
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Role of carboxylates in the phase determination of metal sulfide nanoparticles†
Andrey A.
Shults
 ab,
Guanyu
Lu
bc,
Joshua D.
Caldwell
abcd and
Janet E.
Macdonald
*ab
ab,
Guanyu
Lu
bc,
Joshua D.
Caldwell
abcd and
Janet E.
Macdonald
*ab
aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235, USA. E-mail: janet.macdonald@vanderbilt.edu
bVanderbilt Institute for Nanoscale Science and Engineering, Nashville, TN 37235, USA
cDepartment of Mechanical Engineering, Vanderbilt University, Nashville, TN 37235, USA
dSensorium Technological Laboratories, Nashville, TN 37205, USA
First published on 28th July 2023
Abstract
Techniques are well established for the control of nanoparticle shape and size in colloidal synthesis, but very little is understood about precursor interactions and their effects on the resultant crystalline phase. Here we show that oleate, a surface stabilizing ligand that is ubiquitous in nanocrystal synthesis, plays a large role in the mechanism of phase selection of various metal sulfide nanoparticles when thiourea is used as the sulfur source. Gas and solid-phase FTIR, 13C, and 1H NMR studies revealed that oleate and thiourea interact to produce oleamide which promotes the isomeric shift of thiourea into ammonium thiocyanate, a less reactive sulfur reagent. Because of these sulfur sequestering reactions, sulfur deficient and metastable nanoparticles are produced, a trend seen across four different metals: copper, iron, nickel, and cobalt. At low carboxylate concentrations, powder XRD indicated that the following phases formed: covellite (CuS); vaesite (NiS2); smythite (FeS1.3), greigite (FeS1.3), marcasite (FeS2) and pyrite (FeS2); and cattierite (CoS2). At high sodium oleate concentration, these phases formed: digenite (CuS0.55), nickel sulfide (NiS), pyrrhotite (FeS1.1), and jaipurite (CoS).
New conceptsWe demonstrated that a common ligand (oleate) influences the crystalline phase of colloidal metal sulfides through molecular side reactions with the precursors. We transcend phenomenological observations, and, using techniques more commonly applied to organic chemistry (1H and 13C NMR, IR), we identify the side reactions that are occurring. Such a deep dive is very uncommon in the synthetic colloidal chemistry literature. What also makes it important is that we show the phenomenon does not just occur for the synthesis of one metal sulfide, but rather Fe, Co, Ni and Cu sulfides showing the universality. The work provides direct insight to allow rational and targeted synthesis of metal sulfides. While oleate is commonly used to influence size and shape nanocrystals, it can, and should be considered for its effect on phase. The work also shows how to practically use 1H NMR, 13C NMR, and both gas phase and solid phase IR to illuminate molecular chemistries that are often occluded in nanocrystal synthesis. |
1. Introduction
For many applications of solid state and nanocrystalline materials, the identity and purity of the crystalline phase is essential to function. The phase space is highly complex. For example, in the geological record, there are nine iron sulfides, four cobalt sulfides, seven nickel sulfides, and eight copper sulfides, of differing stoichiometries and polytypes each with their own physical properties.The current and potential applications of the metal sulfides are highly varied even within each metal class. As examples, while pyrite (FeS2) is a paramagnetic iron sulfide and is useful in various environmental oxidative processes,1 greigite (FeS1.3) shows superparamagnetic behavior at small particle sizes which is potentially useful for treatment of cancer through magnetically induced hyperthermia.2 In the copper sulfide family, many of the copper sulfides including digenite (CuS0.55) and covellite (CuS) possess localized surface plasmon resonances (LSPR) in the near IR and so can be used as plasmonic semiconductors in optoelectronic devices.3 Covellite (CuS) has also been used as a catalytic glucose oxidizer for glucose detection.4 In the cobalt sulfide family, cattierite (CoS2) has been used in lithium–sulfur battery cathodes to accelerate the redox reactions of polysulfides5 while jaipurite (Co9S8) has been used as a supercapacitor.6 In the nickel sulfide family, vaesite (NiS2) is an electrocatalyst for the hydrogen evolution reaction (HER)7,8 while nickel sulfide (NiS) can be used as a supercapacitor.9
Rationally synthesizing one phase over the other can be challenging when dealing with transition metal sulfides because of the multiple phases of varying stoichiometry and symmetry. There are many one-off reports of colloidal syntheses of these metal sulfides, but the reasons behind phase selection under certain conditions remain occluded. Common sulfur precursors include thiourea,10,11 elemental sulfur,12 sodium sulfide,13 thioacetamides,14 carbon disulfide,15 oleylamine–sulfur(thioamides),16 dithiocarbamates,17,18 thiobiurets,19 thiols,20 and thioesters,21 among many others.22
Even with such a vast library of sulfur reagents, studies of their decomposition pathways rarely elucidate complete mechanisms. Rhodes et al. were able to select for specific iron sulfide phases based on the strength of the C–S bonds of the chosen thiols, thioethers, and disulfides. Stronger C–S bonds yielded sulfur poor pyrrhotite (FeS) while weaker C–S bonds yielded sulfur rich pyrite (FeS2). While the general trend was straightforward, it was found that the unique decomposition mechanism of diallyl disulfide, facilitated by the oleylamine solvent, was essential to the formation of pyrite (FeS2).21 The Hogarth group has also contributed to our understanding of precursor decomposition pathways and their effect on the synthesized phase.23,24 Most notably, they were able to achieve four various phases of the nickel sulfide family: α-NiS, β-NiS, Ni3S4 and NiS2. Starting out with a series of bis(dithiocarbamate) complexes, [Ni(S2CNR2)2], Hollingsworth et al. and Roffey et al. were able to link decomposition pathways of their precursor to the synthesized phase through temperature and concentration studies. Very few other decomposition routes have been studied.25,26
Thiourea has become one of the more popular sulfur reagents because it is inexpensive, solid at room temperature, has a low vapor pressure compared to other sulfur reagents, and readily reacts at temperatures as low as 150 °C with transition metal cations. In addition, Hendricks et al. showed that changing the N-substitution on a library of thioureas can vary the conversion rate over more than five orders of magnitude, which impacted the nucleation rate of lead sulfide (PbS),10 zinc sulfide (ZnS),27 and cadmium sulfide (CdS) nanoparticles.28
In many of the aforementioned studies, metal carboxylates, whether added directly or formed in situ, are common metal precursors as the carboxylate ligands solubilize the metal ion in a high boiling organic solvent and act as surface stabilizing ligands for the product nanocrystals.29,30 Carboxylates can affect particle nucleation and growth in contradictory ways depending on the synthetic environment. Demortièr et al. showed that increasing the ratio of oleic acid (ligand) to iron–oleate complex (metal precursor) increased the size of the synthesized iron oxide nanoparticles when using di-n-octyl ether as the solvent.31 Baaziz et al. saw the same pattern when using these conditions, but noted that changing the solvent also has an effect on the size of nanoparticles.32 In contrast to di-n-octyl ether, using octadecane as a solvent resulted in a decrease in particle size with an increase in oleic acid concentration. The unique solvent environment of each synthesis likely influences the decomposition of the metal precursor complex and the effect of oleic acid. Solvent and precursor choice are one of the many factors that may change the way the metal precursor decomposes influencing the time of the nucleation process.
Since carboxylate influences nucleation and growth, it comes as no surprise that carboxylate ions are also known to affect phase formation. The most well-known example is that carboxylates such as oleate or stearate will stabilize zinc blende over the wurtzite polytype in CdSe quantum dots.33–35 This particular metal selenide has only two polytypes. In contrast, the phase diagrams of the mid transition metal sulfides are far more diverse with several polytypes and differing stoichiometries. One may hypothesize that oleate, as a strong ligand for first-row transition metal ions, may slow the release of metal precursors for particle formation and yield metal-poor phases. Yet at the high temperatures of nanocrystal synthesis, nucleophilic carboxylate could reasonably be expected to react with some sulfur reagents, especially in the presence of Lewis acidic metal centers. It remains a mystery how oleate will influence phase in transition metal sulfide nanocrystal formation.
Here we carefully examine the role of carboxylate in synthesis of iron, cobalt, nickel, and copper sulfides with thiourea as the sulfur reagent. High concentrations of carboxylates cause the formation of sulfur poor phases indicating that carboxylates parasitically react with thiourea. We will provide evidence that under low carboxylate concentration, the active sulfur source is thiourea, whereas at high carboxylate concentration, the active sulfur source changes to a mixture of carbon disulfide and thiocyanate.
2. Experimental
All nanoparticle synthesis reactions were performed in oven-dried three-neck round-bottom flasks using standard Schlenk techniques under argon atmosphere. A thermocouple was used to monitor the internal temperature of the reaction.2.1 Materials
Copper(II) chloride anhydrous (CuCl2, >98.0%) was purchased from Alfa Aesar; iron(III) stearate (Fe(stearate)3), cobalt(II) stearate (Co(stearate)2, >97.0%), and sodium(I) oleate (Na(oleate), >97.0%) were purchased from TCI Chemicals; nickel(II) stearate (Ni(stearate)2) was purchased from AmBeed; octadecene (CH2CH(CH2)5CH3, 90.0%, technical grade), thiourea (SC(NH2)2, ≥99.0%), ammonium thiocyanate (NH4SCN, ≥98.0), and deuterated dimethyl sulfoxide ((CD3)2SO, >99.0%) were purchased from Sigma-Aldrich; and oleic acid (CH3(CH2)7CHCH(CH2)7COOH) was purchased from EDM™.2.2 Synthesis of copper(II) oleate precursor
The synthesis is adapted from Tappan et al.29 A mixture of sodium oleate (9.85 mmol) and anhydrous copper(II) chloride (4.93 mmol) was added into a 100 mL three-neck round-bottom flask. A solvent mixture of ethanol (10 mL), deionized water (8 mL), and hexanes (17 mL) was then added into the flask. The solution was heated to 70 °C for 25 min after which an additional portion of hexanes (10 mL) was added. The solution was reheated to 70 °C and kept at that temperature for 4 h. After the reaction was cooled to room temperature, the solution was washed three times with deionized water in a separatory funnel. After the separation process, the product was vacuumed to form a dry and teal powder.2.3 Synthesis of transition metal sulfides nanoparticles
A solution of metal precursor (0.50 mmol) in octadecene (ODE) (10 mL) was added to a 25 mL three neck round-bottom flask (Fig. S16, ESI†). Thiourea (3.0 mmol) and ODE (5 mL) were added to an addition funnel, connected to round-bottom flask. The apparatus was placed under vacuum while the three-neck flask was heated at 100 °C for 30 min. After refilling with nitrogen, the three-neck flask was heated 170 °C for 1 h. The flask was then heated to either 210 °C (for nickel and cobalt) or 220 °C (for copper and iron). The addition funnel was warmed with a heat gun to approximately 170 °C (∼5 min) to allow the thiourea to dissolve in the ODE, and then the contents were added swiftly to the round-bottom flask (Scheme 1). The solution was continuously stirred at 1100 rpm and kept in the reaction vessel for 60 min, with aliquots taken at 5, 30, and 60 min. Nanoparticle products were isolated by precipitation with ethanol (10–25 mL), centrifugation (8000–8700 rpm), and resuspension with chloroform (3–10 mL) three times. Higher volumes of washing solvents were used with high-oleate reactions.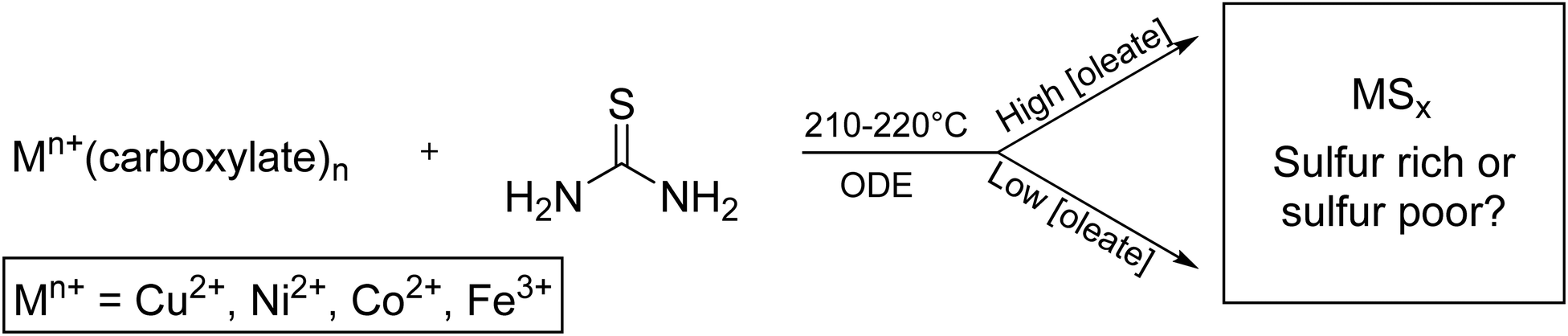 | ||
| Scheme 1 General synthesis of transition metal (Mn+) sulfide nanoparticles to give either sulfur rich or sulfur poor phases. | ||
2.4 Synthesis of transition metal sulfides nanoparticles (NMR scale)
In a nitrogen filled glovebox, an NMR tube was loaded with metal carboxylate (0.0798 mmol) and thiourea (0.136 mmol) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.7 ratio, and additional sodium oleate to obtain final metal:carboxylate ratios of 1:2, 1:3, and 1:4. The tube was capped with a septum and removed from the glove box. A nitrogen-filled balloon with a needle was attached to the NMR tube to allow for safe gas expansion during heating (Fig. S17, ESI†). The tube was held in an oil bath for either 10 or 60 min at 150, 170, 200, or 220 °C. After cooling, DMSO-d6 (0.6 mL) was added for NMR analysis. In DMSO-d6, the organics dissolved, but the particles remained at the bottom of the NMR tube. In control reactions, the same procedure was employed but with a combination of oleic acid (0.32 mmol) and thiourea (0.32 mmol).
:1.7 ratio, and additional sodium oleate to obtain final metal:carboxylate ratios of 1:2, 1:3, and 1:4. The tube was capped with a septum and removed from the glove box. A nitrogen-filled balloon with a needle was attached to the NMR tube to allow for safe gas expansion during heating (Fig. S17, ESI†). The tube was held in an oil bath for either 10 or 60 min at 150, 170, 200, or 220 °C. After cooling, DMSO-d6 (0.6 mL) was added for NMR analysis. In DMSO-d6, the organics dissolved, but the particles remained at the bottom of the NMR tube. In control reactions, the same procedure was employed but with a combination of oleic acid (0.32 mmol) and thiourea (0.32 mmol).
2.5 Preparation of samples for gas FTIR analysis
A 25 mL three-neck round-bottom flask was loaded with (a) thiourea (13.1 mmol) and ODE (10 mL) (b) Nickel stearate (0.5 mmol), sodium oleate (3.0 mmol), and ODE (10 mL), (c) thiourea (13 mmol), oleic acid (13 mmol), and ODE (10 mL) (d) nickel stearate (0.5 mmol), thiourea (3 mmol), and ODE (15 mL) (e) nickel stearate (0.5 mmol), thiourea (3 mmol), sodium oleate (3 mol), and ODE (15 mL). Two gas adapters were attached to the round-bottom flask to allow for the flow of nitrogen gas into and gasses out of the flask. The flask was then placed under vacuum at 100 °C for 30 min before refilling with N2. The gas IR cell outlet was attached to a bubbler and was flushed with nitrogen to eliminate any atmospheric gasses. In IR cell inlet was attached to the flask through a gas adapter. The whole system was flushed one more time with nitrogen until the spectrum reached a steady state, then the nitrogen flow rate reduced to ∼2 bubbles per s. Then, the flask was heated, and the IR spectra of the outflowing gasses were collected approximately every 10 °C from 100 °C to 220 °C.2.6 Characterization
Nanoparticles were characterized with powder X-ray diffraction (pXRD) using a Rigaku Smart Lab Diffractometer with Cu Kα X-ray (λ = 0.154 nm) radiation source set to 40 kV and 44 mA. Before analyzing the sample, it was dissolved in chloroform and drop-cast on a low background pXRD plate. The patterns were matched to the corresponding phase using ICSD database.For decomposition product analysis, nuclear magnetic resonance (NMR) was used. 1H NMR and 13C NMR were collected in a Bruker Advanced HD 400 MHz Spectrometer.
Gas-phase transmission measurements of gas products from the studied chemical reactions were performed using a Bruker Vertex 70v Fourier transform infrared (FTIR) spectrometer along with a Pike short-path gas cell with KRS5 windows. The IR source was globar in the FTIR bench, and detector was liquid-nitrogen-cooled HgCdTe (MCT) detector. The gas cell was placed in the sample compartment of the FTIR bench, which was under a constant N2 purge. The remainder of the FTIR bench was also under a constant N2 purge. The Pike short-path gas cell is equipped with four ports: two external ports for N2 purging of the areas outside of the gas cell windows and two internal (inlet and outlet) ports for supplying gas to the gas cell (Fig. S18, ESI†). The gas cell was initially purged with N2 using inlet and outlet ports, and a background transmission measurement was recorded under N2 purge.
Solid-phase transmission measurements of solid products from the studied chemical reactions were performed using an attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) measurements of solid products were performed using ThermoFisher Scientific Nicolet iS5 FTIR equipped with an iD7 ATR accessory. One drop of the sample was drop-cast on the monolithic diamond crystal window. After complete drying the solid sample was secured with a sample presser. All samples were collected at room temperature and normalized to a background scan taken before collecting experimental data.
3. Results and discussion
Long chain carboxylic acids and carboxylate ions, especially oleic acid and oleate are ubiquitous in nanocrystal synthesis. For many years now, they have been a go-to surface stabilizing ligand, and many studies have used oleate to control both shape and size of nanocrystals, especially metal chalcogenides. This is especially important due to the high solubility of metal carboxylates in oily and high-boiling solvents like octadecene. Traditionally, a metal reagent such as a metal halide or a metal oxide is first heated with an oleate or a stearate to form the metal carboxylate complex that is later used as a precursor for a nanoparticle synthesis.29,36,37The relationship between size and oleate concentration can be complicated. Increasing oleate concentrations allows for lowering of surface energy and stabilization of smaller particles. However, in some cases, oleate is known to bind precursors, slowing reaction rates, causing fewer nuclei to form and resulting in fewer, larger particles. What has not been investigated well is how oleic acid might influence crystalline phase in systems where multiple phases of different stoichiometries are possible. Again, oleate may be affecting the thermodynamics of the growing nuclei and it might influence the precursor kinetics, both of which may impact phase composition.
The role of oleate is well-studied in the canon of CdS and CdSe quantum dot synthesis.33 In addition to being an X-type ligand to surface cations, Cd(oleate)2 acts as a Z-type ligand, terminating surface anions. Oleate has been shown to influence polytypism in CdSe. As an X-type ligand, it especially stabilizes the zinc blende phases, because these present eight charged [111] surfaces, whereas hexagonal wurtzite phases only present two charged [001] facets. How might oleate influence phase when both polytypism and phases of multiple stoichiometries are possible? Will oleate only affect particle size, or crystalline phase as well? Will behaviors transcend across several metals, each with their own unique chemistries and d-electron counts?
To first examine if oleate influences phase in the synthesis of transition metal sulfides, metal sulfides of Fe, Co, Ni, and Cu were synthesized according to a modified one-pot hot-injection method reported by Joo et al.38 In this synthesis, a solution of thiourea solubilized in ODE was injected into a solution of metal carboxylate via an addition funnel at 210–220 °C in ODE for 1 h. Sodium oleate was included to achieve overall oleate:metal ratios of 1:2, 3, 4, 8. Our initial hypothesis was that sodium oleate would shift the equilibrium of oleate/stearate dissociation from the metal complex and cause the formation of metal-poor nanocrystalline phases; however, to our amazement, the very opposite trend was observed.
Characterization with pXRD showed that as the amount of additional sodium oleate was increased, the product became increasingly sulfur poor (Fig. 1 and Table S1, ESI†). For copper, at low concentrations of oleate, covellite (CuS) formed, and as the concentration of oleate was increased, there resulted in an increasing proportion of digenite (Cu1.8S). For nickel and cobalt, the resultant phase changed from MS2 to MS. For iron, low concentrations of oleate gave mostly the two polymorphs of FeS2 (pyrite and marcasite) and the spinel polymorphs greigite and smythite (FeS1.3). High concentrations yielded the low sulfur content pyrrhotite (FeS1.1).
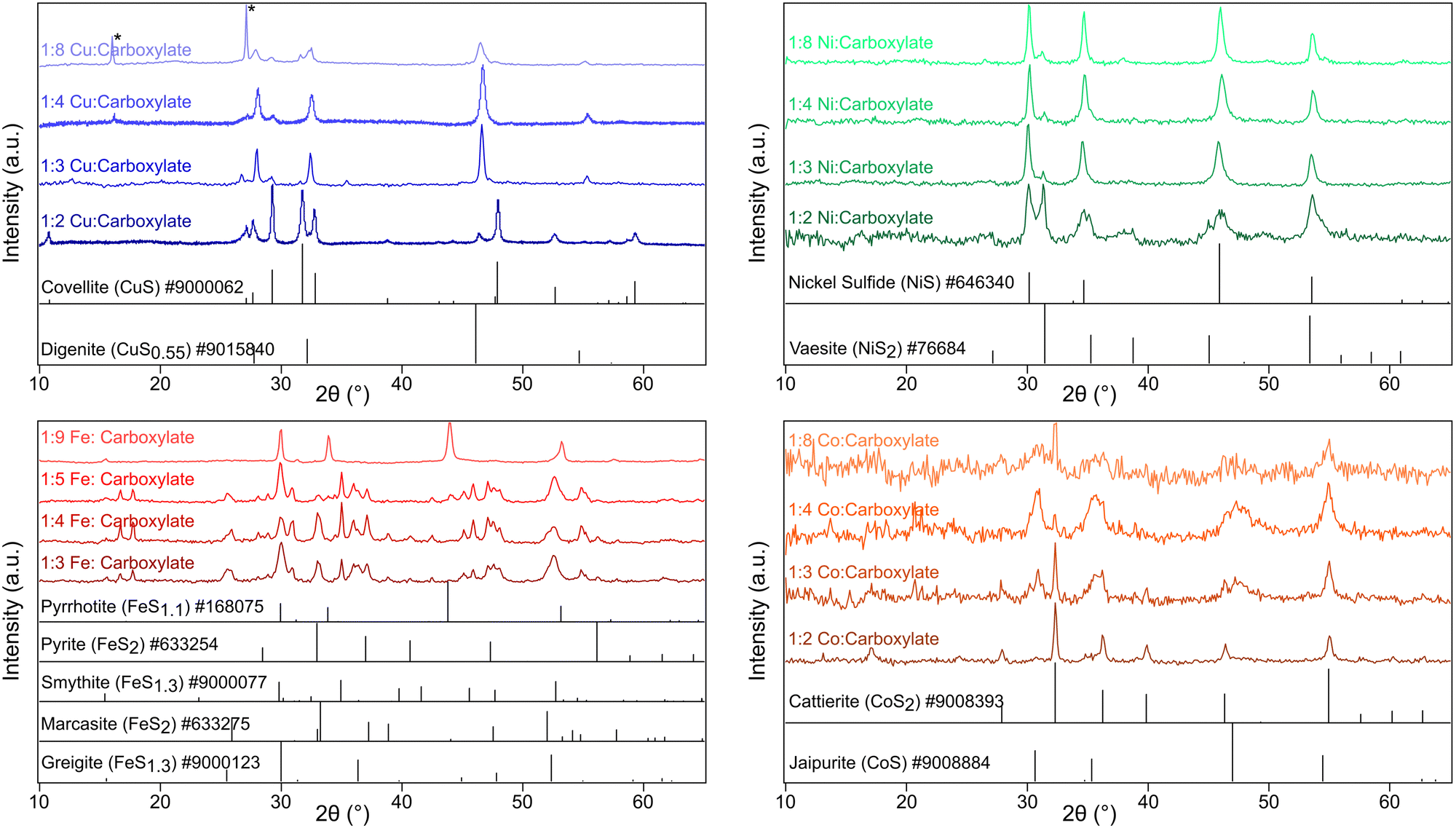 | ||
| Fig. 1 pXRD of transition metal sulfides: copper sulfide (top left), nickel sulfide (top right), iron sulfide (bottom left), and cobalt sulfide (bottom right). Reflections from copper(I) thiocyanate denoted by * (ICSD #24372). | ||
All of the families of metal sulfides studied contain structures with approximate cubic close packed (CCP) or hexagonal close packed (HCP) staking of S2− or S22− anions. There was no visible trend in CCP vs. HCP structures with changing oleate concentration. For example, the copper series saw a shift from hexagonal covellite (CuS) to cubic digenite (Cu1.8S) while nickel series saw the opposite trend from cubic vaesite (NiS2) to hexagonal NiS (Table S1, ESI†) with increasing oleate concentration.
The decrease in sulfur content with increased oleate concentration disproved the initial hypothesis that oleate would bind the metal centers, decreasing metal reactivity. Therefore, it was instead hypothesized that the oleate was parasitically interacting with the sulfur precursor, thiourea.
Thermal decomposition pathways of thiourea by itself and in the presence of other chemicals in both air and inert environments have been widely studied.39–41 Thiourea undergoes two main thermal processes when heated to temperatures higher than 170 °C.39 The first process, which can happen between 171.2 and 187.5 °C is the isomerization of thiourea into ammonium thiocyanate (Scheme 2). In a fully equilibrated melt, the thiocyanate concentration triples that of thiourea.42 Therefore, it was important in the experimental design that thiourea was only briefly and consistently heated before addition to the heated metal solution. Inconsistent preheating may be a source of irreproducibility in literature nanocrystal preparations.
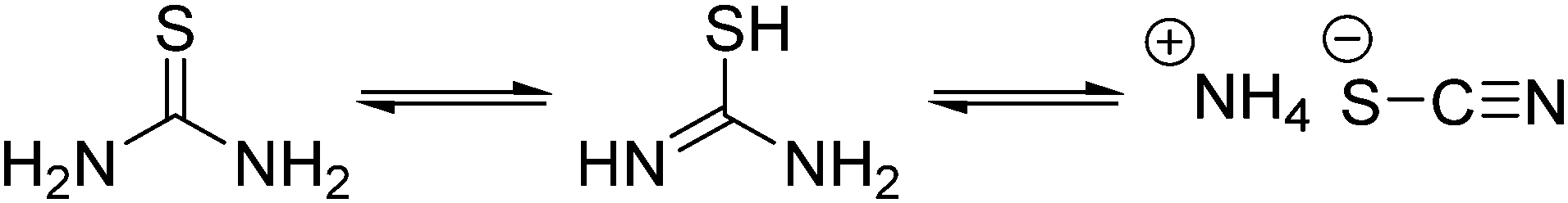 | ||
| Scheme 2 Isomerization of thiourea to thiocyanic acid at temperatures over 170 °C. | ||
The second process in the decomposition of thiourea occurs between 187.5 to 246.2 °C and results in a loss of 80% of the total weight of thiourea. The gaseous decomposition products of this step include carbon disulfide (CS2) and ammonia (NH3). Other studies suggest that at temperatures above 500 °C, cyanamide (H2NCN) and hydrogen cyanide (HCN) are also products.40 Cyanamide has also been detected as one of the decomposition products starting at 200 °C, but likely trimerizes into melamine, so it is never seen in the gas phase at lower temperatures.43
Nuclear magnetic resonance (NMR) was used to analyze the decomposition products of the copper-based reactions in hope of identifying a parasitic side reaction. Copper was chosen for deeper study because it often produces diamagnetic products, which would be easier to study by NMR. (Fig. 2). Reactions were performed on NMR scale (without the solvent ODE) at varying temperature (150, 170, 200, and 220 °C), metal: oleate concentration (1:2, 1:3, 1:4), and time (10 and 60 min). Thiourea was added at a ratio of 1.7:1 thiourea:metal for the metal studies and at a ratio of 1:1 thiourea:oleic acid for the NMR studies without metal present. Immediately after cooling, DMSO-d6 was added to dissolve the product mixture for 1H and 13C NMR (Fig. S1–S5, ESI†).
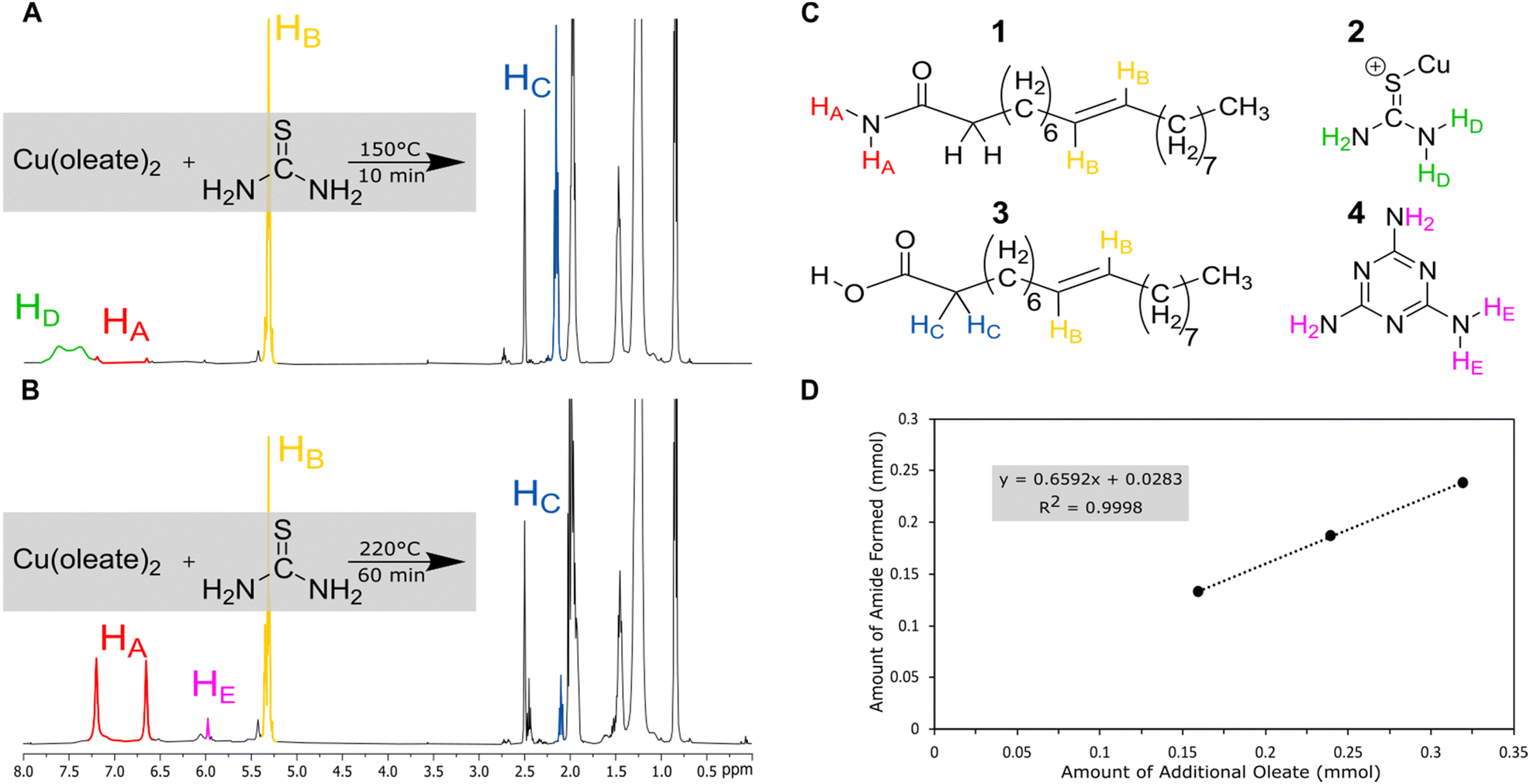 | ||
| Fig. 2 NMR studies of copper series: (A) 1H NMR taken on a 400 MHz instrument of thiourea and copper oleate heated to 220 °C for one hour (DMSO-d6 was injected post-cooling); (B) 1H NMR taken on a 400 MHz instrument of thiourea and copper oleate heated to 150 °C for ten minutes (DMSO-d6 was injected post-cooling); (C) structures of oleamide (1), metal-coupled thiourea (2), oleic acid (3), and melamine (4); (D) relationship between amount of sodium oleate and amount of oleamide produced (see SI for calculation method). | ||
At low temperatures and times (150 °C), the reaction of copper oleate with thiourea showed only minor changes from the starting material. The broad signal at δ = 7.25–7.75 ppm can be ascribed to the protons of thiourea complexed to the copper oleate (2) since it is shifted down-field from a thiourea control (6.75–7.25 ppm, Fig. S7, ESI†). As temperatures and times increased, the thiourea signal decreased and was replaced with a new product with a singlet at 6.00 ppm. This was assigned to melamine (4), which is a known product of thiourea decomposition.39,43 In addition, two new sharp singlets at 6.66 ppm and 7.21 ppm were observed. These protons were identified as resulting from the terminal NH2 protons of a new product, oleamide (1). In control experiments without the presence of the copper, this reaction required temperatures of 220 °C for 60 min to go to completion (Fig. S3, ESI†), whereas with the copper present, the reaction started at 150 °C at 10 min and could go nearly to completion at 200 °C in 10 min (Fig. S1, ESI†). Therefore, the Lewis acidic copper promotes the transformation of oleate to oleamide. Furthermore, the amount of amide that formed was linearly proportional to the amount of oleate added (Fig. 2(D)). To check that the formation of oleamide is not exclusive to copper only, similar temperature and time studies were performed for nickel (Fig. S6, ESI†). The formation of oleamide was apparent for the sample heated to 150 °C for 60 minutes, although, most of the NMR signal was disrupted by magnetic nickel sulfide nanoparticles.
The high temperature reaction of thiourea and carboxylates to give amides is known.44 It is proposed that thiourea undergoes a transformation into ammonium thiocyanate around 170 °C and reacts with the carboxylic acid group of sodium oleate to form oleamide (Scheme 3).44 Thiocyanate signals are also present in the NMR spectra and occur at around 7.0 ppm for 1H NMR (NH4+) and 130.7 ppm for 13C NMR (SCN−)45 (Fig. S1, S2 and S11, ESI†). In 1H NMR, the peak shifts position between 6.9 and 7 ppm depending on the sample, which we suggest is from solvation effects and differing degrees of coordination to carboxylate. Despite the identification of melamine and amide byproducts, that leaves us with the question: where is all the sulfur going, if not to the synthesis of the nanoparticles?
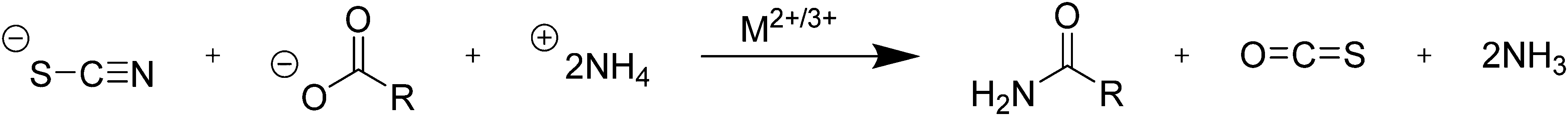 | ||
| Scheme 3 Reaction of thiocyanic acid with the carboxylate ion to produce an amide and carbonyl sulfide gas. | ||
In their study of amide formation, Mittal et al. suggested that as thiourea reacts with a carboxylate to produce an amide, carbonyl sulfide (OCS) gas, hydrogen sulfide (H2S), and ammonium polysulfides are possible sulfur-based byproducts. It was then hypothesized that in the nanocrystal syntheses sulfur may be escaping in the form of a gaseous sulfide (OCS or H2S) or as a polysulfide, thereby lowering the amount of available sulfur for the nanoparticle synthesis.
The gaseous product of the reactions of thiourea in ODE between 100 and 220 °C were monitored using in situ gas-cell IR spectroscopy (Fig. 3). In all spectra, the water signal between 1300 and 2000 cm−1 appears due to either contamination from air or one of the precursors.
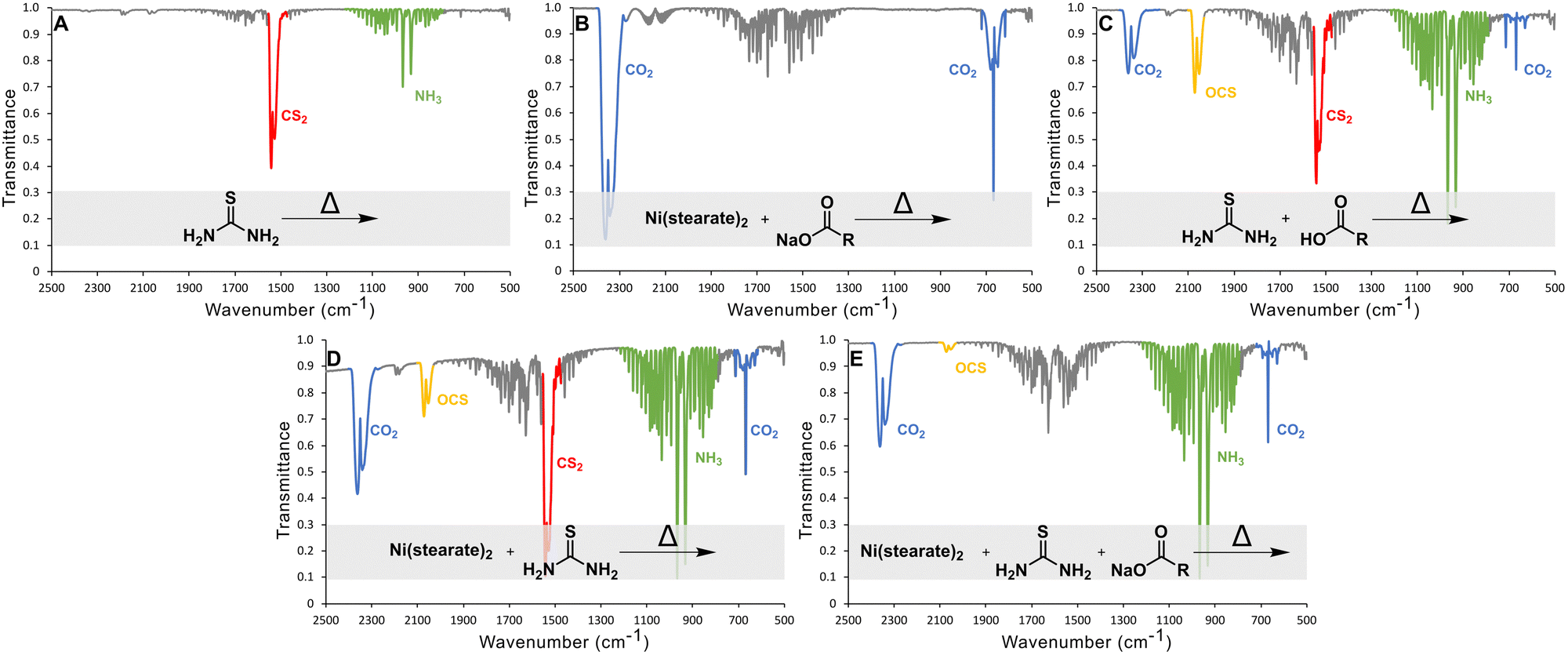 | ||
| Fig. 3 Gas FTIR of thermal decomposition products of (A) thiourea (13.1 mmol) in ODE at 206 °C, (B) nickel stearate (0.5 mmol) and sodium oleate (3 mmol) in ODE at 210 °C, (C) thiourea (13 mmol) and oleic acid (13 mmol) in ODE at 213 °C, (D) nickel stearate (0.5 mmol) and thiourea (3 mmol) in ODE at 212 °C, and (E) nickel stearate (0.5 mmol), thiourea (3 mmol), and sodium oleate (3 mmol) in ODE at 200 °C. | ||
Control studies showed the main gasses produced from thermal decomposition of thiourea (Fig. 3(A)) are carbon disulfide (CS2) and ammonia (NH3) consistent with previous reports.39 No H2S was identified. Nickel stearate heated in the presence of sodium oleate yielded CO2 from decarboxylation (Fig. 3(B)). Thiourea heated in the presence of oleic acid produces substantial amounts OCS as Mittal et al. had predicted.44 CS2 and CO2 arise from direct decomposition of thiourea and carboxylate. Ammonia is also present from the aforementioned thermal decomposition of thiourea. (Fig. 3(C)). OCS is known to disproportionate to CS2 and CO2 based on thermodynamic calculations (Gibbs energy and equilibrium constants) at temperatures above 800 °C; thus it is not a likely path here but may occur in a nanocrystal synthesis if the transformation can be catalyzed by metal ions.46
In nanocrystal syntheses with nickel present, it was found that increasing the oleate concentration did not cause more sulfur-based gases to evolve and instead the opposite was true. When thiourea and nickel oleate were heated together (thiourea:oleate 3:1), NH3, CO2, CS2, and OCS gasses were produced (Fig. 3(D)). However, when additional sodium oleate was added (thiourea:oleate 3:4), it eliminated the evolution of CS2 gas and decreased the amount of OCS gas. (Fig. 3(E)). Therefore, increasing oleate concentration does not yield sulfur poor phases because of a side reaction that produces sulfur-based gaseous species that escape. Moving forward, the alternative explanation should also answer why CS2 does not evolve when high oleate concentrations are used. We propose, therefore, that under high oleate concentrations, CS2 is the active sulfur source in the formation of metal sulfides, rather than thiourea.
Previously, it was mentioned that thiourea isomerizes to [NH4+][−SCN]. At room temperature, the NMR studies showed predominantly thiourea, but heating and the addition of sodium oleate pushes the equilibrium towards the ammonium thiocyanate. The change to thiocyanate is rationalized because of the production of oleamide, and because oleate (pKa ∼ 5) is a stronger base than thiocyanate (pKa ∼ 1.1) and will preferentially coordinate the ammonium in the anhydrous conditions. To test this hypothesis, copper sulfide nanoparticles were synthesized at low and high oleate concentrations employing identical amounts of washing solvents before centrifugation. The supernatant solutions were studied by ATR-FTIR (Fig. 4). In both cases, stretches from the amide byproduct (3353 cm−1 and 3180 cm−1 for N–H and 1658 cm−1 for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches) could be identified along with thiocyanate ion (2060 cm−1). Unreacted carboxylate (CO 1556 cm−1) was present when high oleate concentrations were employed. Most importantly, an increase in sodium oleate concentration in the reaction resulted in a much stronger thiocyanate stretch in the washings. The increase in thiocyanate indicates that the transformation of thiourea to ammonium thiocyanate was promoted by carboxylates.
O stretches) could be identified along with thiocyanate ion (2060 cm−1). Unreacted carboxylate (CO 1556 cm−1) was present when high oleate concentrations were employed. Most importantly, an increase in sodium oleate concentration in the reaction resulted in a much stronger thiocyanate stretch in the washings. The increase in thiocyanate indicates that the transformation of thiourea to ammonium thiocyanate was promoted by carboxylates.
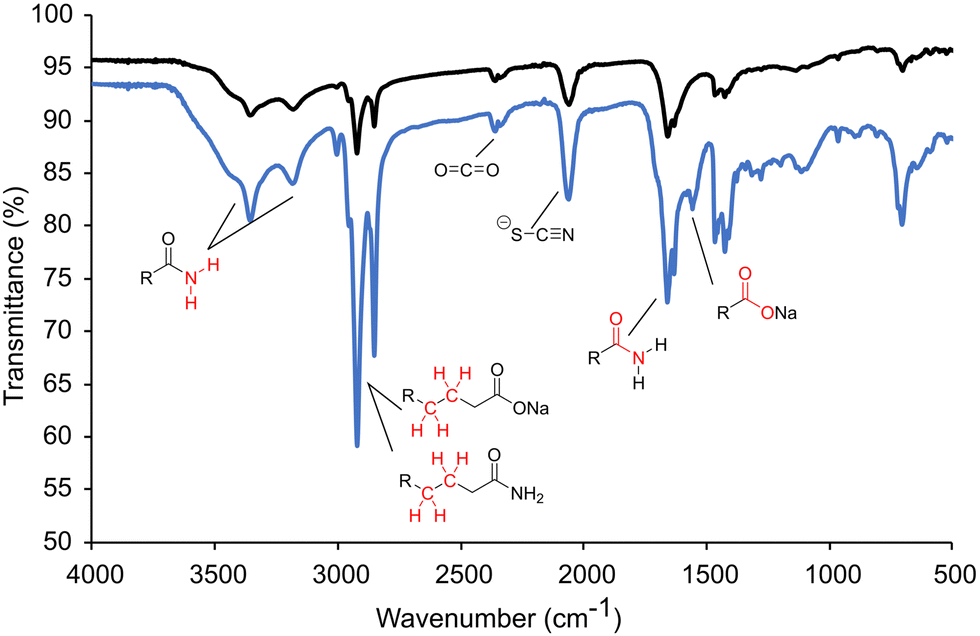 | ||
| Fig. 4 ATR-FTIR of the nanoparticle solution after centrifugation isolated at low oleate (black) and high oleate (blue). | ||
Since oleate forces the transformation of thiourea to thiocyanate, we tested if thiocyanate is an active sulfur precursor for metal sulfide formation. Thiocyanate is known as a sulfur source in nanocrystal reactions. It has been previously reported that when copper thiocyanate (CuSCN) is heated between 180 °C and 280 °C in oleylamine, sulfur poor djurleite (CuS0.52) is formed.47 In our laboratory, under analogous conditions to the above experiments, ammonium thiocyanate was reacted with copper oleate (ammonium thiocyanate: oleate: metal 3:1:0.5) in ODE. The result was sulfur-poor metastable digenite (CuS0.55) nanoparticles (Fig. S13, ESI†), similar to when high oleate conditions are used with thiourea (Fig. 1 and Table S1, ESI†). While it is most likely that thiocyanate is the sulfur source, it is also possible that CS2 is an active sulfur source since CS2 can be released from the thermal decomposition of thiourea (Fig. S14, ESI†). Further evidence for a metal thiocyanate intermediate at high oleate concentrations comes from the aforementioned reaction of copper(II) oleate with thiourea, in the presence of 8:1 oleate: metal. Copper thiocyanate was identified as an impurity (Fig. 1) in the sulfur-poor CuS0.55 product. Regardless of the path, thiocyanate appears to be a more reluctant sulfur source than thiourea, resulting in sulfur-poor metal-sulfide phases throughout the different metals studied.
4. Conclusions
In summary (Scheme 4), our understanding of the system is that on its own, thiourea can thermally decompose into CS2 and NH3 gases as seen in gas-phase IR (I). Under low oleate conditions, thiourea coordinates to metal centers starting at temperatures as low as 150 °C (II). Thiourea decomposition is promoted by the metal center and becomes the preferred and uninhibited sulfur source for metal sulfide formation, yielding sulfur-rich metal phases (III) of Fe, Co, Ni, Cu.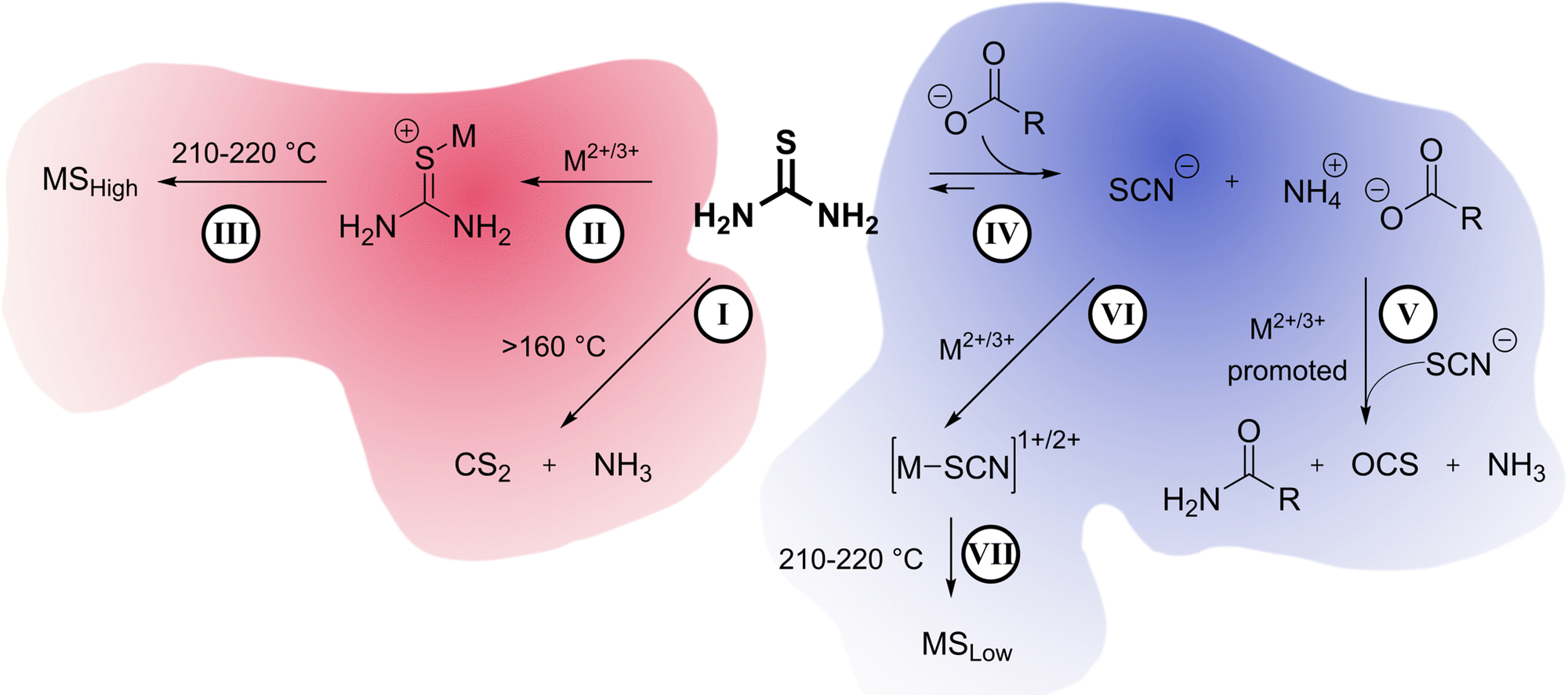 | ||
| Scheme 4 All reactions responsible for the formation of sulfur poor phases (blue, MSLow) at high carboxylate concentrations and sulfur rich phases (red, MSHigh) at low carboxylate concentrations: (I) thermal decomposition of thiourea into CS2 and NH3 gasses; (II) coordination of thiourea to the metal center; (III) direct decomposition of thiourea on the metal; (IV) preferential isomerization of thiourea into ammonium thiocyanate driven by the carboxylate's coordination to ammonium ion; (V) reaction between thiocyanate ion, carboxylate, and ammonium catalyzed by the metal center to produce an amide and carbonyl sulfide and ammonia gas by-products; (VI) coordination of the thiocyanate ion to the metal center; (VII) direct decomposition of thiourea on the metal. | ||
Under high oleate conditions, thiourea isomerizes to ammonium thiocyanate (IV) driven by the coordination of ammonium to oleate. Ammonium thiocyanate and carboxylates produce amides and OCS gas via a reaction that is promoted by the metal centers (V). Ammonium thiocyanate becomes the sulfur source in the formation of metal sulfides (VI, VII). Ammonium thiocyanate is a reluctant sulfur source compared to thiourea, and sulfur-poor metal sulfides of Fe, Co, Ni, Cu result.
These results came contrary to our initial hypothesis that increased oleate concentration would slow metal reactivity and highlight the importance of deep-dives in the molecular transformations that occur in colloidal synthesis.
While these studies have resulted in an explanation for stoichiometric phase control, one detail that has not yet been explained is why these particular reaction conditions had a tendency to produce some rare metastable polymorphs (similar or identical stoichiometry, but different crystal packing) (Table S2, ESI†). Smythite (FeS1.3) and marcasite (FeS2) formed in addition to their more stable counterparts greigite (FeS1.3) and pyrite (FeS2). Jaipurite (CoS) selectively formed over the more stable cobaltpentlandite (CoS0.89). Nickel sulfide (NiS) formed selectively over the more common and stable millerite (NiS). Cubic digenite (CuS0.55) formed over the more stable hexagonal polymorphs chalcocite (CuS0.50) and djurleite (CuS0.52). Polymorphic phase control is a complex field that we are currently studying.
Author contributions
Syntheses, powder X-ray diffraction, solid phase Fourier transform infrared spectroscopy, and nuclear magnetic spectroscopy experiments were performed by AAS. Gas phase Fourier transform infrared spectroscopy was carried out by AAS and GL. AAS and JEM came up with the methodology and visualization; wrote the paper. All authors contributed to editing. Conceptualization, funding acquisition, project administration, and supervision were by JEM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AAS and JEM acknowledge the financial support from NSF (National Science Foundation) project no. CHE-1905265 and CHE-2305161. GL and JDC gratefully acknowledge support for this work by the Office of Naval Research grant N00014-22-12035.Notes and references
- B. Song, Z. Zeng, E. Almatrafi, M. Shen, W. Xiong, C. Zhou, W. Wang, G. Zeng and J. Gong, Water Res., 2022, 211, 118048 CrossRef CAS PubMed.
- J. Moore, E. Nienhuis, M. Ahmadzadeh and J. McCloy, AIP Adv., 2019, 9, 035012 CrossRef.
- A. Villa, M. Telkhozhayeva, F. Marangi, E. Teblum, A. M. Ross, M. Prato, L. Andena, R. Frassine, F. Scotognella and G. D. Nessim, Adv. Opt. Mater., 2023, 11, 2201488 CrossRef CAS.
- Y. J. Yang, W. Li and J. Zi, Electrochem. Commun., 2013, 34, 304–307 CrossRef CAS.
- Z. Yuan, H. J. Peng, T. Z. Hou, J. Q. Huang, C. M. Chen, D. W. Wang, X. B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS PubMed.
- W. Xiong, K. Hu, Z. Li, Y. Jiang, Z. Li, Z. Li and X. Wang, Nano Energy, 2019, 66, 104149 CrossRef CAS.
- M. S. Faber, M. A. Lukowski, Q. Ding, N. S. Kaiser and S. Jin, J. Phys. Chem. C, 2014, 118, 21347 CrossRef CAS.
- M. A. S. Adamson, P. Yox, T. Hernandez, F. Wang and J. Vela, Chem. Mater., 2022, 34, 746–755 CrossRef CAS.
- L. Yu, B. Yang, Q. Liu, J. Liu, X. Wang, D. Song, J. Wang and X. Jing, J. Electroanal. Chem., 2015, 739, 156–163 CrossRef CAS.
- M. P. Hendricks, M. P. Campos, G. T. Cleveland, I. J. La Plante and J. S. Owen, Science, 1979, 2015(348), 1226–1230 Search PubMed.
- L. S. Hamachi, I. Jen-La Plante, A. C. Coryell, J. De Roo and J. S. Owen, Chem. Mater., 2017, 29, 8711–8719 CrossRef CAS.
- R. Karthikeyan, D. Thangaraju, N. Prakash and Y. Hayakawa, CrystEngComm, 2015, 17, 5431–5439 RSC.
- Y. Fazli, S. Mahdi Pourmortazavi, I. Kohsari and M. Sadeghpur, Mater. Sci. Semicond. Process., 2014, 27, 362–367 CrossRef CAS.
- T. Thongtem, A. Phuruangrat and S. Thongtem, Mater. Lett., 2010, 64, 136–139 CrossRef CAS.
- W. Du, X. Qian, M. Xiaodong, Q. Gong, H. Cao and J. Yin, Chem. – Eur. J., 2007, 13, 3241–3247 CrossRef CAS.
- J. W. Thomson, K. Nagashima, P. M. MacDonald and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 5036–5041 CrossRef CAS PubMed.
- A. Roffey, N. Hollingsworth, H. U. Islam, M. Mercy, G. Sankar, C. R. A. Catlow, G. Hogarth and N. H. De Leeuw, Nanoscale, 2016, 8, 11067–11075 RSC.
- N. Hollingsworth, A. Roffey, H. U. Islam, M. Mercy, A. Roldan, W. Bras, M. Wolthers, C. R. A. Catlow, G. Sankar, G. Hogarth and N. H. De Leeuw, Chem. Mater., 2014, 26, 6281–6292 CrossRef CAS.
- A. L. Abdelhady, M. A. Malik, P. O’Brien and F. Tuna, J. Phys. Chem. C, 2012, 116, 2253–2259 CrossRef CAS.
- M. J. Turo and J. E. Macdonald, ACS Nano, 2014, 8, 10205–10213 CrossRef CAS.
- J. M. Rhodes, C. A. Jones, L. B. Thal and J. E. Macdonald, Chem. Mater., 2017, 29, 8521–8530 CrossRef CAS.
- A. Balakrishnan, J. D. Groeneveld, S. Pokhrel and L. Mädler, Chem. – Eur. J., 2021, 27, 6390–6406 CrossRef CAS PubMed.
- A. Roffey, N. Hollingsworth, H. U. Islam, M. Mercy, G. Sankar, C. R. A. Catlow, G. Hogarth and N. H. de Leeuw, Nanoscale, 2016, 8, 11067–11075 RSC.
- N. Hollingsworth, A. Roffey, H. U. Islam, M. Mercy, A. Roldan, W. Bras, M. Wolthers, C. R. A. Catlow, G. Sankar, G. Hogarth and N. H. de Leeuw, Chem. Mater., 2014, 26, 6281–6292 CrossRef CAS.
- A. C. Koziel, R. B. Goldfarb, E. J. Endres and J. E. Macdonald, Inorg. Chem., 2022, 61, 14673–14683 CrossRef CAS PubMed.
- S. Yao, C. Yang, H. Zhao, S. Li, L. Lin, W. Wen, J. Liu, G. Hu, W. Li, Y. Hou and D. Ma, J. Phys. Chem. C, 2017, 121, 5154–5160 CrossRef CAS.
- E. Bennett, M. W. Greenberg, A. J. Jordan, L. S. Hamachi, S. Banerjee, S. J. L. Billinge and J. S. Owen, Chem. Mater., 2022, 34, 706–717 CrossRef CAS.
- L. S. Hamachi, I. Jen-La Plante, A. C. Coryell, J. De Roo and J. S. Owen, Chem. Mater., 2017, 29, 8711–8719 CrossRef CAS.
- B. A. Tappan, G. Barim, J. C. Kwok and R. L. Brutchey, Chem. Mater., 2018, 30, 5704–5713 CrossRef CAS.
- S. H. Choi, K. An, E. G. Kim, J. H. Yu, J. H. Kim and T. Hyeon, Adv. Funct. Mater., 2009, 19, 1645–1649 CrossRef CAS.
- A. Demortière, P. Panissod, B. P. Pichon, G. Pourroy, D. Guillon, B. Donnio and S. Bégin-Colin, Nanoscale, 2011, 3, 225–232 RSC.
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J. M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795–3810 CrossRef CAS.
- J. Cassidy, D. Harankahage, J. Ojile, D. Porotnikov, L. Walker, M. Montemurri, B. S. L. Narvaez, D. Khon, M. D. E. Forbes and M. Zamkov, Chem. Mater., 2022, 34, 2484–2494 CrossRef CAS.
- Y. Gao and X. Peng, J. Am. Chem. Soc., 2014, 136, 6724–6732 CrossRef CAS PubMed.
- B. Mahler, N. Lequeux and B. Dubertret, J. Am. Chem. Soc., 2010, 132, 953–959 CrossRef CAS.
- S. H. Choi, K. An, E. G. Kim, J. H. Yu, J. H. Kim and T. Hyeon, Adv. Funct. Mater., 2009, 19, 1645–1649 CrossRef CAS.
- J. Park, K. H. Lee, J. F. Galloway and P. C. Searson, J. Phys. Chem. C, 2008, 112, 17849–17854 CrossRef CAS.
- J. Joo, H. bin Na, T. Yu, J. H. Yu, Y. W. Kim, F. Wu, J. Z. Zhang and T. Hyeon, J. Am. Chem. Soc., 2003, 125, 11100–11105 CrossRef CAS PubMed.
- S. Wang, Q. Gao and J. Wang, J. Phys. Chem. B, 2005, 109, 17281–17289 CrossRef CAS PubMed.
- J. Madarász and G. Pokol, J. Therm. Anal. Calorim., 2007, 88, 329–336 CrossRef.
- Z. D. Wang, G. Feng, M. G. Garcia and T. Wang, Eur. J. Org. Chem., 2019, 598–604 CrossRef.
- L. Gmelin, Gmelin Handbuch der Anorganischen Chemie, Springer, New York, 8th edn, 1978, vol. D6 Search PubMed.
- P. M. Schaber, J. Colson, S. Higgins, D. Thielen, B. Anspach and J. Brauer, Thermochim. Acta, 2004, 424, 131–142 CrossRef CAS.
- A.-U. Rahman, M. A. Medrano and O. P. Mittal, Recl. Trav. Chim. Pays-Bas, 1960, 79, 188–192 CrossRef CAS.
- M. Hanumantha Rao, R. Pallepogu and K. Muralidharan, Inorg. Chem. Commun., 2010, 13, 622–624 CrossRef.
- O. Taichman, V. Kaplan, E. Wachtel and I. Lubomirsky, Solid Fuel Chem., 2022, 56, 21–28 CrossRef CAS.
- D. Yoon, H. Jin, S. Ryu, S. Park, H. Baik, S. J. Oh, S. Haam, C. Joo and K. Lee, CrystEngComm, 2015, 17, 4627–4631 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Nuclear magnetic resonance spectra, powder X-ray diffraction pattern, Fourier transform infrared spectra, and additional experimental details. See DOI: https://doi.org/10.1039/d3nh00227f |
| This journal is © The Royal Society of Chemistry 2023 |