
Reactive oxygen species-responsive polydopamine nanoparticles for targeted and synergistic chemo and photodynamic anticancer therapy†
Gaole
Dai
 a,
Jacky C. H.
Chu
a,
Cecilia Ka Wing
Chan
bc,
Chung Hang Jonathan
Choi
*b and
Dennis K. P.
Ng
*a
a,
Jacky C. H.
Chu
a,
Cecilia Ka Wing
Chan
bc,
Chung Hang Jonathan
Choi
*b and
Dennis K. P.
Ng
*a
aDepartment of Chemistry, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong, China. E-mail: dkpn@cuhk.edu.hk
bDepartment of Biomedical Engineering, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong, China. E-mail: jchchoi@cuhk.edu.hk
cDepartment of Surgery, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong, China
First published on 9th September 2021
Abstract
A thioketal-linked dimer of 3,4-dihydroxy-L-phenylalanine was prepared which underwent self-polymerisation in the presence of doxorubicin (Dox) in an ethanol/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, v/v) mixture with ammonia. The resulting Dox-encapsulated polydopamine (PDA) nanoparticles were further conjugated with molecules of a zinc(II) phthalocyanine (Pc)-based photosensitiser and a peptide containing the heptapeptide QRHKPRE sequence (labelled as QRH) that can target the epidermal growth factor receptor (EGFR) overexpressed in cancer cells. Upon internalisation into these cells through receptor-mediated endocytosis, these nanoparticles labelled as PDA-Dox-Pc-QRH were disassembled gradually via cleavage of the thioketal linkages by the intrinsic intracellular reactive oxygen species (ROS). The stacked Pc molecules were then disaggregated, resulting in activation of their photosensitising property upon irradiation. The ROS generated by the activated Pc promoted further degradation of the nanoparticles and release of Dox, thereby enhancing cell death by synergistic chemo and photodynamic therapy. Systemic injection of PDA-Dox-Pc-QRH into EGFR-overexpressed tumour-bearing nude mice led to targeted delivery to the tumour, and subsequent light irradiation caused complete tumour ablation without inducing notable toxicity.
:4, v/v) mixture with ammonia. The resulting Dox-encapsulated polydopamine (PDA) nanoparticles were further conjugated with molecules of a zinc(II) phthalocyanine (Pc)-based photosensitiser and a peptide containing the heptapeptide QRHKPRE sequence (labelled as QRH) that can target the epidermal growth factor receptor (EGFR) overexpressed in cancer cells. Upon internalisation into these cells through receptor-mediated endocytosis, these nanoparticles labelled as PDA-Dox-Pc-QRH were disassembled gradually via cleavage of the thioketal linkages by the intrinsic intracellular reactive oxygen species (ROS). The stacked Pc molecules were then disaggregated, resulting in activation of their photosensitising property upon irradiation. The ROS generated by the activated Pc promoted further degradation of the nanoparticles and release of Dox, thereby enhancing cell death by synergistic chemo and photodynamic therapy. Systemic injection of PDA-Dox-Pc-QRH into EGFR-overexpressed tumour-bearing nude mice led to targeted delivery to the tumour, and subsequent light irradiation caused complete tumour ablation without inducing notable toxicity.
Introduction
Polydopamine (PDA) is a promising biomaterial derived from the self-polymerisation of dopamine under alkaline conditions based on covalent bonding, hydrogen bonding and π–π interactions.1,2 With reactive functional groups such as amine, imine and catechol on the surface, PDA supports facile modification with thiol- and amine-containing molecules and coating on different types of materials, such as metal oxides, synthetic polymers, noble metals and carbon nanomaterials.3–5 PDA-based nanomaterials have found a wide range of biomedical applications, such as bioimaging,6 molecular diagnostics,7 gene delivery,8 controlled drug release,9,10 photothermal therapy,11 cancer theranostics12 and antimicrobials,13 partly due to their biocompatibility, high cellular uptake, low cytotoxicity and fluorescence quenching ability. However, their limited biodegradability greatly obstructs their clinical translation. As reported earlier, PDA-coated nanoparticles (NPs) can remain stable in vivo for at least six weeks following an intravenous injection into animals.14 Despite the preponderance of polymeric NP-based drug carriers, such as poly(lactic-co-glycolic acid), polycaprolactone and chitosan that upon intravenous injection can decompose into small metabolic fragments for renal clearance,15–17 biodegradable PDA-based NPs remain infrequently reported.18,19To boost the translational potential of PDA-based nanomedicines, we have been interested in next-generation PDA-based NPs that can be degraded in response to specific biochemical stimuli inside the cells or in vivo. As a proof-of-concept, we constructed a thioketal-linked PDA nanosystem that is degradable upon the action of reactive oxygen species (ROS). These oxygen-containing reactive species, such as superoxide anions, singlet oxygen, hydrogen peroxide and hydroxyl radicals play vital roles in living organisms.20 As a pathological hallmark of tumours, ROS are also over-produced (up to 100 μM) in the tumour microenvironment when compared to the levels in normal tissues (about 20 nM).21,22 Recently, nanomedicines with built-in ROS-cleavable linkers (e.g. sulfide, thioketal, selenium and tellurium)23,24 have been studied with a view to regulating the release of therapeutic components in the tumour. However, most of these nanocarriers are not sensitive enough to respond to the intra-tumoural concentrations of ROS, which greatly hampers their antitumour efficacy.25,26 We report herein a highly sensitive ROS-responsive PDA-based nanocarrier that can release the immobilised chemotherapeutic and photosensitising drugs upon interactions with the intracellular ROS. The release of the latter also leads to activation of its photosensitising property upon light irradiation to generate additional ROS that can accelerate the degradation of the NPs and release of the therapeutic components, resulting in synergistic chemo and photodynamic therapy (PDT). The combination of these anticancer treatment modalities is of great potential and much current interest.27–29
The design, preparation and mechanistic actions of this multifunctional nanosystem are shown in Fig. 1. As a starting material, a dimer of 3,4-dihydroxy-L-phenylalanine (L-DOPA) was prepared by covalently linking two L-DOPA units with a ROS-sensitive thioketal linker. This dimer was mixed with doxorubicin (Dox) and underwent self-polymerisation under alkaline conditions to give Dox-encapsulated PDA-based NPs (labelled as PDA-Dox). The surface of the NPs was then modified sequentially with molecules of a zinc(II) phthalocyanine (Pc) and the peptide sequence QRHKPREGGGSC (labelled as QRH) to give PDA-Dox-Pc and PDA-Dox-Pc-QRH respectively. Zinc(II) phthalocyanines have been well-documented as superior photosensitisers for PDT,30 while the heptapeptide QRHKPRE in QRH can preferentially bind to the epidermal growth factor receptor (EGFR) that is typically overexpressed in cancer cells.31,32 Owing to the self-quenching of the stacked Pc molecules and the quenching effect of PDA, the Pc molecules immobilised on the NPs are photodynamically inactive. Upon selective uptake by EGFR-positive cancer cells via receptor-mediated endocytosis, the initially basal amount of intracellular ROS would trigger the degradation of PDA-Dox-Pc-QRH via cleavage of the thioketal linkages to release Dox and Pc molecules. The disaggregation of the Pc molecules results in activation upon excitation with near-infrared light to generate additional ROS, particularly singlet oxygen, that can cause cellular damage via the photodynamic action and accelerate the degradation of the thioketal-linked NPs and the release of larger amounts of the two therapeutic components. The light-triggered on-demand release of Dox can induce additional cytotoxicity through a different pathway and reduce the undesired side effects. It was expected that this multifunctional nanodrug could localise in tumour in vivo by the active EGFR-targeting and enhanced permeability and retention (EPR) effects and exhibit light-triggered ROS-responsive degradation, subsequently leading to synergistic chemo and photodynamic anticancer therapy.
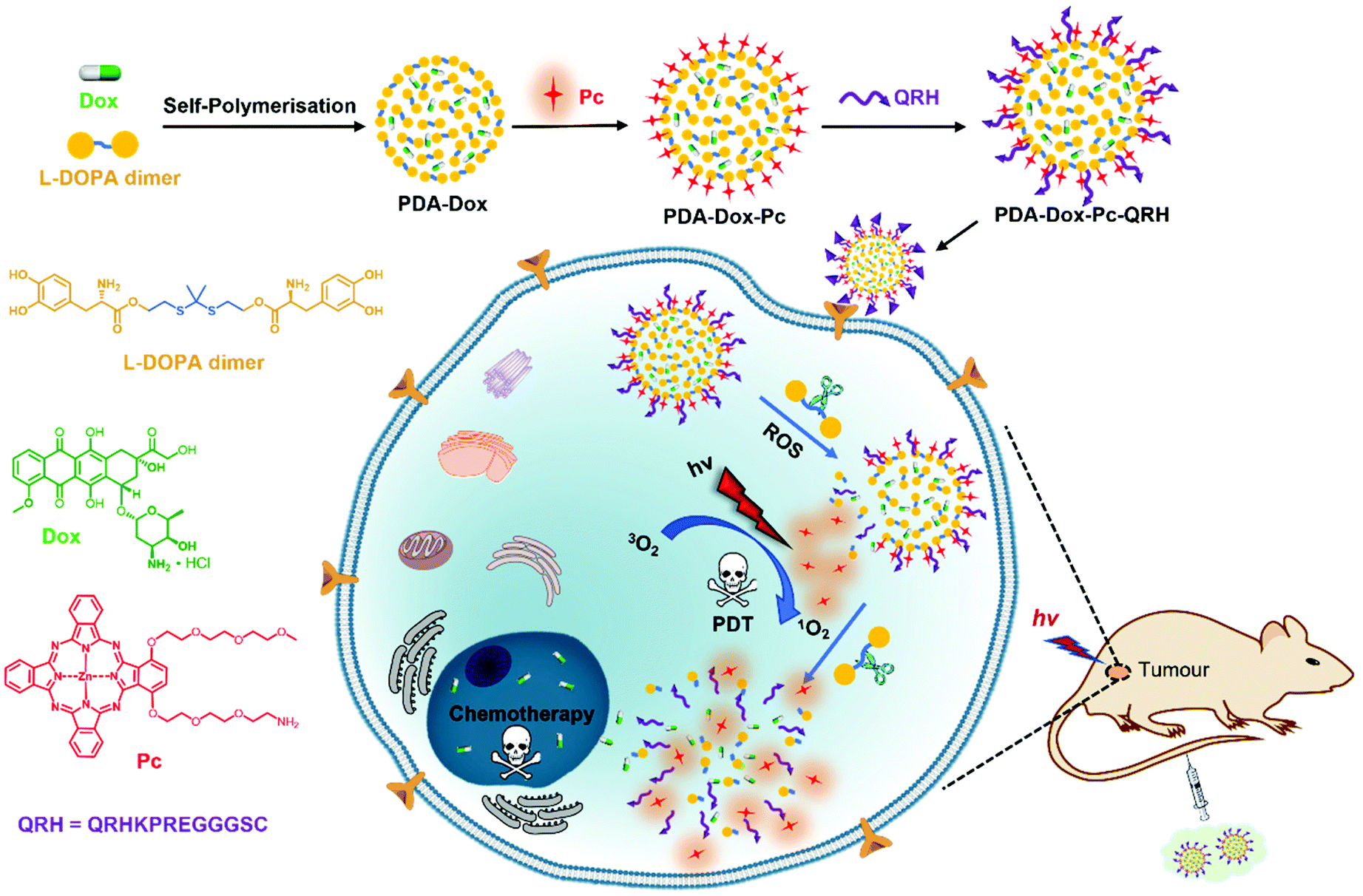 | ||
| Fig. 1 Design, preparation and mechanistic actions of PDA-Dox-Pc-QRH. | ||
Results and discussion
Synthesis and characterisation of raw materials
The synthetic scheme used to prepare this thioketal-linked L-DOPA dimer (see its structure in Fig. 1) is shown in Scheme S1 (ESI†). Treatment of L-DOPA of which the two hydroxyl groups and the amino group were previously protected with tert-butyldimethylsilyl (TBDMS) and tert-butyloxycarbonyl (Boc) moieties respectively (i.e. compound 1)33 with 0.5 equiv. of thioketal-linked diol 234 in the presence of N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) and N,N-diisopropylethylamine (DIPEA) gave the condensed product 3. The TBDMS and Boc protecting groups were then removed sequentially upon treatment with Bu4NF and trifluoroacetic acid (TFA), affording dimer 4 and 5 respectively. The target L-DOPA dimer 5 was obtained in quantitative yield through recrystallisation from diethyl ether.For the phthalocyanine-based photosensitiser simply labelled as Pc (see its structure in Fig. 1), it was prepared according to our previously reported procedure.35 The UV-Vis spectrum of this compound in N,N-dimethylformamide (DMF) showed an intense and sharp Q-band at 689 nm (ε ≈ 2 × 105 M−1 cm−1), a Soret band at 338 nm (ε ≈ 6 × 104 M−1 cm−1) and a vibronic band at 621 nm (ε ≈ 4 × 103 M−1 cm−1). The linear dependence of the Q-band absorbance on the concentration indicated that the compound was essentially non-aggregated under these conditions (Fig. S1a, ESI†). Upon excitation at 610 nm, Pc showed a weak fluorescence emission at 707 nm with a fluorescence quantum yield (ΦF) of 0.18 relative to the unsubstituted ZnPc (ΦF = 0.28). The singlet oxygen generation efficiency of Pc was also evaluated in DMF by a steady-state method using 1,3-diphenylisobenzofuran (DPBF) as the scavenger of singlet oxygen.36 The conversion of this probe to 1,2-dibenzoylbenzene via an unstable peroxide intermediate was monitored by recording the absorbance of DPBF at 415 nm with the irradiation time (Fig. S1b, ESI†), from which the singlet oxygen quantum yield (ΦΔ) was determined to be 0.77, which was significantly higher than that of ZnPc (ΦΔ = 0.56). The weaker fluorescence and higher singlet oxygen generation efficiency of Pc are the general features of di-α-substituted zinc(II) phthalocyanines.37
The QRH peptide sequence was prepared according to the methodology we reported earlier32 with minor modifications. The heptapeptide QRHKPRE was prepared and linked to a non-functional GGGS spacer by using standard 9-fluorenylmethyloxycarbonyl (Fmoc)-mediated solid-phase peptide synthesis, and then a cysteine residue was introduced at the C-terminus to provide a thiol group for immobilisation on the PDA-based NPs. The peptide intermediate was cleaved from the resin and fully deprotected upon treatment with a mixture of TFA, triisopropylsilane (TIPS) and H2O (95:2.5:2.5, v/v/v). The free QRH peptide was purified with high-performance liquid chromatography (HPLC) and characterised with matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrometry, which clearly showed an intense isotopic cluster at m/z 1310.958 due to the molecular ion (Fig. S2, ESI†).
Preparation and characterisation of PDA-based NPs
The Dox-loaded PDA-based NPs (i.e. PDA-Dox) were prepared by a solution oxidation method.38 After separately dissolving Dox (in the range of 4–12 μg mL−1) and L-DOPA dimer (36 mmol) in deionised water, the two aqueous solutions were injected into a mixture of ethanol and deionised water (1:4, v/v) with ammonia introduced to maintain a basic reaction environment. After stirring the mixture at room temperature for 36 h, the NPs formed were collected by centrifugation and washed with deionised water, followed by lyophilisation. By monitoring the fluorescence of Dox in the supernatant (Fig. S3, ESI†), it was found that only when the concentration of Dox reached 12 μg mL−1, a strong fluorescence was observed, which suggested that the saturated concentration of encapsulated Dox was 10 μg mL−1. The Dox loading for this batch of PDA-Dox was estimated to be 9% (by weight).
To immobilise Pc on PDA-Dox, the two components were mixed in Tris buffer (pH 8.5, 10 mM) with 0.5% Tween 20 added to promote the water solubility of Pc. After being stirred at room temperature for 14 h, the mixture was centrifuged to collect the NPs formed. With a nucleophilic amino functionality, Pc could be immobilised readily on the PDA-based NPs through Schiff base reaction or Michael addition.3 Based on the Q-band absorbance of the supernatant, the amount of free Pc could be determined from which the Pc loading was estimated to be 15% (by weight).
The resulting NPs (i.e. PDA-Dox-Pc) were then further modified with the QRH peptide by mixing the NPs with the peptide in deionised water at 4 °C overnight. The thiol group of the cysteine residue of QRH coupled with the reactive functionalities on the PDA surface, such as quinone via Michael addition,3 enabling peptide conjugation on the surface of the resulting PDA-Dox-Pc-QRH.
The size and morphology of PDA-Dox, PDA-Dox-Pc and PDA-Dox-Pc-QRH were studied using transmission electron microscopy (TEM) and dynamic light scattering (DLS). As shown in Fig. 2a, the PDA-Dox NPs were spherical in shape with an average diameter of 127 ± 7 nm. After loading with Pc, a darkened shell appeared surrounding the PDA core, suggesting that the immobilisation occurred on the surface of the NPs. The average diameter was increased to 162 ± 10 nm, giving a shell thickness of ca. 18 nm. The size was further increased to 180 ± 9 nm after further conjugation with QRH. The dimensions of these NPs were generally in accordance with the hydrodynamic diameters determined by DLS, ranging from 165 nm (for PDA-Dox) to 201 nm (for PDA-Dox-Pc) and 220 nm (for PDA-Dox-Pc-QRH) (Fig. 2b, Table 1). The small polydispersity index (PDI) (0.05–0.07) indicated that these NPs were well dispersed in water without significant aggregation.
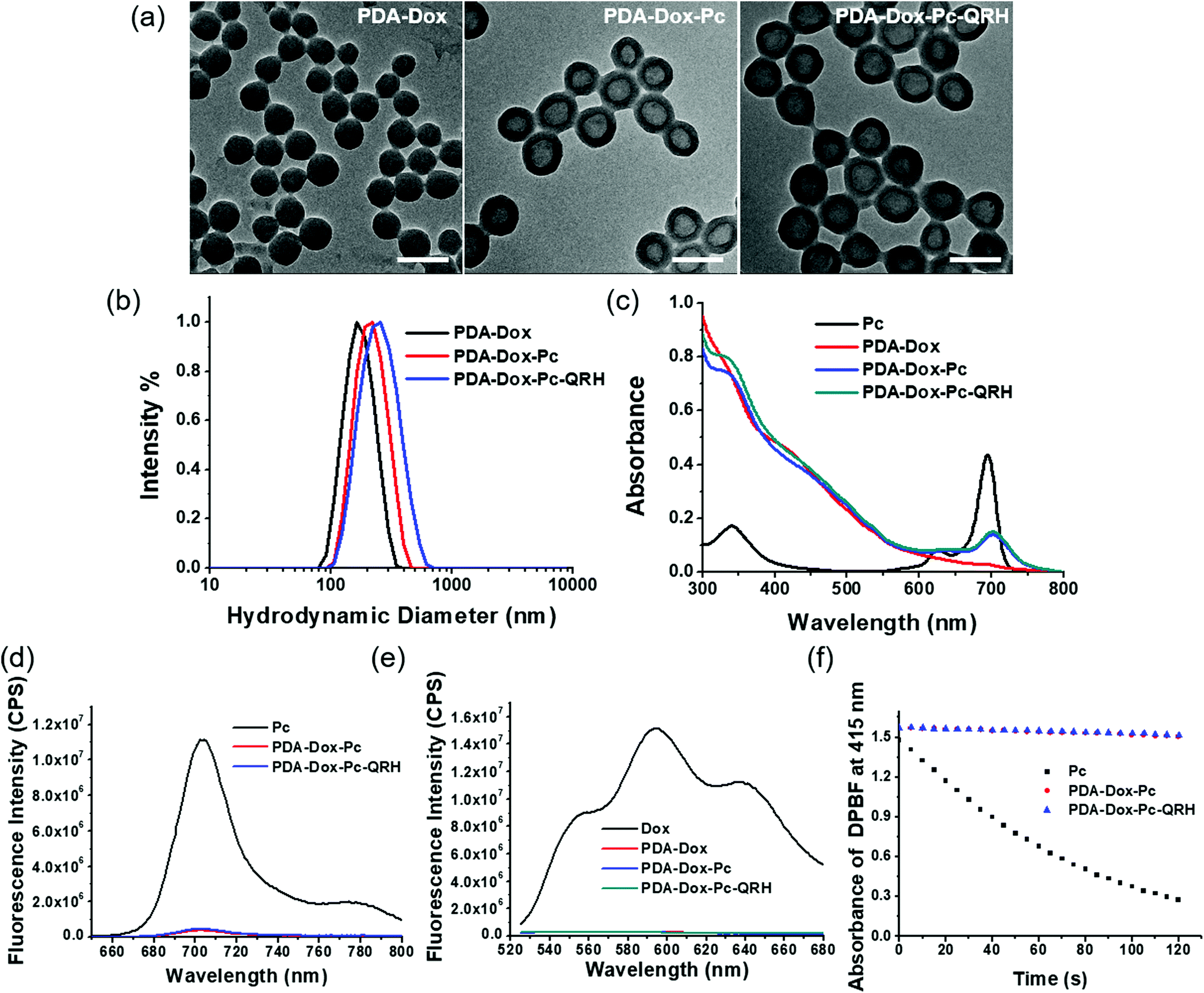 | ||
| Fig. 2 (a) TEM images of PDA-Dox, PDA-Dox-Pc and PDA-Dox-Pc-QRH. Scale bar: 200 nm. (b) Hydrodynamic diameter distribution of these NPs in water measured by DLS. (c) Electronic absorption and (d) fluorescence (λex = 610 nm) spectra of free Pc and these NPs ([Pc] = 2.0 μM) in PBS with 0.5% Tween 20. (e) Fluorescence spectra (λex = 488 nm) of free Dox and these NPs ([Dox] = 1.8 μM) in PBS with 0.5% Tween 20. (f) Comparison of the rates of decay of DPBF sensitised by free Pc, PDA-Dox-Pc and PDA-Dox-Pc-QRH. | ||
| NPs | Physical sizea (nm) | Hydrodynamic diameterb (nm) | Zeta potentialb (mV) | PDIb |
|---|---|---|---|---|
| a The values were obtained from the analysis of 100 particles in the TEM images by ImageJ. b The values are reported as the mean ± standard deviation (SD) of three independent measurements. | ||||
| PDA-Dox | 127 ± 7 | 165 ± 2 | −26.1 ± 0.3 | 0.06 ± 0.02 |
| PDA-Dox-Pc | 162 ± 10 | 201 ± 1 | −20.6 ± 0.6 | 0.05 ± 0.03 |
| PDA-Dox-Pc-QRH | 180 ± 9 | 220 ± 1 | −11.6 ± 0.2 | 0.07 ± 0.03 |
The zeta potentials of these NPs were also determined by DLS (Table 1). It was found that conjugation of QRH led to a less negative value (−11.6 ± 0.2 mV for PDA-Dox-Pc-QRH) compared with the non-peptide-conjugated NPs (−26.1 ± 0.3 mV for PDA-Dox and −20.6 ± 0.6 mV for PDA-Dox-Pc), which could be attributed to the positive charges of the peptide.
Fig. 2c shows the electronic absorption spectra of these NPs in phosphate-buffered saline (PBS) with 0.5% Tween 20, which was added to increase the water solubility of the NPs. The spectrum of free Pc is also included for comparison. Both the Pc-containing PDA-Dox-Pc and PDA-Dox-Pc-QRH showed the Q-band absorption at ca. 700 nm, which was significantly broadened and weaker compared with that of free Pc. This band was not seen for the Pc-free PDA-Dox. Upon excitation at 610 nm, a very weak fluorescence band due to Pc was observed at 705 nm for PDA-Dox-Pc and PDA-Dox-Pc-QRH (Fig. 2d). Again, the intensity was much weaker than that of free Pc. These spectral features strongly indicated that the Pc molecules were significantly stacked in the NPs. We also excited the Dox component in these NPs at 488 nm. Similarly, negligible fluorescence was observed for all the NPs, while strong fluorescence could be seen for free Dox (Fig. 2e). The results indicated that the encapsulated Dox molecules were also severely quenched.
For the Pc-containing NPs, their singlet oxygen generation efficiency was also evaluated and compared with that of free Pc in DMF, using DPBF as the singlet oxygen scavenger. The rate of conversion of this probe to the corresponding peroxide was monitored by recording the absorbance of DPBF at 415 nm along with the irradiation time. As shown in Fig. 2f, while free Pc could efficiently consume DPBF by sensitising the formation of singlet oxygen, both PDA-Dox-Pc and PDA-Dox-Pc-QRH could not generate singlet oxygen to consume the probe due to the significantly stacked and hence quenched Pc molecules.
ROS-responsive properties of PDA-Dox-Pc-QRH
To examine the effect of ROS on the release of Pc from PDA-Dox-Pc-QRH, we monitored the change in fluorescence spectrum of the NPs in the presence of different concentrations of H2O2 (0, 0.1 and 1 mM), a representative type of ROS, at 37 °C over a period of 24 h [Fig. S4 (ESI†) and Fig. 3a]. It was found that the intensity of the fluorescence due to the released Pc increased gradually with time and with concentration of H2O2 added. While the percentage of fluorescence recovery was less than 10% in the absence of H2O2 after 24 h, the value reached ca. 70% when 1 mM of H2O2 was used. The results suggested the cleavage of the thioketal linkers in the NPs by H2O2, triggering the release and disaggregation of the Pc molecules. The released Pc also promoted the formation of singlet oxygen. As shown in Fig. 3b, the rate of decay of DPBF increased with the concentration of H2O2, showing that the singlet oxygen generation efficiency of PDA-Dox-Pc-QRH was also higher at a higher concentration of H2O2. The trend was consistent with that of fluorescence recovery as shown in Fig. 3a. H2O2 alone could not sensitise the photo-decay of DPBF (Fig. S5, ESI†). Hence, ROS such as H2O2 could activate the fluorescence emission and singlet oxygen generation of the Pc immobilised on the NPs through cleavage of the thioketal linkers to disassemble the NPs. As the ROS levels are often elevated in tumour (up to 0.1 mM),21,22 these NPs should also be responsive in the tumour microenvironment.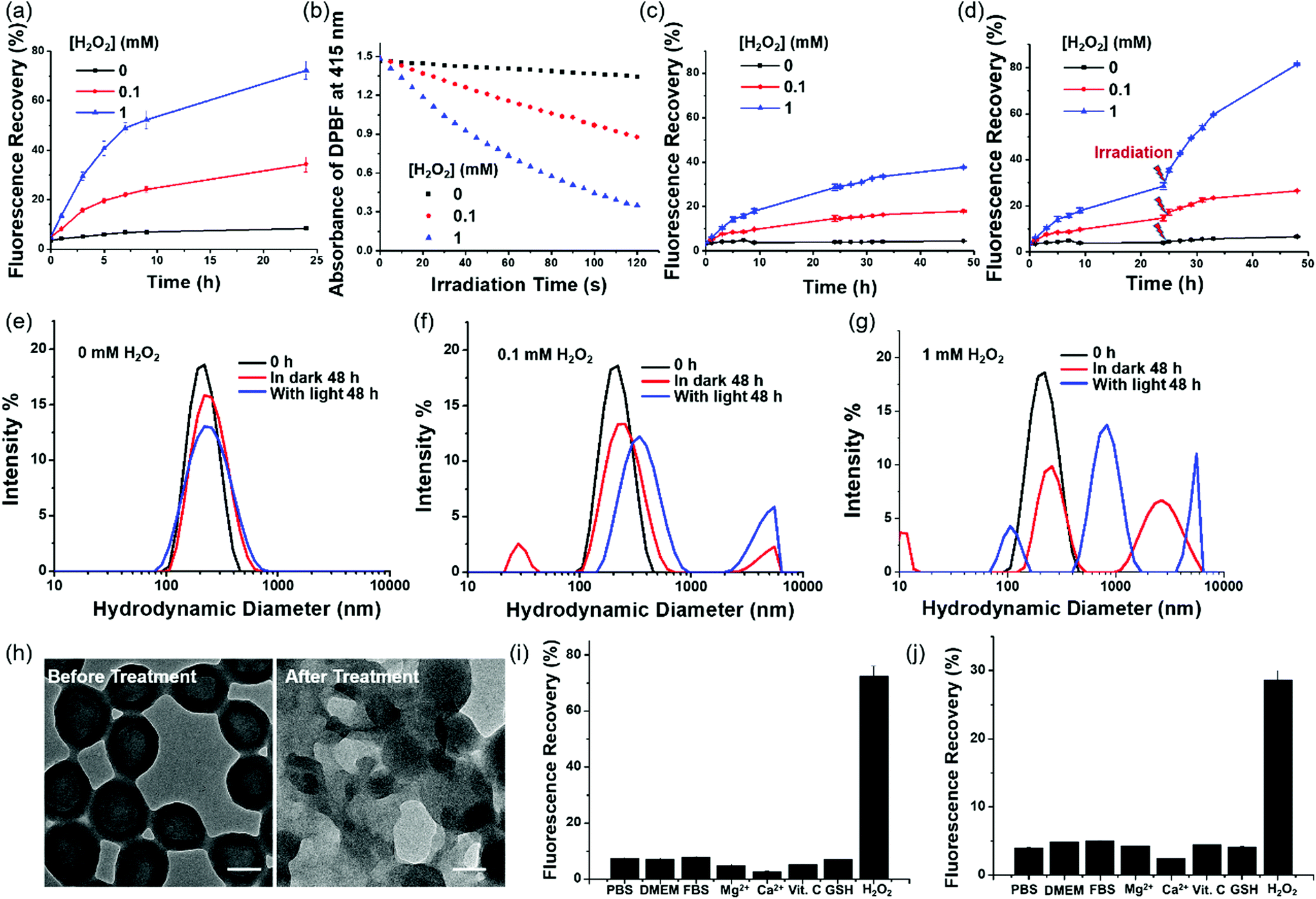 | ||
| Fig. 3 (a) Recovery of Pc fluorescence (λex = 610 nm, λem = 707 nm) upon exposure of PDA-Dox-Pc-QRH to different concentrations of H2O2. (b) Comparison of the rates of decay of DPBF sensitised by PDA-Dox-Pc-QRH after being treated with different concentrations of H2O2 for 24 h. Recovery of Dox fluorescence (λex = 488 nm, λem = 550 nm) upon exposure of PDA-Dox-Pc-QRH to different concentrations of H2O2 (c) without and (d) with light irradiation (λ > 610 nm) for 20 min at 24 h post-treatment with H2O2. Change in hydrodynamic diameter distribution of PDA-Dox-Pc-QRH (e) without and with treatment with (f) 0.1 and (g) 1 mM of H2O2 for 48 h. Light irradiation (λ > 610 nm) was applied for 20 min at 24 h post-treatment with H2O2. (h) TEM images of PDA-Dox-Pc-QRH before and after treatment with 1 mM of H2O2 for 48 h with light irradiation as above. Scale bars: 100 nm. Recovery of (i) Pc and (j) Dox fluorescence for PDA-Dox-Pc-QRH in PBS, DMEM or FBS with 0.5% Tween 20 at 37 °C for 24 h, and after treatment with different species at a concentration of 1 mM in PBS with 0.5% Tween 20 at 37 °C for 24 h. Data are reported as the mean ± SD of three independent experiments. For figure (a) to (g), the measurements were performed in PBS with 0.5% Tween 20 at 37 °C. The concentration of the NPs was fixed at [Pc] = 2.0 μM or [Dox] = 1.8 μM. | ||
The H2O2-triggered release of Dox from PDA-Dox-Pc-QRH was also monitored using fluorescence spectroscopy. As shown in Fig. S6a–c (ESI†), the fluorescence due to Dox was negligible without the treatment with H2O2, but the intensity increased gradually over a period of 48 h when the NPs were treated with H2O2. Again, higher concentration of H2O2 resulted in higher fluorescence intensity, reflecting higher percentage of release of Dox (18% vs. 38% when the concentration of H2O2 was increased from 0.1 to 1 mM) (Fig. 3c). As the concomitantly released Pc could generate singlet oxygen upon irradiation, we believed that light irradiation could promote the degradation of the NPs and release of Pc and Dox. Hence, we applied light irradiation (λ > 610 nm) for 20 min at 24 h post-treatment with H2O2. As expected, the fluorescence intensity of Dox was significantly increased [Fig. S6d–f (ESI†) and Fig. 3d]. The release of Dox was increased to 26% and 81% for the treatment with 0.1 and 1 mM of H2O2 respectively. Without the treatment with H2O2, the release of Dox was lower than 7% even with light irradiation, showing that the NPs are generally stable unless they are specifically triggered to degrade by ROS.
The H2O2 and light-triggered disassembly of PDA-Dox-Pc-QRH in PBS with 0.5% Tween 20 was also studied using DLS. As shown in Fig. 3e, the hydrodynamic diameter distribution was not significantly changed after 48 h in the absence of H2O2 even with light irradiation. It was in accordance with the negligible Pc and Dox release as reported above and reflected the high stability of the NPs. Upon treatment with H2O2 (0.1 and 1 mM), particularly with light irradiation, the NPs disassembled forming several forms of aggregates with different hydrodynamic diameters (Fig. 3f and g). TEM also showed the breakdown of the core–shell structure of these NPs upon the actions of H2O2 and light irradiation (see representative TEM images of the NPs before and after the treatment with 1 mM of H2O2 and light irradiation in Fig. 3h).
To examine the stimuli selectivity of PDA-Dox-Pc-QRH, we also examined the response of these NPs towards various potential interferential species, including Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), Mg2+, Ca2+, L-ascorbic acid (Vit. C) and glutathione (GSH). It was found that in the presence of these species, both the fluorescence intensities of Pc and Dox were not significantly changed compared with those in blank PBS with Tween 20. None of them could significantly increase the fluorescence intensities as H2O2. Fig. 3i and j display the corresponding percentage fluorescence recovery for Pc and Dox, which clearly show that the NPs are susceptible only towards H2O2.
Cellular uptake and intracellular degradation of PDA-Dox-Pc-QRH
To monitor the intracellular stability of PDA-Dox-Pc-QRH after internalisation, A431 human epidermoid carcinoma cells were incubated with the NPs ([Pc] = 0.50 μM or [Dox] = 0.45 μM) for 1 h and then with a NP-free medium for 7 h, followed by exposing the cells to near-infrared light irradiation (λ > 610 nm) for 20 min and further incubation for 2 h. The post-incubation in a NP-free medium was necessary to provide sufficient time for the internalised NPs to degrade inside the cells. As Fig. 3a shows that the fluorescence of Pc could be largely restored in 8 h, we used 7 h as the post-incubation period. As shown by TEM (Fig. 4a), the NPs were accumulated inside the cells instead of adhering on the cell membrane. Interestingly, the TEM images also showed the degradation and deformation of the core–shell morphology of the NPs inside the cells.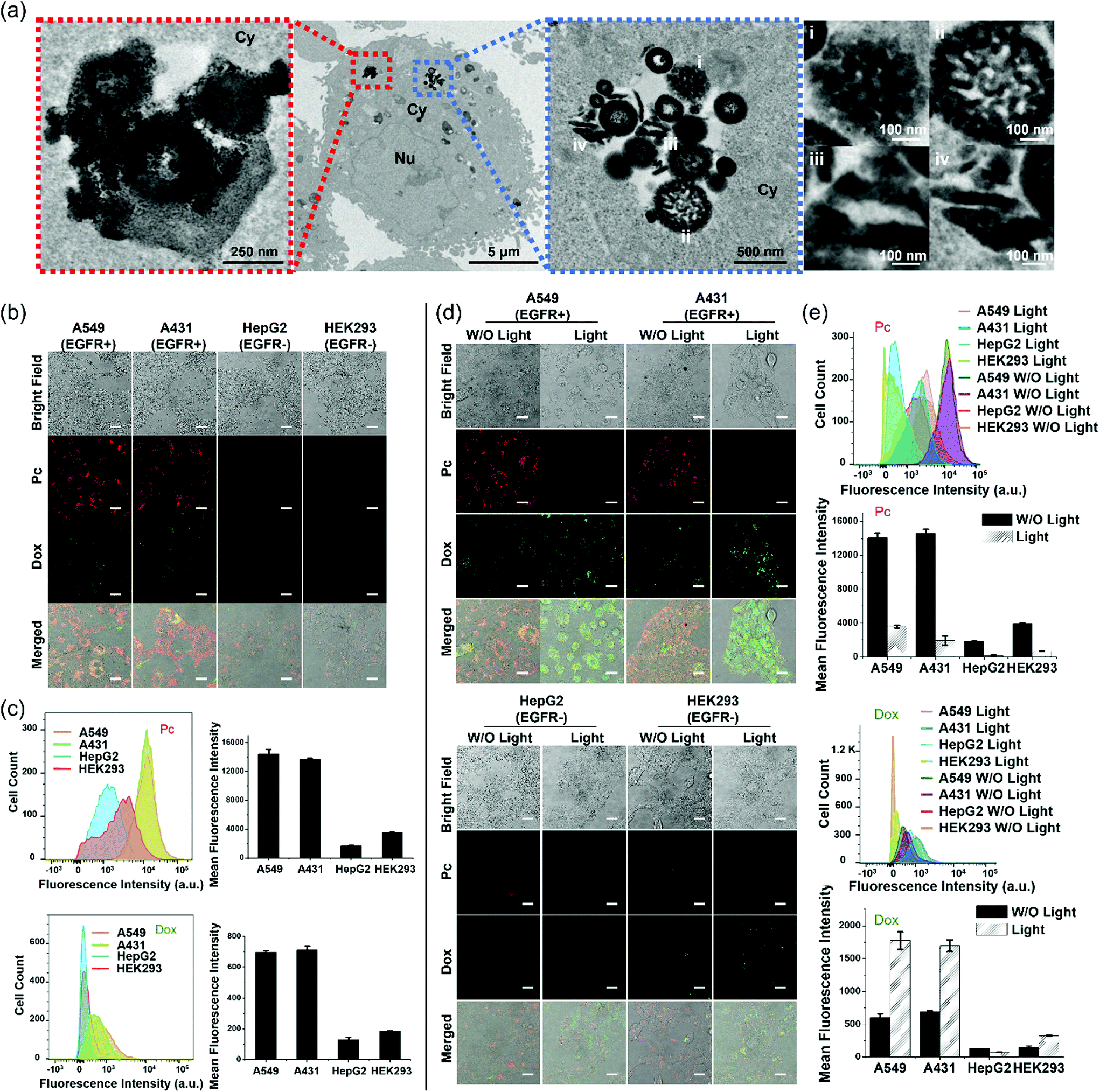 | ||
| Fig. 4 (a) TEM images of a A431 cell upon incubation with PDA-Dox-Pc-QRH ([Pc] = 0.50 μM, [Dox] = 0.45 μM) for 1 h and then with a NP-free medium for 7 h, followed by light irradiation (λ > 610 nm) for 20 min and further incubation for 2 h. The leftmost and the third (counting from the left) images are the magnified images of the second image. The rightmost image shows the enlarged images at four different sites [labelled (i)–(iv)] in the third image (Nu = nucleus, Cy = cytosol). (b) Bright-field, fluorescence (due to Pc and Dox) and the merged images of A549, A431, HepG2 and HEK293 cells after incubation with PDA-Dox-Pc-QRH ([Pc] = 0.50 μM, [Dox] = 0.45 μM) for 1 h and then with a NP-free medium for 7 h. Scale bar: 25 μm. (c) Flow cytometric measurements for the four cell lines after the above treatment. (d) Bright-field, fluorescence (due to Pc and Dox) and the merged images of the four cell lines after the above treatment without or with further light irradiation (λ > 610 nm) for 20 min and incubation for 2 h. Scale bar: 25 μm. (e) Flow cytometric measurements for the four cell lines after the treatment described in figure (d). Data in figure (c) and (e) are reported as the mean ± SD of three independent experiments. | ||
We then studied the tumour-targeting effect of the NPs and whether the intracellular degradation would lead to release of the therapeutic cargoes inside the cells. To this end, a range of cell lines with different expression levels of EGFR was used, including the EGFR-positive A431 and A549 human lung carcinoma cells, as well as the EGFR-negative HepG2 human hepatocarcinoma cells and HEK293 human embryonic kidney normal cells.32,39 The cells were incubated with PDA-Dox-Pc-QRH ([Pc] = 0.5 μM or [Dox] = 0.45 μM) for 1 h and then with a NP-free medium for 7 h before the fluorescence microscopic images were taken. As shown in Fig. 4b, the EGFR-positive A549 and A431 cells showed strong intracellular fluorescence due to Pc and Dox, while the fluorescence of these two species could hardly be seen inside the EGFR-negative HepG2 and HEK293 cells. Similar studies were performed using flow cytometry to quantify the fluorescence intensities of these cells. The results as shown in Fig. 4c were in good agreement with those obtained by confocal fluorescence microscopy. The results showed that PDA-Dox-Pc-QRH could be preferentially taken up by the EGFR-positive cells as a result of the EGFR-binding effect of the QRH peptide. The internalisation was followed by gradual degradation of the NPs and release of the immobilised Pc and Dox inside the cells.
To investigate the effect of light irradiation on the intracellular degradation of the NPs and release of the cargoes, the four cell lines were further irradiated (λ > 610 nm) for 20 min and then incubated for further 2 h after the above treatment. The results obtained by confocal fluorescence microscopy (Fig. 4d) and flow cytometry (Fig. 4e) were compared with those obtained without the extra light treatment. For the two EGFR-positive cell lines, the intracellular Dox fluorescence was significantly increased upon light irradiation. This observation could be attributed to the accelerated degradation of the NPs by the additional ROS generated upon excitation of the released Pc. For the two EGFR-negative cell lines, as the cellular uptake of NPs was low, the effect of irradiation was negligible and the fluorescence intensity remained very weak even after light irradiation. In contrast, the intracellular Pc fluorescence was significantly reduced upon light irradiation, particularly for the two EGFR-positive cell lines. These results might be due to the partial photo-bleaching of the released Pc. In fact, the fluorescence of Pc in PBS with 0.5% Tween 20 was significantly reduced upon light irradiation (λ > 610 nm) for 20 min (Fig. S7, ESI†). Despite the apparently limited photostability, the released Pc, upon light irradiation, could promote the degradation of the NPs and release of the two therapeutic components.
The EGFR-targeting effect of PDA-Dox-Pc-QRH was further examined by a competitive binding assay. The EGFR-positive A549 and A431 cells were pretreated with different concentrations of free QRH peptide (0, 50 and 500 μM) for 1 h, and then incubated with the NPs ([Pc] = 0.5 μM) for 1 h followed by incubation in a NP-free medium for 7 h. The confocal fluorescence images of the cells under different conditions were then captured and compared. As shown in Fig. 5a, the intracellular fluorescence intensity was generally weaker as the concentration of free QRH increased for both the cell lines. Similar results were obtained by flow cytometry (Fig. 5b). The intensity was reduced by ca. 30% when the cells were pretreated with 500 μM of free QRH. The results suggested that the EGFR-targeting effect of PDA-Dox-Pc-QRH was due to the QRH peptide chains immobilised on the surface of the NPs.
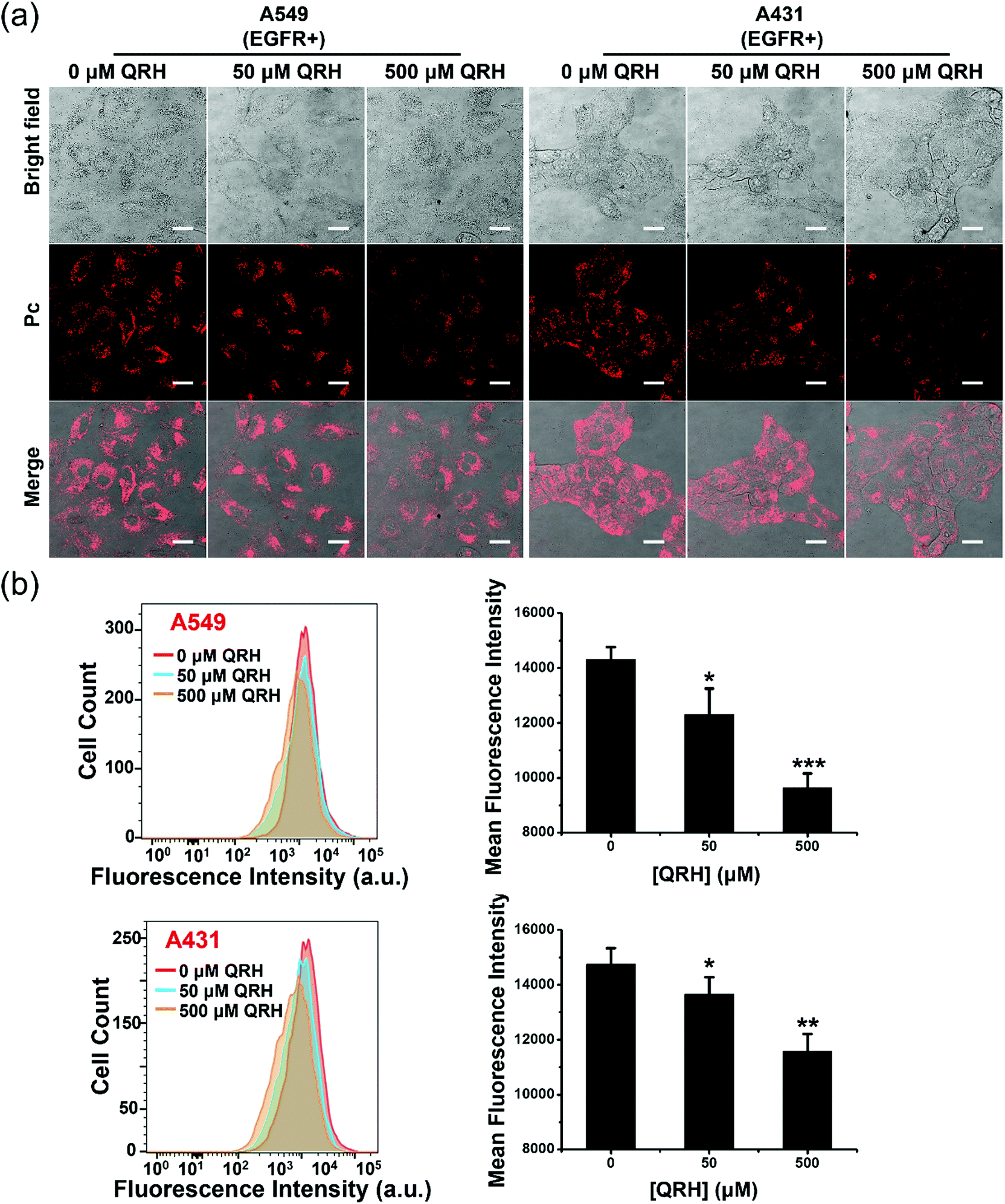 | ||
| Fig. 5 (a) Confocal images of A549 and A431 cells being pretreated with culture medium in the absence or presence of free QRH (50 or 500 μM) for 1 h, and then with PDA-Dox-Pc-QRH ([Pc] = 0.5 μM) for 1 h followed by incubation in a NP-free medium for 7 h. Scale bar: 25 μm. (b) Flow cytometric measurements for the two cell lines after the treatment conditions described above. Data are reported as the mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. | ||
In vitro chemotherapy and PDT
The in vitro anticancer efficacy of PDA-Dox-Pc-QRH was then assessed. To differentiate the chemocytotoxic effect of the Dox from the PDT effect of the Pc in the NPs, a model nanosystem without the encapsulated Dox, labelled as PDA-Pc-QRH, was prepared similarly without adding Dox during the self-polymerisation of L-DOPA dimer. These NPs were characterised with TEM and DLS (Fig. S8, ESI†). The size, morphology, zeta potential and Pc loading were very similar to those of PDA-Dox-Pc-QRH (Table S1, ESI†) so that the results of these two nanosystems can be directly compared.The cytotoxicities of PDA-Pc-QRH and PDA-Dox-Pc-QRH against the four cell lines were studied, both in the dark and upon light irradiation (λ > 610 nm, 40 mW cm−2, 48 J cm−2), using a live/dead double staining protocol based on calcein acetoxymethyl ester (calcein AM) and ethidium homodimer-1 (EthD-1).40 As shown in Fig. 6a, for the EGFR-negative HepG2 and HEK293 cells, after the treatment with PDA-Pc-QRH or PDA-Dox-Pc-QRH ([Pc] = 100 nM or [Dox] = 90 nM), the cells remained largely viable as evidenced by the intense green intracellular calcein fluorescence regardless of whether light irradiation was applied or not. Red fluorescence of EthD-1, which reflects the number of dead cells, remained extremely weak. This was in accordance with the low cellular uptake of the NPs to trigger the release of Dox and Pc. In contrast, for the EGFR-positive A549 and A431 cells, the fluorescence of calcein was significantly reduced upon light irradiation for both the nanosystems, while notable fluorescence of EthD-1 was observed, indicating the presence of less viable cells and more dead cells under these conditions. These results could be explained by the selective uptake of these QRH-modified NPs and the PDT effect of the activated Pc. In the absence of light irradiation, the fluorescence intensity of EthD-1 was higher for PDA-Dox-Pc-QRH than that for PDA-Pc-QRH. The higher dark cytotoxicity of the former could be attributed to the chemocytotoxic effect of the released Dox.
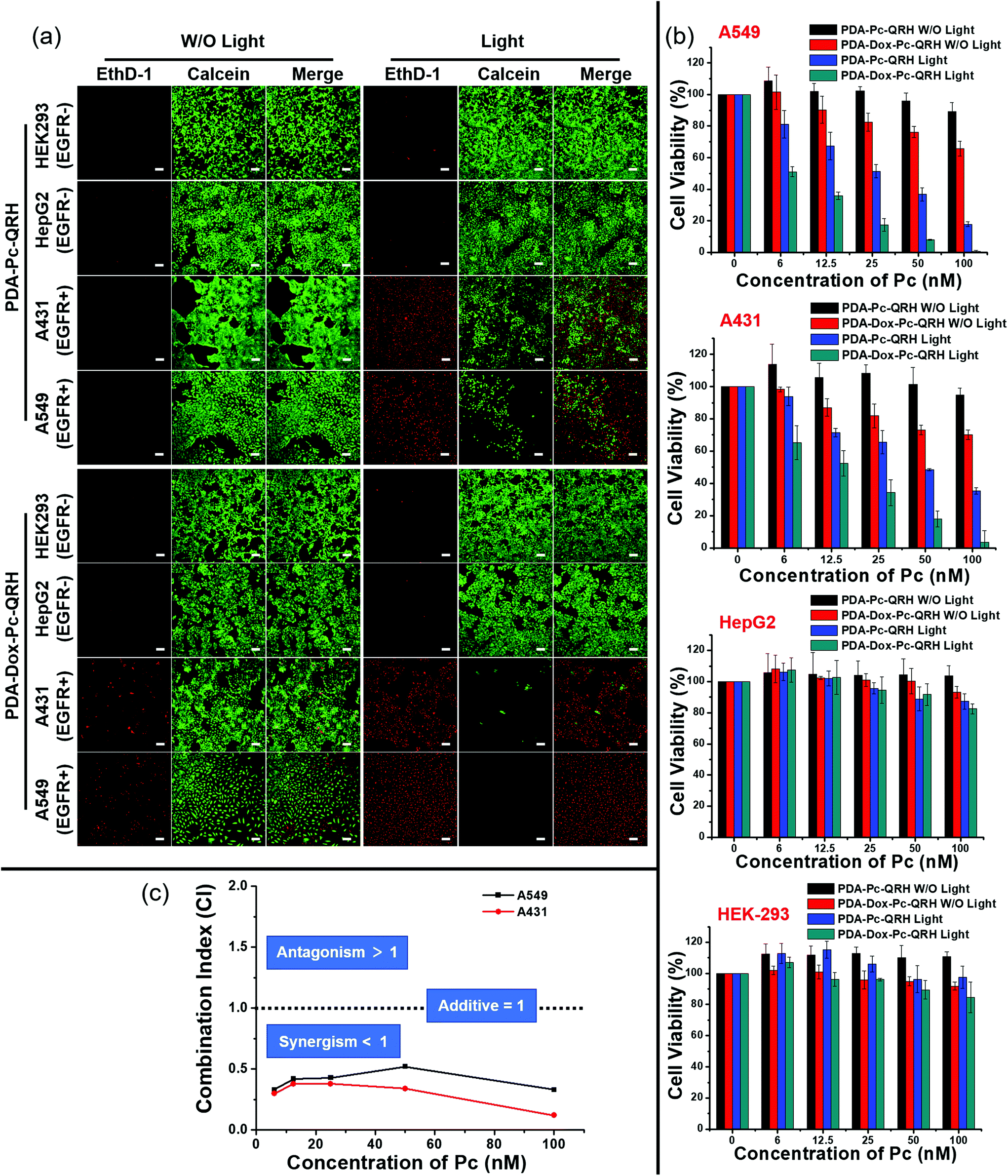 | ||
| Fig. 6 (a) Fluorescence images of A549, A431, HepG2 and HEK293 cells upon incubation with PDA-Pc-QRH or PDA-Dox-Pc-QRH ([Pc] = 100 nM or [Dox] = 90 nM) for 1 h, and then with a NP-free medium for 7 h with or without subsequent light irradiation (λ > 610 nm, 40 mW cm−2, 48 J cm−2). At 14 h post-irradiation, the cells were stained with calcein AM (for viable cells in green) and EthD-1 (for dead cells in red). Scale bar: 100 μm. (b) Flow cytometric measurements for the four cell lines that were treated as described above. Data are reported as the mean ± SD of three independent experiments. (c) Combination indices calculated according to the MTT results in figure (b) at different concentrations of Pc and Dox, keeping the loading mole ratio of Pc to Dox at 10:9. | ||
To quantify the cell viabilities under all these conditions, a MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay was used.41 The results as summarised in Fig. 6b were in good agreement with the live-dead imaging studies described above. For the EGFR-negative HepG2 and HEK293 cells, over 80% of the cells remained alive after the treatment with either PDA-Pc-QRH or PDA-Dox-Pc-QRH regardless of whether light irradiation was applied. This indicated that both nanosystems exhibited low cytotoxicity towards these two cell lines both in the dark and upon irradiation. For the EGFR-positive A549 and A431 cells, the cell viabilities varied significantly under different conditions. While PDA-Pc-QRH was essentially non-cytotoxic in the dark, PDA-Dox-Pc-QRH could kill ca. 30% of the cells at a Pc concentration of 100 nM (equivalent to 90 nM of Dox) even without light irradiation. The cytotoxicity could be attributed to the released Dox in PDA-Dox-Pc-QRH. Upon light irradiation, both nanosystems exhibited higher cytotoxicity due to the PDT effect of the released Pc. In particular, PDA-Dox-Pc-QRH could eradicate essentially all the cells at a Pc concentration of 100 nM. The IC50 values, defined as the dye concentrations required to kill 50% of the cells, were found to be 27.6 nM (for A549) and 47.8 nM (for A431) for PDA-Pc-QRH and 6.5 nM (for A549) and 14.1 nM (for A431) for PDA-Dox-Pc-QRH (all with respect to the concentration of Pc). The higher potency of the latter was clearly due to the additional cytotoxic effect of Dox.
To reveal whether the two cytotoxic effects exerted in a synergistic manner, the combination indices (CI) at different Pc concentrations were calculated based on the dose-dependent survival curves of PDA-Pc-QRH with light irradiation (PDT only), PDA-Dox-Pc-QRH without light irradiation (chemotherapy only) and PDA-Dox-Pc-QRH with light irradiation (both PDT and chemotherapy) for both A549 and A431 cell lines. It is noted that a CI value lower than 1 indicates synergism, a CI value of 1 indicates an additive effect and a CI value higher than 1 indicates antagonism.42 As shown in Fig. 6c, the CI values were consistently lower than 1 at all the Pc concentrations for both cell lines. The results clearly showed that PDA-Dox-Pc-QRH was a very potent therapeutic nanosystem that exhibits synergistic chemotherapy and PDT.
In vivo fluorescence imaging
To evaluate the in vivo tumour-localisation property of PDA-Dox-Pc-QRH, its biodistribution in tumour-bearing nude mice was examined by whole-body fluorescence imaging. A431 cells were used to induce the tumour subcutaneously owing to their high EGFR expression and fast growth rate.43 These A431 tumour-bearing nude mice were administered with an intravenous dose of these NPs ([Pc] = 5 μM, 200 μL). The fluorescence due to Pc at the tumour site was monitored continuously for 120 h (Fig. 7a, upper panel). During the first 8 h, fluorescence could hardly be seen in the tumour possibly because the NPs required a longer time to accumulate and disassemble in the tumour. The intratumoural fluorescence intensity grew steadily and continuously, showing that the NPs were accumulated in the tumour gradually and the immobilised Pc molecules were also released therein. To reveal whether the immobilised QRH peptide contributed to the tumour localisation, the mice were also injected with free QRH peptide (200 μM) as a competitive ligand of EGFR together with the NPs. It was found that the fluorescence intensity at the tumour was significantly reduced over the whole period of time (Fig. 7a, lower panel). After 120 h, the intensity was reduced by ca. 50% in the presence of the competitive free peptide (Fig. 7b). The results suggested that apart from the potential EPR effect arising from the nanoscale nature of the NPs, PDA-Dox-Pc-QRH also exhibited an active EGFR-targeting effect due to the QRH peptide chains on the surface.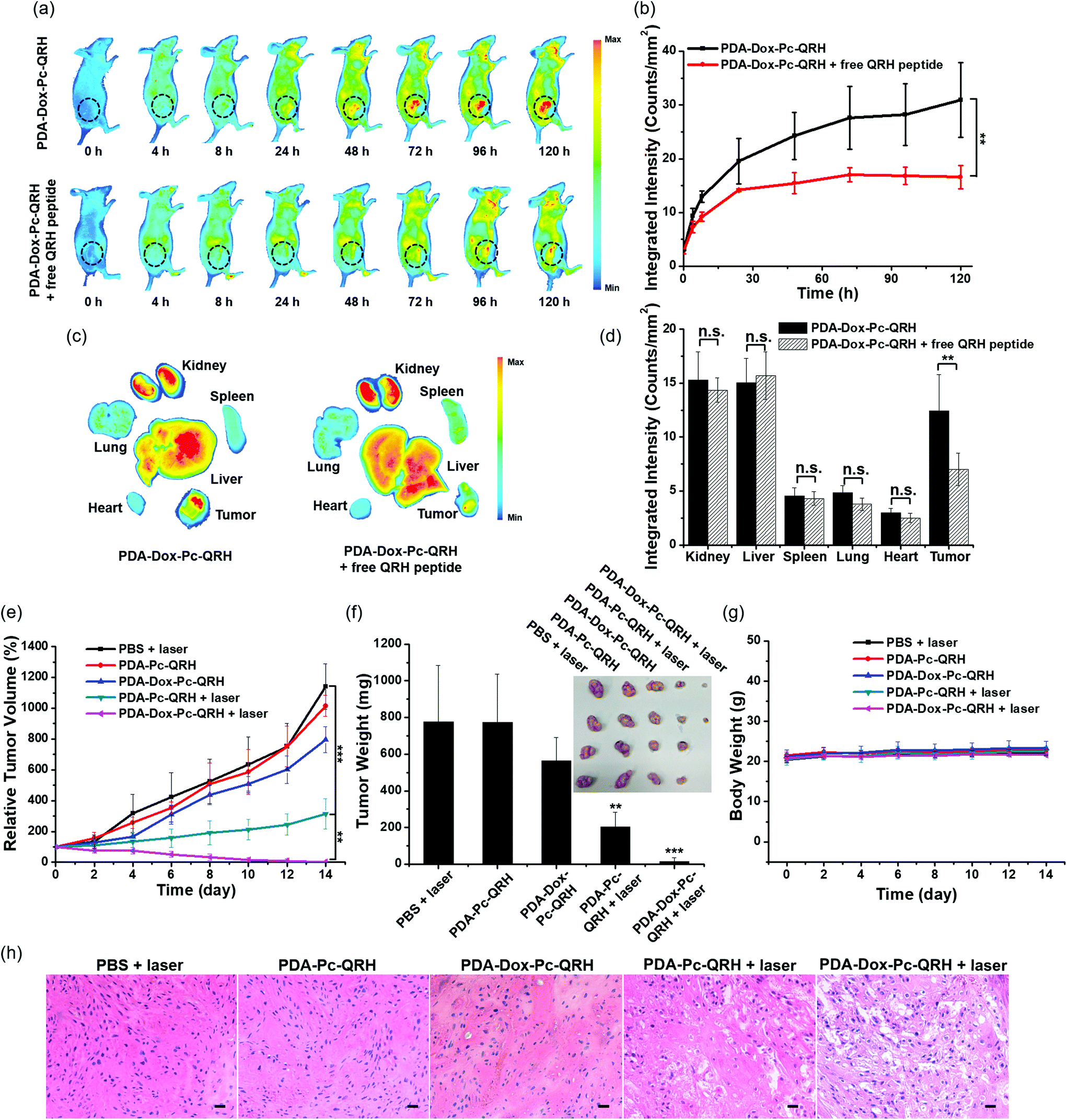 | ||
| Fig. 7 (a) Whole-body fluorescence images of A431 tumour-bearing nude mice before and after intravenous injection with PDA-Dox-Pc-QRH ([Pc] = 5 μM, 200 μL) without (upper panel) or with (lower panel) free QRH peptide (200 μM). The tumours are indicated by black circles. (b) The corresponding fluorescence intensities (per unit area) at the tumour sites at different time points. (c) Ex vivo fluorescence images of the tumour and major organs harvested at 120 h post-administration. (d) Quantification of the fluorescence intensities (per unit area) at the tumour and major organs harvested at 120 h post-administration. (e) Tumour growth curves of tumour-bearing nude mice after different treatments. The mice were intravenously injected with PDA-Dox-Pc-QRH, PDA-Pc-QRH or PBS and the tumour region was irradiated with laser (675 nm, 0.9 W) for 10 min (20 J cm−2) to trigger the PDT effect or without irradiation. (f) The corresponding tumour weights and images collected on Day 14 for different treatment groups. (g) Monitoring the body weights of the mice in different experimental groups. (h) Hematoxylin and eosin (H&E) staining of the tumour sections collected from the mice in different experimental groups. Scale bar: 200 μm. Date in figure (b) and (d)–(g) are reported as the mean ± SD (n = 4). n.s., not significant; **p < 0.01; ***p < 0.001. | ||
In addition, we examined the organ-level distribution of PDA-Dox-Pc-QRH by sacrificing the mice and harvesting the tumour and major internal organs, including kidney, liver, spleen, lung and heart at 120 h post-injection for ex vivo fluorescence imaging (Fig. 7c, left part). It was found that the fluorescence intensity of the tumour was remarkably high and comparable with those of the kidney and liver, while the intensities of spleen, lung and heart were significantly lower (Fig. 7d). The high accumulation of NPs in kidney and liver can be explained by the excretion mechanisms of the renal and hepatobiliary systems for the clearance of NPs.44 Coinjection of PDA-Dox-Pc-QRH with free QRH peptide led to a significant reduction (by 46%) in tumour accumulation, while the effect on all the organs was not significant (Fig. 7c, right part, and Fig. 7d). This observation further supported the in vivo EGFR-targeting effect of the immobilised QRH peptide.
In vivo PDT
Finally, the in vivo therapeutic effect of PDA-Dox-Pc-QRH was investigated and compared with that of PDA-Pc-QRH. A431 tumour-bearing mice were treated with an intravenous dose of these nanosystems ([Pc] = 5 μM, 200 μL). As shown in Fig. 7a, the tumour showed the strongest fluorescence intensity at 120 h after the injection, reflecting the highest drug accumulation at this time point. Hence, the tumour was irradiated at 120 h post-injection with a diode laser at 675 nm for 10 min (20 J cm−2). The tumour size was then monitored continuously for 14 days. The corresponding tumour growth curves as well as those for some control conditions are shown in Fig. 7e. The images of the mice before and after the different treatments are also given in Fig. S9 (ESI†). It can be seen that PDA-Dox-Pc-QRH with laser irradiation showed the highest antitumour effect, which could completely ablate the tumour after about 10 days. For PDA-Pc-QRH, laser irradiation could also suppress the tumour growth effectively. In contrast, while PDA-Dox-Pc-QRH without laser irradiation could still exert a weak tumour-retardation effect, PDA-Pc-QRH without laser irradiation basically had no effect on the tumour growth as in the case of the negative control group by simply injecting PBS into the mice with laser irradiation. The weight and image of the tumours for each group were also determined. As shown in Fig. 7f, the results were in good agreement with the tumour growth curves shown in Fig. 7e. As expected, with two therapeutic components, PDA-Dox-Pc-QRH showed the highest potency. Upon tumour localisation via the potential EPR and EGFR-targeting effects and cellular internalisation, these NPs were disassembled to release the chemotherapeutic Dox and photocytotoxic Pc for eradicating the tumour effectively. The body weights of the mice for each group were also monitored over a period of 14 days. It was found that they were essentially unchanged (Fig. 7g), suggesting that the side effects of all these treatments were not notable.After 14 days of all these treatments, all the mice were sacrificed and the tumour and some major organs, including heart, liver, spleen, lung and kidney of each mouse were harvested for histological examination. For the tumour tissues for the treatment groups of PDA-Pc-QRH and PDA-Dox-Pc-QRH with laser irradiation, severe tissue damage was observed, but not on those for the other three control groups (Fig. 7h). In contrast, there was no major abnormality in the stained organ slides for all the treatment groups (Fig. S10, ESI†), showing again the minimal toxicity of these two nanosystems in the absence of direct laser irradiation.
Conclusion
A novel ROS-responsive PDA-based multifunctional nanosystem has been developed for synergistic chemo and photodynamic therapy of cancer. The preparation of these NPs involves simply mixing of a thioketal-linked L-DOPA dimer with Dox in an alkaline alcohol–water mixture, followed by conjugation with molecules of a Pc-based photosensitiser and a EGFR-targeting peptide on the surface of the resulting NPs. These NPs can be selectively taken up by EGFR-positive cancer cells and degraded in response to the elevated levels of ROS in cancer cells through cleavage of the thioketal linkers, triggering the release of the encapsulated Dox and the activation of the immobilised Pc. Upon light irradiation (λ > 610 nm), the released Pc can generate additional ROS for cell killing and promoting the degradation of the NPs and further release of the two therapeutic components. Both in vitro and in vivo studies showed that the chemotherapeutic effect of Dox and the PDT effect of Pc in PDA-Dox-Pc-QRH work in a synergistic manner, rendering this nanosystem an effective anticancer agent. This work provides insights for the design of smart and efficient ROS-responsive biodegradable PDA-based nanomedicines for various biomedical applications with translational potential.Experimental section
General
All reactions were performed under a nitrogen atmosphere. Chromatographic purification was performed on silica gel (Macherey Nagel, 230–400 mesh) with the indicated eluents. All solvents and reagents were of reagent grade and used as received. 1H and 13C{1H} NMR spectra were recorded on a Bruker Avance III HD 500 spectrometer (1H, 500 MHz; 13C, 125.7 MHz). Spectra were referenced internally using the residual solvent [1H, δ = 7.26 (for CDCl3), δ = 2.50 (for DMSO-d6), δ = 4.79 (for D2O)] or solvent [for 13C, δ = 77.2 (for CDCl3), δ = 39.5 (for DMSO-d6)] resonances relative to SiMe4. Electrospray ionisation (ESI) and MALDI-TOF mass spectra were recorded on a Thermo Finnigan MAT 95 XL mass spectrometer and a Bruker Autoflex speed MALDI-TOF mass spectrometer respectively. UV-Vis absorption spectra and steady-state fluorescence spectra were taken with a Cary 5G UV-Vis-NIR spectrophotometer and a Hitachi F-7000 spectrofluorometer respectively.Measurements of hydrodynamic size and zeta potential were performed with a DelsaMax Pro analyser. For TEM study, 5 μL of NP sample in Milli-Q water was deposited on carbon film-coated copper grids (200 mesh; Beijing Zhongjingkeyi Technology Co. Ltd) and air-dried before the TEM images were taken using a FEI Tecnai G2 Spirit transmission electron microscope operated at 120 kV acceleration voltage.
HPLC separation was performed by using a HPLC system equipped with a Waters 1525 binary HPLC pump, a Waters 2998 photodiode array detector, a Waters 2707 autosampler and a Waters reverse-phase chromatographic column (XBridge-C18; 5 mm, 10 mm × 250 mm).
Preparation of compound 3
Compounds 1 (2.0 g, 3.8 mmol) and 2 (0.3 g, 1.5 mmol) were dissolved in CH2Cl2. HBTU (2.3 g, 6.0 mmol) was then added to the mixture at 0 °C, followed by the addition of DIPEA (1 mL, 5.7 mmol) in dropwise over 10 min. The resulting mixture was stirred at room temperature for 15 h, and then washed with saturated brine. The organic phase was separated, and the aqueous phase was extracted with CH2Cl2. The combined organic phase was dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with a mixture of ethyl acetate and hexane (1:10, v/v) as eluent to yield 3 (1.3 g, 71%). 1H NMR (500 MHz, CDCl3): δ 6.70 (d, J = 8.0 Hz, 2 H, ArH), 6.61 (d, J = 2.0 Hz, 2 H, ArH), 6.56 (dd, J = 2.0, 8.0 Hz, 2 H, ArH), 4.98 (d, J = 8.0 Hz, 2 H, NH), 4.48–4.52 (m, 2 H, CH), 4.17–4.27 (m, 4 H, CH2), 2.90–2.99 (m, 4 H, CH2), 2.80 (t, J = 7.5 Hz, 4 H, CH2), 1.58 (s, 6 H, CH3), 1.40 (s, 18 H, CH3), 0.96 (s, 18 H, CH3), 0.95 (s, 18 H, CH3), 0.16–0.18 (m, 24 H, CH3). 13C{1H} NMR (125.7 MHz, CDCl3): δ 171.6, 155.0, 146.7, 145.9, 128.9, 122.3, 122.2, 121.0, 79.8, 64.2, 56.5, 54.3, 37.4, 31.0, 28.8, 28.3, 26.0, 25.9, 18.4, −4.0, −4.1. HRMS (ESI): m/z calcd for C59H106N2NaO12S2Si4 [M + Na]+: 1233.6162, found: 1233.6156.
Preparation of compound 4
A solution of Bu4NF in THF (1.0 M, 5 mL, 5 mmol) was added to a solution of 3 (1.3 g, 1.1 mmol) in THF (20 mL) at 0 °C. After stirring at this temperature for 1 h, the mixture was quenched with saturated NH4Cl solution (20 mL) and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with a mixture of ethyl acetate and hexane (1:1, v/v) as eluent to give 4 (0.46 g, 55%). 1H NMR (500 MHz, DMSO-d6): δ 8.73–8.76 (m, 4 H, OH), 7.17 (d, J = 8.0 Hz, 2 H, ArH), 6.59–6.62 (m, 4 H, ArH and NH), 6.45 (dd, J = 2.0, 8.0 Hz, 2 H, ArH), 4.09–4.21 (m, 4 H, CH2), 4.01–4.05 (m, 2 H, CH), 2.65–2.81 (m, 8 H, CH2), 1.55 (s, 6 H, CH3), 1.34 (s, 18 H, CH3). 13C{1H} NMR (125.7 MHz, DMSO-d6): δ 172.6, 155.9, 145.4, 144.3, 128.6, 120.2, 116.9, 115.8, 78.8, 64.0, 56.5, 56.1, 36.4, 31.1, 30.1, 28.6. HRMS (ESI): m/z calcd for C35H50N2NaO12S2 [M + Na]+: 777.2703, found: 777.2694.
Preparation of L-DOPA dimer 5
A solution of 4 (0.46 g, 0.6 mmol) in CH2Cl2 (30 mL) was mixed with TFA (3 mL) at 0 °C. The mixture was stirred at room temperature for 4 h, and then evaporated under reduced pressure. The residue was recrystallised from diethyl ether to give 5 as a white precipitate (0.34 g, 100%). 1H NMR (500 MHz, D2O): δ 6.84 (d, J = 8.0 Hz, 2 H, ArH), 6.73 (d, J = 1.5 Hz, 2 H, ArH), 6.64 (dd, J = 1.5, 8.0 Hz, 2 H, ArH), 4.28–4.37 (m, 6 H, CH and CH2), 3.05–3.14 (m, 4 H, CH2), 2.82 (d, J = 6.0 Hz, 4 H, CH2), 1.56 (s, 6 H, CH3). 13C{1H} NMR (125.7 MHz, D2O): δ 169.4, 144.3, 143.7, 125.9, 121.6, 116.8, 116.4, 65.5, 55.6, 53.9, 35.2, 29.8, 28.2. HRMS (ESI): m/z calcd for C25H35N2O8S2 [M + H]+: 555.1835, found: 555.1829.Preparation of QRH
Sieber amide resin (150 mg) was used as the solid support. 1-Hydroxybenzotriazole (HOBt) and HBTU were used as the activating agents of the carboxyl group. Commercially available Fmoc-protected amino acids were coupled to the resin sequentially by a Fmoc-mediated solid-phase peptide synthesis protocol. After coupling of the last amino acid, the resin was washed with DMF and CH2Cl2, and then dried in vacuo. The remaining protecting groups on the peptide and the resin were removed by the treatment with a mixture of TFA (95%), TIS (2.5%) and H2O (2.5%) for 3 h. The resin was removed by filtration and the filtrate was precipitated by adding diethyl ether. The yellow solid obtained was redissolved in H2O and then purified by HPLC. MS (MALDI-TOF): an isotopic cluster with the lowest m/z value calculated for C51H86N22O17S [M]: 1310.626; found: 1310.958.Preparation of PDA-Dox
An ammonia aqueous solution (NH4OH, 0.2 mL) was mixed with ethanol (6 mL) and deionised water (24 mL). Dox (4, 6, 8, 10, or 12 μg mL−1) was added and the resulting mixture was stirred at room temperature for 30 min. The L-DOPA dimer 5 (40 mg) was dissolved in deionised water (2 mL) and then the solution was injected into the above mixture. The colour of this solution changed from purple to pale brown and gradually to dark brown. The reaction was allowed to proceed for 36 h. The NPs of PDA-Dox were obtained by centrifugation at 9000 rpm for 10 min. After washing with water for three times, the precipitate was freeze-dried.Preparation of PDA-Dox-Pc
PDA-Dox (11.2 mg) and Pc (1.6 mg) were mixed in Tris buffer (pH 8.5, 10 mM, 300 mL) with 0.5% Tween 20. After stirring at room temperature for 14 h, the mixture was centrifuged to collect the NPs. The absorbance of the supernatant was determined. The amount of Pc conjugated to PDA-Dox was calculated by subtracting the amount of Pc in the supernatant after centrifugation from the amount of Pc in the initial reaction mixture, from which the loading of Pc in these NPs was determined. The precipitated NPs were washed with water twice and then resuspended in deionised water (300 mL) for further conjugation.Preparation of PDA-Dox-Pc-QRH
The QRH peptide in water (0.1 M, 100 μL) was added to the above PDA-Dox-Pc in water. The mixture was incubated at 4 °C for 14 h followed by centrifugation. The precipitate was collected and washed with water for three times. The resulting NPs were suspended in 10 mL of deionised water for further studies.Preparation of PDA-Pc-QRH
An ammonia aqueous solution (NH4OH, 0.2 mL) was mixed with ethanol (6 mL) and deionised water (24 mL) under mild stirring at room temperature for 30 min. The L-DOPA dimer 5 (40 mg) was dissolved in deionised water (2 mL) and then the solution was injected into the above mixture. The reaction was allowed to proceed for 30 h. The NPs formed were obtained by centrifugation at 9000 rpm for 10 min followed by washing with water for three times. The resulting NPs were then conjugated with Pc and QRH using the aforementioned procedure.Determination of fluorescence quantum yields
Fluorescence quantum yields (ΦF) were determined by the equation: ΦF(s) = (Fs/Fref)(Aref/As)(ns2/nref2)ΦF(ref), where subscript s refers to the sample and ref stands for the reference. F, A and n are the measured fluorescence (area under the emission peak), the absorbance at the excitation position (610 nm) and the refractive index of the solvent respectively.45 Unsubstituted ZnPc in DMF was used as the reference [ΦF(ref) = 0.28].35 To minimise reabsorption of radiation by ground-state species, the emission spectra were obtained in a very dilute solution in which the absorbance at the excitation wavelength was less than 0.05.Determination of singlet oxygen quantum yields
Singlet oxygen quantum yields (ΦΔ) were calculated by using the unsubstituted ZnPc in DMF as the reference (ΦΔ = 0.56) and DPBF as the singlet oxygen scavenger.35 A mixture of DPBF (90 μM) and the photosensitiser (6 μM) in DMF was irradiated with red light from a 100 W halogen lamp after passing through a water tank for cooling and a colour filter (Newport) with a cut-on wavelength at 610 nm. The absorbance of DPBF at 415 nm was monitored along with the irradiation time. The ΦΔ values were calculated according to the equation: ΦΔ(s) = ΦΔ(ref) (Ws × Iabs(ref))/(Wref × Iabs(s)), where ΦΔ(ref) is the ΦΔ of ZnPc in DMF, Ws and Wref are the DPBF photobleaching rates in the presence of the photosensitiser and ZnPc respectively, and Iabs(s) and Iabs(ref) are the rates of light absorption by the photosensitiser and ZnPc respectively.46ROS-responsive fluorescence emission
PDA-Dox-Pc-QRH ([Pc] = 2.0 μM) was mixed with different concentrations of H2O2 (0, 0.1 and 1 mM) in PBS with 0.5% Tween 20. The mixtures were incubated at 37 °C over a period of time. To study the effect of light irradiation, a 100 W halogen lamp was used. The light was passed through a water tank for cooling and a colour glass filter (Newport) cut on at λ = 610 nm and applied to the mixtures for 20 min. The mixtures were then centrifuged, and the supernatant was extracted to obtain the release profile using fluorescence spectroscopy.ROS-responsive singlet oxygen generation
PDA-Dox-Pc-QRH ([Pc] = 2.0 μM) was treated with different concentrations of H2O2 (0, 0.1 and 1 mM) in PBS with 0.5% Tween 20 at 37 °C for 24 h. The supernatant collected (396 μL) was mixed with an aqueous solution of DPBF (9 mM, 4 μL). Before each measurement of the absorbance at 415 nm, the solution was illuminated with red light that was emitted from a 100 W halogen lamp and passed through a water tank for cooling and a colour glass filter (Newport) cut on at λ = 610 nm for 5 s. The decay of DPBF was monitored over a total irradiation time of 120 s.Cell lines and culture conditions
A549 (ATCC, no. CCL-185), A431 (ATCC, no. CRL-1555), HepG2 (ATCC, no. HB-8065) and HEK293 (ATCC, no. CRL-1573) cells were maintained in DMEM supplemented with FBS (10%) and penicillin–streptomycin (100 units per mL and 100 μg mL−1 respectively). All the cells were grown in a humidified incubator with 5% CO2 at 37 °C.TEM imaging of cells
All reagents and accessories for preparing ultrathin sections were purchased from Electron Microscopy Sciences (EMS). Freshly harvested cell pellets were fixed in glutaraldehyde (2.5% in phosphate buffer, pH = 7.2–7.4; EMS) for 2 h and stained with osmium tetroxide (1%; EMS) for another 2 h. The cell pellets were then gradually dehydrated in an increasing ethanol gradient. The samples were then embedded in Epon 812 resins (EMS) and polymerised at 55 °C for 48 h. Ultrathin sections of ∼70 nm thick were prepared with a 45° diamond knife (Diatome), deposited onto 200-mesh copper grids and stained with 4% uranyl acetate (in 50% methanol/water) and Reynolds lead citrate for observation under a transmission electron microscope (Hitachi H7700) at a beam voltage of 100 kV.A431 cells were incubated with PDA-Dox-Pc-QRH ([Pc] = 0.50 μM or [Dox] = 0.45 μM) for 1 h and then with a NP-free medium for 7 h, followed by light irradiation for 20 min and further incubation for 2 h. The light was emitted from a 300 W halogen lamp and passed through a water tank for cooling and a colour glass filter (Newport) cut on at λ = 610 nm. The fluence rate (λ > 610 nm) was 40 mW cm−2. Illumination for 20 min led to a total fluence of 48 J cm−2.
Confocal laser scanning microscopy
Approximately 4 × 105 cells in DMEM (2 mL) were seeded on a confocal dish and incubated overnight at 37 °C with 5% CO2. The cells were treated with the NPs with or without subsequent light irradiation (λ > 610 nm, 40 mW cm−2, 48 J cm−2). After that, the medium was removed, and the cells were rinsed with PBS and examined with a Leica TCS SP8 high speed confocal microscope. The excitation and emission wavelengths of Pc were 638 and 650–750 nm respectively. The excitation and emission wavelengths of Dox were 488 and 500–600 nm respectively.Flow cytometry
Approximately 2 × 105 cells were inoculated into each of the wells in a 12-well plate and incubated in DMEM overnight at 37 °C and 5% CO2. The cells were treated with the NPs with or without subsequent light irradiation (λ > 610 nm, 40 mW cm−2, 48 J cm−2). After that, the cells were rinsed with PBS and harvested by adding 0.5 mL of 0.25% trypsin–ethylenediaminetetraacetic acid (EDTA) (Invitrogen). After adding 0.5 mL of medium to quench the activity of trypsin, the cell mixture was centrifuged at 1500 rpm for 3 min at room temperature. The cell pellet was washed with 1 mL of PBS and subject to centrifugation for three times. After resuspending the cells in 1 mL of PBS, their intracellular fluorescence intensities were measured by using a BD FACSVerse flow cytometer (Becton Dickinson) with 104 cells counted in each sample. For Pc, the excitation wavelength was 640 nm and the emission wavelength was 720–840 nm. For Dox, the excitation wavelength was 488 nm and the emission wavelength was 540–620 nm. Data collected were analysed by using the BD FAC-Suite. All experiments were performed in triplicate.Two-colour fluorescence cell viability assay
Cells were seeded onto a confocal dish at a density of 2 × 105 cells per well and incubated for 24 h. The culture medium was replaced by 1 mL of fresh medium containing PDA-Dox-Pc-QRH ([Pc] = 100 nM or [Dox] = 90 nM) or PDA-Pc-QRH ([Pc] = 100 nM). After incubating for 1 h, the cells were incubated in a NP-free medium for 7 h. The cells were rinsed with 100 μL of PBS twice, replenished with 1 mL of fresh medium and irradiated. The light was emitted from a 300 W halogen lamp and passed through a water tank for cooling and a colour glass filter (Newport) cut on at λ = 610 nm. The fluence rate (λ > 610 nm) was 40 mW cm−2. Illumination for 20 min led to a total fluence of 48 J cm−2. After incubation for further 14 h, the cells were stained with calcein AM and EthD-1 (Invitrogen) in the dark for 30 min to distinguish the live (green) from dead (red) cells. The cells were imaged under a confocal microscope (Leica TCS SP8 MP) to evaluate the drug efficacy.Photocytotoxicity
Approximately 2 × 104 cells were inoculated into each of the wells in a 96-well plate and incubated in DMEM overnight at 37 °C and 5% CO2. The cells were incubated with different concentrations of PDA-Pc-QRH or PDA-Dox-Pc-QRH for 1 h, followed by incubating in a NP-free medium for 7 h. The cells were rinsed with 100 μL of PBS twice and replenished with 100 μL of fresh medium. For the dark cytotoxicity assay, the plate was directly incubated at 37 °C overnight. For the photocytotoxicity test, the cells were illuminated at room temperature for 20 min and then incubated overnight. A solution of MTT (Sigma) in PBS (3 mg mL−1, 50 μL) was added to each well. After incubation for 4 h under the same condition, 100 μL of DMSO was added to each well and the plates were placed on a Bio-Rad microplate reader to detect the absorbance at 490 nm. The average absorbance of the blank wells (not seeded with cells) was subtracted from the measured absorbance values of wells of various treatment groups. Cell viability was determined by the equation: %Viability = [∑(Ai/Acontrol × 100)]/n, where Ai is the absorbance of the ith datum (i = 1, 2, …, n), Acontrol is the average absorbance of control wells in which the nanosystem was absent. The size of treatment group (n) is 4.Animal studies
All experiments involving live animals were performed in strict accordance with the animal experimentation guidelines and were approved by the Animal Experimentation Ethics Committee of The Chinese University of Hong Kong (CUHK) (ref. no. 20-028-GRF). Licence to conduct animal experiments was obtained from the Department of Health, Government of the Hong Kong Special Administrative Region. Female Balb/c nude mice (20–25 g), obtained from the Laboratory Animal Services Centre at CUHK, were kept under pathogen-free conditions with free access to food and water. A431 human epidermoid carcinoma cells (107 cells in 200 μL) were inoculated subcutaneously on the back of the mice. Once the tumours had grown to a size range of 60–100 mm3, the mice were fed on a low-fluorescence diet (TestDiet, AIN-93M) for 4 days.In vivo fluorescence imaging
PDA-Dox-Pc-QRH ([Pc] = 5 μM, 200 μL), with or without free QRH peptide (200 μM), was injected intravenously to tumour-bearing mice via the tail vein. In vivo fluorescence imaging was performed before and after the injection at different time points with an Odyssey infrared imaging system (excitation wavelength: 680 nm, emission wavelength: ≥700 nm). The images were digitised and analysed by the Odyssey imaging system software (no. 9201-500). The animals were sacrificed after the last scan. The tumour and various internal organs were harvested, and their fluorescence intensities were measured with the Odyssey infrared imaging system ex vivo. Four mice were used for each type of nanosystem.In vivo chemo and photodynamic therapy
A431 tumour-bearing mice were randomly divided into five treatment groups: (i) injection of PBS with laser irradiation, (ii) injection of PDA-Pc-QRH without laser irradiation, (iii) injection of PDA-Dox-Pc-QRH without laser irradiation, (iv) injection of PDA-Pc-QRH with laser irradiation and (v) injection of PDA-Dox-Pc-QRH with laser irradiation. Afterwards, PDA-Pc-QRH or PDA-Dox-Pc-QRH ([Pc] = 5 μM, 200 μL) was intravenously injected to the tumour-bearing mice via the tail vein. At 5 day of post-injection, the tumour was irradiated with a diode laser (Biolitec Ceralas) at 675 nm operated at 0.9 W. Illumination on a spot size of 26 cm2 for 10 min led to a total fluence of 20 J cm−2. Tumour size of the nude mice was monitored periodically for the next 14 days. Tumour volumes of all treatment groups were compared with the tumour volume of a control group of mice that did not receive injection of nanosystem but were still exposed to laser irradiation. Tumour volume was calculated using the following formula: Volume (mm3) = (length × width2)/2. At 14 day of post-treatment, the mice were sacrificed, and the internal organs and the tumour were harvested. Tissues were fixed with 4% neutral buffered formalin, dehydrated with an increasing gradient of ethanol and mixed with a solution of xylene and paraffin (1:1, v/v). Finally, the tissues were embedded in paraffin for sectioning and stained by H&E. The stained tissue sections were observed under a Ti-E motorised inverted fluorescence microscope (Nikon).
Statistical analysis
Data shown on figures were presented as the mean with the standard deviation. The data were analysed using Student's t-test with p values < 0.05 considered as significant; *p < 0.05; **p < 0.01; ***p < 0.001. Statistical calculations were performed using a Microsoft Excel spreadsheet (Microsoft Corporation, Redmond, WA, USA).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a General Research Fund from the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. 14306320).Notes and references
- J. Liebscher, Eur. J. Org. Chem., 2019, 4976 CrossRef CAS.
- S. El Yakhlifi and V. Ball, Colloids Surf., B, 2020, 186, 110719 CrossRef CAS.
- W. Cheng, X. Zeng, H. Chen, Z. Li, W. Zeng, L. Mei and Y. Zhao, ACS Nano, 2019, 13, 8537 CrossRef CAS PubMed.
- H. A. Lee, E. Park and H. Lee, Adv. Mater., 2020, 32, 1907505 CrossRef CAS PubMed.
- X. Xie, J. Tang, Y. Xing, Z. Wang, T. Ding, J. Zhang and K. Cai, Adv. Healthcare Mater., 2021, 10, 2002138 CrossRef CAS PubMed.
- P. Yang, S. Zhang, X. Chen, X. Liu, Z. Wang and Y. Li, Mater. Horiz., 2020, 7, 746 RSC.
- G. Dai, C. K. K. Choi, Y. Zhou, Q. Bai, Y. Xiao, C. Yang, C. H. J. Choi and D. K. P. Ng, Nanoscale, 2021, 13, 6499 RSC.
- C. K. K. Choi, J. Li, K. Wei, Y. J. Xu, L. W. C. Ho, M. Zhu, K. K. W. To, C. H. J. Choi and L. Bian, J. Am. Chem. Soc., 2015, 137, 7337 CrossRef CAS PubMed.
- Z. Wang, Y. Duan and Y. Duan, J. Controlled Release, 2018, 290, 56 CrossRef CAS PubMed.
- H. Li, D. Yin, W. Li, Q. Tang, L. Zou and Q. Peng, Colloids Surf., B, 2021, 199, 111502 CrossRef CAS PubMed.
- C. K. K. Choi, Y. T. E. Chiu, X. Zhuo, Y. Liu, C. Y. Pak, X. Liu, Y.-L. S. Tse, J. Wang and C. H. J. Choi, ACS Nano, 2019, 13, 5864 CrossRef CAS PubMed.
- M. Farokhi, F. Mottaghitalab, M. R. Saeb and S. Thomas, J. Controlled Release, 2019, 309, 203 CrossRef CAS PubMed.
- I. Singh, G. Dhawan, S. Gupta and P. Kumar, Front. Microbiol., 2021, 11, 607099 CrossRef PubMed.
- X. Liu, J. Cao, H. Li, J. Li, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 9384 CrossRef CAS PubMed.
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 2012, 64, 72 CrossRef.
- P. Jana, M. Shyam, S. Singh, V. Jayaprakash and A. Dev, Eur. Polym. J., 2021, 142, 110155 CrossRef CAS.
- Y. Arun, R. Ghosh and A. J. Domb, Adv. Funct. Mater., 2021, 31, 2010284 CrossRef.
- L. Dong, C. Wang, W. Zhen, X. Jia, S. An, Z. Xu, W. Zhang and X. Jiang, J. Mater. Chem. B, 2019, 7, 6172 RSC.
- M. Battaglini, A. Marino, A. Carmignani, C. Tapeinos, V. Cauda, A. Ancona, N. Garino, V. Vighetto, G. La Rosa, E. Sinibaldi and G. Ciofani, ACS Appl. Mater. Interfaces, 2020, 12, 35782 CrossRef CAS PubMed.
- B. Yang, Y. Chen and J. Shi, Chem. Rev., 2019, 119, 4881 CrossRef CAS PubMed.
- X. Xu, P. E. Saw, W. Tao, Y. Li, X. Ji, S. Bhasin, Y. Liu, D. Ayyash, J. Rasmussen, M. Huo, J. Shi and O. C. Farokhzad, Adv. Mater., 2017, 29, 1700141 CrossRef PubMed.
- Y. Wu, T. Guo, Y. Qiu, Y. Lin, Y. Yao, W. Lian, L. Lin, J. Song and H. Yang, Chem. Sci., 2019, 10, 7068 RSC.
- C.-C. Song, F.-S. Du and Z.-C. Li, J. Mater. Chem. B, 2014, 2, 3413 RSC.
- C. Tapeinos and A. Pandit, Adv. Mater., 2016, 28, 5553 CrossRef CAS PubMed.
- C. de Gracia Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758 CrossRef CAS PubMed.
- X. Zhang, B. Huang, Y. Shen, C. Yang, Z. Huang, Y. Huang, X. Xu, Y. Jiang, X. Sun, X. Li, M. Yan and C. Zhao, J. Mater. Chem. B, 2018, 6, 2347 RSC.
- M.-R. Ke, S.-F. Chen, X.-H. Peng, Q.-F. Zheng, B.-Y. Zheng, C.-K. Yeh and J.-D. Huang, Eur. J. Med. Chem., 2017, 127, 200–209 CrossRef CAS PubMed.
- C. Liu, Q. Liu, L. Chen, M. Li, J. Yin, X. Zhu and D. Chen, Adv. Healthcare Mater., 2020, 9, 2000899 CrossRef CAS PubMed.
- K. Zheng, X. Liu, H. Liu, D. Dong, L. Li, L. Jiang, M. Huang and C. Ding, ACS Appl. Mater. Interfaces, 2021, 13, 10674–10688 CrossRef CAS.
- P.-C. Lo, M. S. Rodríguez-Morgade, R. K. Pandey, D. K. P. Ng, T. Torres and F. Dumoulin, Chem. Soc. Rev., 2020, 49, 1041 RSC.
- Q. Zhou, Z. Li, J. Zhou, B. P. Joshi, G. Li, X. Duan, R. Kuick, S. R. Owens and T. D. Wang, Photoacoustics, 2016, 4, 43 CrossRef PubMed.
- E. Y. Xue, R. C. H. Wong, C. T. T. Wong, W.-P. Fong and D. K. P. Ng, RSC Adv., 2019, 9, 20652 RSC.
- D. Hong, H. Lee, B. J. Kim, T. Park, J. Y. Choi, M. Park, J. Lee, H. Cho, S.-P. Hong, S. H. Yang, S. H. Jung, S.-B. Ko and I. S. Choi, Nanoscale, 2015, 7, 20149 RSC.
- S. Z. F. Phua, C. Xue, W. Q. Lim, G. Yang, H. Chen, Y. Zhang, C. F. Wijaya, Z. Luo and Y. Zhao, Chem. Mater., 2019, 31, 3349 CrossRef CAS.
- J. C. H. Chu, M. L. Chin, C. T. T. Wong, M. Hui, P.-C. Lo and D. K. P. Ng, Adv. Ther., 2020, 4, 2000204 CrossRef.
- T. Entradas, S. Waldron and M. Volk, J. Photochem. Photobiol., B, 2020, 204, 111787 CrossRef CAS PubMed.
- J.-Y. Liu, X.-J. Jiang, W.-P. Fong and D. K. P. Ng, Org. Biomol. Chem., 2008, 6, 4560 RSC.
- F. Bernsmann, V. Ball, F. Addiego, A. Ponche, M. Michel, J. J. de Almeida Gracio, V. Toniazzo and D. Ruch, Langmuir, 2011, 27, 2819 CrossRef CAS PubMed.
- F. Zhang, S. Wang, L. Yin, Y. Yang, Y. Guan, W. Wang, H. Xu and N. Tao, Anal. Chem., 2015, 87, 9960 CrossRef CAS PubMed.
- A. Kummrow, M. Frankowski, N. Bock, C. Werner, T. Dziekan and J. Neukammer, Cytometry, Part A, 2013, 83A, 197 CrossRef CAS PubMed.
- H. Tada, O. Shiho, K. Kuroshima, M. Koyama and K. Tsukamoto, J. Immunol. Methods, 1986, 93, 157 CrossRef CAS PubMed.
- T.-C. Chou, Pharmacol. Rev., 2006, 58, 621 CrossRef CAS PubMed.
- I. Bortolomai, S. Canevari, I. Facetti, L. De Cecco, G. Castellano, A. Zacchetti, M. R. Alison and S. Miotti, Cell Cycle, 2010, 9, 1194 CrossRef CAS PubMed.
- D. Gao and P.-C. Lo, J. Controlled Release, 2018, 282, 46 CrossRef CAS PubMed.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107 CAS.
- S. E. Maree and T. Nyokong, J. Porphyrins Phthalocyanines, 2001, 5, 782 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr04278e |
| This journal is © The Royal Society of Chemistry 2021 |