
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A DNA origami-based device for investigating DNA bending proteins by transmission electron microscopy†
Ashwin Karthick
Natarajan‡
 *,
Joonas
Ryssy§
and
Anton
Kuzyk
*
*,
Joonas
Ryssy§
and
Anton
Kuzyk
*
Department of Neuroscience and Biomedical Engineering, Aalto University, School of Science, P.O. Box 12200, FI-00076 Aalto, Finland. E-mail: ashwin.natarajan@aalto.fi; anton.kuzyk@aalto.fi
First published on 17th January 2023
Abstract
The DNA origami technique offers precise positioning of nanoscale objects with high accuracy. This has facilitated the development of DNA origami-based functional nanomechanical devices that enable the investigation of DNA–protein interactions at the single particle level. Herein, we used the DNA origami technique to fabricate a nanoscale device for studying DNA bending proteins. For a proof of concept, we used TATA-box binding protein (TBP) to evaluate our approach. Upon binding to the TATA box, TBP causes a bend to DNA of ∼90°. Our device translates this bending into an angular change that is readily observable with a conventional transmission electron microscope (TEM). Furthermore, we investigated the roles of transcription factor II A (TF(II)A) and transcription factor II B (TF(II)B). Our results indicate that TF(II)A introduces additional bending, whereas TF(II)B does not significantly alter the TBP–DNA structure. Our approach can be readily adopted to a wide range of DNA-bending proteins and will aid the development of DNA-origami-based devices tailored for the investigation of DNA–protein interactions.
Introduction
DNA bending proteins play an important role in DNA packaging, transcription, and replication.1–3 The double helical structure of DNA is often distorted by DNA-bending proteins such as transcription factors, restriction endonucleases, operons, and DNA repair proteins such as MutS.4,5 Accurate interpretation of the structural basis of interactions between DNA and DNA bending proteins is imperative for understanding various biological processes. Structural studies based on traditional ensemble approaches6,7 assume synchronous behaviour of the molecules and present an average of the outcomes. However, in reality, the molecular interactions are stochastic and averaging often leads to the loss of important details. Single-molecule techniques, such as optical tweezers, magnetic tweezers, atomic force microscopy (AFM), electron microscopy (EM), and fluorescence microscopy allow monitoring of individual molecules enabling an in-depth understanding of complex biological processes.8 Several single molecule-based approaches for the investigation of DNA bending proteins, such as TATA-box binding protein (TBP),9–11 integration host factor (IHF),12,13 transcription factor SRY-related-HMG-box 2,14 Lac Operon,15 and MutS16 were realized in the past. Most of these approaches rely on single-molecule Förster resonance energy transfer (smFRET). While smFRET is an elegant way to understand the dynamics of bending and provides high spatial and temporal resolution, the smFRET setups are rather complicated and structural information has to be obtained from the analysis of optical responses. In contrast, nanomicroscopy techniques, such as AFM and EM, in principle, enable direct measurement of structural parameters of individual molecular nanostructures. However, observation of small biological structures, e.g., individual dsDNA–protein complexes, remains challenging with conventional nanomicroscopy approaches. This problem can be circumvented by utilizing large molecular scaffolds to which molecules of interest are attached.17,18 DNA origami-based molecular scaffolds provide unique advantage of arranging the molecules of interest with nanometre precision.19–22 Furthermore, the DNA sequences that interact with the protein of interest can be easily