
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-atomic rhenium-assisted 2H-to-1T phase transformation of MoS2 nanosheets boosting electrocatalytic hydrogen evolution†
Jianmin
Yu‡
ac,
Yongteng
Qian‡
 d,
Qing
Wang
a,
Chenliang
Su
c,
Hyoyoung
Lee
*ef,
Lu
Shang
*a and
Tierui
Zhang
*ab
d,
Qing
Wang
a,
Chenliang
Su
c,
Hyoyoung
Lee
*ef,
Lu
Shang
*a and
Tierui
Zhang
*ab
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: lushang@mail.ipc.ac.cn; tierui@mail.ipc.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cInternational Collaborative Laboratory of 2D Materials for Optoelectronic Science and Technology of Ministry of Education, Institute of Microscale Optoelectronics, Shenzhen University, Shen Zhen, 518060, China
dDepartment of Physics and Interdisciplinary Course of Physics and Chemistry, Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea
eCenter for Integrated Nanostructure Physics, Institute for Basic Science (IBS), Sungkyunkwan University, Suwon, 16419, Republic of Korea
fDepartment of Chemistry, Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea. E-mail: hyoyoung@skku.edu
First published on 21st April 2023
Abstract
Metallic (1T) molybdenum disulfide (MoS2) with high electronic conductivity has been recognized as a promising catalytic material for the hydrogen evolution reaction (HER). However, there are still some challenges in obtaining high-degree and stable 1T-MoS2 due to its thermodynamically metastable characteristics. Here, we reported the single-atomic rhenium (Re)-assisted 2H-to-1T phase transformation (ca. 85%) of MoS2 nanosheets using a simple one-pot hydrothermal method. The resulting single-atomic Re doped 1T-2H MoS2 heterostructures delivered small overpotentials of 34 and 38 mV at a current density of 10 mA cm−2 in acid and alkaline media, respectively. Structural characterization and theoretical calculations indicate that the implantation of the Re single atom not only promotes the phase transition of MoS2 from the 2H to 1T phase and charge redistribution through the constructed Re–S–Mo site but also the additional Re single atom acts as the active site to facilitate the dissociation of water and the adsorption of the hydrogen intermediate. All these factors effectively improve the electrocatalytic properties of MoS2 for the HER.
Broader contextSingle-atomic Re doped 1T-2H MoS2 heterostructures were successfully synthesized using a simple hydrothermal method. The as-prepared Re doped 1T-2H MoS2 heterostructures delivered small overpotentials of 34 and 38 mV at a current density of 10 mA cm−2 in acid and alkaline media, respectively. Our work demonstrates that the introduction of the Re single-atom is a facile method to realize the 2H-to-1T phase transition of MoS2, resulting in highly durable and efficient electrocatalysts for hydrogen evolution. |
Introduction
Hydrogen is considered as one of the most promising energy carriers to replace fossil fuels due to its high energy density, cleanliness, and renewability.1 Electrocatalytic water splitting driven by green electricity (such as solar cells) represents a potential environment-friendly approach for large-scale hydrogen generation.2 The hydrogen evolution reaction (HER) is crucial in water electrolysis. Noble-metal-based materials (such as Pt-based materials) with suitable Gibbs-free energies for H adsorption have displayed superior HER performance.3,4 However, it is widely concerning that their high cost and scarcity are seriously impeding their industrial applications. Therefore, it is important to develop HER electrocatalysts with excellent performance at low cost.5During the past several years, two-dimensional (2D) transition metal dichalcogenides (TMD) have been considered as a promising alternative catalytic material due to their unique electronic and physical properties, such as atomic-scale thickness, strong spin–orbit coupling, and direct band gap.6,7 In particular, 2D TMD materials with a metallic 1T phase are usually regarded as favorable for catalyzing electrochemical hydrogen production from water compared with other phases due to the following merits: firstly, the higher electrical conductivity supplied better charge transport capability.8 Secondly, both basal planes and edges of the 1T phase can be activated for hydrogen adsorption rather than just the edges of the 2H phase.9 In recent years, many works have attempted to promote the accessibility of the 2H-to-1T phase transformation. The typical synthesis approaches of metallic 1T-MoS2 involved alkali–ion intercalation,10 external irradiation,11 micromechanical exfoliations,12 and thermal treatment.13 Nevertheless, the thermodynamic instability of the 1T phase of TMD still impedes its practical application.8,14 To overcome this limitation, heteroatom doping as a prevalent strategy has been applied to stabilize the metallic 1T phase. For example, Ir-doped,15 Rh-doped,16 or (Pd, Ru)-co-doped,17 either trigger metallic 1T or activate the inert plane to elevate the catalytic performance of pristine MoS2. Besides that, phase engineering with the incorporation of single atoms (SA) is also another important strategy, similar to heteroatom doping, which speculated that the large quantities of active sites and the intrinsic activity of each existing active site would afford an increase in the performance and stability of the HER.18 For instance, single-atom Ru supported on MoS2 as an alternative electrocatalyst to produce the stable 1T-phase MoS2 with a S-vacancy formation has been proposed for an excellent HER.19 In another example, the synergistic strategy of phase conversion engineering and incorporation of Pt single atoms into the MoS2 lattice was reported, which can convert the 2H-phase to the 1T-phase via a potential cycling method and enhance the performance of inert 2D MoS2 for the HER.20
Rhenium (Re), as a 5d transition metal, is close to the Pt group, but its price is much less than that of the noble metals (Pt, Ir, and Pd).21,22 What's more, Re possesses a similar radius and a different number of valence electrons compared with Mo, which is emerging as a suitable dopant to induce phase transition of 2D TMD.23,24 Herein, the 2H-to-1T phase transformation of MoS2 was successfully realized by introducing Re SA into MoS2 (Re–SA/MoS2) through a facile hydrothermal method. The optimized Re–SA/MoS2 showed excellent catalytic activity for the HER with small overpotentials of 34 and 38 mV at 10 mA cm−2 in acidic and alkaline electrolytes, respectively. Moreover, the optimized Re–SA/MoS2 also presents good stability for hydrogen evolution in both acidic and alkaline electrolytes. The density functional theory (DFT) results demonstrated that the injection of the Re SA into MoS2 can significantly adjust the electronic structure, optimize the adsorption-free energy of the MoS2, and further enhance the catalytic kinetics. This work demonstrates that the introduction of a Re single-atom is a facile method to realize the 2H-to-1T phase transition of MoS2 and thereby produces durable and efficient electrocatalysts for hydrogen evolution.
