
Nanomaterial catalysts for organic photoredox catalysis-mechanistic perspective
Qiushi
Hu
 ,
Xuemeng
Yu
,
Shaokuan
Gong
and
Xihan
Chen
*
,
Xuemeng
Yu
,
Shaokuan
Gong
and
Xihan
Chen
*
Department of Mechanical and Energy Engineering, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China. E-mail: chenxh@sustech.edu.cn
First published on 15th October 2021
Abstract
Solar energy conversions play a vital role in the renewable energy industry. In recent years, photoredox organic transformations have been explored as an alternative way to use solar energy. Catalysts for such photocatalytic systems have evolved from homogeneous metal complexes to heterogeneous nanomaterials over the past few decades. Herein, three important carrier transfer mechanisms are presented, including charge transfer, energy transfer and hot carrier transfer. Several models established by researchers to understand the catalytic reaction mechanisms are also illustrated, which promote the reaction system design based on theoretical studies. New strategies are introduced in order to enhance catalytic efficiency for future prospects.
 Qiushi Hu | Mr Hu Qiushi is currently a PhD student in the SUSTech-CityU (Southern University of Science and Technology-City University of Hong Kong) joint PhD program under the supervision of professor Xihan Chen. He acquired his bachelor's degree of Science at Lanzhou University in 2018. Later, he received his master's degree in Materials Engineering at University of Southern California (USC) in 2020. His major focus is ultrafast spectroscopy in solar energy conversion system. |
 Xuemeng Yu | Mr Xuemeng Yu is currently a master's student at the Department of Mechanical and Energy Engineering, Southern University of Science and Technology. He completed his undergraduate degree in energy and power engineering from the Northeastern University (NEU) in 2021. Now he is working on mechanistic understanding of spin-dynamics and photocatalysis dynamics with ultrafast spectroscopy under the supervision of professor Xihan Chen. |
 Shaokuan Gong | Mr Shaokuan Gong is currently studying for a master's degree in Electronic Science and technology at SUSTech (South University of science and technology). He graduated from NCEPU (North China Electric Power University) with a bachelor's degree in engineering, majoring in new energy science and Engineering (wind power generation). Now he is working on studying solar cell materials with ultrafast spectroscopy under the supervision of professor Xihan Chen. |
 Xihan Chen | Dr Xihan Chen is an assistant professor at the Department of Mechanical and Energy Engineering, Southern University of Science and Technology. He received his bachelor's degree in Chemistry from the Hong Kong University of Science and Technology (HKUST) in 2012 and PhD degree in Chemistry at University of California at Berkeley (UCB) in 2017. His research focuses on developing ultrafast spectroscopy for energy conversions systems including solar to electricity conversion, solar to fuel conversion. He has published over 40 peer-reviewed journal articles in international journals including Science, Nature Catalysis, ACS Energy Letters, Journal of the American Chemical Society, etc. |
1. Introduction
Solar energy is considered one of the most promising renewable energy sources. The conversion of solar energy to heat, electricity and chemical fuels is the most promising technology.1–6 Solar to chemical conversion is especially important as it can create chemicals that can be stored and used later. In general, photoexcitation creates electron–hole pairs that can then subsequently transfer (electron/hole/energy) to reactants to initiate a reaction. A subsequent hole/electron transfer returns the catalyst to its original state, completing the catalytic cycle (if initiated by the charge transfer mechanism). The development of new efficient photocatalytic systems requires knowledge from many areas of chemistry and physics, including design, synthesis, characterizations, and mechanistic understanding, so as to direct the solar energy into desired products with little loss. Apart from most desired water splitting7–10 and CO2 reduction,11 in recent years, the conversion of solar energy to organic chemicals has been gaining attention. Pioneered by Macmillan,12 who utilized organometallic complex Ru(bpy)32+ to trigger the α-alkylation of aldehydes, a crucial reaction in organic synthesis, the field of organic photoredox catalysis grew at a very fast pace. Following the work of Macmillan, Yoon13 also applied Ru(bpy)32+ as photocatalysts and proposed a chain reaction mechanism for the photoredox transformation with over 1800% quantum yield. Yoon14,15 also reported [4 + 2] Diels–Alder cycloaddition reaction regarding catalyst overloading and slow reaction speed for metal complex Ru(bpy)32+ and conducted ligand modifications to produce robust catalysts Ru(bpy)3(PF6)2 and Ru(bpy)3(BArF)2 as substitutes. An improved quantum yield was shown within hours. However, researchers soon ran into obstacles when bringing metal complexes into more important and specific photoredox reactions, such as the functionalization of stronger C–H bonds and radical transmetalation with diastereoselectivity. Alternative choices such as precious metal, dual-catalyst and supramolecular host complexes were not ideal since the capital cost was high. Besides, overloading and poor selectivity in photoredox reactions were also reported for several kinds of metal complexes. Therefore, researchers have been looking for low-cost and high yield photocatalysts to overcome these difficulties and in this perspective, we will be discussing the recent advancements in finding and understanding photoredox catalysis with cheap catalysts. We will first introduce the use of nanocrystals as photoredox catalysts, then we will discuss the initial steps of photo redox catalysis, including charge transfer, energy transfer and hot carrier transfer. Last but not least, we will discuss full reaction mechanisms and future perspectives on catalyst design.2. Nanocrystals for photoredox catalysis
In recent years, with the emergence of functional nanocrystals, the use of colloidal nanomaterials (including quantum dots, nanocrystals and nanoplatelets, etc.) in photoredox reactions provided solutions. Colloidal nanocrystals are solution-grown, nanometre-sized, inorganic particles that are stabilized by a layer of surfactants attached to their surface. The inorganic cores possess useful properties that are controlled by their composition, size and shape, and the surfactant coating ensures that these structures are easy to fabricate and process further into more complex structures.16 Upon light irradiation, an electron of the nanocatalyst is excited to the conduction band (CB), leaving a positively charged hole in the valence band (VB). These photogenerated charge carriers then migrate to the nanocrystal surface and catalyze redox reactions (Fig. 1(a)). For example, early development by Weix et al.17 described a carbon–carbon bond formation reaction catalyzed by CdS quantum dots (QDs). It was also found that CdSe QDs were capable of replacing precious metals such as Ir/Ru with equal photoredox performances (Fig. 1(b)). Besides, because of the excellent visible light absorption ability, the band edge position of CdS or CdSe,18 QDs could be easily tuned through the modification of the particle size and surface functionalization, which was suitable for a variety of organic transformations. Weiss et al.19 studied a C–C coupling reaction between 1-phenylpyrrolidine (PhPyr) and phenyl trans-styryl sulfone using CdS catalysts and discovered that the disordering of the ligand shell could increase the initial reaction rate by a factor of 2.3 by facilitating the hole-transfer step. Weiss et al.20 also explored a [2 + 2] cycloaddition reaction catalyzed by CdSe quantum dots whose catalytic efficiency is comparable to that of a traditional metal complex (Fig. 1(c)). In another example, Feng et al.21 synthesized CF2-containing azaheterocycles with CdS QDs through a single electron transfer mechanism. The pharmaceutical uses of the product could be seen in the respiratory system curing and cancer treatment. Further developments in colloidal QDs, such as PbS,22,23 ZnS24–26 and core–shell structured bicomponent catalysts27,28 resulted in several valuable organic catalytic transformations, such as dehalogenation, reductive deprotection, and polymerization.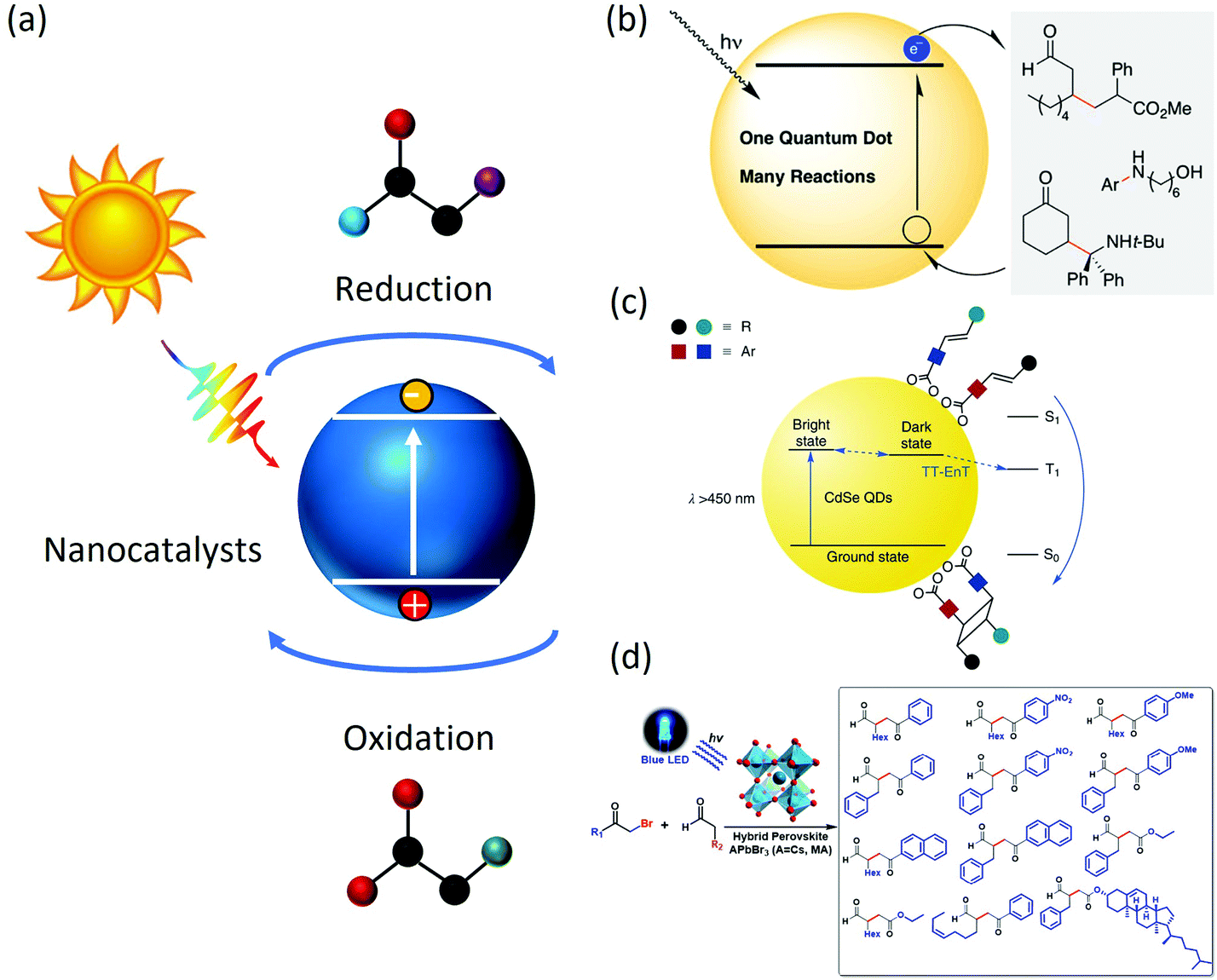 | ||
| Fig. 1 (a) General scheme for organic photocatalysis. Light is absorbed by photocatalysts which generate electron and hole pairs to complete organic transformation; (b) example photoredox C–C coupling reaction catalyzed by CdSe QDs. The scheme was adapted from ref. 17 with permission from American Chemical Society; (c) example photoredox [2 + 2] cycloaddition reaction catalyzed by CdSe QDs. The scheme was adapted from ref. 20 with permission from Springer Nature (d) example photoredox C–C coupling reaction catalyzed by CsPbBr3 nanocrystals. The scheme was adapted from ref. 31 with permission from American Chemical Society. | ||
Apart from commonly used CdSe-based chalcogenide QDs, recent developments in perovskite nanocrystals also lead to several interesting applications in photocatalysis.29,30 Perovskite nanocrystals have superior light absorption ability and desirable band gap, which is suitable for photo redox reactions. For example, Zhu et al.31 synthesized CsPbBr3 nanocrystals and applied them to perform an α-alkylation of aldehyde, a useful C–C bond formation reaction (Fig. 1(d)), resulting in a product yield near unity and an optimum selectivity over 95%. Zhu et al.32 also studied C–C formation, N-heterocyclization and C–O cross-coupling reactions using CsPbBr3 photocatalysts and found a much lower catalyst loading (0.1–0.5 mmol%) is needed compared with metal complexes, which could greatly reduce the cost for such organic synthesis. Chen Qi33 investigated the surface structure of MAPbI3 and its effect on photocatalytic performances and suggested that the positively charged Pb2+ defects central sites played a key role in the catalytic reaction. The surface termination mechanism of MAPbI3 was found to be different under different moisture levels, where a high moisture-level made surface MA+ more soluble and leaves the surface, resulting in an n-type featured surface, and low moisture level confines the MA+ to the surface, resulting in a p-type featured surface. The p-type and n-type surfaces would function as efficient hole/electron donors, respectively. The formation of C–C, C–N, C–O, etc.28,32,34–40 are later developed, among which the production of 2,5-diformylfuran (DFF), an important reagent that could be utilized in organic, pharmaceutical and biomass synthesis, was found feasible through the oxidation reaction from hydroxymethylfuran (HMF) catalyzed by MAPbBr3. A 90% selectivity and near 100% conversion rate was also reported. In addition, the oxidation of stronger C–H bonds was also made possible by a Cs3Bi2Br9-assisted co-catalyst under visible light irradiation.41,42
Ou Chen43 and Beard44 made surface treatments to modify the catalytic potential of perovskite nanocrystal catalysts. It was proven by Wu that the exchange of the nanocrystal ligand shell will not affect the core section electron reactivity and mobility.45 Stereoselective oxidative C–C coupling with an aryl substrate catalyzed by CsPbBr3 was achieved assisted by a conjugated π system.43 The zwitterionic ligand capped CsPbBr3 exhibited outstanding stability and reaction rate. Evidently, the carbon chain length, anchoring groups and coverage ligand density played key roles in catalytic performance. A systematic study of ligand shell exchange is still lacking, which requires further exploration as an important surface modification technique.46
3. Initial steps of photo redox catalysis-charge transfer, energy transfer and hot carrier transfer
The understanding of charge-carrier dynamics and reaction kinetics provides key insights in designing and synthesizing new systems.47–49 The nature of organic photoredox catalysis is the transfer of charge in a reaction system empowered by photon absorption. The photo-generated charge can then be transferred over to the reactant to initiate photoredox reactions, generating transient reaction intermediates. The reaction intermediates then form the desired new chemical bonds. In these processes, carrier transfer plays a key role in photoredox catalysis since it competes with the recombination process, which wastes the absorbed photon energy into light/heat emission other than completing reaction cycles. Besides the bandgap (Eg) structure, there are several elements that could affect the carrier transfer efficiency, including binding energy (Eb), the interaction between the catalyst and reactant molecules (binding geometry), trap state density, which bends the conduction band down and increases the possibility of recombination and the rate of electron/hole scattering, which could influence the charge transport inside the nanocrystals. In general, carrier transfer from nanocrystals to reactants includes charge transfer,50 energy transfer51 or hot carrier transfer.523.1. Charge transfer
The charge transfer (CT) process, which is commonly seen in photoredox reactions, refers to the transfer of electrons or holes to the reaction system separately after being activated (photon absorbed across the bandgap). For this type of carrier transfer, electrons and holes will transfer to different reactants to initiate the reaction, as shown in Fig. 2(b). For CT to happen efficiently, the Highest Occupied Molecular Orbitals (HOMO) of the hole acceptor needs to be above the valence band (VB) of nanocrystal or the Lowest Unoccupied Molecular Orbitals (LUMO) of the electron acceptor needs to be below Conduction Band (CB). In other words, a driving force will be beneficial for the CT to happen efficiently. As CT generally is the first step of photocatalysis, the rate of CT needs to be fast compared with the intrinsic recombination rate.50 In recent years, with the development of transient spectroscopy, CT rate can be directly measured by following the dynamics of charge carriers in nanocrystals, which can range from a few ps to several ns depending on the binding geometry of substrate on the surfaces (inner sphere vs. outer sphere53) and wavefunction overlapping between specific atoms.50,54 For example, Jensen et al. demonstrated the use of CdS quantum dots (QDs) as photocatalyst for the six-electron, six-proton reduction of nitrobenzene (NB) to aniline (AN) through a series of photoinduced one-electron transfers and nitrosobenzene (NSB) and phenylhydroxylamine (PHA) intermediates (Fig. 2(c)).55 The excitonic hole transfer to substrates was found to be within ∼5 ps to form QD˙−, where the electron transfer to nitrobenzene or the intermediates nitrosobenzene and phenylhydroxylamine then occurred on the nanosecond time scale. Such transfer rate constants for the single-electron transfer reactions were found to be correlated with the driving forces for the corresponding proton-coupled electron transfers. A fast hole scavenging from the photoexcited QD was a critical part of this catalytic cycle, which allowed the reducing electron to exist long enough to perform the slow reduction of phenylhydroxylamine to aniline. This fast rate of hole transfer also suggested the existence of a large number of available hole transfer pathways per QD on the surfaces. Wu et al. investigated charge transfer dynamics in CsPbBr3 perovskite QDs.54 The study pointed out that excitons in CsPbBr3 QDs can be efficiently dissociated to free carriers in the presence of electron or hole acceptors. The half-live of electron transfer (ET) to benzoquinone was 65 ± 5 ps and hole transfer (HT) to phenothiazine was 49 ± 6 ps. Such a transfer rate was also a function of the number of surface ligands and the ultrafast electron and hole transferability may enable the development of efficient lead halide perovskite QDs-based light-harvesting devices.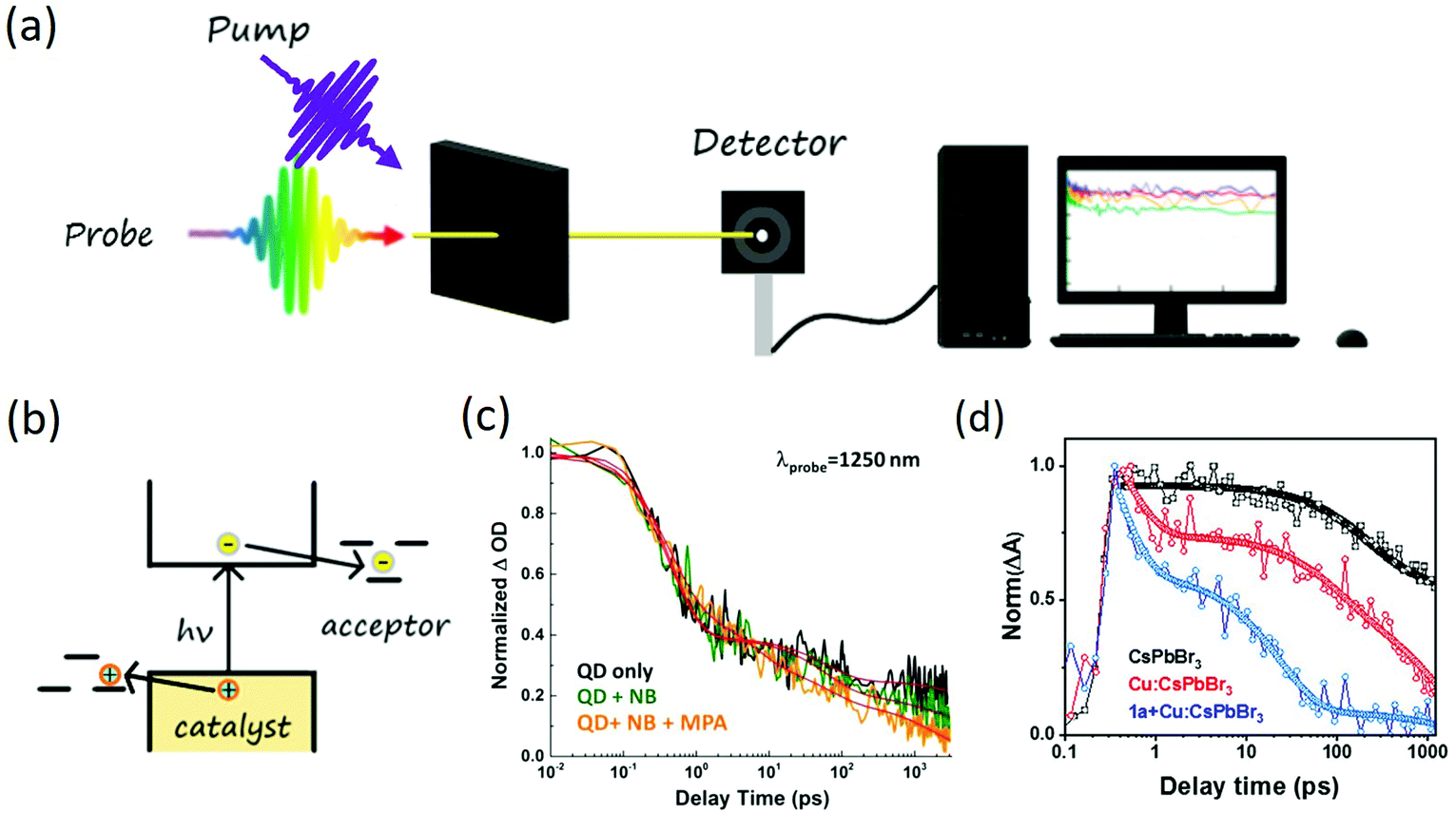 | ||
| Fig. 2 (a) Scheme of transient absorption. The pump pulses create charge carriers inside the photocatalyst and the probe monitor the dynamics of charge carriers. (b) Charge transfer mechanism in nanocrystal based photocatalytic system, the general criteria for charge transfer is the HOMO above VB or the LUMO below CB of photocatalyst; (c) charge transfer dynamics in nitrobenzene reduction reaction to aniline using CdS quantum dots. Excitonic hole transfer of 5 ps is observed. The figure was adapted from ref. 55 with permission from American Chemical Society (d) inner sphere charge transfer dynamics in N–N heterocoupling reaction catalyzed by Cu doped CsPbBr3 nanocrystals. Charge localization of <1 ps and hole transfer around 20 ps is discovered. The figure was adapted from ref. 58 with permission from American Chemical Society. | ||
In another example, Wang et al. investigated the charge transfer process in an α-alkylation of aldehyde catalyzed by CsPbBr3 nanocrystals.56 The study showed 2-bromoacetophenone could be the electron acceptor. With a 0.2 eV driving force, the electron transfer time was determined to be around 50 ± 3 ps with 67% efficiency. The nitrogen-containing cyclohexylamine or enamine could be the hole acceptor with 72 ± 7 ps transfer time, enabling a radical mechanism to form the C–C bonds. Kobosko et al. also probed the excited-state interaction between CsPbBr3 and electron acceptor methyl viologen (MV2+).57 An ultrafast electron transfer time of around 10 ps was discovered. These CT processes seem to happen with the outer sphere pathway where both electrons and hole acceptors are free in the solution. More recently, San Martin et al.58 explored an intramolecular N–N heterocyclization with Cu-doped CsPbBr3 nanocrystals as catalysts. In this reaction, the two N atoms in the substrate would bond to the same surface of the Cu atom, enabling an inner sphere charge transfer mechanism. With transient absorption spectroscopy, an ultrafast charge localization of less than 1 ps was discovered from the valence band of CsPbBr3 to Cu. The localized charge could then be transferred to N atoms on the substrate with ∼20 ps transfer time (Fig. 2(d)). After the first N atom was activated, the second hole transfer could happen, which then forms the new N–N bond. Other charge transfer mechanism can also be found during the formation of C–N, C–O bonds, C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond oxidation, C
C bond oxidation, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, CC, CO bond hydrogenation, as well as alcohol and amine oxidation.59 Such ultrafast transfer of charges to the acceptor is desirable in organic photocatalysis since an intermediate with a higher chance of participating in the reaction will be formed accordingly.
C, CC, CO bond hydrogenation, as well as alcohol and amine oxidation.59 Such ultrafast transfer of charges to the acceptor is desirable in organic photocatalysis since an intermediate with a higher chance of participating in the reaction will be formed accordingly.
3.2. Energy transfer
The energy transfer (ET) process, which represents another common way to initiate photoredox reaction, refers to the whole exciton transfer to the acceptor after the photon is absorbed by the catalyst (Fig. 3(a)). This type of initiation is very useful for substrates whose energy state could not be activated by light. For example, for polycyclic aromatic hydrocarbons (PAHs) with a large HOMO–LUMO gap, triplet energy transfer from photocatalysts to the triplet states of the substrates could initiate catalysis.60 Generally, there are two types of energy transfers, Foster energy transfer (columbic interaction) and Dexter energy transfer (electronic exchange), depending on the primary interaction between donor and acceptors.61 Researchers have demonstrated energy transfer mechanisms in various reactions including epoxidation of the CC bond in an aerobic environment, intermolecular [2 + 2] cycloaddition, and Diels–Alder cycloaddition.62 The rate of energy transfer can also be ultrafast within a few ps to a few ns. Early investigations by Castellano focused on triplet excitons transfer from CdSe quantum dots to surface-anchored 9-anthracenecarboxylic acid.63 Using transient absorption spectroscopy, interfacial Dexter-like triplet–triplet energy transfer was discovered, which could extend the excited-state lifetime by six orders of magnitude. The time for energy transfer was determined to be around 450 ps, enabling various organic transformations. In a more recent example, Weiss20 utilized CdS quantum dots to catalyze an intramolecular [2 + 2] cycloaddition of 4-vinylbenzoic acid and achieved 98% switchable regioselectivity and 98% diastereoselectivity. The energy transfer time in the system was determined to be ∼130 ps using transient absorption spectroscopy (Fig. 3(b)), enabling an efficient [2 + 2] coupling.
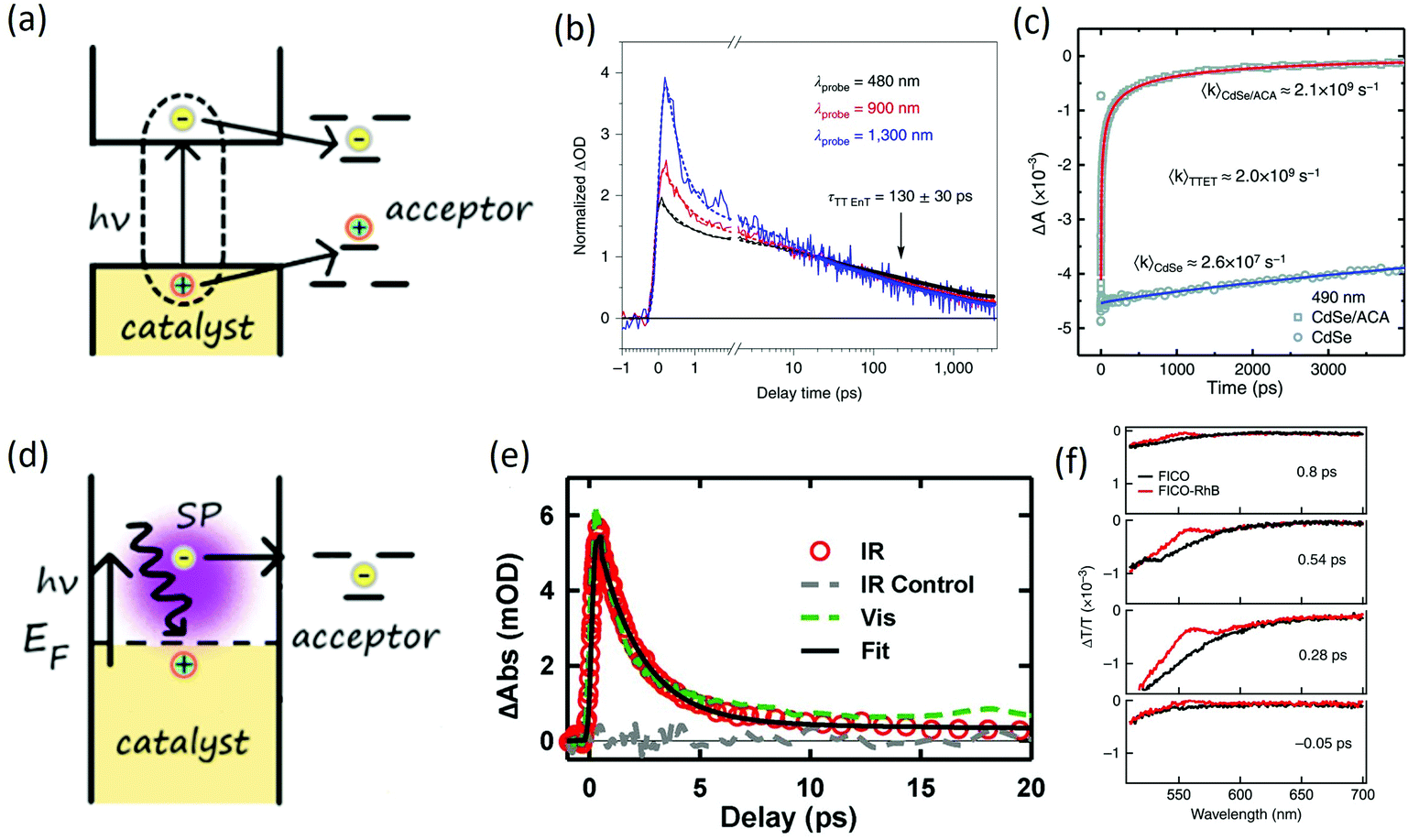 | ||
| Fig. 3 (a) Energy transfer mechanism, HOMO and LOMO or the triplet states of the molecular acceptor needs to lie within the bandgap of photocatalyst; (b) energy transfer dynamics in [2 + 2] cycloaddtion reaction catalyzed by CdS quantum dots. The figure was adapted from ref. 20 with permission from Springer Nature (c) triplet energy transfer dynamics in CdSe/ACA system. The figure was adapted from ref. 63 with permission from AAAS (d) hot carrier transfer mechanism, hot carriers can be generated by plasmon resonance and thus transfer to substrate; (e) dynamics of plasmon induced hot electron transfer in Au–CdS. Plasmon-induced hot electron transfer time of 20 fs is observed with 1.45 ps recombination time. The figure was adapted from ref. 66 with permission from AAAS. (f) Dynamics of hot carrier transfer from CdO quantum dots to surface anchored rhodamine B molecules. Ultrafast hot carrier transfer of <50 fs is observed. The figure was adapted from ref. 67 with permission from Springer Nature. | ||
Wu also studied energy transfer from colloidal CsPbCl3 and CsPbBr3 perovskite nanocrystals to the rhodamine B substrate.64 The study revealed an electron-transfer-mediated triplet energy transfer mechanism. The electron-mediated ET could happen within 20 ps. Compared with conventional one-step triplet transfer, the electron-transfer-mediated mechanism was less demanding in terms of interfacial electronic coupling hence was more generally implementable. Weiss65 reported a competing cycloaddition reaction via Triplet Energy Transfer (TET) mechanism. Due to the singlet–triplet splitting of excited states, the diene–olefin photocycloaddition was driven to the [2 + 2] reaction over the [4 + 2] reaction. The selectivity reached 94% for cyclobutene catalyzed by CdSe. With efficient energy transfer for initiation, the lifetime of the excitons could be lengthened and thus enabling a later chemical bond formation process.
3.3. Hot carrier transfer
The hot carrier transfer process accounts for another type of carrier transfer process. It is less common compared with CT or ET and generally occurs within metal nanocrystals or heavily doped semiconductor nanocrystals. In this type of carrier transfer, the surface plasmon of metal or heavily doped semiconductor nanocrystals is excited by light, which can create electrons (or holes) that are high in energy (or “hot” in nature). Such a hot carrier generation process is quite different from the conventional nanocrystals where bandgap excitation by light generates electrons in the CB and holes in the VB. Metal or heavily doped semiconductor nanocrystals generally have no defined bandgap and surface plasmons are excited. After excitation, high-energy charge carriers then can be transferred to substrate molecules (Fig. 3(d)). However, the surface plasmon resonance is well-localized on the nanoparticles themselves, making the hot-electron transfer a challenging task. Early work by Wu66 described a plasmon-induced interface hot-carrier transfer process. By using Au nanoparticles and CdSe QDs as a bi-component QD on the tips of two CdSe nanorods, the electrons in the Au nanoparticles could be directly excited onto the CdSe nanorod with a quantum yield of 24% (Fig. 3(e)). Recently, Zhou described another system using doped semiconductor nanocrystals, which had a broadly tunable plasmon frequency.67 With infrared light excitation, hot electron transfers happened in ultrafast timescale (<50 fs) with an efficiency of 1.4% from CdO nanocrystals to surface-anchored rhodamine B molecules. In this system, hot electron transfers could happen right after Landau damping before electron thermalization, which provided a new idea towards using hot electrons for photocatalysis (Fig. 3(f)). Further developments in hot carrier-based initiation have suggested other various strategies, including the Schottky barrier that allows hot electrons to irreversibly transport through the interface, localized surface plasmon resonance with amplified chemicurrent, and hybrid nanocatalysts, which contain both metal and semiconductors.68 The manipulation of the flow of hot electrons by changing the electrical characteristics at the interfaces give rise to the intriguing capability of further tuning the catalytic activity.4. Full reaction mechanism-combination of transient spectroscopy and intermediate identification
After initiation by carrier transfer, the reaction then proceeds towards a new chemical bond formation to complete the reaction cycle. To study the full reaction mechanism, one could start with the investigation of several important aspects such as the lifetime of reaction intermediates, chemical structures and the final product formation.69 To identify reaction intermediates and understand their lifetime, transient spectroscopy methods and chemical methods are often used in conjunction with each other, especially for homogeneous catalytic systems.70,71 Transient absorption spectroscopy or time-resolved photoluminescence spectroscopy could follow the decay dynamics of charge carriers inside the nanoparticles and the rise dynamics of reaction intermediates. Therefore, the charge-separated state lifetime after carrier transfer could be identified. On the other hand, chemical radical trapping/quenching with NMR, GC-MS or EPR spectroscopy could provide structure information about chemical identities of reaction intermediates. For example, Simlandy et al. used a combination of steady-state chemical measurements, transient laser spectroscopy, and time-resolved photoluminescence to investigate aerobic oxidation of boronic acids.72 With transient absorption and photoluminescence spectroscopy, the existence of a rapid 350 ps initial electron transfer followed by hole transfer is discovered from CdSe quantum dots to boronic acids. The resulting phenols and aliphatic alcohols are produced in good to high yield with turnover numbers as high as >62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Fig. 4(a)).
000 (Fig. 4(a)).
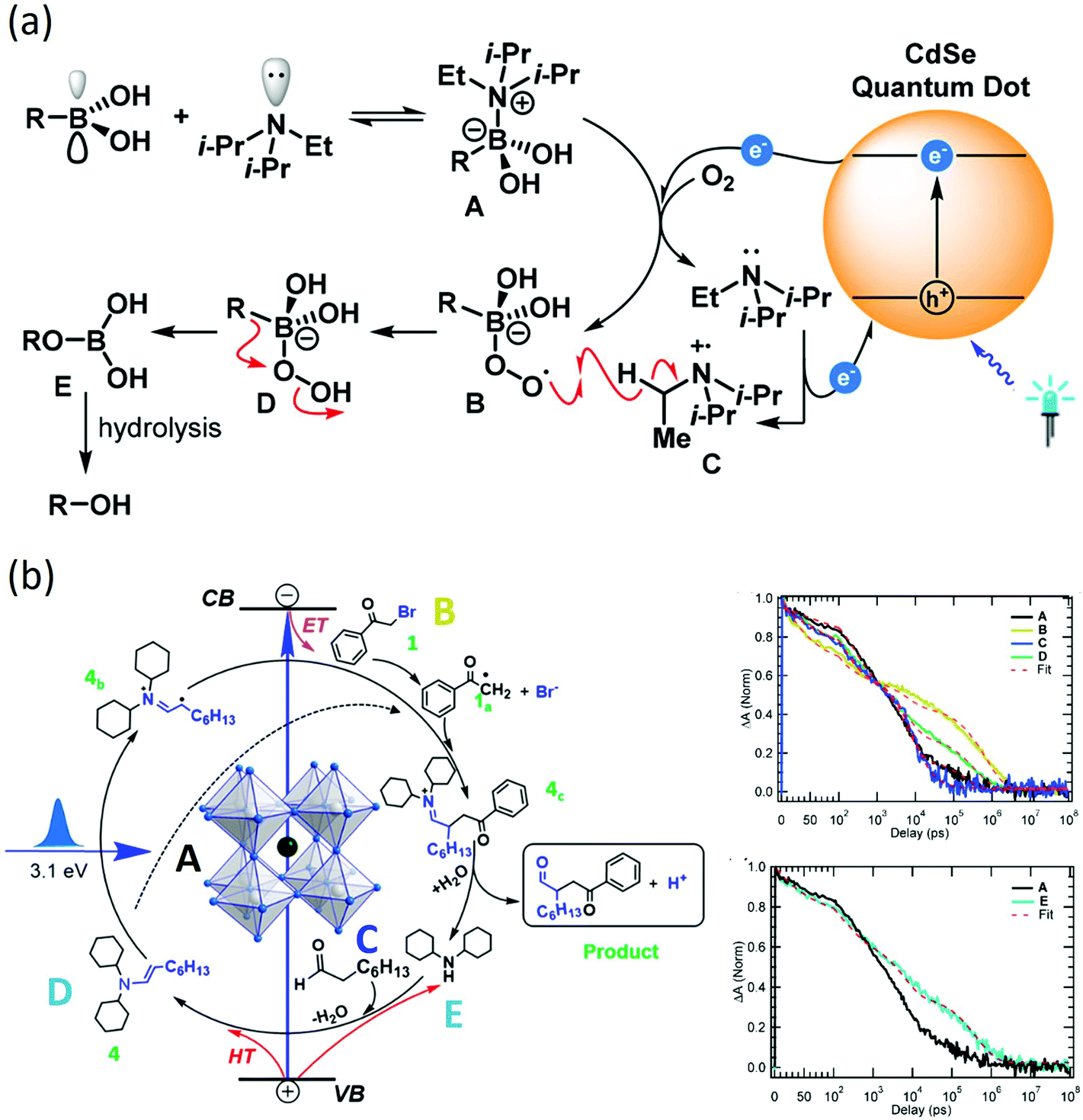 | ||
| Fig. 4 (a) Reaction mechanism for CdSe catalyzed aerobic oxidation of boronic acids probed by transient spectroscopy and steady state chemical measurements. The figure was adapted from ref. 72 with permission from American Chemical Society. (b) Reaction mechanism and dynamics for CsPbBr3 nanocrystal catalyzed C–C coupling probed by transient absorption and radical trapping. A: CsPbBr3B: CsPbBr3 + 2-bromoacetylphenone C: CsPbBr3 + octanal D: CsPbBr3 + enamine E: CsPbBr3 + cyclohexamine. Charge separation lifetime of ~0.8 μs is discovered to facilitate the reaction. The figure was adapted from ref. 56 with permission from American Chemical Society. | ||
Another example is the study of CsPbBr3 catalyzed C–C coupling.56 With transient absorption spectroscopy, the charge separation lifetime was found to be ∼0.8 μs after electron transfer and 0.5 μs after hole transfer. Such a microsecond charge separation time was needed for the formation of C–C bonds. In the meantime, the study incorporated 2,2,6,6-tetramethylpiperidin-1-oxyl as a radical trapping agent and identified 2-bromoacetophenone radical anion and enamine radical cation as reaction intermediates. To go one step further, the study proposed a diffusion–reaction model to clarify the radical coupling mechanism. The main idea was to use the average distance L between reactants (derived from reactant concentration) to represent the interaction possibility. According to the diffusion model, the reaction time τ could be represented as:
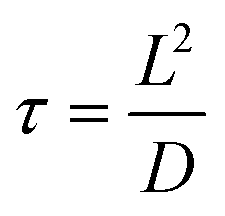 | (1) |
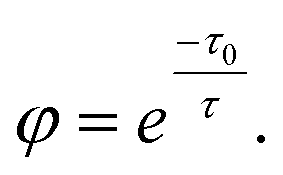 | (2) |
By comparing the actual reaction yield and theoretical reaction yield, the radical reaction mechanism could then be proposed. Together, the full reaction cycle was identified and is shown in Fig. 4(b). He et al. also investigated the photochemical activity in CsPbBr3 nanocrystals.73 The study involved the functionalization of CsPbBr3 with phenanthrene ligands that discovered a triplet energy transfer from photoexcited NCs to phenanthrene. Such a transfer was followed by thermally activated repopulation of NC excitons, leading to delayed NC emission of as long as ∼80 μs at room temperature probed by time-resolved photoluminescence, four orders of magnitude longer than that of unmodified CsPbBr3 NCs (∼5 ns). Such an exceptionally long lifetime enabled phenanthrene-functionalized CsPbBr3 NCs to efficiently drive steady-state photoreduction reactions via diffusion-controlled electron transfer.
5. Lessons learned and future prospective
Functional colloidal nanocrystals have great potential for applications in organic photoredox reactions.74 The low cost in material synthesis could greatly facilitate the production of organic chemicals. From ultrafast and mechanistic studies of nanocrystals driven photoredox catalysis, we have learned several important perspectives. The initiation process, no matter the charge transfer, energy transfer or hot carrier transfer, needs to be fast and efficient. From several ultrafast studies, the transfer rate can range from a few ps to several ns and is highly dependent on the surface acceptor coverage/energy alignment. Ultrafast carrier transfer could compete with the intrinsic recombination process and thus improve the quantum yield of the reaction. After the charge transfer, the lifetime of reaction intermediates also plays a role. Combing transient spectroscopy and chemical methods, the lifetime and chemical structures of organic intermediates could be identified. Studies have suggested that organic reactions are highly dependent on the diffusion of reaction intermediates, and a close to μs (microsecond) lifetime is generally needed for the intermediates to couple efficiently. From what we have learned from current studies, there are two more aspects we can consider for the development of future photocatalytic systems.5.1. Catalytic sites identification
The first aspect is catalytic site identification. Unlike homogeneous catalytic systems with Ru(bpy)3 or Ir(bpy)3, colloidal nanocrystals are intrinsically heterogeneous, such that the surface contains specific reaction sites that are active towards the charge transfer. Identifying such catalytic sites will be essential to improve catalytic efficiency. Imaging techniques such as scanning transmission electron microscopy,75 Kelvin probe force microscopy,76 X-ray absorption spectroscopy77 and theoretical modelling58 could be very useful. Progress has been made by researchers in the field using the combination of experimental and theoretical approaches. In one recent example, San Martin et al. characterized the surface of Cu-doped perovskite nanocrystals for N–N bond coupling.58 Combining the theoretical investigation, the surface bonding mode of the substrate to Cu was proposed and the surface-bounded Cu was identified as the reaction site. Such a study would inspire researchers to utilize advanced imaging strategies to investigate and identify active sites in photocatalytic transformations (Fig. 5). Apart from imaging techniques, vibrational spectroscopy such as IR or Raman spectroscopy could also be quite useful, especially the transient IR/Raman could provide both identity and reaction kinetics on the active sites.78 For example, Kennehan et al. utilized transient mid-infrared absorption spectroscopy to investigate the ligand structure and bonding on the surface PbS QDs.79 The oleate ligands bonded to PbS QD surfaces change markedly when excited and the transient vibrational spectra reveal a net reduction of the Pb–O coordination at the surfaces likely due to an increase in the surface electron density in their excitonic states. Such a binding geometry change could provide useful information towards PbS based catalysis.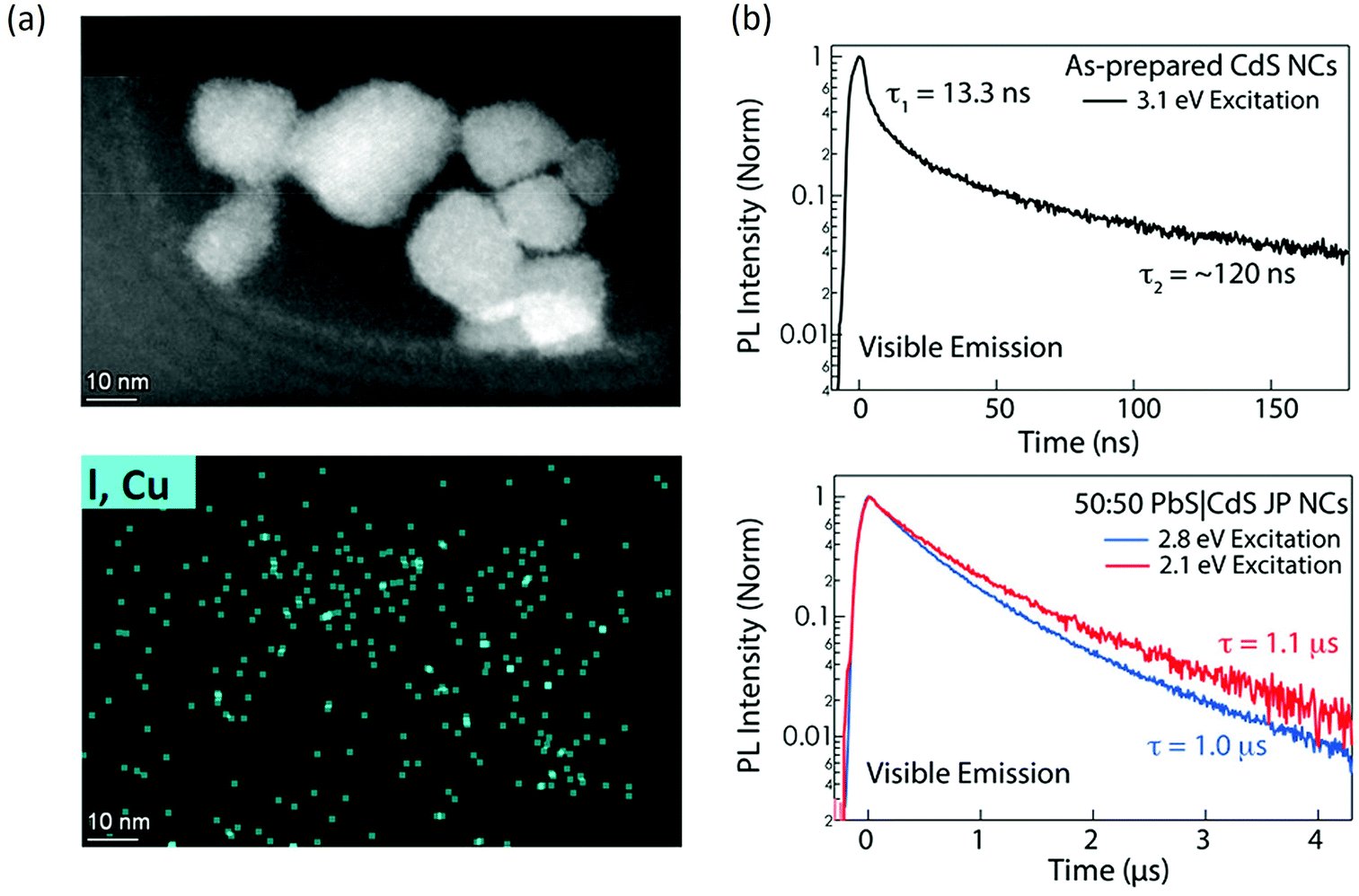 | ||
| Fig. 5 (a) STEM image and EDS image area for Cu doped CsPbBr3 nanocrystal photocatalysts. The figure was adapted from ref. 57 with permission from American Chemical Society. (b) Decay dynamics of photogenerated charge carriers in CdS nanocrystals and CdS/PbS Janus nanocrystals. Elongate charge separation of ~1 μs is observed with Janus structure. The figure was adapted from ref. 80 with permission from American Chemical Society. | ||
5.2. Improve photocatalytic efficiency
The second aspect is how to improve the photocatalytic efficiency from fundamental understanding. The quantum yield for organic transformation using nanocrystals is still low, on the order of 1%–30%. The two key aspects will be increasing charge transfer rate and elongating charge lifetime. To increase the charge transfer rate, one effective strategy will be the direct anchoring of the receptor molecules on the surface of the photocatalyst, this could reduce the physical distance required and create an inner sphere charge transfer pathway.58 For extended carrier lifetime, one can design nanocatalysts with heterostructures that have an internal electric field to facilitate charge separation and suppress recombination (Janus structure could be useful). For example, Kroupa et al. synthesized CdS/PbS Janus particles and discovered an enhanced lifetime from 120 ns to 1.2 μs as a result of enhanced charge separation (Fig. 5).80 Such an extended carrier lifetime could be helpful for later photocatalysis. Another strategy will be designing acceptor molecules with a long lifetime, such as acenes. The triplet exciton lifetime inside a polyaromatic compound could be several microseconds and thus can be used to form products.81 These examples pave the way to expand new paths and platforms for future photocatalysis system design.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China with grant number 22103034.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- A. J. Nozik and J. Miller, Chem. Rev., 2010, 110, 6443–6445 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS PubMed.
- J. Xue, R. Wang, X. Chen, C. Yao, X. Jin, K.-L. Wang, W. Huang, T. Huang, Y. Zhao, Y. Zhai, D. Meng, S. Tan, R. Liu, Z.-K. Wang, C. Zhu, K. Zhu, M. C. Beard, Y. Yan and Y. Yang, Science, 2021, 371, 636 CrossRef CAS PubMed.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- J. Yuan, H. Liu, S. Wang and X. Li, Nanoscale, 2021, 13, 1281–1134 Search PubMed.
- X. Chen, D. J. Aschaffenburg and T. Cuk, Nat. Catal., 2019, 2, 820–827 CrossRef CAS.
- X. Chen, S. N. Choing, D. J. Aschaffenburg, C. D. Pemmaraju, D. Prendergast and T. Cuk, J. Am. Chem. Soc., 2017, 139, 1830–1841 CrossRef CAS PubMed.
- X. Chen, R. T. Pekarek, J. Gu, A. Zakutayev, K. E. Hurst, N. R. Neale, Y. Yang and M. C. Beard, ACS Appl. Mater. Interfaces, 2020, 12, 40339–40346 CrossRef CAS PubMed.
- X. Chen and T. Cuk, J. Phys. Chem. C, 2021, 125, 18204–18209 CrossRef CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS PubMed.
- S. Lin, C. E. Padilla, M. A. Ischay and T. P. Yoon, Tetrahedron Lett., 2012, 53, 3073–3076 CrossRef CAS PubMed.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS PubMed.
- J. A. Caputo, L. C. Frenette, N. Zhao, K. L. Sowers, T. D. Krauss and D. J. Weix, J. Am. Chem. Soc., 2017, 139, 4250–4253 CrossRef CAS PubMed.
- T. Torimoto, K. Maeda, J. Maenaka and H. Yoneyama, J. Phys. Chem., 1994, 98, 13658–13664 CrossRef CAS.
- Z. Zhang, K. Edme, S. Lian and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 4246–4249 CrossRef CAS PubMed.
- Y. Jiang, C. Wang, C. R. Rogers, M. S. Kodaimati and E. A. Weiss, Nat. Chem., 2019, 11, 1034–1040 CrossRef CAS PubMed.
- J. Hu, T.-J. Pu, Z.-W. Xu, W.-Y. Xu and Y.-S. Feng, Adv. Synth. Catal., 2019, 361, 708–713 CrossRef CAS.
- T. Torimoto, H. Uchida, T. Sakata, H. Mori and H. Yoneyama, J. Am. Chem. Soc., 1993, 115, 1874–1880 CrossRef CAS.
- T. Torimoto, T. Sakata, H. Mori and H. Yoneyama, J. Phys. Chem., 1994, 98, 3036–3043 CrossRef CAS.
- H. Inoue, N. Ichiroku, T. Torimoto, T. Sakata, H. Mori and H. Yoneyama, Langmuir, 1994, 10, 4517–4522 CrossRef CAS.
- Y. Wada, T. Kitamura, S. Yanagida and H. Yin, Chem. Commun., 1998, 2683–2684, 10.1039/A806035E.
- H. Yin, Y. Wada, T. Kitamura and S. Yanagida, Environ. Sci. Technol., 2001, 35, 227–231 CrossRef CAS PubMed.
- A. Pal, I. Ghosh, S. Sapra and B. König, Chem. Mater., 2017, 29, 5225–5231 CrossRef CAS.
- K. A. Perez, C. R. Rogers and E. A. Weiss, Angew. Chem., Int. Ed., 2020, 59, 14091–14095 CrossRef CAS PubMed.
- C. Han, X. Zhu, J. S. Martin, Y. Lin, S. Spears and Y. Yan, ChemSusChem, 2020, 13, 4005–4025 CrossRef CAS PubMed.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733–738 CrossRef CAS PubMed.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843–2843 CrossRef PubMed.
- Y. Dong, K. Li, W. Luo, C. Zhu, H. Guan, H. Wang, L. Wang, K. Deng, H. Zhou, H. Xie, Y. Bai, Y. Li and Q. Chen, Angew. Chem., Int. Ed., 2020, 59, 12931–12937 CrossRef CAS PubMed.
- M. Zhang, Z. Li, X. Xin, J. Zhang, Y. Feng and H. Lv, ACS Catal., 2020, 10, 14793–14800 CrossRef CAS.
- L. Tian, N. A. Till, B. Kudisch, D. W. C. MacMillan and G. D. Scholes, J. Am. Chem. Soc., 2020, 142, 4555–4559 CrossRef CAS PubMed.
- Z.-J. Li, S. Li, A. H. Davis, E. Hofman, G. Leem and W. Zheng, Nano Res., 2020, 13, 1668–1676 CrossRef CAS.
- A. Mahdavi-Shakib, J. Sempel, M. Hoffman, A. Oza, E. Bennett, J. S. Owen, A. Rahmani Chokanlu, B. G. Frederick and R. N. Austin, ACS Appl. Mater. Interfaces, 2021, 13, 11793–11804 CrossRef CAS PubMed.
- C. M. Bernt, P. T. Burks, A. W. DeMartino, A. E. Pierri, E. S. Levy, D. F. Zigler and P. C. Ford, J. Am. Chem. Soc., 2014, 136, 2192–2195 CrossRef CAS PubMed.
- C. Huang, X.-B. Li, C.-H. Tung and L.-Z. Wu, Chem. – Eur. J., 2018, 24, 11530–11534 CrossRef CAS PubMed.
- H. Hao and X. Lang, ChemCatChem, 2019, 11, 1378–1393 CrossRef CAS.
- Q. Li, T. Song, Y. Zhang, Q. Wang and Y. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 27323–27333 CrossRef CAS PubMed.
- Y. Dai, C. Poidevin, C. Ochoa-Hernández, A. A. Auer and H. Tüysüz, Angew. Chem., Int. Ed., 2020, 59, 5788–5796 CrossRef CAS PubMed.
- Y. Yuan, H. Zhu, K. Hills-Kimball, T. Cai, W. Shi, Z. Wei, H. Yang, Y. Candler, P. Wang, J. He and O. Chen, Angew. Chem., Int. Ed., 2020, 59, 22563–22569 CrossRef CAS PubMed.
- H. Lu, X. Zhu, C. Miller, J. San Martin, X. Chen, E. M. Miller, Y. Yan and M. C. Beard, J. Chem. Phys., 2019, 151, 204305 CrossRef PubMed.
- X. Wu, S. Xie, C. Liu, C. Zhou, J. Lin, J. Kang, Q. Zhang, Z. Wang and Y. Wang, ACS Catal., 2019, 9, 8443–8451 CrossRef CAS.
- Y. Yuan, N. Jin, P. Saghy, L. Dube, H. Zhu and O. Chen, J. Phys. Chem. Lett., 2021, 12, 7180–7193 CrossRef CAS PubMed.
- C. S. Ponseca, P. Chábera, J. Uhlig, P. Persson and V. Sundström, Chem. Rev., 2017, 117, 10940–11024 CrossRef CAS PubMed.
- X. Chen, K. Wang and M. C. Beard, Phys. Chem. Chem. Phys., 2019, 21, 16399–11647 RSC.
- T. Li, X. Chen, X. Wang, H. Lu, Y. Yan, M. C. Beard and D. B. Mitzi, ACS Energy Lett., 2020, 5, 347–352 CrossRef CAS.
- H. Zhu, Y. Yang, K. Wu and T. Lian, Annu. Rev. Phys. Chem., 2016, 67, 259–281 CrossRef CAS PubMed.
- Z. Huang, Z. Xu, M. Mahboub, X. Li, J. W. Taylor, W. H. Harman, T. Lian and M. L. Tang, Angew. Chem., 2017, 129, 16810–16814 CrossRef.
- T. Tatsuma, H. Nishi and T. Ishida, Chem. Sci., 2017, 8, 3325–3337 RSC.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- K. Wu, G. Liang, Q. Shang, Y. Ren, D. Kong and T. Lian, J. Am. Chem. Soc., 2015, 137, 12792–12795 CrossRef CAS PubMed.
- S. C. Jensen, S. Bettis Homan and E. A. Weiss, J. Am. Chem. Soc., 2016, 138, 1591–1600 CrossRef CAS PubMed.
- K. Wang, H. Lu, X. Zhu, Y. Lin, M. C. Beard, Y. Yan and X. Chen, ACS Energy Lett., 2020, 5, 566–571 CrossRef CAS.
- S. M. Kobosko, J. T. DuBose and P. V. Kamat, ACS Energy Lett., 2020, 5, 221–223 CrossRef CAS.
- J. S. Martin, X. Zeng, X. Chen, C. Miller, C. Han, Y. Lin, N. Yamamoto, X. Wang, S. Yazdi, Y. Yan, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2021, 143, 11361–11369 CrossRef CAS PubMed.
- X. Huang, K. Zhang, B. Peng, G. Wang, M. Muhler and F. Wang, ACS Catal., 2021, 11, 9618–9678 CrossRef CAS.
- X. Luo, R. Lai, Y. Li, Y. Han, G. Liang, X. Liu, T. Ding, J. Wang and K. Wu, J. Am. Chem. Soc., 2019, 141, 4186–4190 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369 CrossRef CAS PubMed.
- X. Luo, G. Liang, Y. Han, Y. Li, T. Ding, S. He, X. Liu and K. Wu, J. Am. Chem. Soc., 2020, 142, 11270–11278 CrossRef CAS PubMed.
- Y. Jiang, M. Yang, Y. Wu, R. López-Arteaga, C. R. Rogers and E. A. Weiss, Chem. Catal., 2021, 1, 106–116 CrossRef PubMed.
- K. Wu, J. Chen, J. R. McBride and T. Lian, Science, 2015, 349, 632 CrossRef CAS PubMed.
- D. Zhou, X. Li, Q. Zhou and H. Zhu, Nat. Commun., 2020, 11, 2944 CrossRef CAS PubMed.
- J. Y. Park, S. M. Kim, H. Lee and I. I. Nedrygailov, Acc. Chem. Res., 2015, 48, 2475–2483 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- L. Tian, N. A. Till, B. Kudisch, D. W. C. MacMillan and G. D. Scholes, J. Am. Chem. Soc., 2020, 142, 4555–4559 CrossRef CAS PubMed.
- B. G. Stevenson, E. H. Spielvogel, E. A. Loiaconi, V. M. Wambua, R. V. Nakhamiyayev and J. R. Swierk, J. Am. Chem. Soc., 2021, 143, 8878–8885 CrossRef CAS PubMed.
- A. K. Simlandy, B. Bhattacharyya, A. Pandey and S. Mukherjee, ACS Catal., 2018, 8, 5206–5211 CrossRef CAS.
- S. He, Y. Han, J. Guo and K. Wu, ACS Energy Lett., 2021, 6, 2786–2791 CrossRef CAS.
- K. S. Schanze, P. V. Kamat, P. Yang and J. Bisquert, ACS Energy Lett., 2020, 5, 2602–2604 CrossRef CAS.
- Y. Zhou, H. Sternlicht and N. P. Padture, Joule, 2019, 3, 641–661 CrossRef CAS.
- R. Chen, S. Pang, H. An, J. Zhu, S. Ye, Y. Gao, F. Fan and C. Li, Nat. Energy, 2018, 3, 655–663 CrossRef CAS.
- J. Ren, F. Weber, F. Weigert, Y. Wang, S. Choudhury, J. Xiao, I. Lauermann, U. Resch-Genger, A. Bande and T. Petit, Nanoscale, 2019, 11, 2056–2064 RSC.
- D. M. Herlihy, M. M. Waegele, X. Chen, C. D. Pemmaraju, D. Prendergast and T. Cuk, Nat. Chem., 2016, 8, 549–555 CrossRef CAS PubMed.
- E. R. Kennehan, K. T. Munson, C. Grieco, G. S. Doucette, A. R. Marshall, M. C. Beard and J. B. Asbury, J. Am. Chem. Soc., 2021, 143, 13824–13834 CrossRef CAS PubMed.
- D. M. Kroupa, G. F. Pach, M. Vörös, F. Giberti, B. D. Chernomordik, R. W. Crisp, A. J. Nozik, J. C. Johnson, R. Singh, V. I. Klimov, G. Galli and M. C. Beard, ACS Nano, 2018, 12, 10084–10094 CrossRef CAS PubMed.
- H. Lu, X. Chen, J. E. Anthony, J. C. Johnson and M. C. Beard, J. Am. Chem. Soc., 2019, 141, 4919–4927 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |