
Heterogeneous photocatalytic reversible deactivation radical polymerization
Zixin
An
 a,
Shilong
Zhu
a and
Zesheng
An
*abc
a,
Shilong
Zhu
a and
Zesheng
An
*abc
aCollege of Chemistry, Jilin University, Changchun 130012, China. E-mail: anzesheng@jlu.edu.cn
bState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, China
cKey Laboratory for Molecular Enzymology and Engineering of Ministry of Education, School of Life Sciences, Jilin University, Changchun 130012, China
First published on 12th March 2021
Abstract
Photocatalytic reversible deactivation radical polymerization (RDRP) permits the use of sustainable solar light for spatiotemporal regulation of radical polymerization under mild conditions. Photocatalysts play a vital role in light absorption, electron/energy transfer, and maintenance of activation/deactivation equilibrium of a specific RDRP process. Recent years have witnessed increasing employment of heterogeneous photocatalysts in RDRP. Heterogeneous photocatalysts may offer additional benefits such as facile preparation, tunable photoelectronic properties, and potential for catalyst recyclization. This review highlights recent advances in photocatalytic RDRP using a diverse range of heterogeneous photocatalysts, including metal semiconductors, metal organic frameworks, carbon-based materials, nanocomposites, and self-assemblies. Remaining challenges and future direction of this field are also briefly discussed.
1. Introduction
Reversible deactivation radical polymerization (RDRP), represented by atom transfer radical polymerization (ATRP),1,2 reversible addition–fragmentation chain transfer polymerization (RAFT)3 and nitroxide-mediated radical polymerization (NMP),4 has undergone impressive development in the past decades.5–16 It has been widely used as a powerful tool for the synthesis of polymers with predictable molecular weight, narrow/adjustable molecular weight distribution, and diverse complex architecture. However, challenges still exist for the development of greener RDRP protocols that can offer effective on-demand control of a polymerization. In recent years, externally regulated RDRP has received significant attention because of the ability to adjust reactivity and kinetics, the ease to control polymerization spatiotemporally, and the promise to conduct polymerization under energy-efficient and mild conditions.17,18 In this regard, light,12,13,16,19 heat,20,21 ultrasound22,23 and voltage24–26 have been explored as individual, orthogonal or combined stimuli to regulate RDRP and demonstrated potential for on-demand synthesis of various polymers.The remarkable achievements in this field have suggested that visible light is a viable candidate among various external stimuli due to its abundance, operational ease, mildness, and non-invasive nature. Photocatalysts play a vital role in a range of photochemical processes, and multidisciplinary research efforts in organic, inorganic and materials chemistry have led to the identification and development of a number of effective photocatalysts, including conjugated organic molecules,27 enzymes,28,29 transition-metal-based coordination compounds,30–32 metal oxide semiconductors,33–35 and carbon-based materials.36,37 Encouraged by the efficiency and richness of the photocatalysts used in related fields, researchers have adopted some of these photocatalysts to the field of RDRP and developed photo-regulated ATRP,27,38,39 RAFT19,40 and NMP.41,42
With the rise of nanotechnology and growing demand for hybrid materials in applications such as drug delivery vehicles and “smart” materials, heterogeneous photocatalytic polymerization (HPP) has emerged as a successful solution.43–45 Since the first report of using ZnO as a heterogeneous photocatalyst for free radical polymerization,46 various semiconductor nanoparticles such as TiO2,47 Bi2O3,48 CdSe,49 perovskites,50 magnetic nanoparticles,51 and upconversion nanoparticles52 have been similarly explored. Application of these nanomaterials to radical photopolymerization is driven by several reasons. These materials are excellent photocatalysts with high quantum efficiency and adjustable band gap energy, allowing for efficient utilization of light of a wide range wavelength. Some of these materials are also very stable and have negligible light scattering. Although some nanomaterials such as CdSe containing toxic metals have been tested as photocatalysts for polymerization, many others can have low toxicity, especially for carbon-based metal-free materials.53–55 Besides, separation of photocatalysts from reactions through centrifugation56 or ultra-centrifugal spin filters57 has been demonstrated, suggesting potential for photocatalyst recycling and reuse. Now the field of heterogeneous photocatalytic RDRP (HP-RDRP) is experiencing a rapid development by incorporation of photocatalysts well beyond those initially tested for traditional radical polymerization; we feel it is necessary to provide a review on this important topic, and the content of which is organized according to the classes of heterogeneous photocatalysts used in RDRP (Fig. 1).
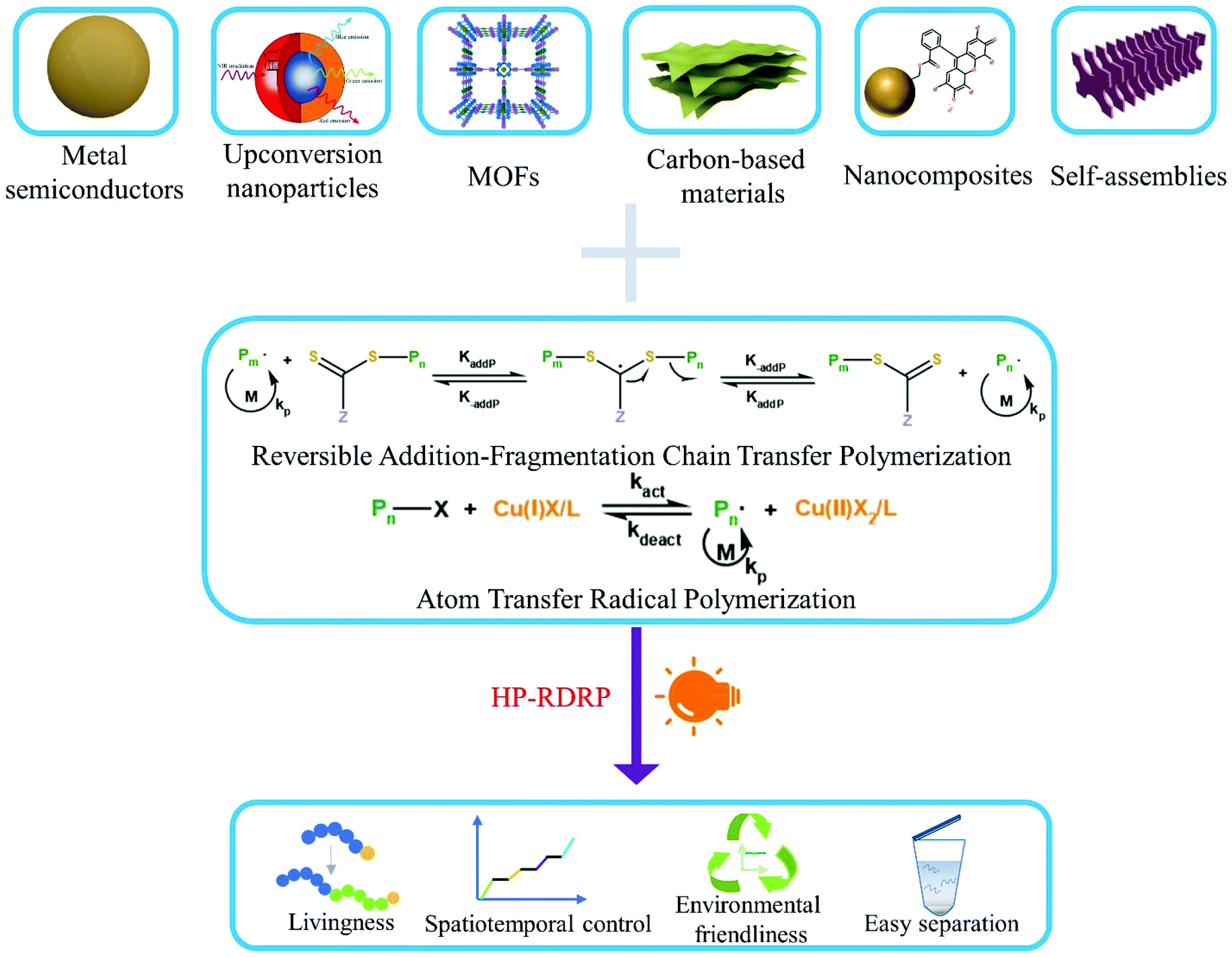 | ||
| Fig. 1 Current development of HP-RDRP. | ||
2. HP-RDRP mechanisms and catalyst selection
2.1. Mechanisms
Before introducing HP-RDRP techniques, it is necessary to briefly clarify the mechanisms of HP-RDRP, mainly photoregulated ATRP and RAFT.In the HP-ATRP process, heterogeneous photocatalysts are used to activate either the ATRP catalyst (e.g., a copper complex) or ATRP initiator. Photoactivation of the photocatalyst generates a hole in the valence band and an electron in the conduction band. Photocatalysts donate electrons from the conduction band to reactive species such as catalysts (e.g. Cu(II)) or alkyl halide initiators (RX, X = Br or Cl), thus initiating a radical polymerization, while holes are filled by sacrificial electron donors such as trialkyl amine, thus leading to NP regeneration and polymerization acceleration. In the case of a dye sensitized semiconductor, the photoexcited dye injects electrons to the conduction band, and then electrons are transferred to active species to initiate the ATRP process.
In HP-RAFT polymerization, catalysts donate electrons to the chain transfer agent (CTA), and the reduced CTA acts as both the initiator to start the traditional RAFT process and the degenerative agent to regenerate the catalyst and accomplish the catalytic cycle, so-called PET-RAFT process. For the localized surface plasmon resonance (LSPR)-induced mechanism, the photoexcitation and self-photoreduction of the catalyst generate noble metal nanoparticles which donate electrons to the CTA, and the nanoparticles are transformed to metal ions to facilitate the regeneration of the catalyst. Under the condition of using upconversion nanoparticles, catalysts can act as internal light sources converting NIR light to UV or visible light to activate alkyl halide initiators in the ATRP process or cleave the CTA in the photoiniferter process.
2.2. Catalyst selection
Many heterogeneous photocatalysts have been developed, and researchers have been always seeking to improve catalytic efficiency through tuning optical and electronic properties.In terms of photocatalyst optical properties, the absorption wavelength and extinction coefficient of photocatalysts are typically the main concerns. Some semiconductor materials have intrinsic absorption peaks in the UV region. UV light can decompose the polymerization control agents and provide more energy to excite the catalytic reaction. Visible and NIR light induced catalytic reactions provide less energy and have a better penetrating ability, which may be of potential interest in biomaterials. Thus, many strategies including element doping and photosensitiser modification have been used to extend the absorption to visible or even NIR range. Meanwhile, catalysts capable of visible-light absorption on their own have been developed including carbon dots, self-assemblies, MOFs, etc. Besides light harvesting ability, fluorescence properties, generated from charge–hole pair recombination, may also play an important role in catalytic reactions, especially for reactions employing the energy transfer process.
Electronic properties significantly influence the catalytic efficiency. As mentioned above, upon light irradiation, charge separation and charge transfer to initiators or catalysts occur, which are two key processes that can affect initiation and polymerization efficiency. The width of the band gap of the semiconductor is affected by the nanoparticle size, surface chemistry and crystal structure of photocatalysts.58 A narrower band gap can provide a longer absorption wavelength, which can avoid side reactions due to the use of high-energy short wavelength. In the charge transfer process, recombination of charges and holes should be inhibited to extend the electron–hole pairs’ lifetime and ensure the efficiency of the catalytic reaction. To this end, avenues including heterojunctions, surface modification and increasing the degree of conjugation are developed to separate electron–hole pairs or localize holes on the surface of nanoparticles.
When selecting a catalyst for HP-RDRP, materials with excellent light absorption, charge separation ability and low fluorescent quantum yield should be preferentially considered. If the tacticity of the polymer needs to be precisely controlled, the surface structure of the catalyst should be delicately designed to create a specific coordination structure. Although catalytic efficiency is the main concern in catalyst selection, solubility, biocompatibility and cost may be considered in some specific applications.
3. Metal semiconductor nanoparticles
Metal semiconductor nanoparticles (NPs), with dielectric constants falling between the insulator and conductor, have shown excellent capability in various photocatalytic transformations such as the hydrogen evolution reaction (HER),59 CO2 reduction60 and polymerization.57,61 Their widespread use in these fields is in part due to the highly tunable band gap and rapid charge separation following photoexcitation. Herein, the use of metal and semiconductor nanoparticles in HP-RDRP is introduced in the following sections (Table 1).| Photocatalyst | Reaction | Size | Absorption peaks | Light source | Monomer | Ref. |
|---|---|---|---|---|---|---|
| a The ATRP initiator is incorporated in the photo-RAFT process. | ||||||
| ZnO | ATRP | 50 nm | 375 nm | 350 nm | MMA | 66 |
| RAFT | 30 nm | 375 nm | 375 nm | MMA | 35 | |
| Fe-ZnO | ATRP | 20 nm | 300, 380 nm | 350 nm | MMA | 66 |
| TiO2 | SI-ATRP | 10–40 nm | 330 nm | 330 nm | SPMA, DMAEMA | 71 |
| RAFT | 10–40 nm | 330 nm | UV light | MMA | 73 | |
| d-TiO2 | SI-ATRP | 10–40 nm | 350 nm | sunlight | SPMA | 34 |
| TiO2/rGO | SI-ATRP | 515 nm | visible light | MMA | 78 | |
| Bi2O3 powder | RAFT | μm scale | 250, 400 nm | white light | VAc, DMA | 48 |
| ATRP@RAFTa | μm scale | 250, 400 nm | CFL light | nBA, MMA | 79 | |
| α-Fe2O3 | ATRP | 14 nm | UV light | MMA | 72 | |
| NbBA | ATRP | 300 nm | visible light | NIPAM | 76 | |
| RAFT | 300 nm | visible light | NIPAM | 77 | ||
| CdSe QDs | ATRP | 3.3 nm | 565 nm | 460–480 nm | MMA, TFEMA | 61 |
| RAFT | 3.8, 5.1, 6.8 nm | 528, 548, 578 nm | 465, 532 nm, white light | MMA, BA, BA | 88 | |
| CPADB-CdSe | RAFT | 3.8 nm | 440, 550 nm | 465, 535 nm | MMA, BA | 33 |
| MPA-CdSe | RAFT | 1.4 nm | 532 nm | DMA | 57 | |
| SiQDs | SI-RAFT | 7.0 nm | <300 nm | 460, 530 nm, white | MMA | 87 |
| CsPbBr3 | RAFT | 11.0 nm | 500 nm | 460, 535,635 nm,800 nm laser | MA | 89 |
| AgNPs(Ag3PO4) | RAFT | 460 nm | 465, 525, 625, 780, 940 nm, sunlight | BzA, MA | 95 | |
3.1. Metal oxides
Metal oxides such as ZnO, TiO2 and Fe2O3 have long been used in photocatalytic radical polymerization for the synthesis of hydrogels62,63 and nanocomposites.64,65 Recently, as RDRP has showed the ability of synthesizing well-defined functional materials from a wide range of monomers, a natural shift in the use of metal oxide semiconductors as photocatalysts is seen from traditional radical polymerization to RDRP, mainly ATRP and RAFT.ZnO is the first metal oxide semiconductor used in radical polymerization46 and it has also been used for initiating RDRP due to its suitable bandgap (ca. 3.37 eV),35 nontoxicity, excellent stability, availability, and recyclability. Yagci and coworkers pioneered the use of ZnO and Fe-doped ZnO NPs in photoinduced ATRP of methyl methacrylate (MMA) using Cu(II)Br2, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), and ethyl α-bromoisobutyrate (EBiB) as the copper source, ligand, and initiator, respectively (Fig. 2).66 The polymerization was conducted under 350 nm UV light with an intensity of 22 mW cm−2. While the polymerizations conducted using both ZnO and Fe-doped ZnO NPs showed pseudo-first-order kinetics and well controlled molecular weights, the latter provided a faster polymerization rate and a lower dispersity possibly due to the higher light absorption of Fe-doped ZnO NPs. The light activation nature was illustrated by switching light on/off intermittently where no polymerization was observed during the dark periods. The livingness of the polymerization was confirmed by successful chain extension from a PMMA macroinitiator using the same monomer to produce a low-dispersity PMMA with a higher molecular weight.
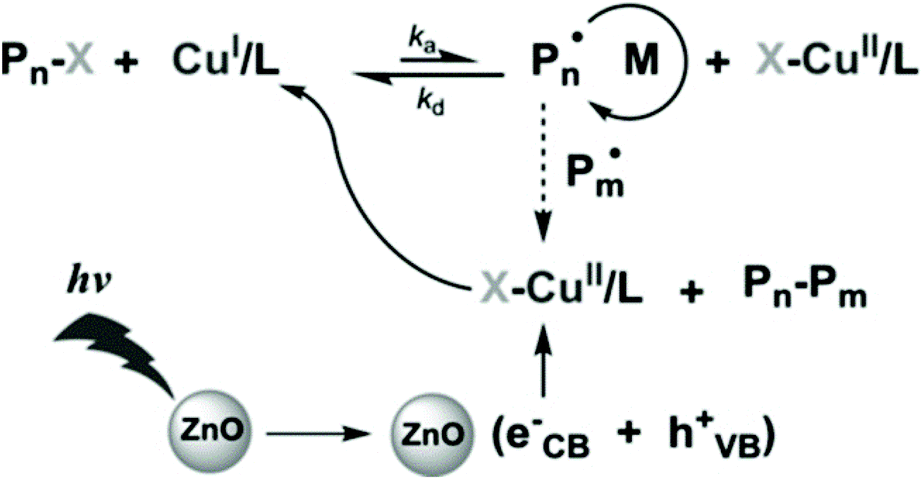 | ||
| Fig. 2 Proposed mechanism of ATRP using ZnO as the photocatalyst. Reproduced from ref. 66 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2014. | ||
As suggested by the authors, electron transfer from the conduction band of nanoparticles reduces Cu(II)/PMDETA to Cu(I)/PMDETA, which activates the ATRP process. However, the mechanism of ZnO regeneration was not discussed. Because the absorption wavelength can be tuned by the element doped in ZnO NPs, it is expected that more kinds of nanoparticles can be developed, through which polymerization activated by visible or infrared light may be achieved.
Wang and coworkers further expanded the use of ZnO NP to induce photo-RAFT polymerization of MMA using 4-cyanopentanoic acid dithiobenzoate (CPADB) as a chain transfer agent (CTA) under UV light (>350 nm, 20 mW cm−2) in N,N-dimethylformamide (DMF).35 The polymerization showed first-order kinetics only in the first 9 hours, and Mn,GPC deviated from Mn,theory. In addition, dispersity increased dramatically for monomer conversions larger than 20%. It was suggested that the loss of polymerization control was caused by photolysis and degradation of CTA under high intensity UV irradiation.
TiO2 is another type of semiconductor widely applied in photoredox catalysis. In particular, TiO2 is highly biocompatible and has been used as biomaterials67 in biomedicine68,69 and bone repair.70 Zhou et al. adopted TiO2 NP as a photocatalyst for surface-initiated ATRP (SI Photo-ATRP) on an Au substrate in water/methanol media, using Cu(bipy)22+, bipyridyl (bipy) and alkyl bromide as the copper source, ligand and initiator, respectively (Fig. 3).71 In agreement with the peak absorption of TiO2 NP, polymer brush growth was conducted under 330 nm UV light at 1.25 mW cm−2. The brush thickness increased with parameters which affected the polymerization rate, including TiO2 NP concentration, light intensity and Cu(I)/Cu(II) ratio. Polymerization livingness was illustrated by successful brush extension from the poly(2-(dimethylamino) ethyl methacrylate) (PDMAEMA) macroinitiator and 3-sulfopropyl methacrylate potassium salt (SPMA) as the second monomer. The authors also applied the strategy for the polymerization of various other monomers, including N-isopropylacrylamide (NIPAM) and oligo(ethylene glycol) methacrylate (OEGMA). As a round of SI Photo-ATRP process only consumed a small fraction of monomers in the solution, it was demonstrated that the solution could be reused 10 times with no differences in the thickness of brushes, suggesting potential for industrial applications.
 | ||
| Fig. 3 Scheme of the reaction setup, initiator, photoinitiation mechanism and brush formation via surface-initiated ATRP using TiO2 NP as the photocatalyst. Reproduced from ref. 71 with permission from the American Chemical Society, copyright 2013. | ||
In addition to ZnO and TiO2 NPs, the use of cheap, readily available Fe2O3 NPs72 was also explored for Photo-ATRP.
TiO2 NPs were also used to induce photoinduced electron/energy transfer (PET)-RAFT polymerization by You et al.73 As with previous work using ZnO NPs, loss of polymerization control occurred after 500 min irradiation. Decomposition of the CTA was suggested and was proved by the decreased characteristic absorption peak of the CTA at 517 nm in the UV-Vis spectra.
Although ZnO and TiO2 nanoparticles have shown promise for use as photocatalysts in RDRP, these semiconductors have large bandgaps and their absorption is limited to the UV region of the solar spectrum. The high-energy UV light may directly initiate polymerization or decompose polymerization control agents, leading to the loss of polymerization control. Other problems associated with the use of UV light include enhanced light scattering and safety concerns. To address these problems, several strategies have been developed to render these photocatalysts more amenable to RDRP under milder conditions, in some cases, with improved polymerization control.
Doping with other elements is a well-established strategy to modify semiconductor properties.74 This strategy has been harnessed by Yagci et al., who used Fe-doped ZnO NPs to enhance light absorption in their photocatalyzed ATRP work.66
Modification of semiconductor NPs with organic molecules is another useful strategy to improve the properties of semiconductor NPs. In one work, an organic dye with a high extinction coefficient in the visible light range is combined with semiconductor NPs to make efficient photosensitizers for RDRP. In an effort to employ visible light for SI Photo-ATRP, Zhou et al. used dye N749-sensitized TiO2 (d-TiO2) as the photocatalyst in H2O/MeOH media under 68 mW cm−2 simulated sunlight.34 In contrast to previous work, this SI Photo-ATRP under sunlight avoided side reactions caused by UV light and utilized sunlight more efficiently. The dye covered on the surface of NPs increased the absorption in the range of UV to infrared. In this system, the amount of d-TiO2 can be used to adjust the ratio of [Cu(II)]/[Cu(I)] to affect the controllability of polymerization. Furthermore, this strategy was applied in photolithography to produce micropatterns and nanostructures using suitable photomasks. In another work, Shishido et al.75 grafted niobium hydroxide (Nb(OH)5) with benzyl alcohol, and the absorption wavelength was increased from 390 nm to more than 420 nm. Building on this knowledge, Liu and co-workers used benzyl-alcohol-modified niobium hydroxide (NbBA) as a photocatalyst in both ATRP and RAFT polymerizations of NIPAM.76,77 They were conducted under visible light with wavelengths of more than 420 nm. It was suggested that the same mechanism was involved for the two polymerizations during the first several steps: light irradiation caused an electron transfer from the oxygen atom of benzyl alcohol to the Nb atom through a ligand-to-metal charge transfer process, resulting in the formation of benzyl alcohol radical cations, which then lost a proton to generate a radical –O–C·H–Ph with high reactivity. This radical was used to initiate RAFT polymerization in the presence of 4-cyano-4-ethyl-trithiopentanoic acid (CETP), and the NbBA was regenerated through decomposition of the NbBA-CTA intermediate in the oligomeric radical reattacking process. This radical mechanism was supported by the decreased yield of poly(N,N-dimethyl acrylamide) (PDMA) after adding 2,2,6,6-tetramethyl-1-piperidine-1-oxyl (TEMPO), a radical scavenger with high selectivity for carbon centered radicals. For ATRP, the radical –O–C·H–Ph reacted with ethyl 2-bromoisobutyrate (EBiB) to generate R˙ which initiated the reaction, and NbBA-Br lost Br˙ to regenerate the NbBA (Fig. 4). The key point of these strategies is the ratio of the grafted benzyl alcoholate to CTA or R–X. If the amount of radical is much less than that of initiators, the reaction rate would be too slow, but if it far exceeds initiators, it would fail to control the polymerization.
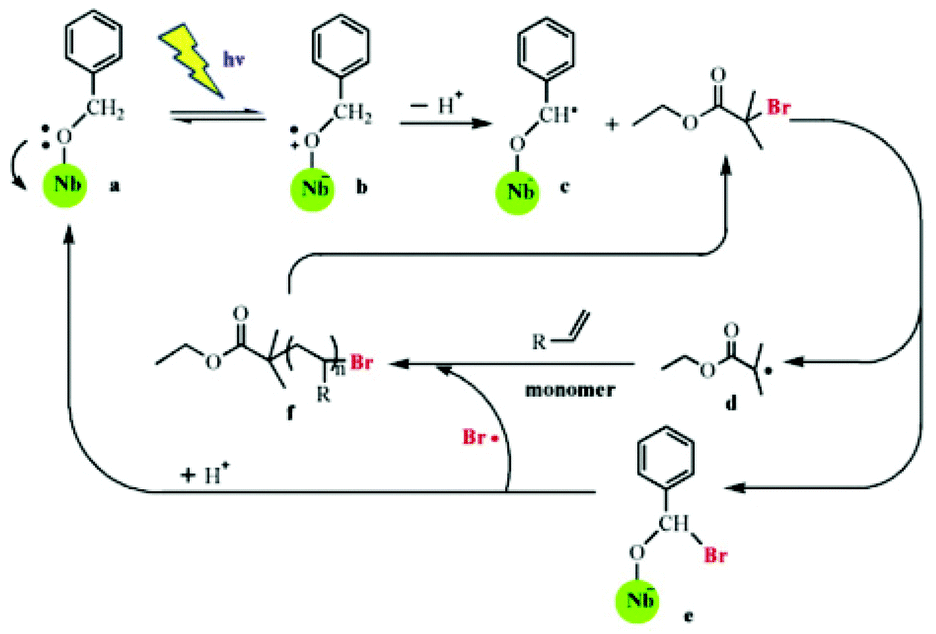 | ||
| Fig. 4 Proposed mechanism of visible-light-mediated ATRP using benzyl-alcohol-modified niobium hydroxide as the catalyst in the presence of ethyl 2-bromoisobutyrate (EBiB). Reproduced from ref. 76 with permission from Elsevier, Copyright 2015. | ||
The properties of semiconductor NPs can also be affected by other semiconductor/conductors. To overcome the low catalytic efficiency brought by fast charge recombination of photoexcited TiO2, Jain and coworkers demonstrated SI Photo-ATRP of MMA catalyzed by the TiO2/reduced graphene oxide (rGO) nanocomposite (TiO2/rGO) under sunlight.78 Because rGO possesses high electron mobility, it was proposed that the heterojunction formed at the interface of rGO and TiO2 slowed the recombination of hole–electron pairs. Supported by the band gap calculation of TiO2/rGO and TiO2 only, 2.4 eV (absorption peak at 515 nm) for TiO2/rGO and 3.2 eV (387 nm) for the latter, the nanocomposite has better ability to harness visible light. Using TiO2/rGO as the photocatalyst, molecular weight increased linearly with the conversion from 42% to 90% for a reaction time of 5 h to 24 h.
While modification of semiconductor NPs such as ZnO and TiO2 with broad band gaps has proved to be successful in improving the efficiency and control of RDRP to some extent, the most direct approach would be using narrow-band-gap semiconductors with a suitable reduction potential. In this direction, the most notable work is the use of Bi2O3 powder by Müllner and coworkers for PET-RAFT/MADIX polymerization.48 Bi2O3 was selected as a photocatalyst because it is nontoxic, cheap, and easily separable by centrifugation (in the form of a powder) and its narrow band gap enables the use of a household light bulb as the white light source. Well-controlled RAFT/MADIX polymerization of several classes of monomers including vinyl acetate (VAc), N-vinylpyrolidone (NVP), DMA, MA, and MMA was successfully achieved in various solvents (DMSO, water, and anisole), with macromolecular characteristics similar to those for common homogeneous polymerizations, by selecting an appropriate CTA for each monomer class. Importantly, chain extension provided block copolymers with low dispersities, which were best showcased with PDMA-b-PVAc and PDMA-b-PNVP, two block copolymers from monomers of more-activated and less-activated monomers.
Building on the success of the use of Bi2O3 as the photocatalyst for PET-RAFT/MADIX, the same team attempted photocatalyzed ATRP which, however, failed to show polymerization control possibly due to its inability to deactivate the propagating radical. The polymerization was then transformed into a RAFT polymerization through the simultaneous use of disulfides as chain transfer agents, which provided polymers with thiodithio-end groups and relatively low dispersities.79
3.2 Quantum dots
Quantum dots (QDs) have attracted much attention in recent years because of their excellent photoelectronic and chemical properties such as high reduction potential (−1.59 V vs. SCE for CdSe QDs)33 and strong and tunable absorption in the UV and visible range owing to the quantum confinement effect. Meanwhile, the ultrasmall size endows QDs with a large surface area and many reactive sites on the surface. These factors all contribute to their wide use in photocatalytic reactions. Besides their applications in organic transformation showing good performance,80 they have also shown promising potential in photocatalytic polymerization. Free-radical polymerization catalyzed by QDs under a one- or two-photon process has been conducted by various research groups.49,50,75,81–85 Moving on from these works, Egap,33,61,86 Pang87,88 and Weiss57 pioneered the use of CdSe QDs as photocatalysts for RDRP.The ATRP of methacrylates, fluorinated and semifluorinated monomers such as 2,2,2-trifluoroethyl methacrylate (TFEMA), and styrene catalyzed by CdSe QDs in THF, C6H6 and DMF was reported by Egap and coworkers,61 in which ethyl α-bromophenylacetate (EBP) and N,N-diisopropylethylamine (DIPEA) served as the initiator and electron donor, respectively. The polymerization was conducted under 460–480 nm blue light at 10 mW cm−2 at 25 °C. The molecular weight distributions of the obtained polymers were generally broad in comparison with well-controlled homogeneous ATRP systems, possibly due to the lack of an effective deactivation pathway for the propagating chains.
Two different mechanisms, oxidative or reductive quenching pathway, were proposed, where the QD acted as a reductant or an oxidant, respectively. The QD was first excited to QD* by light irradiation. In the oxidative quenching pathway, the QD* donated an electron to EBP to generate a QD(˙+) radical cation and an alkyl radical, thus initiating the polymerization. Deactivation was realized through oxidation of the propagating radical by the QD (˙+), accompanied by regeneration of the QD photocatalyst. In contrast, the QD* accepted an electron from trialkylamine in the reductive quenching pathway. The oxidative quenching pathway was proven to be the dominant mechanism as confirmed by the increased fluorescence quenching with increasing EBP concentration in the Stern–Volmer plot (Fig. 5).
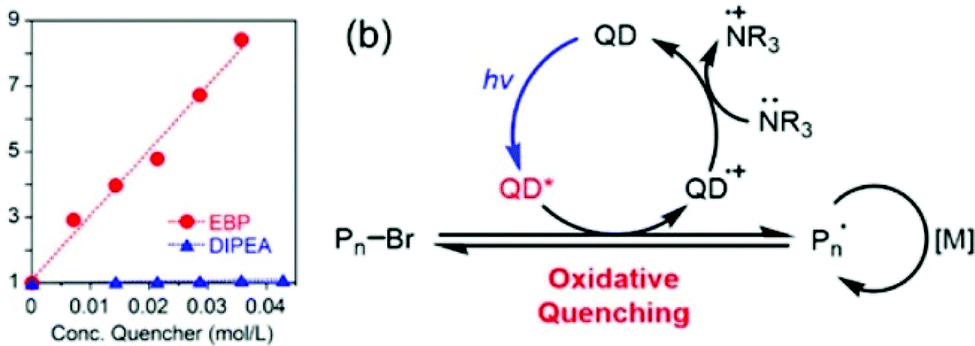 | ||
| Fig. 5 (a) Stern–Volmer plot of QD fluorescence (λex = 460 nm) effectively quenched by EBP but not by DIPEA. (b) Proposed oxidative quenching mechanism of ATRP catalyzed by CdSe QDs. Reproduced from ref. 61 with permission from the American Chemical Society, copyright 2018. | ||
Later, the Egap group reported the first QD-catalyzed RAFT polymerization under 465 nm blue light.33 The CPADB CTA was capped on the CdSe QD surface via Lewis acid–base interaction (Fig. 6) through ligand exchange with oleic acid. Thus, polymer chains can grow from the QD surface through the HP-RDRP process. Thus, various functional monomers were successfully polymerized at high monomer conversions and low-dispersity polymers were achieved using ultralow QD loading (15–45 ppm). In this strategy, the CTA was preorganized on the QD surface, which could enhance the control of polymerization. This grafting-from strategy may be used for facile synthesis of polymer–QD nanocomposites.
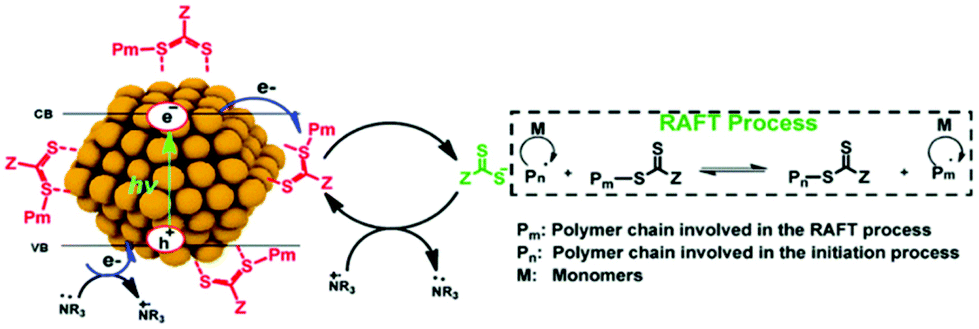 | ||
| Fig. 6 Proposed mechanism of RAFT polymerization catalyzed by CdSe QDs. Reproduced from ref. 33 with permission from the Royal Society of Chemistry, copyright 2020. | ||
Particle size is one of the key parameters that influence the optical and electronic properties of QDs. To explore the QD size effect in HP-RDRP, Pang and coworkers investigated PET-RAFT polymerization of MMA using CdSe QDs of different diameters, 3.8 nm (G), 5.1 nm (Y) and 6.8 nm (R) (the abbreviations represent the fluorescence colour of QDs under 365 nm UV light excitation).88 The polymerization was conducted in DMF under blue (465 nm), green (532 nm) and white light, and sunlight with CPADB serving as the CTA. Under blue light irradiation, the polymerization catalyzed by R showed the highest monomer conversion (72%), highest molecular weight (Mn = 14.6 kg mol−1) and lowest dispersity (Đ = 1.17) in 28 h.
Although CdSe QDs have been successfully used in visible-light-controlled HP-RDRP, their high toxicity may hinder their widespread use. To address this issue, Pang and coworkers adopted silicon quantum dots (SiQDs), an emerging type of QD with low toxicity and broad absorption from UV to visible light, in PET-RAFT polymerization of MMA initiated from the surface of hydroxylated silicon wafer.87 CPADB was anchored on SiQDs through the condensation reaction between the carboxyl group of CTA and the amino group on SiQDs. The polymerization was performed in DMSO under blue light irradiation (λmax = 460 nm, 2 mW cm−2). After 32 h reaction, the thickness of the coarse surface increased from 1.3 ± 0.9 nm to 6.2 ± 3.9 nm according to the AFM image, indicating that successful polymerization occurred on the silicon wafer.
One of the benefits of HP-RDRP is that the catalysts can be potentially recovered and reused for multiple polymerizations. However, QDs are difficult to recover from the polymerization media due to their small size. Weiss et al. reported the use of mercaptopropionic acid (MPA)-capped CdSe QDs as highly efficient photocatalysts for PET-RAFT polymerization of vinyl monomers including DMA, N,N-diethylacrylamide (DEA), 4-acryloylmorpholine (AMP), 2-hydroxyethyl acrylate (HEA), and poly(ethylene glycol) methyl ether acrylate (PEGA), under 532 nm green light at an intensity of 40 mW cm−2 in aqueous media.57 Notably, a DMA polymerization rate of 1.07 h−1 was achieved using an ultralow QD loading (0.43 ppm), which was 6 times faster than that in the presence of 7.5 ppm Eosin Y, a homogeneous catalyst having similar absorption with CdSe QDs at 532 nm. The high polymerization rate may be attributable to the localization of the photogenerated holes by the thiolate groups on the surface of excited QDs. In order to separate the QDs from the polymerization media, the authors used Amicon ultracentrifugal spin filters, which are traditionally used to separate proteins, to purify the product mixture. By adjusting the pore size of the filters from 3 kDa to 30 kDa, it was possible to separate QDs from the polymer solution (Fig. 7). The colloidal stability of MPA-capped QDs was well-maintained after separation and no significant decrease of the QDs’ catalytic ability was observed which was confirmed by the successful chain extension experiments. The successful recovery and reuse of heterogeneous photocatalysts are an important technical benefit of HP-RDRP. However, ultracentrifugation consumes a large amount of energy, and advancing new techniques that can efficiently separate heterogeneous photocatalysts of small size still poses a significant challenge.
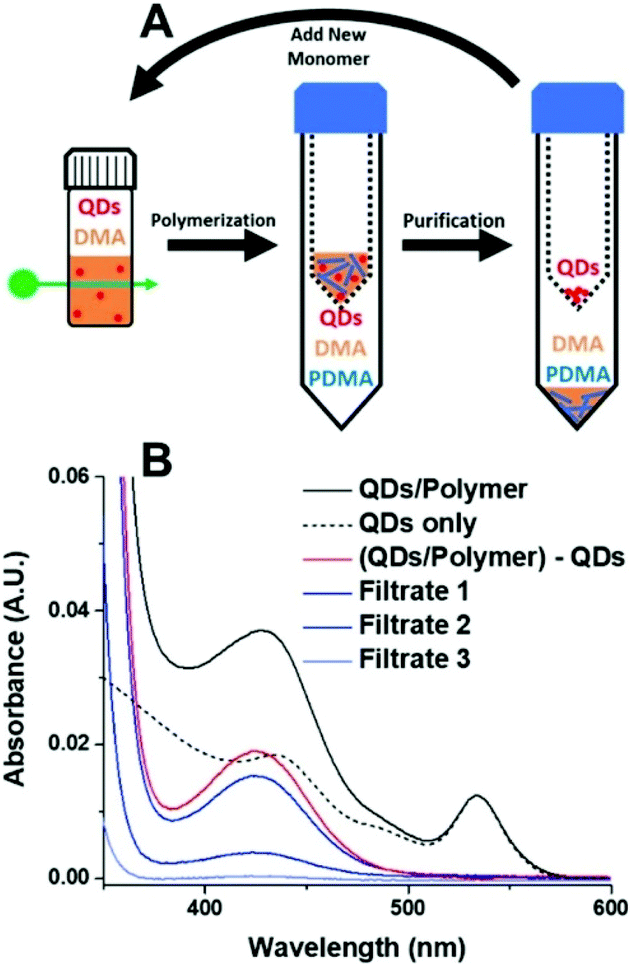 | ||
| Fig. 7 Procedure for the separation of QDs from polymer solution using protein concentrators. Reproduced from ref. 57 with permission from the American Chemical Society, copyright 2019. | ||
3.3 Perovskites
Most recently, CsPbBr3 perovskite was introduced to the field of PET-RAFT polymerization as a photocatalyst by Egap et al., due to its strong light absorption covering the range from UV to near infrared,89 narrow full width at half-maximum (FWFM),90 highly tunable band gap,91 and large two-photon absorption (TPA) cross-section.92 The PET-RAFT polymerizations of MA, BA and TFEA were successfully conducted in toluene using trithiocarbonates as CTAs under 10 mW cm−2 blue light irradiation. Again, the CsPbBr3 nanoparticles could be recycled by centrifugation. Moreover, successful TPA-induced polymerization of MA was demonstrated using 800 nm femtosecond laser pulses at an energy density of 0.6 mJ cm−2. The monomer conversion reached up to 60.2% with a PMA dispersity of 1.07 in 11 h. This was the first example of TPA-induced HP-RDRP. Although it still suffered drawbacks such as low conversion, low molecular weight and long reaction time, it may offer opportunities for localized polymer synthesis and high-precision photolithography. However, efforts are still needed to improve the compatibility of perovskite nanoparticles with polymerization conditions since many of them suffer poor stability in the presence of functional monomers, polar solvents and initiators.Gratifyingly, the metal-containing semiconductors have exhibited excellent performances in photocatalytic controlled radical polymerization in terms of polymerization rate, molecular weight distribution control and range of monomers, and the light source used in HP-RDRP is becoming softer. Meanwhile, these nanoparticles can be separated from the reaction systems and reused in several cycles. But there are still problems such as toxicity and the lack of eco-friendliness, and the difficulty in high-throughput preparation limits the industrial production. Although some of these efficient polymerization methods can be used in the fabrication of hydrogels and other materials for healthcare, microlithography, coatings and so on, the potential danger of toxic transition metal leakage may hinder their adoption in real applications.
