
An antioxidant and antibacterial polydopamine-modified thermo-sensitive hydrogel dressing for Staphylococcus aureus-infected wound healing†
Pengjin
Ge
a,
Shuhua
Chang
a,
Ting
Wang
b,
Quan
Zhao
a,
Gang
Wang
*a and
Bin
He
 *a
*a
aNational Engineering Research Center for Biomaterials, College of Biomedical Engineering, Sichuan University, Chengdu 610065, China. E-mail: wgang@scu.edu.cn; bhe@scu.edu.cn
bDepartment of Ophthalmology, West China Hospital, Sichuan University, No. 37 Guoxue Alley, Chengdu 610041, China
First published on 6th December 2022
Abstract
Bacteria-infected wound healing is a complex and chronic process that poses a great threat to human health. A thermo-sensitive hydrogel that undergoes a sol–gel transition at body temperature is an attractive wound dressing for healing acceleration and infection prevention. In this paper, we present a thermo-sensitive and reactive oxygen species (ROS)-scavenging hydrogel based on polydopamine modified poly(ε-caprolactone-co-glycolide)-b-poly(ethylene glycol)-b-poly(ε-caprolactone-co-glycolide) (PDA/P2) triblock copolymer. The PDA/P2 solution at a concentration of 30 wt% could form a gel at 34–38 °C. The ROS-scavenging ability of PDA/P2 was demonstrated by DPPH and ABTS assays and intracellular ROS downregulation in RAW264.7 cells. Furthermore, silver nanoparticles were encapsulated in the hydrogel (PDA/P2–4@Ag gel) to provide antibacterial activity against E. coli and S. aureus. An in vivo S. aureus-infected rat model demonstrated that the PDA/P2–4@Ag hydrogel dressing could promote wound healing via inhibiting bacterial growth, alleviating the inflammatory response, and inducing angiogenesis and collagen deposition. This study provides a new strategy to prepare temperature-sensitive hydrogel-based multifunctional wound dressings.
1. Introduction
Skin is the largest organ of the human body.1 It offers the first barrier to protect the human body from injuries and microbial invasions.2 Once the skin barrier is disrupted by injuries, a wound healing process is triggered instantly.3 Traditional wound dressings such as cotton bandages and gauzes fail to efficiently promote wound closure,4 and often cause secondary damage during the process of replacing dressings.5 Hydrogels possess a highly three-dimensional network structure and bind more water6 to maintain the moist environment of the wound.7–9 In addition, hydrogels exhibit significant advantages in moisture permeability,10 biocompatibility,11 drug loading capacity,12etc. Thermosensitive hydrogels can remain in the liquid state at room or a low temperature and convert to semi-solid gels at body temperature. They can readily adapt to wounds with irregular margins without damaging wound surfaces.13 Polyethylene glycol (PEG)–polyester14 and PEG–polypeptide15 are typical biodegradable polymers with sol–gel transition ability near body temperature. Specifically, polyester–PEG–polyester triblock copolymers represent a type of excellent thermo-sensitive material due to their convenient one-pot synthesis, and controlled thermo-sensitiveness and biodegradation behaviors.16,17 The gelation mechanism of amphiphilic triblock copolymers is that the copolymers can self-assemble into nanoscale micelles at room temperature and then form hydrogels at an elevated temperature.In addition to their physical isolation and moisture maintenance functions, multifunctional hydrogel dressings that have antibacterial,18,19 adhesive and hemostatic properties to promote wound healing,20,21 and antioxidant/anti-inflammatory ability22 are urgently needed to address the concerns of pathogenic infection and inflammation at wounds. Physically mixing biologically active macromolecules,23 small molecules24 and nanoparticles25 with copolymer solutions is a facile method to endow thermo-sensitive hydrogel dressings with antioxidant and anti-inflammatory functions. Polydopamine (PDA), a polymer formed by the oxidative polymerization of dopamine, has been widely used in the surface modification of materials,26 and PDA-based hydrogels have the properties of preventing bacteria from attachment27 and good tissue adhesiveness,28,29 both of which make them attractive in wound healing. Despite PDA-based hydrogels having been extensively studied in wound treatment, PDA-modified thermo-sensitive hydrogels based on polyester–PEG–polyester triblock copolymers have rarely been reported.
Herein, we synthesized three triblock copolymers, poly(ε-caprolactone-co-glycolide)-b-poly(ethylene glycol)-b-poly(ε-caprolactone-co-glycolide) (P1–P3, Table S1†) by varying the feeding molar ratios of ε-caprolactone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolide:PEG. The physical–chemical properties and temperature sensitivity of the copolymers were investigated in detail and the P2 polymer showed thermo-sensitive gelation behavior. PDA was in situ synthesized on the surface of P2 micelles and the impact of the PDA modification degree on the phase transition temperature of P2 was also studied. PDA and silver nanoparticles30 were introduced to form a hydrogel (PDA/P2–4@Ag) with antioxidant and antibacterial functions (Fig. 1). PDA/P2–4@Ag exhibited good antioxidant capacity, low cytotoxicity and excellent antibacterial performance. The healing effect and biocompatibility of the hydrogels were further evaluated in a Staphylococcus aureus-infected rat model. The inflammation reduction, collagen deposition and angiogenesis promotion at wound tissues were evaluated.
:glycolide:PEG. The physical–chemical properties and temperature sensitivity of the copolymers were investigated in detail and the P2 polymer showed thermo-sensitive gelation behavior. PDA was in situ synthesized on the surface of P2 micelles and the impact of the PDA modification degree on the phase transition temperature of P2 was also studied. PDA and silver nanoparticles30 were introduced to form a hydrogel (PDA/P2–4@Ag) with antioxidant and antibacterial functions (Fig. 1). PDA/P2–4@Ag exhibited good antioxidant capacity, low cytotoxicity and excellent antibacterial performance. The healing effect and biocompatibility of the hydrogels were further evaluated in a Staphylococcus aureus-infected rat model. The inflammation reduction, collagen deposition and angiogenesis promotion at wound tissues were evaluated.
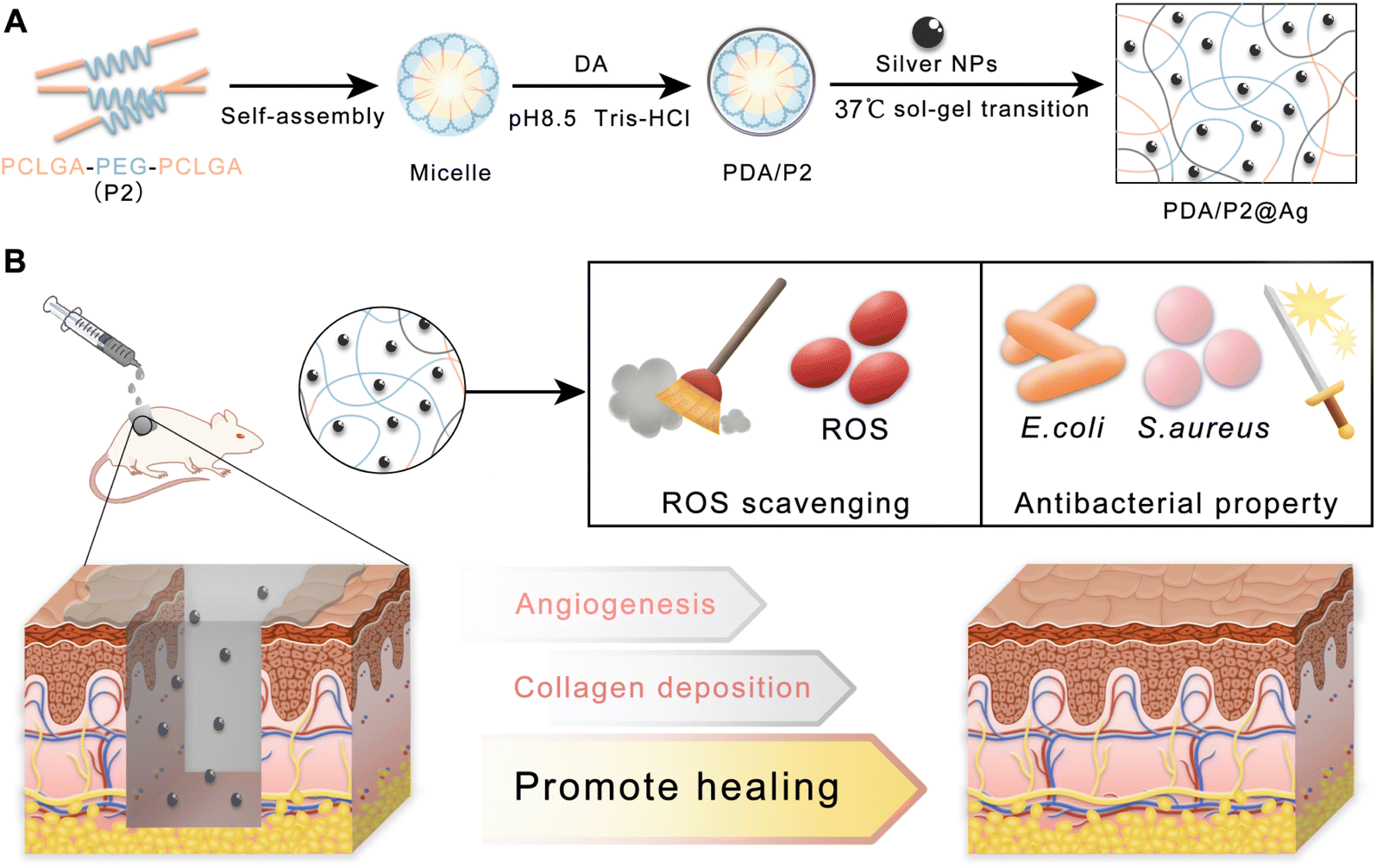 | ||
| Fig. 1 (A) Schematic and mechanism of preparing the PDA/P2@Ag hydrogel. (B) Application of PDA/P2@Ag dressing for infected wound healing. | ||
2. Materials and methods
2.1 Materials and reagents
ε-Caprolactone and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) were purchased from Tianjin Xiens Biochemical Technology Co. Ltd. PEG (Mw 1500) and stannous octoate were purchased from Sigma-Aldrich (USA). Glycolide (GA) was purchased from Purac Biochem Co. (Gorinchem, Netherlands). The ROS kit was purchased from Beyotime Biotechnology Inc. Diphenyl-2-pyridyl hydrazide (DPPH) was purchased from Macklin Biochemical Co., Ltd (Shanghai, China). Dopamine (DA) hydrochloride was obtained from Aladdin Co., Ltd (Shanghai, China). Silver nitrate (AgNO3) and polyvinylpyrrolidone K30 (PVP, K30) were supplied by Chengdu Kelong Chemicals Co., Ltd.2.2 Synthesis of PCLGA–PEG–PCLGA triblock polymers
PCLGA–PEG–PCLGA triblock copolymers were synthesized via a typical ring-opening polymerization of CL and GA using PEG (Mw 1500) as a macroinitiator and stannous octoate as a catalyst.31 Briefly, PEG was added to a polymerization tube and heated at 120 °C under vacuum for 3 h to remove the residual water in the polymer. Certain proportions of GA, CL, and 3‰ stannous octoate were added in sequence. The mixture in the polymerization tube was vacuumed-dried at room temperature to remove the residual solvent and gas. The polymerization tube was sealed and placed in an oil bath at 120 °C for 48 h. The product was poured into excess hot water (85 °C), and the supernatant was discarded to obtain a precipitate. The precipitation was repeated twice and the resulting precipitate was freeze-dried to yield PCLGA–PEG–PCLGA.2.3 Polydopamine modified PCLGA–PEG–PCLGA (PDA/PCLGA–PEG–PCLGA)
PCLGA–PEG–PCLGA (1.0 g) was added into a glass bottle, followed by adding a certain amount of Tris–HCl (pH 8.5) solution. After heating in an oven at 60 °C for 20 min, the mixture was completely mixed and then stood at room temperature overnight to obtain a clear solution. Dopamine hydrochloride (500 mg) was added into the solution and stirred overnight at room temperature, then dialyzed for 2 d and freeze-dried.322.4 Synthesis of silver nanoparticles
Polyvinylpyrrolidone (PVP, K30, 0.5 g) was dissolved in 50 mL of absolute ethanol and stirred at 60 °C for 1 h. Silver nitrate was dissolved in 50 mL of absolute ethanol at a concentration of 2 mg mL−1. The silver nitrate solution was added dropwise to the PVP solution. After refluxing at 95 °C for 12 h, ethanol was removed by rotary evaporation to obtain silver nanoparticles (AgNPs).332.5 Preparation of the hydrogel
An appropriate amount of PDA/PCLGA–PEG–PCLGA and water were added to a glass bottle to form a mixed system of PDA/PCLGA–PEG–PCLGA with a mass fraction of 30%. The glass bottle was heated at 60 °C for 20 min and then cooled at room temperature to obtain a solution. The hydrogel was obtained by placing the polymer solution at 37 °C for 10 min. The AgNPs hydrogel (PDA/P2–4@Ag gel) was prepared by heating a mixed solution of AgNPs (500 μg mL−1) and the copolymer (PDA/P2–4) at 37 °C.2.6 Characterization of polymers
The characterization studies of PCLGA–PEG–PCLGA, PDA/PCLGA–PEG–PCLGA and AgNPs are provided in the ESI.†2.7 Sol–gel transition
The sol–gel transition temperature of the copolymer solution was determined by a vial inversion method with a temperature increment of 1 °C.34 Specifically, vials containing the polymer solution (1 mL) were immersed in a water bath at a specific temperature for 10 min, and the temperature range investigated was from 30 to 50 °C. The physical state of the copolymer solution was recorded at each temperature interval by tilting the vial. If no flow was observed after 30 s of inversion, the sample was considered as a gel. The experiments were performed in triplicate. The results were plotted into phase diagrams after all the sol–gel transition temperatures were obtained.The gelling time of the PDA/P2 polymer solutions was also investigated using a vial inversion method with a time step of 30 s.35 The polymer solution (1 mL, 30 wt%) was incubated in a water bath at a corresponding gelling temperature for a specific time. The state of the polymer solution at each incubation time point was determined by inverting the vial. If no flow was observed within 30 s after inversion, the sample was considered as a gelled state. Each experiment was repeated three times.
For the rheological study, the polymer aqueous solution (500 μL) was analyzed using an MCR 302 rheometer (Anton Pear, Austria) equipped with a PP25 plate (25 mm diameter, 1 mm gap). Polymer solutions were stored at a low temperature and then added to the precooled plates. Measurements were carried out at an oscillatory frequency of 1 Hz and a shearing strain (γ) of 1% at a temperature ranging from 15 to 45 °C. The strain sweep test of polymer solutions (30 wt%) was conducted in which the strain was increased from 0.1% to 1000% at 37 °C, and the angular frequency was held constant at 10 rad s−1. Then the strain step cycling between 1% and 1000% was performed at 37 °C and 10 rad s−1.
2.8 Antioxidative activity
The antioxidative activities of P2 and PDA/P2–4 were investigated using a DPPH-scavenging test.36,37 P2 and PDA/P2–4 were dissolved in DMSO at a concentration of 6 mg mL−1. Different volumes of P2 and PDA/P2–4 solutions were added to the DPPH solution (50 μg mL−1, 2 mL), and anhydrous ethanol was added to make a total volume of 3 mL. After mixing well, the solutions were placed in the dark for 30 min and the absorbance at 517 nm was measured using a UV-vis spectrophotometer (UV 2600 Shimadzu). The DPPH-scavenging efficiency (E) was calculated using the following equation:| E = (A0 − As)/A0 × 100%, |
We also tested the reactive oxygen species scavenging ability of P2 and PDA/P2 using the ABTS method.38 Briefly, a solution of 2.6 mmol L−1 potassium persulfate was mixed with 7.4 mmol L−1 ABTS solution in the dark for 12 h. Then the mixture was diluted 12 times with distilled water to obtain the ABTS working solution. 10 μL of the polymer solutions (10, 20, 40, 50, 80, and 100 mg mL−1) was mixed with 200 μL of the ABTS working solution and placed in the dark for 30 min. Then the UV absorbance at 725 nm was monitored. The ABTS radical scavenging abilities were calculated as follows:
| ABTS radical scavenging rate (%) = (A0 − As)/A0 × 100%, |
2.9 In vitro ROS scavenging
RAW264.7 cells (5 × 105 cells in 1 mL of DMEM) in a Petri dish were cultured with P2 or PDA/P2–4 (1 mg mL−1) in Petri dishes. After incubating for 12 h, the cells were treated with a 10 μM 2′,7′-dichlorofluorescein diacetate (DCFH-DA) probe for 1 h. H2O2 (300 mM, 1 μL) was added to each group. After 20 min, each group was washed with PBS three times. The ROS levels were measured using a confocal laser scanning microscope (CLSM; TCSSP5, Leica, Germany). The cells were treated in the same manner and quantitative analysis was performed by flow cytometry. The cell suspension (2 × 105 viable cells) was collected in each tube and analyzed by flow cytometry (BD LSR Fortessa, BD Biosciences). An unstained control sample was also prepared as a compensation control to determine the background levels. A minimum of 1 × 104 ungated cells were acquired from each sample and the percentage of positive events in the FL1 (FITC) channel (ROS-presenting cells) was recorded. FlowJo software was used to analyze the data.25,392.10 In vitro antibacterial activity
2.11 Blood compatibility assessment
A fixed amount of polymer solutions was mixed with blood cells and incubated at 37 °C for 1 h, and then the absorbance at 540 nm was measured to evaluate the hemolysis. The specific process is shown in the ESI.†20
2.12 Wound healing evaluation in an infective skin trauma model
All animal experiments strictly complied with the Animal Management Rules of the Ministry of Health of the People's Republic of China (document no. 55, 2001) and the institutional guidelines. All experiments were approved by the Animal Care and Use Committee of Sichuan University. Sprague Dawley (SD) rats (8 weeks, 230–250 g, male) were purchased from Chengdu Dashuo Experimental Animal Co., Ltd. All rats were anesthetized via an intraperitoneal injection of chloral hydrate (10%, w/v) and four full-thickness round wounds (diameter = 1.0 cm) were made on the shaved dorsal side of each rat. The Staphylococcus aureus suspension (200 μL, 1 × 109 CFU mL−1) was dripped onto the wound. After infecting for 24 h (day 0), P2, PDA/P2–4, and PDA/P2–4@Ag solutions (30%wt and the concentration of AgNPs at 500 μg mL−1) were injected into the wound to form wound dressings. The wounds were then bandaged with gauzes. The wounds in the control group were bandaged without any treatment. On days 3, 5, 10, and 15, the rats were euthanized and the wounded skins were collected for further analysis. Photographs of the wound area were taken on days 0, 3, 5, 10, 13, and 15. The wound healing rate was the percentage of the healed area relative to the original wound area. The following formula was used to calculate the wound healing rate: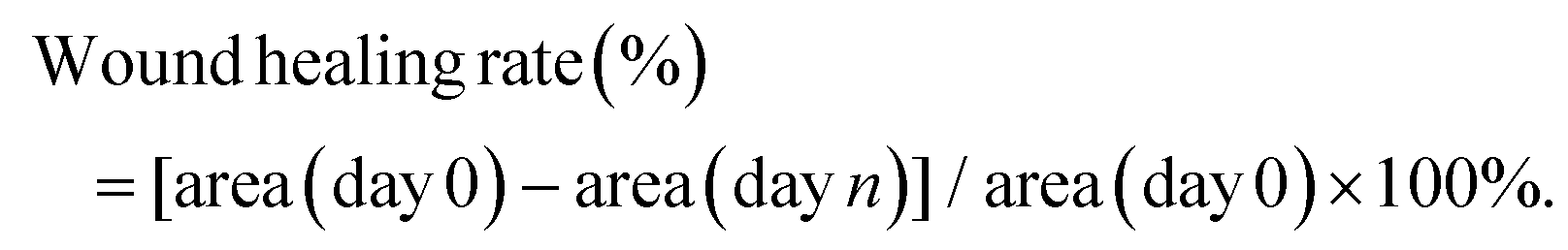
2.13 Histological analysis
On days 3, 5, 10, and 15, the tissue samples were fixed in 4% paraformaldehyde for 48 h, embedded with paraffin, and cut into 7 μm sections. These sections were processed and stained with hematoxylin and eosin (H&E), and Masson. Subsequently, immunostaining was performed using an antibody solution containing anti-CD 31 and anti-IL-6. The images were captured with an optical microscope (Leica, DMI 4000). To further verify the results of immunohistochemistry, the gene expressions of IL-6 and CD31 were detected by RT-PCR.2.14 Antibacterial effect in vivo
On days 5 and 15 after treatment, the wounds were gently wiped with sterile cotton swabs. The cotton swab was placed in 4 mL of sterile normal saline. The saline was diluted 2 times and applied to agar plates. Photographs of bacterial growth were taken after the plates were incubated at 37 °C for 24 h.402.15 Statistical analysis
The data were presented as the means ± standard deviation. Statistical significance was analyzed using a one-way analysis of variance (ANOVA) with a post hoc test. A significant difference was considered when *p < 0.05, **p < 0.01, and ***p < 0.001.3. Results and discussion
3.1 Synthesis and thermo-sensitive sol–gel transition of triblock polymers
Three triblock copolymers PCLGA–PEG–PCLGA were synthesized by varying the CL/GA feeding ratios via a typical ring opening polymerization (Table S1†). The polymers were characterized using 1H NMR (Fig. 2A)41 and gel permeation chromatography (Fig. 2B). The differential scanning calorimetry (DSC) curves showed that the incorporation of the GA segment into PCL blocks greatly hindered the crystallization (Fig. 2C). The higher GA content had a greater effect on the conventional crystallization of PCL, resulting in a shift of the two peaks of the PCLGA fragment of P1 to a lower temperature compared with those of P2 and P3.42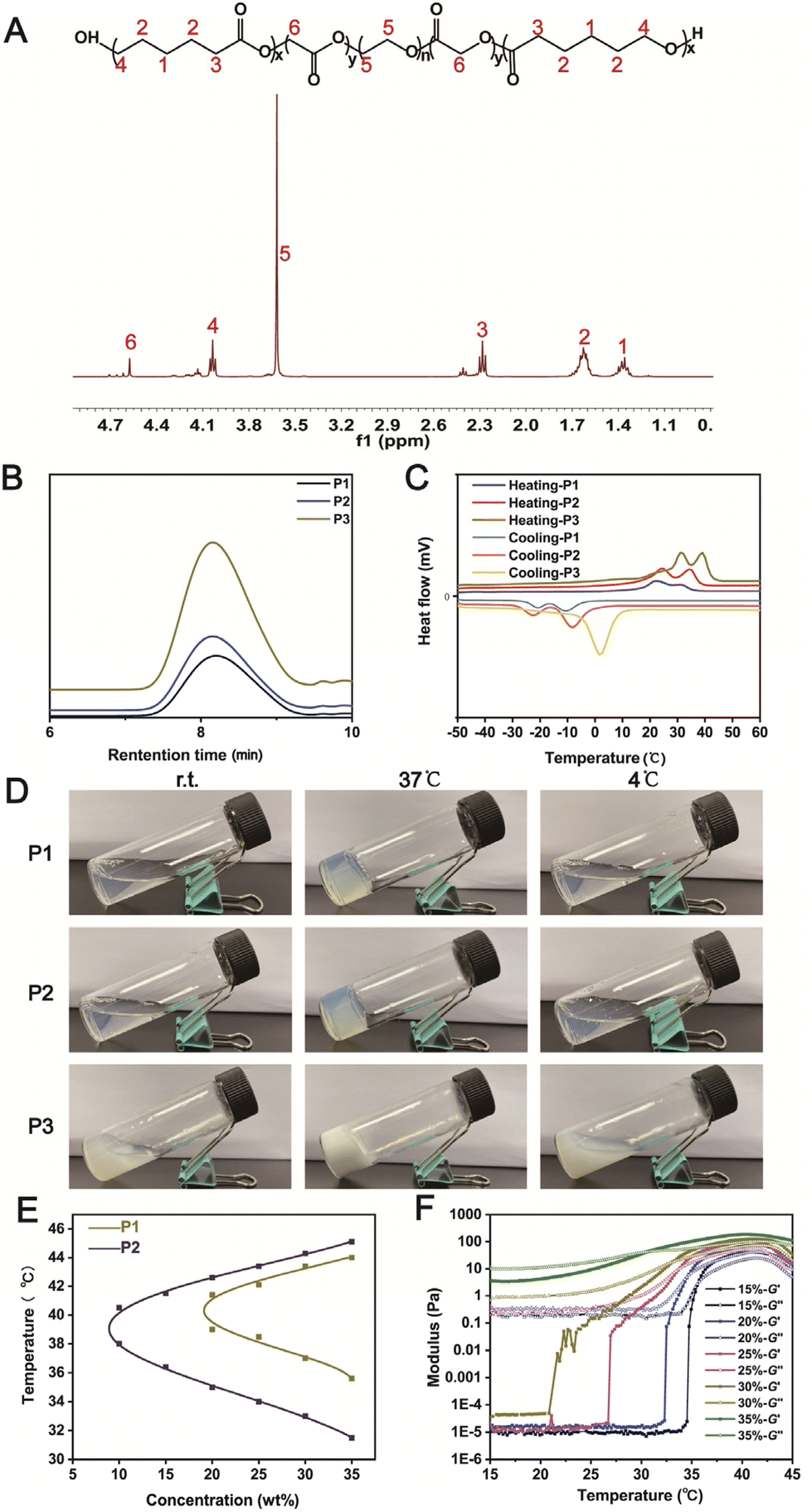 | ||
| Fig. 2 (A) 1H NMR spectrum of P2. (B) GPC curves of P1, P2 and P3. (C) DSC thermograms of P1, P2 and P3 at a heating or cooling rate of 20 °C min−1. (D) Photographs of P1, P2, and P3 solutions at different temperatures with a polymer concentration of 30% (wt%). (E) Phase diagram of P1 and P2 solutions with different concentrations in a water bath plotted by a vial inversion method at different temperatures. (F) Storage modulus G′ and loss modulus G′′ of P2 solutions with different concentrations as a function of temperature. | ||
The thermo-sensitive gelation ability of the three polymers was then studied (Fig. 2D). P1 and P2 solutions formed gels at 37 °C for 10 min, which returned to transparent solutions after cooling at 4 °C for 10 min, indicating that P1 and P2 had reversible temperature-sensitive sol–gel transition properties. In contrast, a white opaque suspension of P3 was observed, suggesting that the polymer was less water-soluble at a 30 wt% concentration. According to the above results (Table S1† and Fig. 2C and D), we speculated that the water solubility and gelation behavior of the triblock polymer were not only related to the length of the hydrophobic chain segment (PCLGA) and crystallization behavior, but also associated with the molecular weight distribution (Mw/Mn) of the copolymer.
We then studied the sol–gel transition behavior of P1/P2 via flask inversion and phase transition diagrams were plotted (Fig. 2E). The sol–gel transition temperature of the polymers was close to body temperature (37 °C), indicating that these injectable thermo-sensitive hydrogels could be formed in vivo for potential biomedical applications. However, the phase transition temperature range of P2 was larger than that of P1. Taking the stability of the gel in considering the application environment, P2 was selected for the follow-up study. The thermo-sensitive sol–gel transition of P2 was further confirmed by dynamic rheological study, where the crossover points of the storage modulus G′ and loss modulus G′′ of P2 approximated the corresponding temperatures in the phase transition diagram (Fig. 2F).
3.2 Physicochemical characterization of polydopamine-modified P2
Polydopamine (PDA) was coated on P2 to endow the thermo-sensitive hydrogel with ROS-scavenging property. We synthesized a series of PDA/P2 polymers by adjusting the volume of the reaction system (Table S2†). The polydopamine coating was performed by the oxidative self-polymerization of dopamine. In the UV-vis spectrum of PDA/P2–4, a typical absorption of PDA at 395 nm was observed (Fig. 3A),43 indicating the successful PDA functionalization. The introduction of PDA was further verified by X-ray photoelectron spectroscopy (XPS) analysis. Fig. S2† showed the XPS spectrum with the presence of C, O, and N in PDA/P2–4. The binding energies of C 1s peaks at 284.5, 285.9, and 288.6 eV were ascribed to C–C, C–N/C–O, and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–OH from the polymer,44 respectively (Fig. S2B†). In the FT-IR spectra of P2 and PDA/P2–4, the absorption bands at 1542 and 1374 cm−1 were attributed to the CC and phenolic C–O–H stretching vibrations, respectively (Fig. S3†).29
C–OH from the polymer,44 respectively (Fig. S2B†). In the FT-IR spectra of P2 and PDA/P2–4, the absorption bands at 1542 and 1374 cm−1 were attributed to the CC and phenolic C–O–H stretching vibrations, respectively (Fig. S3†).29
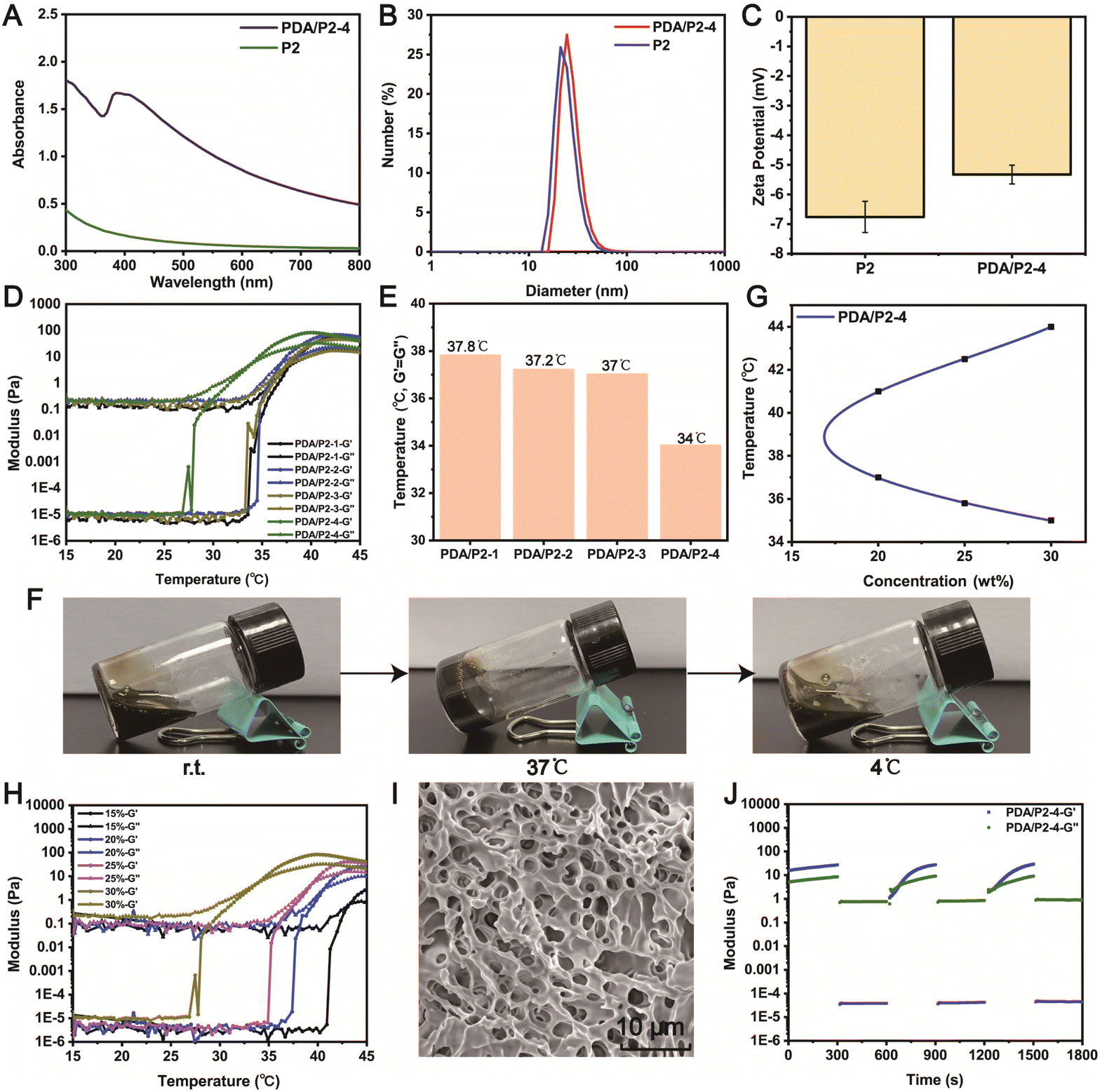 | ||
| Fig. 3 (A) UV-vis spectra of PDA/P2–4 and P2 solutions. DLS (B) and zeta (C) analyses of PDA/P2–4 and P2 solutions (1 mg mL−1). (D) Storage modulus (G′) and loss modulus (G′′) of different PDA-modified P2 solutions (30 wt%) as a function of temperature. (E) The temperature at the intersection of the storage modulus G′ and loss modulus G′′ of different PDA/P2 solutions (30 wt%). (F) Photographs of PDA/P2–4 solution (30 wt%) at different temperatures. (G) Phase diagram of PDA/P2–4 with different concentrations plotted by a vial inversion method at different temperatures. (H) Storage and loss modulus of PDA/P2–4 solutions with different concentrations as a function of temperature. (I) SEM analysis of the PDA/P2–4 gel. (J) Repeated dynamic strain step testing (γ = 1% or 1000%, 10 rad s−1) of the PDA/P2–4 solution (30 wt%) at 37 °C. | ||
Then we tested the influence of polydopamine coating modification on the crystallization behavior of the P2 polymer, and the results are shown in Fig. S4.† Compared with that of P2, the XRD pattern of PDA/P2–4 still showed two strong crystallization peaks at 21.4° and 23.8°, which were attributed to the crystallization of PCL segments. But the weak crystallization peak of 19.4° that was attributed to the crystallization of PEG segment disappeared in the XRD pattern of PDA/P2–4.45 It is possible that the XRD pattern of polydopamine itself was responsible for this result.46
Then, we investigated whether the modification of PDA coating had an impact on the self-assembly behavior of the amphiphilic polymer in aqueous solution.47 The TEM results suggested that the micelles of PDA/P2–4 had a larger particle size (29.53 ± 2.37 nm) than those of P2 (24.30 ± 2.78 nm) at a concentration of 1 mg mL−1 (Fig. S5†), implying that PDA modification did not affect the self-assembly behavior of the amphiphilic polymer. This result was further confirmed by the DLS results (Fig. 3B), and PDA/P2–4 showed a higher zeta potential than P2 owing to the introduction of positively charged PDA (Fig. 3C).48 The self-assembly behaviors of these polymers were further investigated by measuring critical micelle concentrations (CMCs, Fig. S6†). P2 and PDA/P2–4 showed comparable CMC values of 1.39 and 0.447 μg mL−1, respectively. Together, these results indicated that the PDA/P2–4 polymer could self-assemble into micelles similar to P2.
To explore the impact of PDA modification on the gel formation ability, the changes in the storage modulus G′ and loss modulus G′′ of four PDA/P2 solutions (30%, wt%) with increasing temperature were monitored. As shown in Fig. 3D, the PDA/P2–4 polymer solution had a crossover point of storage modulus (G′) and loss modulus (G′′) at about 34 °C, indicating that the sol–gel transition occurred at this temperature. The sol–gel transition temperatures of the other three PDA/P2 polymers were much higher than 34 °C (Fig. 3E). The gelling times of the different PDA/P2 polymers at their corresponding gelation temperatures are summarized in Table S3.† According to the results of Fig. S7,† the PDA content of PDA/P2–4 was the lowest among these PDA/P2 polymers. Combined with the results of Fig. 3E, S6 and S7,† this showed that the impact of PDA contents on the thermo-sensitive gelation ability was ascribed to the hydrophilicity change of polymer micelles.49,50 The PDA/P2–4 solution (30 wt%) showed thermo-sensitive gelation and reversible sol–gel transition (Fig. 3F). The sol–gel transition diagram, examined using the flask inversion method, showed that PDA/P2–4 showed a sol–gel transition temperature range close to body temperature (Fig. 3G). Fig. 3H revealed that PDA/P2–4 showed a weight-dependent gelation temperature. 30 wt% PDA/P2–4 had a gelation temperature of 34 °C, which was slightly lower than the skin temperature, and was more conducive to practical use. According to the above results, PDA/P2–4 was selected for further investigations.
The SEM image (Fig. 3I) of the PDA/P2–4 hydrogel showed a three-dimensional porous structure, indicating that the functional coating modification of polydopamine did not change the microstructure of the P2 hydrogels (Fig. S1†). According to the SEM–EDS analysis (Fig. S8†) of the PDA/P2–4 hydrogel, the contents of C, O, and N in this polymer were 71.85%, 23.57%, and 4.59%. Previous studies have shown that the modification of polydopamine could endow hydrogels with certain self-healing properties.51–53 In our study, 1000% strain significantly disrupted the hydrogel network and the storage modulus (G′) was smaller than the loss modulus (G′′) at 37 °C (Fig. S9†). Then, with the aim of testing the self-healing properties of the hydrogels, repeated dynamic strain step tests (γ = 1% or 1000%) were carried out. As shown in Fig. S10,† after the strain scanning of 1000% strain, the storage modulus and loss modulus of the P2 polymer were far less than their initial values at 1% strain, but the storage modulus was still greater than the loss modulus, indicating that the P2 hydrogel had poor self-healing properties. Fig. 3J showed that the modulus of the PDA/P2–4 hydrogel could recover to its initial value at 1% strain after the repeated two-cycle scanning, suggesting that the PDA/P2–4 hydrogel had excellent self-healing property. For amphiphilic polymers such as PCLGA–PEG–PCLGA, the sol–gel transition is mainly caused by the aggregation of hydrophobic fragments at the sol–gel transition temperature.54 At the same time, the hydrophobic fragment can act as a reversible cross-linking point and can freely flow to form new aggregates again after the scanning of 1000% strain, thus endowing the P2 hydrogels with a certain self-healing ability.55 For the PDA/P2–4 hydrogel, in addition to the hydrophobic chain segment, polydopamine also was a reversible crosslinking point.56 So the PDA/P2–4 hydrogel has excellent self-healing performance.
In addition, we found that the introduction of PDA improved the adhesion ability of the hydrogels (Fig. S11†), where the P2 gel was readily peeled off from a PP25 plate rotor while PDA/P2–4 showed a certain adhesion ability. The adhesion and self-healing properties enhanced by PDA are conducive to the application of hydrogels as wound dressings.57 Together, P2 was successfully modified by polydopamine, and the PDA/P2–4 polymer still reserved the body-temperature sensitive sol–gel transition properties.
3.3 Reactive oxygen species (ROS)-scavenging evaluation
A PDA coating is a powerful strategy to endow materials with ROS-scavenging ability. The diphenyl-2-picrylhydrazyl (DPPH) free radical scavenging assay was conducted to investigate the antioxidative property of PDA/P2–4. The absorption peak of DPPH at 517 nm significantly decreased in the UV-vis spectrum of PDA/P2–4 in comparison with P2 (Fig. 4A), demonstrating the PDA-mediated ROS depletion. Furthermore, the ROS scavenging of PDA/P2–4 is proportional to the polymer concentration (Fig. 4B). The clearance rate of DPPH by PDA/P2–4 was 70% at a polymer concentration of 0.6 mg mL−1. In stark contrast, P2 alone cannot deplete the ROS at the same polymer concentration. We also tested the ROS scavenging ability of P2 and PDA/P2 using the ABTS method (Fig. S13†). The radical scavenging ability of PDA/P2–4 was positively correlated with the polymer concentration, whereas P2 cannot scavenge ROS. Although PDA/P2–4 showed the lowest DPPH scavenging capacity among the four PDA/P2 polymers owing to the lower PDA content (Fig. S7, S12, and S13†), it was still used for further studies owing to the appropriate sol–gel transition temperature and acceptable ROS-scavenging capacity.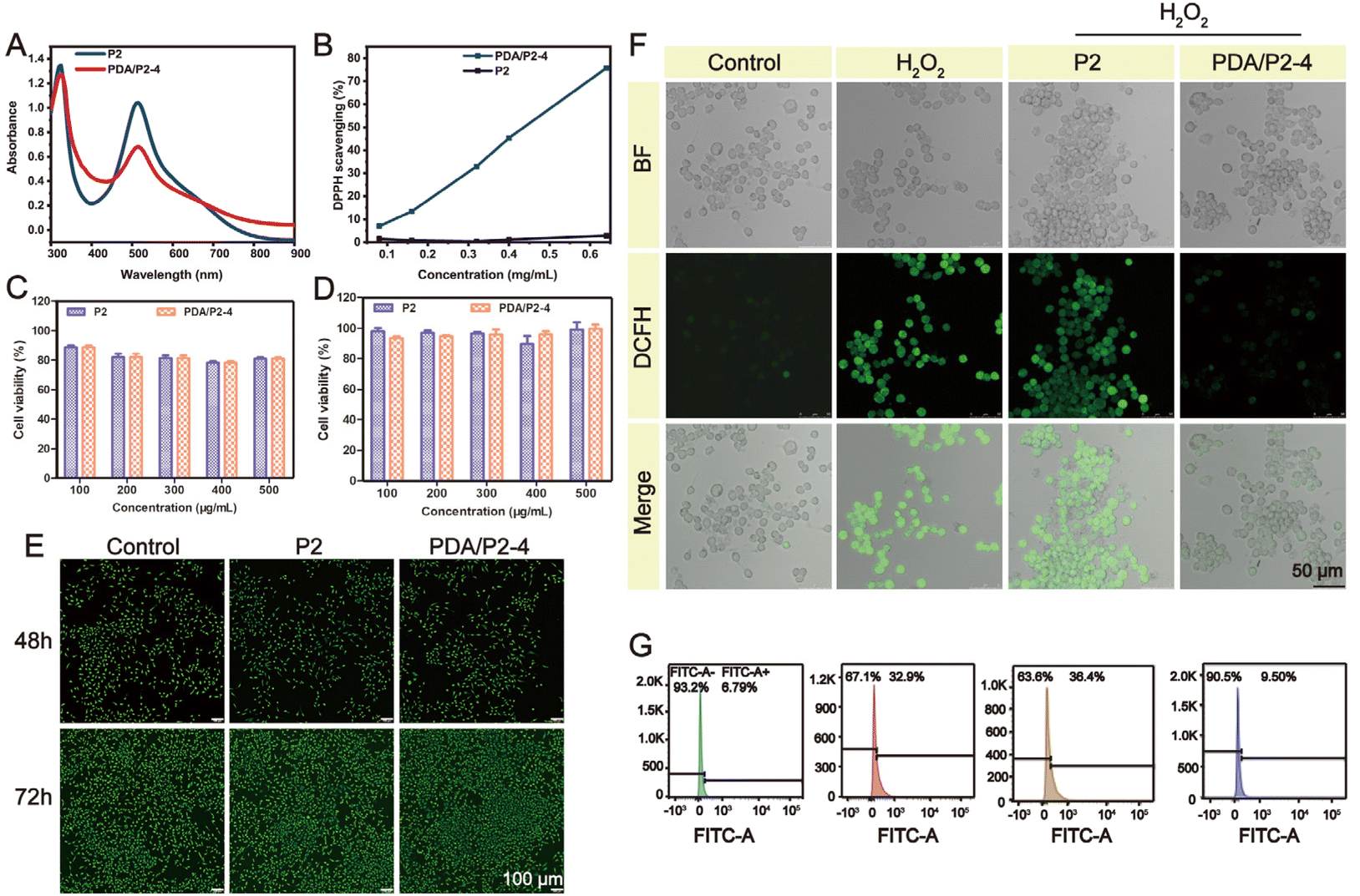 | ||
| Fig. 4 (A) UV-vis spectra of DPPH after treatment with P2 and PDA/P2–4. (B) ROS-scavenging efficiency of P2 and PDA/P2–4. Viability of L929 cells after co-incubation with P2 and PDA/P2–4 for 48 (C) and 72 h (D). (E) AO/EB staining of L929 cells incubated with polymers. (F) Intracellular ROS-scavenging performance of P2 and PDA/P2–4 solutions. (G) Intracellular ROS level obtained by the flow cytometry analysis of DCFH-DA labeled cells in a fluorescein isothiocyanate FITC-A channel on different groups. The bars on the left represent a subpopulation of cells lacking DCFH fluorescence, while the bars on the right spanning the stained ROS-presenting subpopulation. The percentages of both sides are also shown. | ||
We then paid attention to the intracellular ROS-scavenging ability of PDA/P2–4 considering that excessive ROS is notorious in wound healing.19,58 First, PDA/P2–4 and P2 showed good cytocompatibility, where more than 80% of L929 cells still alive after the 48 or 72 h of incubation at a concentration up to 500 μg mL−1 (Fig. 4C and D). AO/EB staining (Fig. 4E) showed that the number of cells increased with the prolongation of incubation time at a polymer concentration of 500 μg mL−1, and no cells with red fluorescence were found, indicating that the polymers were not cytotoxic. Thereafter, the ROS-scavenging potential of PDA/P2–4 in RAW264.7 cells was evaluated. The intracellular ROS level was upregulated by exogeneous H2O2 and monitored using a 2′,7′-dichlorofluorescein diacetate (DCFH-DA) probe.59,60 The fluorescence intensity of the PDA/P2–4 group was the lowest, similar to that of the control group, while the fluorescence intensity of the P2 group was similar to that of the H2O2 group (Fig. S14†). Quantitative flow cytometry analysis also confirmed the effectiveness of PDA/P2–4 in scavenging ROS (Fig. 4G), where the fluorescence-positive rates of the cells were 9.50% and 36.4% in the PDA/P2–4 and P2 groups, respectively. Overall, these results clearly demonstrated that PDA-modified P2 exhibited ROS-scavenging ability.
3.4 In vitro antibacterial evaluation and blood compatibility assessments
With the ROS-scavenging thermo-sensitive hydrogel in hand, we incorporated antibacterial silver nanoparticles into the hydrogel (PDA/P2–4@Ag gel) to address the pathogenic infection issue, which is generally encountered in clinical wound treatments. Silver nanoparticles (AgNPs) with a size of 9.02 ± 0.56 nm (n = 30) and an approximately spherical shape were prepared by ethanol reduction (Fig. S15 and S16†).61 The SEM–EDS results clearly suggest the homogeneous distribution of AgNPs in the PDA/P2–4@Ag gel (Fig. S17†). The silver content in the hydrogel determined by SEM–EDS analysis was found to be 0.07%. The Ag release behaviors of the PDA/P2–4@Ag hydrogel were studied by detecting the released amount of Ag with ICP. As shown in Fig. S18,† the cumulative release concentration of Ag increased with time. The Ag release of the PDA/P2–4@Ag hydrogel was approximately 40% at 96 h, suggesting the slow release of Ag from the hydrogel, thus avoiding the side effects of rapid Ag release. The antibacterial activity of the PDA/P2–4@Ag gel against Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus) was then studied by the inhibition zone method (Fig. 5A and B). The P2 and PDA/P2–4 hydrogels had no observable antibacterial activity; however, the PDA/P2–4@Ag gel showed significant bacterial inhibition and inhibition zones of 0.715 and 0.741 cm were observed in the E. coli and S. aureus plates, respectively. These results indicated that the addition of AgNPs allowed for excellent antibacterial function. The antibacterial activity of AgNPs-doped PDA/P2–4 was further confirmed in the spread plate assay (Fig. S19†). Meanwhile, according to the results of Fig. S20,† more than 80% of L929 cells survived after co-incubation with the medium containing PDA/P2–4@Ag (500 μg mL−1 and the concentration of AgNPs at 8.3 μg mL−1) for 48 h. The above results indicated that PDA/P2–4@Ag has good biocompatibility and excellent antibacterial properties.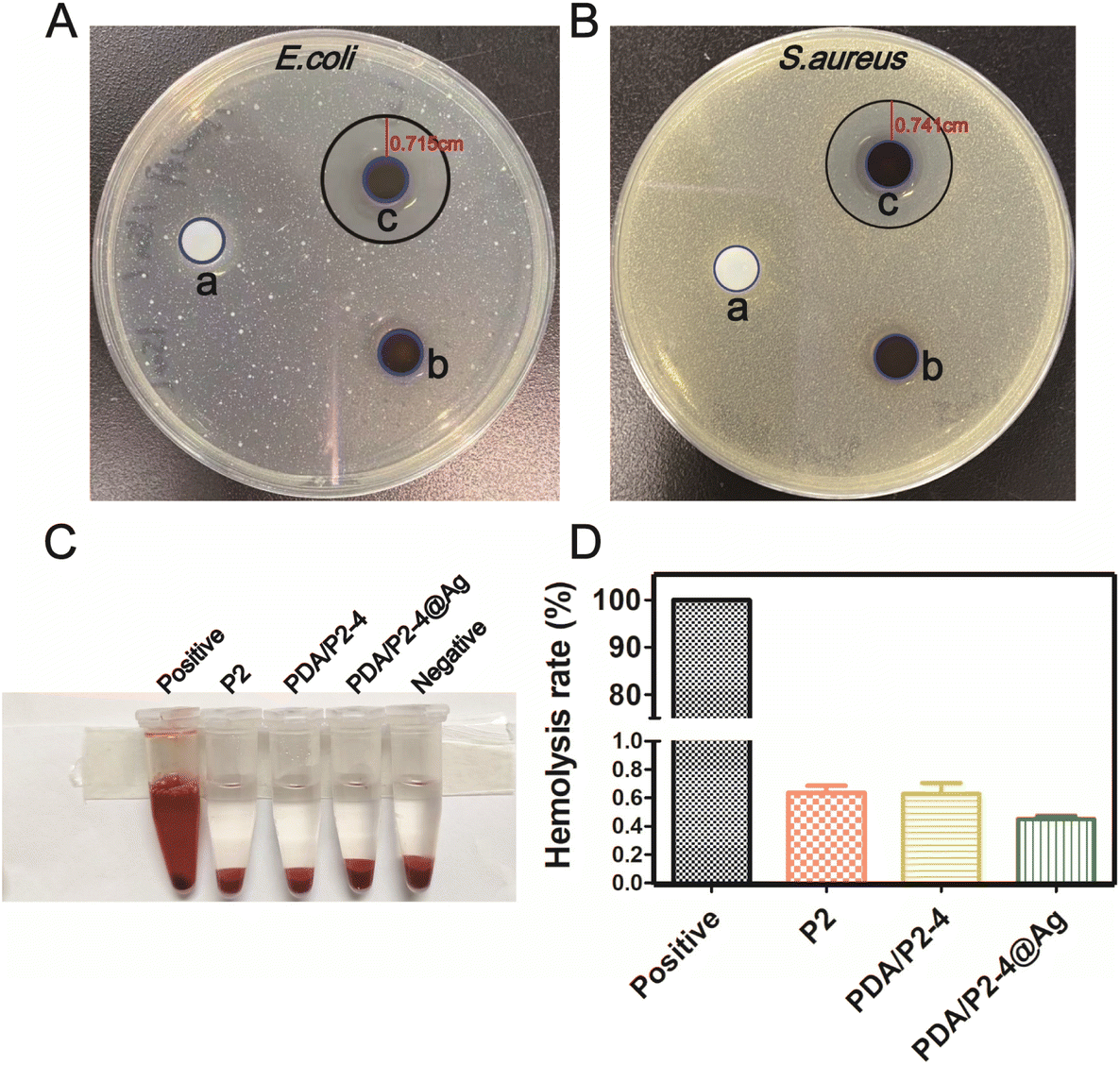 | ||
| Fig. 5 (A and B) Bacteriostatic study of gels for 24 h. (a): P2 gel; (b): PDA/P2–4 gel; and (c): PDA/P2–4@Ag gel. (C) Photographs of the blood compatibility results of different treatment groups. (D) The percentage of the hemolysis rate of different treatment groups. | ||
Good blood compatibility is a prerequisite for the wound healing application of biomaterials. The blood compatibilities of P2, PDA/P2–4 and PDA/P2–4@Ag were evaluated by an in vitro hemolysis test. Hemolysis was hardly observed in the three groups (Fig. 5C) and the quantitative results showed that the hemolysis ratios of all polymers were less than 1% (Fig. 5D), proving that all polymers have good blood compatibility.
3.5 In vivo infected wound healing assay
The in vivo infected wound healing ability of the PDA/P2–4@Ag gel was investigated in a rat wound model with S. aureus infection. Polymer solutions were injected at the wound sites and converted into gels within 10 min (Fig. S21†). The wounds were photographed at different time points and the results were shown in Fig. 6A. Wound healing occurred in all groups with different rates. After 15 days, the wounds treated with the PDA/P2–4@Ag gel were almost closed. The wounds treated with the PDA/P2–4 and P2 gels were also nearly closed with only a small amount of scarring. In contrast, the wound of the control group remained open. The wound closure rate was further calculated to quantitatively compare the wound healing performances of different groups (Fig. 6B and C). The wound healing rate of the PDA/P2–4@Ag gel group was the fastest during the entire treatment period, followed by the PDA/P2–4 gel group (Fig. 6B and C). Although the wound healing rate of the P2 gel was lower than that of the PDA/P2–4 gel, it was still higher than that of the control group, which was attributed to that the P2 gel could keep the wound surface moist and physically isolate the wound from the external environment to prevent potential bacterial infection, thus promoting wound healing.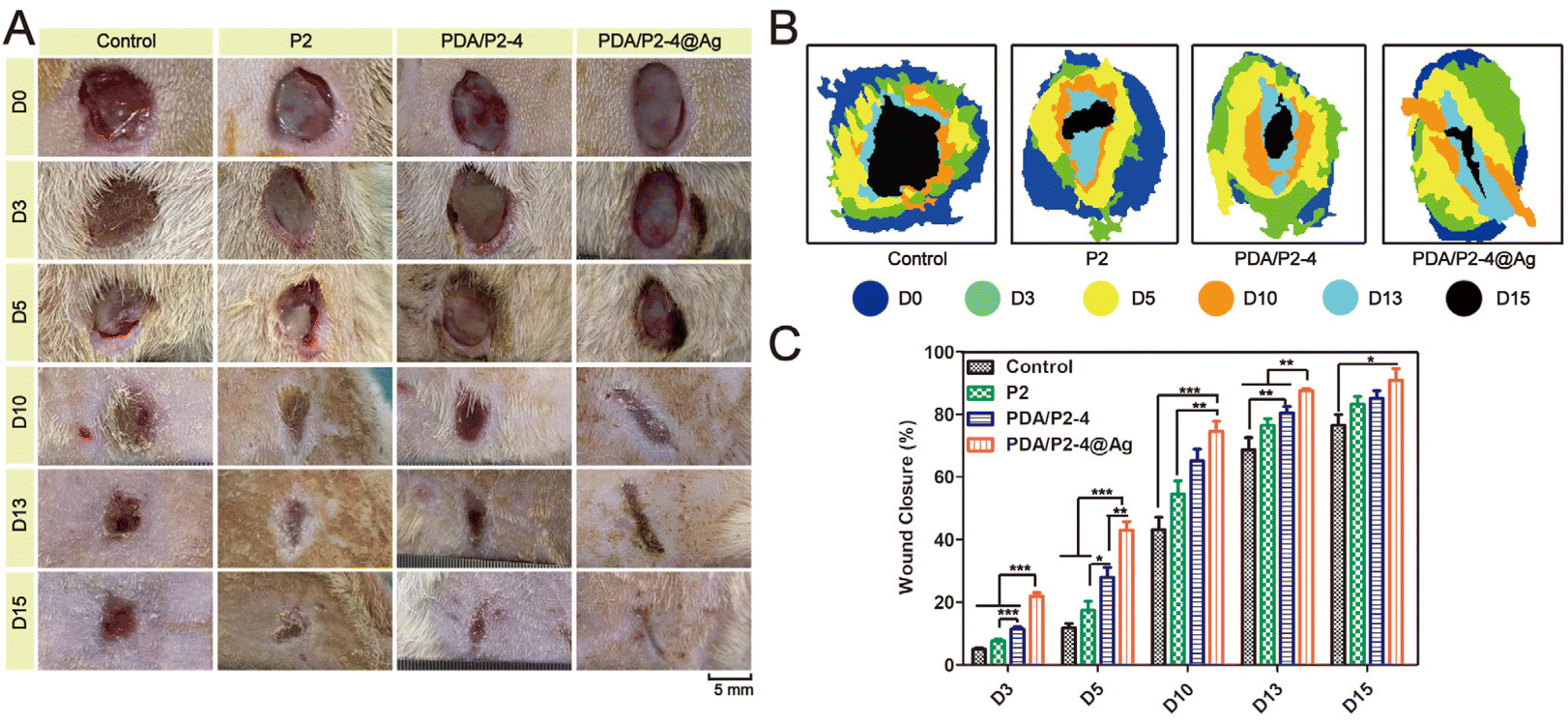 | ||
| Fig. 6 In vivo healing study of different hydrogel dressings on the wound healing of S. aureus infection. (A) Wound size measurements. (B) Area diagrams of S. aureus infected wounds in different groups. (C) Statistical analysis of the wound healing of S. aureus in different treatment groups (n = 5, *p < 0.05, **p < 0.01, and ***p < 0.001). | ||
Meanwhile, we examined the antibacterial effects of different hydrogels on infected wounds, and the results were shown in Fig. 7A. On the 5th day after treatment, the PDA/P2–4@Ag gel showed the best antibacterial effect, where only a few bacterial colonies were observed in the wounds of the PDA/P2–4@Ag gel treatment group, clearly indicating that AgNPs endowed the hydrogel with excellent in vivo antibacterial performance.
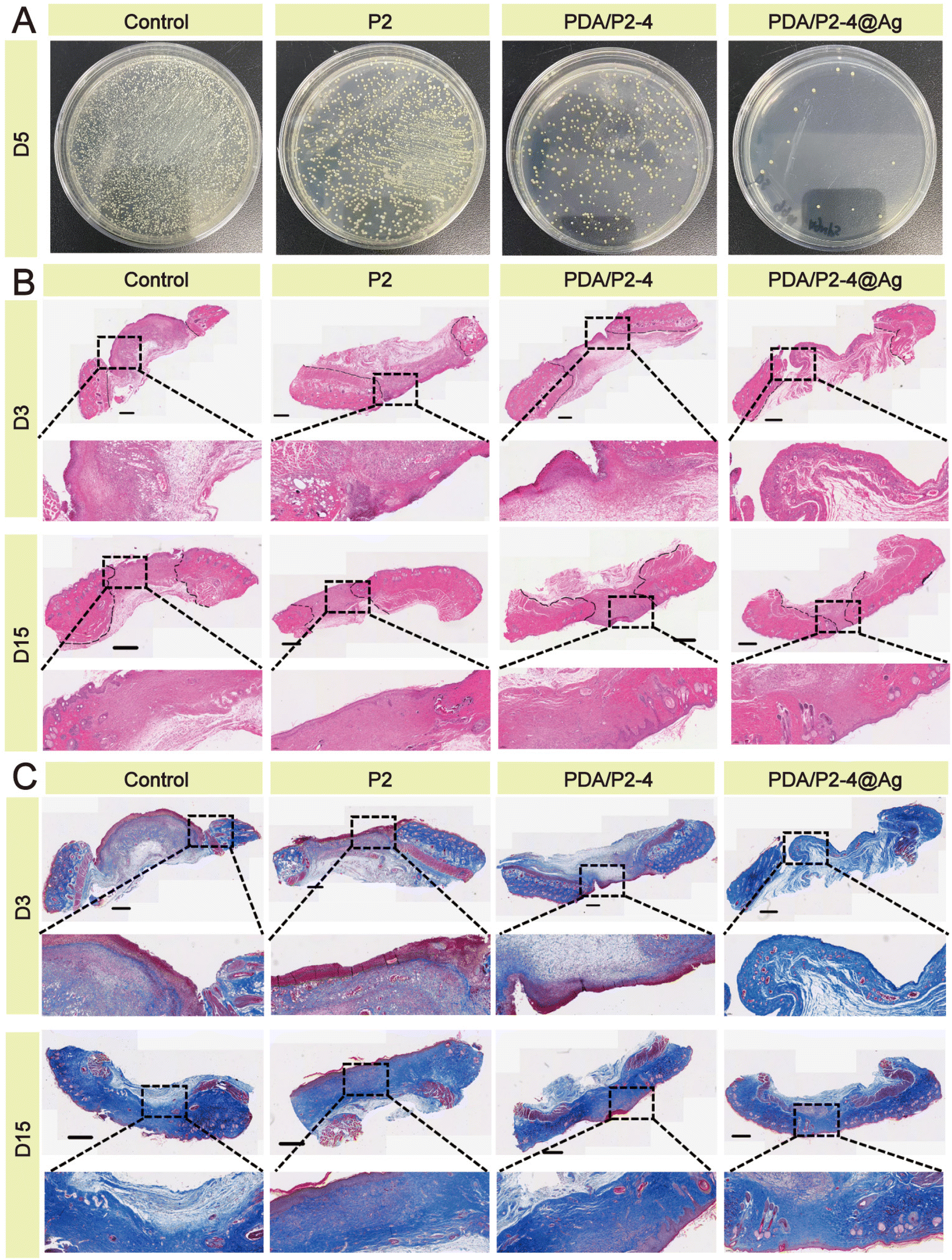 | ||
| Fig. 7 (A) Antibacterial activity against S. aureus infection in vivo. (B) H&E staining of the wound tissue; scale bar 800 μm. (C) Masson staining of the wound tissue; scale bar 800 μm. | ||
H&E staining of wound tissues was carried out to evaluate the skin regeneration effect. Many inflammatory cells gathered at the wounds except for the PDA/P2–4@Ag group (Fig. 7B). On the 15th day, formed hair follicles were visible in the wounds of the PDA/P2–4@Ag gel group, indicating that these wounds had recovered to a similar state to normal skin. A small number of incomplete hair follicles were also observed in the wounds of the PDA/P2–4 gel group. The Masson staining results (Fig. 7C) further suggested that collagen deposition was remarkably visible in the wounds of the PDA/P2–4@Ag gel group on the 3rd day after treatment, and the wound tissue was close to the normal level at day 15.
The inflammation level and angiogenesis in the wound microenvironment were studied using IL-6 and CD31 immunohistochemical staining, respectively. IL-6 is an important pro-inflammatory cytokine and the PDA/P2–4@Ag gel caused the most significant down-regulation of IL-6 on days 5 and 15 (Fig. 8A and C). In addition, the PDA/P2–4 gel showed a remarkable IL-6 decrease in comparison with the P2 gel, which was possibly attributed to the ROS-scavenging effect of PDA.62 In addition, angiogenesis is a vital hallmark of skin repair. Up-regulated CD31 levels were observed in the PDA/P2–4 and PDA/P2–4@Ag groups on day 5, showing that there was a large amount of neovascularization at these wounds. It is well established that blood vessels can transport substances for cell proliferation, and the formation of a lot of blood vessels can further promote wound healing. As the wound healed gradually, a lot of angiogenesis was no longer required in the later stages to transport the nutrients for cell proliferation.63 On the 15th day, the wounds of the PDA/P2–4@Ag gel group showed the lowest vessel density, indicating the late stage of wound healing.
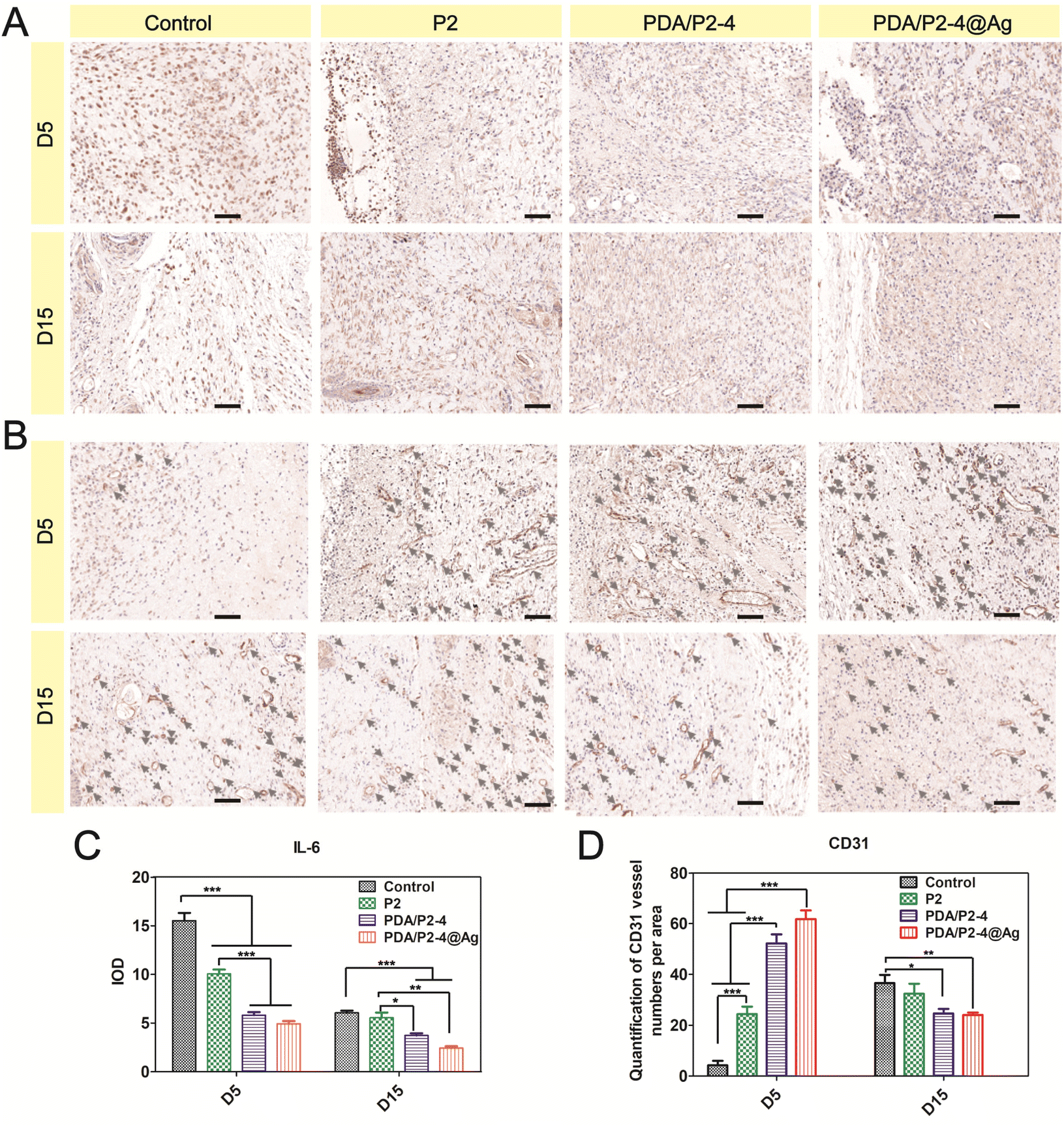 | ||
| Fig. 8 IL-6 (A) and CD31 (B) immunohistochemical staining of wound tissues in different treatment groups on days 5 and 15. Scale bar 50 μm. Quantitative statistical results of IL-6 (C) and CD31 (D); n = 5, *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
In order to further verify the reliability of immunohistochemical staining, the gene expression levels of IL-6 and CD31 were investigated by RT-PCR (Fig. 9). The expression of IL-6 in the PDA/P2–4@Ag group was the lowest on days 5 and 15. The expression of CD31 in the PDA/P2–4@Ag group was the highest, followed by the PDA/P2–4 group on day 5. But after treatment for 15 days, the expression of CD31 of the PDA/P2–4@Ag group was the lowest, followed by the PDA/P2–4 group. These results were consistent with those of immunohistochemical staining (Fig. 8). Combined with the above analysis results, the PDA/P2–4@Ag gel could accelerate wound healing by reducing inflammation and promoting angiogenesis.
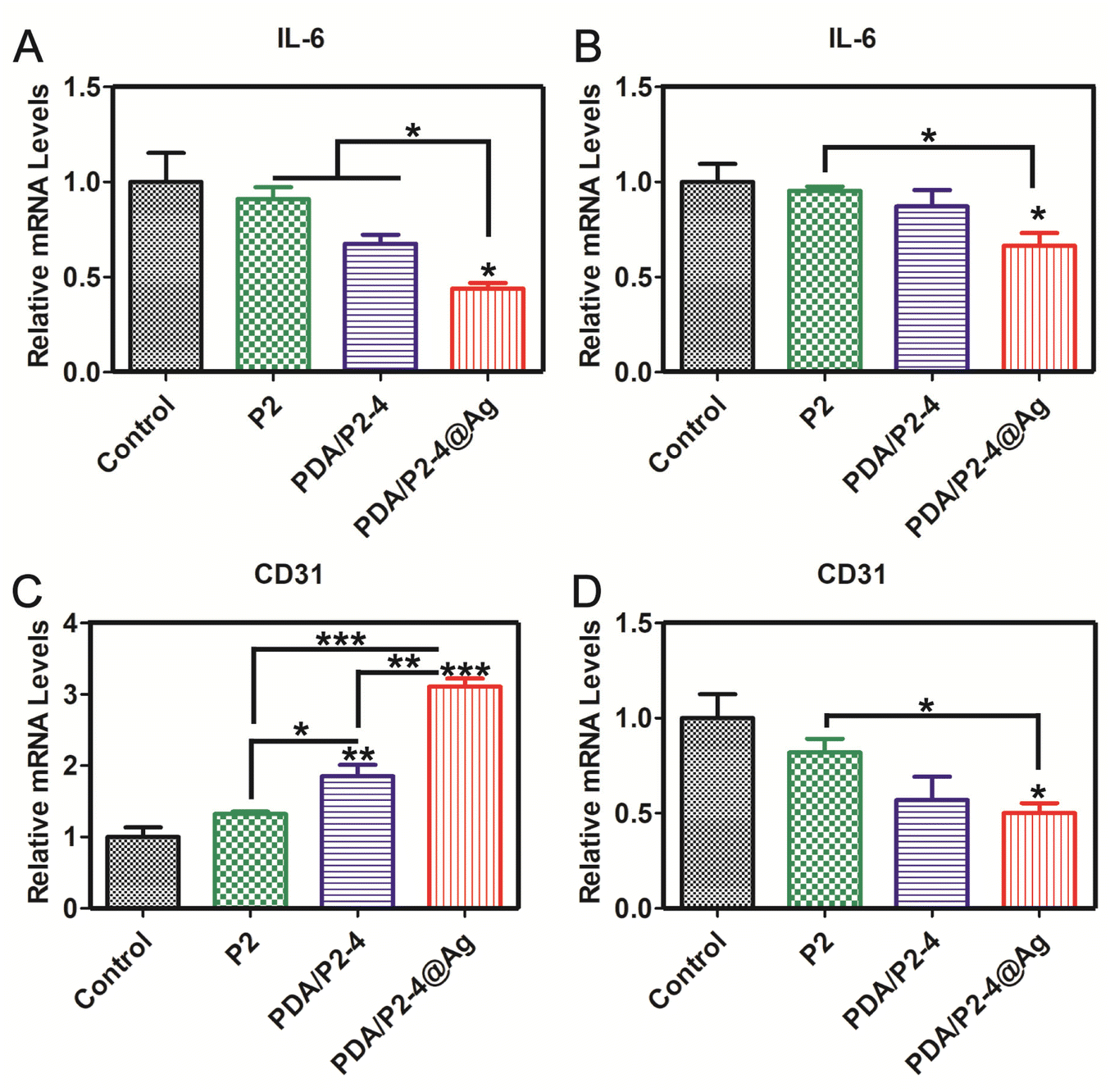 | ||
| Fig. 9 mRNA levels of IL-6 determined by RT-PCR after the treatment of 5 (A) and 15 days (B). mRNA levels of CD31 after the treatment of 5 (C) and 15 days (D) (n = 3, *p < 0.05, **p < 0.01, and ***p < .001). | ||
4. Conclusion
In summary, we developed a polydopamine modified thermo-sensitive PCLGA–PEG–PCLGA triblock copolymer hydrogel with antioxidant and antibacterial functions as a novel wound dressing. PDA modification not only maintained the thermo-sensitive gel formation ability of PCLAG–PEG–PCLGA but also provided good ROS-scavenging properties and increased the adhesion and self-healing properties, which played a positive role in wound healing. Antibacterial silver nanoparticles were further efficiently loaded into the PDA/P2–4 gel to inhibit the proliferation of E. coli and S. aureus. In a full-layer S. aureus infected model on rats, the PDA/P2–4@Ag gel significantly accelerated the wound healing process via reducing the inflammatory response, inhibiting bacterial growth and promoting collagen deposition and angiogenesis.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors greatly acknowledge the financial support from the National Natural Science Foundation of China (51773130 and 31971390).References
- M. Ke, Z. Wang, Q. Dong, F. Chen, L. He, C. Huselstein, X. Wang and Y. Chen, Nanoscale, 2021, 13, 15743–15754 RSC.
- R. Xu, G. Luo, H. Xia, W. He, J. Zhao, B. Liu, J. Tan, J. Zhou, D. Liu, Y. Wang, Z. Yao, R. Zhan, S. Yang and J. Wu, Biomaterials, 2015, 40, 1–11 CrossRef CAS PubMed.
- Z. Xu, S. Han, Z. Gu and J. Wu, Adv. Healthcare Mater., 2020, 9, e1901502 CrossRef PubMed.
- Y. Li, Z. Ma, X. Yang, Y. Gao, Y. Ren, Q. Li, Y. Qu, G. Chen and R. Zeng, Int. J. Biol. Macromol., 2021, 182, 311–320 CrossRef CAS PubMed.
- A. Zhang, Y. Liu, D. Qin, M. Sun, T. Wang and X. Chen, Int. J. Biol. Macromol., 2020, 164, 2108–2123 CrossRef CAS PubMed.
- A. Maleki, J. He, S. Bochani, V. Nosrati, M. A. Shahbazi and B. Guo, ACS Nano, 2021, 15, 18895–18930 CrossRef CAS PubMed.
- S. Cascone and G. Lamberti, Int. J. Pharm., 2020, 573, 118803 CrossRef CAS PubMed.
- J. E. Cun, X. Fan, Q. Pan, W. Gao, K. Luo, B. He and Y. Pu, Adv. Colloid Interface Sci., 2022, 305, 102686 CrossRef CAS PubMed.
- Y. Dong, W. U. Hassan, R. Kennedy, U. Greiser, A. Pandit, Y. Garcia and W. Wang, Acta Biomater., 2014, 10, 2076–2085 CrossRef CAS PubMed.
- E. Zhao, H. Liu, Y. Jia, T. Xiao, J. Li, G. Zhou, J. Wang, X. Zhou, X. J. Liang, J. Zhang and Z. Li, Acta Biomater., 2022, 140, 302–313 CrossRef CAS PubMed.
- Y. Guo, X. An and Z. Fan, J. Mech. Behav. Biomed. Mater., 2021, 118, 104452 CrossRef CAS PubMed.
- H. Cheng, Z. Shi, K. Yue, X. Huang, Y. Xu, C. Gao, Z. Yao, Y. S. Zhang and J. Wang, Acta Biomater., 2021, 124, 219–232 CrossRef CAS PubMed.
- B. Yang, J. Song, Y. Jiang, M. Li, J. Wei, J. Qin, W. Peng, F. L. Lasaosa, Y. He, H. Mao, J. Yang and Z. Gu, ACS Appl. Mater. Interfaces, 2020, 12, 57782–57797 CrossRef CAS PubMed.
- X. Li, J. Ding, Z. Zhang, M. Yang, J. Yu, J. Wang, F. Chang and X. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 5148–5159 CrossRef CAS PubMed.
- M. Patel, H. J. Moon, Y. Ko du and B. Jeong, ACS Appl. Mater. Interfaces, 2016, 8, 5160–5169 CrossRef CAS PubMed.
- M. Boffito, P. Sirianni, A. M. Di Rienzo and V. Chiono, J. Biomed. Mater. Res., Part A, 2015, 103, 1276–1290 CrossRef PubMed.
- L. Chen, T. Ci, L. Yu and J. Ding, Macromolecules, 2015, 48, 3662–3671 CrossRef CAS.
- X. Zhang, M. Qin, M. Xu, F. Miao, C. Merzougui, X. Zhang, Y. Wei, W. Chen and D. Huang, Eur. Polym. J., 2021, 146, 110268 CrossRef CAS.
- C. Liu, H. Zeng, Z. Chen, Z. Ge, B. Wang, B. Liu and Z. Fan, Int. J. Biol. Macromol., 2022, 202, 418–430 CrossRef CAS PubMed.
- Y. Yang, Y. Liang, J. Chen, X. Duan and B. Guo, Bioact. Mater., 2022, 8, 341–354 CrossRef CAS PubMed.
- X. Zhang, Z. Yin, Y. Guo, H. Huang, J. Zhou, L. Wang, J. Bai and Z. Fan, New J. Chem., 2020, 44, 20776–20784 RSC.
- L. Mao, L. Wang, M. Zhang, M. W. Ullah, L. Liu, W. Zhao, Y. Li, A. A. Q. Ahmed, H. Cheng, Z. Shi and G. Yang, Adv. Healthcare Mater., 2021, 10, e2100402 CrossRef PubMed.
- Y. Dong, H. Zhuang, Y. Hao, L. Zhang, Q. Yang, Y. Liu, C. Qi and S. Wang, Int. J. Nanomed., 2020, 15, 1939–1950 CrossRef CAS PubMed.
- M. Zakerikhoob, S. Abbasi, G. Yousefi, M. Mokhtari and M. S. Noorbakhsh, Carbohydr. Polym., 2021, 271, 118434 CrossRef CAS PubMed.
- Z. Xu, Y. Liu, R. Ma, J. Chen, J. Qiu, S. Du, C. Li, Z. Wu, X. Yang, Z. Chen and T. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 14059–14071 CrossRef CAS PubMed.
- W. Cheng, X. Zeng, H. Chen, Z. Li, W. Zeng, L. Mei and Y. Zhao, ACS Nano, 2019, 13, 8537–8565 CrossRef CAS PubMed.
- L. Garcia-Fernandez, J. Cui, C. Serrano, Z. Shafiq, R. A. Gropeanu, V. S. Miguel, J. I. Ramos, M. Wang, G. K. Auernhammer, S. Ritz, A. A. Golriz, R. Berger, M. Wagner and A. del Campo, Adv. Mater., 2013, 25, 529–533 CrossRef CAS PubMed.
- E. Chalmers, H. Lee, C. Zhu and X. Liu, Chem. Mater., 2019, 32, 234–244 CrossRef.
- H. Huang, D. He, X. Liao, H. Zeng and Z. Fan, Mater. Sci. Eng., C, 2021, 129, 112395 CrossRef CAS PubMed.
- K. Li, D. Li, C. H. Li, P. Zhuang, C. Dai, X. Hu, D. Wang, Y. Liu, X. Mei and V. M. Rotello, Nanoscale, 2021, 13, 6531–6537 RSC.
- L. Yu, H. Hu, L. Chen, X. Bao, Y. Li, L. Chen, G. Xu, X. Ye and J. Ding, Biomater. Sci., 2014, 2, 1100–1109 RSC.
- L. Cheng, H. Wu, J. Li, H. Zhao and L. Wang, Corros. Sci., 2021, 2021, 109064 CrossRef.
- A. Pal, S. Shah and S. Devi, Mater. Chem. Phys., 2009, 114, 530–532 CrossRef CAS.
- Z. Guo, Y. Bai, Z. Zhang, H. Mei, J. Li, Y. Pu, N. Zhao, W. Gao, F. Wu, B. He and J. Xie, J. Mater. Chem. B, 2021, 9, 3874–3884 RSC.
- J. Zhao, L. Cui, X. Wang and C. Deng, Colloid Polym. Sci., 2022, 300, 1075–1086 CrossRef CAS.
- Y. Liu, X. Zhang, T. Wu, B. Liu, J. Yang and W. Liu, Nano Today, 2021, 41, 101306 CrossRef CAS.
- M. Li, Y. Liang, J. He, H. Zhang and B. Guo, Chem. Mater., 2020, 32, 9937–9953 CrossRef CAS.
- X. Li, T. Wang, J. Liu, Y. Liu, J. Zhang, J. Lin, Z. Zhao and D. Chen, Arabian J. Chem., 2020, 13, 184–192 CrossRef CAS.
- C. Zhu, S. Han, X. Zeng, C. Zhu, Y. Pu and Y. Sun, J. Nanobiotechnol., 2022, 20, 221 CrossRef CAS PubMed.
- T. Wang, Y. Zheng, Y. Shi and L. Zhao, Drug Delivery Transl. Res., 2019, 9, 227–239 CrossRef CAS PubMed.
- Y. Zhuang, X. Yang, Y. Li, Y. Chen, X. Peng, L. Yu and J. Ding, ACS Appl. Mater. Interfaces, 2019, 11, 29604–29618 CrossRef CAS PubMed.
- Y. Chen, J. Luan, W. Shen, K. Lei, L. Yu and J. Ding, ACS Appl. Mater. Interfaces, 2016, 8, 30703–30713 CrossRef CAS PubMed.
- H. Zhang, X. Sun, J. Wang, Y. Zhang, M. Dong, T. Bu, L. Li, Y. Liu and L. Wang, Adv. Funct. Mater., 2021, 31, 2100093 CrossRef CAS.
- Z. Jia, J. Gong, Y. Zeng, J. Ran, J. Liu, K. Wang, C. Xie, X. Lu and J. Wang, Adv. Funct. Mater., 2021, 31, 2010461 CrossRef CAS.
- L. Yu, W. Sheng, D. Yang and J. Ding, Macromol. Res., 2012, 21, 207–215 CrossRef.
- H. Luo, C. Gu, W. Zheng, F. Dai, X. Wang and Z. Zheng, RSC Adv., 2015, 5, 13470–13477 RSC.
- X. Xiao, H. Cai, Q. Huang, B. Wang, X. Wang, Q. Luo, Y. Li, H. Zhang, Q. Gong, X. Ma, Z. Gu and K. Luo, Bioact. Mater., 2023, 19, 538–549 CrossRef CAS PubMed.
- B. Poinard, S. Kamaluddin, A. Q. Q. Tan, K. G. Neoh and J. C. Y. Kah, ACS Appl. Mater. Interfaces, 2019, 11, 4777–4789 CrossRef CAS PubMed.
- P. L. Lai, D. W. Hong, K. L. Ku, Z. T. Lai and I. M. Chu, Nanomedicine, 2014, 10, 553–560 CrossRef CAS PubMed.
- S. Cui, L. Yu and J. Ding, Macromolecules, 2018, 51, 6405–6420 CrossRef CAS.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- X. Jing, H. Y. Mi, Y. J. Lin, E. Enriquez, X. F. Peng and L. S. Turng, ACS Appl. Mater. Interfaces, 2018, 10, 20897–20909 CrossRef CAS PubMed.
- X. Cao, H. Liu, X. Yang, J. Tian, B. Luo and M. Liu, Compos. Sci. Technol., 2020, 191, 108071 CrossRef CAS.
- D. P. Huynh, W. S. Shim, J. H. Kim and D. S. Lee, Polymer, 2006, 47, 7918–7926 CrossRef CAS.
- D. L. Taylor and M. In Het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS PubMed.
- L. Han, M. Liu, B. Yan, YueS. Li, Ji Lan, L. Shi and R. Ran, Mater. Sci. Eng., C, 2020, 110567 CrossRef CAS PubMed.
- T. Chen, Y. Chen, H. U. Rehman, Z. Chen, Z. Yang, M. Wang, H. Li and H. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 33523–33531 CrossRef CAS PubMed.
- Z. Deng, W. Wang, X. Xu, Y. Nie, Y. Liu, O. E. C. Gould, N. Ma and A. Lendlein, ACS Appl. Mater. Interfaces, 2021, 13, 10748–10759 CrossRef CAS PubMed.
- Z. Li, H. Cai, Z. Li, L. Ren, X. Ma, H. Zhu, Q. Gong, H. Zhang, Z. Gu and K. Luo, Bioact. Mater., 2023, 21, 299–312 CAS.
- Z. Zhang, Y. Pan, J. E. Cun, J. Li, Z. Guo, Q. Pan, W. Gao, Y. Pu, K. Luo and B. He, Acta Biomater., 2022, 151, 480–490 CrossRef CAS PubMed.
- S. Zhang, Y. Tang and B. Vlahovic, Int. J. Nanosci., 2017, 16, 1750008 CrossRef.
- Y. Li, L. Yang, Y. Hou, Z. Zhang, M. Chen, M. Wang, J. Liu, J. Wang, Z. Zhao, C. Xie and X. Lu, Bioact. Mater., 2022, 18, 213–227 CrossRef CAS PubMed.
- L. Pang, P. Tian, X. Cui, X. Wu, X. Zhao, H. Wang, D. Wang and H. Pan, ACS Appl. Mater. Interfaces, 2021, 13, 29363–29379 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr04908b |
| This journal is © The Royal Society of Chemistry 2023 |