Results and discussion
Synthesis and structural characterization of Re–SA/MoS2 nanosheets
As depicted in Fig. 1a, the Re–SA/MoS2 nanosheets were synthesized on carbon cloth using a hydrothermal method. Phase identification and crystallinity in pristine MoS2 and Re–SA/MoS2 were investigated by the corresponding X-ray diffraction (XRD) patterns (Fig. 1b). The prominent diffraction peaks at 14.5° and 33.5° could be ascribed to the (002) and (101) facets of the 2H-phase MoS2 (JCPDS No. 65-0160). With the addition of Re ions, the (002) facets of Re–SA/MoS2 shifted to lower 2θ values compared with the pristine MoS2 (Fig. S1, ESI†), which indicates the Re SA were successfully doped into the lattice of MoS2.25 Scanning electron microscopy (SEM) images (Fig. S2, ESI†) and transmission microscopy (TEM) images (Fig. 1c and Fig. S3a–c, ESI†) revealed that both MoS2 and Re–SA/MoS2 possessed a typical flower-like nanosheet structure. Moreover, compared with MoS2, the surfaces of Re–SA/MoS2 were rougher and the size of the Re–SA/MoS2 nanosheets had a significant reduction in the lateral diameter, implying that the nanosheet morphology of Re–SA/MoS2 was retained after the introduction of the Re SA.25 From the high-resolution TEM (HRTEM) images (Fig. 1d), the lattice fringe distance of 0.69 nm corresponding to the (002) planes of Re–SA/MoS2 is slightly larger than that of pristine MoS2 (0.65 nm) (Fig. S3d, ESI†), reflecting a lattice expansion after incorporation of the Re SA, which is consistent with the XRD observation. The images of energy dispersive X-ray spectrometry (EDS) elemental mapping (Fig. 1e and Fig. S3e, ESI†) proved a homogeneous distribution of the Re SA over the MoS2 nanosheets.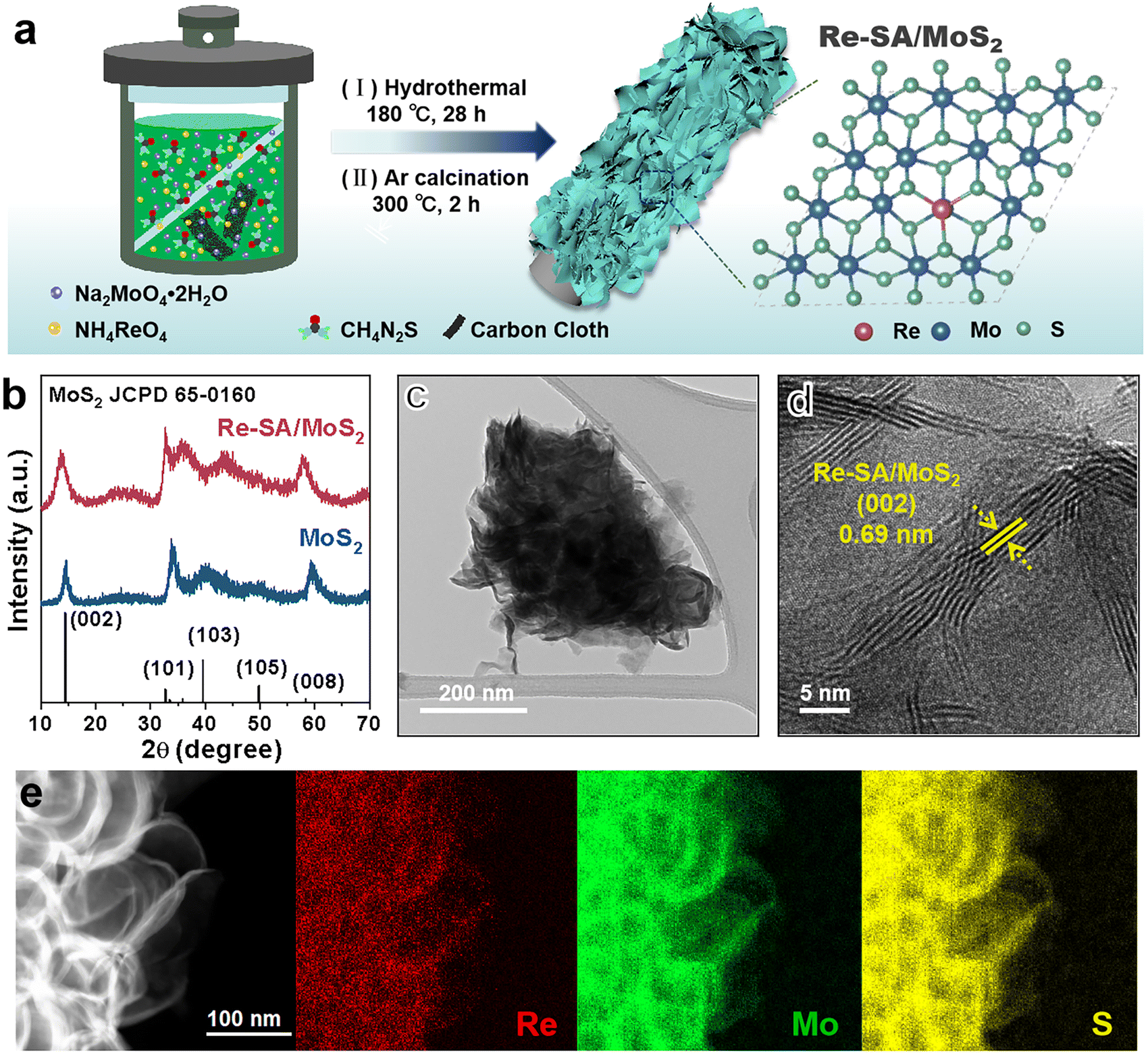 | ||
| Fig. 1 (a) Schematic illustration of the fabrication of the Re–SA/MoS2 catalysts. (b) XRD patterns of the fabricated MoS2 and Re–SA/MoS2 catalysts. (c and d) TEM and HRTEM images of the Re–SA/MoS2 catalyst. (e) EDS mapping of the Re–SA/MoS2 catalyst. | ||
To further validate the 2H to 1T phase conversion of MoS2 and the atomic dispersion of Re sites in Re–SA/MoS2, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was performed. As illustrated in Fig. 2a and b, the average (101) interlayer spacing with ∼0.27 nm and a unique honeycomb-like structure are displayed in MoS2. However, the averaged (101) interlayer spacing of Re–SA/MoS2 increased to 0.29 nm (Fig. 2c), corresponding to the probable existence of the surface tensile strain from the ambient Re atoms.26 The magnified HAADF-STEM images (Fig. 2b and d) combined with the indicated line 1 and 2 profiles (Fig. 2e) exhibited that a natural atomic pattern of 2H-phase MoS2 is Mo–S–Mo, but 1T-phase MoS2 shows a distinct atomic pattern of the two S atoms between neighboring Mo atoms, confirming that the crystal phase of MoS2 was transformed into the 1T-phase in Re–SA/MoS2. Importantly, the existence of the isolated bright dots was marked by yellow circles (Fig. 2c and d), indicating that the Re sites were dispersed in the MoS2 in the form of single atoms.27 Further direct evidence was corroborated by the intensity profile collected at line 3 (Fig. 2e), where the higher HAADF intensity of the third atom is observable, indicating that the heavier Re SA (Z = 75) were possibly anchored to the surface of MoS2 (referred to as “adsorption”) or substituted the lighter Mo atom (Z = 42).28,29 Raman spectroscopy was also applied to further distinguish the lattice structure of the 1T-phase and 2H-phase of MoS2.30 As shown in Fig. 2f, the bands located at 378 cm−1 and 404 cm−1 could be indexed as the in-plane E2g1 vibration mode and the out-of-plane A1g vibration mode of 2H-MoS2. In contrast, the new prominent bands at 146 cm−1, 196 cm−1, 283 cm−1, and 337 cm−1 were attributed to the J1, J2, E1g, and J3 of 1T-phase of MoS2 in the Re–SA/MoS2 nanosheets, which confirmed that spontaneous phase transition of MoS2 from the 2H to the 1T phase could be achieved with the assistance of the Re SA.31 Notably, the low vibrational mode intensity of E2g1 and 2H-A1g peak vanished in Re–SA/MoS2, indicating that a high degree of the 1T-phase of MoS2 was obtained. X-ray photoelectron spectroscopy (XPS) was also conducted to analyze the content of the 2H-to-1T crystal phase and surface chemical states of different elements. The full XPS spectra (Fig. S4a and b, ESI†) exhibited the existence of Re in the structure of MoS2, and the surface Re concentration was calculated as 8.80 at% for Re–SA/MoS2, in line with the EDS results (8.45 at% Re). As illustrated in Fig. S5a and b, ESI,† an apparent negative shift of 0.7 and 0.5 eV in the high-resolution XPS spectra of Mo 3d and S 2p of the Re–SA/MoS2 nanosheets compared to the pristine MoS2 moiety, suggests the formation of the 1T phase when the Re atom became accessed. The high-resolution Mo 3d spectra (Fig. 2g) of the Re–SA/MoS2 and MoS2 samples were divided into four typical peaks in terms of the standard deconvolution principles. The two peaks at 229.1 and 232.2 eV were attributed to Mo4+ 3d5/2 and Mo4+ 3d3/2 in 2H-phase MoS2, respectively32 while the two distinct peaks located at about 228.5 and 231.6 eV were present and attributed to 1T-phase MoS2 according to previous works.33 From the XPS results, the content of the 2H-phase-derived 1T phase of MoS2 in Re–SA/MoS2 was calculated to be about 85%, which can be ascribed to the Re atoms serving as electron donors within the lattice of MoS2 and thereby resulting in the conversion of the 2H-phase to the 1T-phase via gliding of the atomic plane of S.24,34 Besides, the two small peaks at 233.2 and 235.7 eV can be assigned to Mo6+ 3d5/2 and Mo6+ 3d3/2, implying that the MoS2 surface undergoes partial oxidation in air.35 Similarly, in the S 2p XPS spectra (Fig. 2h) of the Re–SA/MoS2 and MoS2 samples, two apparent peaks located at 163.1 eV and 161.9 eV correspond to S2− 2p1/2 and S2− 2p3/2, respectively. With the introduction of the Re species, the Re–SA/MoS2 nanosheets revealed that the S 2p peaks change to a lower binding energy of about 162.7 eV for S2− 2p1/2 and 161.4 eV for S2− 2p3/2. This result also confirmed that the increase in the electronic density in MoS2 was caused by the abundance of the 1T metallic phase.36 Meanwhile, the Re 4f XPS spectrum of the Re–SA/MoS2 sample exhibited a relatively high content of Re4+ 4f5/2 and Re4+ 4f7/2 oxidation states (Fig. 3a), implying that the local charge transfer occurred from the isolated Re atom to the adjacent S atom.37
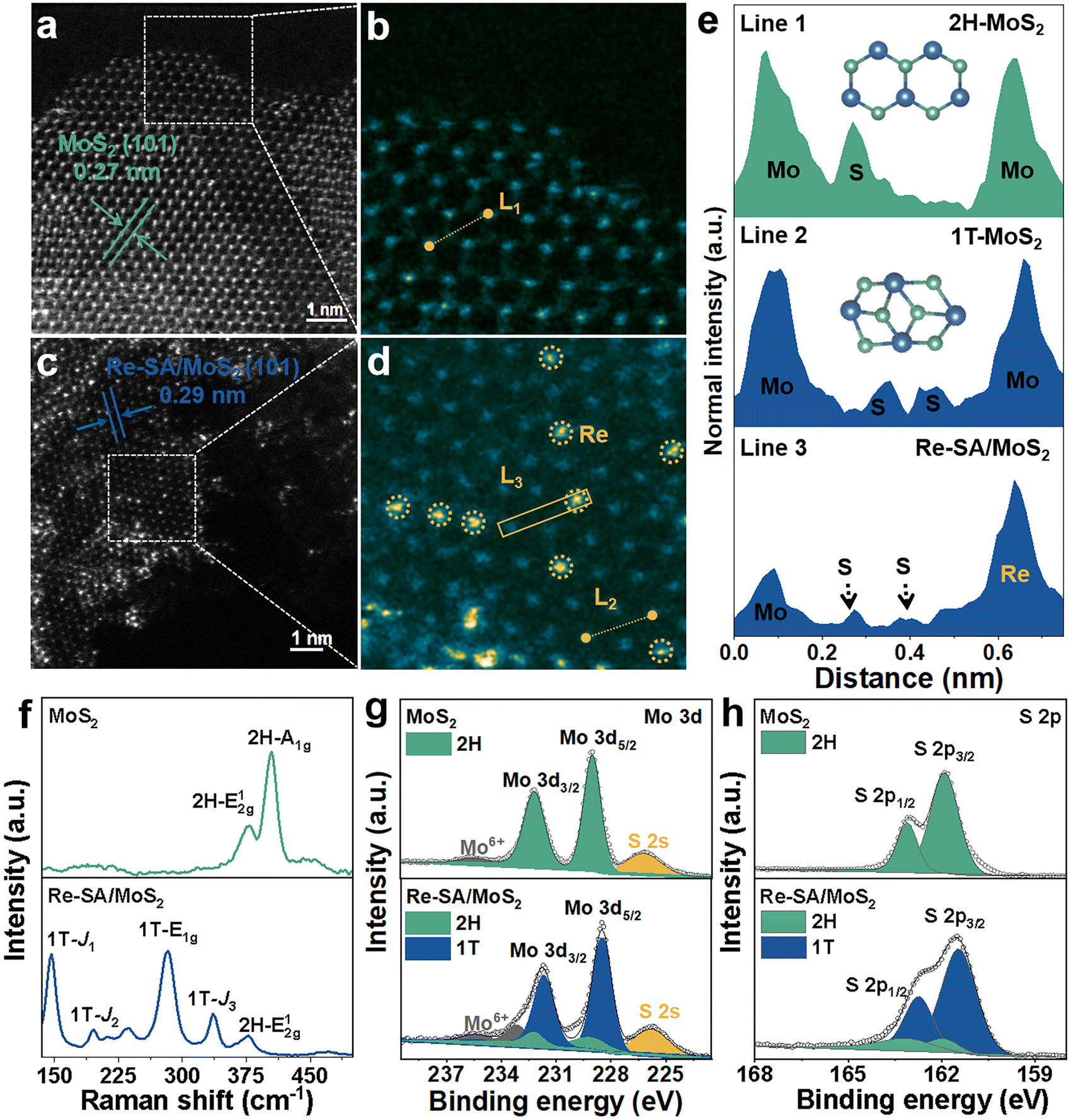 | ||
| Fig. 2 (a and c) HAADF and (b and d) enlarged HAADF images of the 2H-MoS2 and Re–SA/MoS2 catalysts. (e) HAADF intensity line profiles taken along the numbered lines indicated in b and d. (f) Raman spectra of the fabricated MoS2 and Re–SA/MoS2 catalysts. (g and h) XPS spectra of the Mo 3d and S 2p of the fabricated MoS2 and Re–SA/MoS2 catalysts. | ||
 | ||
| Fig. 3 (a) XPS spectrum of Re 4f in the Re–SA/MoS2 catalyst. (b) XANES spectra of Re L3-edge of Re foil, ReO2 and Re–SA/MoS2. (c) k3-weighted Re L3-edge Fourier-transformed EXAFS spectra for Re foil, ReO2 and Re–SA/MoS2. (d) EXAFS spectra (experimental and fitting) of Re L3-edge of Re–SA/MoS2. (e and f) Wavelet-transformation for Re L3-edge of Re foil and Re–SA/MoS2 in R space. | ||
To verify the electronic state and local coordination environment of the Re atoms and Mo atoms in Re–SA/MoS2, the X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements were further performed. In the Re L3-edge XANES spectra (Fig. 3b), the intensity of the white line peak for Re–SA/MoS2 is higher than that of the reference Re foil but lower than that of standard ReO2, suggesting that the isolated Re atom has a valence between Re0 (Re foil) and Re4+ (ReO2), agreeing with the XPS results. Also the k3-weighted Re L3-edge Fourier-transformed EXAFS spectra (Fig. 3c) showed that the peak at 2.02 Å was attributed to the first Re–S shell in the spectrum of Re–SA/MoS2. In comparison, the absence of a scattering peak at about 2.62 Å, derived from the Re–Re bond in the spectrum of Re foil, indicated the existence of the Re individual atoms rather than Re clusters.38 Additionally, the EXAFS fitting results (Fig. 3d and Table S1, ESI†) revealed the average coordination number about the central Re atoms to be 4.9 with a bond length of 2.37 Å for the Re–S scattering path, implying that Re atoms may exist as adsorption atoms or substitution within the MoS2 lattice.23,28 To observe the coordination information of Re sites in both k and R spaces, the wavelet transform (WT) of the Re L3-edge EXAFS spectra (Fig. 3e and f) was explored. The maximum intensity of Re foil was 12.3 Å−1 in the k space, corresponding