3.4 Noble metal nanoparticles
HP-RDRP excited by ultraviolet and visible light has been extensively explored; however, research studies using NIR light sources are still rare, and it remains a challenge to develop efficient photocatalysts with broadband light especially NIR light harvesting ability. Boyer and coworkers contributed a lot towards this field including the first NIR light excited RAFT polymerization using a bacteriochlorophyll photocatalyst.93 And the Yagci group firstly developed the NIR excited ATRP using zwitterionic polymethine as the PC.94Sharp absorption at specific wavelengths occurs when noble metal nanostructures interact with light, known as localized surface plasmon resonance (LSPR), which also has potential to be applied in HP-RDRP under NIR light. Ye, chen and Matyjaszewski explored the localized surface plasmon resonance (LSPR) effect of Ag nanoparticles (AgNPs) generated by in situ photoreduction of Ag3PO4 in HP-RDRP.95 The RAFT polymerization of MA was conducted in the presence of Ag3PO4 with various morphologies including rhombic dodecahedral (RD), tetrahedral (TH), and tetrapodal (TP) under 465 nm blue light with an intensity of 2 mW cm−2. TH-Ag3PO4 showed the best catalytic performance, with the monomer conversion reaching up to 82.6% in 3.7 h, owing to its highly exposed {1,1,1} facet with high surface energy. It is well known that the high surface energy is beneficial to the surface reaction rate.96 And efficient RAFT polymerization catalyzed by RD-Ag3PO4 of MA and BzA was also investigated under green (525 nm, 2 mW cm−2) and red (625 nm, 2 mW cm−2) light. The generation of AgNPs during polymerization was observed and the LSPR absorption intensity of the nanocomposites around 800 nm increased with time. Inspired by this, the authors conducted the RAFT polymerization of BzA under 780 nm (6 mW cm−2) and 940 nm (16 mW cm−2) light, and the >99% monomer conversion, dispersity of 1.24 and Mn close to the theoretical value were obtained in 18.8 h under 940 nm light, the longest wavelength used to induce the RARP until now. And the unique penetrating ability of NIR light was demonstrated by successful polymerization either in the PMMA tube (thickness ≈ 5 nm) or with opaque paper (thickness ≈ 0.1 mm) as the barrier.
The polymerization mechanism was discussed in detail. Upon light illumination, AgNPs generated from the self-photoreduction process, and the LSPR effect enabled the efficient charge transfer from AgNPs towards RAFT agents where the RAFT agent acts as both the initiator and CTA. After donating an electron, the AgNPs transformed to Ag+ to regenerate the Ag3PO4. Furthermore, this polymerization system can be performed without deoxygenation in the presence of a singlet oxygen (1Δ) quencher such as 9,10-dimethylanthracene (DMAn), and the solvent DMSO can also eliminate the singlet oxygen to generate sulfone (DMSO2) (Fig. 8).
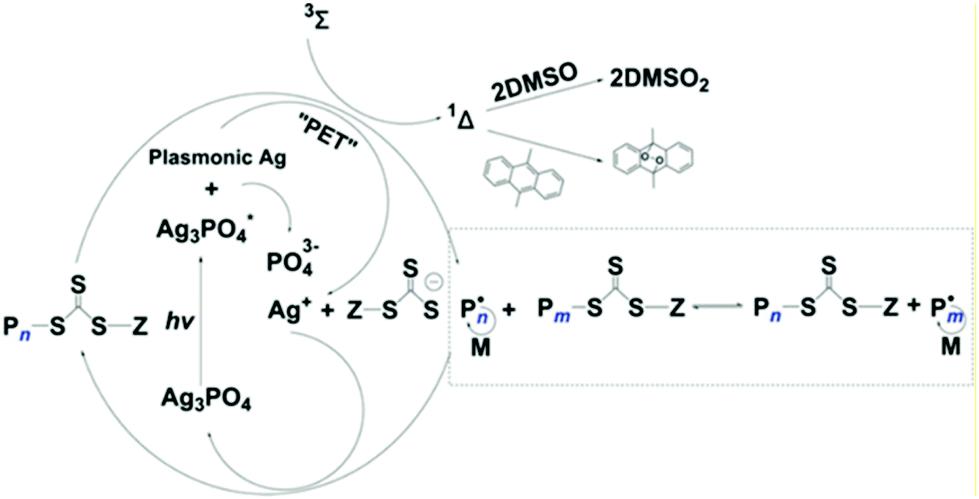 | ||
| Fig. 8 Proposed mechanism of PET-RAFT catalyzed by Ag3PO4 and the singlet oxygen quenching pathway. Reproduced from ref. 95 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2019. | ||
There have been many protocols to synthesize noble metal nanoparticles absorbing light from UV to NIR. For example, the strong absorption peak of the gold nanorod shifts to longer wavelength by increasing its aspect ratio. Thus, more efficient catalysts are expected to be found in this big toolbox.
4. Upconversion nanoparticles
Upconversion nanoparticles (UCNPs) are composed of lanthanide dopants and matrix hosts. It is known that rare-earth materials present both down conversion (Stokes) and excellent upconversion (anti-Stokes) luminescence. Upconversion is a nonlinear process, which is characterized by the absorption of two or more photons and leads to the emission of a single photon with shorter wavelength and higher energy. Lanthanide-doped UCNPs were first reported in 2000.97 Due to lanthanide ions’ ladder-like energy levels and long excited state lifetime, UCNPs can convert NIR light to UV or visible light,98 which greatly enhances the potential of UCNPs in photo-controlled reactions. It is well known that using long wavelength and low energy light, such as NIR, can suppress the possible cleavage of compounds with weak bonds and the possible side reaction. In 2009, Branda et al. reported the first example of UCNP-assisted photoswitching.99 Benefitted by the highly modifiable surface and efficient upconversion performance, the application of UCNPs has been greatly expanded in the biological field in the past few years, such as cancer cell imaging100 and drug delivering.101 These developments are mainly attributed to the polymer layer modification of the hydrophobic surface of UCNPs. However, there are few studies applying the efficient upconversion characteristics of UCNPs in polymerization. In 2006, Soga et al. reported the first example of UCNP-assisted polymerization by infrared-to-visible upconversion emission.102 Nevertheless, the dispersity and molecular weight of the polymers were not well-regulated. Although RDRP has received extensive attention in the past decades, studies about UCNP-assisted RDRP are rare (Table 2).| Photocatalyst | Reaction | Size | Light source | Emitting light | Monomer | Ref. |
|---|---|---|---|---|---|---|
| NaYF4:Yb/Tm | RAFT | 980 nm laser | 325–380 nm, 425–500 nm | MMA, BA, VAc | 103 | |
| NaYF4:Yb/Tm@NaYbF4:Gd@NaNdF4:Yb@NaYF4 | RAFT | 45 nm | 808 nm laser | 290 nm, 335–370 nm, 440–490 nm | AA, DEGMA, PEGMA | 101 |
| β-NaYF4:Yb/Tm | RAFT | 32 nm | 980 nm laser | 325–380 nm, 425–500 nm | MMA | 104 |
| β-NaYF4:30% Yb3+, 1% Tm3+ | ATRP | 15.7 nm | 980 nm laser | 365 nm, 340–480 nm, 625–750 nm | MA | 105 |
In 2016, Zhu et al.103 reported the first NIR light-induced RDRP using UCNPs as internal light sources (Fig. 9). It was confirmed by UV-vis absorption spectra that NaYF4:Yb/Tm nanoparticles emitted light at 325–380 nm and 425–500 nm under 980 nm light irradiation, matching well with the typical absorption of xanthates (2-((phenoxycarbonothioyl)thio) ethyl propanoate (PXEP), 2-(ethoxycarbonothioyl)sulfanyl propanoate (EXEP)) and trithiocarbonate 3-((((2-methoxy-2-oxo-1-phenylethyl)thio)carbonothioyl)thio) propanoate (AMP). The UCNP-assisted polymerizations of butylacrylate (BA), vinyl acetate (VAc) and methyl methacrylate (MMA) were conducted in the presence of PXEP, EXEP, and AMP, respectively, using 980 nm NIR light. All the obtained polymers showed well-controlled molecular weight and narrow dispersity (Đ < 1.3).
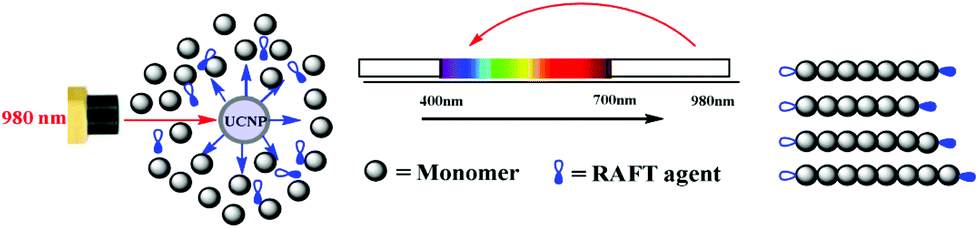 | ||
| Fig. 9 Proposed mechanism of RAFT polymerization catalyzed by UCNPs. Reproduced from ref. 103 with permission from the Royal Society of Chemistry, copyright 2016. | ||
The NIR light-induced RDRP using UCNPs as internal light sources shows a brand-new solution to combine low energy and high penetrability with photo-controlled RDRP, particularly for systems containing sensitive bonds. The biological tissue penetrability of NIR light and mild conditions of RDRP may greatly expand the UCNP-assisted RDRP application, particularly in the biomedicine field.
UCNPs have characteristics of high chemical and physical stability, low background fluorescence, and low toxicity, which make UCNPs promising materials in theranostics and cell imaging. Lin et al.101 reported a surface NIR-light-initiated RAFT polymerization on core–shell UCNPs (NaYF4:Yb/Tm@NaYbF4:Gd@NaNdF4:Yb@NaYF4). The amino group containing alendronate replaced oleic acid through the ligand exchange process on the surface of UCNPs. Then carboxyl-ended CTA molecules were anchored on the surface by amide conjugation. Under irradiation of 808 nm NIR light, the UCNP upconversion emission at 290 nm, 350 nm and 365 nm was efficiently absorbed by CTA molecules, leading to energy transfer from UCNPs to CTA molecules. Polymerization livingness was illustrated by successful synthesis of diblock copolymer brushes composed of poly(acrylic acid) and poly(oligo(ethylene oxide)methacrylate-co-2-(2-methoxy-ethoxy)ethyl methacrylate) under 808 nm laser irradiation. Moreover, the targeted Arg-Gly-Asp (RGD) molecules were modified at the end of polymer chains to promote the specific cancer therapy. Furthermore, the final nanohybrids carried pH-responsive anticancer drug doxorubicin (DOX) hydrochloride by Coulomb force to kill U87MG cancer cells. This report shows that UCNPs are excellent photocatalysts for RAFT polymerization; however, few polymerization kinetic studies have been conducted and the CTA anchoring process leads to complicated synthesis and purification steps.
Qiao and Pang et al.104 recently reported a UCNP-assisted NIR photo-controlled RAFT polymerization on the surface of UCNPs through an efficient in situ ligand exchange between CTA molecules and the ligands on the surface of UCNPs. The NIR-initiated polymerization of MMA was conducted in DMSO in the presence of NOBF4, β-NaYF4:Yb/Tm UCNPs and CTA (CDTPA) under laser irradiation (1.5 W cm−2, 980 nm) at room temperature and well-controlled molecular weight and narrow molecular weight distribution (Đ < 1.30) were achieved. Pseudo-first order kinetics and temporal control were confirmed by the kinetic analysis and “switch on/off” experiments, respectively. Furthermore, the polymerization of MMA with a 1.2 mm thick chicken skin barrier was successfully conducted, and well-defined polymers with molecular weights of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and 15300 g mol−1 were achieved after 24 h and 36 h, respectively, and showed narrow molecular weight distributions (Đ < 1.30), which verified the tissue penetration ability of NIR light.
000 g mol−1 and 15300 g mol−1 were achieved after 24 h and 36 h, respectively, and showed narrow molecular weight distributions (Đ < 1.30), which verified the tissue penetration ability of NIR light.
Recently, Pan et al.105 reported a UCNP-assisted NIR photo-induced ATRP. Under irradiation at 980 nm, the emission of UCNPs (β-NaYF4:30% Yb3+, 1% Tm3+) at 365 nm matched well with the absorption of the ATRP system, which contained ethyl α-bromoisobutyrate (EBiB) as the initiator and tris(2-pyridylmethyl)amine (TPMA) as the ligand (Fig. 10). The polymerization of MA was conducted successfully with 58% monomer conversion in 24 h. The switching “on/off” experiment confirmed the excellent temporal control of the UCNP-assisted NIR photo-induced ATRP and provided the final polymer with low dispersity (Đ = 1.17). The “living” chain-end of the final polymer was confirmed by the successful chain extension experiment from the PMA-Br macro-initiator; the molecular weight increased from 5200 to 15300 and the dispersity decreased from 1.20 to 1.13. Furthermore, the polymerization of MA under 980 nm irradiation for 36 h with pig skin (1.2 mm) and A4 paper (0.2 mm) was conducted with 88% and 64% monomer conversion, respectively. The polymerizations of other hydrophobic and hydrophilic monomers were also conducted with predictable molecular weight and narrow molecular weight distribution, which indicated broad applicability of the UCNP-assisted NIR photo-induced ATRP system. However, the polymerization efficiency needs to be improved since long polymerization time is generally needed to achieve moderate monomer conversions.
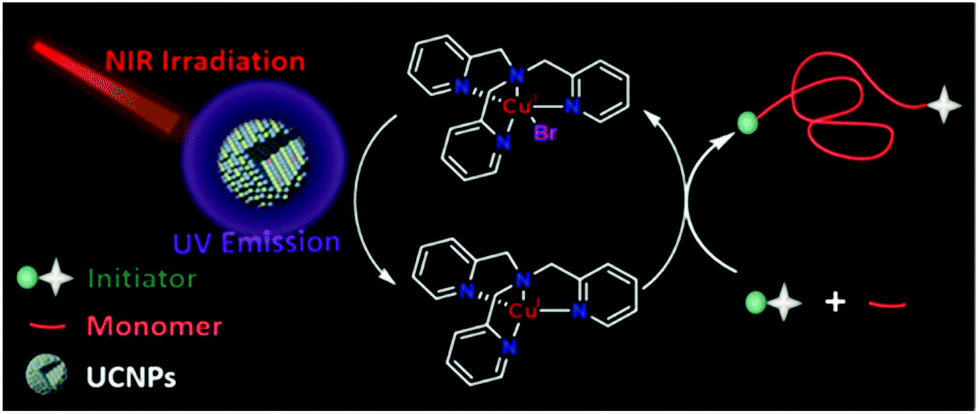 | ||
| Fig. 10 The proposed mechanism for photoATRP under NIR irradiation assisted by UCNPs. Reproduced from ref. 105 with permission from the American Chemical Society, copyright 2020. | ||
UCNPs can have broad emission spectra, from visible light to UV, by tuning the doped lanthanide. Moreover, UCNPs have great physical and chemical stability so that they can be easily recycled and reutilized. These properties make UCNPs very promising in HP-RDRP. However, single-doped UCNPs are limited in the upconversion efficiency and breadth of emission wavelength. Multi-doped UCNPs can overcome these limitations but the synthesis of core–shell multi-doped UCNPs is complicated. Therefore, more efforts are needed to tailor UCNP photoelectronic and surface properties for application in HP-RDRP.
5. Metal–organic frameworks (MOFs)
Metal–organic frameworks (MOFs), a class of organic–inorganic hybrid materials, have experienced a rapid development since the first report by Yaghi et al.106 Because of their structural versatility and precise pore size, they have attracted extensive interest in various research fields such as gas storage107 and separation,108 oxygen109 and hydrogen110,111 evolution and biomedicine.112 MOFs have also been explored as photocatalysts for free-radical polymerization113 and RDRP,114–116 due to the potential of metal complexes to serve as photocatalysts for the photoinduced electron transfer process (Table 3). Their porous nature endows them with confined space to control the tacticity of polymers.117,118 In contrast to polymerization catalyzed by nanoparticles, metal complexes are periodically arranged in the porous MOF, which may potentially provide more reactive sites and may accelerate the polymerization rate. However, a large fraction of metal complexes may be inaccessible for large polymer chains because of their complex structure and small pore diameter, making special designs of MOFs of high importance.| Photocatalyst | Composition | Reaction | Absorption peaks | Light source | Monomer | Ref. |
|---|---|---|---|---|---|---|
| NNU-28 | [Zr6O4(OH)4(L)6]·6DMF | ATRP | 490 nm | 520 nm | MMA | 119 |
| NNU-32 | (Me2NH2)[InL]·3(H2O)·0.5DMF | ATRP | 510 nm | 521 nm | i-BMA | 120 |
| NNU-35 | [Zn(bdc)(L1)]2·DMF | ATRP | 480 nm | 522 nm | MMA | 121 |
| MOF-545(H2) | {Zr6(μ3-OH)8(OH)8(H2-TCPP)2} | ATRP | 540 nm | 420 nm < λ < 800 nm | MMA | 122 |
| MOF-545(Zn) | {Zr6(μ3-OH)8(OH)8(Zn-TCPP)2} | ATRP | 605 nm | 420 nm < λ < 800 nm | MMA | 122 |
| MOF-525(Zn) | Zr clusters, Zn-TCPP | RAFT | 445, 570, 600 nm | 405, 470, 565, 595, 680 nm | MA, DMA, MMA | 56 |
| Cu(II) MOF | [Cu2(bdc)2(dabco)]n | ATRP | 270 nm | 420 nm < λ < 800 nm | 4VP, 2VP, DMAEMA, MMA | 125 |
Considering the composition of MOFs, either the organic linkers or the metal ions can be altered to endow MOFs with appropriate photocatalytic properties to induce effective RDRP.
The Xing group prepared MOFs with anthracene derivatives or tetracarboxylic porphyrin (TCPP) as the organic linkers, including NNU-28,119 NNU-32,120 NNU-35,121 MOF-545(H2) and MOF-545(Zn)122 (incorporating Zr cluster and H2-TCPP or Zn-TCPP), and used them as photocatalysts for ATRP of MMA, n-butyl methacrylate (n-BMA) and iso-butyl methacrylate (i-BMA) under 520 nm visible light irradiation (25 mW cm−2). The Zn-MOF (NNU-35)121 has a pillared-layer structure, in which the layers are composed of Zn metal ions linked by terephthalate ligands, and layers are pillared by anthracene-derived bipyridines. It showed a broad absorption in the visible range as a result of ligand–metal charge transfer interaction. The polymerization was conducted in the presence of EBiB as the initiator and [CuBr2]/[PMDETA] as the ATRP catalyst. Under light irradiation, supported by the electron paramagnetic resonance (EPR) signal, a radical was generated within the MOF through the excitation of the anthracene bipyridine pillar ligand, which reduced Cu(II) to Cu(I), activating the alkyl halide initiator and initiating the polymerization. Meanwhile, the positively charged MOF oxidized amine to restore the charge-neutral state of the MOF (Fig. 11). The utility of anthracene derivative-based MOFs in photocatalytic RDRP was further explored by using In120 and Zr119 in place of Zn.
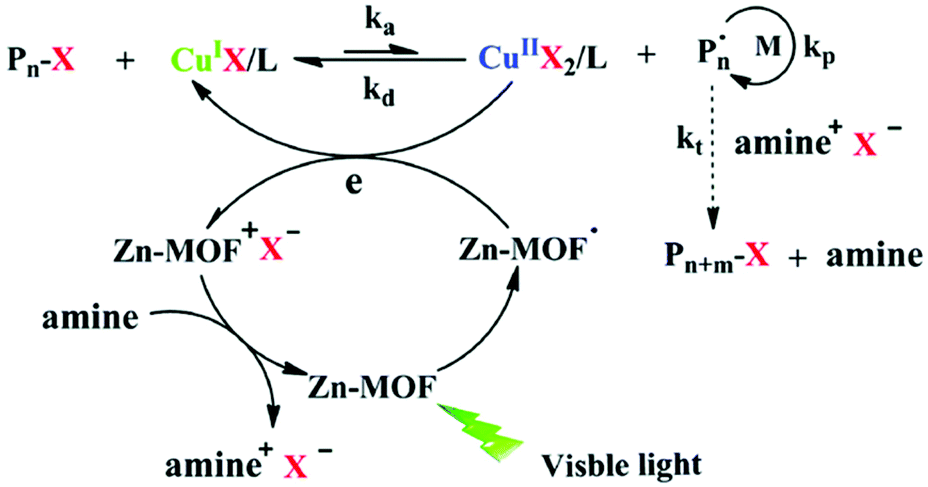 | ||
| Fig. 11 Proposed mechanism of ATRP catalyzed by NUU-35 under visible light. Reproduced from ref. 121 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Recently, oxygen-tolerant PET-RAFT polymerization of MMA, DMA and MA catalyzed by porphyrinic Zr-MOFs (Zn) in DMSO under visible light (λmax = 405, 470, 565, 595 and 680 nm, 9 mW cm−2) was investigated by the Boyer group.56 Zr-MOFs (Zn) are all composed of Zr clusters and Zn-TCPP complexes. The polymerization occurred in the solution rather than inside the MOF. Because porphyrinic Zr-MOFs (Zn) can photo-reduce triplet oxygen to singlet oxygen, oxygen-tolerant PET-RAFT polymerization of MA was successfully performed. The robustness of the MOF photocatalysts was demonstrated by 5 cycles of polymerization where no obvious difference in monomer conversion, molecular weight dispersity and MOF morphology was observed. Interestingly, these MOF photocatalysts were further explored in stereolithography using a stoichiometry of [PEGDA]:[BTPA]:[TEOA] = 400:1:20 to make a cross-shaped object in air, where the PEGDA served as the crosslinker (PEGDA: poly(ethylene glycol) diacrylate; BTPA: 2-(n-butyltrithiocarbonate)-propionic acid; TEOA: triethanolamine). The storage modulus (G′) of the object can be enhanced from 90 MPa to 428 MPa at 20 °C by introduction of 0.01 equivalent Erythrosin B as the cocatalyst.
The optical properties of MOFs can be tuned by altering organic linkers or metal atoms as mentioned above.123,124 For instance, the band gap decreases with increasing degree of conjugation of the organic linkers. In addition, the unsaturated metal ions on MOFs’ surface can act as the Lewis-acid center, coordinating with electron-rich atoms or vinyl groups of monomers, which may be explored to adjust the band gap of MOFs.
In this regard, Schmidt et al. reported ATRP of vinylpyridines and methacrylates catalyzed by Cu(II) MOF [Cu2(bdc)2(dabco)]n (with Cu2 segments, terephthalic acid (H2bdc) and 1,4-diazabicyclo[2.2.2]octane (dabco) serving as a dication, 2D linker and pillar, respectively) upon visible light using EBiB as the initiator (Fig. 12).125 In this system, nitrogen atom-containing monomers such as 2-vinylpyridine (2VP) and 4-vinylpyridine (4VP) can interact with copper atoms and form coordination bonds on the surface of the MOF. A red shift of MOF's absorption from around 250–350 nm to visible range (300–600 nm) was reported after adding 4VP and 2VP, indicating that the band gap decreased from around 3.5 eV to 2.6 eV. However, this red shift was not obvious in the case of adding MMA or DMAEMA, in accordance with the strength of the coordination bond formed by the monomers 4VP > 2VP > DMAEMA > MMA. Upon irradiation, Cu(II) was reduced to Cu(I), which then activated EBiB and regulated ATRP. Interestingly, the isotactic triads of P4VP increased from 13% to 27%, which was attributed to the coordination effect between nitrogen and orderly aligned copper ions on the surface of MOFs. However, because of the steric hindrance of the heteroaromatic ring of 2VP and the relatively weak affinity of methacrylates towards Cu, the tacticity of their corresponding polymers was not improved.
