
Peptide functionalized DNA hydrogel enhances neuroblastoma cell growth and differentiation†
Pravin
Hivare‡
 a,
Ankit
Gangrade‡
a,
Gitanjali
Swarup
a,
Krishna
Bhavsar
a,
Ankur
Singh
a,
Ratnika
Gupta
b,
Prachi
Thareja
b,
Sharad
Gupta
*ac and
Dhiraj
Bhatia
*ac
a,
Ankit
Gangrade‡
a,
Gitanjali
Swarup
a,
Krishna
Bhavsar
a,
Ankur
Singh
a,
Ratnika
Gupta
b,
Prachi
Thareja
b,
Sharad
Gupta
*ac and
Dhiraj
Bhatia
*ac
aBiological Engineering Discipline, Indian Institute of Technology Gandhinagar, Palaj, Gujarat 382355, India. E-mail: dhiraj.bhatia@iitgn.ac.in; sharad@iitgn.ac.in
bChemical Engineering Discipline, Indian Institute of Technology Gandhinagar, Palaj, Gujarat 382355, India
cCenter for Biomedical Engineering, Indian Institute of Technology Gandhinagar, Palaj, Gujarat 382355, India
First published on 18th May 2022
Abstract
Designing programmable biomaterials that could act as extracellular matrices and permit functionalization is a current need for tissue engineering advancement. DNA based hydrogels are gaining significant attention owing to their self-assembling properties, biocompatibility, chemical robustness and low batch to batch variability. The real potential of DNA hydrogels in the biomedical domain remains to be explored. In this work, a DNA hydrogel was coated on a glass surface and coupled to a synthetic IKVAV peptide by a chemical crosslinker. We observe enhanced neuronal differentiation, prolonged neurite length, dynamic movement of microtubules and cytoskeleton, and altered endocytic mechanisms in neuroblastoma-based stem cells for the peptide modified DNA hydrogel compared to the unmodified DNA hydrogel and controls. We anticipate that a peptide-modified DNA hydrogel could emerge as a promising scaffold coating material to develop nerve tissue conduits in the future for application in neuroscience and neuroregeneration.
 Dhiraj Bhatia | Dr Dhiraj Bhatia obtained his PhD from NCBS-TIFR in Bangalore, India, in DNA nanotechnology. Post PhD he went to Curie Institute in Paris to join the team of Ludger Johannes initially as a Curie fellow and later as an HFSP long-term fellow where he learnt the cellular and biological applications of DNA nanodevices. In 2018, he moved to India to start his own laboratory at the Indian Institute of Technology Gandhinagar where he is an Assistant Professor and Ramanujan fellow. His lab focusses on translational aspects of DNA nanotechnology to develop tools to program biological systems for biomedical applications. |
1. Introduction
One of the key challenges in neuroscience is the access to inner tissues in neural injuries that are hard to treat by surgery. Researchers aim to develop bioactive scaffolds that could support the neural cell viability, induce differentiation, promote migration, and elongate axonal/neurite growth in vitro, which could later be implanted in vivo as a functional tissue graft.1 The precursor stem cells or neuroblastoma cells (such as SH-SY5Y cells) can differentiate into mature neurons by providing various supplements in the form of growth factors, drugs, and peptides.2 Laminin is a crucial support element of the brain ECM.3 Several motifs/short peptides have been identified from laminin to be responsible for neural cell attachment, and enhanced differentiation, including Ile-Lys-Val-Ala-Val (IKVAV), Arg-Gly-Asp (RGD), etc.4 These short peptides could easily be synthesized in the lab and have numerous applications in scaffold decoration. Previously, researchers have functionalized different biomaterials with short peptides for tissue engineering applications and are reviewed elsewhere.5–8Deoxyribose nucleic acid (DNA) is a block copolymer known for carrying forward genetic information from one generation to the next generation.9 Recently, its application as a structural biomaterial has been getting massive attention globally.10,11 The precise Watson and Crick base pairing between the building block nucleotides such as Adenine (A) with Thymine (T) and Guanine (G) with Cytosine (C) makes DNA a choice of material for various biomedical applications as this feature provides minimal batch-to-batch variation and maximum reproducibility. Compared to many synthetic biomaterials, DNA is a non-toxic and degradable natural polymer depicting its high biocompatibility and resorbability. Also, a rich toolkit of enzymes/crosslinkers is available to manipulate or modify the DNA biopolymer at a single base resolution.12 Restriction enzymes, for example, are capable of cleaving the DNA at a specific site in contrast to ligases that can join the two strands by forming a phosphodiester linkage. Similarly, the crosslinkers like EDC-NHS (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide) and sulfo-MBS (m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester) could be used to modify the DNA strands with particular dyes or peptides. DNA nanotechnology applies these enzymatic/non-enzymatic (metal ions, biomolecules) toolkits to prepare multiple stimuli-responsive DNA hydrogels and nanocarriers.13
Hydrogels are scaffold materials that swell in water and mimic the extracellular matrix (ECM) and hence are employed by tissue engineers worldwide for cell reprogramming and other biomedical applications.10,11 Various natural and synthetic materials can be used to prepare hydrogels; however, DNA hydrogels have recently been explored in biomedical applications.10,11,14 DNA hydrogels are a programmable self-assembled network of multiple DNA strands.15 We previously demonstrated that DNA hydrogels act as a cushion for cell attachment, cell growth, and cell spreading. Additionally, the adhesive properties of DNA hydrogels reprogramme the cellular membrane for enhanced receptor expression and endocytosis mechanisms.12
Herein, we decorated native DNA hydrogels by linking the peptide with an X1 primer strand using sulfo-MBS crosslinking chemistry. The Ile-Lys-Val-Ala-Val (IKVAV) peptide was chemically synthesized in our lab and is responsible for the enhanced attachment and differentiation of neural cells.4 We hypothesize that the decoration of DNA hydrogels with the IKVAV peptide (XP) could further improve the capability of DNA hydrogels. Our results demonstrate strongly the ability of DNA-peptide based scaffolds as potential materials to develop nerve tissue conduits for neural tissue engineering applications in the future.
2. Results and discussion
A DNA hydrogel (HGX) was prepared by mixing four synthetic primers X1-NH2, X2, X3, and X4, in equal proportions, followed by temperature gradient PCR. Furthermore, a peptide was linked to HGX by sulfo-MBS chemistry and XP was prepared (Fig. 1a).12 The gel retardation assay confirmed the assembly of all the four primers and the formation of a hydrogel. The single oligo primer (X1) exhibited higher mobility on the gel; however, the HGX, due to aggregation and network formation, retarded and remained in the well (Fig. 1b). The dry morphology of the HGX obtained using a scanning electron microscope (SEM) at different magnifications suggests that it is a porous material (Fig. 1c) that might swell upon the addition of water and could act as a cushion material for cell growth. The peptide was synthesized and characterized by a peak at a retention time (RT) of 6.7 min in the total ion chromatogram which corresponds to the peptide peak at 844.58 m/z (M), with a (M + Na) peak at 866.58 m/z (Fig. S1†). Moreover, the optical microscopy image showed the network formation of HGX on a glass coverslip. The HGX morphology changed after IKVAV peptide modification. The hydrogel may get compacted on the peptide linkage (Fig. 1d), probably due to the hydrophobicity of the peptides decorated on the DNA surface. MALDI TOF/TOF data further confirmed the successful coupling of DNA-NH2 with the IKVAV peptide, where we observed the m/z peak shifting from 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 880 of X-NH2 to 13272 of X-P (Fig. 1e).
880 of X-NH2 to 13272 of X-P (Fig. 1e).
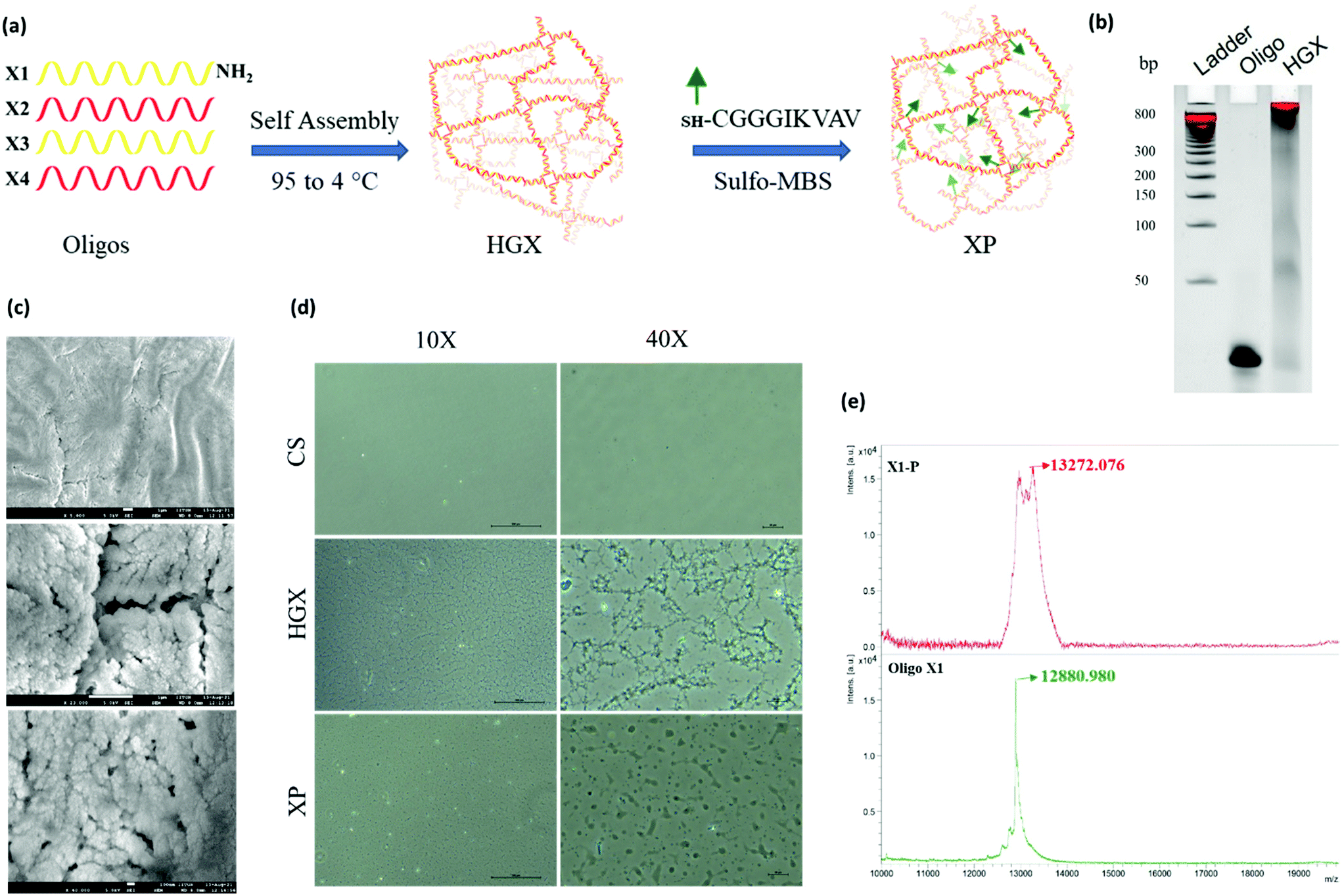 | ||
| Fig. 1 DNA hydrogel synthesis and characterization (a) schematic representation for the preparation of a DNA-based hydrogel (HGX) and its peptide modification (XP) through sulfo-MBS. (b) Gel retardation assay to confirm the formation of the DNA hydrogel. Lane 1 was loaded with a 50 bp DNA ladder, lane 2 with X1 oligo and lane 3 with HGX in 10% native polyacrylamide gel. (c) Field emission scanning electron microscopy image at increasing magnifications showing the porosity and cushion-like network of HGX. The scale bar in (c) is equal to 1 μm. (d) phase-contrast light microscopy image at 10× and 40× magnifications for the HGX and XP hydrogel morphology comparison where CS was the uncoated glass surface control. (e) Oligo X1 (bottom black, peak value 12880.980) and oligo X1 modification by the IKVAV peptide (top red, peak value 13272.076) were confirmed by MALDI TOF/TOF m/z peak shifting. | ||
SH-SY5Y neuroblastoma cells were cultured on glass coverslips (CS), DNA hydrogel (HGX), and IKVAV peptide modified DNA hydrogel (XP) in two separate differentiation media (DM) called DM#A (Fig. 2a) and DM#B (Fig. 2b). These differentiation media vary in their composition. DM#A contains DMEM/F12 media with 2.5% fetal bovine serum (FBS) and 1% pen–strep; however, DM#B is a neurobasal medium supplemented with 1% B-27, 20 mM KCl, 2 mM glutamax1, and 1% pen–strep without FBS. We quantified at day 3 and day 7 the neurite length of differentiated neurons cultured in both the media and compared. The DM#B medium showed a drastic increase in the size of the neurite/axon from day 3 to day 7 (Fig. S2†). At the same time, on day 7 alone, the neurite length of DM#B medium was more extended than DM#A (Fig. 2c). Therefore, we selected DM#B for all our further experiments. Moreover, we also noticed a significant change in the neurite length in CS, HGX, and XP. The neurite length of SH-SY5Y was significantly long over XP compared to HGX and CS, independent of the DM used (Fig. 2d and e). However, the HGX showed elongated neurites than CS, suggesting that DNA hydrogel supports the growth of the SH-SY5Y cells; however, its modification with peptide IKVAV further elongated the processes and offered the advantage of DNA hydrogel modification with peptides.