to a Re–Re scattering path. However, the maximum intensity for Re–SA/MoS2 was observed around 6.6 Å−1 in the k space, demonstrating the formation of Re SA surrounded by S atoms, and Re atoms in Re–SA/MoS2 were atomically dispersed. Furthermore, the enlarged Mo K-edge XANES spectra for Re–SA/MoS2 (the inset in Fig. S6a, ESI†) were negatively shifted compared to those of MoS2, manifesting the strong interaction between MoS2 and Re atoms in Re–SA/MoS2. This was also reflected by the EXAFS spectroscopy and WT-EXAFS analysis of the Mo K-edge of MoS2 and Re–SA/MoS2 (Fig. S6 and Table S2, ESI†), which showed the two distinct peaks located at 1.99 and 2.88 Å attributed to Mo–S and Mo–Mo bonds (Fig. S6b, ESI†), respectively. Compared to MoS2, the peak intensity of Mo–Mo coordination of the Re–SA/MoS2 was reduced significantly, thus indicating a typical characteristic of 1T-phase MoS2.39 Based on these results, it could be speculated that the phase transformation and the electronic structure adjustment that occurred by incorporating the Re species into the lattice.15
Electrocatalytic performance toward the HER
The HER electrocatalytic activities were first performed in an alkaline media (1.0 M KOH), while MoS2, Re–SA/MoS2, and commercial Pt/C (20 wt%) were used as control samples. As depicted in Fig. 4a and b and Fig. S7a and b, ESI,† the iR-compensated linear sweep voltammetry (LSV) polarization curves displayed that the activity of the optimized Re–SA/MoS2 only required overpotentials of 38 mV and 161 mV to reach the −10 mA cm−2 and −100 mA cm−2, respectively, which are remarkably better than those observed for MoS2 (191 and 323 mV), commercial Pt/C (54 and 251 mV), and comparable to that of the state-of-the-art MoS2-based HER electrocatalysts (Table S3, ESI†). Additionally, the Tafel slope (Fig. 4c) of the MoS2 without the incorporation of the Re SA exhibited a value of 104 mV dec−1, much inferior to that of 66 mV dec−1 for the optimized Re–SA/MoS2 (Fig. S7b and c, ESI†), implying that the HER typically followed the Volmer–Heyrovsky mechanism with water dissociation to H* and OH* as the rate-determining step.40 Electrochemical impedance spectroscopy (EIS) was further conducted to probe the charge-transfer dynamics in the HER process. As illustrated in Fig. S7d and Table S4, ESI,† the optimized Re–SA/MoS2 catalysts possessed faster interfacial electron-transfer kinetics between the electrode and electrolyte, according to the smaller charge transfer resistance (Rct) than the MoS2 samples. Furthermore, the electrochemical HER performance was also measured in an acid media (0.5 M H2SO4). The LSV curves of the optimized Re–SA/MoS2 catalysts delivered a significantly lower overpotential of 34 and 120 mV to reach −10 mA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−2 and −100 mA cm−2 (Fig. 4d, e and Fig. S8a, b, ESI†), much superior to those of MoS2 (166 and 317 mV) and commercial Pt/C (43 and 200 mV), and surpassing those of most of the previously reported MoS2-based materials (Table S5, ESI†). The promising acidic HER activity of Re–SA/MoS2 was further verified by the lower charge transfer resistance (Rct) (Fig. S8c and Table S6, ESI†) with a more downward Tafel slope of 34 mV dec−1 compared to Pt/C (42 mV dec−1) (Fig. 4f and Fig. S8d, ESI†), suggesting that the HER process on Re–SA/MoS2 was accelerated by the rapid electron transfer kinetics and reduced energy barrier of the Volmer–Tafel step.41 The electrochemical surface area (ECSA) of catalysts under acidic media was investigated through the use of double-layer capacities (Cdl) in Fig. S9a and b, ESI;† the favorable Re–SA/MoS2 catalysts exhibited a higher ECSA value (Fig. S9c and d, ESI†) of 63 mF cm−2 than that of MoS2 (11 mF cm−2), confirming that the more plentiful active site was exposed through the implanted of Re SA for boosting the HER activity. The Re–SA/MoS2 catalysts exhibited superior durability without any noticeable degradation after 25 h at ∼20 mA cm−2 and 23 mA cm−2 for HER in both alkaline and acidic electrolytes, respectively (Fig. 4g and h). In addition, there were no significant changes in the TEM, HRTEM, and EDS mapping results for the verification of the morphology (Fig. S10 and S11, ESI†), and the lattice structure and the content of 1T-phase, were maintained after the stability test (Fig. S12 and S13, ESI†).
cm−2 and −100 mA cm−2 (Fig. 4d, e and Fig. S8a, b, ESI†), much superior to those of MoS2 (166 and 317 mV) and commercial Pt/C (43 and 200 mV), and surpassing those of most of the previously reported MoS2-based materials (Table S5, ESI†). The promising acidic HER activity of Re–SA/MoS2 was further verified by the lower charge transfer resistance (Rct) (Fig. S8c and Table S6, ESI†) with a more downward Tafel slope of 34 mV dec−1 compared to Pt/C (42 mV dec−1) (Fig. 4f and Fig. S8d, ESI†), suggesting that the HER process on Re–SA/MoS2 was accelerated by the rapid electron transfer kinetics and reduced energy barrier of the Volmer–Tafel step.41 The electrochemical surface area (ECSA) of catalysts under acidic media was investigated through the use of double-layer capacities (Cdl) in Fig. S9a and b, ESI;† the favorable Re–SA/MoS2 catalysts exhibited a higher ECSA value (Fig. S9c and d, ESI†) of 63 mF cm−2 than that of MoS2 (11 mF cm−2), confirming that the more plentiful active site was exposed through the implanted of Re SA for boosting the HER activity. The Re–SA/MoS2 catalysts exhibited superior durability without any noticeable degradation after 25 h at ∼20 mA cm−2 and 23 mA cm−2 for HER in both alkaline and acidic electrolytes, respectively (Fig. 4g and h). In addition, there were no significant changes in the TEM, HRTEM, and EDS mapping results for the verification of the morphology (Fig. S10 and S11, ESI†), and the lattice structure and the content of 1T-phase, were maintained after the stability test (Fig. S12 and S13, ESI†).
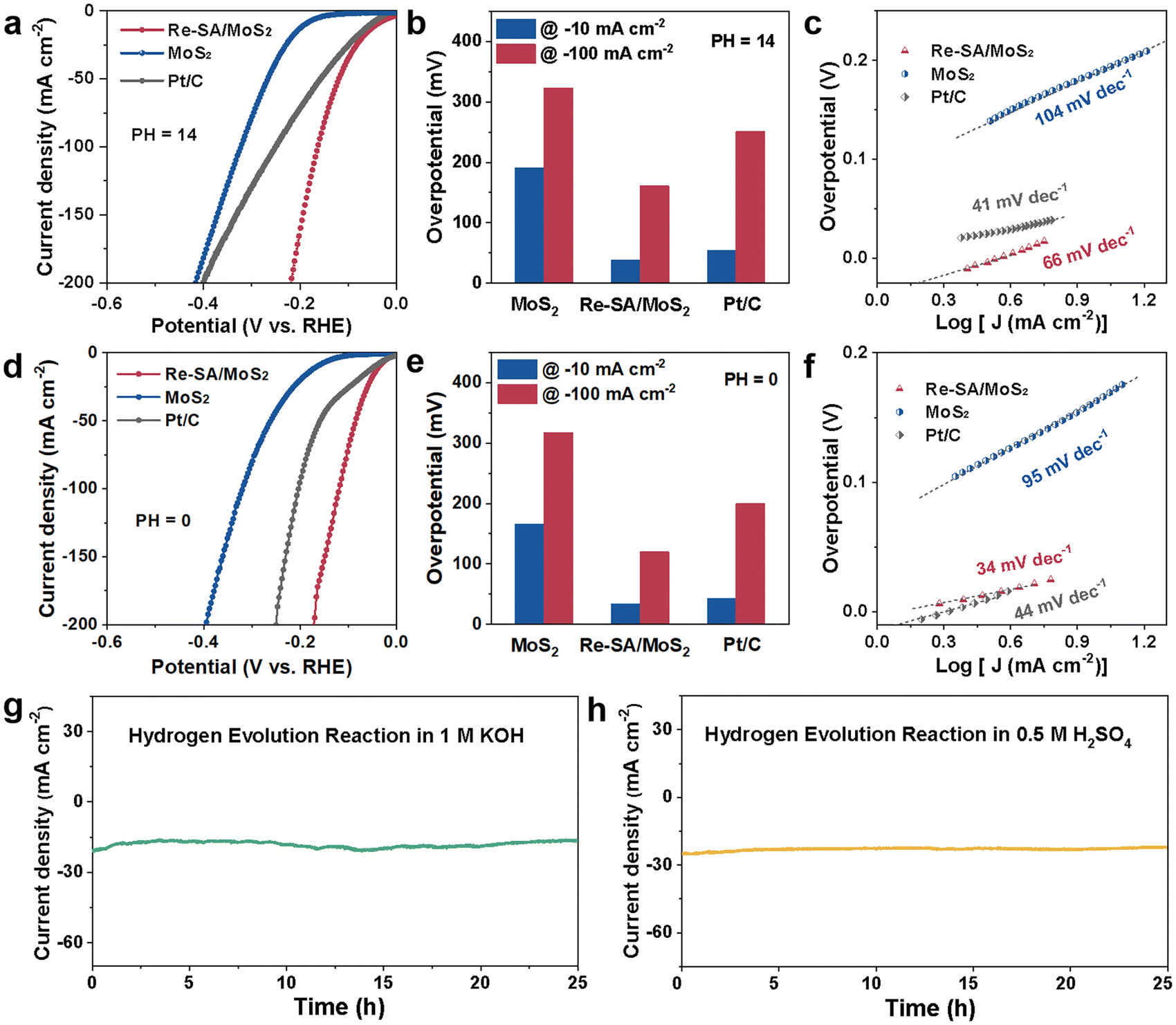 | ||
| Fig. 4 HER performances of the Re–SA/MoS2 electrocatalysts and control samples in H2-saturated 1.0 M KOH: (a) LSV curves, (b) overpotentials at current densities of −10 and −100 mA cm−2, and (c) Tafel plots toward HER. The HER performances of Re–SA/MoS2 electrocatalysts and control samples in H2-saturated 0.5 M H2SO4: (d) LSV curves, (e) overpotentials at current densities of −10 and −100 mA cm−2, (f) Tafel plots toward HER. (g and h) Long-term cycling stability test of Re–SA/MoS2 for HER in 1.0 M KOH and 0.5 M H2SO4, respectively. | ||
Catalytic mechanisms
To unravel the underlying origin of superior HER catalytic activities with the Re atoms incorporated into MoS2, the reaction Gibbs free energies profiles on HER of pure MoS2 and Re–SA/MoS2 have been deliberately assessed through first-principles DFT calculations. The constructed slab models for DFT calculations are illustrated in Fig. S14, ESI.† As shown, the Re atom was substituted at the Mo site or adsorbed on the Mo site via covalent bonds with S atoms, which are denoted as “Resub–SA/MoS2” and “Reads–SA/MoS2”. Moreover, the pristine MoS2 (2H-MoS2) and the reference 1T-phase MoS2 (1T-MoS2) were also constructed for comparison. The optimized geometry structures with the adsorption intermediates (H2O*, OH* + H*, and H*) of HER on 2H-MoS2, 1T-MoS2, Reads–SA/MoS2, and Resub–SA/MoS2 are shown in Fig. 5a and Fig. S15, ESI.† As indicated in Fig. 5b, the introduction of Re atoms into MoS2 triggers the water hydrolysis reaction with a significantly lower energy barrier (−0.22 eV) than pure MoS2 (3.52 eV), thereby accelerating the water splitting. This much easier water association on newly implanted Re atoms of Re–SA/MoS2 facilitates faster proton supply for the subsequent HER process. Calculated free energy changes of adsorbed hydrogen (ΔGH*) on possible active sites as key descriptors for evaluating HER electrocatalysis are illustrated in Fig. 5c, from which it is apparent that the highest HER activity is strongly associated with the absolute value of ΔGH* close to zero.42 Our results reveal that the ΔGH* values of Reads–SA/MoS2 on the S site (−0.2 eV) are much closer to the optimal value than those of pure 2H-MoS2 on the S site (2.1 eV), pure 1T-MoS2 on S site (0.8 eV), and Resub–SA/MoS2 on S site (−0.8 eV), implying that the integration of Re atoms could redistribute electronic structures and thus optimize the H adsorption/desorption on the MoS2 surface.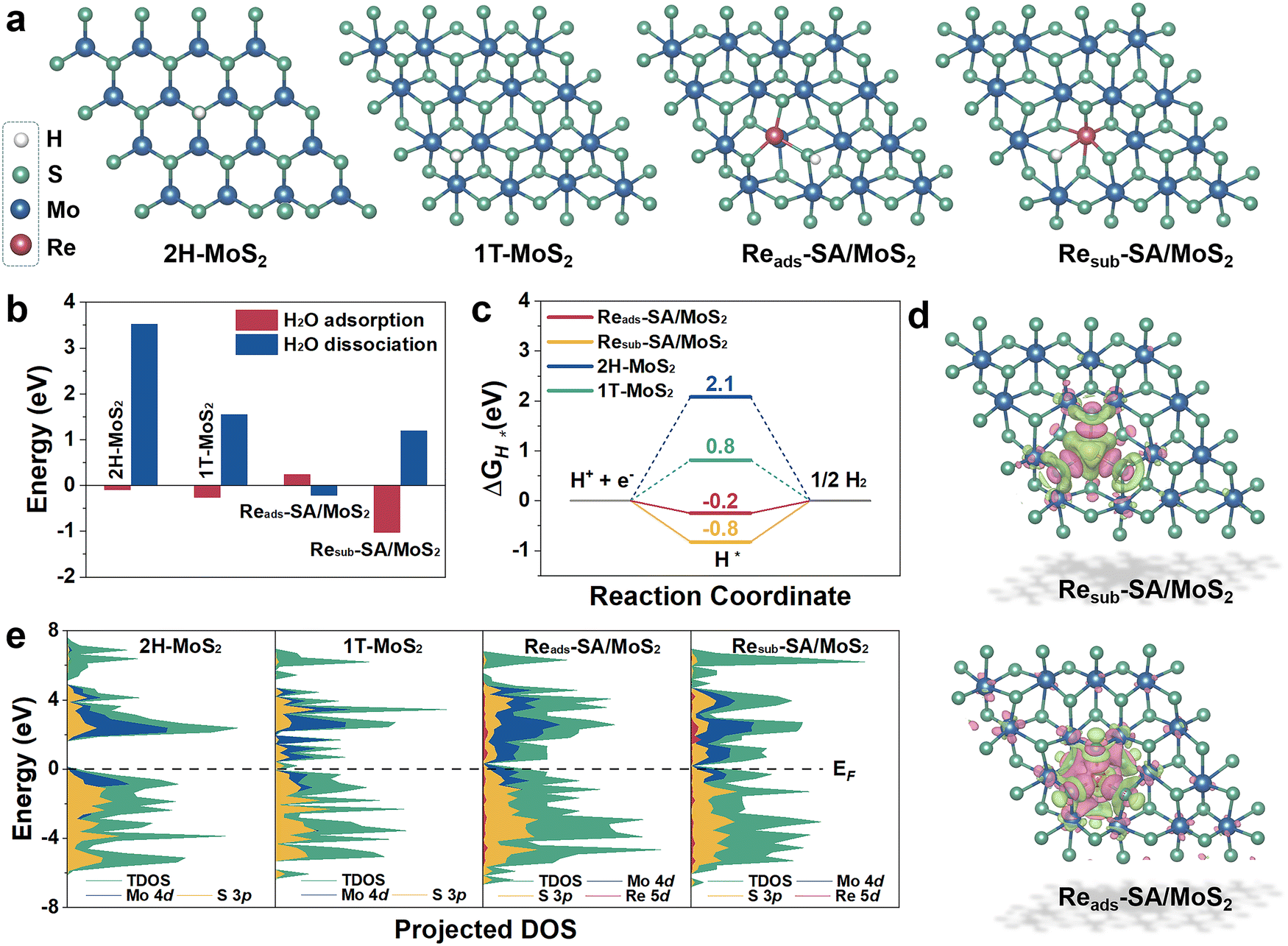 | ||
| Fig. 5 Theoretical calculations on the HER. (a) Atomic structures (top views) of the optimized slabs in 2H-MoS2, 1T-MoS2 and two Re doping configurations. (b) Calculated adsorption/dissociation energy of H2O for hydrogen evolution reaction on the 2H-MoS2, 1T-MoS2, Reads–SA/MoS2 and Resub–SA/MoS2 surface. (c) Free energy diagram of 2H-MoS2, 1T-MoS2, Reads–SA/MoS2 and Resub–SA/MoS2 for HER. (d) Differential charge density distributions of Reads–SA/MoS2 and Resub–SA/MoS2. The green and purple areas represent electron accumulation and depletion, respectively. (e) Projected DOS of 2H-MoS2, 1T-MoS2, Reads–SA/MoS2 and Resub–SA/MoS2. | ||
Furthermore, the corresponding differential charge density distribution and density of state (DOS) were simulated for investigating the intrinsic activity of Re–SA/MoS2 and elucidating the effect of distinct electronic structures and activity in the HER. As exhibited in Fig. 5d, the significant differential charge density occurs around Re–S bonds after the Re atoms substitute/adsorption in the Mo atoms, meaning that introducing Re atoms into MoS2 can remarkably adjust the sample's electronic structure. Additionally, the electronic characteristic of the different samples is further indicated according to the corresponding DOS (Fig. 5e). The pristine 1T-MoS2 and Re–SA/MoS2 possess an inherent metallic nature. Intriguingly, the introduction of Re atoms into MoS2 triggers the computing DOS to become more pronounced around the Fermi level (Ef) as compared to MoS2 due to the hybridization of Re 5d with Mo 4d and S 3p orbitals, indicating more increased metallic features and higher electrical conductivity on the Re–SA/MoS2 catalysts than the pure 2H-MoS2, leading to more efficient electrochemical reactions. This electronic behavior thus reveals that introducing the Re SA could result in local charge redistribution and modulate the electronic structure of 2H-MoS2 to improve the hydrogen evolution performance.
Conclusions
In summary, we have successfully demonstrated that the introduction of single-atom Re on MoS2 induced the phase transition (ca. 85%) of MoS2 from the 2H-phase to the 1T-phase for improving the catalytic HER performance. The optimized Re–SA/MoS2 exhibited a Pt-like HER performance, with an overpotential of 34 mV and 38 mV at a current density of −10 mA cm−2 in both acidic and alkaline media, much superior to that of pristine MoS2 and comparable to that of the state-of-the-art Pt/C. The considerable enhancement of catalytic activity for the Re–SA/MoS2 nanosheets could be ascribed to the following reasons. First, the conductivity properties of MoS2 were improved by an electron injection effect of the single atom Re for modulating the lattice structure of 2H-MoS2 to promote phase conversion to the 1T phase. Second, single-atom Re acted as a catalytic site, effectively accelerating the water dissociation kinetics. Third, the adsorption of the H intermediate with the energy barrier of the rate-determining step was reduced by the strong synergistic between Re–SA and MoS2, further facilitating the subsequent H2 generation. In short, this work's phase transition with a single-atom engineering strategy will enrich the design of advanced HER catalysts and has broad applicability for other catalytic systems.Author contributions
Conceptualization: J. Y., T. Z., L. S., and H. L. Methodology: J. Y., Y. Q., Q. W., and L. S. Formal analysis: J. Y. and Y. Q. Investigation: J. Y., Y. Q., and Q. W. Visualization: J. Y. and L. S. Supervision: C. S., L. S., and T. Z. Writing – original draft: J. Y., and L. S. Writing – review and editing: J. Y., C. S., H. Y., L. S., and T. Z.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Projects for Fundamental Research and Development of China (2021YFA1500803), the National Natural Science Foundation of China (51825205, 52120105002, 22088102, and 22279150), the Beijing Natural Science Foundation (2222080), the Youth Innovation Promotion Association of the CAS (Y2021011) and the Institute for Basic Science of Korea (IBS-R011-D1). The 1W1B beamline of the Beijing Synchrotron Radiation Facility (BSRF) is acknowledged for XAFS experiments.Notes and references
- X. Wang, Y. Zhang, J. Wu, Z. Zhang, Q. Liao, Z. Kang and Y. Zhang, Chem. Rev., 2022, 122, 1273–1348 CrossRef CAS PubMed.
- J. Yu, T. A. Le, N. Q. Tran and H. Lee, Chem. – Eur. J., 2020, 26, 6423–6436 CrossRef CAS PubMed.
- W.-H. Lai, L.-F. Zhang, W.-B. Hua, S. Indris, Z.-C. Yan, Z. Hu, B. Zhang, Y. Liu, L. Wang, M. Liu, R. Liu, Y.-X. Wang, J.-Z. Wang, Z. Hu, H.-K. Liu, S.-L. Chou and S.-X. Dou, Angew. Chem., Int. Ed., 2019, 58, 11868–11873 CrossRef CAS PubMed.
- Y. Li, Y. Sun, Y. Qin, W. Zhang, L. Wang, M. Luo, H. Yang and S. Guo, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS.
- S. Zhang, X. Zhang, Y. Rui, R. Wang and X. Li, Green Energy Environ., 2021, 6, 458–478 CrossRef CAS.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 17033 CrossRef CAS.
- Q. Fu, J. Han, X. Wang, P. Xu, T. Yao, J. Zhong, W. Zhong, S. Liu, T. Gao, Z. Zhang, L. Xu and B. Song, Adv. Mater., 2021, 33, 1907818 CrossRef CAS PubMed.
- Y. Cao, ACS Nano, 2021, 15, 11014–11039 CrossRef CAS PubMed.
- Q. Tang and D. Jiang, ACS Catal., 2016, 6, 4953–4961 CrossRef CAS.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- J. Zhu, Z. Wang, H. Yu, N. Li, J. Zhang, J. Meng, M. Liao, J. Zhao, X. Lu, L. Du, R. Yang, D. Shi, Y. Jiang and G. Zhang, J. Am. Chem. Soc., 2017, 139, 10216–10219 CrossRef CAS PubMed.
- Z. Liu, K. Nie, X. Qu, X. Li, B. Li, Y. Yuan, S. Chong, P. Liu, Y. Li, Z. Yin and W. Huang, J. Am. Chem. Soc., 2022, 144, 4863–4873 CrossRef CAS PubMed.
- S. Wang, D. Zhang, B. Li, C. Zhang, Z. Du, H. Yin, X. Bi and S. Yang, Adv. Energy Mater., 2018, 8, 1801345 CrossRef.
- S. Jiménez Sandoval, D. Yang, R. F. Frindt and J. C. Irwin, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 3955–3962 CrossRef PubMed.
- S. Wei, X. Cui, Y. Xu, B. Shang, Q. Zhang, L. Gu, X. Fan, L. Zheng, C. Hou, H. Huang, S. Wen and W. Zheng, ACS Energy Lett., 2018, 4, 368–374 CrossRef.
- X. Meng, C. Ma, L. Jiang, R. Si, X. Meng, Y. Tu, L. Yu, X. Bao and D. Deng, Angew. Chem., Int. Ed., 2020, 59, 10502–10507 CrossRef CAS PubMed.
- Z. Luo, H. Zhang, Y. Yang, X. Wang, Y. Li, Z. Jin, Z. Jiang, C. Liu, W. Xing and J. Ge, Nat. Commun., 2020, 11, 1116 CrossRef CAS PubMed.
- T. Rao, H. Wang, Y.-J. Zeng, Z. Guo, H. Zhang and W. Liao, Adv. Sci., 2021, 8, 2002284 CrossRef CAS PubMed.
- J. Zhang, X. Xu, L. Yang, D. Cheng and D. Cao, Small Methods, 2019, 3, 1900653 CrossRef CAS.
- Y. Li, Q. Gu, B. Johannessen, Z. Zheng, C. Li, Y. Luo, Z. Zhang, Q. Zhang, H. Fan, W. Luo, B. Liu, S. Dou and H. Liu, Nano Energy, 2021, 84, 105898 CrossRef CAS.
- K. Zhang, B. M. Bersch, J. Joshi, R. Addou, C. R. Cormier, C. Zhang, K. Xu, N. C. Briggs, K. Wang, S. Subramanian, K. Cho, S. Fullerton-Shirey, R. M. Wallace, P. M. Vora and J. A. Robinson, Adv. Funct. Mater., 2018, 28, 1706950 CrossRef.
- S.-Z. Yang, Y. Gong, P. Manchanda, Y.-Y. Zhang, G. Ye, S. Chen, L. Song, S. T. Pantelides, P. M. Ajayan, M. F. Chisholm and W. Zhou, Adv. Mater., 2018, 30, 1803477 CrossRef PubMed.
- X. Tian, D. S. Kim, S. Yang, C. J. Ciccarino, Y. Gong, Y. Yang, Y. Yang, B. Duschatko, Y. Yuan, P. M. Ajayan, J. C. Idrobo, P. Narang and J. Miao, Nat. Mater., 2020, 19, 867–873 CrossRef CAS PubMed.
- A. N. Enyashin, L. Yadgarov, L. Houben, I. Popov, M. Weidenbach, R. Tenne, M. Bar-Sadan and G. Seifert, J. Phys. Chem. C, 2011, 115, 24586–24591 CrossRef CAS.
- Y. Sun, Y. Zang, W. Tian, X. Yu, J. Qi, L. Chen, X. Liu and H. Qiu, Energy Environ. Sci., 2022, 15, 1201–1210 RSC.
- A. Kumar, X. Liu, J. Lee, B. Debnath, A. R. Jadhav, X. Shao, V. Q. Bui, Y. Hwang, Y. Liu, M. G. Kim and H. Lee, Energy Environ. Sci., 2021, 14, 6494–6505 RSC.
- Y. C. Lin, D. O. Dumcenco, H. P. Komsa, Y. Niimi, A. V. Krasheninnikov, Y. S. Huang and K. Suenaga, Adv. Mater., 2014, 26, 2857–2861 CrossRef CAS PubMed.
- I. S. Kwon, I. H. Kwak, S. Ju, S. Kang, S. Han, Y. C. Park, J. Park and J. Park, ACS Nano, 2020, 14, 12184–12194 CrossRef CAS PubMed.
- J. Deng, H. Li, J. Xiao, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- X. Geng, W. Sun, W. Wu, B. Chen, A. Al-Hilo, M. Benamara, H. Zhu, F. Watanabe, J. Cui and T. Chen, Nat. Commun., 2016, 7, 10672 CrossRef CAS PubMed.
- Y. Fang, J. Pan, J. He, R. Luo, D. Wang, X. Che, K. Bu, W. Zhao, P. Liu, G. Mu, H. Zhang, T. Lin and F. Huang, Angew. Chem., Int. Ed., 2018, 57, 1232–1235 CrossRef CAS PubMed.
- Z. Li, C. Li, J. Chen, X. Xing, Y. Wang, Y. Zhang, M. Yang and G. Zhang, J. Energy Chem., 2022, 70, 18–26 CrossRef CAS.
- Y. Yu, G.-H. Nam, Q. He, X.-J. Wu, K. Zhang, Z. Yang, J. Chen, Q. Ma, M. Zhao, Z. Liu, F.-R. Ran, X. Wang, H. Li, X. Huang, B. Li, Q. Xiong, Q. Zhang, Z. Liu, L. Gu, Y. Du, W. Huang and H. Zhang, Nat. Chem., 2018, 10, 638–643 CrossRef CAS PubMed.
- Y.-C. Lin, D. O. Dumcenco, Y.-S. Huang and K. Suenaga, Nat. Nanotechnol., 2014, 9, 391–396 CrossRef CAS PubMed.
- G. Wang, G. Zhang, X. Ke, X. Chen, X. Chen, Y. Wang, G. Huang, J. Dong, S. Chu and M. Sui, Small, 2022, 18, 2107238 CrossRef CAS PubMed.
- M. Liu, J.-A. Wang, W. Klysubun, G.-G. Wang, S. Sattayaporn, F. Li, Y.-W. Cai, F. Zhang, J. Yu and Y. Yang, Nat. Commun., 2021, 12, 5260 CrossRef CAS PubMed.
- Y. Shi, Z.-R. Ma, Y.-Y. Xiao, Y.-C. Yin, W.-M. Huang, Z.-C. Huang, Y.-Z. Zheng, F.-Y. Mu, R. Huang, G.-Y. Shi, Y.-Y. Sun, X.-H. Xia and W. Chen, Nat. Commun., 2021, 12, 3021 CrossRef PubMed.
- Z. Li, X. Yan, D. He, W. Hu, S. Younan, Z. Ke, M. Patrick, X. Xiao, J. Huang, H. Wu, X. Pan and J. Gu, ACS Catal., 2022, 12, 7687–7695 CrossRef CAS.
- Q. Liu, X. Li, Q. He, A. Khalil, D. Liu, T. Xiang, X. Wu and L. Song, Small, 2015, 11, 5556–5564 CrossRef CAS PubMed.
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang, Z. Zhang, P. Zhang, X. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS PubMed.
- Z. Zheng, L. Yu, M. Gao, X. Chen, W. Zhou, C. Ma, L. Wu, J. Zhu, X. Meng, J. Hu, Y. Tu, S. Wu, J. Mao, Z. Tian and D. Deng, Nat. Commun., 2020, 11, 3315 CrossRef CAS PubMed.
- X. Zhang, A. Chen, L. Chen and Z. Zhou, Adv. Energy Mater., 2022, 12, 2003841 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section. See DOI: https://doi.org/10.1039/d3ey00037k |
| ‡ J. Y. and Y. Q. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |