
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring the MOF structure via ligand optimization afforded a dandelion flower like CoS/Co–Nx/CoNi/NiS catalyst to enhance the ORR/OER in zinc–air batteries†
Mohan
Gopalakrishnan
 a,
Mohamad
Etesami
a,
Jayaraman
Theerthagiri
b,
Myong Yong
Choi
b,
Suttipong
Wannapaiboon
c,
Mai Thanh
Nguyen
d,
Tetsu
Yonezawa
d and
Soorathep
Kheawhom
*aef
a,
Mohamad
Etesami
a,
Jayaraman
Theerthagiri
b,
Myong Yong
Choi
b,
Suttipong
Wannapaiboon
c,
Mai Thanh
Nguyen
d,
Tetsu
Yonezawa
d and
Soorathep
Kheawhom
*aef
aDepartment of Chemical Engineering, Faculty of Engineering, Chulalongkorn University, Bangkok 10330, Thailand. E-mail: soorathep.k@chula.ac.th
bCore-Facility Center for Photochemistry & Nanomaterials, Department of Chemistry (BK21 FOUR), Research Institute of Natural Sciences, Gyeongsang National University, Jinju 52828, Republic of Korea
cSynchrotron Light Research Institute, 111 University Avenue, Muang District, Nakhon Ratchasima 30000, Thailand
dDivision of Materials Science and Engineering, Faculty of Engineering, Hokkaido University, Hokkaido 060-8628, Japan
eBio-Circular-Green-economy Technology & Engineering Center (BCGeTEC), Faculty of Engineering, Chulalongkorn University, Bangkok 10330, Thailand
fCenter of Excellence on Advanced Materials for Energy Storage, Chulalongkorn University, Bangkok 10330, Thailand
First published on 7th November 2022
Abstract
Due to their affordability and good catalytic activity for oxygen reactions, MOF-derived carbon composites containing metal alloys have piqued interest. However, during synthesis, MOFs have the disadvantage of causing significant carbon evaporation, resulting in a reduction of active sites and durability. This study proposes tailoring the molecular structure of MOFs by optimizing bipyridine and flexible 4-aminodiacetic terephthalic acid ligands, which have numerous coordination modes and framework structures, resulting in fascinating architectures. MOF frameworks having optimized N and O units are coordinated with Co and Ni ions to provide MOF precursors that are annealed at 700 °C in argon. The MOF-derived Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 catalyst exhibits excellent catalytic activity, revealing an ORR half-wave potential of 0.86 V and an overpotential (OER) of 196 mV at 10 mA cm−2, a potential gap of 0.72 V and a Tafel slope of 79 mV dec−1. The proposed strategy allows for the rational design of N-coordinated Co and CoNi alloys attached to ultrathin N, S co-doped graphitic carbon sheets to enhance bifunctional activity and sufficient active sites. Consequently, the zinc–air battery using the synthesized catalyst shows a high peak power density of 206.9 mW cm−2 (Pt/C + RuO2 116.1 mW cm−2), a small polarization voltage of 0.96 V after 370 h at 10 mA cm−2, and an outstanding durability of over 2400 cycles (400 h). The key contributions to the superior performance are the synergetic effects of the CoNi alloys plus the N,S-incorporated carbon skeleton, due to the small charge transfer resistances and enhanced active sites of CoNi, metal–S, and pyridinic N.
1. Introduction
In a wide range of energy conversion and storage technologies, the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER) both play crucial roles in hydrogen electrolyzers, fuel cells, and rechargeable metal–air batteries.1–4 However, enhancing the ORR and OER activities remains a significant problem due to complex mechanisms, intermediates, and huge energy barriers.5,6 Noble metal catalysts i.e. ORR (Pt/C) and OER (RuO2/IrO2) are limited in their use for commercial applications due to high cost and lack of resources.7,8 Furthermore, none of them have enough bifunctional activity for both the ORR and OER making them unsuitable for thebifunctional activities of rechargeable Zn–air batteries.9,10 It is important for these electrochemical devices to develop multifunctional catalysts with highly efficient ORR and OER kinetics.11 Recently, alternative non-noble metal based electrocatalysts (Fe, Co, Ni, Mn, and Mo),12,13 metal alloys,14 carbon-based materials,15–18 transition metal oxides19,20/hydroxides21/MOFs22–24 and their hybrids25 have been extensively investigated as highly efficient and low-cost oxygen catalysts for bifunctional activities. As a result, finding a suitable material for non-noble metal electrocatalysts with remarkable stability, high specific surface areas, large pores, and lots of active sites is important.26Metal–organic frameworks (MOFs) with excellent architecture and large surface areas capable of enabling reactions via electron transport in energy storage or conversion process are promising new platforms for electrocatalysis.27 A combination of rich porosity and intrinsic nanopores (less than 100 nm diameter pores) provides a favorable environment for the development and preparation of exceptionally well-organized oxygen electrocatalysts.28,29 MOFs also contain inorganic components such as layers, clusters, chains, or three-dimensional arrangements that are linked to organic linkers via strong bonds with complexing groups such as phosphonates, carboxylates, and N-containing chemicals.30,31 Prior studies in the electrochemical field have focused on element doping and size modifcation of catalysts using MOF precursors.32 Existing research suggested that changing the morphologies can change catalytic performance.33 It is therefore still highly desirable but challenging to develop novel methods for the synthesis of morphology-controlled MOF templates to get customized active components and structures with high electrocatalytic performance.34,35 In practice, controlling coordination geometry like oxidation state of metal cations, binding modes of ligands and solvent molecules is critical for the creation of MOFs with morphological and structural diversity.36 Ligands can effectively adopt the topology of coordination nodes, various binding modes and certain solvent molecules to compete for unoccupied metal sites.37 Due to the favorable rearrangement of electronic structures to minimize kinetic energy barriers, a multi-metallic species frequently generates synergistically improved catalytic activity.38 Furthermore, during the calcination of MOFs, the inclusion of a secondary metal can inhibit the total agglomeration kinetics of each metal (MOFs).38
Lately, MOF-derived nonprecious bimetallic alloy materials, particularly using transition metals, have received much attention as oxygen electrocatalysts, because of their highly active catalytic activities in the ORR and OER. Several findings have shown that metal alloy catalysts such as FeCo,39,40 FeNi,32,41 Fe–Mn,42 and NiCo14,15,43 have better activity than their single metal materials and that the intrinsic polarity that can be provided by a connection between two metals might help catalytic activities. It is noted that metal alloys without a carbon support cannot achieve outstanding ORR/OER performance. The combination of metal alloy nanoparticles with hetero atoms and graphitic carbon that have high stability, superior conductivity. A large active surface area is preferable for enhancing the catalytic activity of alloy-based materials, thus favors increasing the number of active sites, as well as protecting metal alloy nanoparticles from aggregation. For example, Sheng et al. (2021) reported an efficient oxygen electrocatalyst made of NiCo alloy nanoparticles anchored on a carbon nanorod decorated with CNTs.44 Tan et al. (2022) observed that the electronic structure of the encapsulated CoNi alloy exhibited good reversible oxygen catalytic activity and stability.45 Thangasamy et al. (2022) investigated CoPi–NPC nanosheets for the ORR in ZABs.46 Furthermore, Sim et al. (2022) demonstrated that iron–cobalt/cobalt–nickel oxide dispersed on a reduced graphene oxide electrode exhibited superior stability at a high current density and high-rate rechargeable ZABs.12
During synthesis, MOFs have the drawback of carbon evaporation causing significantly reduced active sites and catalytic durability. However, this work suggests tailoring the molecular architectures of MOFs using the optimization of flexible multi-carboxylate and 4,4-bpy ligands. The electrocatalysts were derived from bimetallic MOFs having different ratios of ligands i.e. 4,4-bipyridine (4,4-bpy) and 4-aminodiacetic terephthalic acid (H4adtp) in the presence of polar solvents. It is acknowledged that electrochemical activities (ORR and OER) and ZAB performance can be controlled by varying the ratios of 4,4-bpy and H4adtp ligands. The corresponding electrochemical performance of the heterostructure built of bimetallic sulfide and alloy composite materials was enhanced in terms of electron transport, adsorption optimization, and durability. The method is simple, rapid, and low-cost, and it sheds light on the synthesis of high-efficiency reversible oxygen electrocatalysts for large-scale production of a variety of energy devices such as electrolytes, fuel cells, and metal–air batteries.
2. Experimental section
2.1. Chemicals and materials
Commercially available materials such as nickel(II) sulfamate tetrahydrate, cobalt(II) acetate tetrahydrate, and 4,4-bipyridine, purchased from Merck, were used as received. For all experiments, analytical grade solvents: ethanol, dimethylformamide and deionized water were used. As described previously in the literature, 4-aminodiacetic terephthalic acid was synthesized (Scheme S1†).47 CoNi alloy nanoparticles were prepared by the method reported previously.48 Ni foam, 1 mm thick, was employed as a substrate for an air electrode and as a current collector for a negative electrode (Qijing Trading Co., Ltd). The Ni foam was cleaned with 37% hydrochloric acid (Qchemical Co., Ltd). Both zinc acetate (Merck) and potassium hydroxide pellets (CT Chemical Co., Ltd) were used to make the electrolyte. For the anode, a 2 mm thick Zn sheet (Zn 99.995 percent) was purchased from Sirikul Engineering Ltd.2.2. Synthesis of bimetallic MOF precursors
An ethanolic solution of (10 ml) of 4,4-bipyridine (0.156 g, 2 mmol) was added dropwise to a stirred solution of Ni(SO3NH2)2·4H2O (0.325 g, 1 mmol) and Co(CH3COO)2·4H2O (0.249 g, 1 mmol), containing dimethylformamide (10 ml); H4adtp (0.253 g, 1 mmol) in water (10 ml) was added dropwise to the mixture. As a result, the solution color changed from green to light blue. The mixture was stirred continuously for 6 h at ambient temperature and pH was adjusted to 8 using concentrated HCl. After the solution was transferred to a 100 ml Teflon lined autoclave, it was kept in an oil bath for 72 h (120 °C). After cooling, the obtained blue precipitate of the MOF precursors was washed several times with distilled water and ethanol before being dried in a vacuum oven (60 °C for 6 h).2.3. Synthesis of electrocatalysts
The resulting precursors were pyrolyzed in a tube furnace at 700 °C for 2 hours in an argon environment at a heating rate of 3 °C min−1. Following that, a dandelion flower-like hierarchical catalyst made of N and S co-doped carbon and carbon-encapsulated CoNi alloy nanoparticles was prepared. The catalysts viz. Co9S8/CoNi/Ni3S2@CNS-1, Co9S8/Co–Nx/CoNi/Ni3S2@CNS-2, Co9S8/CoNi/Ni3S2@CNS-3, and Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 were obtained by optimizing the ligand ratios. Supporting documentation contains details about electrocatalyst characterization, electrochemical measurement, electrode fabrication of the air cathode and assembly of ZABs.3. Results and discussion
3.1. Synthesis
The hierarchical electrocatalyst was prepared using a reaction of 4,4-bpy and 4-aminodiacetic terephthalic acid ligands and Ni and Co metal ions in the presence of DMF/ethanol/water. The use of neutral N-donor ligands (4,4-bpy), as pillars to space 2D layers based on polycarboxylate ligands, is an effective and controllable method of generating hierarchical architectures.49 Architectures are frequently built with mixed ligands as linkers; single linkers are uncommon because most linkers are difficult to build in one mode while spacing them in the other. Fig. 1 shows the distinct function of both linkers viz. 4,4-bipyridine and 4-aminodiacetic terephthalic acid. Compared with aromatic rigid carboxylates (BTC),50 the flexible multidentate iminodiacetate functionality can easily chelate and bridge metals by adopting diverse bonding modes. Nickel–cobalt MOF clusters were generated homogeneously and remodeled into carbon frameworks after drying and subsequent carbonization at 700 °C for 2 h under an Ar atmosphere. The ligands in the MOF structures (4,4-bpy and H4adtp) can be easily optimized with a constant molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for Ni2+ and Co2+, as shown in Fig. 2. Controlled calcination can produce CoNi alloy nanoparticle/carbon composites derived from MOFs with unique structures, tailorable porosity, and large surface areas suitable for electrocatalytic applications (OER/ORR).51 During the heat treatment process, the formation of alloy nanoparticles and carbon materials occurs simultaneously, and the CoNi-based nanoparticles may be uniformly dispersed in carbon frameworks, which is still extremely difficult to achieve via other methods. Furthermore, carbonaceous materials used in three-dimensional porous hierarchical designs have been proven as an effective way to enhance electrocatalytic performance.52,53 Thus, a variety of interconnected nanostructures are created when metal-based nanoparticles are incorporated into three-dimensional carbon frameworks with voids.38 The entire synthetic process is quite simple and facile, requiring only an oil bath and subsequently a tubular furnace.
:1 for Ni2+ and Co2+, as shown in Fig. 2. Controlled calcination can produce CoNi alloy nanoparticle/carbon composites derived from MOFs with unique structures, tailorable porosity, and large surface areas suitable for electrocatalytic applications (OER/ORR).51 During the heat treatment process, the formation of alloy nanoparticles and carbon materials occurs simultaneously, and the CoNi-based nanoparticles may be uniformly dispersed in carbon frameworks, which is still extremely difficult to achieve via other methods. Furthermore, carbonaceous materials used in three-dimensional porous hierarchical designs have been proven as an effective way to enhance electrocatalytic performance.52,53 Thus, a variety of interconnected nanostructures are created when metal-based nanoparticles are incorporated into three-dimensional carbon frameworks with voids.38 The entire synthetic process is quite simple and facile, requiring only an oil bath and subsequently a tubular furnace.
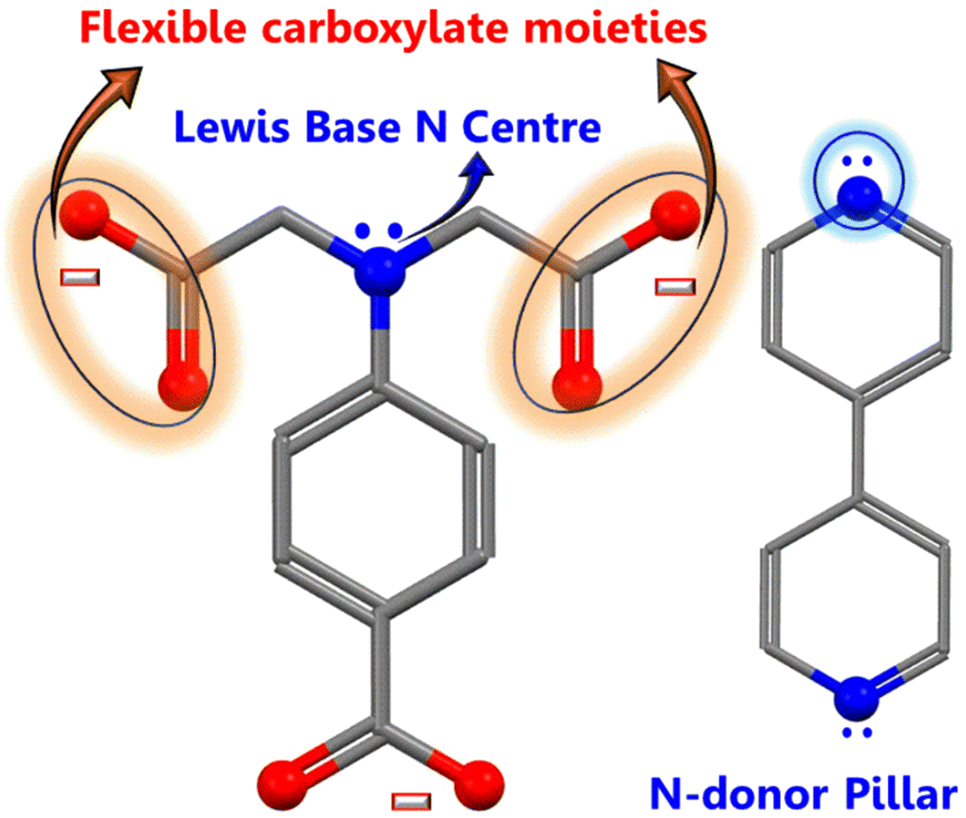 | ||
| Fig. 1 Ligand structures demonstrating their distinct functionalities. | ||
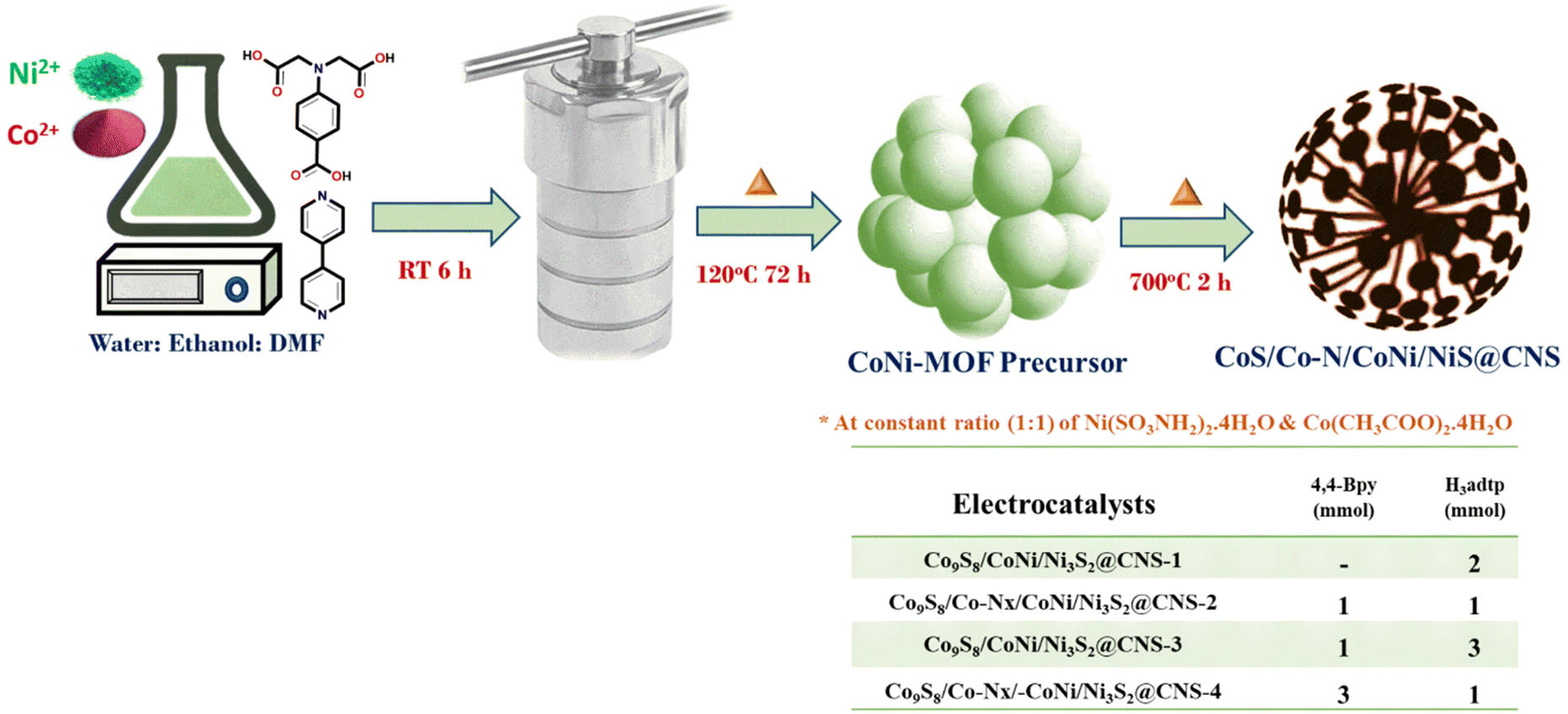 | ||
| Fig. 2 Scheme for the synthesis of electrocatalysts 1–4 (CoS/CoN/CoNi/Co–N@CNS) optimizing the ligand concentration. | ||
3.2. Morphological and structural analysis
Images from field-emission scanning electron microscopy (FE-SEM) reveal polyhedral structures with multilayered structures. In Fig. 3a, the prepared MOFs display homogeneous spherical surfaces, indicating that multi-carboxylate and 4,4-bpy were uniformly coordinated on the Ni–Co ion center. In Fig. 3b, after pyrolysis, dandelion flower-like structures (3 μm) with a rough surface are observed; their edges still had a similar spheroid morphology. The Ni–Co elements are largely found in the particles and overlap with the N and S elements, indicating the formation of CoNi alloy nanoparticles. Consequently, the diameters of the carbon nanofibers are seen to differ due to the optimization of the ligands (50–100 nm). In Fig. 3c–f, the catalyst morphology was investigated further using TEM and EDS. The hierarchical structure of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 with extended graphitic carbon networks on the surface of carbon polyhedra is confirmed via TEM. In Fig. 3c, CoNi alloys show a polyhedron-like shape with less than 10 nm diameter nanoparticles hanging on the surface and edges. In Fig. 3d and S2,† the metal sulfides (Co9S8 and Ni3S2) appear homogeneous on the surface carbon structures.54 The creation of the heterojunction55 is further confirmed by high-resolution TEM (HRTEM), which also exhibits distinct lattice fringes and close contact between Co9S8 (440) and Ni3S2 (110) as shown in Fig. 3d and e. In Fig. 3f (inset), the (111) plane of CoNi and the (002) plane of carbon are represented by lattice spacings of 0.521 and 0.316 nm in the HRTEM image of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4. A few layers of graphitic carbon can be seen on the surface of CoNi and metal sulfides as a result of the electronic interaction between the carbon shells and metal nanoparticles. Such carbon layers enhance electrocatalytic activity and the carbon shells on the CoNi alloy and metal sulfides (Co9S8 and Ni3S2) can control aggregation, thus enhancing the stability of the catalyst. In comparison to the as-prepared CoNi/Ni3S2@CNS-1 and Co9S8/Co–Nx/CoNi/Ni3S2@CNS-3 catalyst (Fig. S5†), the surface of the carbon polyhedrons does not generate hierarchical structures. However, similar structures can be observed in Co9S8/Co–Nx/CoNi/Ni3S2@CNS-2, indicating that the optimization of ligands is required for the formation of a good graphitic carbon structure. Several studies asserted that particle size has a direct influence on catalyst mass activity due to its effect on the specific surface area, which is the total surface area per unit mass of the catalytic material (m2 g−1).56 In comparison to conventional catalysts, which frequently contain a wide range of metal particle sizes, only a small proportion of metal particles of the appropriate size can exhibit significant electrocatalytic activity.57 Furthermore, variations in particle size associated with extensively conductive carbon networks encourage the rapid diffusion or transfer of metal ions and electrons and improve the electrical conductivity and rate performance of the electrode. Hierarchically ordered architectures made of different-sized structures prevent the serious agglomeration of alloy nanoparticles caused by the high durability of electrode and surface roughness.58,59 | ||
| Fig. 3 (a) FE-SEM images of MOF precursor-4 and that (b) after calcination of the corresponding dandelion flowers like Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 catalyst, (c–e) TEM images of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4, Co9S8/Ni3S2 and their heterojunction, (f) TEM image of CoNi alloy nanoparticle; inset show derived fast Fourier transform SAED patterns, and (g) EDS mappings for carbon (C), nitrogen (N), sulphur (S), cobalt (Co) and nickel (Ni). | ||
To understand the solid-state thermolytic transformations, MOF materials were studied by thermogravimetric analysis (TGA) under inert (N2) conditions. In Fig. 4a, TGA traces for Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 under an atmosphere of N2 are shown. The decomposition of MOFs began at around 200 °C and was followed by a precipitous mass loss in the MOF precursor. At 278.6 °C, the TG curve shows the first weight loss of 10.8 percent, indicating the decomposition of the lattice sulfamate and phenyl group attached to the Co and Ni atoms. The second stage mass loss of 28% occurred at 377.6 °C, corresponding to the removal of the metal coordinating oxygen and sulfur. The final maximum rate of mass loss i.e. 37% at 490–800 °C indicates that CoNi alloy nanoparticles, as well as N and S co-doped graphitic carbon networks, are formed. The ceramic yield (21.5 percent) of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 caused the decomposition of MOF precursor-4.
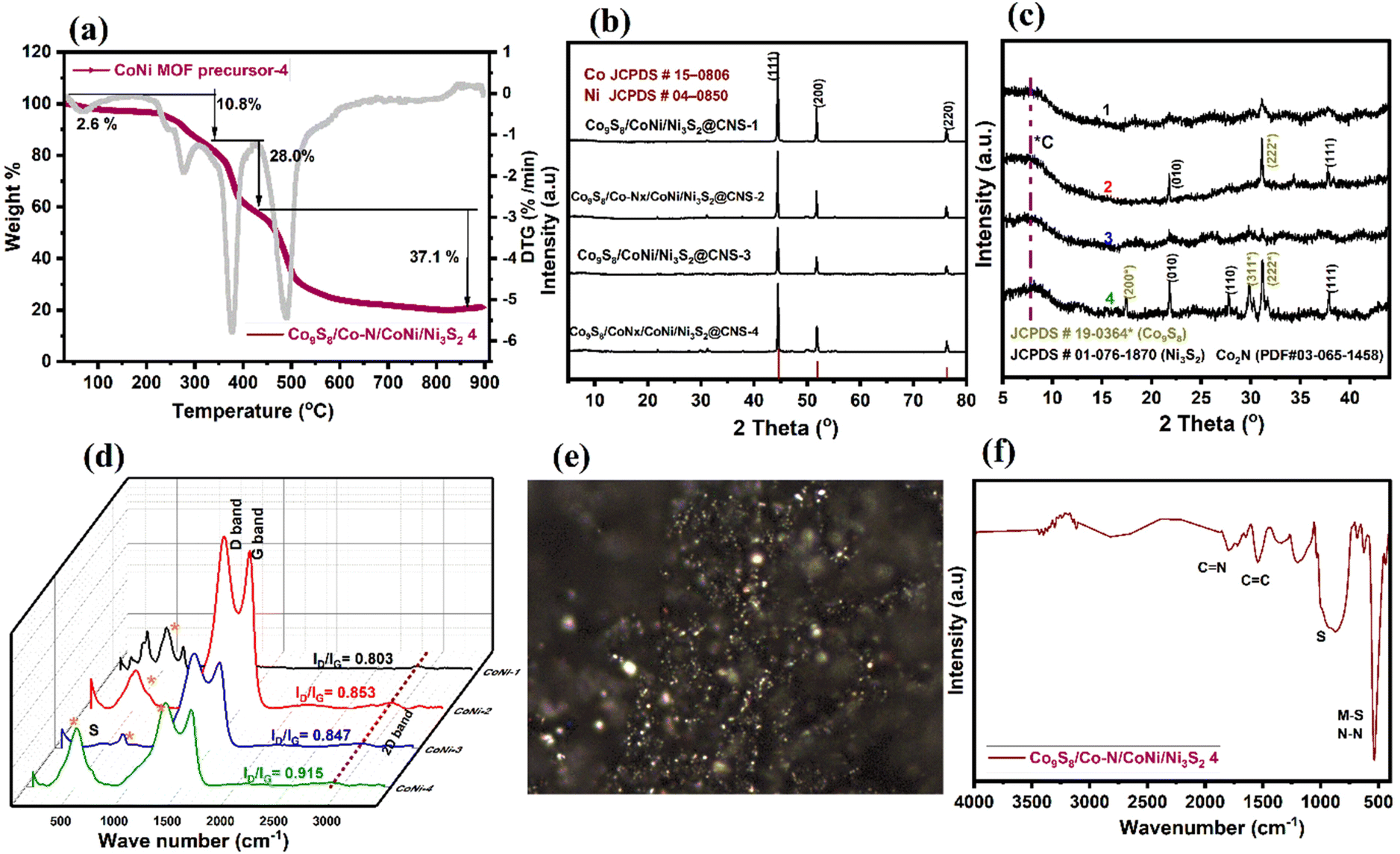 | ||
| Fig. 4 (a) TGA spectrum of MOF precursor-4 conversion to catalyst Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4, (b and c) XRD patterns of electrocatalysts CoS/Co–Nx/CoNi/NiS@CNS, (d) Raman spectra of catalysts 1–4, (e) Raman image (laser at 532 nm) of a filter showing the distribution of graphitic carbon, and (f) the FT-IR spectrum of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4. | ||
In Fig. 4b and c, the X-ray diffraction (XRD) patterns of four different catalysts are shown. At 44.55, 51.79, and 76.23°, three distinct peaks with strong intensities were observed, which correspond to the NiCo alloy in all catalysts. The three peaks, which are sandwiched between Co (PDF#15-0806) and Ni (PDF#04-0850), correspond to the (111), (200), and (220) lattice planes for the metallic Co and the Ni fcc structure, indicating that the metal alloys are attached to the heteroatom doped graphitic carbon structures.60 In Fig. 4c, due to the optimization of the ligands, catalysts Co9S8/Co–Nx/CoNi/Ni3S2@CNS-2 and Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 display consistent diffraction peaks at 17.3, 29.8 and 37.8° for the corresponding (200), (311) and (222) crystal planes, which match the standard data of Co9S8 (PDF#19-0364) and Co2N (PDF#03-065-1458). Other peaks at 21.8, 27.8 and 37.8° are well indexed to the (010) (110) and (111) planes of Ni3S2 (PDF#01-076-1870); some relatively weak peaks match with CoC, Ni–N and NS diffraction.15,16 In Fig. 4d, the Raman spectra of all the electrocatalysts are shown. Two major peaks are observed: one between 1348 and1354 cm−1 (D-band) and another between 1579 and 1591 cm−1 (G-band), which have slightly shifted from the reported CoNi electrocatalyst.14,43 The D-band is associated with the A1g mode and finite-sized graphite crystals. The G-band is matched to permissible E2g mode vibrations in a graphitic network and can be seen in all sp2 bonds. The Co9S8/Co–Nx/CoNi/Ni3S2@CNS-2, Co9S8/CoNi/Ni3S2@CNS-3, and Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 catalysts have ID/IG values of 0.853, 0.847 and 0.915, indicating that electrocatalysts have the most organized graphitic carbon structures.61 Due to the absence of the 4,4-bpy ligand, the catalyst Co9S8/CoNi/Ni3S2@CNS-1 has both a lower ID/IG value and less N and S doping. The characteristic peaks of 486, 525, 675 and 2750 cm−1 are found in electrocatalysts 1–5, which correspond to Eg, F2g and A1g nodes of the metal sulfides and 2D bands, respectively.15 The ID/IG values allow for the generation of Raman images from specific areas of the filters with high concentrations of graphitic carbon particles. Fig. 4e shows a Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 Raman spectroscopy image, clearly representing the five and six membered rings of the particles and the zonification among them. The Raman results demonstrate that the hierarchical Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 catalyst possesses a well-organized graphitic carbon network. In Fig. 4f, the FT-IR spectrum shows how the structure of the material can change after calcination. The two strong intense peaks at 1779 and 1540 cm−1 correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretching. The strong peaks at 1185 and 540 cm−1 are representative of the vibrations of the M cation on the surface of the metal sulfides and metal nitrides (Co9S8, Ni3S2 and Co2N). All the above findings imply that the CoS/Co–Nx/CoNi/NiS@NCS catalysts: 1–5 are multicomponent nanocomposite materials made of CoNi alloys and both N,S-elements incorporated into graphitic carbons.
N and CC stretching. The strong peaks at 1185 and 540 cm−1 are representative of the vibrations of the M cation on the surface of the metal sulfides and metal nitrides (Co9S8, Ni3S2 and Co2N). All the above findings imply that the CoS/Co–Nx/CoNi/NiS@NCS catalysts: 1–5 are multicomponent nanocomposite materials made of CoNi alloys and both N,S-elements incorporated into graphitic carbons.
In Fig. 5a, the presence of a micro/meso-porous structure is confirmed by the Brunauer–Emmett–Teller (BET) surface area of catalysts and a hysteresis loop in the N2 adsorption and desorption isotherm in the P/P0 range between 0.2 and 1.0. It is noted that the BET surface areas of the as-prepared catalyst increase due to ligand optimization with the increase in pore volumes (24 to 92 cm3 g−1); larger porosity makes it easier for the electrolyte to access the interior of the pores, which speeds up the diffusion of catalytic reactants.62 Both the surface area and pore volume of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 (369.5; 91.72 cm3 g−1) are significantly greater than those of Co9S8/CoNi/Ni3S2@CNS-1 (99.86; 35.18 cm3 g−1), which is due to optimization of the ratio (3:1) of the ligands 4,4-bpy and 4-aminodiacetic terephthalic acid. In Fig. 5a (inset), the pore-size distribution curve verifies the mesoporous structure; the size of the mesopore is in the range of 2–10 nm. Therefore, the superior catalytic activity of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 can be attributed to the high intrinsic catalytic activity of CoNi, the heterojunction of metal sulfides, and the well-dispersed state of the catalyst in the composite electrode. The Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 rich mesoporous structure and large specific surface area not only expose more active sites, but also make it easier for ions and electrons to move around, improving electrochemical kinetic performance of the catalyst throughout the ORR and OER processes.63
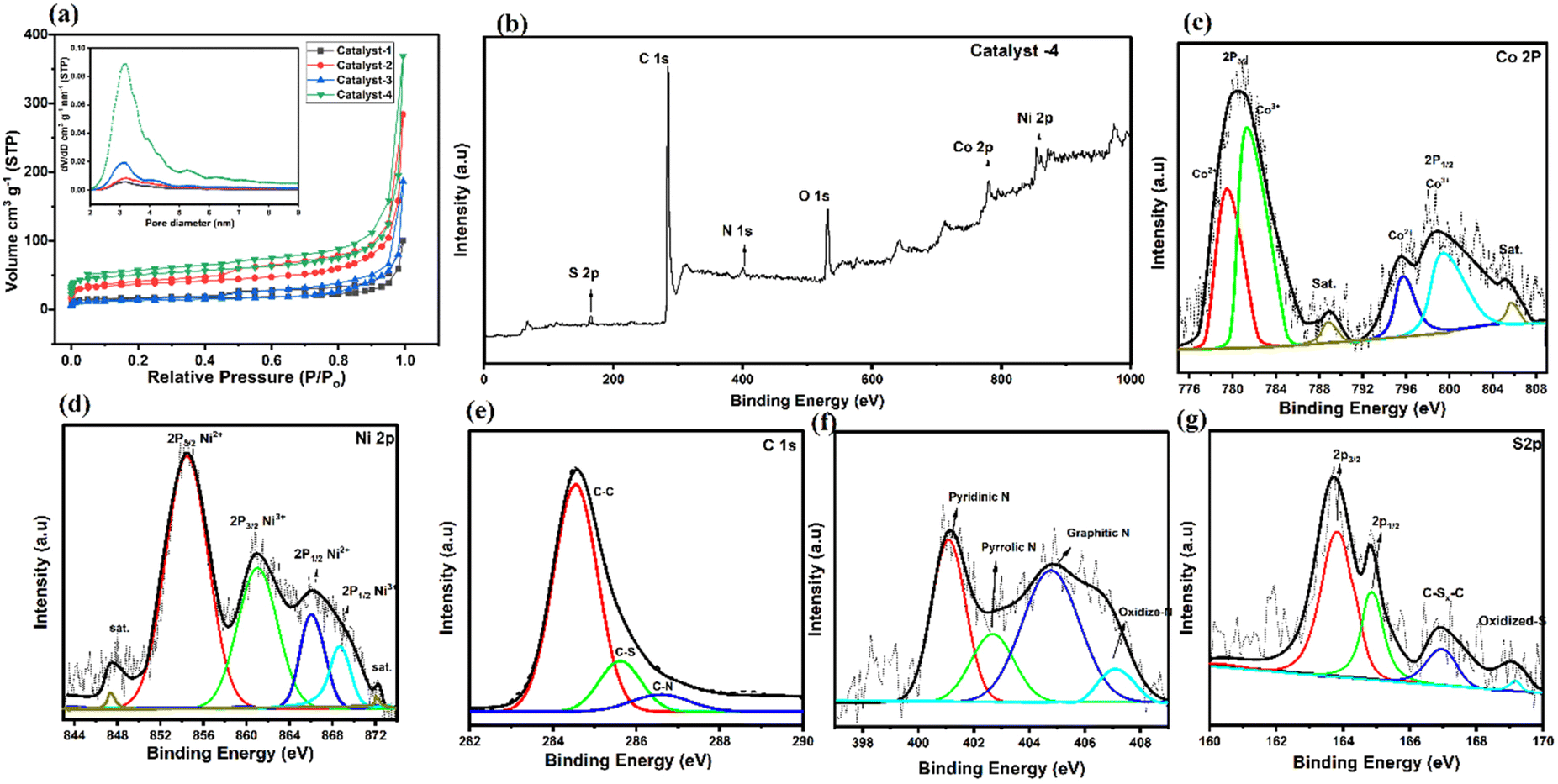 | ||
| Fig. 5 (a) Nitrogen adsorption–desorption isotherm curves of catalysts 1–4; inset pore size distribution image of catalysts 1–4, (b) XPS wide spectrum of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4, (c) Co 2p, (d) Ni 2p, (e) C 1s, (f) N 1s, and (g) S 2p spectra. | ||
XPS measurements were carried out to extend the investigation of the elemental composition of the prepared catalysts 1–4. In Fig. 5b, the survey-scan confirms the existence of Co, Ni, C, N and S in the Co9S8/CoNx/CoNi/Ni3S2-4. This spectrum is consistent with evidence obtained via EDS and XRD. Deconvoluting the Co 2p core level spectra (Fig. 5c) yields binding energies of 780.6 and 798.3 eV, indicating that Co3+ and Co2+ are present in Co9S8–Ni3S2/CNS. The Co 2p3/2/Co 2p1/2 spin–orbit coupling of Co2+ cations bound to N (N–Co)64 or S65 elements. At binding energies of 788.9 and 804.8 eV, corresponding satellite peaks were observed. The high-resolution Ni 2p spectrum exhibits four peaks at 854.4, 861.1, 866.1 and 869.6 eV, matching the Ni3+ and Ni2+ cations: the two peaks at 854.4 eV and 861.1 eV focus on Ni3+ and Ni2+ 2p3/2 respectively.16,66 Additionally in Fig. 5d the two peaks of Ni 2p in Ni3S2/NCS shift to 861.1 and 869.6 eV, 2p1/2 respectively. The positive shift of both Co and Ni ions in the Co9S8/CoNx/CoNi/Ni3S2-4 suggests the presence of interfaces, implying that the four active sites of the CoNi alloy, Co–Nx, Ni3S2 and Co9S8 interact with each other.67 Moreover, in Fig. 5e, the four primary types that can be seen in the N 1s spectrum are pyridinic N (401.1 eV), pyrrolic N (402.6 eV), graphitic N (404.7 eV), and oxidized N (406.9 eV). When compared to the other catalysts, it is significant that the pyridinic N and graphitic N are seen to enhance the ORR and OER quite considerably. The Co9S8/Co–Nx/CoNi/Ni3S2-4 catalyst shows that the binding energies of the N 1s peaks are clearly higher than those of catalysts 1–3 and reported in the literature,68 which can be the optimization result of a CoNi alloy and Co–N electron moving to the N atom. The C 1s spectrum can be deconvoluted into three independent peaks at 284.6, 286.8, and 289.1 eV, which correspond to C–C/CC, CS, and CN, respectively (Fig. 5f). The fine spectrum of S 2p is shown in Fig. 5g: the peaks at 168.7 and 164.8 eV are attributed to the 2p3/2 and 2p1/2 orbitals (metal sulfur bonds/Co9S8–Ni3S2). Furthermore, the peaks at 167.4 eV representing C–Sx–C, indicate that sulfur atoms have been successfully incorporated into the carbon grid during the process of calcination. Meanwhile, the presence of nickel and cobalt ions having mixed valences can increase the conductivity of metal sulfide.69,70 It is important to note that non-metallic heteroatoms (N and S) are known to increase the effectiveness of electron transport and significantly increase the catalyst conductivity. The above results confirm the successful combination of metal sulfides, metal alloys and the N and S doped graphitic carbon matrix, thereby closely binding each other in Co9S8/CoN/CoNi/Ni3S2-4. Catalyst 4, due to its strong electronic coupling, can effectively regulate the charge distribution of the active sites, thereby promoting the electrocatalytic process. With its 2D structural design, active site charge control, and quick reaction kinetics, the Co9S8/Co–Nx/CoNi/Ni3S2-4 catalyst is anticipated to increase the bifunctional activities. The high resolution XPS spectra of other catalysts 1–3 are shown in Fig. S8 and S9.†
3.3. Electrochemical oxygen reduction reaction (ORR)
In Fig. 6, the catalytic performance of ORR is demonstrated by linear sweep voltammograms (LSVs), obtained in O2-saturated 0.1 M KOH at a rotating speed of 1600 rpm on a rotating disk from 0.06 to 0.96 V (vs. RHE: reversible hydrogen electrode) with scan rates of 10 mV s−1. Under identical conditions, the ORR activity of the commercial Pt/C (20 wt% Pt on Vulcan XC-72, Merck) was also examined. In Fig. 6a, it is seen that the limiting current density of Co9S8/CoNi/Ni3S2@CNS-1 cannot reach -4 mA cm−2, whereas the other catalysts clearly exceeded this value. The corresponding half-wave potentials of the catalysts Co9S8/Co–Nx/CoNi/Ni3S2@CNS (2–4) are found to be 0.83, 0.79, and 0.86 V (vs. RHE). In comparison to the commercial Pt/C and PtC + RuO2 (0.83 V vs. RHE), Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 showed a greater cathodic peak at 0.86 V, which was higher than the commercial catalysts. However, under a saturated N2 atmosphere, no such noticeable peak was detected, showing that the cathodic peak was caused by the ORR (Fig. S10b†). The ORR performance of catalyst Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 proved to be better in comparison to recently reported catalysts, in the alkaline electrolyte (Table S1†).12,45,46 In Fig. 6b, to further investigate catalytic performance, LSV was conducted applying different rotations (400–2000 rpm) using RRDE; the limiting current densities of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 were found to be 2.02 to 7.05 mA cm-2, respectively. It is noted that electrocatalyst 4 had a much higher half-wave potential (E1/2) and limiting current density (id) than other transition metals and N,S-doped graphitic carbon catalysts.18,71 In Fig. 6c, for a more quantitative assessment of ORR kinetics, the Tafel slopes derived from the LSVs are shown. As for catalyst Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4, the smallest Tafel value (40.87120 mV dec−1) outperformed the benchmark Pt/C (45.93 mV dec−1). The Tafel values of catalysts Co9S8/CoNi/Ni3S2@CNS-1 (111.99 mV dec−1), Co9S8/Co–Nx/CoNi/Ni3S2@CNS-2 (49.62 mV dec−1), and Co9S8/CoNi/Ni3S2@CNS-3 (100.57 mV dec−1) are better when compared with the recent literature.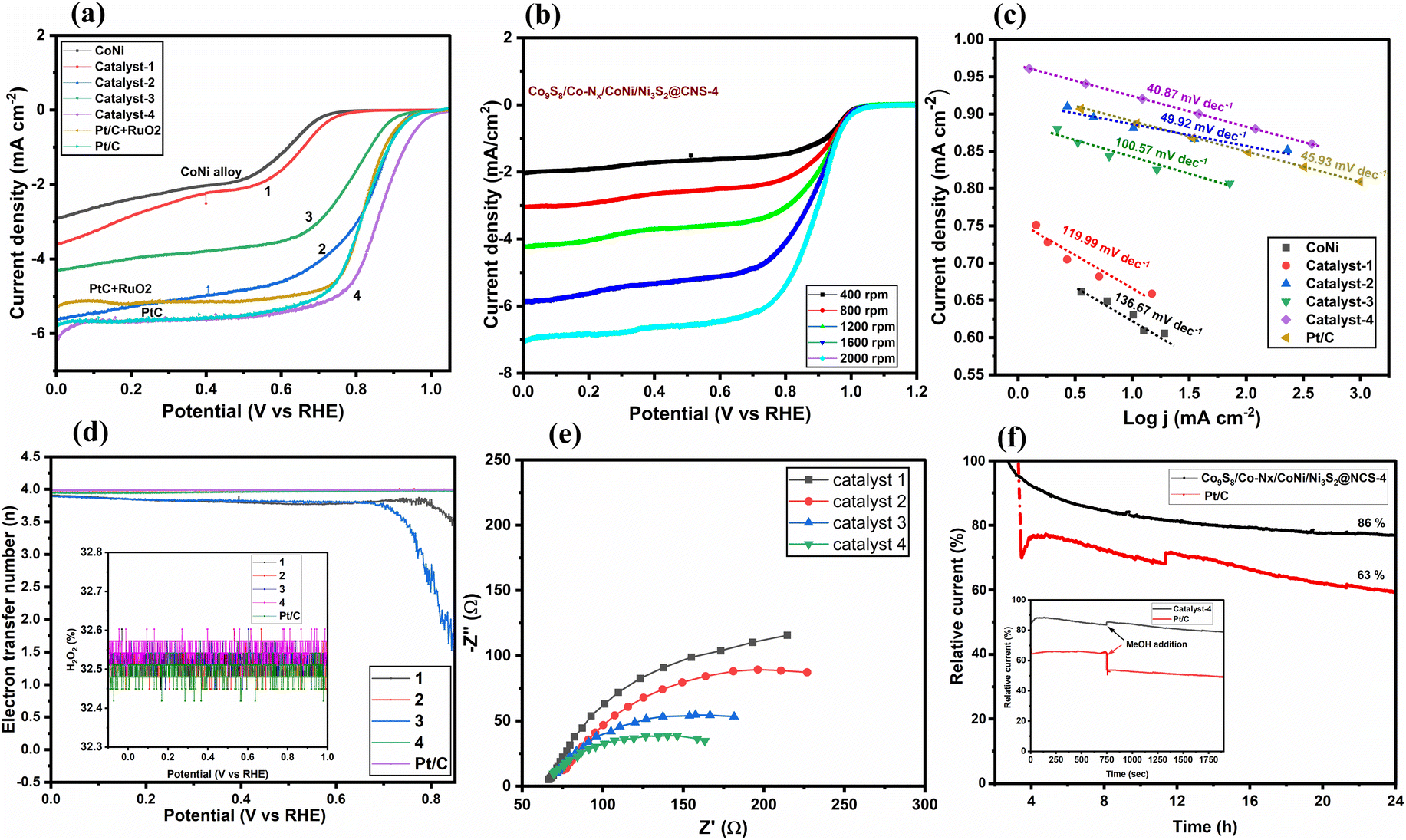 | ||
| Fig. 6 Electrocatalytic ORR performances: (a) LSV curves of CoS/Co–Nx/CoNi/NiS@CNS 1–4, commercial 20% Pt/C and Pt/C + RuO2 in O2-saturated 0.1 M KOH, (b) LSV curves of CoS/Co-Nx/CoNi/NiS@CNS-4, at various rotation rates, (c) Tafel plots of catalysts 1-4 and Pt/C, (d) Electron transfer number (n) of catalysts 1–4, and Pt/C, inset: the corresponding peroxide yields, (e) EIS spectra of catalysts 1–4, and (f) chronoamperometric responses of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4, and commercial 20% Pt/C in O2-saturated 0.1 M KOH solution at a rotation rate of 1600 rpm at ∼0.4 V vs. RHE. | ||
One of the most important aspects of the electrocatalytic ORR in aqueous electrolytes is the electron transfer number (n), which not only represents the efficiency of oxygen conversion but also provides information about the mechanism of the ORR. The ORR can involve two reactions: a four-electron reaction (O2 + 2H2O + 4e− → 4OH−) and a two-electron reaction (O2 + H2O + 2e− → HO2− + OH−). The fractions of HO2− produced and the predicted electron transfer numbers are shown in Fig. 6d. In Fig. 6d (inset), even though the accompanying LSVs are significantly different, all the catalysts such as Co9S8/Co–Nx/CoNi/Ni3S2@CNS-2 (3.96), Co9S8/CoNi/Ni3S2@CNS-3 (3.83), Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 (3.98) and Pt/C (3.99) show better electron transfer numbers than the metallic catalyst Co9S8/CoNi/Ni3S2@CNS-1 (∼3.69). The electrochemical impedance spectroscopy (EIS) tests provide more evidence in favor of this. In Fig. 6e, according to the Nyquist plot, the catalyst Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 exhibits the lowest charge transfer resistance, among the three catalysts 1–3, having a noticeably smaller semicircle in the intermediate frequency region (Table S2†). As shown in Fig. 6f, the long-term durability of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 and commercial Pt/C electrocatalyst was investigated using chronoamperometric experiments. Only 16% catalytic activity loss was discovered on the surface of the Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 electrode after more than 24 h of continuous chronoamperometric analysis, demonstrating the superior stability of catalyst 4 to the commercial Pt/C electrocatalyst. This outcome was brought about by the stronger incorporation of metal sulfides, nitrogen-coordinated Co, and CoNi alloys into the porous graphitic carbon framework, which increased the electrochemical ORR activity and endurance. In Fig. 6f (inset), the catalyst was tested against methanol poisoning to prove its applicability in direct methanol fuel cells (DMFCs). The catalyst Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 showed strong resistance to methanol because there was no current density loss when 2.0 mL of methanol (CH3OH) was added to the 0.1 M KOH solution. On the other hand, the commercial Pt/C electrocatalyst exhibit a considerable decrease in cathodic current density during the addition of CH3OH because of the methanol oxidation process being facilitated on the Pt/C catalytically active surface sites. These findings indicate that Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 can be exploited as a possible cathodic material in the exciting field of ZABs since catalyst 4 has excellent ORR kinetics, outstanding durability, and strong resistance to methanol poisoning. It is noted that the unique hierarchical porous structure and interfacial synergy between the CoNi, CoN, Ni3S2, and Co9S8 nanoparticle structures with rich defects can increase mass transfer and graphitization to a degree that is advantageous for electron transport.
3.4. Oxygen evolution reaction (OER)
As shown in Fig. 7a, OER performance of the electrocatalysts and the commercial RuO2/PtC + RuO2 catalysts were evaluated. Results were compared with the well-developed transition metal alloy catalysts reported recently (Table S1†). It is seen that Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 (1.58 V) and Co9S8/Co–Nx/CoNi/Ni3S2@CNS-2 (1.61 V) both possessed low overpotentials of 196 and 201 mV at 10 mA cm−2, which are smaller than the commercial RuO2 (203 mV) and associated with the synergy between Co9S8 and Ni3S2 in catalyst-4. In contrast, Co9S8/CoNi/Ni3S2@CNS-1 (205 mV) and Co9S8/Co–Nx/CoNi/Ni3S2@CNS-3 (211 mV) catalysts have high overpotential of catalysts. However, the OER performance of prepared catalysts 1–4 proved to be superior to the recently reported catalysts (Table S1†). The results indicated that bimetallic alloys and sulfides acted positively in promoting the performance of the OER.54 As shown in Fig. 7b, the Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 catalyst had the smallest Tafel value (79.51 mV dec−1) among all as-prepared catalysts: Co9S8/CoNi/Ni3S2@CNS-1 (130.74 mV dec−1), Co9S8/Co–Nx/CoNi/Ni3S2@CNS-2 (119.38 mV dec−1) and Co9S8/CoNi/Ni3S2@CNS-3 (91 mV dec−1); all indicating fast OER kinetics. As shown in Fig. 7c, the EIS measurements based on the Nyquist plot revealed that of the three samples, Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 had the shortest semicircle. The potential gap ΔE between the ORR and OER (E = Ej10 − E1/2) has been used to evaluate an electrocatalyst's total bifunctional activity. Consequently, Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 had a tiny ΔE value of 0.72 V, which is comparable to the most recently described metal-involving oxygen electrocatalysts (Fig. S10d†).12,18 In contrast to benchmark Pt/C and RuO2, which decreased after the 24 h tests, the performance of catalyst 4 did not significantly decline after continuous operation for 24 hours at 10 mA cm−2 according to the long-term durability test, representing that the material had good bifunctional catalytic stability, as shown in Fig. 7d. The superior ORR and OER activities of the Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 catalyst can be attributed to the following factors based on the above results: firstly, the optimized ligand concentration in catalyst-4 had a high surface area, hierarchical pores, and defect rich structures, which helped to increase the number of electroactive sites, enhancing reactions and electrocatalytic performance. Secondly, the 2D graphitic carbon polyhedron heterogeneous structure with N and S elements was able to boost catalytic processes at the interface and provide long-range conductivity. Finally, the porous multi-compound system of the Co9S8, Co–Nx, CoNi alloy nanoparticles, and Ni3S2 enhanced the electrochemical kinetics by synergistically facilitating charge/ion diffusion and increasing the interfacial contact between the electrolyte and the electrode. A comparison of ORR/OER catalytic activity with that of a previously published transition metal and heteroatom-doped carbon composite catalyst, as shown in Table S1,† further supports the exceptional bifunctional activity of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 in our work.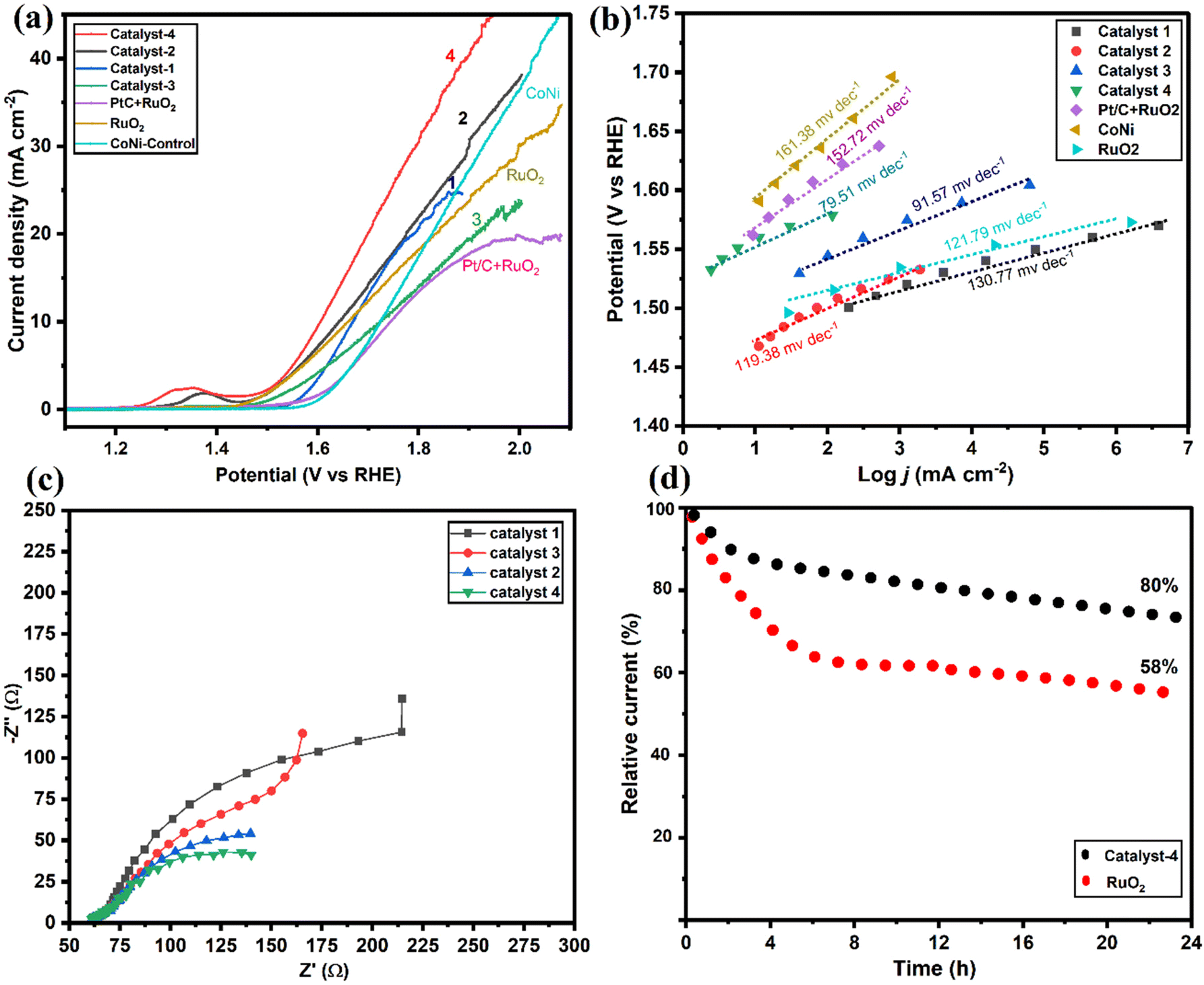 | ||
| Fig. 7 Electrocatalytic OER performances: (a) LSV curves of CoS/Co–Nx/CoNi/NiS@CNS 1–4, commercial RuO2 and Pt/C + RuO2 in N2-saturated 0.1 M KOH, (b) Tafel plots of LSV curves of catalysts 1–4, RuO2 and Pt/C + RuO2, (c) EIS spectra of catalysts 1–4, and (d) chronoamperometric responses of Co9S8/Co–N/CoNi/Ni3S2@CNS-4, and commercial RuO2 in N2-saturated 0.1 M KOH solution. | ||
3.5. Zinc–air battery performance
The Co9S8/Co–Nx/Co–Ni/Ni3S2@CNS-4 catalyst utilized in real ZABs (Fig. 8a) as an air cathode was properly evaluated based on its better ORR/OER activities. Thus, an aqueous rechargeable Zn–air battery was prepared using catalyst Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 as the air cathode. Comparisons were made between its performance and that of the catalyst used in commercial air cathodes (Pt/C + RuO2, mixed catalyst, 1:1 ratio). It was found that the ZAB based on Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 had an open circuit potential (OCP) of 1.59 V (Fig. 8b), which is greater than that of the Pt/C + RuO2 catalyst (1.47 V) and recently published bifunctional catalysts (Table S3†). In Fig. 8c, both discharge and charge polarization curves of Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 are observed to display a smaller voltage gap than Pt/C + RuO2 due to its strong bifunctional activities. In Fig. 8d, catalyst 4 based ZABs exhibited a peak power density of 206.9 mA cm−2 at 325 mA cm−2, outperforming Pt/C + RuO2 (116.1 mWcm−2). The presence of micro- and mesopore structures in the Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 was found to be primarily responsible for the high electrochemically active surface areas, which facilitated the transport of mass electrons during performance. A specific capacity of 801 mA h g−1 for the collectively exhaustive Co9S8/Co–N/CoNi/Ni3S2@CNS-4 was obtained from the galvanostatic discharge curve at a current density of 10 mA cm−2, as shown in Fig. 8e. This was somewhat higher than the ZAB using the Pt/C + RuO2 (762 mA h g−1) air cathode. Furthermore, under different discharge current densities, Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 demonstrated a significant rate capability (Fig. 8f). In Fig. 8g, long-term cyclability testing at 10 mA cm−2 revealed that Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 had a narrow initial voltage gap of 0.96 V and no significant voltage attenuation after cycling for more than 370 h (Fig. S11†), which was significantly better than the cycle time of Pt/C + RuO2-based ZABs (only 50 h). In Fig. 8h, two catalyst-4-based ZABs are connected in series facilitating green light-emitting diodes (LEDs, 2.2 V). In Table S3,† the catalytic behavior (ORR/OER) of the Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 catalyst is compared to that of recent CoNi-based electrocatalysts in the literature. It is important that the facile synthesis of ligand-optimized Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 outperforms the as-assembled ZABs in terms of open-circuit voltage, specific capacity, and power density.
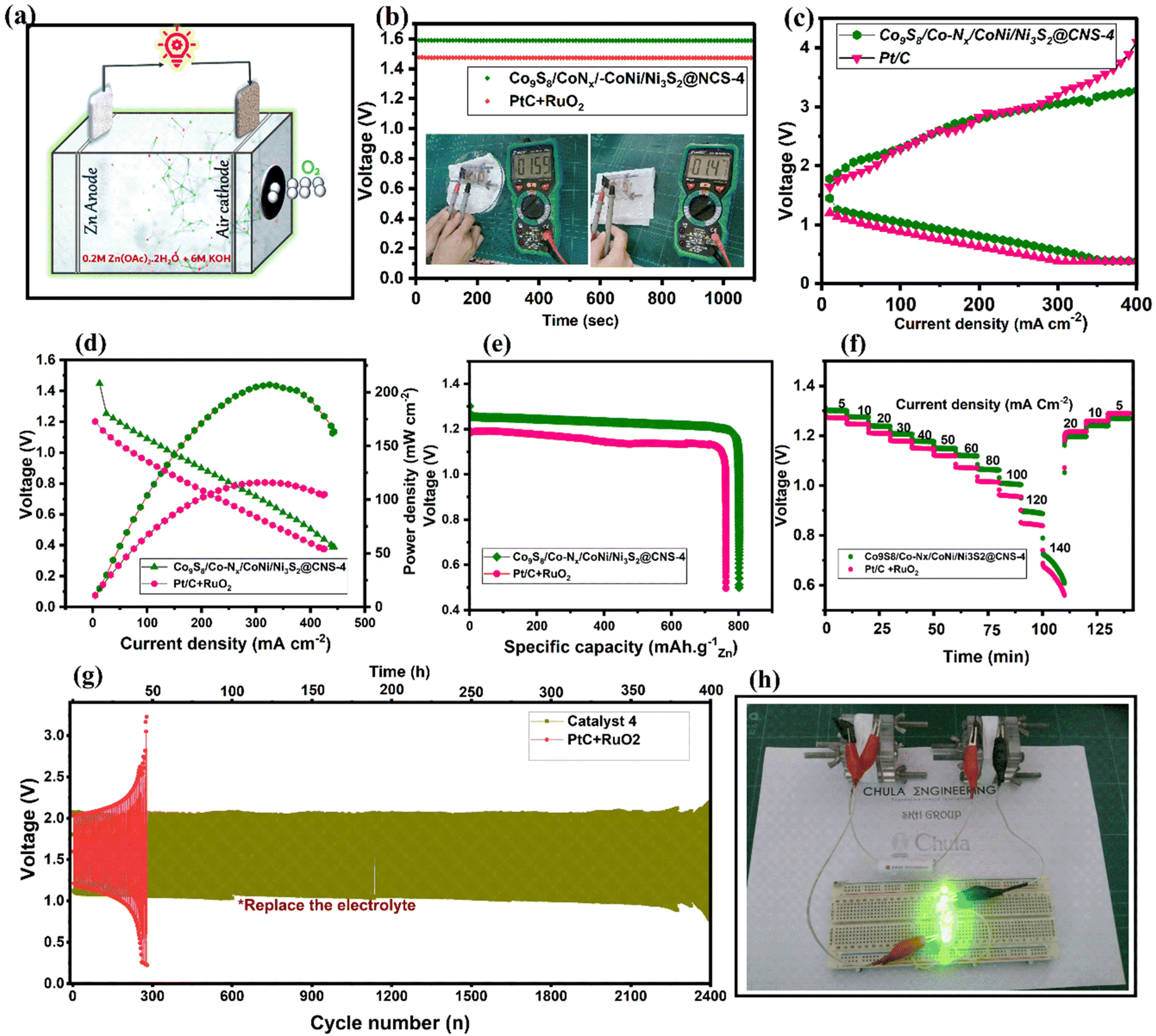 | ||
| Fig. 8 Zn–air battery (ZABs) performances: (a) schematic representation of the assembled rechargeable ZABs (b) open-circuit voltage curves of ZABs using Co9S8/Co–N/CoNi/Ni3S2@CNS-4 and Pt/C + RuO2 air cathodes, (c and d) charge and discharge polarization curves and the corresponding power density curves, (e) specific capacity plots at 10 mA cm−2, (f) discharge curves at different current densities of Co9S8/Co–N/CoNi/Ni3S2@CNS-4 and Pt/C + RuO2; (g) galvanostatic cycling stability tests of the zinc–air battery with Co9S8/Co–N/CoNi/Ni3S2@CNS-4 and Pt/C + RuO2 and (h) the photos of two ZABs connected in series to drive green LED lights. | ||
4. Conclusion
In this study, a convenient and controllable ligand optimization (4-aminodiacetic terephthalic acid and 4,4-bipyridine) approach to synthesize CoS/Co–Nx/CoNi/NiS@CNS by tailoring graphitic carbon under a moderate temperature of 700 °C was developed. The Co9S8/Co–Nx/CoNi/Ni3S2@CNS-4 catalyst displayed a dandelion flower like structure and exhibited excellent bifunctional activity, robust stability, and a potent resistance to methanol. The catalyst 4 fabricated air cathode displayed the highest power density and specific capacity when compared to the ZAB performance utilizing Pt/C + RuO2 electrocatalysts. The long-term cyclability test on catalyst 4 under 10 mA cm−2 demonstrated that it had a small initial voltage gap and the air cathode used in the practical ZABs can be circulated for more than 350 h. The exceptional electrocatalytic performances are attributed to the abundant CoNi alloys and N and S heteroatoms serving as active sites, which interlace and loosely pack the active components and partially graphitized carbonaceous matrix to ensure efficient electron/charge transport. In conclusion, the stated synthesis approach can be used to generate effective electrocatalysts for electrochemical energy applications by utilizing various metal-coordination complexes with different carboxylate and N-donor ligands.Data availability
Due to technical or time constraints, the raw/processed data required to reproduce these findings cannot be shared at this time.Author contributions
M. G.: Conceptualization: equal, investigation: lead, data curation: lead, formal analysis: equal, writing – original draft: lead, and writing – review & editing: supporting. M. E.: formal analysis: supporting and writing – review & editing: supporting. J. T.: formal analysis: supporting and writing – review & editing: supporting. M. Y. C.: formal analysis: supporting and writing – review & editing: supporting. S. W.: formal analysis: supporting and writing – review & editing: supporting. M. T. N.: formal analysis: supporting and writing – review & editing: supporting. T. Y.: formal analysis: supporting; and writing – review & editing: supporting. S. K.: conceptualization: equal, formal analysis: equal, funding acquisition: lead, supervision: equal, writing – original draft: supporting, and writing – review & editing: lead.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The Program Management Unit for Human Resources & Institutional Development, Research and Innovation (B16F630071), and Ratchadapisek Sompoch Endowment Fund, Chulalongkorn University are acknowledged. M. G. thanks the Chulalongkorn Academic Advancement into its 2nd Century Project for Postdoctoral Fellowship.References
- Y. P. Zhu, C. Guo, Y. Zheng and S.-Z. Qiao, Acc. Chem. Res., 2017, 50, 915–923, DOI:10.1021/acs.accounts.6b00635.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289, DOI:10.1038/s41560-018-0108-1.
- W. Lao-atiman, S. Olaru, A. Arpornwichanop and S. Kheawhom, Sci. Data, 2019, 6, 168, DOI:10.1038/s41597-019-0178-3.
- A. Abbasi, S. Hosseini, A. Somwangthanaroj, R. Cheacharoen, S. Olaru and S. Kheawhom, Sci. Data, 2020, 7, 196, DOI:10.1038/s41597-020-0539-y.
- M.-I. Jamesh, P. Moni, A. S. Prakash and M. Harb, Mater. Sci. Energy Technol., 2021, 4, 1–22, DOI:10.1016/j.mset.2020.12.001.
- T. Li, Z. He, X. Liu, M. Jiang, Q. Liao, R. Ding, S. Liu, C. Zhao, W. Guo, S. Zhang and H. He, Surf. Interfaces, 2022, 102270, DOI:10.1016/j.surfin.2022.102270.
- M. Etesami, S. Mehdipour-Ataei, A. Somwangthanaroj and S. Kheawhom, Int. J. Hydrogen Energy, 2021 DOI:10.1016/j.ijhydene.2021.09.133.
- Y. Sun, X. Zhang, M. Luo, X. Chen, L. Wang, Y. Li, M. Li, Y. Qin, C. Li, N. Xu, G. Lu, P. Gao and S. Guo, Adv. Mater., 2018, 30, 1802136, DOI:10.1002/adma.201802136.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201, 10.1039/C4CS00484A.
- M. Etesami, A. A. Mohamad, M. T. Nguyen, T. Yonezawa, R. Pornprasertsuk, A. Somwangthanaroj and S. Kheawhom, J. Alloys Compd., 2022, 889, 161738, DOI:10.1016/j.jallcom.2021.161738.
- M. Etesami, R. Khezri, A. Abbasi, M. T. Nguyen, T. Yonezawa, S. Kheawhom and A. Somwangthanaroj, J. Alloys Compd., 2022, 922, 166287, DOI:10.1016/j.jallcom.2022.166287.
- W. J. Sim, M. T. Nguyen, Z. Huang, S. Kheawhom, C. Wattanakit and T. Yonezawa, Nanoscale, 2022, 14, 8012–8022, 10.1039/D2NR01258H.
- M. D. Bhatt and J. Y. Lee, Energy Fuels, 2020, 34, 6634–6695, DOI:10.1021/acs.energyfuels.0c00953.
- Y. Fu, H.-Y. Yu, C. Jiang, T.-H. Zhang, R. Zhan, X. Li, J.-F. Li, J.-H. Tian and R. Yang, Adv. Funct. Mater., 2018, 28, 1705094, DOI:10.1002/adfm.201705094.
- Z. Cai, I. Yamada and S. Yagi, ACS Appl. Mater. Interfaces, 2020, 12, 5847–5856, DOI:10.1021/acsami.9b19268.
- W. Wan, X. Liu, H. Li, X. Peng, D. Xi and J. Luo, Appl. Catal., B, 2019, 240, 193–200, DOI:10.1016/j.apcatb.2018.08.081.
- J.-C. Li, P.-X. Hou, S.-Y. Zhao, C. Liu, D.-M. Tang, M. Cheng, F. Zhang and H.-M. Cheng, Energy Environ. Sci., 2016, 9, 3079–3084, 10.1039/C6EE02169G.
- Z. Huang, M. T. Nguyen, W. J. Sim, M. Takahashi, S. Kheawhom and T. Yonezawa, Sustainable Energy Fuels, 2022, 6, 3931–3943, 10.1039/D2SE00606E.
- Y. Wang, R. Gan, Z. Ai, H. Liu, C. Wei, Y. Song, M. Dirican, X. Zhang, C. Ma and J. Shi, Carbon, 2021, 181, 87–98, DOI:10.1016/j.carbon.2021.05.016.
- Z. Bai, J. Heng, Q. Zhang, L. Yang and F. Chang, Adv. Energy Mater., 2018, 8, 1802390, DOI:10.1002/aenm.201802390.
- H. Yi, S. Liu, C. Lai, G. Zeng, M. Li, X. Liu, B. Li, X. Huo, L. Qin, L. Li, M. Zhang, Y. Fu, Z. An and L. Chen, Adv. Energy Mater., 2021, 11, 2002863, DOI:10.1002/aenm.202002863.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006, DOI:10.1038/nenergy.2015.6.
- H.-F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448, 10.1039/C9CS00906J.
- Z. Li, R. Gao, M. Feng, Y.-P. Deng, D. Xiao, Y. Zheng, Z. Zhao, D. Luo, Y. Liu, Z. Zhang, D. Wang, Q. Li, H. Li, X. Wang and Z. Chen, Adv. Energy Mater., 2021, 11, 2003291, DOI:10.1002/aenm.202003291.
- L. Zou, Y.-S. Wei, C.-C. Hou, C. Li and Q. Xu, Small, 2021, 17, 2004809, DOI:10.1002/smll.202004809.
- Z. Liang, T. Qiu, S. Gao, R. Zhong and R. Zou, Adv. Energy Mater., 2022, 12, 2003410, DOI:10.1002/aenm.202003410.
- C.-C. Hou and Q. Xu, Adv. Energy Mater., 2019, 9, 1801307, DOI:10.1002/aenm.201801307.
- C. Li, S. Dong, P. Wang, C. Wang and L. Yin, Adv. Energy Mater., 2019, 9, 1902352, DOI:10.1002/aenm.201902352.
- G. Li, S. Zhao, Y. Zhang and Z. Tang, Adv. Mater., 2018, 30, 1800702, DOI:10.1002/adma.201800702.
- W.-H. Li, W.-H. Deng, G.-E. Wang and G. Xu, EnergyChem, 2020, 2, 100029, DOI:10.1016/j.enchem.2020.100029.
- H. Wu, M. Almalki, X. Xu, Y. Lei, F. Ming, A. Mallick, V. Roddatis, S. Lopatin, O. Shekhah, M. Eddaoudi and H. N. Alshareef, J. Am. Chem. Soc., 2019, 141, 20037–20042, DOI:10.1021/jacs.9b11446.
- D. Xie, D. Yu, Y. Hao, S. Han, G. Li, X. Wu, F. Hu, L. Li, H.-Y. Chen, Y.-F. Liao and S. Peng, Small, 2021, 17, 2007239, DOI:10.1002/smll.202007239.
- S. N. Khan, S. Ge, Y. Huang, H. Xu, W. Yang, R. Hong, Y. Mai, E. Gu, X. Lin and G. Yang, Sci. China Mater., 2022, 65, 612–619, DOI:10.1007/s40843-021-1769-7.
- T. Guo, L. Chen, Y. Li and K. Shen, Small, 2022, 18, 2107739, DOI:10.1002/smll.202107739.
- Y. Xin, F. Wang, L. Chen, Y. Li and K. Shen, Green Chem., 2022, 24, 6544–6555, 10.1039/D2GC02426H.
- Z. Zhang and M. J. Zaworotko, Chem. Soc. Rev., 2014, 43, 5444–5455, 10.1039/C4CS00075G.
- B. Zheng, X. Sun, G. Li, A. J. Cairns, V. C. Kravtsov, Q. Huo, Y. Liu and M. Eddaoudi, Cryst. Growth Des., 2016, 16, 5554–5562, DOI:10.1021/acs.cgd.6b01031.
- M. K. Lee, M. Shokouhimehr, S. Y. Kim and H. W. Jang, Adv. Energy Mater., 2022, 12, 2003990, DOI:10.1002/aenm.202003990.
- C.-Y. Su, H. Cheng, W. Li, Z.-Q. Liu, N. Li, Z. Hou, F.-Q. Bai, H.-X. Zhang and T.-Y. Ma, Adv. Energy Mater., 2017, 7, 1602420, DOI:10.1002/aenm.201602420.
- Y. Xiong, Y. Yang, F. J. DiSalvo and H. D. Abruña, J. Am. Chem. Soc., 2019, 141, 10744–10750, DOI:10.1021/jacs.9b03561.
- X. Xie, L. Shang, R. Shi, G. I. N. Waterhouse, J. Zhao and T. Zhang, Nanoscale, 2020, 12, 13129–13136, 10.1039/D0NR02486D.
- Z. Dong, G. Liu, S. Zhou, Y. Zhang, W. Zhang, A. Fan, X. Zhang and X. Dai, ChemCatChem, 2018, 10, 5475–5486, DOI:10.1002/cctc.201801412.
- X. Xu, F. Ran, Z. Fan, Z. Cheng, T. Lv, L. Shao and Y. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 17870–17880, DOI:10.1021/acsami.0c01572.
- K. Sheng, Q. Yi, A. L. Chen, Y. Wang, Y. Yan, H. Nie and X. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 45394–45405, DOI:10.1021/acsami.1c10671.
- Y. Tan, Z. Zhang, Z. Lei, L. Yu, W. Wu, Z. Wang and N. Cheng, Appl. Catal., B, 2022, 304, 121006, DOI:10.1016/j.apcatb.2021.121006.
- P. Thangasamy, S. Oh, H. Randriamahazaka, S. Nam and I.-K. Oh, Appl. Catal., B, 2022, 316, 121656, DOI:10.1016/j.apcatb.2022.121656.
- R. Maity, D. Chakraborty, S. Nandi, A. K. Yadav, D. Mullangi, C. P. Vinod and R. Vaidhyanathan, ACS Appl. Nano Mater., 2019, 2, 5169–5178, DOI:10.1021/acsanm.9b01047.
- W. Lu, D. Sun and H. Yu, J. Alloys Compd., 2013, 546, 229–233, DOI:10.1016/j.jallcom.2012.08.063.
- X. Meng, X.-Z. Song, S.-Y. Song, G.-C. Yang, M. Zhu, Z.-M. Hao, S.-N. Zhao and H.-J. Zhang, Chem. Commun., 2013, 49, 8483–8485, 10.1039/C3CC44106G.
- M. F. Sanad, A. R. Puente Santiago, S. A. Tolba, M. A. Ahsan, O. Fernandez-Delgado, M. Shawky Adly, E. M. Hashem, M. Mahrous Abodouh, M. S. El-Shall, S. T. Sreenivasan, N. K. Allam and L. Echegoyen, J. Am. Chem. Soc., 2021, 143, 4064–4073, DOI:10.1021/jacs.1c01096.
- J. Ren, Y. Huang, H. Zhu, B. Zhang, H. Zhu, S. Shen, G. Tan, F. Wu, H. He, S. Lan, X. Xia and Q. Liu, Carbon Energy, 2020, 2, 176–202, DOI:10.1002/cey2.44.
- X. Chu, F. Meng, T. Deng and W. Zhang, Nanoscale, 2021, 13, 5570–5593, 10.1039/D1NR00160D.
- X. Wang, L. Li, M. Shi, Y. Wang, G. Xu, K. Yuan, P. Zhu, M. Ding and Y. Chen, Chem. Sci., 2022, 13, 11639–11647, 10.1039/D2SC03585E.
- X. Wang, Y. Yang, R. Wang, L. Li, X. Zhao and W. Zhang, Langmuir, 2022, 38, 7280–7289, DOI:10.1021/acs.langmuir.2c00805.
- H. Chang, L.-N. Shi, Y.-H. Chen, P.-F. Wang and T.-F. Yi, Coord. Chem. Rev., 2022, 473, 214839, DOI:10.1016/j.ccr.2022.214839.
- Y. Shi, Z. Lyu, M. Zhao, R. Chen, Q. N. Nguyen and Y. Xia, Chem. Rev., 2021, 121, 649–735, DOI:10.1021/acs.chemrev.0c00454.
- S. Li, E. Li, X. An, X. Hao, Z. Jiang and G. Guan, Nanoscale, 2021, 13, 12788–12817, 10.1039/D1NR02592A.
- F. Zeng, C. Mebrahtu, L. Liao, A. K. Beine and R. Palkovits, J. Energy Chem., 2022, 69, 301–329, DOI:10.1016/j.jechem.2022.01.025.
- S. Kurungot, A. Pandikassala, G. Singla, T. S. Khan, M. A. Haider, V. Prabhakaran and V. Kashyap, Nanoscale, 2022, 14, 15928–15941, 10.1039/D2NR04170G.
- Z. Zhu, Q. Xu, Z. Ni, K. Luo, Y. Liu and D. Yuan, ACS Sustainable Chem. Eng., 2021, 9, 13491–13500, DOI:10.1021/acssuschemeng.1c04259.
- K. Ghosh, M. Kumar, T. Maruyama and Y. Ando, Carbon, 2009, 47, 1565–1575, DOI:10.1016/j.carbon.2009.02.007.
- J. Qin, Z. Liu, D. Wu and J. Yang, Appl. Catal., B, 2020, 278, 119300, DOI:10.1016/j.apcatb.2020.119300.
- L. Cao, Y. Wang, Q. Zhu, L. Fan, Y. Wu, Z. Li, S. Xiong and F. Gu, ACS Appl. Mater. Interfaces, 2022, 14, 17249–17258, DOI:10.1021/acsami.2c00163.
- P. Yu, L. Wang, F. Sun, Y. Xie, X. Liu, J. Ma, X. Wang, C. Tian, J. Li and H. Fu, Adv. Mater., 2019, 31, 1901666, DOI:10.1002/adma.201901666.
- Y. Li, J. Yin, L. An, M. Lu, K. Sun, Y.-Q. Zhao, D. Gao, F. Cheng and P. Xi, Small, 2018, 14, 1801070, DOI:10.1002/smll.201801070.
- Q. Zhang, X. L. Li, B. X. Tao, X. H. Wang, Y. H. Deng, X. Y. Gu, L. J. Li, W. Xiao, N. B. Li and H. Q. Luo, Appl. Catal., B, 2019, 254, 634–646, DOI:10.1016/j.apcatb.2019.05.035.
- L. Wu, J. Li, C. Shi, Y. Li, H. Mi, L. Deng, Q. Zhang, C. He and X. Ren, J. Mater. Chem. A, 2022, 10, 16627–16638, 10.1039/D2TA03554E.
- C. Tang, B. Wang, H.-F. Wang and Q. Zhang, Adv. Mater., 2017, 29, 1703185, DOI:10.1002/adma.201703185.
- D. Lyu, S. Yao, A. Ali, Z. Q. Tian, P. Tsiakaras and P. K. Shen, Adv. Energy Mater., 2021, 11, 2101249, DOI:10.1002/aenm.202101249.
- N. Radenahmad, R. Khezri, A. A. Mohamad, M. T. Nguyen, T. Yonezawa, A. Somwangthanaroj and S. Kheawhom, J. Alloys Compd., 2021, 883, 160935, DOI:10.1016/j.jallcom.2021.160935.
- A. Wang, C. Zhao, M. Yu and W. Wang, Appl. Catal., B, 2021, 281, 119514, DOI:10.1016/j.apcatb.2020.119514.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr04933c |
| This journal is © The Royal Society of Chemistry 2022 |