
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A large, square-shaped, DNA origami nanopore with sealing function on a giant vesicle membrane†
Shoji
Iwabuchi
a,
Ibuki
Kawamata
ab,
Satoshi
Murata
a and
Shin-ichiro M.
Nomura
 *a
*a
aDepartment of Robotics, Tohoku University, Miyagi 980-8579, Japan. E-mail: Shinichiro.nomura.b5@tohoku.ac.jp
bNatural Science Division, Ochanomizu University, Tokyo 112-8610, Japan
First published on 1st February 2021
Abstract
Intaking molecular information from the external environment is essential for the normal functioning of artificial cells/molecular robots. Herein, we report the design and function of a membrane nanopore using a DNA origami square tube with a cross-section of 100 nm2. When the nanopore is added to a giant vesicle that mimics a cell membrane, the permeation of large external hydrophilic fluorescent molecules is observed. Furthermore, the addition of up to four ssDNA strands enables size-based selective transport of molecules. A controllable artificial nanopore should facilitate the communication between the vesicle components and their environment.
Bottom-up manufacturing from the molecular scale has been found to be attractive for creating artificial chemical systems such as artificial cells1 and molecular robots.2 Giant unilamellar vesicles (GUVs), which are micrometre-sized closed membranes, have been actively adopted to perform various biochemical reactions.3 The vesicle's lipid bilayer membrane, which is similar to a live cell membrane, separates its internal solution from the external environment and does not allow the exchange of molecules through the membrane. Therefore, it is essential to design and implement a system that permits molecular transport through the membrane. To achieve molecular transportation, many types of reconstituted membrane proteoliposomes have been developed.4 However, in the case of the pore-forming membrane proteins, the cross-sectional area of the pore is approximately 1–4 nm2, thus limiting the size of molecules that can be transported.5 Although de novo artificially designed membrane proteins have been reported,6 realising a flexible design of membrane proteins, such as variable pore size and complex functions, remains a major challenge.7
In contrast, advanced bottom-up nanostructure fabrication techniques have been developed by utilising self-assembly based on the complementarity of synthetic DNA in the past two decades.8 This technique has been used to synthesize artificial nanopores that mimic membrane proteins.9 In particular, three-dimensional DNA origami10 was used to synthesize synthetic nanopores.9g–k The reported DNA origami nanopores allow the transport of differently sized molecules by expanding their pore size9g–i and further modifying the molecules inside the pores to enable size-based selective molecular transport.9k Artificial nanopores that allow the passage of certain chemical species through GUV membranes are a realisation of molecular system compartments where chemical information can be exchanged, and they pass the information to macroscopic devices.
In this report, we describe a nanopore with a large diameter, built by DNA origami that can selectivity transport molecules based on a new approach. The fabricated artificial nanopore has a cross-section of approximately 100 nm2, which is about 60% larger than that of previously reported DNA origami nanopores that penetrate lipid membranes.9g–k As an additional functionality, we implemented a mechanism to block molecular permeation using four DNA strands, thereby improving the selectivity of molecular permeation by artificial nanopores.
The DNA origami artificial nanopore was designed using caDNAno11 software (Fig. S1, ESI†). M13mp18 DNA was used as the scaffold and assembled in a square lattice pattern. The nanopore is composed of two parts: a pore-penetrating region and a region that interacts with the lipid membrane (Fig. 1a). The pore has a diameter of 10 nm, and the region interacting with the lipid membrane consists of a double-stranded DNA with a length of 42 nm. In the region that interacts with the lipid membrane, a total of 47 DNA strands that can hybridise with cholesterol-modified single-stranded DNA (Anchor-DNA) are exploited to overcome the energy barrier for membrane insertion (Fig. 1b). Anchor-DNA is inserted into the lipid membrane owing to the hydrophobicity of cholesterol, and hybridisation with the artificial nanopores promotes the binding and penetration of the nanopores into the lipid membranes.
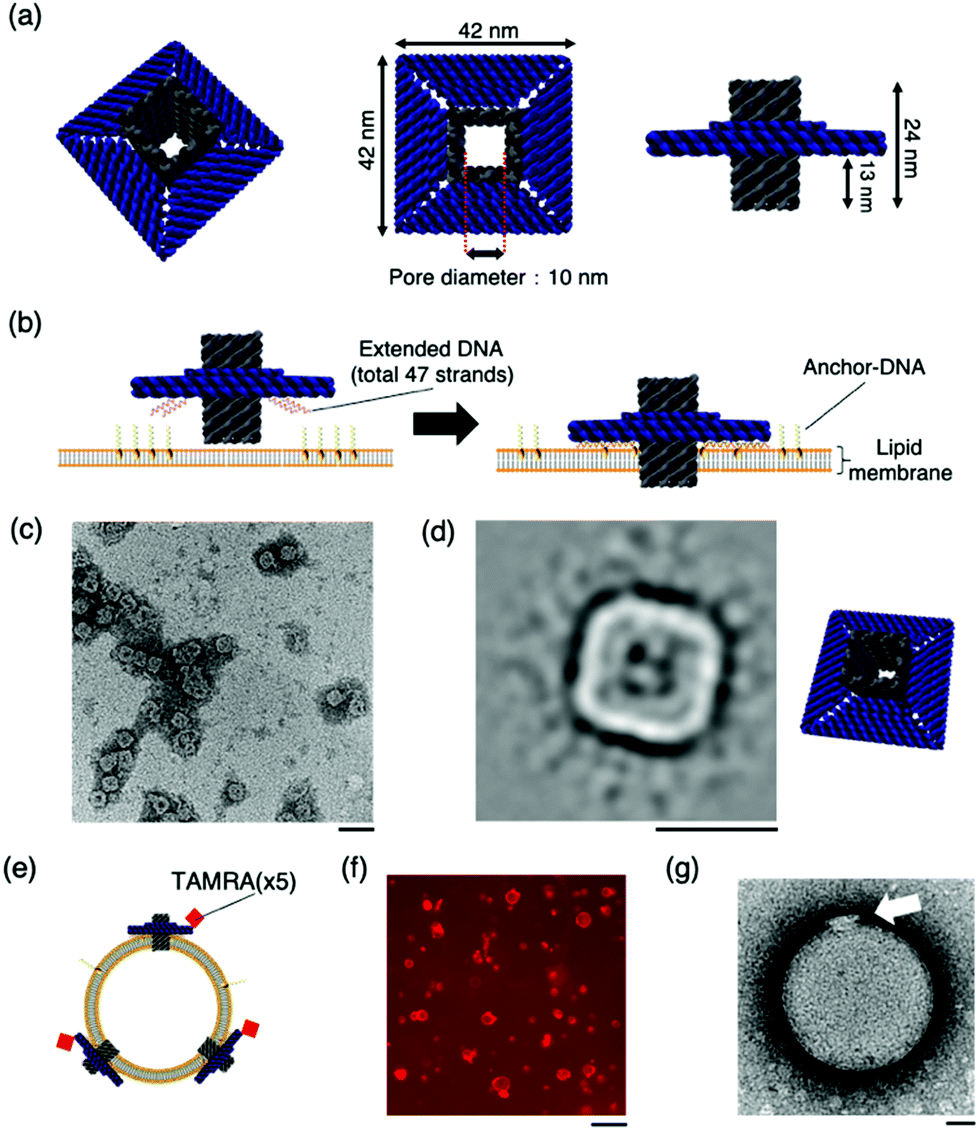 | ||
| Fig. 1 The design and structural confirmation of DNA origami artificial nanopores. (a) A schematic of the DNA origami nanopore. (b) A schematic of the insertion of the nanopore into the lipid bilayer. A total of 47 strands are present at the bottom of the nanopore, complementary to the Anchor-DNA, to achieve the binding and insertion of the nanopores into the lipid membrane. 47 Anchor-DNA strands are considered sufficient for the nanopore to penetrate the membrane.9g,k (c) A raw nsTEM image of the nanopore. Scale bar: 100 nm. (d) A class-average image of the nanopore (N = 1013). Scale bar: 50 nm. The figure on the right is the schematic view. (e) Schematic of nanopores with TAMRA-DNA binding to the vesicle membrane. (f) A representative confocal microscopy image of the sample incubated with TAMRA-modified nanopores and GUVs. Scale bar: 50 μm. (g) A representative nsTEM image of the nanopore incubated with LUV. Scale bar: 50 nm. | ||
The structure of the nanopore was confirmed by electrophoresis and negative-staining transmission electron microscopy (nsTEM). Agarose gel electrophoresis showed a shift in the band position compared to that of the scaffold DNA, indicating the formation of the desired structures (Fig. S2, ESI†). The nsTEM images provided a detailed view of the geometry of the structure. The square-planar section that interacts with the lipid membrane can be clearly observed from the raw data (Fig. 1c and Fig. S3, ESI†). The acquisition of two-dimensional class average images also confirm the formation of the pore in the centre of the structure (Fig. 1d). The size of the nanopore was also measured, and the total length was calculated to be 41.6 ± 1.2 nm (N = 20), which is comparable to the design value of 42 nm. These results substantiated the formation of the nanopore, as designed.
The binding of the artificial nanopore to the lipid membrane was confirmed using nsTEM and fluorescence microscopy images of the samples where the nanopore and Anchor-DNA strands were incubated with large unilamellar vesicles or GUVs with an average particle size of ∼200 nm and tens of micrometres, respectively. The binding of the nanopore to the vesicles was achieved by incubating the vesicles with Anchor-DNA, before mixing them with the nanopore solution (see methods). Fluorescence microscopy images show the binding of the nanopores to the GUV membrane surface (Fig. 1f). The details of the binding of the nanopore to the vesicle membrane surface were observed by nsTEM; images of the nanopore bound to the vesicle were obtained (Fig. 1g and Fig. S4, ESI†). The final concentration of the Anchor-DNA (500 nM) was chosen from the binding experiments with multiple concentrations of Anchor-DNA (200–2000 nM) (Fig. S5 and supporting note 1, ESI†).
To evaluate the transport of molecules through the lipid membrane via artificial nanopores, a dye influx assay with a hydrophilic fluorescent molecule (calcein, MW: 622.55) was conducted (Fig. 2a). Time-lapse images of the sample, when calcein was mixed with Anchor-DNA and the nanopore outside the GUVs, were obtained. Immediate binding of the artificial nanopores was observed under a confocal microscope when the nanopores were mixed with the GUV solution containing Anchor-DNA. The confocal microscopy time-lapse observation revealed the influx of calcein fluorescent molecules in multiple liposomes (Fig. 2b and c). Even in the presence of the nanopores, no remarkable change in the shape of the vesicles was observed within the observation period of ∼750 min. In contrast, a smaller fraction of the influx occurred in GUVs without the nanopores, as the negative control (Fig. 2d and e). This result shows that the number of GUVs that allow the influx of dye molecules increases by the mixing of GUVs with the artificial nanopores (Fig. 2f). This indicates that the passage of molecules occurs via the nanopores on the GUV membrane surface. A majority of the pores did not show any function, perhaps because they could not penetrate the membrane even when they were attached or because lipids were stuck in the pore.
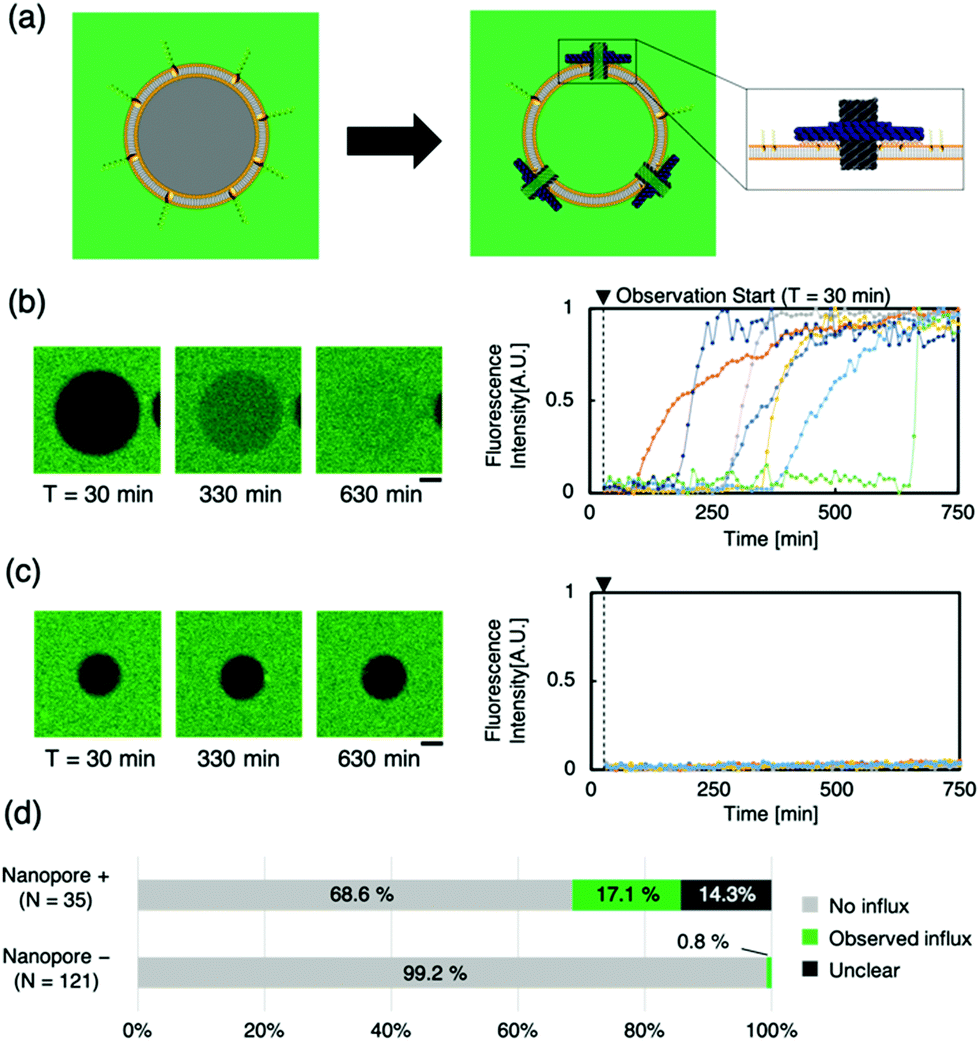 | ||
| Fig. 2 Evaluation of nanopore activity on the GUV through a calcein influx assay. (a) A schematic of the dye molecule influx assay. When the nanopore penetrates the GUV membrane surface and forms a molecular transport pathway, calcein outside the liposome flows inside. (b) and (c) show, respectively, the representative confocal microscopy time-lapse images and fluorescence intensity of GUVs with and without nanopores (N = 7 and 7, respectively). Scale bars: 10 μm. (d) The ratio of the GUVs with and without the influx of calcein in the presence and absence of nanopores. | ||
Next, we attempted to regulate molecular transport using “the lid”— ssDNA (Blocker-DNA) that binds across the pore. We modified the design of the nanopore to bind Blocker-DNA by extending the staple DNA strands from the upper part of the pore (Fig. 3a). Up to four Blocker-DNAs can bind to the pore, and it is expected that size-based selective molecular transport will be achieved.
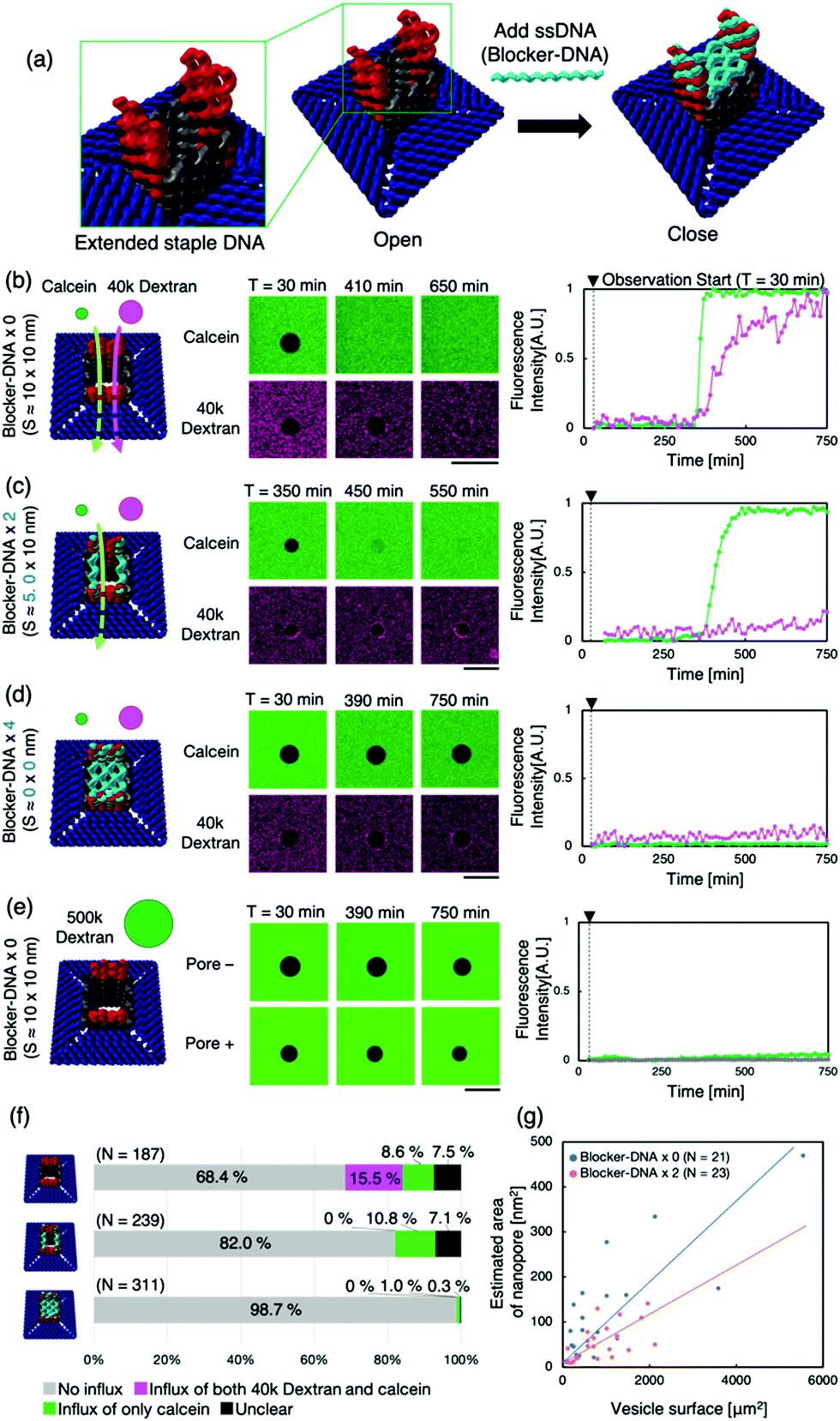 | ||
| Fig. 3 The change in the dye influx into the GUVs via the nanopore with the Blocker-DNA. (a) Schematic representation of the closure of the pore by the Blocker-DNA. The lid, four ssDNAs (Blocker-DNA, blue) that can bind to the staple DNA present on the upper part of the pore, allows the pore to be closed. (b), (c), and (d) show the schematic figure of the passage/blockage of the dye molecules by the Blocker-DNA, representative confocal microscope time-lapse images and fluorescence intensity of GUVs without Blocker-DNA, with two units of Blocker-DNA and with four units of Blocker-DNA, respectively. Scale bars: 50 μm. (e) Shows the results obtained when 500k dextran was used without Blocker-DNA. (f) The ratio of the GUVs with and without the influx of each dye molecule. (g) The plot of the surface area of the GUV versus the estimated area of the nanopore obtained from the data of calcein with different units of Blocker-DNA. | ||
The binding of the Blocker-DNA to the staple DNA was confirmed by polyacrylamide gel electrophoresis (PAGE) (Fig. S8, ESI†). To verify the selectiveness in molecular transport by the Blocker-DNA, the dye influx assay described earlier was performed by adding a larger molecule, dextran (MW = 40k, Rh ≈ 4.8 nm12), using the nanopore with 0 (“open” state), 2 (“semi-closed” state) or 4 (“closed” state) Blocker-DNA units. When the Blocker-DNA was absent (open state), an influx of both dye molecules, 40k dextran and calcein, into the GUVs was observed (Fig. 3b). In contrast, the influx of 40k dextran into GUVs was not observed whereas that of calcein was observed when two units of Blocker-DNA were used (Fig. 3c). Furthermore, when four units of Blocker-DNA were added, almost no influx of dye molecules occurred (Fig. 3d). We performed a similar influx assay using 500k dextran (Rh ≈ 15.9 nm12), which has a larger molecular weight and hydrodynamic radius than that of the nanopore, as a control experiment. As expected, no influx of fluorescent molecules was observed, regardless of the presence of the nanopore (Fig. 3e).
To investigate the influx phenomenon, the number and cross-sectional area of nanopores penetrating the GUV membrane surface was estimated using Fick's law (supporting note 2, ESI†). The actual area of the nanopores penetrating each GUV was calculated from the influx velocity–time constant and the estimated cross-sectional area of the single nanopore. The area of the nanopores formed correlated with the surface area of the GUVs under both conditions of 0 and 2 Blocker-DNAs (Fig. 3g). Importantly, the slope of the area of nanopores decreased with 2 Blocker-DNAs. These results show that the control of influx and size-selective molecular transport by Blocker-DNA was achieved, as planned.
Furthermore, we attempted to dissociate Blocker-DNA from the nanopore using a strand displacement reaction in situ (supporting note 3, ESI†). The dye influx assay (Fig. S14c, ESI†) showed a result similar to that with 2 Blocker-DNAs (Fig. 3f, middle), though all four Blocker-DNAs were added. It appears that the pore partly opened due to a signal. However, we are sure that another study is needed to study the critical conditions for the complete and accurate reaction.
In this work, we fabricated an artificial nanopore with a diameter of 10 nm through DNA origami and evaluated its molecular-permeation ability by observing the influx of fluorescent molecules using a confocal microscope. There are a few reports about successful dye transport through a synthetic DNA origami nanopore; previous reports presented square- and honeycomb-shaped nanopores with pore diameters of 4 nm9h and 9 nm,9k respectively. Our newly designed nanopore achieved dye molecule transportation as effectively as in these previous studies. The large nanopores were created in the membrane by pre-mixing cholesterol-modified DNA with GUV. We attempted to physically block the pore with ssDNA (Blocker-DNA), which can bind to the inlet of the square tube, as a means of introducing selectivity into the molecular transport via artificial nanopores. Blocker-DNA shields the inlet by binding across the pore. Since Blocker-DNAs are single-stranded DNAs, it is predicted that a non-negligible free space of 1–2 nm or less exists between DNAs. However, the existence of Blocker-DNAs resulted in a change in the influx of dye molecules into GUVs. This result suggests that the shielding of pores by ssDNA is effective for the transport of molecules larger than a certain size. Thomsen et al. also reported the shielding of pores by PEG-attached ssDNA. Our design suggests that this shielding can be achieved using only ssDNA strands. Burns et al. attempted to block pores using ssDNA in a non-origami 6HB DNA synthetic nanopore and observed that conductance decreased in the presence of ssDNA (i.e., through its blocked ion current).9d Through techniques, such as those using dsDNA, it may be possible to control the permeation of smaller molecules reversibly. The development of pairing nanopores might establish a molecular communication network by bridging liposomes.
A selective molecular machinery intaking information from the external environment will realise molecular communication between molecular robots and artificial cells.13 It would expand their ability for information processing for applications, such as drug delivery and artificial cell-based medicines.
We thank Dr Yuki Suzuki for constructive comments, Mr Koichiro Katayama for designing the proto-type nanopore, Dr Takamichi Miyazaki for TEM observation support, and Mrs Kaori Tanabe for confocal microscopy experiment support.
This work was supported by the JSPS and MEXT KAKENHI (grant numbers JP20H05701, JP20H00619, 20H00618, 18K18144, 19KK0261, and 17H00769); the FRIS Program for Creative Interdisciplinary Research; and AMED-CREST 20gm0810001h0106.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. L. Luisi, F. Ferri and P. Stano, Naturwissenschaften, 2006, 93(1), 1–13 CrossRef CAS; (b) P. Stano, Life, 2019, 9(1), 3.
- S. Murata, A. Konagaya, S. Kobayashi, H. Saito and M. Hagiya, New Gener. Comput., 2013, 31, 27–45 CrossRef.
- (a) P. Walde, K. Cosentino, H. Engel and P. Stano, ChemBioChem, 2010, 11, 848–865 CrossRef CAS; (b) P. Walde, R. Wick, M. Fresta, A. Mangone and P. L. Luisi, J. Am. Chem. Soc., 1994, 116(26), 11649–11654 CrossRef CAS; (c) W. Yu, K. Sato, M. Wakabayashi, T. Nakaishi, E. P. Ko-Mitamura, Y. Shima, I. Urabe and T. Yomo, J. Biosci. Bioeng., 2001, 92(6), 590–593 CrossRef CAS; (d) S. M. Nomura, K. Tsumoto, T. Hamada, K. Akiyoshi, Y. Nakatani and K. Yoshikawa, ChemBioChem, 2003, 4(11), 1172–1175 CrossRef CAS; (e) K. Kurihara, Y. Okura, M. Matsuo, T. Toyota, K. Suzuki and T. Sugawara, Nat. Commun., 2015, 6, 8352 CrossRef CAS; (f) S. Fujii, T. Matsuura, T. Sunami, Y. Kazuta and T. Yomo, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(42), 16796–16801 CrossRef CAS; (g) P. Van Nies, I. Westerlaken, D. Blanken, M. Salas, M. Mencía and C. Danelon, Nat. Commun., 2018, 9(1), 1583 CrossRef.
- (a) E. Racker and W. Stoeckenius, J. Biol. Chem., 1974, 249(2), 662–663 CrossRef CAS; (b) S. M. Nomura, S. Kondoh, W. Asayama, A. Asada, S. Nishikawa and K. Akiyoshi, J. Biotechnol., 2008, 133(2), 190–195 CrossRef CAS; (c) V. Noireaux and A. Libchaber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(51), 17669–17674 CrossRef CAS; (d) M. Kaneda, S. M. Nomura, S. Ichinose, S. Kondo, K. Nakahama, K. Akiyoshi and I. Morita, Biomaterials, 2009, 30, 3971–3977 CrossRef CAS; (e) K. Okamura, S. Matsushita, Y. Kato, H. Watanabe, A. Matsui, T. Oka and T. Matsuura, J. Biosci. Bioeng., 2019, 127(5), 544–548 CrossRef CAS; (f) K. Y. Lee, S.-J. Park, K. A. Lee, S.-H. Kim, H. Kim, Y. Meroz, L. Mahadevan, K.-H. Jung, T. K. Ahn, K. K. Parker and K. Shin, Nat. Biotechnol., 2018, 36, 530–535 CrossRef CAS; (g) S. Berhanu, T. Ueda and Y. Kuruma, Nat. Commun., 2019, 10, 1325 CrossRef; (h) Z. Chen, J. Wang, W. Sun, E. Archibong, A. R. Kahkoska, X. Zhang, Y. Lu, F. S. Ligler, J. B. Buse and Z. Gu, Nat. Chem. Biol., 2018, 14, 86–93 CrossRef CAS.
- B. C. Buddingh’, J. Elzinga and J. C. M. Van Hest, Nat. Commun., 2020, 11, 1652 CrossRef.
- S. E. Boyken, M. A. Benhaim, F. Busch, M. Jia, M. J. Bick, H. Choi, J. C. Klima, Z. Chen, C. Walkey, A. Mileant, A. Sahasrabuddhe, K. Y. Wei, E. A. Hodge, S. Byron, A. Quijano-Rubio, B. Sankaran, N. P. King, J. Lippincott-Schwartz, V. H. Wysocki, K. K. Lee and D. Baker, Science, 2019, 364, 658–664 CrossRef CAS.
- (a) A. Mariam and H. Bayley, Curr. Opin. Chem. Biol., 2016, 34, 117–126 CrossRef; (b) K. Brian and P. Bradley, Nat. Rev. Mol. Cell Biol., 2019, 20, 681–687 CrossRef.
- (a) N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS; (b) E. Winfree, F. Liu, L. A. Wenzler and N. C. Seeman, Nature, 1998, 394, 539–544 CrossRef CAS; (c) P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS; (d) N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2018, 3, 17068 CrossRef CAS.
- (a) J. R. Burns, K. Göpfrich, J. W. Wood, V. V. Thacker, E. Stulz, U. F. Keyser and S. Howorka, Angew. Chem., Int. Ed., 2013, 52, 12069–12072 CrossRef CAS; (b) A. Seifert, K. Göpfrich, J. R. Burns, N. Fertig, U. F. Keyser and S. Howorka, ACS Nano, 2015, 9, 1117–1126 CrossRef CAS; (c) K. Göpfrich, T. Zettl, A. E. C. Meijering, S. Hernández-Ainsa, S. Kocabey, T. Liedl and U. F. Keyser, Nano Lett., 2015, 15, 3134–3138 CrossRef; (d) J. R. Burns, A. Seifert, N. Fertig and S. Howorka, Nat. Nanotechnol., 2016, 11, 152–156 Search PubMed; (e) J. R. Burns, E. Stulz and S. Howorka, Nano Lett., 2013, 13, 2351–2356 CrossRef CAS; (f) K. Göpfrich, C. Y. Li, I. Mames, S. P. Bhamidimarri, M. Ricci, J. Yoo, A. Mames, A. Ohmann, M. Winterhalter, E. Stulz, A. Aksimentiev and U. F. Keyser, Nano Lett., 2016, 16, 4665–4669 CrossRef; (g) M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS; (h) S. Krishnan, D. Ziegler, V. Arnaut, T. G. Martin, K. Kapsner, K. Henneberg, A. R. Bausch, H. Dietz and F. C. Simmel, Nat. Commun., 2016, 7, 1–7 Search PubMed; (i) K. Göpfrich, C. Y. Li, M. Ricci, S. P. Bhamidimarri, J. Yoo, B. Gyenes, A. Ohmann, M. Winterhalter, A. Aksimentiev and U. F. Keyser, ACS Nano, 2016, 10, 8207–8214 CrossRef; (j) T. Diederichs, G. Pugh, A. Dorey, Y. Xing, J. R. Burns, Q. Hung Nguyen, M. Tornow, R. Tampé and S. Howorka, Nat. Commun., 2019, 10, 1–11 CrossRef; (k) R. P. Thomsen, M. G. Malle, A. H. Okholm, S. Krishnan, S. S. R. Bohr, R. S. Sørensen, O. Ries, S. Vogel, F. C. Simmel, N. S. Hatzakis and J. Kjems, Nat. Commun., 2019, 10, 1–10 CrossRef; (l) K. Göpfrich and U. F. Keyser, in Adv. Exp. Med. Biol., Springer, 2019, vol. 1174, pp. 331–370 Search PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS.
- S. M. Douglas, A. H. Marblestone, S. Teerapittayanon, A. Vazquez, G. M. Church and W. M. Shih, Nucleic Acids Res., 2009, 37, 5001–5006 CrossRef CAS.
- J. K. Armstrong, R. B. Wenby, H. J. Meiselman and T. C. Fisher, Biophys. J., 2004, 87, 4259–4270 Search PubMed.
- (a) Y. Sato, K. Komiya, I. Kawamata, S. Murata and S. I. M. Nomura, Chem. Commun., 2019, 55, 9084–9087 RSC; (b) Y. Sato, Y. Hiratsuka, I. Kawamata, S. Murata and S. M. Nomura, Sci. Robot., 2017, 2, eaal3735 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/d0cc07412h |
| This journal is © The Royal Society of Chemistry 2021 |