inserted at the desired locations into the DNA origami structures. In the past, DNA origami molecular scaffolds were utilized to investigate DNA–protein interaction in AFM,23–25 transmission electron microscopy (TEM),26–28 and cryo-EM settings.29–31 Among the existing approaches, TEM provides the benefits of relatively straightforward sample preparation, operation, and fast data acquisition. Here, we use the DNA origami technique to build a molecular device that translates DNA bending induced by DNA-bending proteins into spatial reconfiguration that can be readily observed with conventional TEM. Specifically, we investigated DNA bending by TBP and associated transcription factor II A (TF(II)A) and transcription factor II B (TF(II)B). Due to its crucial role in the formation of the eukaryotic initiation complex,32–35 TBP is, perhaps, the most intensively studied DNA-bending protein and TBP–DNA interactions are well understood. The influence of transcription factors on TBP–DNA interaction has been investigated to a much lesser extent, and, in the case of TF(II)A, the published results are contradictory at times.9,36–38Experimental
Materials
The DNA origami scaffold (p7650) was purchased from Tilibit Nanosystems. The staple strands, bridge strands and proteins were purchased from Sigma-Aldrich. Uranyl formate for negative TEM staining was purchased from Telubio, Denmark. The TEM grids were purchased from Science Services, Germany. Other chemicals were purchased either from Sigma-Aldrich or VWR.Design and folding of origami
The DNA origami devices were designed in caDNAno39 and the design was adopted from the previous studies.40–43 The DNA origami structure consists of two fourteen double helix DNA bundles (14HBs) attached in the centre by a flexible scaffold linker. Staple sequences for the DNA origami template are listed in ESI Table S1.† The DNA scaffold strands (10 nM) were mixed with core staple strands in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 in a 1× TE (10 mM Tris, 1 mM EDTA, pH 8) buffer containing 20 mM MgCl2. The concentration of bridge strands was varied to obtain optimized yields of structures with desired angle between the bundles. The scaffold, staples, and bridge strands were thermally annealed from 80 °C to 20 °C over 30 hours. The origami structures were purified from staples9 using the spin filtering technique.44
:10 in a 1× TE (10 mM Tris, 1 mM EDTA, pH 8) buffer containing 20 mM MgCl2. The concentration of bridge strands was varied to obtain optimized yields of structures with desired angle between the bundles. The scaffold, staples, and bridge strands were thermally annealed from 80 °C to 20 °C over 30 hours. The origami structures were purified from staples9 using the spin filtering technique.44
Design of the bridge
The angle between the two 14HBs was tuned by using a double-stranded (ds) DNA bridge that connects the two bundles and defines the initial configuration. The dsDNA bridge connects the two bundles of the origami either through (i) the hybridization of two individual staple strand extensions, one from each 14HB (design 1, ESI Fig. S1 and S3†) or via (ii) a single staple strand that is hybridized with the scaffold in both 14HBs (design 2, ESI Fig. S2 and S3†). In design 2, an additional DNA strand was added to form the DNA duplex. Three bridge sequences were investigated: (a) an adenovirus major late promoter (AdMLP) consensus sequence (CTATAAAAG) (S1),45 (b) a consensus sequence, where the first and last bases in S1 are switched to its complement (GTATAAAAC) (S2), and (c) a scramble sequence (ACTTCTCGG) to which TBP should not bind (S3). The bridge-forming strands were incorporated into the DNA origami structures during folding.Bending of the TATA box by TBP
DNA origami devices at 1 nM concentration containing the respective bridge sequences were incubated with various concentrations of TBP, TF(II)A, and TF(II)B in the modified HEPES buffer containing 1× TE, 10 mM NaCl, 10 mM MgCl2, 150 mM KCl, 10 mM HEPES, and 12% glycerol.9 The mixture was incubated at 25 °C for 2 hours followed by imaging using TEM.Transmission electron microscopy and angle measurement
The samples containing DNA origami devices incubated with the respective proteins were used for TEM sample preparation without further purification. The TEM grids were prepared as follows: 5 μl of the sample was deposited on a freshly glow discharged carbon/formvar TEM grid and the liquid was blotted away after 5 minutes. The grids were negatively stained with 1% uranyl formate solution. The grids were imaged with either Tecnai F12 operating at 120 kV or with Tecnai F20 operating at 200 kV. The acute angles between the bundles of the origami devices were obtained by manual analysis of the TEM images in Corel Draw (2018). Considerations taken while measuring the angle manually are provided in ESI Note 1.† The histograms of relative frequencies were plotted in Origin 2020b.Results and discussion
Design of DNA origami-based devices
To study DNA bending, we used the DNA origami technique to fabricate molecular devices comprising two 14HBs connected in the middle by a flexible join point (Fig. 1A). To define the initial spatial configuration of the device with a specific angle, the two 14HBs were connected by a dsDNA bridge (Fig. 1A). We tested two dsDNA bridge designs: design 1 included two complementary extensions from the two bundles of the device to form the dsDNA bridge; design 2 included a single-stranded DNA connecting the two bundles and a complementary strand to form a dsDNA bridge (ESI Fig. S3†). In both designs, the initial angle between the bundles was fixed at 80° (Fig. 1B). We varied the concentration of the bridge strands from 2-fold to 10-fold molar excess to the origami scaffold concentration and evaluated the yield of correctly formed devices with the intended initial angle between the two bundles by analyzing individual structures in the TEM micrographs (ESI Fig. S4–7†). The considerations taken while measuring the angle between the two origami bundles are detailed in ESI Note 1.† Notably, only the acute angle between the origami bundles was considered during angle measurement. Further, the structures landing sidewise on the TEM grid, structures on the edge of the image, and broken origami structures were not considered for angle measurement (ESI Note 1 and ESI Fig. S8†). The histograms for the relative frequency of the angle between the origami bundles were calculated based on the measurements from at least 200 origami structures for each sample.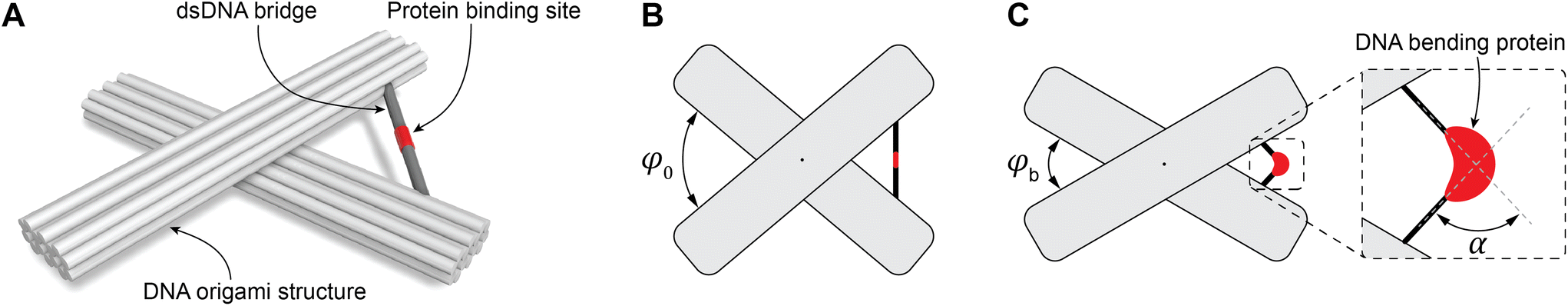 | ||
| Fig. 1 Schematics of the DNA origami-based device. (A) Representation of the DNA origami structure with the TATA box (colored in red) incorporated into the bridge. (B) The origami structures are designed to form an angle ∼80° (φ0 = 80°) between the origami bundles. (C) The angle between the origami bundles (φb) changes due to the distortion of the TATA box by TBP resulting in bend angle α in the bridge. | ||
The relative frequencies plots (ESI Fig. S9†) show that 4-fold molar excess of bridge strands (40 nM) to scaffold concentration (10 nM) had the highest yield of devices with 80° between the bundles (∼40%) for design 1. For design 2, the highest yield (∼41%) was obtained with 8-fold molar excess of bridge strands (ESI Fig. S10†). Although the yields of origami with desired initial angle were similar for designs 1 and 2, there was a difference in the concentration of bridge strands required to obtain the maximum yield in the two tested designs. These results show that the design of such bridges requires careful optimization and fine-tuning of parameters to achieve the optimal yield. For further experiments, we chose DNA origami devices with design 1 bridge. We would like to note that the yield of devices with the desired angle between the bundles might be improved by engineering the TATA-box sequence into the scaffold strand and routing it to form the bridge.46,47
TATA box bending by TBP
To evaluate the general applicability of our approach, we first used our DNA origami-based devices to investigate the DNA bending by well-characterized TBP protein.45,48–50 For this, the consensus sequence CTATAAAAG (S1) from adenovirus major late promoter (AdMLP) was inserted in the middle of the dsDNA bridge. Upon TBP binding, the bridge is expected to bend ∼90°,45,48 thereby causing an angle change in the DNA origami devices (Fig. 1C). The change of the angle can be directly detected using TEM. Fig. 2 shows the TEM images of DNA devices with acute angles marked and their corresponding relative frequency histograms of devices without a bridge (structures with undefined angle, Fig. 2A and B), devices with a bridge and intended initial 80° between the bundles (Fig. 2C and D), and devices with a bridge and intended initial 80° between the bundles incubated with 100 nM TBP (Fig. 2E and F). For brevity, we call the structures without bridging staples, i.e., structures with undefined angles, open structures, as opposed to the structures designed to be closed at a particular angle by the bridging strands. For all the experiments, the concentration of origami devices was kept at 1 nM unless stated otherwise. The dissociation constant of human TBP to TATA box is 2.7 nM,51 which implies that at 100 nM concentration TBP should be bound to most of the devices with the bridge. As expected,43,52 open devices had a broad distribution of relative frequency over angles (Fig. 2B). In the absence of TBP, the devices with a dsDNA bridge (S1 sequence) had a yield of ∼40% around the desired 80° angle (Fig. 2D). In the presence of TBP, the maximum frequency in the histogram shifted from around 80° to around 65° in the histogram (Fig. 2F) implying dsDNA bridge bending by TBP. The dsDNA bridge bending angle α (Fig. 1C) can be obtained from the angle φ between the device bundles using simple trigonometrical relations (ESI Note 2 and Fig. S11†). As shown in ESI,† the observed maximum of angle distribution at φ ≈ 65° corresponds to α ≈ 83°, which is in good agreement with the consensus DNA bending angle by TBP.45,48 We also investigated the influence of TBP concentration on the angle distribution (ESI Fig. S12†). As the concentration increased from 25 nM to 100 nM, the occurrence of structures with ∼65° angle increased and corroborated with the decrease in relative frequency around 80°. There was no further change in angle distribution when the TBP concentration was increased to 150 nM (ESI Fig. S12†) implying that almost all the origami devices were bound by TBP.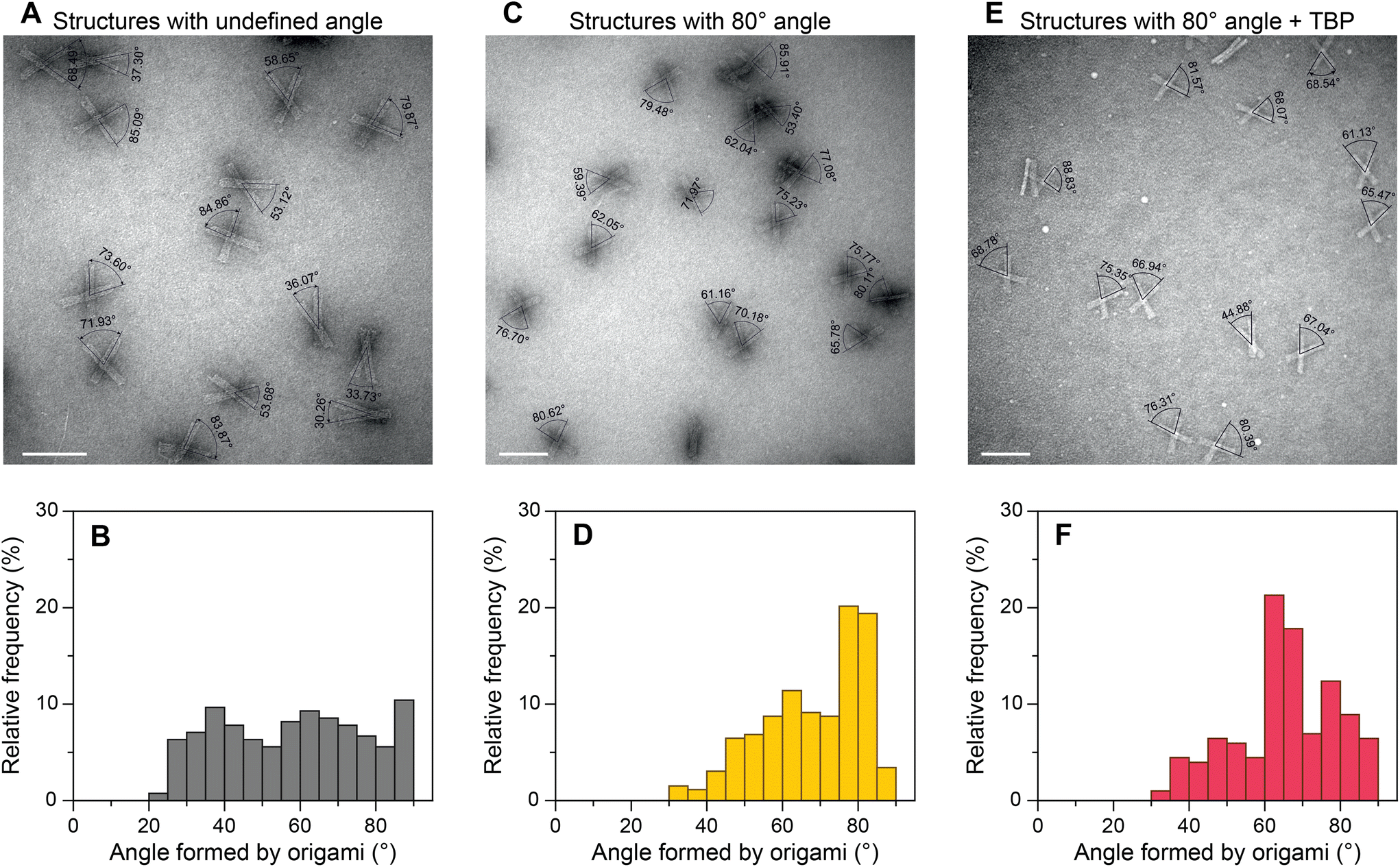 | ||
| Fig. 2 TEM angle measurement and relative frequency histograms of origami structures with undefined angle (open structures) (A and B), origami structures forming 80° angle between the bundles (C and D), and origami structures forming 80° angle between the bundles incubated with 100 nM TBP (E and F). Scale bars: 100 nm. | ||
Transcription factors (TFIIA and TFIIB) and TBP–DNA bending
As mentioned above, the influence of various transcription factors on DNA bending by TBP has been explored to a lesser extent than DNA bending by TBP alone. Although the crystal structures of TFIIA–TBP–DNA and TFIIB–TBP–DNA complexes have been available since 1990s,53,54 the biophysical studies produced contradictory results at times.9,36–38 Binding of transcription factors to the TBP–DNA complex can influence the stability of the complex and/or alter its structure. TFIIA binds to the upstream region of the TATA box and the underside of the TBP protein through its two conserved domains, a 4-helix bundle, and a 12-stranded β-barrel.34,35,55 It has been reported that TFIIA binding results in the stabilization of the TBP–DNA complex38 without altering the TBP–DNA structure.54 And that the stability TFIIA–TBP–DNA complex is dependent on the DNA sequence.56 Another report, however, suggested that TFIIA most likely changes the conformation of DNA in TBP–DNA complexes along with increasing the kinetic stability.36 To investigate whether TFIIA causes increased stability of the TBP–DNA complex, we incubated our devices with 50 nM TBP and various concentrations of TFIIA (Fig. 3). As the concentration of TFIIA in the system increased from 0 nM to 100 nM the angle distribution maximum shifted from ∼65° to ∼55°. At 25 nM TFIIA concentration, the angle distribution was broad, as not all TATA-box sequences occupied by TBP were supplemented with TFIIA. At 100 nM TFIIA concentration, the distribution was similar to 50 nM TFIIA, which indicates the saturation of TFIIA–TBP–DNA complexes. To evaluate further, we investigated equimolar concentrations of TBP and TFIIA from 25 nM to 100 nM (ESI Fig. S13†) and observed the increase in the relative frequency of angles around 55° as the concentration increased. In contrast to some previous studies,9,38 our results cannot be explained only by TFIIA mediated increase of TBP–DNA complex stability, as modulation of stability alone would influence the amplitude of angle distribution maximum but not its position. Our results strongly indicate that TFIIA alters the structure and one possible mechanism might be TFIIA binding induced DNA twisting in addition to bending by TBP.36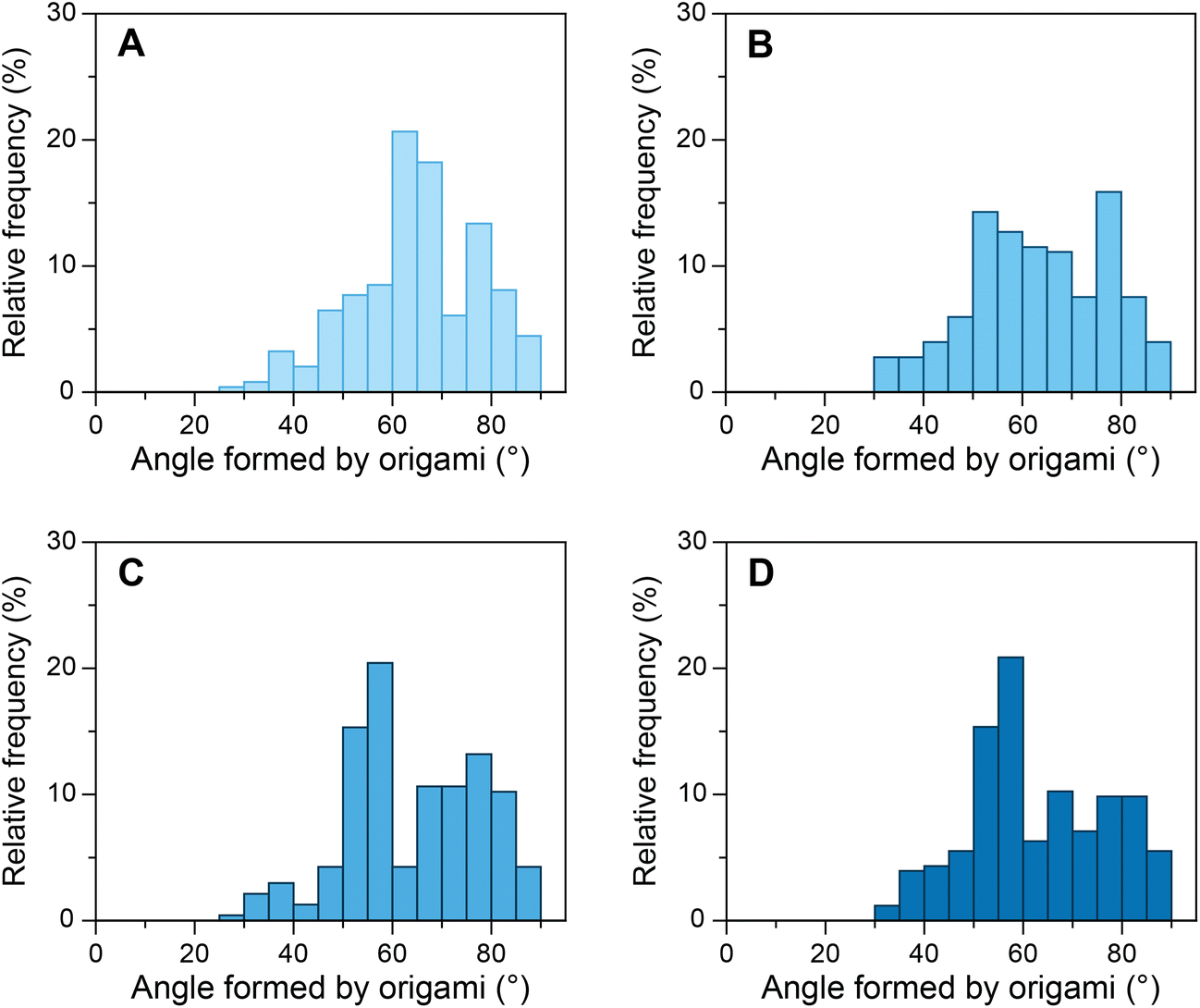 | ||
| Fig. 3 Relative frequency histograms of origami structures with S1 sequence forming 80° angle incubated with constant 50 nM TBP and increasing concentrations of TFIIA. (A) 0 nM TFIIA; (B) 25 nM TFIIA; (C) 50 nM TFIIA; and (D) 100 nM TFIIA. The maximum of the relative frequency distribution of angles shifts from around 65° to 55° with an increase in TFIIA concentration. | ||
Furthermore, we evaluated the role of TFIIB with our device. It has been hypothesized that TFIIB either induces the intermediate unbent TBP–DNA complex to reach the final bent state or decreases the bend angle upon association with the TBP–DNA complex.37,57–59 We incubated our device containing consensus S1 sequence with equimolar concentrations of TBP and TFIIB and equimolar concentrations of TBP, TFIIA, and TFIIB. At 25 nM TBP and TFIIB concentration, the observed distribution was broad, similar to what we observed with TFIIA. We then increased the concentration up to 100 nM of TBP and TFIIB and the distribution had a peak at ∼65° as observed in the presence of TBP alone (ESI Fig. S14A–C†). The results with equimolar concentrations of TBP, TFIIA, and TFIIB were comparable to the samples incubated with TBP and TFIIA (ESI Fig. S14D–F†). The configurations of the TBP–DNA or TFIIA–TBP–DNA complex were not altered further by TFIIB. These results imply that TFIIB does not cause significant structural changes to TBP–DNA or TFIIA–TBP–DNA complexes.
Bending of TATA box with S2 sequence
We then investigated a mutated sequence (S2), where the consensus sequence from AdMLP was taken and the bases before and after the TATA box were mutated to their complements (GTATAAAAC). S2 allowed us to study whether these bases affect the bending of the TATA box by TBP. The yield of origami device with the intended 80° angle between the bundles formed by design 1 bridging staples containing S2 sequence was ∼39% (Fig. 4A). We performed experiments with the same ratios of TBP and TFIIA with origami devices and imaged the samples in TEM (ESI Fig. S15†). Fig. 4B–D presents the histograms of origami incubated with various concentrations of TBP. The results show that as the concentration of TBP increased the structures shifted from their designed 80° configuration to ∼65° configuration. The relative frequency shift was very similar to that of the consensus sequence, and the decrease in relative frequency around 80° corroborated with the increase in frequency around 65°. In the presence of equimolar TFIIA, the histogram shifted further to ∼55° configuration with the same trend as seen with the consensus sequence (ESI Fig. S16†). These results indicate that the base pairs before and after the TATA box do not influence the structural changes in the TATA box caused by TBP and that TBP binds and predominantly bends the TATA-box sequence.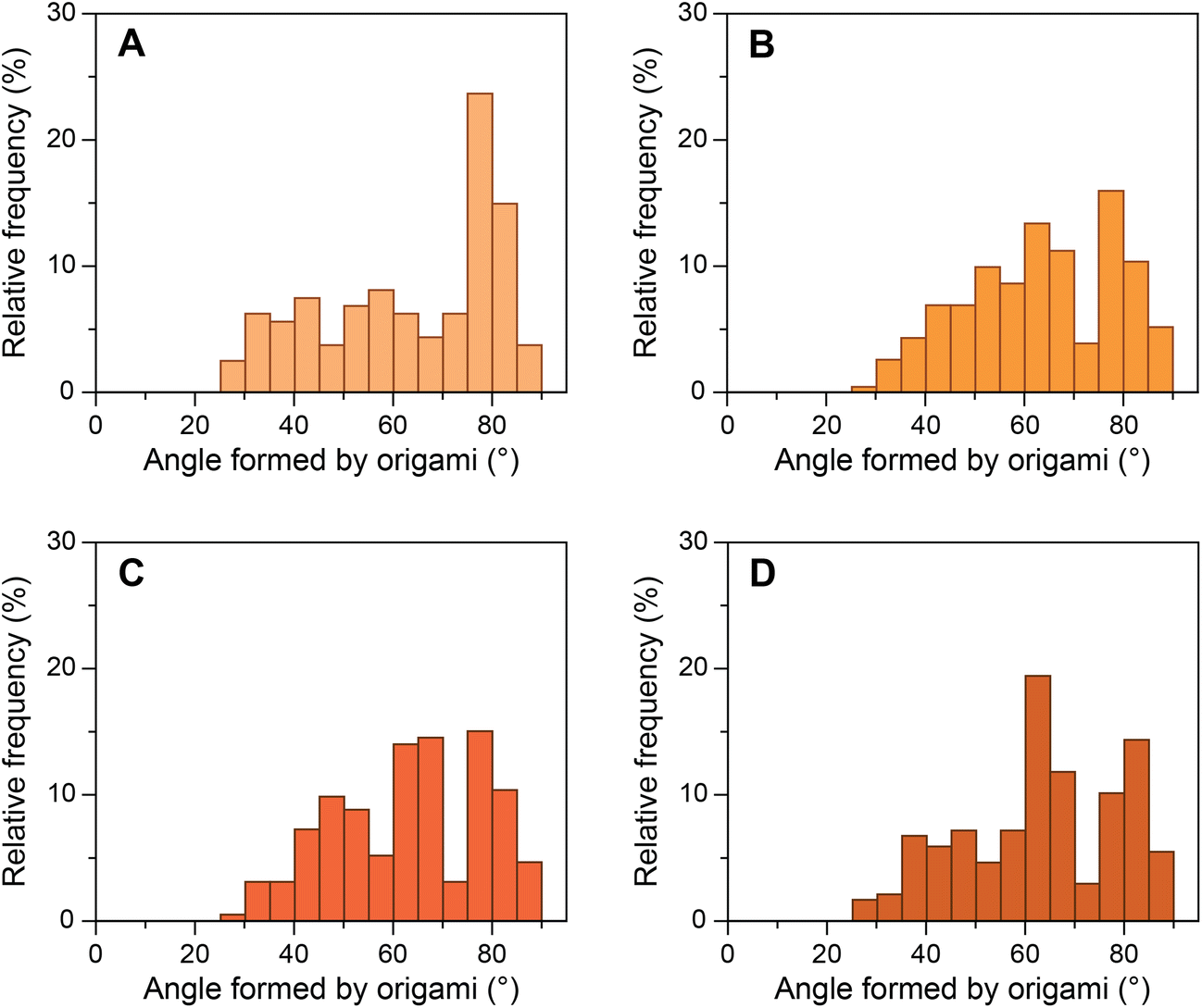 | ||
| Fig. 4 Relative frequency histograms of origami structures with S2 sequence. (A) Control S2 structures forming 80° angle; S2 structures incubated with (B) 25 nM TBP; (C) 50 nM TBP; and (D) 100 nM TBP. | ||
Control experiments
Two control experiments were performed to evaluate the validity of our approach. First, we incubated the origami devices forming an 80° angle with S1 sequence with either 50 nM TFIIA or 50 nM TFIIB without TBP (ESI Fig. S17†). Second, we replaced the TATA-box sequence in the bridge with a random sequence (S3) to which TBP should not bind, and we incubated the origamis devices containing the S3 sequence with 50 nM TBP. We further evaluated the origami containing scramble S3 sequence incubated with 50 nM TBP and 50 nM of either TFIIA or TFIIB. ESI Fig. S18† shows the representative TEM micrographs and corresponding angle measurements and ESI Fig. S19† shows the relative frequency histogram of origami with S3 sequence incubated with TBP, TBP with TFIIA, and TBP with TFIIB. As expected in all the test cases, the angle distribution did not change suggesting that the origami devices are bent by TBP binding to TBP-specific TATA-box sequence. Furthermore, TFIIA and TFIIB do not cause bending by themselves as seen from the control experiments (ESI Fig. S17†). The summary of all the designs and experimental conditions and the corresponding angle distribution frequency peaks is provided in ESI Table S2.†Conclusions
The results of this study highlight utility of spatially reconfigurable DNA origami structures for the investigation of DNA–protein interaction. Our approach facilitates direct observation of the changes caused by DNA bending proteins using conventional and generally accessible TEM. The design of origami-based devices can be expanded to study a range of DNA-bending proteins such as IHF (E. coli), HU (B. stearothermophilus), TF1 (B. subtilis) etc. The results of such studies can be used to guide the selection of experimental conditions for more advanced and accurate techniques, e.g., cryo-EM. The design of our devices can be easily modified and tailored to specific needs and the data analysis can be, in principle, semi-automated.60Author contributions
AK conceived and supervised the research. AKN designed and performed the experiments. AKN and JR imaged the samples using TEM. AKN measured the angles, constructed the histograms, and analysed the data. AK and AKN wrote the manuscript and the supplementary section. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the provision of facilities and technical support by Aalto University at OtaNano Nanomicroscopy Center (Aalto-NMC). The authors thank Joshua Johnson for valuable discussions and Robin Silva for their contributions to the preliminary experiments. The authors would also like to thank Arttu Lehtonen and Minh-Kha Nguyen for their helpful suggestions. The authors acknowledge the financial support from the Academy of Finland (grant 308992). J. R. acknowledges the financial support from the Academy of Finland Flagship Programme, Photonics Research, and Innovation (PREIN) (decision number: 320167).References
- P. C. van der Vliet and C. P. Verrijzer, BioEssays, 1993, 15, 25–32 CrossRef CAS PubMed.
- S. Harteis and S. Schneider, Int. J. Mol. Sci., 2014, 15, 12335–12363 CrossRef PubMed.
- G. Vámosi and D. Rueda, Biophys. J., 2018, 114, 2253–2254 CrossRef PubMed.
- J. Pérez-Martín, F. Rojo and V. de Lorenzo, Microbiol. Rev., 1994, 58, 268–290 CrossRef PubMed.
- S. Jones, P. van Heyningen, H. M. Berman and J. M. Thornton, J. Mol. Biol., 1999, 287, 877–896 CrossRef CAS PubMed.
- C. Liu, E. Kim, B. Demple and N. C. Seeman, Biochemistry, 2012, 51, 937–943 CrossRef CAS PubMed.
- W. Shen, M. F. Bruist, S. D. Goodman and N. C. Seeman, Angew. Chem., Int. Ed., 2004, 43, 4750–4752 CrossRef CAS PubMed.
- W.-Q. Wu, X. Zhu and C.-P. Song, New Phytol., 2019, 223, 508–510 CrossRef PubMed.
- R. H. Blair, J. A. Goodrich and J. F. Kugel, Biochemistry, 2012, 51, 7444–7455 CrossRef CAS PubMed.
- P. C. Nickels, B. Wünsch, P. Holzmeister, W. Bae, L. M. Kneer, D. Grohmann, P. Tinnefeld and T. Liedl, Science, 2016, 354, 305–307 CrossRef CAS PubMed.
- K. Kramm, T. Schröder, J. Gouge, A. M. Vera, K. Gupta, F. B. Heiss, T. Liedl, C. Engel, I. Berger, A. Vannini, P. Tinnefeld and D. Grohmann, Nat. Commun., 2020, 11, 2828 CrossRef CAS PubMed.
- A. Brunet, S. Chevalier, N. Destainville, M. Manghi, P. Rousseau, M. Salhi, L. Salomé and C. Tardin, Nucleic Acids Res., 2015, 43, e72–e72 CrossRef PubMed.
- S. Le, H. Chen, P. Cong, J. Lin, P. Dröge and J. Yan, Sci. Rep., 2013, 3, 3508 CrossRef PubMed.
- H. Morimura, S.-I. Tanaka, H. Ishitobi, T. Mikami, Y. Kamachi, H. Kondoh and Y. Inouye, ACS Nano, 2013, 7, 10733–10740 CrossRef CAS PubMed.
- O. K. Wong, M. Guthold, D. A. Erie and J. Gelles, PLoS Biol., 2008, 6, e232 CrossRef PubMed.
- I. Tessmer, Y. Yang, J. Zhai, C. Du, P. Hsieh, M. M. Hingorani and D. A. Erie, J. Biol. Chem., 2008, 283, 36646–36654 CrossRef CAS PubMed.
- Q. Yao, S. J. Weaver, J.-Y. Mock and G. J. Jensen, Structure, 2019, 27, 1148–1155 CrossRef CAS PubMed.
- Y. Liu, S. Gonen, T. Gonen and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3362–3367 CrossRef CAS PubMed.
- T. Tørring, N. V. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636–5646 RSC.
- W. Engelen and H. Dietz, Annu. Rev. Biophys., 2021, 50, 469–492 CrossRef CAS PubMed.
- N. Stephanopoulos and P. Šulc, Appl. Sci., 2021, 11, 2802 CrossRef CAS.
- S. Dey, C. Fan, K. V. Gothelf, J. Li, C. Lin, L. Liu, N. Liu, M. A. D. Nijenhuis, B. Saccà, F. C. Simmel, H. Yan and P. Zhan, Nat. Rev. Methods Primers, 2021, 1, 1–24 CrossRef.
- M. Endo and H. Sugiyama, Acc. Chem. Res., 2014, 47, 1645–1653 CrossRef CAS PubMed.
- A. Rajendran, M. Endo and H. Sugiyama, Chem. Rev., 2014, 114, 1493–1520 CrossRef CAS PubMed.
- S. Yamamoto, D. De, K. Hidaka, K. K. Kim, M. Endo and H. Sugiyama, Nano Lett., 2014, 14, 2286–2292 CrossRef CAS PubMed.
- J. V. Le, Y. Luo, M. A. Darcy, C. R. Lucas, M. F. Goodwin, M. G. Poirier and C. E. Castro, ACS Nano, 2016, 10, 7073–7084 CrossRef CAS PubMed.
- J. J. Funke, P. Ketterer, C. Lieleg, S. Schunter, P. Korber and H. Dietz, Sci. Adv., 2016, 2, e1600974 CrossRef PubMed.
- J. J. Funke, P. Ketterer, C. Lieleg, P. Korber and H. Dietz, Nano Lett., 2016, 16, 7891–7898 CrossRef CAS PubMed.
- T. G. Martin, T. A. M. Bharat, A. C. Joerger, X.-C. Bai, F. Praetorius, A. R. Fersht, H. Dietz and S. H. W. Scheres, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7456–E7463 CAS.
- T. Aksel, Z. Yu, Y. Cheng and S. M. Douglas, Nat. Biotechnol., 2021, 39, 378–386 CrossRef CAS PubMed.
- N. Aissaoui, J. Lai-Kee-Him, A. Mills, N. Declerck, Z. Morichaud, K. Brodolin, S. Baconnais, E. Le Cam, J. B. Charbonnier, R. Sounier, S. Granier, V. Ropars, P. Bron and G. Bellot, ACS Nano, 2021, 15, 4186–4196 CrossRef CAS PubMed.
- N. Hernandez, Genes Dev., 1993, 7, 1291–1308 CrossRef CAS PubMed.
- R. J. White and S. P. Jackson, Trends Genet., 1992, 8, 284–288 CrossRef CAS PubMed.
- S. Sainsbury, C. Bernecky and P. Cramer, Nat. Rev. Mol. Cell Biol., 2015, 16, 129–143 CrossRef CAS PubMed.
- F. T. Tsai and P. B. Sigler, EMBO J., 2000, 19, 25–36 CrossRef CAS PubMed.
- A. R. Hieb, W. A. Halsey, M. D. Betterton, T. T. Perkins, J. F. Kugel and J. A. Goodrich, J. Mol. Biol., 2007, 372, 619–632 CrossRef CAS PubMed.
- S. F. Tolić-Nørrelykke, M. B. Rasmussen, F. S. Pavone, K. Berg-Sørensen and L. B. Oddershede, Biophys. J., 2006, 90, 3694–3703 CrossRef PubMed.
- A. N. Imbalzano, K. S. Zaret and R. E. Kingston, J. Biol. Chem., 1994, 269, 8280–8286 CrossRef CAS PubMed.
- S. M. Douglas, A. H. Marblestone, S. Teerapittayanon, A. Vazquez, G. M. Church and W. M. Shih, Nucleic Acids Res., 2009, 37, 5001–5006 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, H. Zhang, A. O. Govorov, T. Liedl and N. Liu, Nat. Mater., 2014, 13, 862–866 CrossRef CAS PubMed.
- Y. Huang, M.-K. Nguyen, A. K. Natarajan, V. H. Nguyen and A. Kuzyk, ACS Appl. Mater. Interfaces, 2018, 10, 44221–44225 CrossRef CAS PubMed.
- Y. Huang, M.-K. Nguyen and A. Kuzyk, J. Visualized Exp., 2019, e59280 CAS.
- J. Ryssy, A. K. Natarajan, J. Wang, A. J. Lehtonen, M.-K. Nguyen, R. Klajn and A. Kuzyk, Angew. Chem., Int. Ed., 2021, 60, 5859–5863 CrossRef CAS PubMed.
- V. Linko, B. Shen, K. Tapio, J. J. Toppari, M. A. Kostiainen and S. Tuukkanen, Sci. Rep., 2015, 5, 15634 CrossRef CAS PubMed.
- D. B. Nikolov, H. Chen, E. D. Halay, A. Hoffman, R. G. Roeder and S. K. Burley, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4862–4867 CrossRef CAS PubMed.
- P. M. Nafisi, T. Aksel and S. M. Douglas, Synth. Biol., 2018, 3, ysy015 CrossRef CAS PubMed.
- F. A. S. Engelhardt, F. Praetorius, C. H. Wachauf, G. Brüggenthies, F. Kohler, B. Kick, K. L. Kadletz, P. N. Pham, K. L. Behler, T. Gerling and H. Dietz, ACS Nano, 2019, 13, 5015–5027 CrossRef CAS PubMed.
- J. L. Kim, D. B. Nikolov and S. K. Burley, Nature, 1993, 365, 520–527 CrossRef CAS PubMed.
- Y. Kim, J. H. Geiger, S. Hahn and P. B. Sigler, Nature, 1993, 365, 512–520 CrossRef CAS PubMed.
- P. F. Kosa, G. Ghosh, B. S. DeDecker and P. B. Sigler, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6042–6047 CrossRef CAS PubMed.
- O. V. Arkova, N. A. Kuznetsov, O. S. Fedorova, N. A. Kolchanov and L. K. Savinkova, Acta Nat., 2014, 6, 36–40 CrossRef CAS.
- A. Kuzyk, Y. Yang, X. Duan, S. Stoll, A. O. Govorov, H. Sugiyama, M. Endo and N. Liu, Nat. Commun., 2016, 7, 10591 CrossRef CAS PubMed.
- D. B. Nikolov, H. Chen, E. D. Halay, A. A. Usheva, K. Hisatake, D. K. Lee, R. G. Roeder and S. K. Burley, Nature, 1995, 377, 119–128 CrossRef CAS PubMed.
- J. H. Geiger, S. Hahn, S. Lee and P. B. Sigler, Science, 1996, 272, 830–836 CrossRef CAS PubMed.
- M. Bleichenbacher, S. Tan and T. J. Richmond, J. Mol. Biol., 2003, 332, 783–793 CrossRef CAS PubMed.
- J. J. Stewart and L. A. Stargell, J. Biol. Chem., 2001, 276, 30078–30084 CrossRef CAS PubMed.
- X. Zhao and W. Herr, Cell, 2002, 108, 615–627 CrossRef CAS PubMed.
- A. Gietl, P. Holzmeister, F. Blombach, S. Schulz, L. V. von Voithenberg, D. C. Lamb, F. Werner, P. Tinnefeld and D. Grohmann, Nucleic Acids Res., 2014, 42, 6219–6231 CrossRef CAS PubMed.
- R. M. Buratowski, J. Downs and S. Buratowski, Mol. Cell Biol., 2002, 22, 8735–8743 CrossRef CAS PubMed.
- J. A. Johnson, A. Dehankar, J. O. Winter and C. E. Castro, Nano Lett., 2019, 19, 8469–8475 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05366g |
| ‡ Present address: Max-Delbrück-Center for Molecular Medicine in the Helmholtz Association, Structural Biology, Robert-Rössle-Straße 10, Berlin, Germany. |
| § Present address: Department of Chemistry, KTH Royal Institute of Technology, Teknikringen 30, 10044 Stockholm, Sweden. |
| This journal is © The Royal Society of Chemistry 2023 |