
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasmonic photothermal activation of an organosilica shielded cold-adapted lipase co-immobilised with gold nanoparticles on silica particles†
Carolina I.
Giunta
a,
Seyed Amirabbas
Nazemi
a,
Magdalena
Olesińska
a and
Patrick
Shahgaldian
 *ab
*ab
aInstitute of Chemistry and Bioanalytics, School of Life Science, University of Applied Sciences and Arts Northwestern Switzerland, Hofackerstrasse 30, Muttenz CH-4132, Switzerland. E-mail: patrick.shahgaldian@fhnw.ch
bSwiss Nanoscience Institute, Klingelbergstrasse 82, Basel CH-4056, Switzerland
First published on 21st October 2022
Abstract
Gold nanoparticles (AuNPs), owing to their intrinsic plasmonic properties, are widely used in applications ranging from nanotechnology and nanomedicine to catalysis and bioimaging. Capitalising on the ability of AuNPs to generate nanoscale heat upon optical excitation, we designed a nanobiocatalyst with enhanced cryophilic properties. It consists of gold nanoparticles and enzyme molecules, co-immobilised onto a silica scaffold, and shielded within a nanometre-thin organosilica layer. To produce such a hybrid system, we developed and optimized a synthetic method allowing efficient AuNP covalent immobilisation on the surface of silica particles (SPs). Our procedure allows to reach a dense and homogeneous AuNP surface coverage. After enzyme co-immobilisation, a nanometre-thin organosilica layer was grown on the surface of the SPs. This layer was designed to fulfil the dual function of protecting the enzyme from the surrounding environment and allowing the confinement, at the nanometre scale, of the heat diffusing from the AuNPs after surface plasmon resonance photothermal activation. To establish this proof of concept, we used an industrially relevant lipase enzyme, namely Lipase B from Candida Antarctica (CalB). Herein, we demonstrate the possibility to photothermally activate the so-engineered enzymes at temperatures as low as −10 °C.
Introduction
Besides their potential in catalysis and nano-electronics,1,2 gold nanoparticles (AuNPs) also display a great potential for biological and biomedical applications owing to their high (bio)chemical stability and non-toxicity.3 These applications include diagnostics,4 drug delivery5,6 bio-imaging7 and therapy.8,9 One of the main stumbling blocks for the implementation of AuNPs in industrial processes, however, is their poor colloidal stability under operational conditions. For example, in a recent report, Yu et al. demonstrated that, upon visible light irradiation, AuNPs catalyse multielectron, multiproton reduction of CO2 to methane and ethane.10 It was observed, however, that these AuNPs showed a propensity to aggregation causing a drastic loss in catalytic efficiency after 10 hours of reaction. It is therefore of great importance to develop methods allowing to increase the stability of colloidal AuNPs without hampering their plasmonic properties.11–13 In this context, immobilised AuNPs have gained substantial interest since their immobilisation often suppresses aggregation.14–16 A range of carrier materials have been used for AuNP immobilisation including TiO2,17 SiO2,18 CeO219 and zeolites.20Surface immobilisation is also a well-established strategy for the stabilisation of biomolecules such as enzymes.21 A large variety of carriers for enzyme stabilisation have been explored; among many are gold nanoparticles.22 For example, Gherardi et al. have recently demonstrated that an α-amylase, immobilised on AuNPs, displays higher stability and efficiency than the soluble enzyme and applied the nanosystems produced to the cleaning of heritage textiles.23 Breger et al., reporting on the immobilisation of a phosphotriesterase, demonstrated that the size of AuNP influences the activity of the immobilised enzyme.24 The photothermal properties of AuNPs have also been exploited for immobilised enzymes with the aim of increasing their activity at higher temperatures. For example, Tadepalli et al. carried out the immobilisation of horseradish peroxidase onto the surface of AuNPs and nanorods (AuNRs) followed by the encapsulation of the enzyme within either an organosilica material25 or a metal organic framework (MOF).26 The authors showed an increase of enzymatic activity at 25 °C of 10% on AuNPs and of 110% on AuNRs after laser exposure compared to the enzyme activity without laser irradiation. Enzyme activity, stability and substrate promiscuity can also be improved by controlling the enzyme nano-environment.27–31 In this context, we have focused our efforts on the supramolecular engineering of enzymes immobilised on the surface of silica and shielded in organosilica layers of controlled thickness and chemical composition.32–36
Among enzymes of interest for industrial applications, cold active enzymes (CAEs) are of particular significance as they exhibit catalytic activity at low temperatures.37–39 Produced by psychrophiles, CAEs typically display a relatively high conformational mobility at the active site, which allows reducing the free energy of activation required to convert the substrate to the corresponding transition state.39,40 CAES find applications in different industries including food, bioremediation, cosmetic and pharmaceutical.37,41,42 Consequently, there is substantial effort invested in the discovery of new natural and engineered CAEs mainly by means of modern metagenomic methods.43 Besides, scientists have been applying protein engineering methods to endow mesophilic enzymes with cryophilic properties.44
Herein, we report a design strategy allowing for the plasmonic photoactivation of enzymes co-immobilised with AuNPs on the surface of silica particles (SPs) and shielded in an organosilica layer of controlled thickness. To that end, we developed a method that allows the efficient immobilisation of gold nanoparticles on the surface of SPs. The hybrid systems produced combine the advantages of the photothermal properties and the enhanced stability of immobilised AuNPs along with an improved endurance of the shielded enzyme at low temperatures. This work represents, to the best of our knowledge, the first example of a synthetic strategy allowing to chemically enhance the cryophilic properties of a natural biocatalyst. Besides, it provides an efficient method for the immobilisation of AuNPs on oxide surfaces.
Results and discussion
Nanbiocatalyst synthesis and characterisation
To design a nanobiocatalyst that can be photothermally activated at low temperatures, our proposed design is as follows. Enzyme and AuNPs are co-immobilised onto silica particles (SPs) and embedded in an organosilica shell of controlled thickness. This shell is expected to provide a structural stabilisation of the enzyme and to allow confining photothermal energy emitted by AuNPs upon light excitation. The shielding layer is transparent to visible light and is expected to allow SPR activation of embedded AuNPs. As a model enzyme, we decided to use Lipase B from Candida Antarctica (CalB), which is an enzyme of interest for a variety of applications.45Our synthetic strategy is presented in Fig. 1a. First, SPs were synthesized using a modified protocol46 derived from the Stöber method.47 Scanning electron micrographs of the SPs produced revealed homogeneous particles, spherical in shape and with an average diameter of 284 ± 8 nm (statistical analysis carried out on at least 100 SPs; ESI, Fig. S3†). AuNPs with citrate as a capping agent were synthetized following the Turkevich method.48 Those nanoparticles were fully characterised by atomic force microscopy (AFM), scanning electron microscopy (SEM), transmission electron microscopy (TEM) and UV-vis spectroscopy (ESI, Fig. S1†). A statistical analysis of the AuNPs diameter was performed measuring ca. 600 nanoparticles from TEM micrographs; the results revealed an average diameter of 18 ± 1 nm and a polydispersity index of 0.06.
 | ||
| Fig. 1 General synthetic strategy (a) allowing the sequential co-immobilisation of AuNPs (SP–Au) and CalB enzyme (SP–Au–CalB) on the surface of SPs and shielding in an organosilica layer produced via the hydrolysis and polycondensation of aminopropyl-triethoxysilane (APTES) and tetraethylorthosilicate (TEOS) to yield SP–Au–CalBOS. Amino-modification of silica with APTES (b) highlighting the formation of amine-rich islands at the surface of silica. Covalent strategy (c) allowing the sequential conjugation of AuNPs and CalB on ATPES-MPTES-modified silica particles, prior to protection layer growth (not shown). | ||
Taking into consideration that the AuNPs will be eventually entrapped within an organosilica shield, we initially explored the possibility to avoid covalent cross-linking and to immobilise the AuNPs via electrostatic interactions. Considering the negative charge of citrate-capped AuNPs, we decided to chemically modify the surface of the silica to introduce positively charged (at neutral pH) primary amino functions. This was achieved by reacting silica particles with (3-aminopropyl)triethoxysilane (APTES) at increasing concentrations (2.6, 7.8 and 26 mM) for 90 min. The ζ-potential of the so-modified particles (ESI, Table S1†), showed an increase of the charge on the surface of the nanoparticles from −55 mV for the bare SPs to −46.0, −19.2 and −9.9 mV when the concentration of APTES was 2.6, 7.8 and 26 mM, respectively. It is known that surface silanisation typically occurs through a sequential process, namely: (i) silane hydrolysis and polycondensation (yielding mainly linear oligomers in acidic or neutral conditions), (ii) adsorption of the newly formed oligomer onto the silica surface (mainly through hydrogen bonding) and (iii) polycondensation with the surface silanol functions (Fig. 1b).49 It is therefore expected that even if the net charge of the SPs produced remained negative, it displayed enough positively charged domains to allow for electrostatic adsorption of negatively charge AuNPs. This was confirmed by SEM investigations carried out on the amino-modified SPs produced further incubated for 15 h with AuNPs (0.310 mg mL−1; Fig. 2a–c). Indeed, the electron micrographs revealed an increasing surface coverage of AuNPs at the surface of SPs displaying increasing ζ-potential values. While this electrostatic strategy was successful to immobilise AuNPs on the surface of SPs, it fell short when it came to co-immobilise the enzyme on the surface of the SPs. Indeed, applying an established crosslinking reaction using glutaraldehyde, the surface coverage drastically decreased (Fig. 2d). The release of the AuNPs was attributed to the suppression of positive charges, upon the formation of imine bonds with surface amines, resulting in the release of the electrostatically bound AuNPs. This prompted us to explore a different surface modification approach.
 | ||
| Fig. 2 Representative scanning electron micrographs of SPs, reacted with increasing concentrations of APTES [2.6 (a), 7.8 (b) and 26 (c) mM] and reacted with AuNPs at a concentration of 0.310 mg mL−1. In (d), are shown SPs reacted with 26 mM of AuNPs and with glutaraldehyde (0.1%, v/v). All scale bars represent 200 nm. | ||
In the second immobilisation approach, we explored a covalent conjugation approach. It is based on the introduction, at the surface of SPs, of both amines, for further enzyme bioconjugation, and thiol moieties for further covalent anchoring of AuNPs through S–Au bonds. To that end, bare SPs were reacted with (3-mercaptopropyl)-trimethoxysilane (MPTES, 23 mM) and APTES (26 mM) for increasing reaction durations (1.5, 3.5 and 20 h). Reactions were stopped by centrifugation and the so-modified SPs were washed with water and further reacted with AuNPs-citrate for 15 hours; representative micrographs are shown in Fig. 3.
 | ||
| Fig. 3 Covalent AuNPs immobilisation on SPs reacted first with a mixture of APTES and MPTES for 1.5 (a), 3.5 (b) and 20 hours (c); and consecutively reacted with AuNPs during All scale bars represent 200 nm. | ||
From Fig. 3, for a silanisation reaction of 1.5 h, the systems produced displayed a high density of AuNPs without any major change in the morphology of the SPs. After 3.5 h reaction, the surface of the SPs appeared somewhat inhomogeneous; this effect is more pronounced after 20 h of reaction. This morphology change is attributed to polycondensation of MPTES and APTES forming an organosilica layer on the surface of silica. From this set of results, we decided to proceed with SP–Au produced with MPTES and APTES (1.5 h reaction). The AuNPs immobilisation yield was quantified by measuring, spectrophotometrically, the amount of AuNPs left in the liquid phase of the reaction mixture after reaction. It was as high as 97%, corresponding to 94 μg of AuNPs per mg of SPs (ESI, Fig. S2a†). UV-vis measurements carried out on SP–Au revealed a bathochromic (red) shift of the SPR band from 520 to 534 nm, suggesting a drastic change experienced by AuNPs upon immobilisation (ESI, Fig. S2b†).25,26 An apparent increase of absorbance was also measured; this was explained by light adsorption of SPs in the visible range.
Next, enzyme immobilisation was performed by reaction of SP–Au with glutaraldehyde (0.1%, v/v) in water for 30 min and subsequent reaction (after SP–Au washing) with CalB (0.1 mg mL−1) in potassium phosphate buffer (pH 6.2) supplemented with polysorbate 80. The cross-linking reaction was stopped by centrifugation; the immobilisation yield, measured by means of a bicinchoninic acid assay (BCA) performed on the liquid phase of the reaction, was 66% (ESI, Table S2†). We also confirmed spectrophotometrically the absence of AuNPs loss in the liquid phase; the UV-vis adsorption spectra of SP–Au with immobilised CalB (referred to as SP–Au–CalB) did not display any significant change when compared to SP–Au (ESI, Fig. S2†). Similarly, SP–CalB were produced omitting the addition of AuNPs. The last synthetic step was the growth of protective organosilica layer on the surface of SP–Au–CalB to produce SP–Au–CalBOS. This was achieved by reaction with a mixture of APTES and tetraethylorthosilicate (TEOS) following a protocol previously published.33 Similarly, as control systems, were produced SP–CalBOS applying the same synthetic procedure. The particles produced were characterised by SEM and cryogenic transmission electron microscopy; Fig. 4.
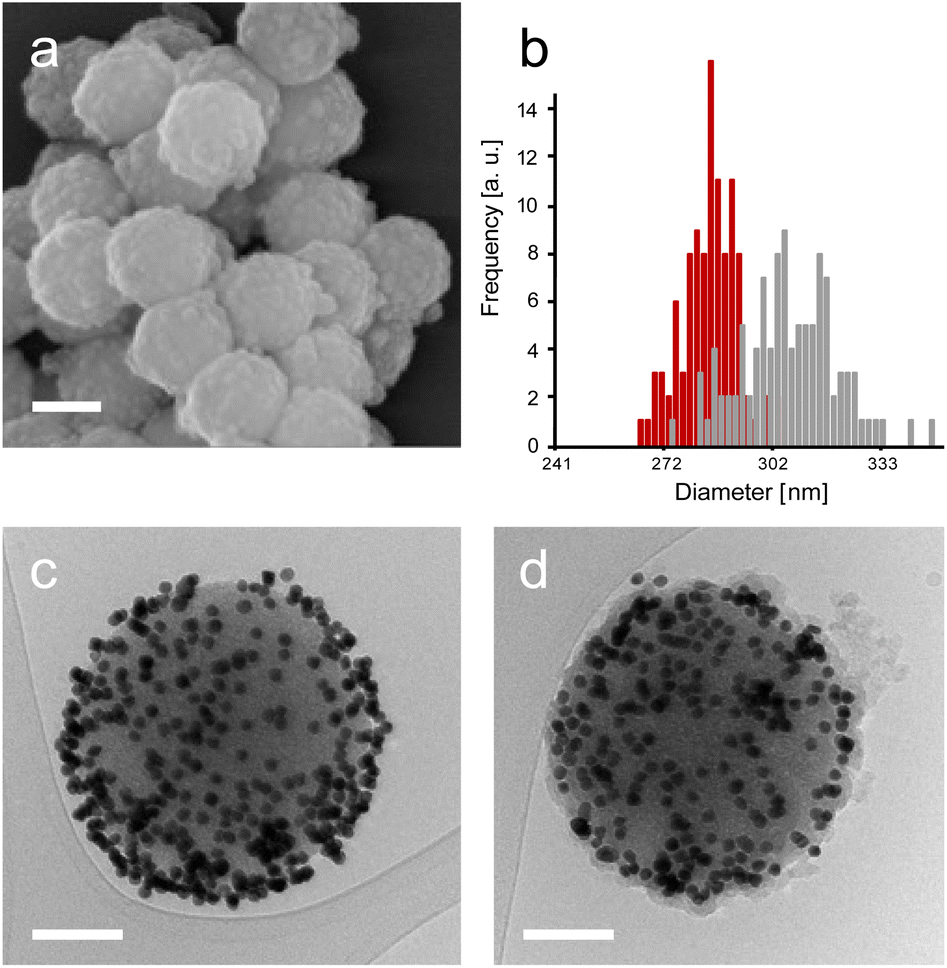 | ||
| Fig. 4 SEM micrographs of SP–Au–CalBOS (a), size distribution of bare SPs (red bars) and SP–Au–CalBOS (grey bars) measured on 100 particles (b). Cryo-EM micrographs of SP–Au–CalB (c) and SP–Au–CalBOS (d). Scale bars represent 200 nm (a) and 100 nm (c and d). | ||
The SP–Au–CalBOS had a diameter of 320 nm consistent with the growth of an organosilica layer. This value corresponds to a protection layer thickness of 18 nm, confirming the shielding of the immobilised CalB and AuNPs. Moreover, the AuNPs on the surface of the SPs were no longer visible after shielding (SP–Au–CalBOS) and the particles surface appeared rough. The presence of the AuNPs underneath the layer is also confirmed from cryo-EM microscopy experiments, where a uniform coverage of gold nanoparticles on the surface of SP–Au–CalB and SP–Au–CalBOS was observed. This confirmed that the immobilisation of CalB and the growth of the layer did not displace the covalently immobilised AuNPs. As a control for the enzymatic activity experiments, SP–CalBOS particles were prepared omitting the AuNPs (ESI, Fig. S3 and Table S3†).
Biocatalytic activity and photothermal activation
To demonstrate the possibility to photoactivate immobilised CalB through SPR excitation, we carried out a thorough activity study of SP–Au–CalBOS. It was expected that, upon light irradiation at the SPR wavelength (535 nm), the AuNPs converted light into heat, thus increasing the temperature in their surrounding where the enzyme was shielded. Therefore, the enzyme would experience a higher temperature in its local environment while being placed inside a bulk solution at low temperature. The experimental set-up used for laser irradiation at controlled temperature included a laser (532 nm, 500 mW) connected, through an optical fiber (diameter 600 μm), to a thermostatic cuvette holder where the sample was magnetically stirred. The esterase activity of CalB was determined via the enzymatic hydrolysis of p-nitrophenyl butyrate (pNPB). Reaction kinetics were measured at −10, −5, 0 and 10 °C; Fig. 5 and 6. We also confirmed the stability of the substrate under the reaction conditions (ESI, Fig. S4†).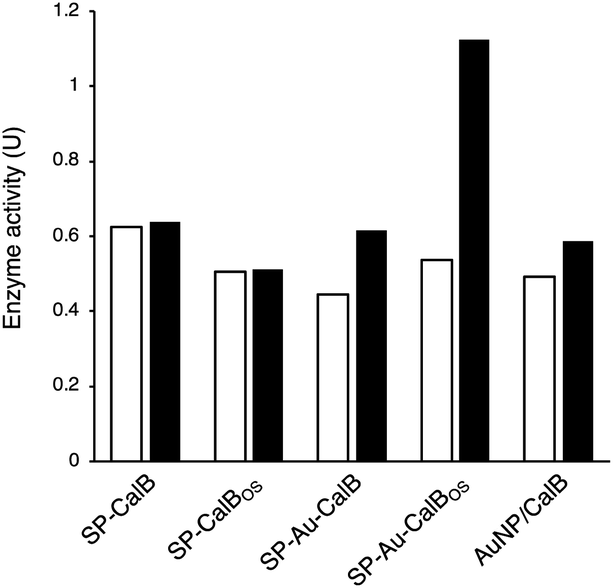 | ||
| Fig. 5 Enzymatic activity (pNPB hydrolysis) measured spectrophotometrically after reactions carried out in dark conditions (white bars) or under irradiation (535 nm, black bars) for SP–CalB, SP–CalBOS, SP–Au–CalB, SP–Au–CalBOS and a physical mixture of AuNPs and CalB at 10 °C. Values shown are obtained from single measurements. | ||
From Fig. 5, activities of both SP–CalB and SP–CalBOS remained mainly unchanged upon light excitation. When AuNPs were co-immobilised on the surface of SPs (SP–Au–CalB); an activity increase of 37% was measured. This effect was also observed when both soluble enzyme and AuNPs were mixed in solution, with a moderate increase in activity of 18%. In the presence of the organosilica shield, the enzyme co-immobilised with AuNPs (SP–Au–CalBOS) showed an activity increase as high as 107%. This result confirmed that the organosilica shield limited heat transfer to the buffer and allowed for a stronger photothermal activation. From Fig. 6, it can be observed that the activity of SP–Au–CalBOS for all the measured temperatures (at −10, −5, 0 and 10 °C) has increased with the most highlighted values at −10 °C with 300% and at 10 °C with 107% enhancement.
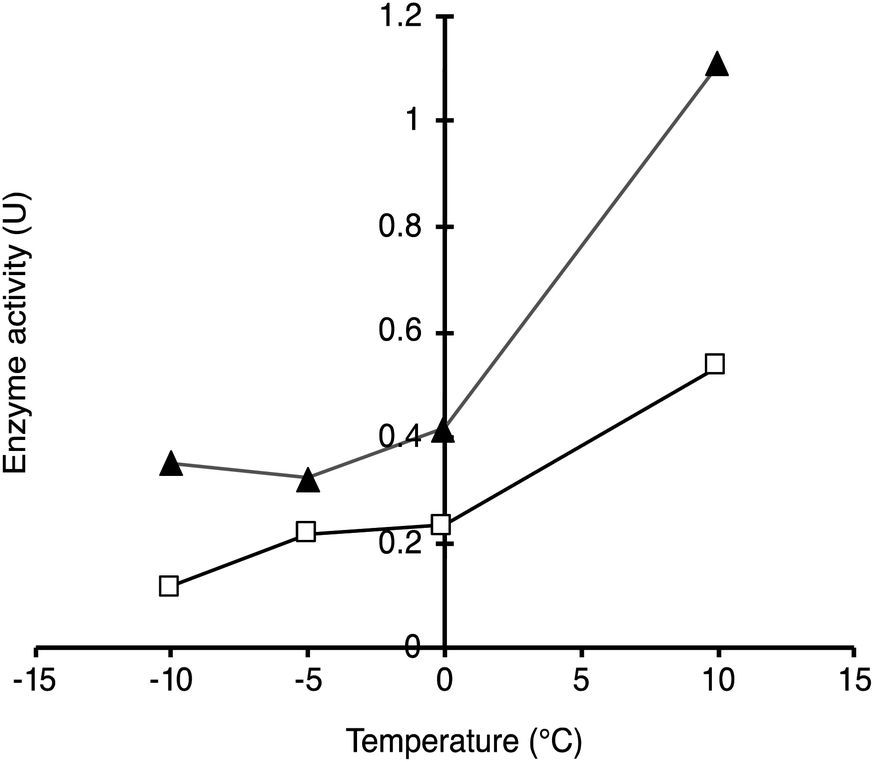 | ||
| Fig. 6 Enzymatic activity of SP–Au–CalBOS measured in dark conditions (white squares) and under 615 mW irradiation (black triangles) at increasing temperatures (−10 to 10 °C). Values shown are obtained from single measurements. | ||
Conclusions
In this work, we reported a convenient method for the stable immobilisation of AuNPs onto silica nanoparticles. This method was used to co-immobilise AuNPs and CalB enzyme molecules on the surface of SPs and shielded with an organosilica layer. Photoactivation experiments confirmed the possibility to photoactivate such immobilised enzymes, through the plasmonic photoactivation of immobilised AuNPs. The shielding layer, in those systems, allows confining thermal energy in the surrounding of the enzyme. We expect our approach to be versatile in that it can be applied to a large variety of enzymes.Experimental
Materials
Tetraethyl orthosilicate (TEOS, ≥ 99%), (3-aminopropyl)triethoxysilane (APTES, ≥ 98%), ammonium hydroxide (ACS grade, 28–30%), ethanol (ACS grade, anhydrous), glutaraldehyde (grade I, 25% in water), 4-nitrophenyl butyrate (pNPB, ≥ 98%) and gold(III) chloride hydrate were purchased from Merck (Switzerland) and used as received. BCA protein assay kit was purchased from Bio-Rad (Switzerland). 3-Mercaptopropyltriethoxysilane 97% (MPTES) was purchased from ABCR (Germany). SPs were prepared using the procedure previously published.33Gold nanoparticles synthesis
AuNPs were synthetized following the Turkevich method.48 Briefly, sodium citrate tribasic dihydrate (2.5 mL, 34 mM) was added in water (100 mL) at 90 °C. After 5 min was a solution of gold(III) chloride hydrate (1 mL, 25 mM) added and reacted until the colour of the solution changed from yellow to dark red. The solution was left to cool down to 20 °C. Sucrose was added to the AuNPs solution (final concentration 5% (w/v)); the solution was stirred until full sucrose dissolution. The solution was consequently snap-frozen in liquid nitrogen and freeze-dried. The AuNPs were subsequently resuspended in water (10 mL) and stored at room temperature. The AuNPs before and after freeze drying were analysed by UV-vis spectroscopy (ESI, Fig. S1†).SPs surface modification and AuNPs immobilisation
In order to electrostatically immobilise AuNPs, the surface of SPs was chemically modified to introduce primary amine functions. The silica particles (18 mL, 3.2 mg mL−1, diameter of 284 ± 8 nm) where reacted during 90 min with (3-aminopropyl)triethoxysilane (APTES) at increasing concentrations (2.6, 7.8 and 26 mM). After removing unreacted APTES, the amino-modified SPs were further incubated with 0.310 mg mL−1 of AuNPs for 15 h and characterised by SEM. An aliquot of the solution was taken (1.5 h, 3.5 h and 15 h), centrifuged (3220 x g, 5 min) and the supernatant was analysed by UV-vis spectroscopy (ESI, Fig. S5†) to evaluate the AuNPs immobilisation yield.For the covalent immobilisation of AuNPs, SPs (18 mL, 3.2 mg mL−1) were reacted under magnetic stirring (20 °C, 400 rpm) with MPTES (23 mM, 10 min) and APTES (26 mM) for 1.5 h. The SPs so modified were then centrifuged (3220 × g, 10 min), the supernatant was discarded, and the white pellet obtained was resuspended in ethanol. After two similar washing cycles in ethanol, the modified SPs were washed once and eventually resuspended in water (18 mL). The suspension was stored in a water bath (20 °C, 20 h) prior to use. Mercapto- and amino-modified SPs suspension (10 mL) was incubated with the AuNPs solution (0.310 mg mL−1) under magnetic stirring at room temperature (100 rpm, 20 h). The SP–Au solution was then centrifuged (3220 x g, 5 min), the supernatant was kept for determining the AuNPs immobilisation yield via UV-vis measurement and the particles were resuspended in water (10 mL). The washing cycle in water was repeated trice.
CalB immobilisation on SPs–Au and on SPs
CalB was co-immobilised on the SPs–Au using glutaraldehyde as cross-linker. Amino-modified SPs–Au (10 mL) where reacted with glutaraldehyde (0.1% (v/v)) under stirring (20 °C, 30 min, 400 rpm). The suspension was then centrifuged (3220 x g, 5 min), the supernatant was discarded, and the pellet was resuspended in water. After two washing cycles in water, the particles were resuspended in a potassium phosphate buffer (10 mM, 9 mL, pH 6.2) containing polysorbate 80 (8 mg L−1). The same procedure was followed for the immobilisation of CalB on SPs omitting the AuNPs.A solution of CalB (1 mL, 1 mg mL−1) was added to the SPs–Au and incubated under stirring (20 °C, 1 h, 400 rpm). After the enzyme immobilisation an aliquot (1 mL) was removed from the solution, it was centrifuged (350 × g, 10 min), the supernatant was kept for protein quantification assay by means of bicinchoninic acid assay (BCA) assay and the pellet was resuspended in potassium phosphate buffer (10 mM, pH 7.4). The concentration of the unbound enzyme was extrapolated from the bovine serum albumin (BSA) calibration curve (ESI, Table S2†).
SP–Au–CalB and SP–CalB shielding
CalB (63 μg mL−1) was immobilised according to the procedures described above. Consequently, the shielding was performed following the method previously published.33 TEOS (0.450 mmol) was added to the CalB–SPs and to the SP–Au–CalB suspension and allowed to react in a water bath (20 °C, 1 h, 400 rpm). Subsequently, APTES (0.090 mmol) was added to the reaction mixture. The polycondensation reaction was stopped after 18 h by washing the particles three times in potassium phosphate buffer (10 mM, pH 7.4) and stored at 20 °C for 18 h. The particles suspension was finally stored (4 °C) for further analysis.CalB activity assay and irradiation of Gold nanoparticles
The enzymatic activity of CalB was determined using a commercial colorimetric assay (Sigma – enzymatic assay of lipoprotein lipase (EC 3.1.1.34)) with some modifications. A solution of p-nitrophenyl butyrate (pNPB) (30 mM, 900 μL) in phosphate buffer (10 mM, pH 7.4), Triton X-100 (5% (v/v)) and isopropanol (10% (v/v)) was added in a poly(methyl methacrylate) (PMMA) cuvette and incubated at the used temperatures (5 min at 10, 0, −5, −10 °C) under stirring. The cuvette was placed inside a qpod device (Ocean Optics, qpod) connected with a temperature controller. Afterwards the enzyme solution (63 μg mL−1, 100 μL) was added to the substrate solution, the laser (MGL-H-532nm-500mW-17111070). The laser was connected to the cuvette holder with an optical fiber (Ocean Optics, UV-vis, 600 μm). The reaction was stopped (every 10 min) by centrifugation (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 x g, 1 min, 200 μL). The amount of produced p-nitrophenol (pNP) was determined by measuring the absorbance of the solution (410 nm, 200 μL) in a 96 well-plate using a Synergy H1 (BioTek). The catalytic activities were calculated using the molar extinction coefficient of p-nitrophenol (ε410nm = 4469 M−1 cm−1) measured by preparing a standard curve using the same buffer. The activity assay was carried out at pH7.4 to avoid spontaneous hydrolysis of the ester in alkaline conditions (in which the extinction coefficient of p-nitrophenol is higher).
100 x g, 1 min, 200 μL). The amount of produced p-nitrophenol (pNP) was determined by measuring the absorbance of the solution (410 nm, 200 μL) in a 96 well-plate using a Synergy H1 (BioTek). The catalytic activities were calculated using the molar extinction coefficient of p-nitrophenol (ε410nm = 4469 M−1 cm−1) measured by preparing a standard curve using the same buffer. The activity assay was carried out at pH7.4 to avoid spontaneous hydrolysis of the ester in alkaline conditions (in which the extinction coefficient of p-nitrophenol is higher).
The activity of the soluble enzymes, of the immobilised and of the protected enzymes were tested as described under laser irradiation and in dark conditions (without laser irradiation) as control experiments using the same concentration of enzyme. For all the reactions carried at lower temperature than 0 °C the qpod holder was connected to a flux of air to avoid humidity formation on the walls of the cuvette.
Scanning electron microscopy and particle size measurement
Particles were imaged using a Zeiss SUPRA® 40VP scanning electron microscope. A drop (2 μL) of each sample was spread on silicon wafer substrates and sputter-coated with a gold–platinum alloy for 15 s at 15 mA. Micrographs were acquired using the InLens mode with an accelerating voltage of 10 kV. Particle sizes were measured on micrographs acquired at a magnification of 150000× using the ®AnalySIS software package. More than 100 measurements were carried out for each sample.
Cryo-transmission electron microscopy (Cryo-TEM)
A 4 μL aliquot of sample was adsorbed onto a holey carbon-coated grid (Lacey, USA), blotted with Whatman 1 filter paper and vitrified into liquid ethane at −178 °C using a Leica GP plunger (Leica, Austria). Frozen grids were transferred onto a Talos electron microscope (FEI, USA) using a Gatan 626 cryo-holder. Electron micrographs were recorded at an accelerating voltage of 200 kV and a nominal magnification of 73000x, using a low-dose system (20 e− Å−2) and keeping the sample at low temperature. Micrographs were recorded on a CETA camera.
Transmission electron microscopy (TEM)
2 μL of a 0.03 mg mL−1 AuNP solution was placed onto standard 3.5 mm copper grids (200 mesh) with Formvar coating and dried at room temperature overnight. Images were taken with a Zeiss EM 900 transmission electron microscope, equipped with an AMT XR280 cMOS camera at an acceleration voltage of 50 kV.Author contributions
CIG: methodology, investigation, writing original draft; SAN: investigation, review and editing; MO: investigation, writing original draft; PS: conceptualization, methodology, funding acquisition, writing-review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the FHNW research fund and by the FuturEnzyme Project funded by the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreement No. 10100032.Notes and references
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Chem. Rev., 2020, 120, 464–525 CrossRef CAS PubMed.
- Y. X. Du, H. T. Sheng, D. Astruc and M. Z. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- C. Carnovale, G. Bryant, R. Shukla and V. Bansal, ACS Omega, 2019, 4, 242–256 CrossRef CAS.
- W. Zhou, X. Gao, D. B. Liu and X. Y. Chen, Chem. Rev., 2015, 115, 10575–10636 CrossRef CAS.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS.
- W. Li, X. Zhao, B. Du, X. Li, S. Liu, X. Y. Yang, H. Ding, W. Yang, F. Pan, X. Wu, L. Qin and Y. Pan, Sci. Rep., 2016, 6, 30619 CrossRef CAS PubMed.
- M. Morita, T. Tachikawa, S. Seino, K. Tanaka and T. Majima, ACS Appl. Nano Mater., 2018, 1, 355–363 CrossRef CAS.
- E. C. Dreaden, A. M. Alkilany, X. H. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- Y. Jiang, S. Huo, T. Mizuhara, R. Das, Y. W. Lee, S. Hou, D. F. Moyano, B. Duncan, X. J. Liang and V. M. Rotello, ACS Nano, 2015, 9, 9986–9993 CrossRef CAS PubMed.
- S. Yu, A. J. Wilson, J. Heo and P. K. Jain, Nano Lett., 2018, 18, 2189–2194 CrossRef CAS PubMed.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- A. Carattino, M. Caldarola and M. Orrit, Nano Lett., 2018, 18, 874–880 CrossRef CAS.
- Y. C. Zhang, S. He, W. X. Guo, Y. Hu, J. W. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS.
- X. Bokhimi, R. Zanella and A. Morales, J. Phys. Chem. C, 2007, 111, 15210–15216 CrossRef CAS.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef CAS PubMed.
- S. Naya, M. Teranishi, K. Kimura and H. Tada, Chem. Commun., 2011, 47, 3230–3232 RSC.
- J. M. C. Soares, P. Morrall, A. Crossley, P. Harris and M. Bowker, J. Catal., 2003, 219, 17–24 CrossRef CAS.
- L. Guczi, A. Beck and K. Frey, Gold Bull., 2009, 42, 5–12 CrossRef CAS.
- P. Lakshmanan, P. Upare, N.-T. Le, K. Y. Lee, D. Hwang, U. H. Lee, H. Kim and J.-S. Chang, Appl. Catal., A, 2013, 468, 260–268 CrossRef CAS.
- J. Lu, C. Aydin, N. D. Browning and B. C. Gates, Angew. Chem., Int. Ed., 2012, 51, 5842–5846 CrossRef CAS PubMed.
- R. A. Sheldon, A. Basso and D. Brady, Chem. Soc. Rev., 2021, 50, 5850–5862 RSC.
- G. Plascencia-Villa and R. Mendoza-Cruz, in Nanomaterials for Biocatalysis, ed. G. R. Castro, A. K. Nadda, T. A. Nguyen, X. Qi and G. Yasin, Elsevier, 2022, pp. 377–434 Search PubMed.
- F. Gherardi, L. Turyanska, E. Ferrari, N. Weston, M. W. Fay and B. J. Colston, ACS Appl. Bio Mater., 2019, 2, 5136–5143 CrossRef CAS PubMed.
- J. C. Breger, E. Oh, K. Susumu, W. P. Klein, S. A. Walper, M. G. Ancona and I. L. Medintz, Bioconjugate Chem., 2019, 30, 2060–2074 CrossRef CAS PubMed.
- S. Tadepalli, J. Yim, K. Madireddi, J. Y. Luang, R. R. Naik and S. Singamaneni, Chem. Mater., 2017, 29, 6308–6314 CrossRef CAS.
- S. Tadepalli, J. Yim, S. Cao, Z. Wang, R. R. Naik and S. Singamaneni, Small, 2018, 14, 1702382 CrossRef.
- R. A. Sheldon, A. Basso and D. Brady, Chem. Soc. Rev., 2021, 50, 5850–5862 RSC.
- J. M. Bolivar, J. M. Woodley and R. Fernandez-Lafuente, Chem. Soc. Rev., 2022, 51, 6251–6290 RSC.
- Q. Yang, B. C. Wang, Z. Zhang, D. S. Lou, J. Tan and L. C. Zhu, RSC Adv., 2017, 7, 38028–38036 RSC.
- L. Lancaster, W. Abdallah, S. Banta and I. Wheeldon, Chem. Soc. Rev., 2018, 47, 5177–5186 RSC.
- Y. F. Cao, X. Y. Li, J. R. Xiong, L. C. Wang, L. T. Yan and J. Ge, Nanoscale, 2019, 11, 22108–22117 RSC.
- M. L. Briand, R. Gebleux, F. Richina, M. R. Correro, Y. Grether, Y. Dudal, S. Braga-Lagache, M. Heller, R. R. Beerli, U. Grawunder, P. F. X. Corvini and P. Shahgaldian, Chem. Commun., 2020, 56, 5170–5173 RSC.
- M. R. Correro, N. Moridi, H. Schützinger, S. Sykora, E. M. Ammann, E. H. Peters, Y. Dudal, P. F. X. Corvini and P. Shahgaldian, Angew. Chem., Int. Ed., 2016, 55, 6285–6289 CrossRef CAS.
- M. R. Correro, M. Takacs, S. Sykora, P. F. X. Corvini and P. Shahgaldian, RSC Adv., 2016, 6, 89966–89971 RSC.
- C. I. Giunta, I. Cea-Rama, S. Alonso, M. L. Briand, R. Bargiela, C. Coscolín, P. F. X. Corvini, M. Ferrer, J. Sanz-Aparicio and P. Shahgaldian, ACS Nano, 2020, 14, 17652–17664 CrossRef CAS.
- S. A. Nazemi, M. Olesinska, C. Pezzella, S. Varriale, C. W. Lin, P. F. X. Corvini and P. Shahgaldian, Chem. Commun., 2021, 57, 11960–11963 RSC.
- M. Mangiagalli, S. Brocca, M. Orlando and M. Lotti, New Biotechnol., 2020, 55, 5–11 CrossRef CAS PubMed.
- A. A. Al-Ghanayem and B. Joseph, Appl. Microbiol. Biotechnol., 2020, 104, 2871–2882 CrossRef CAS.
- G. Feller and C. Gerday, Nat. Rev. Microbiol., 2003, 1, 200–208 CrossRef CAS PubMed.
- M. Santiago, C. A. Ramirez-Sarmiento, R. A. Zamora and L. P. Parra, Front. Microbiol., 2016, 7, 1408 Search PubMed.
- J. C. Marx, V. Blaise, T. Collins, S. D'Amico, D. Delille, E. Gratia, A. Hoyoux, A. L. Huston, G. Sonan, G. Feller and C. Gerday, Cell. Mol. Biol., 2004, 50, 643–655 CAS.
- F. Roulling, F. Piette, A. Cipolla, C. Struvay and G. Feller, in Extremophiles Handbook, ed. K. Horikoshi, Springer Japan, Tokyo, 2011, pp. 891–913 Search PubMed.
- M. Ferrer, O. Golyshina, A. Beloqui and P. N. Golyshin, Curr. Opin. Microbiol., 2007, 10, 207–214 CrossRef CAS PubMed.
- C. Struvay and G. Feller, Int. J. Mol. Sci., 2012, 13, 11643–11665 CrossRef CAS PubMed.
- R. R. C. Monteiro, J. J. Virgen-Ortiz, A. Berenguer-Murcia, T. N. da Rocha, J. C. S. dos Santos, A. R. Alcantara and R. Fernandez-Lafuente, Catal. Today, 2021, 362, 141–154 CrossRef CAS.
- A. Cumbo, B. Lorber, P. F. X. Corvini, W. Meier and P. Shahgaldian, Nat. Commun., 2013, 4, 1503 CrossRef PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- J. Turkevich, Gold Bull., 1985, 18, 125–131 CrossRef CAS.
- G. T. Hermanson, in Bioconjugate Techniques, 3rd edn, Academic Press, London, 2013, pp. 537–548 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00605g |
| This journal is © The Royal Society of Chemistry 2023 |