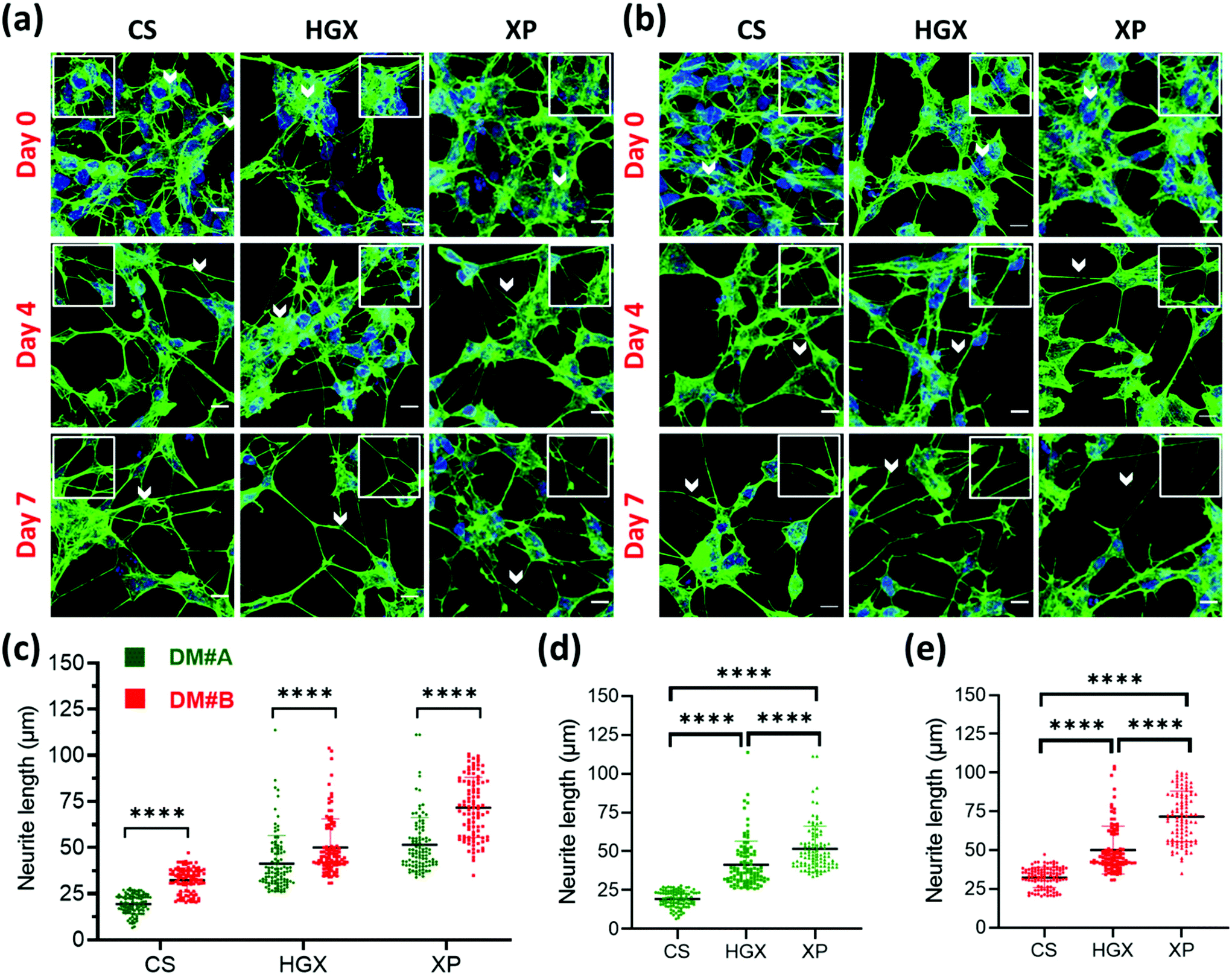 | ||
| Fig. 2 Optimization of differentiation media. The SH-SY5Y cells were cultured on CS, HGX, and XP from day 0 to day 7 in (a) differentiation media A (DM#A, shown in green) and (b) differentiation media B (DM#B, shown in red). The representative composite images have actin filaments stained with phalloidin-488 (green) and the nucleus with Hoechst (blue). The neurite length was measured using ImageJ software and the values were plotted for (c) both DM#A and DM#B for media comparison and individually (d) DM#A and (e) DM#B for materials comparison. For the quantification a total of n = 30 cells were selected randomly from 3 independent images. The statistical significance was tested by one-way ANOVA in the Prism software and is represented as **** when p < 0.0001. | ||
Our observation that HGX and XP favour SH-SY5Y cell growth laid the ground for studying further the change in neural cell pathways such as endocytosis. Galectin-3 (Gal-3) and transferrin (Tf) are standard molecules used to explore clathrin-independent and clathrin-dependent endocytic pathways.16 Fluorescently labelled Gal-3 (Alexa 647) and Tf (NHS rhodamine 123) were used to mark the endocytic pathways in SH-SY5Y cells cultured on CS, HGX, and XP at day 0 (Fig. 3a), day 3 (Fig. 3b), and day 7 (Fig. 3c), and imaged through a confocal microscope. The quantification of Gal-3 in the cell body indicates a more or less nonsignificant difference in CS, HGX and XP at day 0, which by day 7 has significantly enhanced in XP than CS and HGX (Fig. 3d). We calculated the intensity/unit area, which is defined as the fluorescence intensity per unit area for Gal 3 at day 0, and to our surprise XP showed a significantly low Gal-3 intensity/unit area. However, by day 7 the intensity/unit area increased for XP compared to CS and HGX, which could be due to neural cell differentiation where the cell body size reduced and various axonal processes occurred (Fig. S3a†). The axonal Gal-3 fluorescence intensity (Fig. 3g) was identical in CS, HGX, and XP at days 3 and 7; however, due to the longer length of axons in the XP matrix, we found a low Gal-3 intensity/unit area (Fig. S3c†).
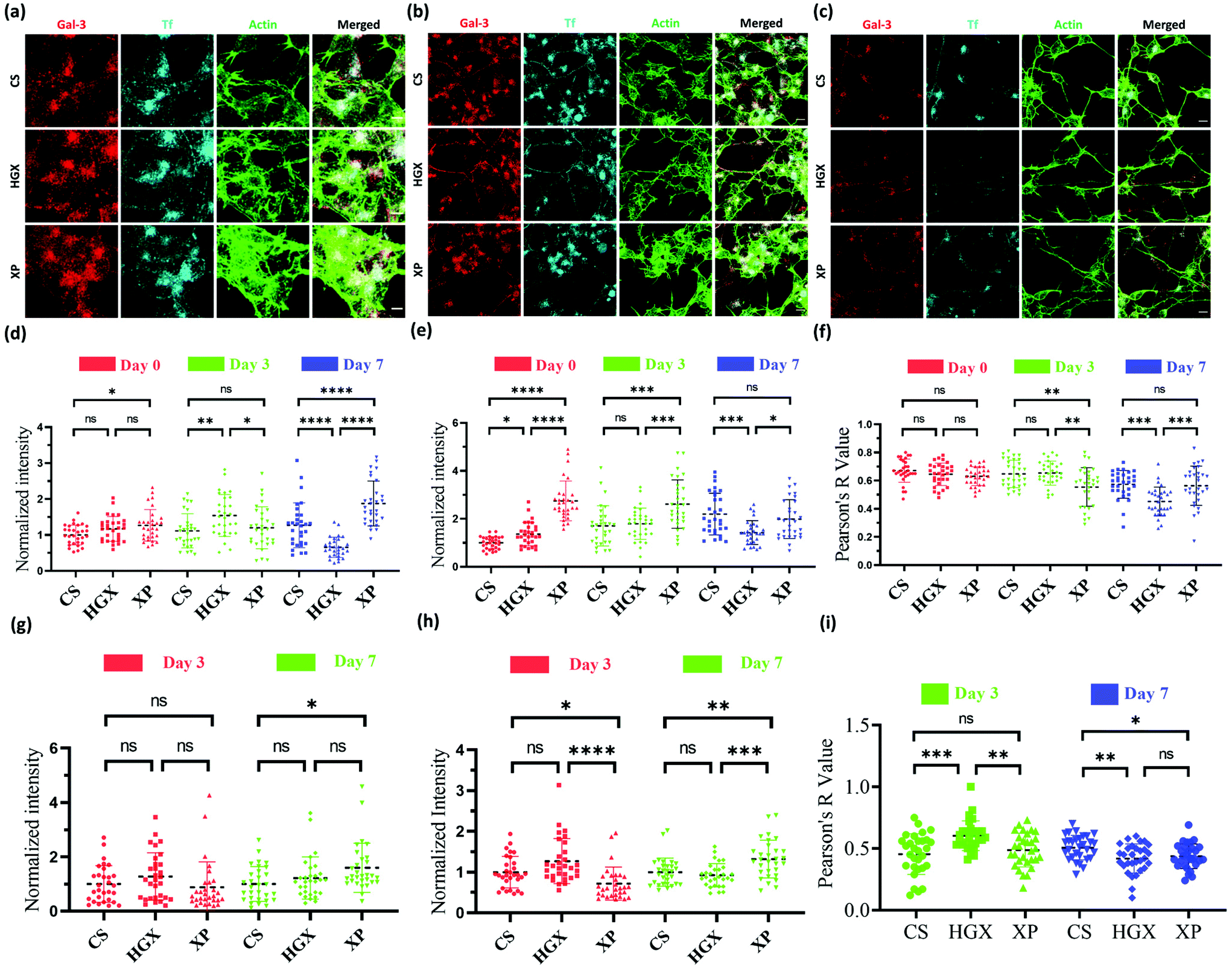 | ||
| Fig. 3 Cellular uptake of Gal-3 and Tf during SH-SY5Y neuroblastoma cell differentiation from day 0 to day 7. SH-SY5Y neuroblastoma cells were cultured on CS, HGX and XP for differentiation and incubated with Gal-3 and Tf endocytic probes and the cytoskeleton was stained with actin phalloidin at (a) day 0, (b) day 3 and (c) day 7. The quantified cellular fluorescence intensity of (d) Gal-3 and (e) Tf and the axonal fluorescence intensity of (g) Gal-3 and (h) Tf were plotted. (f) Cellular and (i) axonal colocalization of Gal-3 and Tf. For colocalization, Pearson's-R-value was calculated using the ImageJ software and plotted. For the quantification a total of n = 30 cells were selected randomly from 3 independent images. The statistical significance was tested by one-way ANOVA in the Prism software and is represented as **** when p < 0.0001, *** when p < 0.001, ** when p < 0.01, * when p < 0.05 and ns when there was no significant difference. | ||
The SH-SY5Y cell body fluorescence intensity of Tf was higher in XP from day 0 to day 3; however, it reduced further on day 7 (Fig. 3e). In contrast, we observed significantly enhanced axonal Tf fluorescence intensity at day 7 (Fig. 3h). Moreover, the cell body Tf intensity/unit area (Fig. S3b†) was significantly enhanced for XP and axonal Tf intensity/unit area (Fig. S3d†) was more or less the same from day 0 to day 7. In summary, our data indicate the distribution of Tf from cell bodies to the axons after differentiation.
We quantified the overlap between uptaken Gal3 and Tf by measuring the Pearson R-value for Gal-3 and Tf in both the cell body (Fig. 3f) and axons (Fig. 3i). The Pearson R-value above 0.3 indicates the statistically significant colocalization of Gal-3 and Tf. In the cell body of undifferentiated SH-SY5Y cells at day 0 Gal-3 and Tf were colocalized equally in all CS, HGX, and XP matrices. However, on day 3 the colocalization was reduced in the cell body of XP but remained the same in the axons compared to CS. On day 7 the colocalization in the cell body of XP did not change but was reduced in the axon. In summary, there was an alteration in colocalization of Gal-3 and Tf on the maturation of neurons, which is also in agreement with our previously published data.16
Microtubule-associated protein-2 (MAP2) and Tau family proteins are the markers of differentiated neurons.17,18 These proteins stabilize and regulate the activity of microtubule networks in axons and dendrites.19 SH-SY5Y cells grown over CS, HGX, and XP were immunostained using primary antibodies against MAP2 (Fig. 4a) and Tau (Fig. 4b) proteins on day 7 to check for neuronal differentiation. We recorded more numbers of axons and dendrites over XP compared to HGX and CS. The quantified fluorescence intensity also followed the same pattern suggesting that the DNA hydrogel modified by the IKVAV peptide could be a suitable material for enhanced neuronal differentiation (Fig. 4c and d).
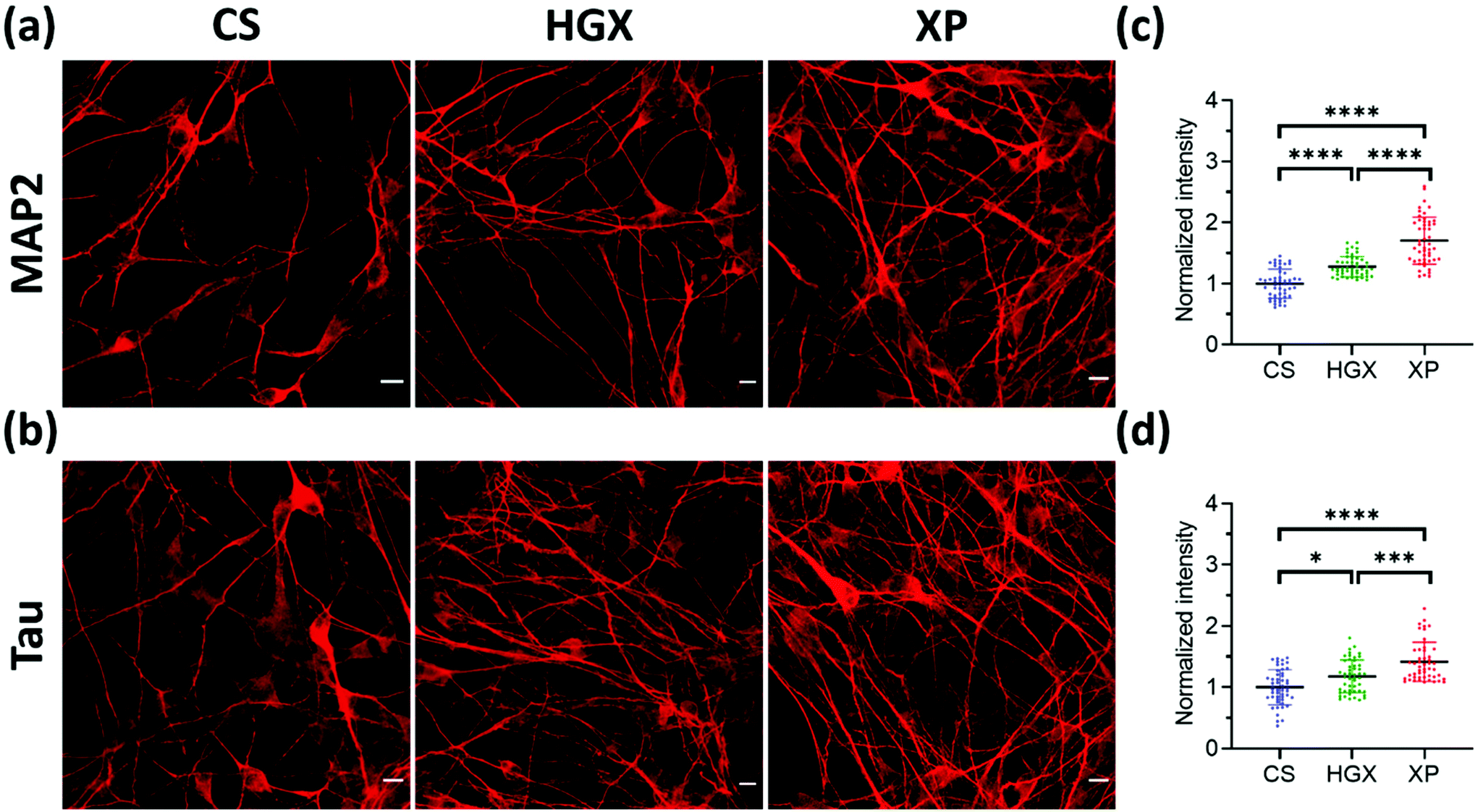 | ||
| Fig. 4 MAP2 and Tau staining in differentiated SH-SY5Y cells. SH-SY5Y neuroblastoma cells were grown over CS, HGX, and XP for seven days and stained for differentiation markers (a) MAP2 and (b) Tau protein. The images were obtained using a Leica confocal microscope. The fluorescence intensity of the (c) MAP2 and (d) Tau expression in the axons is quantified and plotted. For the quantification a total of n = 100 axonal regions were selected randomly from 3 independent images. The statistical significance was tested by one-way ANOVA in the Prism software and is represented as **** when p < 0.0001, *** when p < 0.001, and * when p < 0.05. | ||
To further understand the differentiated neural physiology and its adherence properties over HGX and XP, we immunostained them with alpha-tubulin, cadherin, vinculin, and paxillin antibodies. Alpha tubulin is a fundamental component for forming dynamic cytoskeleton microtubules involved in many crucial cell functions, from providing structural support to transport, cell division, cell migration, and neuronal axon/dendrite growth.20 We observed an enhanced alpha-tubulin fluorescence intensity of SH-SY5Y cells cultured over HGX and XP on day 0 (Fig. 5a and c). However, the neuronal processes in all cases at day 7 showed an insignificant difference but were rich in tubulin expression (Fig. 5b and e). Our observation indicates that the DNA hydrogel matrix might be an activator of the dynamic microtubules of SH-SY5Y cells. Therefore, from day 0 onwards, enhanced cellular growth causes longer neurite processes that we recorded and are shown in Fig. 2. Moreover, we could also track the active dividing cells characterized by microtubules that facilitate the segregation of chromosomes during cell division with alpha-tubulin antibody immunostaining (Fig. S4† inset). We could see the high growth rate and a greater number of neuroblastoma cells in the XP matrix. However, we counted only the dividing cells (inset) using the ImageJ software, and to our surprise, in a similar area under observation, we found 32 dividing cells in the XP matrix compared to 24 on HGX and 12 on CS. It makes a huge difference and aids in our claim about the potentiality of the XP matrix for neural tissue engineering.
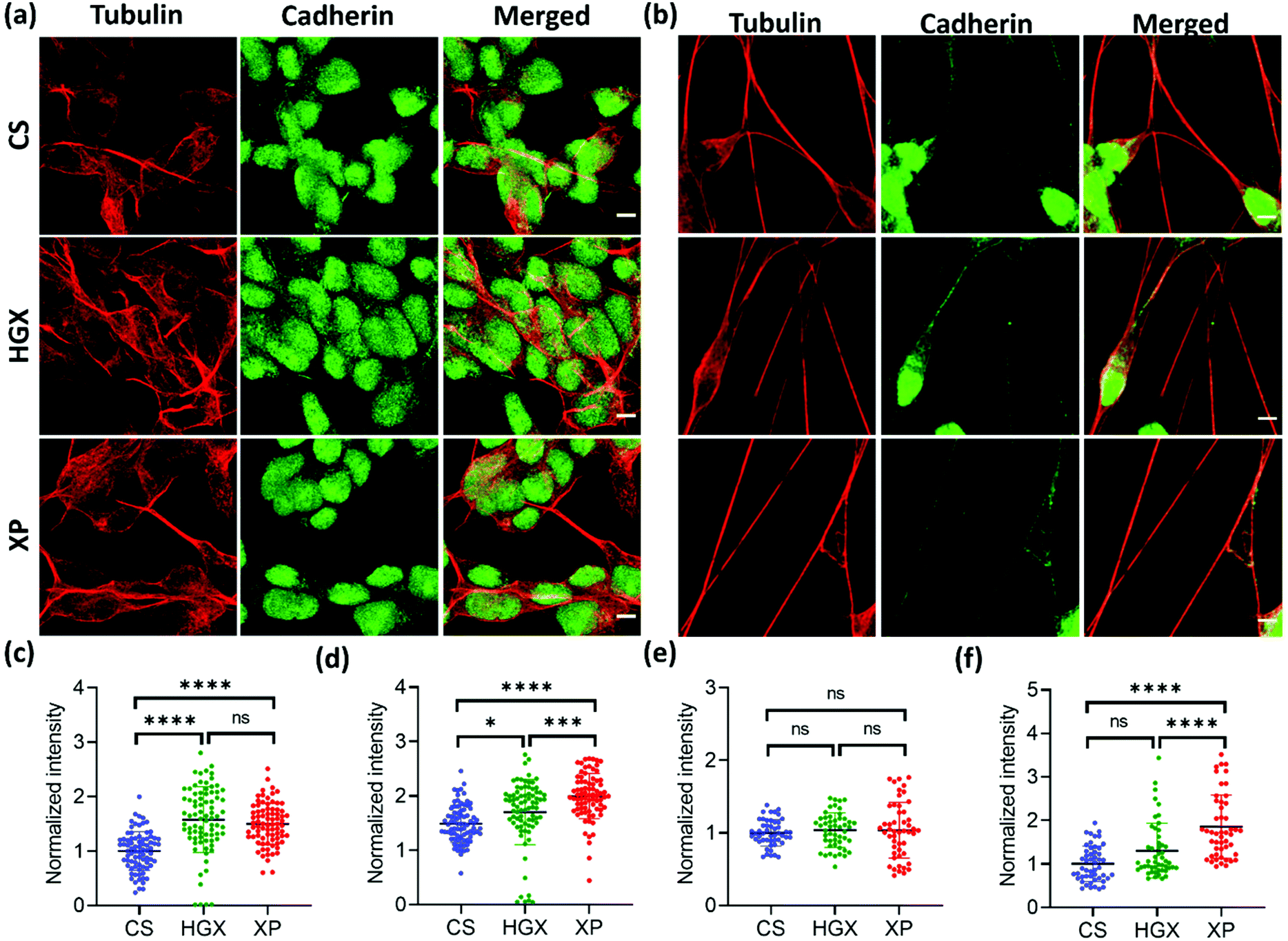 | ||
| Fig. 5 Tubulin and cadherin immunostaining in SH-SY5Y cells during differentiation. The SH-SY5Y cells grown over CS, HGX and XP were immunostained with tubulin and cadherin antibodies followed by the respective secondary antibodies at (a) day 0 and (b) day 7 and fluorescence images were obtained using a confocal microscope. The fluorescence intensity was quantified for both tubulin and cadherin at (c and d) day 0 and (e and f) day 7, respectively. For the quantification a total of n = 30 cells were selected randomly from 3 independent images. The statistical significance was tested by one-way ANOVA in the Prism software and is represented as **** when p < 0.0001, *** when p < 0.001, * when p < 0.05 and ns when there was no significant difference. | ||
Cadherin is a family of transmembrane proteins involved in calcium-dependent cell–cell adhesion. Cadherin is usually present on the plasma membrane of cells; however, we found its unusual localization in the nuclear region (Fig. 5a and b). Also, the cadherin fluorescence intensity remained high in the XP matrix from day 0 to day 7 (Fig. 5d and f). To our knowledge, this is the first report to show the nuclear localization of cadherin in neuroblastoma cells. However, previously, researchers have reported the nuclear localization of cadherin in lung cancer cells,21 solid pseudopapillary tumors of the pancreas,22 pulmonary adenocarcinomas,23etc. The reason behind the nuclear localization of cadherin is unknown; however, it could be a characteristic feature of neuroblastoma cells.
Next, we immunostained the SH-SY5Y cells with a vinculin antibody. Vinculin is a cytoskeletal protein involved in cell–cell and cell–ECM adhesion. We have recorded a high fluorescence intensity indicating high vinculin expression on XP seeded SH-SY5Y cells at day 0 (Fig. 6a and e). The vinculin expression also remained high on differentiated neuronal projections as obtained and quantified on day 7 (Fig. 6c and g). Furthermore, the paxillin antibody was used to immunostain the focal adhesion points of the SH-SY5Y cells on CS, HGX, and XP that appeared as puncta in the images (Fig. 6b and d). We counted significantly high numbers of puncta in undifferentiated SH-SY5Y cells cultured in the XP matrix compared to HGX and CS at day 0 (Fig. 6b and f). The high puncta numbers indicate the crucial role of the IKVAV peptide in cell adhesion and consequent signalling pathways. The number of puncta in differentiated neuronal projection was further increased, which we quantified by the fluorescence intensity presented in Fig. 6d and h.
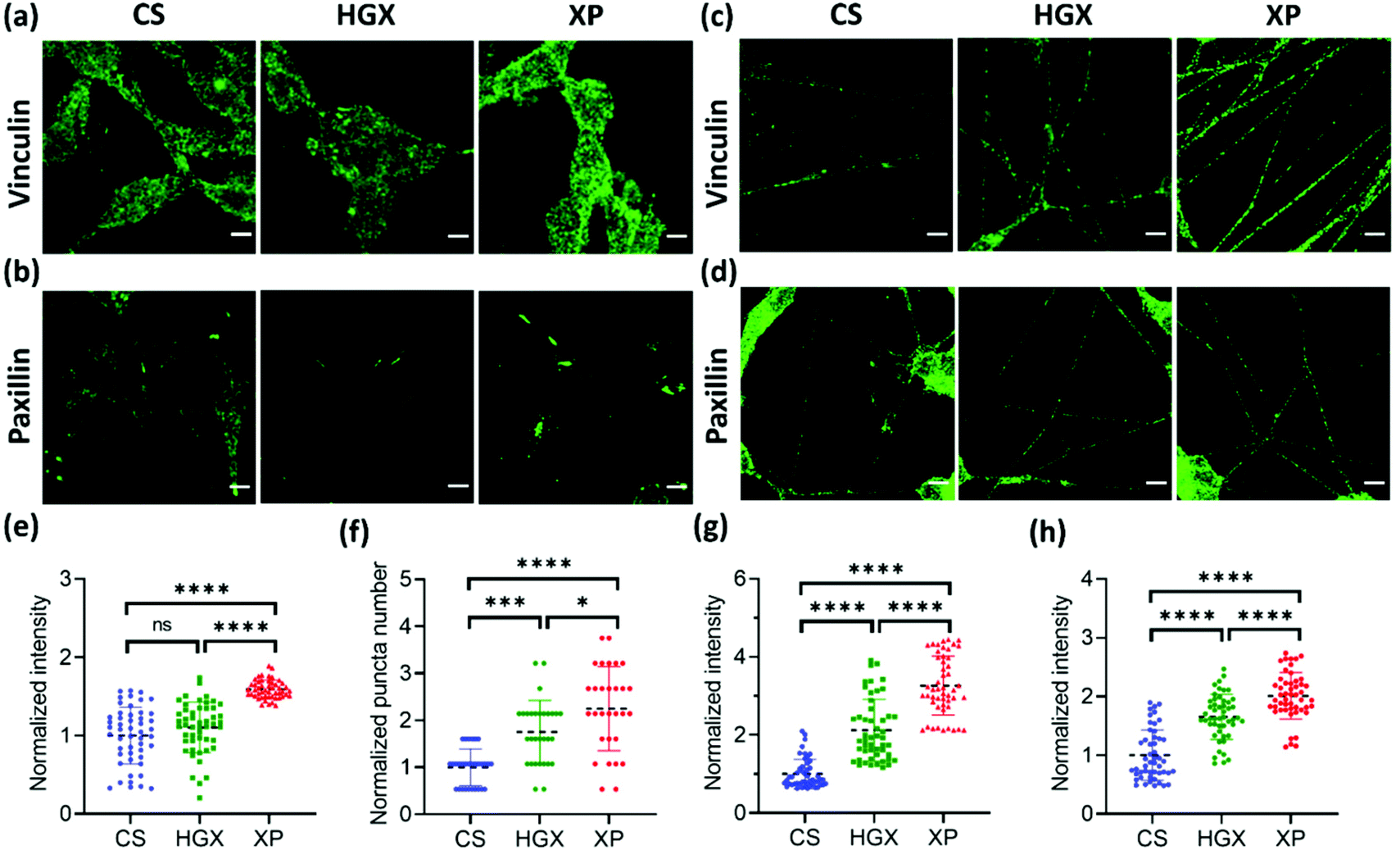 | ||
| Fig. 6 Vinculin and paxillin immunostaining in SH-SY5Y cells during differentiation. The SH-SY5Y cells grown over CS, HGX, and XP were immunostained with vinculin and paxillin antibodies followed by the respective secondary antibodies at (a and b) day 0 and (c and d) day 7, and fluorescence images were obtained using a confocal microscope. The fluorescence intensity was quantified for vinculin and paxillin at (e and f) day 0 and (g and h) day 7, respectively. For the quantification a total of n = 30 cells were selected randomly from 3 independent images. The statistical significance was tested by one-way ANOVA in the Prism software and is represented as **** when p < 0.0001, *** when p < 0.001, * when p < 0.05 and ns when there was no significant difference. | ||
3. Conclusion
DNA hydrogels have started to emerge as a novel class of biomaterials that have recently been studied for their numerous biomedical applications.24 Our work demonstrates that surface coating with a DNA hydrogel matrix (HGX) supports and promotes the growth of SH-SY5Y neuroblastoma cells. Moreover, when the DNA hydrogel was modified and linked with the peptide IKVAV, the growth rate of these neuroblastoma cells is further enhanced, and its differentiation is accelerated compared to HGX and CS. Similar culture conditions, including neurobasal media and equal incubation time, were provided to all three comparative groups of cells. However, we observed multiple physiological changes in XP cultured undifferentiated neuroblastoma cells from day 0, resulting in an accelerated differentiation process of neurons on the matrix by day 7. The longer neural projections, enhanced endocytic pathways, overexpression of neural cell differentiation markers MAP2/Tau family protein, and improved expression of cytoskeletal marker proteins namely tubulin, cadherin, vinculin, and paxillin indicate the superiority of HGX modification with the IKVAV peptide. Taken together, we present an entirely new role of DNA based scaffolds which have the ability to trigger neuronal regeneration which we hope to metamorphose into real world applications of creating organs on chips with applications in neuroscience and neurodegenerative therapeutics.4. Materials and methods
Reagents
Fetal bovine serum (FBS), Dulbecco's Modified Eagle's Medium/F12 (DMEM/F12), penicillin–streptomycin, trypsin-EDTA (0.25%)–phenol red, phosphate buffered saline (PBS), penicillin–streptomycin, B-27™ Plus Supplement, and Neurobasal™ Medium were purchased from Gibco. MAP2 (Product# 13-1500), Hoechst (Product# 62249), phospho-Vinculin (Tyr100) polyclonal antibody (Catalog # 44-1074G), Pan-cadherin Polyclonal Antibody (Catalog # 71-7100), alpha Tubulin Monoclonal Antibody (DM1A) (Catalog # 62204), Paxillin Polyclonal Antibody (Catalog # PA5-111334), Alexa Fluor™ 488 phalloidin (Product # A12379), Goat anti-Mouse IgG (H+L), Superclonal™ recombinant secondary antibody, Alexa Fluor 647 (Catalog # A21235), Goat anti-Rabbit IgG (H+L), Superclonal™ secondary antibody, and Alexa Fluor® 488 (Catalog # A27034) were purchased from ThermoFisher Scientific. Tau (5A6) monoclonal antibody (Catalog # DSHB-C1-130) against tau protein was purchased from DSHB. Retinoic acid and transferrin were purchased from Sigma. The Gal-3-His plasmid was a gift from the Ludger Johannes team at Institut Curie, Paris, and was used as provided and labeled with rhodamine dye.Preparation of the DNA hydrogel (HGX)
The sequences of ssDNA primers X1-NH2, X2, X3 and X4 with sticky ends were taken from previously published reports.12,25 The primers were mixed at 100 μM concentration with 2 mM MgCl2 followed by a gradient temperature PCR reaction.12 The formed HGX was stored at 4 °C until further use. The gel retardation assay was performed to check the polymerization of X-monomers. The 10% native polyacrylamide gel was prepared as previously done12 and loaded in the wells containing 2 μL (ladder, oligo, and HGX) with 3 μL of DNA loading dye and 3 μL of TAE buffer. The gel was run at 90 V for 90 min, followed by ethidium bromide staining. The gel image was obtained using a Syngene Gel documentation system. The DNA hydrogel was vacuum dried over a mica sheet and subjected to field emission scanning electron microscopy (FESEM) and phase-contrast light microscopy for morphology assessment.IKVAV peptide synthesis
The 9 amino acid peptide C-G-G-G-I-K-V-A-V was synthesized using our in-house Fmoc based solid-phase peptide synthesis protocol.26 All amino acids (AA) and reagents were purchased from Sigma Aldrich unless specified otherwise. The peptide sequence was assembled on Rink amide aminomethyl polystyrene resin (loading 0.6 mmol g−1) in a microwave-assisted peptide synthesizer Initiator + SP wave (Biotage, Uppsala, Sweden) using the Fmoc method. All compositions are reported as % volume unless specified otherwise.The resin was weighed and taken in a 12 mL polypropylene fritted syringe and swelled in anhydrous DCM (2 mL) for 2.5 hours. After draining, the resin was washed with DCM (3 × 2 mL) and subjected to Fmoc removal followed by coupling of the first Fmoc–AA as described below. Each subsequent Fmoc–AA was coupled to the peptidyl resin using the same two-step procedure:
1. The Fmoc group from the peptidyl resin was removed by incubation with 2 mL of deprotection solution (5% piperazine + 2% DBU in DMF unless specified) for 3 min and 12 min at ambient temperature with continuous vertexing at 900 rpm. The resin was additionally washed with 2 mL of deprotection solution followed by alternate washes with DMF (3 × 2 mL) and DCM (3 × 2 mL).
2. All Fmoc–AA complexes were coupled twice by the DIC/Oxyma method. Briefly, Fmoc–AA (5 equiv., 0.1 M in dry DMF) was mixed with Oxyma (5 equiv.), preactivated with DIC (5 equiv.) for 3 min with mild shaking and added to the peptidyl resin manually. After incubation for 5 min at rt in the MW reactor, the reaction temperature was increased to 65 °C (∼40-watt output) and maintained for 15 min with continuous vortexing at 700 rpm. The resin was drained and washed with DMF (2 mL) and the coupling reaction was repeated one additional time to ensure completion. Post-coupling, the resin was washed with DMF (3 × 2 mL) and DCM (3 × 2 mL) alternately.
N-Terminal acetylation: The fully deprotected resin was reacted with triethyl amine (5 eq of resin) and acetic anhydride (10 eq. of resin) solution in 2 ml of DMF, and incubated for 1.5 h with continuous vortexing at 700 rpm for the reaction to proceed. The above step was repeated twice to ensure complete acetylation.
Peptide cleavage
Post-synthesis the peptidyl resin was washed with methanol (3 × 2 mL) and DCM (3 × 2 mL) alternately and dried under reduced pressure at 4 °C for 2 h. The peptide was released from the resin with 92.5% TFA, 2.5% water, 2.5% DoDT, and 2.5% Tis, at rt for 3 h with continuous vortexing at 900 rpm. The resin was removed by filtration and washed with TFA. All filtrates were combined and evaporated under nitrogen flow to nearly 1/5th of the volume. A fully unprotected peptide was precipitated from ice-cold diethyl ether and pelleted by centrifugation. After decanting ether, the peptide pellet was dried under nitrogen flow and stored at −80 °C until further characterization.The peptide sample was analysed using a UHPLC-MS (Thermo Scientific™, LTQ XL™ Linear Ion Trap Mass Spectrometer) equipped with a reverse phase C18 column (Hypersil GOLD TM, 150* 2.1 mm dia, 3 μ particle size, Thermo Scientific™) using the ESI positive method. Peptides bound to the column were eluted with the method in 15 min, with a linear gradient from 0 to 100% acetonitrile over 10 min and 5 min of reverse gradient, at a flow rate of 0.3 mL min−1 and monitored at 220 nm. The solvents used were acetonitrile and water with 0.1% formic acid.
Preparation of XP
The X1 oligo primer with a primary amine group attached at the 5′ end (X1-NH2) was purchased from Sigma Aldrich. The peptide C-G-G-G-I-K-V-A-V contains the sulfhydryl (SH) group in the cysteine. For XP synthesis, the HGX was first prepared as mentioned above. Once the HGX-NH2 formed, it was spread over the glass coverslip. The NH2 functional group-containing HGX was submerged in an aqueous sulfo-MBS (m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester) chemical crosslinker at room temperature for 2 h to maleimide for activating HGX-NH2. Next, the SH group-containing peptide was spread over the activated HGX to form stable thioether bonds between HGX and the peptide (XP). The oligo-peptide linkage was confirmed by MALDI-TOF/TOF MS, where 2 μL samples of each oligo X1 and oligo X1-P were mixed with the 3-hydroxypicolinic acid matrix and subjected to laser desorption.Cell culture
Immunofluorescence assay
:250, in 1% BSA staining solution and incubated for 3 h at room temperature, and labelled with goat anti-mouse IgG (H+L), Superclonal™ recombinant secondary antibody, Alexa Fluor 647 (Catalog # A21235) at a dilution of 1:1000 for 30 min at room temperature. The samples were mounted and stored at 4 °C until microscopy was performed.
:100, in 1% BSA staining solution and incubated for 3 h at room temperature. Vinculin, cadherin and paxillin primary antibodies were labelled with Goat anti-Rabbit IgG (H+L) Superclonal™ Secondary Antibody, Alexa Fluor® 488 at a dilution of 1:1000 for 30 min at room temperature. However, alpha Tubulin primary antibody was labelled with Goat anti-Mouse IgG (H+L) Superclonal™ Secondary Antibody, Alexa Fluor®633 at the same dilution and incubation time as before. The final slide was placed under a confocal microscope (Leica TCS SP8), and images were obtained at 63× magnification. The image analysis was done on particular parameters (as indicated in the figure legends) in the ImageJ software, where random 50 or 100 cells were selected from 3 different sets of images.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 8.0.2 software. An ordinary one-way ANOVA test was performed for P-value significance. The P-value summary indicates the significance. * indicates the significance as * = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001, and ns = nonsignificant and indicated in the respective figures wherever applicable.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We sincerely thank all the members of the D. B. and S. G. groups for critically reading the manuscript and for their valuable feedback. P. H., A. G., and K. B. thank the IITGN–MHRD, Government of India, for the fellowship. G. S. and A. S. thank the CSIR and DBT, Government of India, for the respective research fellowships. D. B. thanks the SERB, Government of India, for the Ramanujan Fellowship and the DBT-EMR, GUJCOST, GSBTM and BRNS–BARC for research funding. S. G. thanks the SERB for the Core research grant. The work in host laboratories was funded by the MHRD, DST-SERB and GSBTM, Government of India.References
- L. R. Doblado, C. Martínez-Ramos and M. M. Pradas, Front. Nanotechnol., 2021, 3, 21 CrossRef.
- S. G. Marapureddy, P. Hivare, S. Kumar, S. Gupta and P. Thareja, Biomed. Mater., 2021, 16(4), 045019 CrossRef CAS PubMed.
- K. J. Hamill, K. Kligys, S. B. Hopkinson and J. C. R. Jones, J. Cell Sci., 2009, 122, 4409 CrossRef CAS PubMed.
- R. Patel, M. Santhosh, J. K. Dash, R. Karpoormath, A. Jha, J. Kwak, M. Patel and J. H. Kim, Polym. Adv. Technol., 2019, 30, 4–12 CrossRef CAS.
- N. Stephanopoulos, R. Freeman, H. A. North, S. Sur, S. J. Jeong, F. Tantakitti, J. A. Kessler and S. I. Stupp, Nano Lett., 2015, 15, 603–609 CrossRef CAS PubMed.
- K. Luder, K. Kulkarni, H. W. Lee, R. E. Widdop, M. P. Del Borgo and M. I. Aguilar, Chem. Commun., 2016, 52, 4549–4552 RSC.
- S. Chen, M. Zhang, X. Shao, X. Wang, L. Zhang, P. Xu, W. Zhong, L. Zhang, M. Xing and L. Zhang, J. Mater. Chem. B, 2015, 3, 6798–6804 RSC.
- R. Jain and S. Roy, ACS Biomater. Sci. Eng., 2020, 6, 2832–2846 CrossRef CAS PubMed.
- A. J. F. Griffiths, J. H. Miller, D. T. Suzuki, R. Lewontin and W. M. Gelbart, Biol. Plant., 2002, 45(1), 50 CrossRef.
- A. Gangrade, N. Stephanopoulos and D. Bhatia, Nanoscale, 2021, 13, 16834–16846 RSC.
- V. Morya, S. Walia, B. B. Mandal, C. Ghoroi and D. Bhatia, ACS Biomater. Sci. Eng., 2020, 6, 6021–6035 CrossRef CAS PubMed.
- S. Walia, V. Morya, A. Gangrade, S. Naskar, A. G. Teja, S. Dalvi, P. K. Maiti, C. Ghoroi and D. Bhatia, bioRxiv, 2021, 2021.07.31.454582.
- F. Li, J. Tang, J. Geng, D. Luo and D. Yang, Prog. Polym. Sci., 2019, 98, 101163 CrossRef CAS.
- G. Janani, M. Kumar, D. Chouhan, J. C. Moses, A. Gangrade, S. Bhattacharjee and B. B. Mandal, ACS Appl. Bio Mater., 2019, 2, 5460–5491 CrossRef CAS PubMed.
- P. Hivare, C. Panda, S. Gupta and D. Bhatia, ACS Chem. Neurosci., 2021, 12, 363 CrossRef CAS PubMed.
- P. Hivare, A. Rajwar, S. Gupta and D. Bhatia, ACS Appl. Bio Mater., 2021, 4(4), 3350–3359 CrossRef CAS PubMed.
- S. Geetha Marapureddy, P. Hivare, A. Sharma, J. Chakraborty, S. Ghosh, S. Gupta and P. Thareja, Carbohydr. Polym., 2021, 269, 118254 CrossRef PubMed.
- M. M. Shipley, C. A. Mangold and M. L. Szpara, J. Visualized Exp., 2016, 2016, 1–11 Search PubMed.
- L. Dehmelt and S. Halpain, Genome Biol., 2005, 6, 1–10 Search PubMed.
- J. Aiken, G. Buscaglia, E. A. Bates and J. K. Moore, J. Dev. Biol., 2017, 5, 8 CrossRef PubMed.
- Y. J. Su, Y. W. Chang, W. H. Lin, C. L. Liang and J. L. Lee, Oncogenes, 2015, 4, e157–e157 CrossRef CAS PubMed.
- S. Serra, S. Salahshor, M. Fagih, F. Niakosari, J. M. Radhi and R. Chetty, J. Pancreas, 2007, 8, 296–303 Search PubMed.
- K. C. Moon, S. Y. Cho, H. S. Lee, Y. K. Jeon, J. H. Chung, K. C. Jung and D. H. Chung, Arch. Pathol. Lab. Med., 2006, 130, 1320–1325 CrossRef CAS PubMed.
- M. Maeda, T. Kojima, Y. Song and S. Takayama, Adv. Healthcare Mater., 2019, 8, 1801243 Search PubMed.
- S. H. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and D. Luo, Nat. Mater., 2006, 5, 797–801 CrossRef CAS PubMed.
- K. Ralhan, V. G. KrishnaKumar and S. Gupta, RSC Adv., 2015, 5, 104417–104425 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1nr07187d |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |