 ± 0.1/64.0 ± 1.3 | 6.7 ± 4.1 |
Below we interpret the results of the KRM modeling. The hypothesis for these observations will be delineated in the discussion sections. The two relative slip angles, θs1 and θs2, are defined as the angles between each TDM and the separation vector, R, and generally relate to aggregate packing type. H-type aggregates will have slip angles near 90°, whereas J-like aggregates will have slip angles close to 0°. The twist angle, θt gives the rotation about the separation vector, R, and generally correlates with the magnitude of the observed CD signal.54 The dyes in this study tend to pack in a somewhat planar arrangement, and thus the twist angles tend to be close to zero. To simplify the analysis, we will discuss α as a global representation of the angular alignment of the TDMs. Relative to H-type aggregates—and when comparing oblique with H-type aggregates—smaller values of alpha are associated with smaller dye–dye separations and higher values of Jm,n. We note that in general, when comparing two oblique aggregates, the subtleties of the extended dipole approximation can complicate this trend.
We begin by reporting on the Cy5Cl and Cy5H, which are consistently oblique type aggregates, independent of the adjacent or transverse conformation, while Cy5tBu is consistently an H-type aggregate. The respective dyes also represent the extremes of the coupling and packing. The Cy5tBu has the largest Jm,n with the smallest R and α, while the Cy5Cl and Cy5H have the inverse. For both dyes, the transverse conformation increases the Jm,n. Although the Cy5tBu observes slightly tighter packing of the already favored H-type dimer; the Cy5Cl and Cy5H have considerably tighter packing in the transverse conformation. For example, R decreases by a factor of 2 for transverse Cy5Cl compared to the adjacent Cy5Cl, resulting in a 50–60% increase in Jm,n.
The KRM modeling of Cy5hex generally gives parameters that are closest to ideal H-dimers, but there are strong proportion of oblique like characteristics in the adjacent dimers. In fact the adjacent HJ BC structure we would characterize as an oblique aggregate. Although the boundaries between H-type, oblique, and J-type aggregates are somewhat ambiguous,32 we consider aggregates to be oblique when α > 30° for small twist angles.
Cy5Peg demonstrates the most variability; the adjacent conformation strongly favors an oblique dimer, while the transverse conformations are strongly coupled H-type dimers. If we compare the adjacent oblique dimers to the transverse dimers of Cy5Peg a nearly 2-fold increase in the Jm,n from 69 ± 18 to 128 ± 4 meV is observed.
The clearest trend from the KRM modeling is that the transverse conformation strongly favors sandwich type π–π stacking and cofacial H-like dimers much more than the adjacent conformation, which favors a T-type π–π stacking (i.e. the indole ring of one dye is perpendicular to that of the second Cy5-R), independently of the inherent properties of the modified Cy5-R. This is seen in the larger Jm,n and smaller R and α values for transverse conformations while the adjacent conformations result in α as high as 83 ± 10° and the subsequent increase in R. This correlates to the results of the HJ melting experiments in which the melting temperature of the transverse AC structure was higher than the adjacent AB structure when comparing within the same dyes.
Discussion
As we have analyzed our particular system, DNA HJ nanostructures with 5,5′-modified Cy5 dyes as dimers at the crossover point, we have observed that the coupling and packing of the dimers is both conformation and dye dependent. The question remains: what are the variables that best predict how the dimers will form? As with any real world system, the modified Cy5 molecules' variables are inherently intertwined. In this case our initial variable of interest was dye hydrophobicity, but the electrostatics and the steric components were also modified. We therefore compared the packing parameters provided by the KRM model to the three dye characteristics: hydrophobicity (log(P)), electron donating capability (σP), and steric component (A-value). The results can be seen in Fig. 6. We note that no evidence for higher order structures was observed for HJ dimers in the Mg2+ experiments nor in the melting curves. Furthermore, due to the greater DNA proportion and increased steric hindrance of the dyes within the HJ conformation the likelihood of higher order structures is small.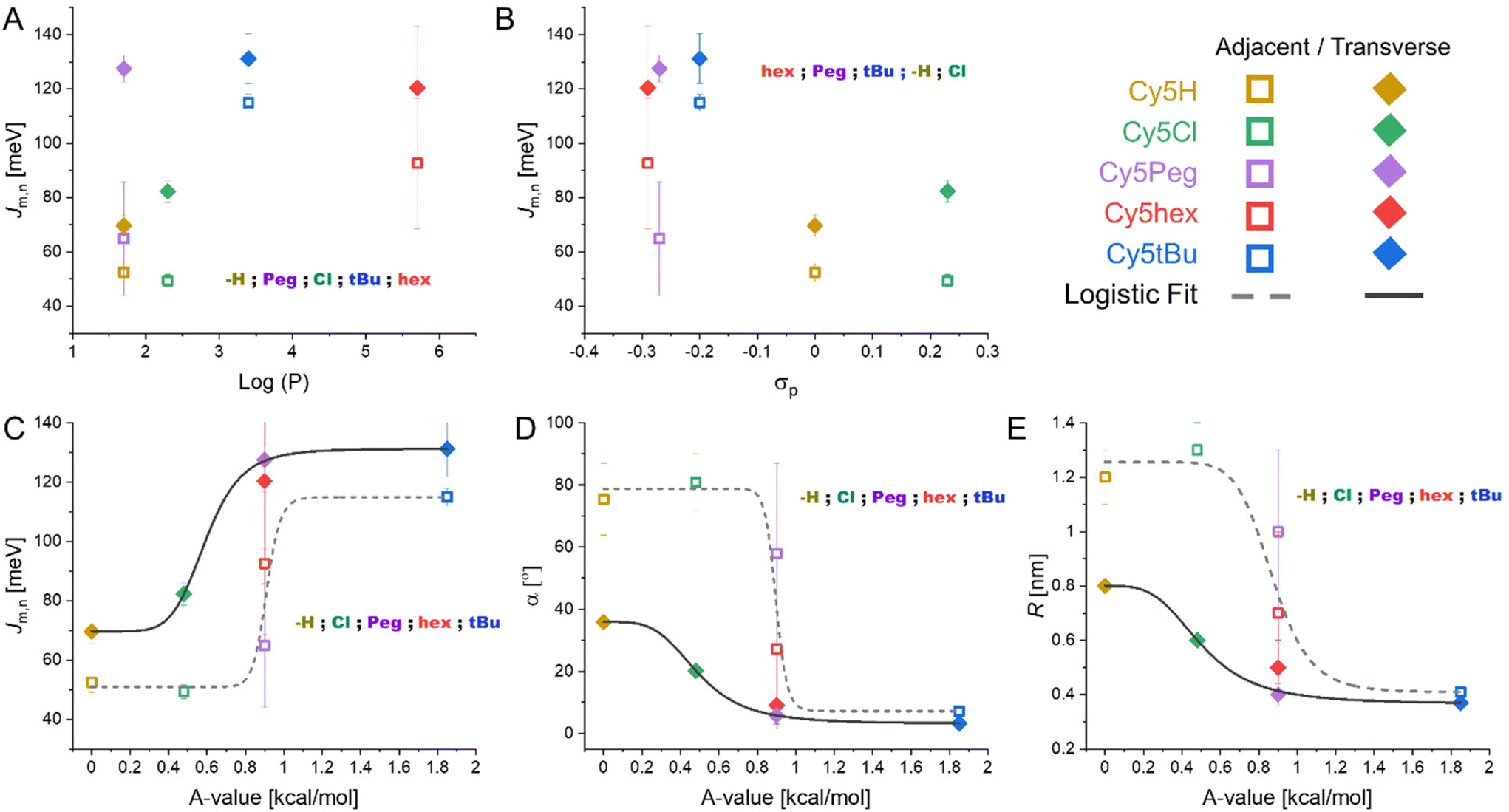 | ||
| Fig. 6 KRM determined packing parameters as function of Cy5-R properties. (A) Jm,n as a function of dye hydrophobicity. (B) Jm,n as a function of electron donating capability of the substituent. (C) Jm,n as a function of steric hindrance of the substituent. (D) α as a function of steric hindrance of the substituent. (E) R as a function of steric hindrance of the substituent. Additional graphs of α and R as functions of dye hydrophobicity and electron donating capability can be found in the ESI (Fig. S10†) along with the fitting details of the curves found in the A-value graphs (Table S2†). | ||
We have chosen to separate the transverse and adjacent HJ conformation results as it appears clear that the placement within the scaffold results in unique interactions in all cases except for Cy5tBu. Observing the data presented in Fig. 6, the parameter with the clearest trend is the correlation of increasing A-value to increasing cofacial H-type dimer packing. This in turn correlates to greater Jm,n and smaller R and α values (see Fig. 6C–E). Our interpretation arises from the fact that only A-value graphs can be fit by simple continuous regression, while the hydrophobicity and electron donating graphs cannot be fit beyond complex polynomials that do not correlate with any physical explanations. The logistic fits used in Fig. 6C–E (R2 ≥ 0.97, except for adjacent fit in Fig. 6C where R2 = 0.90, see Table S2 in the ESI†) provide a bit more information. Logistic functions are generally used to model the probability of a binary outcome as a function of an independent variable.55 This approach is most common in dose–response curves, but is also used in describing molecular states.56,57 In our system it models the probability of the dimers taking an oblique like or H-type packing, with this packing then correlating with the Jm,n, R, and α values. Pertinently, the transition value, i.e. the center of the fit, is unique for the adjacent and transverse dimers yet similar throughout Fig. 6C–E. For adjacent dimers the transition from oblique to H-type packing being more likely when the substituents have A-values greater than 0.89 ± 0.04 kcal mol−1, yet for the transverse dimers the value is only 0.53 ± 0.06 kcal mol−1. Hydrophobicity and electron donating strength likely play a more subtle role. Particularly hydrophobicity appears to drive the distinction between Cy5hex and Cy5Peg, which are very similar in σP and A-value, into H-type and oblique dimers, respectively, in the adjacent HJ conformations.
We hypothesize that the sterics play a key role in the packing as they strongly favor sandwich type π–π stacking (H-like) over the T-type π–π stacking (oblique) of the dyes with smaller substituents. This is particularly notable in the unmodified Cy5. The π–π stacking interactions are relatively weak (<10 kcal mol−1)58 with shallow potential energy landscapes that can be tuned through substituents. The traditional concept of π–π stacking is the Hunter-Sanders model; in this model the aromatic ring is considered a positively charged σ framework and a negatively charged π cloud.59 As such, the more electron withdrawing a substituent group is the more it minimizes electrostatic repulsion of the π-clouds and therefore the greater the interaction. If the model holds true for our system we would expect Jm,n to correlate with σp. As we see in Fig. 6B Cy5Cl has a greater Jm,n than Cy5H as would be predicted by the model, but it is the electron donating groups that have the greater Jm,n by approximately 2-fold in contraposition to the model. Other experimental reports have shown that the Hunter-Sanders model is often in disagreement with the experiment and does not account for significant steric interactions.60,61
Pertinent to our work, the Sherrill lab recently published studies comparing sandwich and T-style π–π stacking utilizing a more complex symmetry-adapted perturbation theory, which considers the electrostatic, London dispersion forces (LDF), induction, and exchange–repulsion components of the interaction energy.62 A key conclusion was that LDF, i.e. induced dipole-induced dipole or van der Waals forces, are the predominant force that determines interaction strength.62 This is particularly important in T-shaped stacking, where distances between aromatic rings are already further apart, as LDF decay with distance to the sixth power. In other words, the possibility of stabilizing T-shaped π–π stacking is proportional to how close the aromatic rings can approach each other. It then correlates that the greater the steric repulsion, i.e. larger A-values, the less favorable T-shaped stacking is. In this case inhibition of T-shaped stacking favors sandwich type π–π stacking which has greater Jm,n. This trend was observed clearly in Fig. 6C. This results in the first design rule for template dimers: substituents with larger A-values hinder the formation of oblique aggregates.
We then look at Cy5Peg vs. Cy5hex, where both the σp and A-value were almost identical but with orders of magnitude difference in hydrophobicity. Here we see that the hydrophobic dye has a greater propensity to interact, avoiding oblique like dimers. This is seen by looking at the adjacent dimers, where Cy5hex has more H-like character and greater Jm,n. It would appear that Cy5Peg also has a greater monomer proportion due to its lower hydrophobicity seen in the lower Jm,n and lower quenching proportion in both adjacent and transverse conformations. This suggests that relative hydrophobicity is beneficial to maximize dimer formation, and subsequently plays a considerable, yet secondary, role in the conformation of the dimer.
The third, perhaps intuitive, rule: the initial position of the dyes relative to each other within the template helps determine the final dimer conformation. It appears clear from the data that transverse HJs favor cofacial interactions that lead to H-type aggregates while adjacent dimers favor T-shaped π–π stacking and oblique dimers. This was supported by the fits that resulted in transition points where the H-type is favored over oblique dimers if the substituents have A-values greater than 0.53 ± 0.06 kcal mol−1 for transverse dimers while the value increased to 0.89 ± 0.04 kcal mol−1 for adjacent dimers. Some evidence has been reported that for unmodified Cy3 dyes, in the case of dsDNA,63 even the particular sequence can determine the coupling orientation of the dye aggregate, which is supported by the variance observed between the four adjacent conformations.
We proceed to compare this work to previous studies and compare the hypothesis stated by their conclusions. We begin by quickly distinguishing the work of modifying freely-diffusing cyanines, in those cases the hydrophobicity of the dyes was key to driving aggregation.26,27,64 In our work the Cy5-R are conjugated to ssDNA which dictates the overall solubility of the system, and the hybridization of the DNA strands is what in fact drives aggregation. Clearly, within this context dye hydrophobicity plays a more subtle role. The work of Stadler, Armitage, and colleagues also looked at the modified cyanines in combination with DNA as a non-covalent nucleation site.26 They reported that increased hydrophobicity did enhance dimerization on the DNA, but also that steric considerations created by the hydrophobic modifications (mainly methyl substituents) did limit the range of extended helical aggregates. Though their system was non-covalent, their hypothesis that hydrophobicity can drive dimerization proportions, but also that the sterics created by the modifications inherently limit some forms of aggregates (e.g. extended helical aggregates)26 is in line with what we observed in our covalent system. Albeit, they did not provide insight on how to exploit the sterics for designer structures. We found that more hydrophobic dyes (tBu, hex) have greater dimer proportions than the more hydrophilic substituents, this is seen most clearly in the zero Mg2+ experiments. Yet, as we discussed above, it is the sterics which principally drive the H-type or oblique like packing.
The most pertinent comparison is to the work of Mass and colleagues looking at the effect of hydrophobicity on DNA HJ templated squaraine dye dimers.35 We highlight three differences, the first being of course the choice of dye, second, the range of hydrophobic variation in the squaraines was smaller than our study (the R = hex or tBu is orders of magnitude more hydrophobic than the most hydrophobic Cl they synthesized), and, importantly, the approach to dye conjugation. The last point is particularly important, though both the squaraine dyes and Cy5-R dyes in this study were covalently bound to the nucleotides, the squaraines utilized a flexible single-point attachment (fully extended length of 2.1 nm) while the Cy5-R were attached using a short 3C two-point attachment. The long linker and subsequent flexibility allows for greater degrees of freedom of approach between the squaraine dyes and also that the initial positioning of the dyes can be much further apart than in the case of the Cy5-R in our system. In fact, in their system the transverse dimers generally had smaller Jm,n than the adjacent dimers. This result correlates with dye attachment points for transverse dimers being further apart than for adjacent dimers, thus, reducing the probability of the coming together of dyes with lower hydrophobicity. Resultantly, hydrophobicity played a more important role in the dimer formation of the squaraines. Due to the shorter linker and two-point conjugation, this was not a crucial variable in our system. In addition, we performed an analysis of their results by considering the A-values of their substituents (we used 1.0 and 1.2 for –CH3 and SO3H, respectively) and did not observe any correlation as we did in the Cy5-R system. Overall, the results of these two studies are consistent, once more, in that hydrophobicity is key to driving the proportion of dimer formation, but we hypothesize that due to the conjugation approach of the dye the rules for determining the dimer properties are not translatable from one system to the other. It would be of interest to invert the conjugation approaches of the dyes and to validate this hypothesis.
Finally, we mention that for close range dye packing (Rm,n = 3.5–4.0 Å) one anticipates the possibility of relaxed excited states such as excimers, exciplexes, and/or charge transfer species. However, we have not observed the signatures of relaxed excited states from fluorescence spectra of Cy5-R dimers or in transient absorption studies that our groups have performed on Cy5 dimers on DNA.24,29 There could be several reasons for not observing relaxed excited states in Cy5-R dimers at close range, including that charge transfer states occur at energies that are too high/low to allow strong mixing with the molecular exciton wavefunctions, or that these relaxed excited state species do occur, but they are beyond the wavelength range that we are able to measure, or that at room temperature the relaxed excited states decay too rapidly to be observed by standard fluorescence detection.
Conclusion
In conclusion, we have begun to elucidate how three factors can be used to direct coupling strength and packing parameters of DNA scaffolded Cy5-R dimers. The principal conclusion is that sterics, as estimated by A-values, play a key role in orienting the dimers and therefore the coupling strength. Additionally, the hydrophobic properties of the Cy5-R play a role in determining the percentage of dimers formed (though within our HJ template system the effect is small), and when steric considerations are equal can determine the packing characteristics of the dimers. Consistent with other reports, the attachment position of the dye within the DNA HJ is very important, comparing both adjacent to transverse HJ dimers results in distinct coupling that favored either H-type or oblique dimers.32,33 Overall it is important to remember that no single dye property is changed in a vacuum and that attempting to change one, for example hydrophobicity, also changes the electrostatics and the steric considerations of the Cy5-R and these must be considered. In order to purposefully control dye coupling and packing on DNA scaffolds, more studies are required to better understand the interplay between dye molecule properties, and the interactions between dyes and DNA.We highlight that though we formed principally H-type and oblique dimers, there is some evidence that some systems may be heterogeneous with a portion of J-type aggregates as well (part of a follow-up study). Cofacial π–π stacking, observed in H-aggregates, are avenues for charge transport and increased charge carrier mobility.13 These properties would have direct application to developing organic dye based electronic devices. There are also reports of H-aggregates being applied as photothermal therapy and photoacoustic imaging agents.65,66 Oblique packing aggregates have also shown some optimal fluorescent properties within crystals, such as the work of the Tian group with 9,10-bis-(2,2-diphenylvinyl) anthracene, where amplified spontaneous emission and electroluminescence were observed.67 Finally, the ability to control exciton coupling in larger dye aggregates on DNA scaffolds is central to the development of exciton devices and circuits68,69 for a variety of photonic applications including light harvesting and nanoscale computing.
The final question that arises is how the steric hypothesis can be further tested and exploited. It would be of interest to test Cy5-R options with A-values between the transition point of the adjacent and transverse dimers (0.5–0.9 kcal mol−1). For example, O-acetyl or O-tosyl substituents have A-values of ∼0.7 kcal mol−1,43 which by our hypothesis should form H-like dimers in transverse conformations but oblique dimers in adjacent conformations. As part of a follow-up study we are designing heterogeneous dimers to test our theory. From our above results, we would expect that the formation of oblique dimers would be favored by the inclusion of a low A-value Cy5-R (e.g. Cy5H or Cy5Cl) even in combination with a large A-value Cy5-R. Furthermore, we imagine that the specific ordering of the two dyes should be deterministic of the final packing characteristic. In other words, a heterogeneous AB dimer should be distinct from a BA dimer. This also opens up the question of utilizing non-symmetrical cyanine dyes to further tune the coupling and packing. The Würthner group demonstrated that dipolar effects of non-symmetrical merocyanine dyes could be used to enhance aggregation in freely diffusing dyes in organic solvents.64 Work with charged cyanine dyes found a minimal effect in similar conditions. The question is whether DNA templated cyanine dyes would prove to resemble either, or demonstrate unique properties.
Methods
The unmodified ssDNA oligos were acquired from Integrated DNA Technologies (Coralville, IA, USA) as lyophilized samples which were reconstituted to 100 μM solutions in molecular biology grade water for use. Cy5-R ssDNA oligos were synthesized following previously published protocol and were also reconstituted into 100 μM solutions in molecular biology grade water for use.36 DNA HJ structures were assembled by combining 10 μM equimolar of A, B, C, D DNA strands or their dye analogues (see Table S1†) in 1× TAE + 15 mM MgCl2 buffer. The samples were assembled via thermal annealing using the previously reported protocol.32,51 Samples were used for downstream analysis without any further purification and were stored at 4 °C otherwise.Polyacrylamide gel electrophoresis (PAGE) analysis shown in Fig. S4† was performed on a 10% pre-cast PAGE gel in 1× Tris Borate EDTA buffer. Each sample was prepared by combining 4 μL of sample (at 10 μM) with 10× GelRed stain and non-SDS loading buffer (New England Biolabs, Ipswich, MA, USA). A 50 bp DNA Ladder (New England Biolabs) was used as a reference molecular weight marker. Electrophoresis conditions were set at 12 V cm−1 at room temperature for 25 min. Gel was imaged using BioRad Gel dock under UV trans-illumination.
Absorption spectra were measured at 20 °C using a Cary 60 UV-Vis spectrophotometer for all samples. Measurements were performed within the spectral range of 200–900 nm, with 1 nm step intervals and a 0.1 second integration time. 2 μM solution of each sample in a 10 mm path length quartz spectrophotometer cell was used for the measurements.
Circular dichroism measurements (CD) were performed at 20 °C using a JASCO J-1500 CD spectrophotometer. Measurements were performed within the spectral range of 200–850 nm, with 1 nm step intervals, at 100 nm min−1, 4 s digital integration time, at 20 °C. 2 μM solution of each sample in a 10 mm path length quartz spectrophotometer cell was used for the measurements.
Fluorescence emission spectra were measured at 20 °C using a TECAN plate reader exciting from above. An excitation wavelength of 615 nm was used to excite the sample and the fluorescence emission was measured from 630–900 nm with 1 nm steps. 100 μl of a 1 μM solution was added to a 96 well black plate, resulting in a 1.25 mm pathlength and a maximum OD of 0.05 avoiding inner filter effects.
Fluorescence QYs of Cy5H were obtained against the reference dye Oxazine 720 (excite at 615 nm with QY = 0.63 ± 0.05) in 1× TAE with 15 mM MgCl2.32 The fluorescence spectra were collected on a Fluoromax 4 (Horiba) fluorescence spectrometer using an orthogonal excitation/detection geometry. All solutions were dilute to a peak optical density ≤ 0.1 to ensure a uniform excited volume, and the linearity of the fluorescence spectra with concentration was confirmed. The Cy5-R QYs were determined by utilizing the Cy5H as a secondary standard in the above mentioned fluorescence conditions. The QYs are reported as an average of three measurements with an uncertainty given by the standard deviation.
The absorption and circular dichroism spectra of the Cy5-R homodimers were used to extract relative orientation information based on Kühn−Renger−May (KRM) theory41 using the KRM Model Simulation Tool (version 13.5). A complete description of the Tool has recently been published elsewhere.31 Briefly, the Tool uses key parameters extracted from experimental single-dye (monomer) spectra and the excitonic hopping parameter – J(m,n), determined using an extended dipole approximation – to populate a Holstein-like Frenkel Hamiltonian.70,71 The Hamiltonian is diagonalized on an appropriate Hilbert space and the results are used to compute theoretical absorbance and CD spectra arising from a given orientation. The tool uses an iterative approach to evaluate spectra from many orientations to find the best match between calculated and experimental spectra.
Author contributions
SAD, AM, MC, KS, DM, ILM prepared, purified, and characterized the dye-labeled DNA oligos and DNA nanostructures (Investigation). GP, LKP, SKR, WBK, BY, JL curated the data and analyzed the spectra for KRM modeling (data curation and formal analysis). SAD, PDC, ILM, JSM designed experimental protocols for spectroscopy (methodology) and realized initial analysis (formal analysis). WBK, BY, JL, ILM, JSM (conceptualization). SAD, GP, LKP, AM, PDC, ILM, JL, JSM (original draft). All authors (writing – review & editing)Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Research at Boise State was supported by the Department of the Navy, Office of Naval Research (ONR) via ONR award no. N00014-19-1-2615. Research at the U.S. Naval Research Laboratory (NRL) was supported by NRL base funding, the NRL Institute for Nanoscience, and ONR award # N0001419WX01811. D. M. was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award Number R00EB030013. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- E. E. Jelley, Spectral absorption and fluorescence of dyes in the molecular state, Nature, 1936, 138, 1009–1010 CrossRef CAS
.
- G. Scheibe, Über die veränderlichkeit der absorptionsspektren in lösungen und die nebenvalenzen als ihre ursache, Angew. Chem., 1937, 50, 212–219 CrossRef CAS
-
T. H. James, The theory of the photographic process, Macmillan Pub Co, 4th edn, 1977 Search PubMed
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, J-aggregates: From serendipitous discovery to supramolecular engineering of functional dye materials, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed
- Y. Hara, T. Fujii, H. Kashida, K. Sekiguchi, X. Liang, K. Niwa, T. Takase, Y. Yoshida and H. Asanuma, Coherent quenching of a fluorophore for the design of a highly sensitive in-stem molecular beacon, Angew. Chem., Int. Ed., 2010, 49, 5502–5506 CrossRef CAS PubMed
- H. Asanuma, T. Fujii, T. Kato and H. Kashida, Coherent interactions of dyes assembled on DNA, J. Photochem. Photobiol., C, 2012, 13, 124–135 CrossRef CAS
- F. Fassioli, D. G. Oblinsky and G. D. Scholes, Designs for molecular circuits that use electronic coherence, Faraday Discuss., 2013, 163, 341–351 RSC
- M. Sarovar, A. Ishizaki, G. R. Fleming and K. B. Whaley, Quantum entanglement in photosynthetic light-harvesting complexes, Nat. Phys., 2010, 6, 462–467 Search PubMed
- S. K. Saikin, A. Eisfeld, S. Valleau and A. Aspuru-Guzik, Photonics meets excitonics: Natural and artificial molecular aggregates, Nanophotonics, 2013, 2, 21–38 Search PubMed
- R. J. Cogdell, A. T. Gardiner and L. Cronin, Learning from photosynthesis: How to use solar energy to make fuels, Philos. Trans. R. Soc., A, 2012, 370, 3819–3826 CrossRef CAS PubMed
- M. A. Castellanos, A. Dodin and A. P. Willard, On the design of molecular excitonic circuits for quantum computing: The universal quantum gates, Phys. Chem. Chem. Phys., 2020, 22, 3048–3057 RSC
- R. R. Frontiera, Quantum coherent phenomena in energy harvesting and storage, J. Phys. Chem. B, 2022, 126, 5727–5729 CrossRef CAS PubMed
- S. Ma, S. Du, G. Pan, S. Dai, B. Xu and W. Tian, Organic molecular aggregates: From aggregation structure to emission property, Aggregate, 2021, 2, e96 Search PubMed
- A. Liess, A. Lv, A. Arjona-Esteban, D. Bialas, A.-M. Krause, V. Stepanenko, M. Stolte and F. Würthner, Exciton coupling of merocyanine dyes from H- to J-type in the solid state by crystal engineering, Nano Lett., 2017, 17, 1719–1726 CrossRef CAS PubMed
- F. Nicoli, M. K. Roos, E. A. Hemmig, M. Di Antonio, R. de Vivie-Riedle and T. Liedl, Proximity-induced H-aggregation of cyanine dyes on DNA-duplexes, J. Phys. Chem. A, 2016, 120, 9941–9947 CrossRef CAS PubMed
- M. Chiriboga, S. A. Diaz, D. Mathur, D. A. Hastman, J. S. Melinger, R. Veneziano and I. L. Medintz, Understanding self-assembled pseudoisocyanine dye aggregates in DNA nanostructures and their exciton relay transfer capabilities, J. Phys. Chem. B, 2022, 126, 110–122 CrossRef CAS PubMed
- D. Mathur, A. Samanta, M. G. Ancona, S. A. Díaz, Y. Kim, J. S. Melinger, E. R. Goldman, J. P. Sadowski, L. L. Ong, P. Yin and I. L. Medintz, Understanding Förster resonance energy transfer in the sheet regime with DNA brick-based dye networks, ACS Nano, 2021, 15, 16452–16468 CrossRef CAS PubMed
- W. P. Klein, B. S. Rolczynski, S. M. Oliver, R. Zadegan, S. Buckhout-White, M. G. Ancona, P. D. Cunningham, J. S. Melinger, P. M. Vora, W. Kuang, I. L. Medintz and S. A. Díaz, DNA origami chromophore scaffold exploiting homoFRET energy transport to create molecular photonic wires, ACS Appl. Nano Mater., 2020, 3, 3323–3336 CrossRef CAS
- S. A. Díaz, S. M. Oliver, D. A. Hastman, I. L. Medintz and P. M. Vora, Increased transfer efficiency from molecular photonic wires on solid substrates and cryogenic conditions, J. Phys. Chem. Lett., 2018, 9, 3654–3659 CrossRef PubMed
- K. Cervantes-Salguero, A. Biaggne, J. M. Youngsman, B. M. Ward, Y. C. Kim, L. Li, J. A. Hall, W. B. Knowlton, E. Graugnard and W. Kuang, Strategies for controlling the spatial orientation of single molecules tethered on DNA origami templates physisorbed on glass substrates: Intercalation and stretching, Int. J. Mol. Sci., 2022, 23, 7690 CrossRef CAS PubMed
- D. Mathur, Y. C. Kim, S. A. Díaz, P. D. Cunningham, B. S. Rolczynski, M. G. Ancona, I. L. Medintz and J. S. Melinger, Can a DNA origami structure constrain the position and orientation of an attached dye molecule?, J. Phys. Chem. C, 2021, 125, 1509–1522 CrossRef CAS
- J. W. Fothergill, A. C. Hernandez, W. B. Knowlton, B. Yurke and L. Li, Ab initio studies of exciton interactions of Cy5 dyes, J. Phys. Chem. A, 2018, 122, 8989–8997 CrossRef CAS PubMed
- J. S. Huff, P. H. Davis, A. Christy, D. L. Kellis, N. Kandadai, Z. S. D. Toa, G. D. Scholes, B. Yurke, W. B. Knowlton and R. D. Pensack, DNA-templated aggregates of strongly coupled cyanine dyes: Nonradiative decay governs exciton lifetimes, J. Phys. Chem. Lett., 2019, 10, 2386–2392 CrossRef CAS PubMed
- J. S. Huff, D. B. Turner, O. A. Mass, L. K. Patten, C. K. Wilson, S. K. Roy, M. S. Barclay, B. Yurke, W. B. Knowlton, P. H. Davis and R. D. Pensack, Excited-state lifetimes of DNA-templated cyanine dimer, trimer, and tetramer aggregates: The role of exciton delocalization, dye separation, and DNA heterogeneity, J. Phys. Chem. B, 2021, 125, 10240–10259 CrossRef CAS PubMed
- B. S. Rolczynski, S. A. Díaz, Y. C. Kim, I. L. Medintz, P. D. Cunningham and J. S. Melinger, Understanding disorder, vibronic structure, and delocalization in electronically coupled dimers on DNA duplexes, J. Phys. Chem. A, 2021, 125, 9632–9644 CrossRef CAS PubMed
- A. L. Stadler, B. R. Renikuntla, D. Yaron, A. S. Fang and B. A. Armitage, Substituent effects on the assembly of helical cyanine dye aggregates in the minor groove of a DNA template, Langmuir, 2011, 27, 1472–1479 CrossRef CAS PubMed
- D. Takahashi, H. Oda, T. Izumi and R. Hirohashi, Substituent effects on aggregation phenomena in aqueous solution of thiacarbocyanine dyes, Dyes Pigm., 2005, 66, 1–6 CrossRef CAS
- B. L. Cannon, D. L. Kellis, L. K. Patten, P. H. Davis, J. Lee, E. Graugnard, B. Yurke and W. B. Knowlton, Coherent exciton delocalization in a two-state DNA-templated dye aggregate system, J. Phys. Chem. A, 2017, 121, 6905–6916 CrossRef CAS PubMed
- P. D. Cunningham, S. A. Díaz, B. Yurke, I. L. Medintz and J. S. Melinger, Delocalized two-exciton states in DNA scaffolded cyanine dimers, J. Phys. Chem. B, 2020, 124, 8042–8049 CrossRef CAS PubMed
- B. L. Cannon, L. K. Patten, D. L. Kellis, P. H. Davis, J. Lee, E. Graugnard, B. Yurke and W. B. Knowlton, Large davydov splitting and strong fluorescence suppression: An investigation of exciton delocalization in DNA-templated holliday junction dye aggregates, J. Phys. Chem. A, 2018, 122, 2086–2095 CrossRef CAS PubMed
- S. K. Roy, O. A. Mass, D. L. Kellis, C. K. Wilson, J. A. Hall, B. Yurke and W. B. Knowlton, Exciton delocalization and scaffold stability in bridged nucleotide-substituted, DNA duplex-templated cyanine aggregates, J. Phys. Chem. B, 2021, 125, 13670–13684 CrossRef CAS PubMed
- A. U. Chowdhury, S. A. Díaz, J. S. Huff, M. S. Barclay, M. Chiriboga, G. A. Ellis, D. Mathur, L. K. Patten, A. Sup, N. Hallstrom, P. D. Cunningham, J. Lee, P. H. Davis, D. B. Turner, B. Yurke, W. B. Knowlton, I. L. Medintz, J. S. Melinger and R. D. Pensack, Tuning between quenching and energy transfer in DNA-templated heterodimer aggregates, J. Phys. Chem. Lett., 2022, 13, 2782–2791 CrossRef CAS PubMed
- O. A. Mass, C. K. Wilson, S. K. Roy, M. S. Barclay, L. K. Patten, E. A. Terpetschnig, J. Lee, R. D. Pensack, B. Yurke and W. B. Knowlton, Exciton delocalization in indolenine squaraine aggregates templated by DNA holliday junction scaffolds, J. Phys. Chem. B, 2020, 124, 9636–9647 CrossRef CAS PubMed
- P. Verma and H. Pal, Intriguing H-aggregate and H-dimer formation of coumarin-481 dye in aqueous solution as evidenced from photophysical studies, J. Phys. Chem. A, 2012, 116, 4473–4484 CrossRef CAS PubMed
- O. A. Mass, C. K. Wilson, G. Barcenas, E. A. Terpetschnig, O. M. Obukhova, O. S. Kolosova, A. L. Tatarets, L. Li, B. Yurke, W. B. Knowlton, R. D. Pensack and J. Lee, Influence of hydrophobicity on excitonic coupling in DNA-templated indolenine squaraine dye aggregates, J. Phys. Chem. C, 2022, 126, 3475–3488 CrossRef CAS PubMed
- A. Meares, K. Susumu, D. Mathur, S. H. Lee, O. A. Mass, J. Lee, R. D. Pensack, B. Yurke, W. B. Knowlton, J. S. Melinger and I. L. Medintz, Synthesis of substituted Cy5 phosphoramidite derivatives and their incorporation into oligonucleotides using automated DNA synthesis, ACS Omega, 2022, 7, 11002–11016 CrossRef CAS PubMed
- R. J. Mazuski, S. A. Díaz, R. E. Wood, L. T. Lloyd, W. P. Klein, D. Mathur, J. S. Melinger, G. S. Engel and I. L. Medintz, Ultrafast excitation transfer in Cy5 DNA photonic wires displays dye conjugation and excitation energy dependency, J. Phys. Chem. Lett., 2020, 11, 4163–4172 CrossRef CAS PubMed
- P. D. Cunningham, A. Khachatrian, S. Buckhout-White, J. R. Deschamps, E. R. Goldman, I. L. Medintz and J. S. Melinger, Resonance energy transfer in DNA duplexes labeled with localized dyes, J. Phys. Chem. B, 2014, 118, 14555–14565 CrossRef CAS PubMed
- C. R. Simmons, T. MacCulloch, M. Krepl, M. Matthies, A. Buchberger, I. Crawford, J. Šponer, P. Šulc, N. Stephanopoulos and H. Yan, The influence of holliday junction sequence and dynamics on DNA crystal self-assembly, Nat. Commun., 2022, 13, 3112 CrossRef CAS PubMed
- M. Kasha, Energy transfer mechanisms and the molecular exciton model for molecular aggregates, Radiat. Res., 1963, 20, 55–70 CrossRef CAS PubMed
- O. Kühn, T. Renger and V. May, Theory of exciton-vibrational dynamics in molecular dimers, Chem. Phys., 1996, 204, 99–114 CrossRef
- G. S. Remya and C. H. Suresh, Quantification and classification of substituent effects in organic chemistry: A theoretical molecular electrostatic potential study, Phys. Chem. Chem. Phys., 2016, 18, 20615–20626 RSC
-
J. A. Hirsch, in Topics in stereochemistry, 1967, pp. 199–222, DOI:10.1002/9780470147108.ch4
- S. E. Boiadjiev and D. A. Lightner, Steric size in conformational analysis. Steric compression analyzed by circular dichroism spectroscopy, J. Am. Chem. Soc., 2000, 122, 11328–11339 CrossRef CAS
- N. Kretschy and M. M. Somoza, Comparison of the sequence-dependent fluorescence of the cyanine dyes Cy3, Cy5, DyLight dy547 and DyLight dy647 on single-stranded DNA, PLoS One, 2014, 9, e85605 CrossRef PubMed
- N. Kretschy, M. Sack and M. M. Somoza, Sequence-dependent fluorescence of Cy3- and Cy5-labeled double-stranded DNA, Bioconjugate Chem., 2016, 27, 840–848 CrossRef CAS PubMed
- E. Sebastian, A. M. Philip, A. Benny and M. Hariharan, Null exciton splitting in chromophoric greek cross (+) aggregate, Angew. Chem., Int. Ed., 2018, 57, 15696–15701 CrossRef CAS PubMed
- J. R. Widom and J. E. Hoeher, Base-stacking heterogeneity in RNA resolved by fluorescence-detected circular dichroism spectroscopy, J. Phys. Chem. Lett., 2022, 13, 8010–8018 CrossRef CAS PubMed
- V. Czikklely, H. D. Forsterling and H. Kuhn, Extended dipole model for aggregates of dye molecules, Chem. Phys. Lett., 1970, 6, 207–210 CrossRef
- D. Heussman, J. Kittell, L. Kringle, A. Tamimi, P. H. von Hippel and A. H. Marcus, Measuring local conformations and conformational disorder of (Cy3)2 dimer labeled DNA fork junctions using absorbance, circular dichroism and two-dimensional fluorescence spectroscopy, Faraday Discuss., 2019, 216, 211–235 RSC
- J. S. Huff, S. A. Díaz, M. S. Barclay, A. U. Chowdhury, M. Chiriboga, G. A. Ellis, D. Mathur, L. K. Patten, S. K. Roy, A. Sup, A. Biaggne, B. S. Rolczynski, P. D. Cunningham, L. Li, J. Lee, P. H. Davis, B. Yurke, W. B. Knowlton, I. L. Medintz, D. B. Turner, J. S. Melinger and R. D. Pensack, Tunable electronic structure via DNA-templated heteroaggregates of two distinct cyanine dyes, J. Phys. Chem. C, 2022, 126, 17164–17175 CrossRef CAS PubMed
- X. Wang, R. Sha, W. B. Knowlton, N. C. Seeman, J. W. Canary and B. Yurke, Exciton delocalization in a DNA-templated organic semiconductor dimer assembly, ACS Nano, 2022, 16, 1301–1307 CrossRef CAS PubMed
- M. S. Barclay, C. K. Wilson, S. K. Roy, O. A. Mass, O. M. Obukhova, R. P. Svoiakov, A. L. Tatarets, A. U. Chowdhury, J. S. Huff, D. B. Turner, P. H. Davis, E. A. Terpetschnig, B. Yurke, W. B. Knowlton, J. Lee and R. D. Pensack, Oblique packing and tunable excitonic coupling in DNA-templated squaraine rotaxane dimer aggregates, ChemPhotoChem, 2022, 6, e202200039 CrossRef CAS
- N. Berova, L. D. Bari and G. Pescitelli, Application of electronic circular dichroism in configurational and conformational analysis of organic compounds, Chem. Soc. Rev., 2007, 36, 914–931 RSC
- L. J. Reed and J. Berkson, The application of the logistic function to experimental data, J. Phys. Chem., 1929, 33, 760–779 CrossRef CAS
- H. Prinz, Hill coefficients, dose–response curves and allosteric mechanisms, J. Chem. Biol., 2010, 3, 37–44 CrossRef PubMed
- M. F. Hamissa, P. Niederhafner, H. Šestáková, M. Šafařík, R. Hadravová and J. Šebestík, Neutral and charged forms of inubosin b in aqueous solutions at different pH and on the surface of Ag nanoparticles, J. Mol. Struct., 2022, 1250, 131828 CrossRef CAS
- E. M. Cabaleiro-Lago and J. Rodríguez-Otero, On the nature of σ–σ, σ–π, and π–π stacking in extended systems, ACS Omega, 2018, 3, 9348–9359 CrossRef CAS PubMed
- C. A. Hunter and J. K. M. Sanders, The nature of π-π interactions, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS
- K. Carter-Fenk and J. M. Herbert, Electrostatics does not dictate the slip-stacked arrangement of aromatic π–π interactions, Chem. Sci., 2020, 11, 6758–6765 RSC
- L.-J. Riwar, N. Trapp, B. Kuhn and F. Diederich, Substituent effects in parallel-displaced π–π stacking interactions: Distance matters, Angew. Chem., Int. Ed., 2017, 56, 11252–11257 CrossRef CAS PubMed
- A. L. Ringer, M. O. Sinnokrot, R. P. Lively and C. D. Sherrill, The effect of multiple substituents on sandwich and T-shaped π–π interactions, Chem. – Eur. J., 2006, 12, 3821–3828 CrossRef CAS PubMed
- P. D. Cunningham, Y. C. Kim, S. A. Díaz, S. Buckhout-White, D. Mathur, I. L. Medintz and J. S. Melinger, Optical properties of vibronically coupled Cy3 dimers on DNA scaffolds, J. Phys. Chem. B, 2018, 122, 5020–5029 CrossRef CAS PubMed
- F. Würthner, S. Yao, T. Debaerdemaeker and R. Wortmann, Dimerization of merocyanine dyes. Structural and energetic characterization of dipolar dye aggregates and implications for nonlinear optical materials, J. Am. Chem. Soc., 2002, 124, 9431–9447 CrossRef PubMed
- A. St Lorenz, E. R. Buabeng, O. Taratula, O. Taratula and M. Henary, Near-infrared heptamethine cyanine dyes for nanoparticle-based photoacoustic imaging and photothermal therapy, J. Med. Chem., 2021, 64, 8798–8805 CrossRef CAS PubMed
- S. Atchimnaidu, D. Perumal, K. S. Harikrishanan, H. V. P. Thelu and R. Varghese, Phototheranostic DNA micelles from the self-assembly of DNA-bodipy amphiphiles for the thermal ablation of cancer cells, Nanoscale, 2020, 12, 11858–11862 RSC
- J. Zhang, B. Xu, J. Chen, S. Ma, Y. Dong, L. Wang, B. Li, L. Ye and W. Tian, An organic luminescent molecule: What will happen when the “butterflies” come together?, Adv. Mater., 2014, 26, 739–745 CrossRef PubMed
- N. P. D. Sawaya, D. Rappoport, D. P. Tabor and A. Aspuru-Guzik, Excitonics: A set of gates for molecular exciton processing and signaling, ACS Nano, 2018, 12, 6410–6420 CrossRef CAS PubMed
- B. Yurke and W. Kuang, Passive linear nanoscale optical and molecular electronics device synthesis from nanoparticles, Phys. Rev. A, 2010, 81, 033814 CrossRef
- D. Abramavicius, B. Palmieri and S. Mukamel, Extracting single and two-exciton couplings in photosynthetic complexes by coherent two-dimensional electronic spectra, Chem. Phys., 2009, 357, 79–84 CrossRef CAS PubMed
- N. J. Hestand and F. C. Spano, Molecular aggregate photophysics beyond the Kasha model: Novel design principles for organic materials, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Oligonucletide table, additional steady-state absorption, circular dichroism, and fluorescence spectroscopy, and additional theoretical calculation and simulation (PDF). See DOI: https://doi.org/10.1039/d2nr05544a |
| This journal is © The Royal Society of Chemistry 2023 |