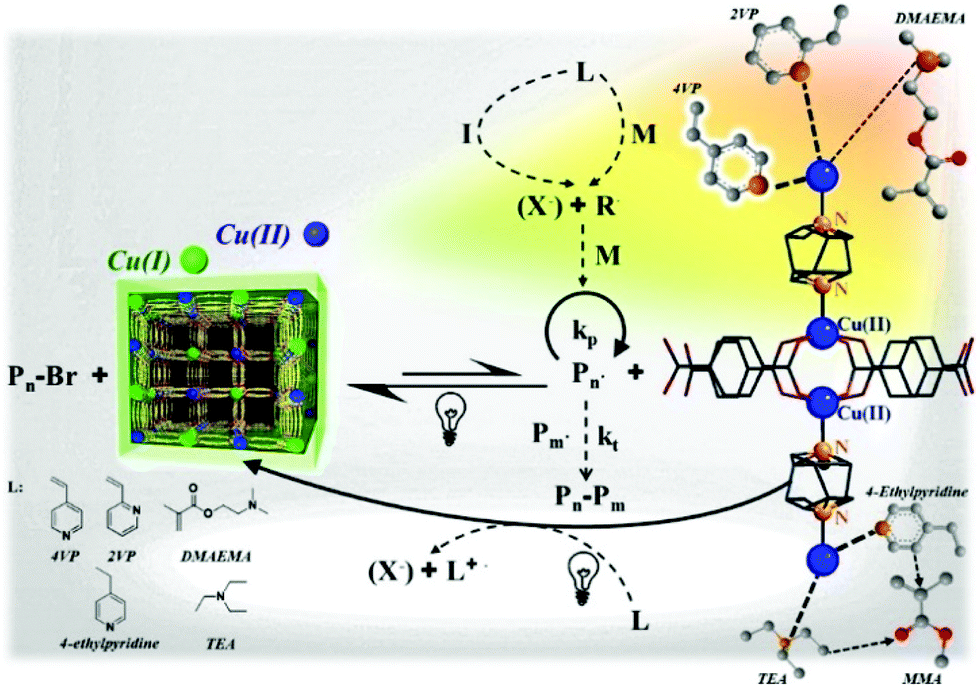 | ||
| Fig. 12 Proposed mechanism of ATRP polymerization catalyzed by the Cu(II) MOF and the coordination structure of monomers and copper. Reproduced from ref. 125 with permission from the American Chemical Society, copyright 2017. | ||
Perhaps, it is noteworthy that MOFs as HP-RDRP photocatalysts have been demonstrated to achieve some degree of tacticity control through specific interactions between coordinating polymers and MOF metal ions. However, current problems associated with MOFs as photocatalysts in HP-RDRP are relatively complex MOF synthesis, low polymerization rate and low monomer conversion. The ability to control molecular weight distributions also needs to be improved.
6. Carbon-based materials
Carbon-based materials have excellent electronic and optical properties and have been well studied in photocatalysis. For instance, graphitic carbon nitride (g-C3N4) with a bandgap of 2.7 eV was first reported by Antonietti et al. as a photocatalyst in water splitting.126 Subsequently, g-C3N4 has been employed in free radical polymerization,55 Pickering emulsion polymerization45,127 and hydrogel synthesis.128–131 Herein, we focus on recent advances of its applications in HP-RDRP (Table 4).| Photocatalyst | Reaction | Size | Absorption peaks | Light source | Monomer | Ref. |
|---|---|---|---|---|---|---|
| a CDs doped by the S element. | ||||||
| mpg-C3N4 | ATRP | 325, 375 nm | 350 nm, sunlight | MMA, MA | 55 | |
| g-C3N4 | RAFT | 370 nm | UV light | MA | 36 | |
| TCA-g-C3N4 | RAFT | 285 nm | 465 nm | MMA | 37 | |
| CD(sodium alginate, EDA) | ATRP | <10 nm | <300 nm | 405 nm | MMA | 134 |
| CD(S)a | RAFT | 6 nm | 270 nm | 465 nm | MMA, MA | 133 |
The first uses of g-C3N4 as photocatalysts in ATRP and RAFT polymerization were reported by Yagci55 and Qiao,36 respectively. Yagci and coworkers reported ATRP of MMA, MA and St catalyzed by mesoporous graphitic carbon nitride under 350 nm light irradiation at an intensity of 25 mW cm−2. PMMA with a monomer conversion of ∼50% and a low dispersity (Đ < 1.2) was obtained in 3 h in the presence of 50 ppm g-CN loading.
In Qiao's work, g-C3N4 was used to catalyze RAFT polymerization of MA, BA and DMA using UV light at an intensity of 3.5 mW cm−2 (Fig. 13). Trithiocarbonates (TTCs) were used as CTAs and triethanolamine (TEOA) served as the electron donor and deoxygenation agent. TTCs were considered to have the ability to facilitate the electron transfer from tertiary amines (TAs) to oxygen132 and the propagation rate was higher with TTCs in comparison that with dithioesters. This was verified by the decrease of the induction period used to remove oxygen from 3 h to 30 min. PMA was obtained within 5 h with a dispersity lower than 1.2 at high conversion (>90%).
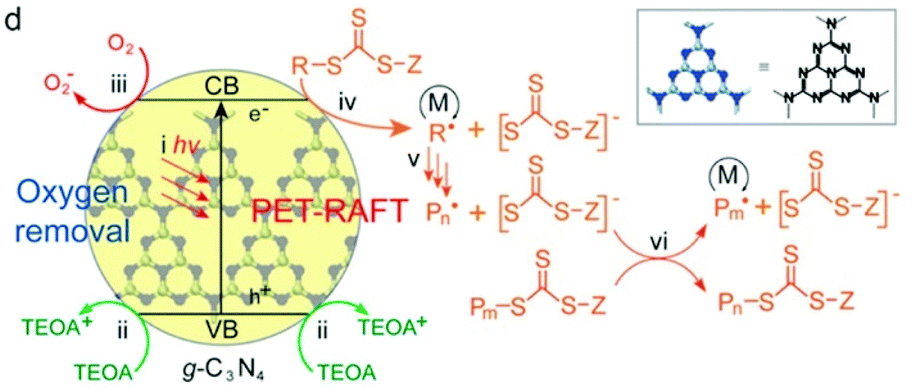 | ||
| Fig. 13 Proposed mechanism of PET-RAFT polymerization using g-C3N4 as the photocatalyst in the presence of TEOA. Reproduced from ref. 36 with permission from the American Chemical Society, copyright 2017. | ||
Besides its 3D form, g-C3N4 2D nanosheets were studied as photocatalysts. Recently, Chen and Matyjaszewski et al. reported the use of g-C3N4 as both heterogeneous and homogeneous catalysts for PET-RAFT polymerization of MMA in DMSO under 465 nm blue light.37
The potential of g-C3N4 as photocatalysts for HP-RDRP has been shown in the aforementioned works; however, the main absorption of g-C3N4 was limited to lower than 400 nm. Harnessing longer visible light for RDRP awaits to be seen using g-C3N4, which may potentially be realized through heteroatom doping or modification with conjugated molecules.
Besides g-C3N4, carbon dots (CDs) have also been investigated as photocatalysts in polymerization, mainly by Chen and Matyjaszewski,133 Strehmel,134 Gedanken,53 and Kang54et al. Attracting features of CDs include facile fabrication, low toxicity, high biocompatibility, high photo- and chemical stability135 and good solubility in water and polar organic solvents.
Chen and Matyjaszewski et al. reported the first PET-RAFT polymerization of MMA using CDs doped by nitrogen, phosphorus or sulfur as photocatalysts under blue (465 nm, 2 mW cm−2) and red (635 nm, 2 mW cm−2) light at room temperature.133 In comparison with the N-doped CDs, the P- and S-doped CDs showed lower photoluminescence intensity but higher catalytic efficiency, which was explained by a higher quantum yield of electron/energy transfer to the CTA to initiate the PET-RAFT process. Upon light irradiation, the excited CD reduced the CTA directly; meanwhile, the hole was sacrificed by TEA. Then the polymerization was initiated and the CTA can be deactivated by oxidized CDs during the chain growth (Fig. 14). The use of heteroatom-doped CDs as photocatalysts opens the window for the exploration of a broad range of carbon-based materials in HP-RDRP since doping is an effective strategy for tuning the optoelectronic properties of these materials.
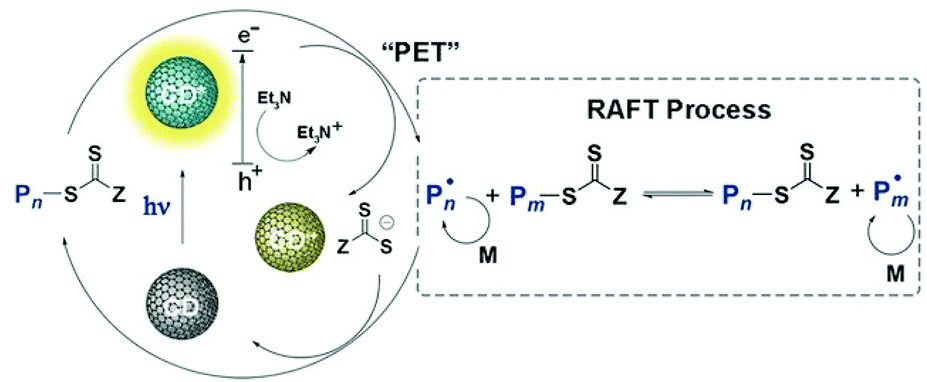 | ||
| Fig. 14 Proposed mechanism of PET-RAFT polymerization catalyzed by CD. Reproduced from ref. 133 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2018. | ||
Most recently, CDs were utilized by Strehmel and coworkers134 as photocatalysts for ATRP of MMA in DMSO using CuBr2 and ethyl α-bromophenylacetate as the copper source and initiator. The polymerization showed first-order kinetics but the dispersity was around 1.5, suggesting that some chain termination had occurred.
Despite the achievements, there remain some problems such as low monomer conversion, long reaction time, low polymerization efficiency under sunlight, and in some cases, high molecular weight distribution. Nevertheless, CDs are very promising photocatalysts for conducting RDRP in cases where high biocompatibility is required because water-soluble CDs with low toxicity are readily available.
7. Nanocomposites
Nanocomposites are composed of two or more components of distinct properties, which are combined together through covalent or noncovalent interactions. In many cases, nanocomposites can combine the useful properties or have synergistic effects of the individual components and show better performance than the individual materials when used alone. Nanocomposites combining nanoparticles and homogeneous photocatalysts such as 5-(4′-propargyloxyphenyl)-10,15,20-triphenyl-21H,23H-porphine zinc (ZnPTPP), ruthenium(II) bipyridine and Eosin Y have been investigated in HP-RDRP. Boyer and coworkers attached Eosin Y to silica nanoparticles (EY-SNPs), which was used as the recyclable photocatalyst in PET-RAFT polymerization using green light (515 nm, 3.6 mW cm−2) in water and DMSO.136 Cai and coworkers reported a series of nanocomposites for RAFT polymerization.137–143 For example, nanocomposites of SiO2@PGM-N3142 (azide-modified poly(glycidyl methacrylate-co-methacrylic acid)), Fe3O4@SiO2-N3143 and SiO2@HPGE-PFPPN141 (hyperbranched polyglycerol-poly[(9,9-bihexylfluorene)-alt-(5,5′-(2,2′-bipyridine))]-graft-poly(N-isopropylacrylamide)) were fabricated and organic photosensitisers were linked to them by the alkyne–azide click reaction or coordination interaction. The temporal control of these systems was shown by regulating light and temperature or magnetic field orthogonally with first-order kinetics and high livingness.
8. Self-assembled photocatalysts
The exploration of supramolecular chemistry to construct photocatalysts is intriguing as diverse supramolecular interactions can be employed to tune the structure and properties. The Zhang group, who pioneers the research of supramolecular polymerization,144,145 has reported several interesting supramolecular catalysts for use in alcohol oxidation,146 Fenton reaction147,148 and photoreduction of Cytochrome c.149,150 Inspired by this supramolecular strategy, An and coworkers adopted supramolecular catalysts cucurbit[7]uril@Zn(II) meso-tetra(4-naphthalylmethylpyridyl) porphyrin (CB[7]@ZnTPOR151) and perylene diimide/cucurbit[7]uril152 for efficient PET-RAFT polymerization. Significantly, the use of only 1 ppm of the latter resulted in successful synthesis of PDMA with an ultrahigh molecular weight. However, the employment of supramolecular chemistry in HPRDR is rare.Most recently, Qiao and coworkers reported an interesting NIR photocatalyst for heterogeneous PET-RAFT.153 The rodlike NIR photocatalyst, with a broad absorption spectrum from 300 to 950 nm, was self-assembled from carboxylated porphyrin (SA-TCPP) (Fig. 15).154 Polymerization of DMA was conducted in the presence of TTC, TEOA and SA-TCPP (5 mg). 90% monomer conversion was achieved in 90 min, 3 h or 4 h using white, blue or red light, respectively. Significantly, 53% monomer conversion was obtained in 66 h under 850 nm NIR light. To demonstrate the potential of this avenue to be used in vivo, the authors studied PET-RAFT polymerization of PEG methacrylate (PEGMA) in the presence of mammalian fibroblast cells in 96 well plates. The polymerization was conducted in cell culture media under 630 nm red light (4 mW cm−2) irradiation at 37 °C in open air. After 45 min reaction, PPEGMA (Đ = 1.52) with 11% conversion was obtained and the cells retained 46% viability. These preliminary results indicated the potential of long-wavelength-absorbing heterogeneous photocatalysts for in vivo applications, though the efficiency and cell viability need to be further improved.
 | ||
| Fig. 15 Self-assembly of rod-like SA-TCPP and its use in PET-RAFT polymerization. Reproduced from ref. 153 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2020. | ||
Self-assembly provides an attractive route to construct long-wavelength-absorbing photocatalysts without complex synthetic procedures. Since there exists a rich library of self-assembled photocatalysts in the literature,155 it is reasonable to expect that their use in HP-RDRP will quickly increase in the near future.
9. Conclusion and perspective
Heterogeneous photocatalysts, in the form of nanoparticles, microparticles, or even bulk (e.g., powder), have been demonstrated as useful alternatives to conventional molecular photocatalysts. These heterogeneous photocatalysts have been studied mainly in ATRP and RAFT with a varied degree of success. Excellent molecular weight control, high livingness and effective spatiotemporal modulation of polymerization kinetics have been achieved for some visible-light photocatalysts with appropriate redox potentials. Perhaps, the most valuable merits of heterogeneous photocatalysts, when compared to molecular photocatalysts, lie in their convenient preparation, readily tunable photoelectronic properties, and recyclability. As a result, this area has undergone rapid progress as evidenced by the breadth of materials, wavelength and polymerization conditions that have been investigated.Despite impressive progress in HP-RDRP, challenges still remain. Many modern technologies demand the use of advanced polymeric materials with exquisite control over composition (monomer scope), tacticity, molecular weight, molecular weight distribution, and architecture, and these requirements equally apply to HP-RDRP. Some heterogeneous photocatalysts are only compatible with a limited number of monomer families due to the interaction of heteroatoms with transition metals, though in few cases heteroatom transition metal interactions can be harnessed to impact tacticity. There is significant current interest in modulation of polymer dispersity in homogeneous solution polymerization but no such research activity in HP-RDRP has been witnessed. To date only modest molecular weights have been achieved; the ability to achieve ultrahigh molecular weight has yet to be realized via HP-RDRP. Undoubtedly, heterogeneous photocatalysts play a central role in the development of HP-RDRP. Ideally, heterogeneous photocatalysts should have a high absorption coefficient in the visible and NIR range, suitable redox potentials to reduce RDRP initiators and regulate the reversible deactivation process, and a balanced size to ensure both a high surface area and recyclability. Considering the diverse range of heterogeneous photocatalysts, RDRP initiators and monomers, it is a grand challenge to efficiently design and optimize such materials for HP-RDRP. It is expected that machine learning and artificial intelligence will be the emerging technology to aid the design of efficient photocatalysts for HP-RDRP. Flow photopolymerization has been successfully applied in homogeneous photocatalytic RDRP156 and its adoption to HP-RDRP can be naturally expected in the near future. Similarly, other emerging technologies that have been actively investigated will equally have an impact in HP-RDRP. For instance, polymerization-induced self-assembly (PISA) has been widely recognized as an efficient method for the preparation of block copolymer nanoobjects with controlled morphologies.157–160
Photopolymerization approaches have been demonstrated to facilitate the polymerization rate and especially for targeting worm-like morphology. Heterogeneous photocatalysts may behave differently in PISA as the nanoparticles may provide an additional interfacial stabilizing effect. HP-RDRP is a rapidly developing field and much effort is needed to develop a new catalyst, improve polymerization control and integrate this technology with emerging research directions (Fig. 16).
 | ||
| Fig. 16 The challenges for HP-RDRP. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21871175) and the Fundamental Research Funds for the Central Universities is acknowledged.Notes and references
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- S. Dadashi-Silab, M. Atilla Tasdelen and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2878–2888 CrossRef CAS.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, Macromol. Rapid Commun., 2017, 38, 1700143 CrossRef PubMed.
- Y.-N. Zhou, J.-J. Li, Y.-Y. Wu and Z.-H. Luo, Chem. Rev., 2020, 120, 2950–3048 CrossRef CAS PubMed.
- S. Li, G. Han and W. Zhang, Polym. Chem., 2020, 11, 1830–1844 RSC.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- D. Konkolewicz, A. J. D. Magenau, S. E. Averick, A. Simakova, H. He and K. Matyjaszewski, Macromolecules, 2012, 45, 4461–4468 CrossRef CAS.
- X.-h. Liu, J. Wang, F.-j. Zhang, S.-l. An, Y.-l. Ren, Y.-h. Yu, P. Chen and S. Xie, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4358–4364 CrossRef CAS.
- Z. Wang, X. Pan, L. Li, M. Fantin, J. Yan, Z. Wang, Z. Wang, H. Xia and K. Matyjaszewski, Macromolecules, 2017, 50, 7940–7948 CrossRef CAS.
- X. Pan, M. Fantin, F. Yuan and K. Matyjaszewski, Chem. Soc. Rev., 2018, 47, 5457–5490 RSC.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- M. Fantin, A. A. Isse, A. Venzo, A. Gennaro and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 7216–7219 CrossRef CAS PubMed.
- F. Lorandi, M. Fantin, S. Shanmugam, Y. Wang, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2019, 52, 1479–1488 CrossRef CAS.
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- F. Zhou, R. Li, X. Wang, S. Du and Z. An, Angew. Chem., Int. Ed., 2019, 58, 9479–9484 CrossRef CAS PubMed.
- R. Li and Z. An, Angew. Chem., Int. Ed., 2020, 59, 22258–22264 CrossRef CAS PubMed.
- N. V. Alfredo, N. E. Jalapa, S. L. Morales, A. D. Ryabov, R. Le Lagadec and L. Alexandrova, Macromolecules, 2012, 45, 8135–8146 CrossRef CAS.
- X. Pan, N. Malhotra, J. Zhang and K. Matyjaszewski, Macromolecules, 2015, 48, 6948–6954 CrossRef CAS.
- M. Ciftci, M. A. Tasdelen and Y. Yagci, Polym. Chem., 2014, 5, 600–606 RSC.
- Y. Zhu and E. Egap, Polym. Chem., 2020, 11, 1018–1024 RSC.
- B. Li, B. Yu and F. Zhou, Macromol. Rapid Commun., 2014, 35, 1287–1292 CrossRef CAS PubMed.
- E. Liang, M.-s. Liu, B. He and G.-X. Wang, Adv. Polym. Technol., 2018, 37, 2879–2884 CrossRef CAS.
- Q. Fu, Q. Ruan, T. G. McKenzie, A. Reyhani, J. Tang and G. G. Qiao, Macromolecules, 2017, 50, 7509–7516 CrossRef CAS.
- L. Zhang, G. Ye, X. Huo, S. Xu, J. Chen and K. Matyjaszewski, ACS Omega, 2019, 4, 16247–16255 CrossRef CAS PubMed.
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
- X. Liu, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2016, 7, 689–700 RSC.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- Y. Guillaneuf, D. Bertin, D. Gigmes, D.-L. Versace, J. Lalevée and J.-P. Fouassier, Macromolecules, 2010, 43, 2204–2212 CrossRef CAS.
- J. Morris, S. Telitel, K. E. Fairfull-Smith, S. E. Bottle, J. Lalevée, J.-L. Clément, Y. Guillaneuf and D. Gigmes, Polym. Chem., 2015, 6, 754–763 RSC.
- M. Liu, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nature, 2014, 517, 68–72 CrossRef PubMed.
- Y. S. Kim, M. Liu, Y. Ishida, Y. Ebina, M. Osada, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nat. Mater., 2015, 14, 1002–1007 CrossRef CAS.
- Q. Cao, T. Heil, B. Kumru, M. Antonietti and B. V. K. J. Schmidt, Polym. Chem., 2019, 10, 5315–5323 RSC.
- J. C. Kuriacose and M. C. Markham, J. Phys. Chem., 1961, 65, 2232–2236 CrossRef CAS.
- X. Wang, X. Song, M. Lin, H. Wang, Y. Zhao, W. Zhong and Q. Du, Polymer, 2007, 48, 5834–5838 CrossRef CAS.
- K. Hakobyan, T. Gegenhuber, C. S. P. McErlean and M. Müllner, Angew. Chem., Int. Ed., 2019, 58, 1828–1832 CrossRef CAS.
- A. Barichard, T. Galstian and Y. Israeli, Phys. Chem. Chem. Phys., 2012, 14, 8208–8216 RSC.
- K. Chen, X. Deng, G. Dodekatos and H. Tuysuz, J. Am. Chem. Soc., 2017, 139, 12267–12273 CrossRef CAS PubMed.
- J. Wang, M. Rivero, A. Muñoz Bonilla, J. Sanchez-Marcos, W. Xue, G. Chen, W. Zhang and X. Zhu, ACS Macro Lett., 2016, 5, 1278–1282 CrossRef CAS.
- S. Beyazit, S. Ambrosini, N. Marchyk, E. Palo, V. Kale, T. Soukka, B. Tse Sum Bui and K. Haupt, Angew. Chem., Int. Ed., 2014, 53, 8919–8923 CrossRef CAS PubMed.
- M. Moorthy, V. B. Kumar, Z. e. Porat and A. Gedanken, New J. Chem., 2018, 42, 535–540 RSC.
- H. Huang, S. Yang, Y. Liu, Y. Yang, H. Li, J. A. McLeod, G. Ding, J. Huang and Z. Kang, ACS Appl. Bio Mater., 2019, 2, 5144–5153 CrossRef CAS.
- B. Kiskan, J. Zhang, X. Wang, M. Antonietti and Y. Yagci, ACS Macro Lett., 2012, 1, 546–549 CrossRef CAS.
- C. Boyer, L. Zhang, X. Shi, Z. Zhang, R. P. Kuchel, R. Namivandi-Zangeneh, N. Corrigan, K. Jung and K. Liang, Angew. Chem., Int. Ed., 2021, 60, 5489–5496 CrossRef PubMed.
- K. P. McClelland, T. D. Clemons, S. I. Stupp and E. A. Weiss, ACS Macro Lett., 2019, 9, 7–13 CrossRef.
- S. Chen and L.-W. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- C. Liu, L. Wang, Y. Tang, S. Luo, Y. Liu, S. Zhang, Y. Zeng and Y. Xu, Appl. Catal., B, 2015, 164, 1–9 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- Y. Huang, Y. Zhu and E. Egap, ACS Macro Lett., 2018, 7, 184–189 CrossRef CAS.
- D. Zhang, J. Yang, S. Bao, Q. Wu and Q. Wang, Sci. Rep., 2013, 3, 1399 CrossRef.
- X. Zheng, D. Wu, T. Su, S. Bao, C. Liao and Q. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 19840–19849 CrossRef CAS PubMed.
- M. Dule, M. Biswas, Y. Biswas and T. K. Mandal, Polymer, 2017, 133, 223–231 CrossRef CAS.
- C. Liao, Q. Wu, T. Su, D. Zhang, Q. Wu and Q. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 1356–1360 CrossRef CAS PubMed.
- S. Dadashi-Silab, M. Atilla Tasdelen, A. Mohamed Asiri, S. Bahadar Khan and Y. Yagci, Macromol. Rapid Commun., 2014, 35, 454–459 CrossRef CAS PubMed.
- K. Kubo, N. Tsukimura, F. Iwasa, T. Ueno, L. Saruwatari, H. Aita, W.-A. Chiou and T. Ogawa, Biomaterials, 2009, 30, 5319–5329 CrossRef CAS PubMed.
- Z. Fei Yin, L. Wu, H. Gui Yang and Y. Hua Su, Phys. Chem. Chem. Phys., 2013, 15, 4844–4858 RSC.
- M. Kulkarni, A. Mazare, E. Gongadze, Š. Perutkova, V. Kralj-Iglič, I. Milošev, P. Schmuki, A. Iglič and M. Mozetič, Nanotechnology, 2015, 26, 062002 CrossRef CAS PubMed.
- K. S. Brammer, S. Oh, C. J. Cobb, L. M. Bjursten, H. v. d. Heyde and S. Jin, Acta Biomater., 2009, 5, 3215–3223 CrossRef CAS PubMed.
- J. Yan, B. Li, F. Zhou and W. Liu, ACS Macro Lett., 2013, 2, 592–596 CrossRef CAS.
- L.-c. Liu, M. Lu, Z.-H. Hou, G.-X. Wang, C.-A. Yang, E.-X. Liang, H. Wu, X.-L. Li and Y.-X. Xu, J. Appl. Polym. Sci., 2015, 132, 42389 Search PubMed.
- B.-F. Cheng, L.-H. Wang and Y.-Z. You, Macromol. Res., 2016, 24, 811–815 CrossRef CAS.
- J. Hu, Y. Lu, X.-L. Liu, C. Janiak, W. Geng, S.-M. Wu, X.-F. Zhao, L.-Y. Wang, G. Tian, Y. Zhang, B.-L. Su and X.-Y. Yang, CCS Chem., 2020, 2, 1573–1581 CrossRef CAS.
- T. Shishido, T. Miyatake, K. Teramura, Y. Hitomi, H. Yamashita and T. Tanaka, J. Phys. Chem. C, 2009, 113, 18713–18718 CrossRef CAS.
- Y. Cao, Y. Xu, J. Zhang, D. Yang and J. Liu, Polymer, 2015, 61, 198–203 CrossRef CAS.
- J. Zhang, A. Li, H. Liu, D. Yang and J. Liu, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2715–2724 CrossRef CAS.
- A. Bansal, A. Kumar, P. Kumar, S. Bojja, A. K. Chatterjee, S. S. Ray and S. L. Jain, RSC Adv., 2015, 5, 21189–21196 RSC.
- K. Hakobyan, C. S. P. McErlean and M. Müllner, Macromolecules, 2020, 53, 10357–10365 CrossRef CAS.
- J. Wang, Y.-X. Feng, M. Zhang, C. Zhang, M. Li, S.-J. Li, W. Zhang and T.-B. Lu, CCS Chem., 2020, 2, 81–88 CrossRef CAS.
- A. J. Hoffman, G. Mills, H. Yee and M. R. Hoffmann, J. Phys. Chem., 1992, 96, 5546–5552 CrossRef CAS.
- L. Verbitsky, N. Waiskopf, S. Magdassi and U. Banin, Nanoscale, 2019, 11, 11209–11216 RSC.
- N. C. Strandwitz, A. Khan, S. W. Boettcher, A. A. Mikhailovsky, C. J. Hawker, T.-Q. Nguyen and G. D. Stucky, J. Am. Chem. Soc., 2008, 130, 8280–8288 CrossRef CAS PubMed.
- T. Nakashima, M. Sakashita, Y. Nonoguchi and T. Kawai, Macromolecules, 2007, 40, 6540–6544 CrossRef CAS.
- E. Buz, F. Morlet-Savary, J. Lalevée and H. Y. Acar, Macromol. Chem. Phys., 2018, 219, 1700365 CrossRef.
- Y. Zhu, Y. Liu, K. A. Miller, H. Zhu and E. Egap, ACS Macro Lett., 2020, 9, 725–730 CrossRef CAS.
- Q. Wang, L. Hu, Z. Cui, P. Fu, M. Liu, X. Qiao and X. Pang, ACS Appl. Mater. Interfaces, 2020, 12, 42161–42168 CrossRef CAS PubMed.
- Y. Liang, H. Ma, W. Zhang, Z. Cui, P. Fu, M. Liu, X. Qiao and X. Pang, Polym. Chem., 2020, 11, 4961–4967 RSC.
- J. Maes, L. Balcaen, E. Drijvers, Q. Zhao, J. De Roo, A. Vantomme, F. Vanhaecke, P. Geiregat and Z. Hens, J. Phys. Chem. Lett., 2018, 9, 3093–3097 CrossRef CAS PubMed.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733–738 CrossRef CAS PubMed.
- J. Chen, K. Žídek, P. Chábera, D. Liu, P. Cheng, L. Nuuttila, M. J. Al-Marri, H. Lehtivuori, M. E. Messing, K. Han, K. Zheng and T. Pullerits, J. Phys. Chem. Lett., 2017, 8, 2316–2321 CrossRef CAS PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 CrossRef CAS PubMed.
- C. Kütahya, C. Schmitz, V. Strehmel, Y. Yagci and B. Strehmel, Angew. Chem., Int. Ed., 2018, 57, 7898–7902 CrossRef PubMed.
- J. Jiang, G. Ye, F. Lorandi, Z. Liu, Y. Liu, T. Hu, J. Chen, Y. Lu and K. Matyjaszewski, Angew. Chem., 2019, 131, 12224–12229 CrossRef.
- D. J. Martin, N. Umezawa, X. Chen, J. Ye and J. Tang, Energy Environ. Sci., 2013, 6, 3380–3386 RSC.
- J. A. Capobianco, F. Vetrone, T. D'Alesio, G. Tessari, A. Speghini and M. Bettinelli, Phys. Chem. Chem. Phys., 2000, 2, 3203–3207 RSC.
- X. Li, F. Zhang and D. Zhao, Chem. Soc. Rev., 2015, 44, 1346–1378 RSC.
- C.-J. Carling, J.-C. Boyer and N. R. Branda, J. Am. Chem. Soc., 2009, 131, 10838–10839 CrossRef CAS PubMed.
- W. Zhang, B. Peng, F. Tian, W. Qin and X. Qian, Anal. Chem., 2014, 86, 482–489 CrossRef CAS PubMed.
- Z. Xie, X. Deng, B. Liu, S. Huang, P. Ma, Z. Hou, Z. Cheng, J. Lin and S. Luan, ACS Appl. Mater. Interfaces, 2017, 9, 30414–30425 CrossRef CAS PubMed.
- K. Soga, A. Okada and M. Yamada, J. Photopolym. Sci. Technol., 2006, 19, 45–48 CrossRef CAS.
- C. Ding, J. Wang, W. Zhang, X. Pan, Z. Zhang, W. Zhang, J. Zhu and X. Zhu, Polym. Chem., 2016, 7, 7370–7374 RSC.
- L. Hu, Q. Hao, L. Wang, Z. Cui, P. Fu, M. Liu, X. Qiao and X. Pang, Polym. Chem., 2021, 12, 545–553 RSC.
- W. Zhang, J. He, C. Lv, Q. Wang, X. Pang, K. Matyjaszewski and X. Pan, Macromolecules, 2020, 53, 4678–4684 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés i Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef CAS PubMed.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- S. Tao and D. Jiang, CCS Chem., 2021, 3, 2003–2024 CrossRef CAS.
- M. Giménez-Marqués, T. Hidalgo, C. Serre and P. Horcajada, Coord. Chem. Rev., 2016, 307, 342–360 CrossRef.
- B. Schmidt, Macromol. Rapid Commun., 2020, 41, e1900333 CrossRef PubMed.
- A. Reyhani, O. Mazaheri, M. S. Alivand, K. A. Mumford and G. Qiao, Polym. Chem., 2020, 11, 2838–2846 RSC.
- A. Reyhani, H. Ranji-Burachaloo, T. G. McKenzie, Q. Fu and G. G. Qiao, Macromolecules, 2019, 52, 3278–3287 CrossRef CAS.
- Q. Fu, H. Ranji-Burachaloo, M. Liu, T. G. McKenzie, S. Tan, A. Reyhani, M. D. Nothling, D. E. Dunstan and G. G. Qiao, Polym. Chem., 2018, 9, 4448–4454 RSC.
- H.-C. Lee, J. Hwang, U. Schilde, M. Antonietti, K. Matyjaszewski and B. V. K. J. Schmidt, Chem. Mater., 2018, 30, 2983–2994 CrossRef CAS.
- J. Hwang, H.-C. Lee, M. Antonietti and B. V. K. J. Schmidt, Polym. Chem., 2017, 8, 6204–6208 RSC.
- H. Xing, D. Chen, X. Li, Y. Liu, C. Wang and Z. Su, RSC Adv., 2016, 6, 66444–66450 RSC.
- X. Li, D. Chen, Y. Liu, Z. Yu, Q. Xia, H. Xing and W. Sun, CrystEngComm, 2016, 18, 3696–3702 RSC.
- Y. Liu, D. Chen, X. Li, Z. Yu, Q. Xia, D. Liang and H. Xing, Green Chem., 2016, 18, 1475–1481 RSC.
- Y. Zhang, D. Chen, Z. Guo, Z. Wei, X. Zhang and H. Xing, New J. Chem., 2020, 44, 5235–5242 RSC.
- H. L. Nguyen, T. T. Vu, D. Le, T. L. H. Doan, V. Q. Nguyen and N. T. S. Phan, ACS Catal., 2016, 7, 338–342 CrossRef.
- H. L. Nguyen, F. Gandara, H. Furukawa, T. L. Doan, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 4330–4333 CrossRef CAS PubMed.
- H.-C. Lee, M. Fantin, M. Antonietti, K. Matyjaszewski and B. V. K. J. Schmidt, Chem. Mater., 2017, 29, 9445–9455 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Q. Cao, Q. Cui, Y. Yang, J. Xu, C. Han and L. Li, Chemistry, 2018, 24, 2286–2291 CrossRef CAS PubMed.
- B. Kumru, M. Shalom, M. Antonietti and B. V. K. J. Schmidt, Macromolecules, 2017, 50, 1862–1869 CrossRef CAS.
- B. Kumru, V. Molinari, M. Shalom, M. Antonietti and B. Schmidt, Soft Matter, 2018, 14, 2655–2664 RSC.
- J. Liu, T. An, Z. Chen, Z. Wang, H. Zhou, T. Fan, D. Zhang and M. Antonietti, J. Mater. Chem. A, 2017, 5, 8933–8938 RSC.
- B. Kumru, V. Molinari, R. Dunnebacke, K. G. Blank and B. Schmidt, Macromol. Rapid Commun., 2019, 40, e1800712 Search PubMed.
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Polym. Chem., 2017, 8, 1519–1526 RSC.
- J. Jiang, G. Ye, Z. Wang, Y. Lu, J. Chen and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 12037–12042 CrossRef CAS.
- C. Kütahya, P. Wang, S. Li, S. Liu, J. Li, Z. Chen and B. Strehmel, Angew. Chem., Int. Ed., 2020, 59, 3166–3171 CrossRef PubMed.
- H. Li, S. Ye, J. Guo, H. Wang, W. Yan, J. Song and J. Qu, Nano Res., 2019, 12, 3075–3084 CrossRef CAS.
- S. Shanmugam, S. Xu, N. N. M. Adnan and C. Boyer, Macromolecules, 2018, 51, 779–790 CrossRef CAS.
- Y. Zhao, S. Shao, J. Xia, Y. Huang, Y. C. Zhang, X. Li and T. Cai, J. Mater. Chem. A, 2020, 8, 9825–9831 RSC.
- X. Li, Y. C. Zhang, Y. Zhao, H. P. Zhao, B. Zhang and T. Cai, Macromolecules, 2020, 53, 1550–1556 CrossRef CAS.
- X. Li, S. Ye, Y. C. Zhang, H. P. Zhao, Y. Huang, B. Zhang and T. Cai, Nanoscale, 2020, 12, 7595–7603 RSC.
- X. Li, S. Ye, Y. Huang, J. L. Li and T. Cai, J. Mater. Chem. A, 2019, 7, 6173–6179 RSC.
- Y. Huang, X. R. Zhang, S. Ye, J. L. Li, X. Li and T. Cai, Nanoscale, 2019, 11, 13502–13510 RSC.
- Y. Huang, X. Li, J. Le Li, B. Zhang and T. Cai, Macromolecules, 2018, 51, 7974–7982 CrossRef CAS.
- X. Li, J. L. Li, W. G. Huang, X. Z. Zhang, B. Zhang and T. Cai, Nanoscale, 2018, 10, 19254–19261 RSC.
- Z. Yin, G. Song, Y. Jiao, P. Zheng, J.-F. Xu and X. Zhang, CCS Chem., 2019, 1, 335–342 CrossRef CAS.
- H. Wang, Y.-Q. Yan, Y. Yi, Z.-Y. Wei, H. Chen, J.-F. Xu, H. Wang, Y. Zhao and X. Zhang, CCS Chem., 2020, 2, 739–748 CrossRef CAS.
- Y. Jiao, B. Tang, Y. Zhang, J.-F. Xu, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6077–6081 CrossRef CAS PubMed.
- B. Tang, W.-L. Li, Y. Jiao, J.-B. Lu, J.-F. Xu, Z. Wang, J. Li and X. Zhang, Chem. Sci., 2018, 9, 5015–5020 RSC.
- Y. Jiao, W.-L. Li, J.-F. Xu, G. Wang, J. Li, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 8933–8937 CrossRef CAS.
- Y. Jiao, J.-F. Xu, Z. Wang and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 22635–22640 CrossRef CAS PubMed.
- Y. Yang, H. Hu, Y. Guo, A. Xia, J.-F. Xu and X. Zhang, Macromol. Rapid Commun., 2020, 41, 2000080 CrossRef CAS.
- L. Shen, Q. Lu, A. Zhu, X. Lv and Z. An, ACS Macro Lett., 2017, 6, 625–631 CrossRef CAS.
- Y. Yang and Z. An, Polym. Chem., 2019, 10, 2801–2811 RSC.
- S. Allison-Logan, Q. Fu, Y. Sun, M. Liu, J. Xie, J. Tang and G. G. Qiao, Angew. Chem., Int. Ed., 2020, 59, 21392–21396 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, X. Chen, H. Zhang and J. Wang, Adv. Mater., 2019, 31, 1806626 CrossRef PubMed.
- Q. Zuo, K. Feng, J. Zhong, Y. Mai and Y. Zhou, CCS Chemistry, 2020, 2, 1963–1971 CrossRef.
- Z.-R. Zhong, Y.-N. Chen, Y. Zhou and M. Chen, Chin. J. Polym. Sci., 2021 DOI:10.1007/s10118-021-2529-8.
- J. Cornel Erik, J. Jiang, S. Chen and J. Du, CCS Chem., 2020, 2, 2104–2125 Search PubMed.
- F. Lv, Z. An and P. Wu, CCS Chem., 2020, 2, 2211–2222 CrossRef.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |