
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShape dependent sensing potential of gold nanoparticles in etching based multicolorimetric plasmonic-ELISA†
Sangeeta
Yadav
 a and
Jitendra
Satija
*b
a and
Jitendra
Satija
*b
aSchool of Biosciences and Technology, Vellore Institute of Technology (VIT), Vellore-632014, Tamilnadu, India
bCentre for Nanobiotechnology, Vellore Institute of Technology (VIT), Vellore-632014, Tamilnadu, India. E-mail: jsatija11@gmail.com; jsatija@vit.ac.in
First published on 17th August 2022
Abstract
In the present study, a systematic investigation has been carried out for the first time to assess the potential of three different shapes of gold nanoparticles (AuNPs), viz. nanorods (AuNRs), nanotriangles (AuNTs), and nanospheres (AuNSs), to develop a horseradish peroxidase (HRP) enzyme-mediated etching-based plasmonic ELISA (p-ELISA) strategy. The etching of the AuNPs in ELISA is achieved by 3′-3-5′-5-tetramethylbenzidine (TMB2+), which is produced by the biocatalytic conversion of chromogenic TMB via HRP. All three types of AuNPs were interacted with varying concentrations of TMB2+ (7–131 μM) (product of HRP enzyme reaction) and characterized for visible color change and by UV-Vis spectroscopy and transmission electron microscopy (TEM). From the comparative analysis of all three shapes of AuNPs, AuNRs exhibited vivid visible color change and absorbance intensity change compared to spherical and triangle-shaped nanoparticles. The TEM analysis of the etched nanoparticles revealed the gradual etching pattern of AuNRs compared to AuNTs which resulted in multicolor generation as opposed to AuNTs where the etching was relatively very fast and thus shows a faster shape transformation and poor color discrimination. Further, the potential of the AuNR etching-based optimized strategy was successfully demonstrated to develop an indirect competitive p-ELISA for human IgG detection. The developed p-ELISA showed an ultra-low visual limit of detection of 1 fg mL−1 (∼6.54 aM) without the aid of any sophisticated instruments. In the future, the developed competitive p-ELISA strategy can be easily employed to develop cost-effective, portable, and point-of-care assays for the detection of various disease biomarkers with ultra-high sensitivity.
Introduction
Plasmonic-enzyme linked immunosorbent assay (p-ELISA) has garnered immense interest in the last decade towards the development of ultrasensitive visual biosensors for various disease biomarkers, environmental pollutants, and food adulterants.1–6 This strategy relies upon the modulation in the localized surface plasmon resonance (LSPR) properties of the noble metal nanoparticles (MNPs) through a biocatalytic-triggered reaction that transforms their shape, size, composition, or agglomeration state.7–10 Since the LSPR property of the nanoparticles is size/shape and composition-dependent, a very minute structural change can give rise to a noticeable signal. Another important advantage of p-ELISA is the high extinction coefficient of the metal nanoparticles (e.g. gold nanoparticles (AuNPs) ε = 108–109 M−1.cm−1) compared to the traditionally used chromogenic substrate, i.e. 3,3′,5,5′-tetramethylbenzidine (TMB) (ε = 5.9 × 104 M−1.cm−1), which make them a prudent choice to produce an intense color change even at a very low concentration of the analyte.11,12To date, the p-ELISA strategy has been realized by inducing the (i) aggregation of the MNPs,13,14 (ii) metallization of the MNPs,15,16 and (iii) etching-mediated shape transformation of MNPs.17,18 In the first approach, the enzyme-catalyzed reaction induces the aggregation of MNPs via altering their surface charges. The degree of MNPs aggregation depends upon the analyte concentration that correspondingly results in a change in characteristic LSPR peak and colloidal solution color which is observed through the naked eye and ELISA plate optical reader. For instance, Nie et al. reported a p-ELISA strategy based on AuNPs aggregation for the detection of syphilis by utilizing the biocatalytic reaction involving acetylcholinesterase mediated generation of cationic thiocholine (TCh) in relation to the analyte concentration.19 The TCh interacts strongly with citrate-capped AuNPs due to thiol groups and thereby alters the surface charges which eventually triggers the aggregation of AuNPs, causing a visual change in color from red to blue.
The second strategy is typically based on the biocatalytic reaction mediated reduction of metal salts which eventually forms a thin metal film (i.e. metallization) in a heteroepitaxial manner on the template nanoparticles to produce core–shell type nanostructure.20,21 A small difference in the lattice constant of the deposited metal and the template MNPs is an important requirement for the uniform deposition of metal on template nanoparticles. For example, in the case of silver and gold, the lattice constant is as low as 0.25%, thus AuNPs can be easily utilized as a template to grow silver to produce a core–shell nanostructure.22 Using this approach, Yang et al. formulated a p-ELISA strategy by depositing silver on the gold nanorods (AuNRs) in a core/shell manner to detect prostate-specific antigen (PSA).23 The biocatalytic reaction involving alkaline phosphatase (ALP) mediates the generation of dephosphorylated p-aminophenol in relation to the PSA concentration, which eventually reduces silver salt into metallic silver that gets deposited on AuNRs. Accordingly, a significant change in the longitudinal LSPR peak and color of the AuNRs solution from pale red to green, purple and brown was observed. The third strategy is based on the enzyme-mediated generation of the etchant that results in the morphological transformation of MNPs and a visually detectable color change. For example, Yuan et al. reported a p-ELISA strategy by oxidative etching of silver nanoprism (AgNPRs) for the qualitative and quantitative determination of danofloxacin (DAN).24 In this indirect competitive p-ELISA, glucose oxidase enzyme-mediated generation of hydrogen peroxide (H2O2) triggers the oxidative etching of AgNPRs, which results in a prominent color change from dark blue to colorless.
All three p-ELISA strategies have demonstrated good sensitivity and selectivity without the aid of an optical reader, however, the MNP etching-based color development approach is the most suitable for the field deployable point-of-care assays. This is based on the fact that the predefined multicolor change in the reaction solution depends upon the shape transformation of MNPs that can be easily correlated with the concentration of the analyte molecules in the sample. Unlike, the aggregation and metallization-based p-ELISA, the effect of reaction conditions and environmental factors such as temperature, is negligible and thus minimizes the chances of false-positive or false-negative results in the case of etching-based p-ELISA strategies.25–28 The sensitivity of the etching-based p-ELISAs can be further enhanced by many folds by precise control of the LSPR peak (or the shape) of the MNPs.
To date, various anisotropic MNPs such as nanorods, nanocages, nanocubes, nanoprisms, and nanoflowers have been explored for the development of LSPR based colorimetric assays.29–37 Among all other etching processes, the oxidative etching of AuNPs via TMB2+ has garnered more attention due to their controlled nature of etching and futuristic convenient translation possibilities. Therefore, the reshaping-induced LSPR peak shift can serve as a probe for the development of multi-colorimetric p-ELISA strategies. Although several studies have utilized any single type of AuNPs for the development of TMB2+-mediated etching reaction based on p-ELISA, a detailed comparative analysis of TMB2+ mediated re-shaping of different shapes of AuNPs and to answer which shape of the AuNPs shows better sensor performance both in terms of UV-Vis spectral shift and naked-eye detection have not been investigated.
In this study, for the first time, we have reported the systematic comparison of selective etching of three different shapes of AuNPs, viz. gold nanospheres (AuNSs), gold nanorods (AuNRs), and gold nanotriangles (AuNTs), by TMB2+ (Scheme 1). The selection of TMB2+ as an etchant is based on the fact that most of the commercial ELISA kits employ horseradish peroxidase (HRP) and TMB as enzyme–substrate pair where the TMB (colorless) hydrolyzed into TMB2+ (yellow) via HRP.38 The TMB2+ oxidizes Au(0) into Au(I) in the presence of an optimum concentration of CTAB at room temperature resulting in their shape transformation causing a visual color change and spectral shift.39 The etching process is tracked via observing the visual color change in the colloidal solution of AuNPs, by recording the UV-Vis absorption spectra to measure the wavelength shift, and TEM micrograph of AuNPs before and after interaction with TMB2+ to assess the level of etching. Amongst the three different shapes, AuNRs showed a gradual change both in the LSPR peak and colorimetric change as a function of etchant concentration. Further, the potential of the AuNRs-etching based optimized strategy was demonstrated by developing an indirect competitive multicolorimetric p-ELISA strategy capable of detecting human IgG (as a model antigen) with a visual detection limit of 1 fg mL−1 (∼6.54 aM).
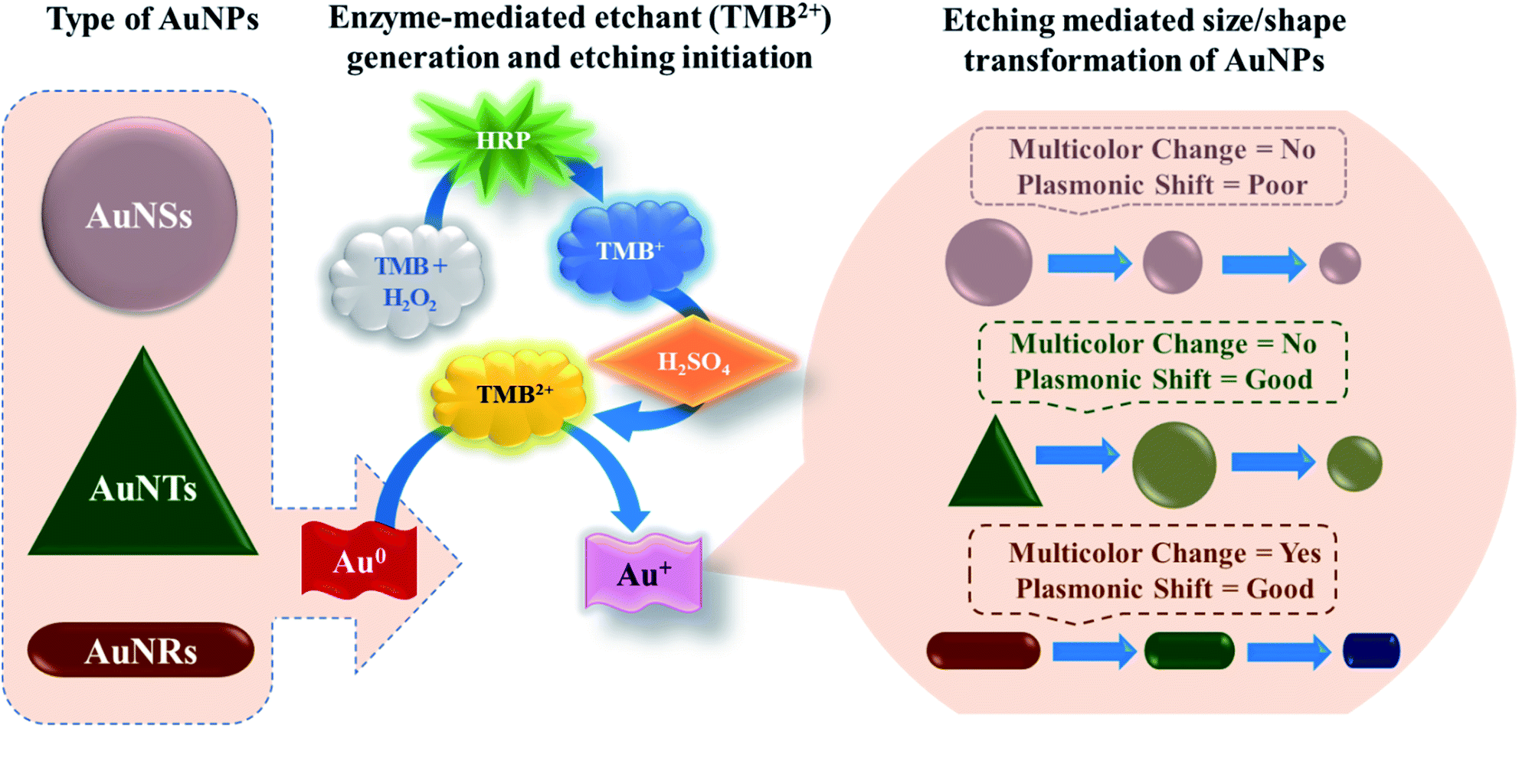 | ||
| Scheme 1 Schematic illustration of etching-based p-ELISA strategy developed by employing gold nanospheres (AuNSs), gold nanorods (AuNRs), and gold nanotriangles (AuNTs). The enzyme horseradish peroxidase (HRP) hydrolyses 3,3′,5,5′-tetramethylbenzidine (TMB) into TMB+, which is oxidized into TMB2+ upon H2SO4 acid treatment. TMB2+ acts as an etchant for the gold nanoparticles and leads to their size and shape transformation. | ||
Materials and methods
Materials
Silver nitrate (AgNO3), gold(III) chloride solution (HAuCl4), phosphate-buffered saline (PBS, pH 7.4), horseradish peroxidase (HRP), sodium borohydride (NaBH4), cetyl-trimethyl ammonium bromide (CTAB), 3,3,5,5-tetramethylbenzidine (TMB), Corning® 96 well microtiter plate, ascorbic acid (AA) and tween20 were procured from Sigma-Aldrich, India. Trisodium citrate (TSC) was procured from Sisco Research Laboratories Pvt. Ltd., India. Human immunoglobulin G (HIgG) and HRP conjugated goat anti-HIgG (GaHIgG@HRP) antibodies were obtained from Bangalore Genei Pvt Ltd., India. Bovine serum albumin (BSA) was purchased from Himedia Labs Ltd., India. Hydrogen peroxide (H2O2) (30 wt%) was purchased from Merck Chemicals Ltd. (India). All the other reagents and chemicals used were of analytical grade and utilized as received. Prior to any experiment, all glassware were carefully cleaned to remove any trace of metallic contaminants with aqua regia (3HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1HNO3). All the aqueous solutions used in the experiments were prepared with ultrapure water (resistivity = 18.2 MΩ cm).
:1HNO3). All the aqueous solutions used in the experiments were prepared with ultrapure water (resistivity = 18.2 MΩ cm).
Characterization of gold nanoparticles
The absorption spectra were recorded using Jasco V-630 UV-Vis spectrophotometer (Japan) and Bio-Rad xMark™ microplate absorbance spectrophotometer (India). The transmission electron microscopy (TEM) images were recorded on FEI Tecnai G2 F20 X-Twin transmission electron microscope (USA) and zeta-potential analysis was performed using Nanobrook 90Plus Zeta Brookhaven Instruments (USA). X-ray diffraction (XRD) pattern was recorded on Bruker D8 Advance diffractometer (India).Synthesis of gold nanoparticles
Preparation of TMB2+ stock solution
To prepare the TMB2+ stock solution, 500 μL of HRP (100 mU) was mixed with TMB substrate solution which was prepared by mixing 5 mL of TMB (0.83 mM) with 1.23 μL of H2O2 (9.7 mM). An immediate change in the solution color from colorless to blue was observed which indicates the oxidation of TMB into TMB+ by HRP. The reaction was continued for about 10 min to ensure the complete conversion of reactant to the product (i.e. TMB to TMB+). Thereafter, to stop the HRP-mediated oxidation reaction and to initiate the acid-triggered oxidative conversion of TMB+ to TMB2+, 1.25 mL of H2SO4 solution (2 M) was added. An instantaneous change in solution color from blue to yellow was observed which indicates the successful conversion of TMB+ (blue) to TMB2+ (yellow). The concentration of TMB2+ was determined using UV-Vis spectroscopy and Beer–Lambert law by measuring the absorbance at 450 nm (Table S1†).11Interaction of AuNPs with TMB2+
To investigate the interaction behavior of different types of AuNPs with TMB2+, the working solutions of all three types of nanoparticles were prepared separately. For this, 2 mL of the as-prepared AuNPs were centrifuged (8000 rpm × 10 min) followed by discarding of the supernatant and re-dispersion of the settled nanoparticles in 1 mL of CTAB solution (0.06 M). For interaction study, 100 μL of working solution of all three types of AuNPs were incubated separately with 150 μL of varying concentrations of TMB2+ solution (7 μM to 131 μM) in separate vials. A control sample was prepared by mixing 100 μL of working solution of AuNPs with 150 μL of DI water. All the samples were incubated at room temperature for 10 min on a rotary mixer, observed for any visible color change, and then characterized using UV-Vis spectrophotometer.Validation of developed strategy for visual quantification of biomolecules
To realize the potential of the developed strategy towards a biosensor development, an indirect competitive p-ELISA assay was adopted using HIgG as a target antigen and HRP conjugated goat anti-HIgG antibody (GaHIgG@HRP) as the detector molecule (Scheme 2). First, the wells of the microtiter plate were coated with 100 μL of competitor HIgG solution (conc. = 10 μg mL−1, prepared in 10 mM PBS) overnight at 4 °C. Afterward, the wells were washed thrice with PBST washing buffer (PBS + 0.01% tween 20) to remove any unbound or loosely bound HIgG. This was followed by blocking of the free reactive sites on the polystyrene surface of the wells by treating with 200 μL of BSA solution (conc. = 5 mg mL−1, prepared in PBS) for 1 h as this helps in the preventing of any kind of non-specific interaction. Thereafter, the wells were incubated with 100 μL of premix of sample HIgG and GaHIgG@HRP for 2 h at 37 °C. This premix was prepared by mixing the 100 μL of GaHIgG@HRP detector antibody with 100 μL of sample HIgG of different concentrations ranging from 1 to 1011 fg mL−1 for 2 h, individually in separate vials. The washing process was repeated after each interaction step until the enzymatic reaction with TMB. After the interaction, the wells were incubated with 100 μL of TMB solution for 10 min. Thereafter, 50 μL of H2SO4 (2 M) was injected into the reaction mixture to stop the enzymatic reaction for successful conversion of TMB+ to TMB2+ which turned the solution color yellow. Afterward, 100 μL of working solution of AuNRs was added to each well which resulted in a visual color change. The change in the optical density [at 450 nm (λmax for TMB2+) and 688 nm (λmax for AuNRs)] and wavelength shift of the reaction mixture was measured using an ELISA plate reader. To assess the specificity of the developed assay, systematic experiments were conducted with negative and positive controls. For negative control, wells were coated with BSA (instead of competitor HIgG) followed by 2 h interaction with 100 μL of GaHIgG@HRP (1:15000 in PBS) detector antibody. For positive control, competitor HIgG pre-coated wells were interacted with 100 μL GaHIgG@HRP (1:15000 in PBS) antibody for 2 h in two different wells and followed by AuNRs addition only in one well, while the other well was kept undistubred. The rest of the experimental procedure was the same as followed for the sample analyte.
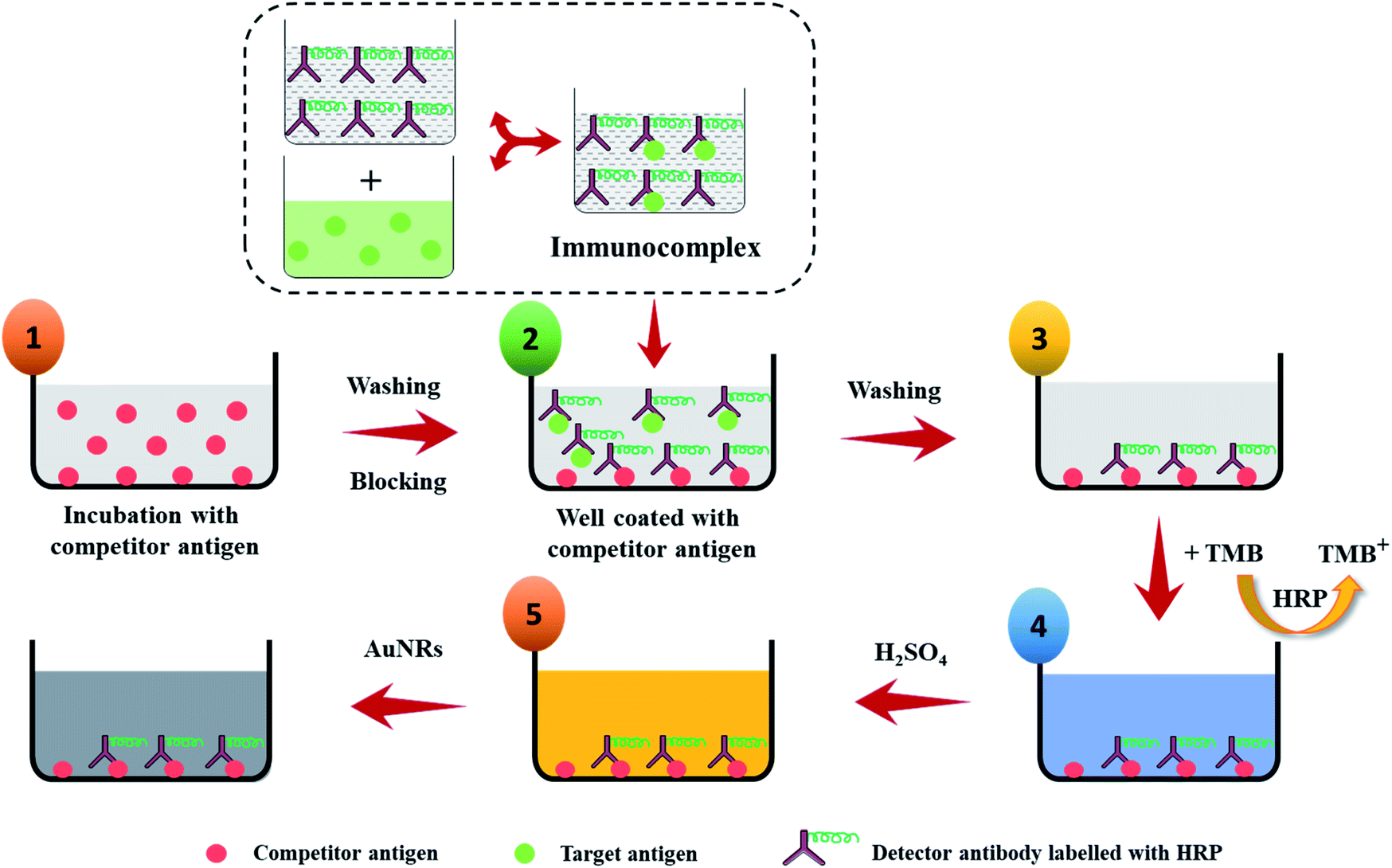 | ||
| Scheme 2 Schematic illustration of the p-ELISA strategy for the detection of HIgG. Step 1: microtiter well plate is immobilized with competitor antigen for overnight and then washed and followed by blocking of active sites using BSA to prevent any non-specific interactions; step 2: the target antigen is incubated with HRP enzyme labelled detector antibody for 2 h at room temperature and then the antibody–antigen complexes are added to the wells of the microtiter plates that have been pre-coated with competitor antigen; step 3: wells are washed to remove unbound antigen–antibody complexes and free target antigen; step 4: enzyme substrate is added to the wells and incubated for 10 min at room temperature; step 5: to stop the enzyme-mediated substrate hydrolysis, H2SO4 is added and followed by addition of the AuNRs to each well of the plate and then observed for any color change. | ||
Results and discussion
Characterization of AuNPs
All types of CTAB-capped AuNPs were characterized using UV-Vis spectroscopy, TEM imaging, X-ray powder diffraction (XRD) and zeta potential (Fig. 1). The characteristic extinction spectrum of AuNSs exhibits the extinction peak at 527 nm (Fig. 1a). On the other hand, the extinction spectrum of AuNRs shows two major plasmon peaks at 513 nm and 684 nm, which are associated with the electron oscillation along the transverse and longitudinal direction, respectively (Fig. 1e). The UV-Vis spectrum of AuNTs displays quadrupole and dipole LSPR modes at 872 nm and 1305 nm, respectively (Fig. 1i). For further experiments, we focused on the longitudinal and dipole LSPR modes of AuNRs and AuNTs, respectively. TEM micrographs revealed the formation of uniformly distributed AuNSs (average size = 21.66 ± 1.20 nm), AuNRs (aspect ratio = 2.89 ± 0.30, average length = 63.27 ± 4.23 nm, average width = 21.96 ± 1.69 nm) and AuNTs (edge length = 157.98 ± 14.65 nm) (Fig. 1b, f and j). The XRD and selected area electron diffraction (SAED) analysis of the AuNPs revealed the crystalline nature of all three AuNPs. The XRD pattern displays four prominent peaks at 38°, 44°, 64°, and 77°, corresponding to (111), (200), (220), and (311) Bragg reflection planes of the face-centered cubic lattice of Au (Fig. 1d, h and l). This indicates that all three shapes of AuNPs consist of pure crystalline Au (JCPDS card no. 04-0784) and the high intensity at (111) reflection plane compared to other planes suggests the predominance of (111) orientation. The clear rings in the SAED patterns, due to sharp reflection from the lattice planes, further confirm the crystalline nature of the AuNPs (Fig. 1b, f and j inset). The zeta potential values of AuNPs were found to be +65.17 ± 2.74 mV, +20.24 ± 8.35 mV, and +9.31 ± 0.92 mV for AuNSs, AuNRs, and AuNTs, respectively, which strongly evidence the presence of positively charged CTAB as capping agent on all types of nanoparticles. | ||
| Fig. 1 UV-Vis absorption spectra, transmission electron micrographs, particle size distribution histogram and X-ray diffraction pattern of AuNSs (a–d), AuNRs (e–h), and AuNTs (i–l). The insets of (a, e and i) and (b, f and j) are the photographic images of the corresponding colloidal solution and selected area electron diffraction (SAED) pattern of AuNPs. | ||
Optimization of AuNP dose and interaction time towards the etching of nanoparticles
To obtain the maximum response and prominent color change, the AuNPs dose and interaction time were optimized using AuNRs as representative plasmonic nanoparticles. For the dose optimization, a series of AuNRs solutions of varying concentrations in the range of 0.13 to 0.55 nM were prepared in the individual wells (in both lines A and B) by diluting 25, 50, 75, and 100 μL of AuNRs with DI water to the final volume of 100 μL (Fig. 2a). This was followed by the addition of 150 μL of TMB2+ (50 μM) solution in the wells of line A, while for line B (i.e. control experiment), 150 μL of DI water was added. A gradual increase in the solution color intensity was observed with an increase in the dose of AuNRs (well 1–4 in Fig. 2a). A prominent color change was observed between the TMB2+ treated AuNRs (line A) and control AuNRs (line B, without TMB2+ treatment) at all the dose levels of AuNRs, however, the color intensity was quite evident at high AuNR dose (i.e. AuNR conc. = 0.55 nM). This is due to the fact that at a lower dose of AuNRs (i.e. AuNR conc. = 0.13 and 0.28 nM), the TMB2+ present in the reaction mixture can completely etch the AuNRs and the solution appears yellow due to the presence of residual TMB2+ in the reaction mixture. In contrast, as the concentration of AuNRs increased to 0.40 nM and 0.55 nM, the color of the reaction mixture changed from brown to blue indicating that the TMB2+ present in the reaction mixture is not sufficient enough to completely etch the AuNRs and it can only etch them to produce NRs with lower aspect ratios (Fig. 3b). Further, the color intensity of the blue color was found to be relatively more for 0.55 nM of AuNR dose compared to 0.40 nM dose. This is purely associated with an increase in the AuNRs concentration or the number of AuNRs available for TMB2+ mediated etching reaction which results in more intense color (Fig. 3a). This is in good agreement with the spectral shift observed after the interaction of 0.55 nM of AuNRs with TMB2+ compared to the untreated AuNRs sample (Fig. 2b). | ||
| Fig. 2 Optimization of the AuNRs dose; (a) photograph image of the AuNRs sample of varying concentration (0.13, 0.28, 0.40 and 0.55 nM) interacted with 150 μL of 50 μM of TMB2+ (A1–A4), and 150 μL of water (B1–B4), respectively (total volume = 250 μL with water); (b) UV-Vis spectra of the AuNRs (0.55 nM) after interaction with 150 μL of each water (black line) and TMB2+ (red line) and (c) optimization of the etching time; AuNRs (0.55 nM) were interacted with 150 μL of water (black dotted line), 28 μM TMB2+ (red dotted line), and 110 μM TMB2+ (blue dotted line). | ||
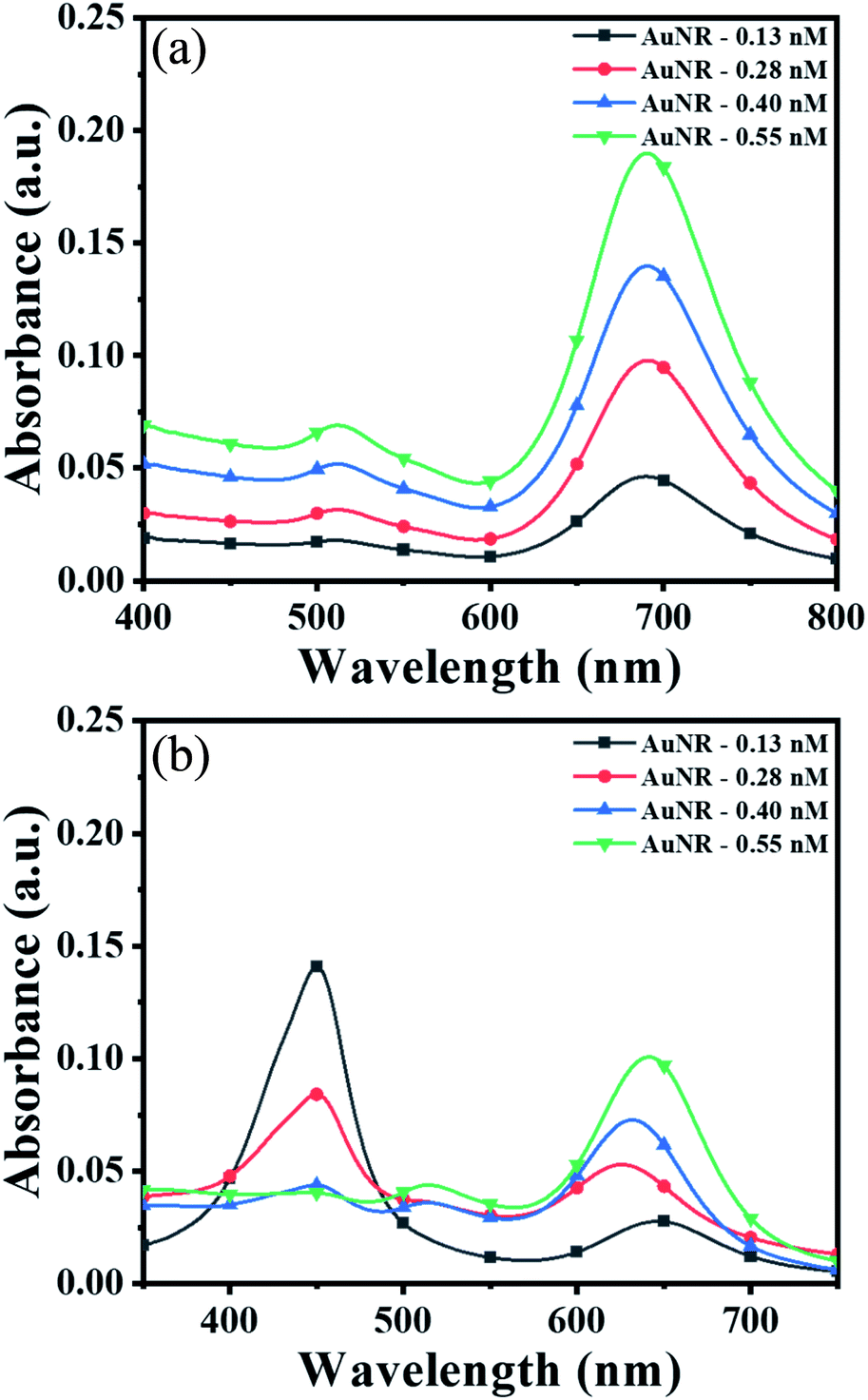 | ||
| Fig. 3 UV-Vis spectra of (a) AuNRs of 0.13, 0.28, 0.40 and 0.55 nM after interaction with 150 μL of water and (b) 150 μL of 50 μM conc. of TMB2+ (for all the samples, total volume was made up to 250 μL with water). | ||
To optimize the reaction time, 150 μL of two different concentrations (28 μM and 110 μM) of TMB2+ mixed with 100 μL of AuNRs solution. For the control study, 150 μL of water was mixed with 100 μL of AuNRs solution. Fig. 2c shows a time-dependent blue-shift in the longitudinal LSPR peak of AuNRs, which reaches a saturation level at 10 min for both the concentration levels indicating that the etching reaction kinetics has reached to a balanced state. However, in the absence of TMB2+ (control experiment), no change in the plasmonic peak was observed suggesting the essential requirement of TMB2+ for the etching of the AuNRs. Hence, 100 μL of AuNRs dose and 10 min interaction time was selected as optimum parameters for TMB2+ mediated etching.
Interaction analysis of different AuNPs with TMB2+
Prior to the interaction study, it is important to establish that enzyme and residual H2O2 (present in the TMB2+ stock solution) have neither direct interaction with AuNPs nor interfere with the etching reaction. To assess this, 100 μL of colloidal AuNRs solution interacted with 150 μL of each of H2O2 (1.2 mM), 10 mU of HRP enzyme, and TMB2+ (110 μM) for 10 min at room temperature. Both H2O2 and HRP interacted samples did not show any shift in the LSPR peak indicating that the chosen concentrations of both H2O2 and HRP neither affect the stability of AuNRs nor cause their etching (Fig. 4). In contrast, TMB2+ treated sample showed a prominent blue shift in the longitudinal plasmon band of AuNRs suggesting their geometrical transformation. Hence, the obtained results established that the HRP and residual H2O2, present in the TMB2+ solution, have no direct effect on AuNRs and also their presence does not influence the TMB2+ mediated etching process.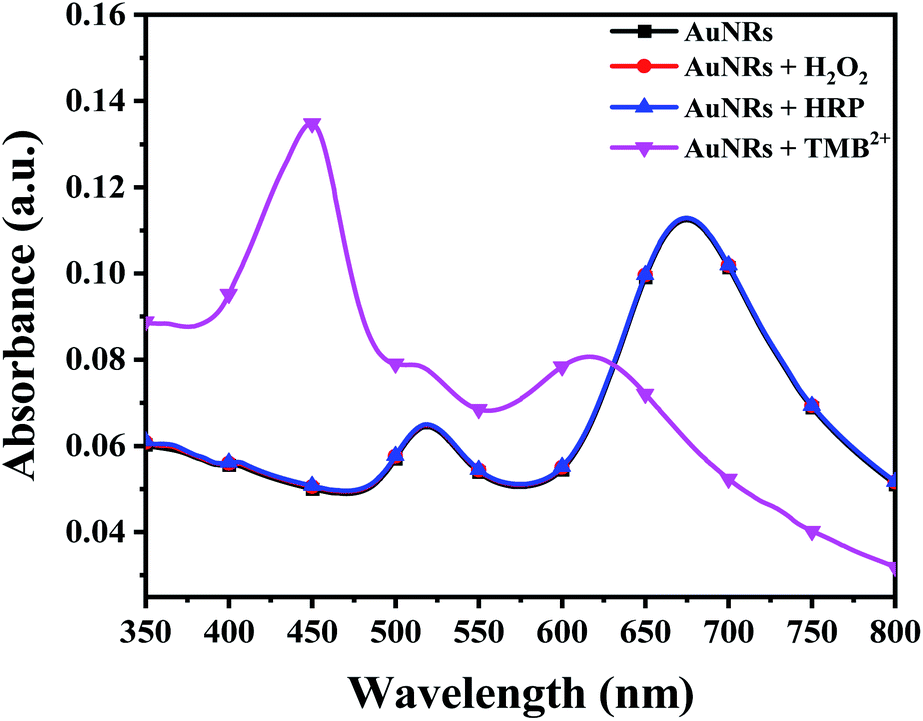 | ||
| Fig. 4 UV-Vis spectra AuNRs interacted with DI water (black curve), H2O2 (red curve), HRP (blue curve) and TMB2+ (pink curve) to study the effect of different reagents on TMB2+ mediated oxidation of AuNRs. (Note: black, blue and red curves are difficult to discern due to overlapping). | ||
To investigate the effect of nanoparticle shape on their etching pattern, TMB2+ solution of varying concentrations (0–131 μM) interacted with all three types of AuNPs individually. The UV-Vis spectra of the interacted samples, the percentage shift in their respective LSPR peak and absorbance ratio (Aplamonic peak/A450 nm) as a function of TMB2+ concentrations are displayed in Fig. 5. The LSPR peak of the interacted AuNPs appeared significantly blue-shifted with a prominent decrease in the absorbance intensity at their respective plasmonic peak as a function of TMB2+ concentration (Fig. 5a–c and S1†). However, the pattern and magnitude of LSPR peak shifts were found to be different for all three shapes of AuNPs. For instance, in the case of AuNSs, at a lower concentration (≤21 μM) of TMB2+, the LSPR peak did not show any considerable shift in the wavelength, however, a prominent change in the absorbance intensity (λmax = 527 nm) was observed due to their homogeneous etching without any shape-transformation (Fig. 5d and g).43 This can be easily correlated with the visual change in the color of the AuNSs solution from dark pink to light pink after interaction with ≤21 μM concentrations of TMB2+. In contrast, at a higher concentration of the TMB2+ (>21 μM), the plasmonic peak gradually disappeared with a significant drop in the absorbance intensity at peak plasmonic wavelength, and a new peak at 450 nm was observed corresponding to TMB2+ (Fig. 5d and g). This was evident also from the change in color of the reaction solution from pink to yellow. This is due to the fact that at high concentrations of TMB2+, the AuNSs (Au0) are completely etched into Au+ resulting in their depletion from the reaction mixture and thereby disappearance of their plasmonic peak.44 Simultaneously, the unreacted or remaining yellow color TMB2+ level increases, which exhibits its characteristic peak at 450 nm.
 | ||
| Fig. 5 AuNPs and TMB2+ interaction analysis. The photographic image, UV-Vis spectra, percentage shift in the LSPR peak wavelength and absorbance ratio (absorbance at LSPR peak to absorbance at 450 nm) of AuNSs (a, d and g), AuNRs (b, e and h), and AuNTs (c, f and i) after interaction with different concentrations of TMB2+ (0–131 μM). (Note: The LSPR plasmonic peak of AuNSs completely disappeared at higher concentrations of the TMB2+ (>21 μM) and a new peak at 450 nm was observed corresponding to TMB2+, therefore %Δλ calculated at those concentrations is zero in fig (d)). | ||
In contrast to AuNSs, both AuNRs and AuNTs showed a TMB2+ concentration-dependent blue-shift along with a decrease in the absorbance intensity at their respective plasmonic peaks even at lower concentrations of TMB2+ (i.e. ≤21) (Fig. 5b and c). Similar to AuNSs, in both cases, the appearance of a new peak at 450 nm with a gradual increase in its intensity was observed with an increase in the TMB2+ concentration to 43 μM onwards, indicating the presence of unreacted TMB2+ in the reaction mixture. As presented in Fig. 5(e and f), %Δλ was found to increase from 0.73–7.87% and 19.54–42.14% for AuNRs and AuNTs, respectively as the concentration of TMB2+ increased from 7–131 μM. This indicates that the LSPR peak shift value was observed to be more in the case of AuNTs than AuNRs. This can be easily correlated with a multicolor and single-color change observed for AuNRs and AuNTs, respectively (Fig. 5). This might be due to the presence of sharp curved tips/features of the AuNTs at the corners (∼60°) which are possibly hot regions towards the etching owing to lesser capping coverage.45 Additionally, as a result of the finite thickness of the AuNTs, a little change in the edge length of AuNTs by means of TMB2+ mediated etching induces a large change in their aspect ratios and thus a sharper LSPR shift.46 Conversely, in the case of AuNRs, the etching reaction is not isotropic in all directions and these are preferentially etched along the axial direction. This can be ascribed to lesser CTAB capping density at the tips as compared to the sides of the AuNRs which favors the greater interaction with TMB2+ at tips, resulting in AuNRs with different aspect ratios.17
In addition to the sharp surface feature of AuNPs, differences in the crystal facets of the nanoparticles may also have a significant role towards different surface binding and reactivities, contributing to variations in etching.47 For instance, the surface energies of metal crystal planes are in order of (110) > (100) > (111) and depending on this, they possess different reactivities and stability. In the case of AuNTs, the bottom and side edge surfaces are composed of (111) and (110) crystal facets, respectively. The atoms at the corner areas and (110) facet possess higher energy than those at the (111) facet due to their lower coordination numbers. Therefore, the corners and side edges of the NTs are more prone to be etched compared to other areas. As the etching reaction continues, morphological changes occur in NTs starting from sharp triangle prisms and progressing to “snipped” triangular, hexagonal, and eventually, disk-like nanoplates. On the other hand, in the case of AuNRs, the side surface is surrounded by (110) and the tip is enclosed by (111) and (001) crystal facets.47 As the (111) facet is considered to be the highly stable surface and least reactive, the (110) facet is extremely reactive and the most prone to reconstructions. However, owing to the presence of CTAB molecules, the (110) and (100) facets of AuNRs become more stable than (111) planes, therefore, the tip of AuNRs with (111) facets might be potentially etched during the reaction.48,49 This is reflected by the gradual change in the color of AuNRs and AuNTs solution during the etching reaction. To further support, TEM analysis of the AuNTs and AuNRs before and after interaction with TMB2+ was carried out which revealed a significant decrease in the aspect ratio of AuNRs (AR = from 2.89 ± 0.30 to 2.16 ± 0.23) and edge length (from 157.98 ± 14.65 nm to 57.84 ± 5.59 nm) of AuNTs (Fig. 6). These results strongly support the crystal facet-dependent preferential etching of the AuNRs and AuNTs.
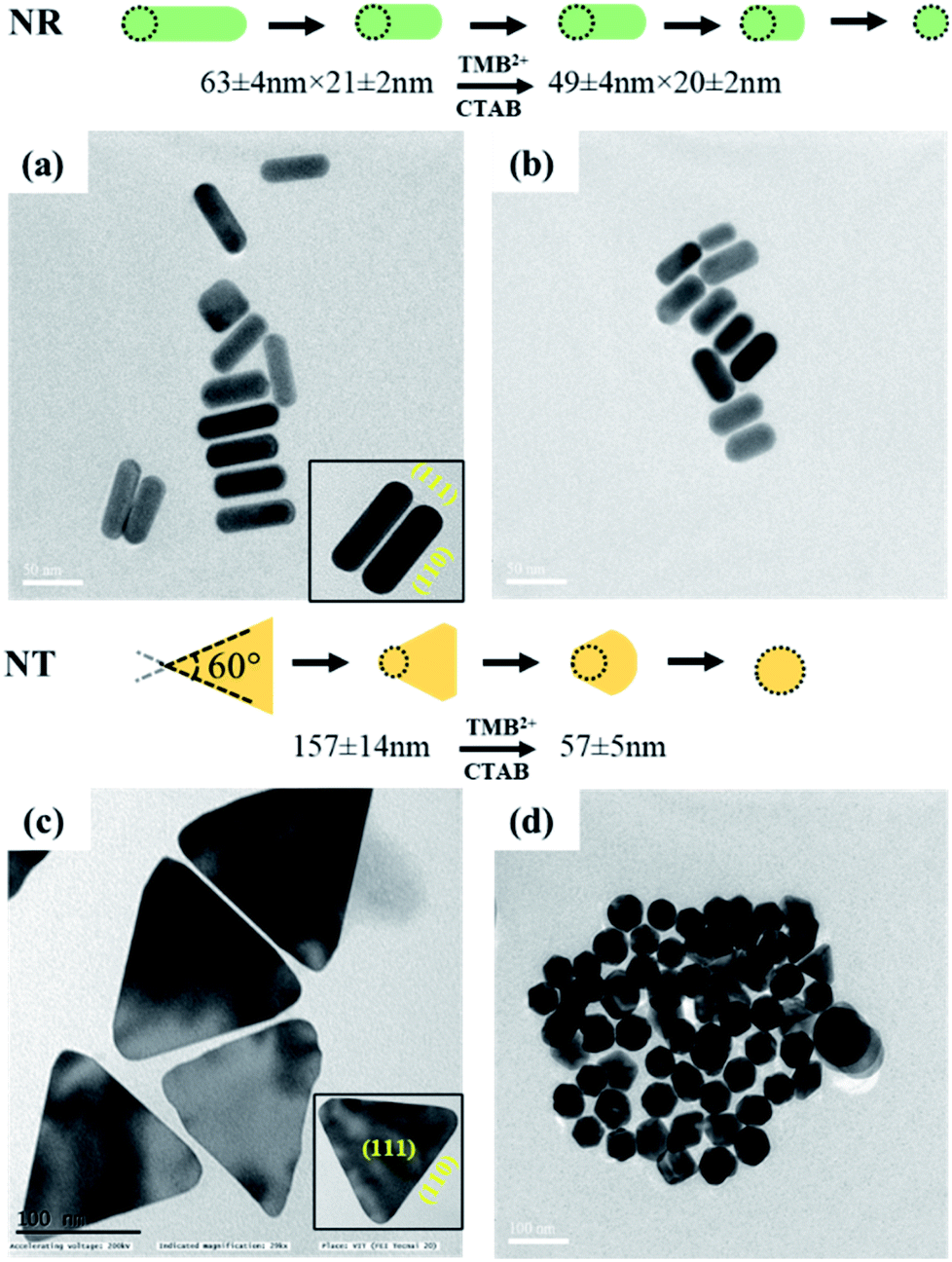 | ||
| Fig. 6 TEM images of AuNRs (top) and AuNTs (bottom) (left, a and c) before and (right, b and d) after interaction with TMB2+. | ||
Conclusively, all three shapes of AuNPs show geometrical transformation in their shape due to etching by TMB2+, however, the change in the plasmonic peak is more prominent for AuNRs and AuNTs. Further, the AuNRs displayed a gradual change in LSPR peak shift, absorbance intensity ratio and visually detectable multicolor change compared to the AuNSs and AuNTs, which showed only single-color transformation (from pink to yellow for AuNSs and green to yellow for AuNTs). Hence, these results indicate that the TMB2+ mediated etching of AuNPs is significantly influenced by their shape which would also reflect during visual detection-based biosensor development when such etching reaction is employed in the reaction.
Immunoassay and detection principle
To demonstrate the feasibility of the AuNRs-etching mediated biosensing, we applied this method to develop an indirect competitive p-ELISA for visual detection of HIgG. The samples containing known concentrations of HIgG (conc. = 1–1011 fg mL−1) interacted with GaHIgG@HRP (enzyme-linked specific detector anybody) in separate vials which form a stable HIgG-GaHIgG@HRP immunocomplex in proportion to the HIgG concentration present in the sample. Thereafter, this pre-mix was introduced in the microtiter plate wells pre-coated with competitor HIgG so that the free GaHIgG@HRP can interact with the competitor HIgG, i.e. pre-coated on the microplate. Subsequently, HRP catalyzed the conversion of TMB into TMB+ which is followed by the addition of concentrated H2SO4 to obtain TMB2+. The generated TMB2+ mediates the oxidative etching of AuNRs, resulting in distinct color changes which are accompanied by a blue shift in their longitudinal plasmonic peak. The solution displays a multicolor change as the concentration of HIgG decreases in the sample (Fig. 7a). As the HIgG concentration in the sample increases, lesser GaHIgG@HRP will be available for binding to the pre-coated competitor HIgG. As a result, lesser or no enzymatic conversion of TMB to TMB+ takes place and thereby no etching of the AuNRs and therefore the solution color remained unchanged.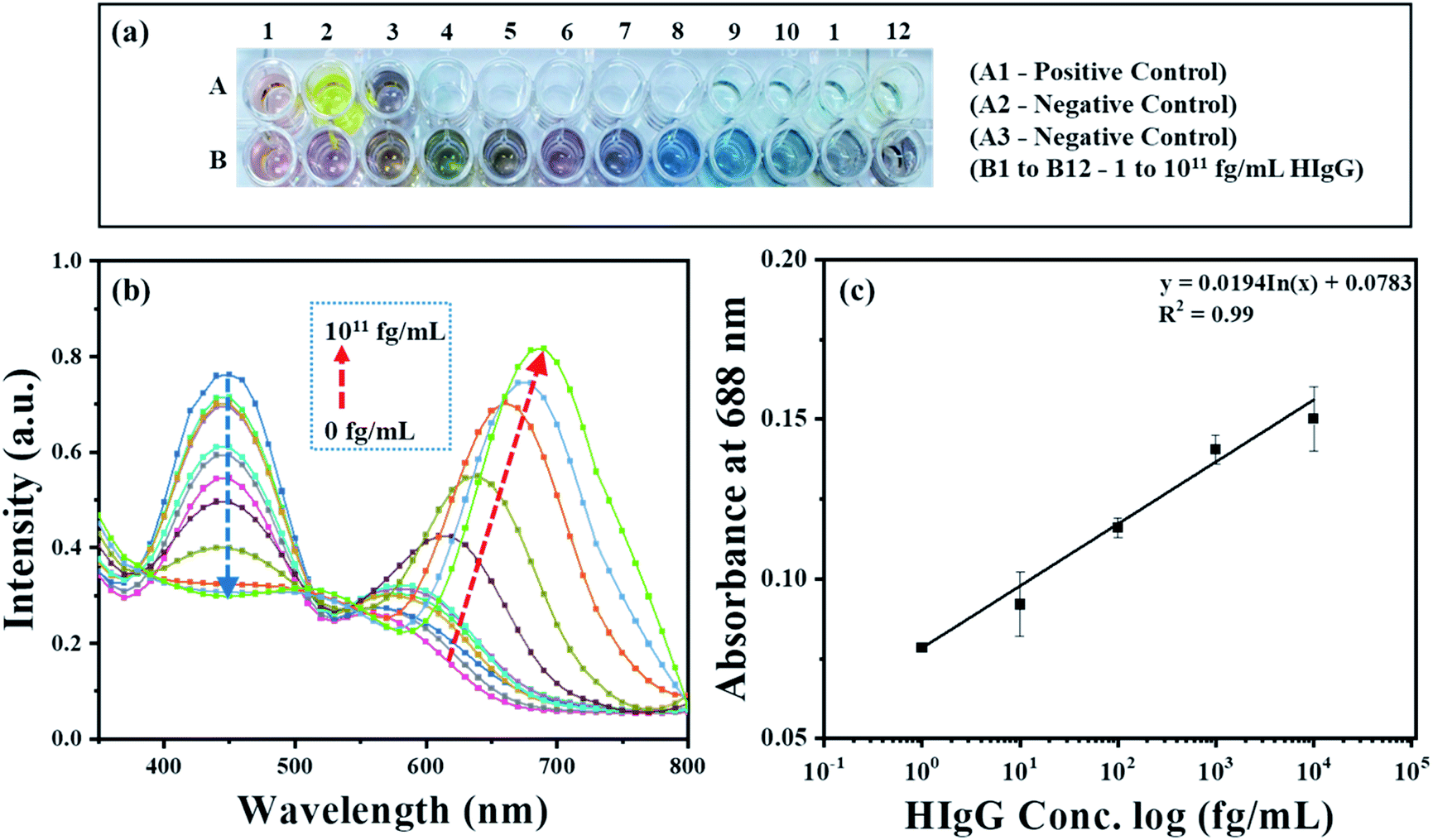 | ||
| Fig. 7 (a) Photos of microtiter plate wells containing AuNRs interacted with TMB2+, produced as a product of the enzymatic reaction in developed competitive p-ELISA and (b) UV-Vis absorption spectra of AuNRs after interaction with different concentrations of HIgG (0 to 1011 fg mL−1). (c) The linear relationship between absorbance change (λmax = 688 nm) and log concentration of HIgG. | ||
The instrumental analysis of the interacted samples corroborated the visual detection findings. The absorbance spectra of the developed competitive p-ELISA showed an increase in the absorbance intensity at 688 nm and a corresponding decrease in the absorbance intensity at 450 nm as a function of HIgG concentration in the sample (Fig. 7b). This is due to the fact that with an increase in the concentration of HIgG, the number of detector antibodies available to interact with pre-immobilized competitor antigen reduces concurrently and this consequently minimizes the conversion of TMB into TMB2+via HRP and thereby the etching of the AuNRs.
To evaluate the selectivity of the developed sensing strategy, both positive and negative control studies were carried and the results are shown in Fig. 7a and S2.† For positive control studies (i.e. in the absence of HIgG in the sample), two different experiments were performed. In the well A1, AuNRs were added to the biocatalytically produced TMB2+ reaction mixture, while in the well A2, AuNRs were not added and thus exhibited the characteristic yellow color of the TMB2+. The absorbance at 450 nm was observed to be the highest for both the controls, which is due to the fact that in the absence of HIgG in the sample, the maximum number of standard HIgG-GaHIgG@HRP binding events took place. This resulted in a greater number of HRP molecules available for the conversion of TMB to TMB2+ (Fig S2,† black curve). In addition, we observed a prominent change in the color of AuNRs solution from brown to light pink (Fig. 7a, well A1), indicating the maximum etching of the nanorods as the GaHIgG@HRP was freely available to form the immunocomplex with the standard immobilized HIgG on the well. For the negative control experiment, the standard HIgG (i.e. pre-coated on the wells) was replaced with BSA and then interacted with the GaHIgG@HRP. In this case, the AuNRs solution did not show any spectral shift and color change (Fig. 7a, well A3 and Fig S2,† red curve). This suggests the no binding of the GaHIgG@HRP towards the BSA and thereby no catalytic conversion of TMB into TMB2+and thus no etching and/or color change. This indicates the highly specific nature of the developed plasmonic immunoassay.
For the calibration curve, the absorbance values (λmax = 688 nm) were plotted against the logarithm concentration of sample HIgG. As shown in Fig. 7c, the calibration curve exhibits good linearity in ranges from 1 to 104 fg mL−1 of HIgG (Table S2 in ESI†). In contrast, a gradual decrease in the absorbance intensity at 450 nm was observed as a logarithm concentration of HIgG (Fig. S3†). This evidences that visual color changes at fg mL−1 concentration level of the analyte are in accordance with the instrumental results and proves the potential of AuNRs-etching based naked eye detection-based ultra-sensitive biosensor. The etching of AuNRs via TMB2+ produced multicolor changes in proportion to the concentration of sample HIgG (Fig. 7a). The naked eye detection limit of the developed immunosensing strategy was found to be as low as 1 fg mL−1 (∼6.54 aM), which is comparable or better than other instrumentation based biosensing platforms developed by employing different AuNPs (Table 1).50–54 In addition, compared to the other reported nanoparticle-based ELISA sensing strategies, the developed method provides several other advantages including (i) easy integration with conventional ELISA schemes, wherein HRP-labeled detector antibodies are used,55,56 (ii) elimination of false positive and negative signal due to autoaggregation of the nanoparticles,57,58 (iii) the greater stability of the AuNRs towards temperature, light and the biological thiols as compared to silver nanoprisms,59 and (iv) easy and simple as it does not require any conjugation or surface functionalization of the AuNRs.60,61
| Technique/Instrument | AuNPs Type | Target analyte | LoD (aM) | Ref. |
|---|---|---|---|---|
| SPR | AuCNPs | B-type natriuretic peptide | 10 | 50 |
| Colorimetric/microplate reader | AuNSs | Prostate-specific antigen (PSA) | 93 | 51 |
| Plasmonic fiber optic sensor | AuNSs | PSA | 3 × 103 | 52 |
| SERS/Raman microscope | AuNSs | C-reactive protein | 10 | 53 |
| Plasmonic fiber optic sensor | AuNSs | HIgG | 7 | 54 |
| p-ELISA/naked eye | AuNRs | HIgG | 6.54 | This study |
Conclusion
In the present work, we have reported the oxidative etching of different shapes of AuNPs via TMB2+, a product of the enzymatic reaction that is most commonly utilized for ELISA-based detection platforms. All three shapes of the AuNPs showed a spectral shift and change in absorbance at their respective LSPR peak after interaction with different concentrations of TMB2+. Amongst all three shapes of AuNPs, TMB2+-mediated etching of AuNRs led to a vivid color response in relation to the concentration of the etchant. This was due to the selective etching at the tips of the nanorods that led to the formation of AuNRs of different aspect ratios as a function of TMB2+ concentration. This optimized strategy was further realized into a multicolorimetric visual detection system for the sensing of HIgG. The competitive p-ELISA could detect the HIgG with a visual detection limit of 1 fg mL−1 through the naked eye without the aid of any sophisticated instrument. Although AuNR-etching based p-ELISA has the potential for visual detection at a very low level of the analyte, it suffers from manual intervention in order to perform the complete bioassay. It may also pose the risk of manual error and thus may lead to a false signal. Hence, to minimize these risks and reduce manual intervention, efforts must be made to integrate the p-ELISA technique with microfluidic or chip-based technology and realize it into a point-of-care diagnostic system for various disease detection.Author contributions
Sangeeta Yadav: investigation, methodology, writing – original draft. Jitendra Satija: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Vellore Institute of Technology (VIT) for providing Seed Grant (SG20210170) for carrying out this research work.References
- R. de la Rica and M. M. Stevens, Nat. Nanotechnol., 2012, 7, 821–824 CrossRef CAS PubMed.
- R. Chen, X. Huang, H. Xu, Y. Xiong and Y. Li, ACS Appl. Mater. Interfaces, 2015, 7, 28632–28639 CrossRef CAS PubMed.
- C. Yao, S. Yu, X. Li, Z. Wu, J. Liang, Q. Fu, W. Xiao, T. Jiang and Y. Tang, Anal. Bioanal. Chem., 2017, 409, 1093–1100 CrossRef CAS PubMed.
- Q. Yang, R. Cai, W. Xiao, Z. Wu, X. Liu, Y. Xu, M. Xu, H. Zhong, G. Sun, Q. Liu, Q. Fu and J. Xiang, Nanoscale Res. Lett., 2018, 13, 397 CrossRef PubMed.
- L. Wu, G. Li, X. Xu, L. Zhu, R. Huang and X. Chen, Trends Anal. Chem., 2019, 113, 140–156 CrossRef CAS.
- K. Kołątaj, J. Krajczewski and A. Kudelski, Environ. Chem. Lett., 2020, 18, 529–542 CrossRef.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed.
- E. Petryayeva and U. J. Krull, Anal. Chim. Acta, 2011, 706, 8–24 CrossRef CAS PubMed.
- Z. Zhang, Z. Chen, S. Wang, F. Cheng and L. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 27639–27645 CrossRef CAS PubMed.
- J. Li, Y. Y. Wei, X. P. Liu and Z. R. Xu, Sens. Actuators, B, 2022, 353, 131139 CrossRef CAS.
- P. D. Josephy, T. Eling and R. P. Mason, J. Biol. Chem., 1982, 257, 3669–3675 CrossRef CAS.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS PubMed.
- J. Liao, Y. Zhang, W. Yu, L. Xu, C. Ge, J. Liu and N. Gu, Colloids Surf., A, 2003, 223, 177–183 CrossRef CAS.
- K. Sato, K. Hosokawa and M. Maeda, J. Am. Chem. Soc., 2003, 125, 8102–8103 CrossRef CAS PubMed.
- J. Tharion, J. Satija and S. Mukherji, Plasmonics, 2014, 10, 753–763 CrossRef.
- J. Satija, J. Tharion and S. Mukherji, RSC Adv., 2015, 5, 69970–69979 RSC.
- R. Zou, X. Guo, J. Yang, D. Li, F. Peng, L. Zhang, H. Wang and H. Yu, CrystEngComm, 2009, 11, 2797–2803 RSC.
- S. A. Alex, J. Satija, M. A. Khan, G. M. Bhalerao, S. Chakravarty, B. Kasilingam, A. Sivakumar, N. Chandrasekaran and A. Mukherjee, Anal. Methods, 2015, 7, 5583–5592 RSC.
- X. M. Nie, R. Huang, C. X. Dong, L. J. Tang, R. Gui and J. H. Jiang, Bioelectron, 2014, 58, 314–319 CrossRef CAS PubMed.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC.
- S. S. Shankar, A. Rai, A. Ahmad and M. Sastry, J. Colloid Interface Sci., 2004, 275, 496–502 CrossRef CAS PubMed.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- X. Yang and Z. Gao, Chem. Commun., 2015, 51, 6928–6931 RSC.
- M. Yuan, Q. Xiong, G. Zhang, Z. Xiong, D. Liu, H. Duan and W. Lai, J. Mater. Chem. B, 2020, 8, 3667–3675 RSC.
- M. R. Ivanov, H. R. Bednar and A. J. Haes, ACS Nano, 2009, 3, 386–394 CrossRef CAS PubMed.
- R. Pamies, J. G. H. Cifre, V. F. Espín, M. Collado-González, F. G. D. Baños and J. G. de la Torre, J. Nanopart. Res., 2014, 16, 2376 CrossRef.
- N. Bizmark and M. A. Ioannidis, Langmuir, 2015, 31, 9282–9289 CrossRef CAS PubMed.
- J. Satija, N. Punjabi, D. Mishra and S. Mukherji, RSC Adv., 2016, 6, 85440–85456 RSC.
- E. Hao, G. C. Schatz and J. T. Hupp, J. Fluoresc., 2004, 14, 331–341 CrossRef CAS PubMed.
- C. Yu and J. Irudayaraj, Anal. Chem., 2007, 79, 572–579 CrossRef CAS PubMed.
- N. Jana and T. Pal, Adv. Mater., 2007, 19, 1761–1765 CrossRef CAS.
- K. M. Mayer, S. Lee, H. Liao, B. C. Rostro, A. Fuentes, P. T. Scully, C. L. Nehl and J. H. Hafner, ACS Nano, 2008, 2, 687–692 CrossRef CAS PubMed.
- B. Tang, J. Wang, S. Xu, T. Afrin, W. Xu, L. Sun and X. Wang, J. Colloid Interface Sci., 2011, 356, 513–518 CrossRef CAS PubMed.
- Z. He, J. Zhu, G. J. Weng, J. J. Li and J. W. Zhao, Nanotechnology, 2020, 31, 335505 CrossRef CAS PubMed.
- Z. Zhang, Z. Chen, C. Qu and L. Chen, Langmuir, 2014, 30, 3625–3630 CrossRef CAS PubMed.
- Z. Zhang, Z. Chen, F. Cheng, Y. Zhang and L. Chen, Analyst, 2016, 141, 2955 RSC.
- Z. Zhang, H. Wang, Z. Chen, X. Wang, J. Choo and L. Chen, Biosens. Bioelectron., 2018, 114, 52–65 CrossRef CAS PubMed.
- T. Porstmann and S. T. Kiessig, J. Immunol. Methods, 1992, 150, 5–21 CrossRef CAS PubMed.
- L. Guo, S. Xu, X. Ma, B. Qiu, Z. Lin and G. Chen, Sci. Rep., 2016, 6, 32755 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- J. S. DuChene, et al. , Chem. Mater., 2013, 25, 1392–1399 CrossRef CAS.
- S. Sun, M. Gao, G. Lei, H. Zou, J. Ma and C. Huang, Nano Res., 2016, 9, 1125–1134 CrossRef CAS.
- X. Ma, Y. Lin, L. Guo, B. Qiu, G. Chen, H.-h. Yang and Z. Lin, Biosens. Bioelectron., 2017, 87, 122–128 CrossRef CAS PubMed.
- J. E. Millstone, W. Wei, M. R. Jones, H. Yoo and C. A. Mirkin, Nano Lett., 2008, 8, 2526–2529 CrossRef CAS PubMed.
- H. Yuan, K. P. F. Janssen, T. Franklin, G. Lu, L. Su, X. Gu, H. Uji-i, M. B. J. Roeffaers and J. Hofkens, RSC Adv., 2015, 5, 6829–6833 RSC.
- T. S. Sreeprasad, A. K. Samal and T. Pradeep, Langmuir, 2007, 23, 9463–9471 CrossRef CAS PubMed.
- Z. L. Wang, R. P. Gao, B. Nikoobakht and M. A. El-Sayed, J. Phys. Chem. B, 2000, 104, 5417–5420 CrossRef CAS.
- C. J. Johnson, E. Dujardin, S. A. Davis, C. J. Murphy and S. Mann, J. Mater. Chem., 2002, 12(6), 1765–1770 RSC.
- H. R. Jang, A. W. Wark, S. H. Baek, B. H. Chung and H. J. Lee, Anal. Chem., 2014, 86, 814–819 CrossRef CAS PubMed.
- D. Liu, J. Yang, H.-F. Wang, Z. Wang, X. Huang, Z. Wang, G. Niu, A. R. H. Walker and X. Chen, Anal. Chem., 2014, 86, 5800–5806 CrossRef CAS PubMed.
- M. Sanders, Y. Lin, J. Wei, T. Bono and R. Lindquist, Biosens. Bioelectron., 2014, 61, 95–101 CrossRef CAS PubMed.
- A. Hwang, E. Kim, J. Moon, H. Lee, M. Lee, J. Jeong, E.-K. Lim, J. Jung, T. Kang and B. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 18960–18967 CrossRef CAS PubMed.
- R. Bandaru, M. Divagar, S. Khanna, C. G. Danny, S. Gupta, V. Janakiraman and V. V. R. Sai, Sens. Actuators, B, 2020, 321, 128463 CrossRef CAS.
- N. Feng, J. Shen, Y. Chen, C. Li, Y. Hu, L. Zhang, S. Chen, Q. Fan, W. Huang and L. Wang, Nano Res., 2020, 13(12), 3364–3370 CrossRef CAS.
- Z. Chen, H. wang, Z. Zhang and L. Chen, Anal. Chem., 2019, 91, 1254–1259 CrossRef CAS PubMed.
- Y. Xianyu, Z. Wang and X. Jiang, ACS Nano, 2014, 8(12), 12741–12747 CrossRef CAS PubMed.
- Y. Xianyu, Y. Chen and X. Jiang, Anal. Chem., 2015, 87, 10688–10692 CrossRef CAS PubMed.
- B. Tang, S. Xu, X. Hou, J. Li, L. Sun, W. Xu and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 646–653 CrossRef CAS PubMed.
- Y. Wu, Y. Xiong, X. Chen, D. Luo, B. Goa, J. Chen, X. Huang, Y. Leng and Y. Xiong, J. Dairy Sci., 2019, 102, 10877–10886 CrossRef CAS PubMed.
- Y. Y. Ozawa, N. Lobsiger, Y. Muto, T. Mori, K. Yoshimura, Y. Yano, W. J. Stark, M. Maeda, T. Asahi, A. Ogawa and T. Zako, RSC Adv., 2021, 11, 11984–11991 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00266c |
| This journal is © The Royal Society of Chemistry 2022 |