
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent polarity driven helicity inversion and circularly polarized luminescence in chiral aggregation induced emission fluorophores†
Qiang
Ye‡
ab,
Feng
Zheng‡
cd,
Enqi
Zhang
a,
Hari Krishna
Bisoyi
 b,
Shuyuan
Zheng
a,
Dandan
Zhu
d,
Qinghua
Lu
*d,
Hailiang
Zhang
a and
Quan
Li
*b
b,
Shuyuan
Zheng
a,
Dandan
Zhu
d,
Qinghua
Lu
*d,
Hailiang
Zhang
a and
Quan
Li
*b
aKey Laboratory of Polymeric Materials and Application Technology of Hunan Province, Key Laboratory of Environmentally Friendly Chemistry and Applications of Ministry of Education, School of Chemistry, Xiangtan University, Xiangtan 411105, China
bAdvanced Materials and Liquid Crystal Institute, Chemical Physics Interdisciplinary Program, Kent State University, Kent, OH 44242, USA. E-mail: quanli3273@gmail.com
cSchool of Chemical Science and Engineering, Tongji University, Shanghai 200092, China
dSchool of Chemistry and Chemical Engineering, State Key Laboratory of Metal Matrix Composite, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: qhlu@sjtu.edu.cn
First published on 17th August 2020
Abstract
Development of functional materials capable of exhibiting chirality tunable circularly polarized luminescence (CPL) is currently in high demand for potential technological applications. Herein we demonstrate the formation of both left- and right-handed fluorescent helical superstructures from each enantiomer of a chiral tetraphenylethylene derivative through judicious choice of the solution processing conditions. Interestingly, both the aggregation induced emission active enantiomers exhibit handedness inversion of their supramolecular helical assemblies just by varying the solution polarity without any change in their molecular chirality. The resulting helical supramolecular aggregates from each enantiomer are capable of emitting circularly polarized light, thus enabling both right- and left-handed CPL from a single chiral material. The left- and right-handed supramolecular helical aggregates in the dried films have been characterized using spectroscopy, scanning electron microscopy, and transmission electron microscopy techniques. These new chiral aggregation induced emission compounds could find applications in devices where CPL of opposite handedness is required from the same material and would facilitate our understanding of the formation of helical assemblies with switchable supramolecular chirality.
Introduction
Controlling handedness is a fascinating challenge that has attracted much attention over several decades worldwide.1 Thus, much effort has been made to precisely control the chirality inversion of self-assemblies as well as to gain an understanding of the inversion mechanisms.2 Primarily, scientists focused on handedness inversion originating from the breaking of the molecular chiral balance and simultaneous reestablishment of a new chiral balance with preferred handedness upon the action of stimuli such as heat,3 solvent,4 electrons,5 and redox.6 Adding an ionic7 or achiral matrix8 was also observed to induce chirality inversion of constructions. Afterwards, researchers found that light driving cis–trans isomerization,9 open–closed cyclization,10etc. can induce chirality inversion as well. So far most reported studies on chirality inversion, which originated from configurational transformation, have application prospects in life science,11 nonlinear optics,12 asymmetric catalysis,13 photoelectric devices,14 and so on.It is known that the chiral supermolecules resulting from molecular or bulky group asymmetric stacking are among the most promising chiral materials due to their dynamic,15 stimuli-responsive16 and amplified chiroptical properties.17 In recent reports, supramolecular helicity inversion was found in chiral molecules with cis–trans configuration18 or multiple chiral centers.19 However, it is a challenge to control the handedness of the self-assembled supramolecular structures by regulating the molecular packing model especially from single chiral center molecules, for example, by modulating external parameters without changing the molecular chirality. In addition, chiroptical properties from chiral supramolecular assemblies are much stronger and more stable.20 To induce or enhance the chiroptical properties, especially the circularly polarized luminescence (CPL), constructing a helical supramolecular assembly from fluorescent molecules is an attractive and efficient strategy.21
Here we disclose the formation of both left- and right-handed fluorescent helical superstructures from each enantiomer of a chiral tetraphenylethylene (TPE) derivative just by varying the solution polarity (Fig. 1). The molecular design is such that a chiral phenylethylamine (PEA) is linked to the fluorescent TPE scaffold as the chiral moiety by an amide linkage through an additional phenyl (Ph) group. Such unique structural feature endowed the enantiomers with conformational flexibility of rotation about the biphenyl single bond, which would be able to vary the molecular orientation and arrangement under different solvent conditions, thus inducing the variation of chirality and CPL at the supramolecular level. Indeed, we found that both R and S enantiomers of aggregation induced emission chiral molecules could form helical assemblies with switchable handedness by only regulating the assembly conditions without changing molecular chirality. By carefully exploring the assembly process and analyzing molecular packing information, it was discovered that the C–C bond rotation between two phenyl groups (ϕ1) plays an important role in simultaneous chirality switching of both supramolecular assembly and the corresponding CPL.
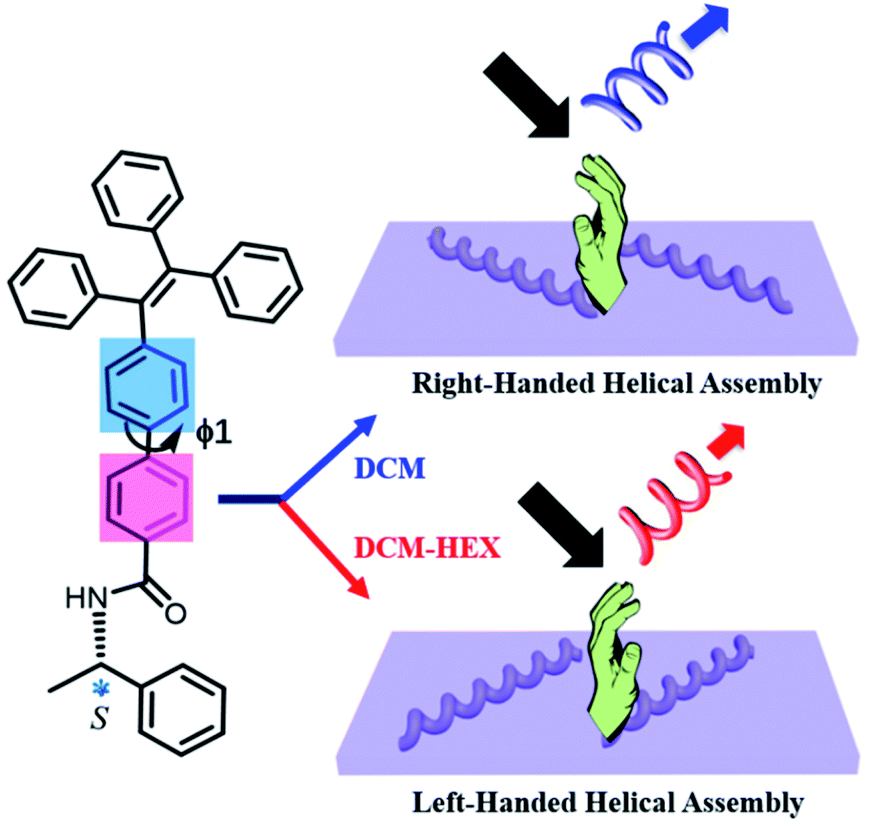 | ||
| Fig. 1 Schematic representation of solvent polarity driven helical inversion and circularly polarized luminescence in S-TPE–Ph–PEA assemblies. | ||
Results and discussion
The S- and R-enantiomers of the chiral aggregation induced emission active compounds based on TPE and PEA were synthesized and characterized according to the procedure illustrated in the ESI (Scheme S1†), with the given names of S-TPE–Ph–PEA and R-TPE–Ph–PEA, respectively. Both S- and R-TPE–Ph–PEA molecules self-assemble when processed from a series of dichloromethane–hexane (DCM/HEX) mixtures with gradually decreased polarity, named as S-assembly and R-assembly. The DCM/HEX mixture is chosen for studies on the assemblies since DCM is found to be a good solvent while HEX is found to be a nonsolvent for these compounds during their synthesis and workup process. The UV/Vis spectra of S- and R-TPE–Ph–PEA in solution presented absorption bands at 252 nm, 277 nm and 341 nm, corresponding to the π–π* transition of the chiral PEA moiety, acylamino substituent phenyl and TPE moiety, respectively (Fig. 2a and S4†). The S- and R-TPE–Ph–PEA compounds successfully inherited aggregation-induced emission properties from the TPE moieties (Fig. S2†), presenting a high fluorescence quantum yield of 40% and 40.5% in the solid state, respectively.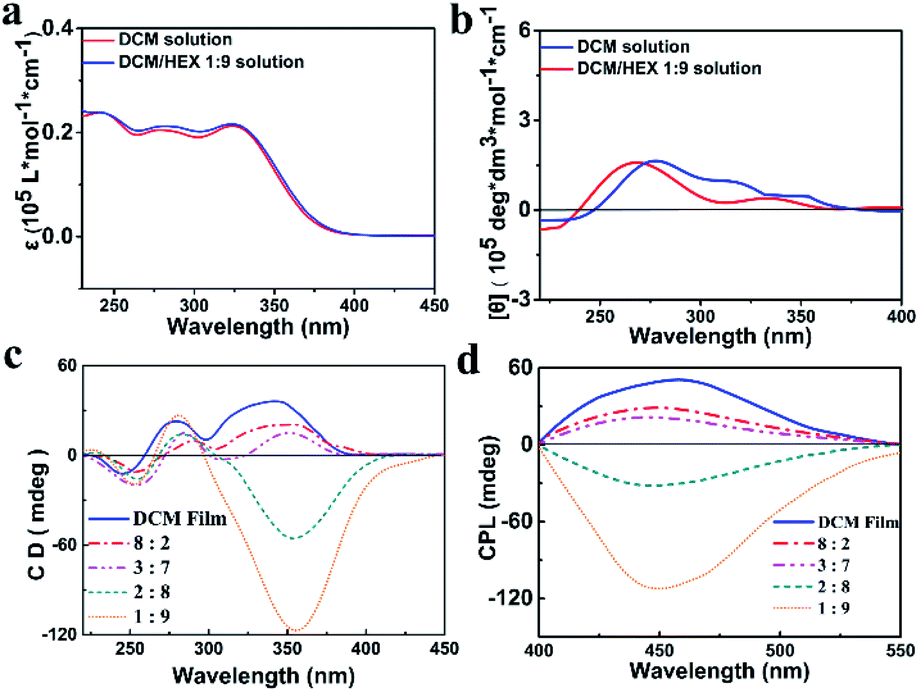 | ||
Fig. 2 Absorption spectra (5 × 10−6 M) (a); CD spectra in solution (b). CD spectra from solid films (c) and circularly polarized luminescence spectra (d) of S-TPE–Ph–PEA assemblies obtained from different solutions: DCM (blue); DCM/HEX 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 (cyan); DCM/HEX 2:8 (pink) and DCM/HEX 1:9 (red). :7 (cyan); DCM/HEX 2:8 (pink) and DCM/HEX 1:9 (red). | ||
The circular dichroism spectra of the S-enantiomer in DCM and HEX solutions are depicted in Fig. 2b. The corresponding spectra of the R-isomer is shown in Fig. S2.† The S-assemblies produced by drying the solution cast films of the chiral compounds from DCM and a DCM–HEX (3:7) mixture showed a Cotton effect with positive peaks at 360 nm and 278 nm, as well as a negative peak at 253 nm. The positive peaks are assigned to the absorption band of TPE and an acylamino substituted phenyl group, respectively, while the negative peak corresponds to the chiral centers of PEA (Fig. 2c). The CD spectra of the above S-assemblies indicated right-handed helicity. Upon further reducing the solvent polarity by increasing the volume ratio of hexane up to 80%, a helical assembly was revealed by the negative CD absorption around 360 nm, which indicated left-handed helicity (Fig. 2c). Thus, supramolecular chirality inversion was achieved from a single enantiomer. The same phenomena were observed for the R-assemblies (Fig. S4c†). Notably, the molecular chirality, i.e., typical CD absorption signals of PEA around 253 nm, is almost identical for both S-/R-assemblies and DCM solution in all cases, suggesting that the molecular chirality did not change during the self-assembly process. Moreover, the CD spectra of S- and R-TPE–Ph–PEA in HEX showed the same chirality as those in the breaking of the molecular chiral balance, but only attributed to different types of molecular packing induced by the solvent environment. Here we note that preliminary investigations on other solvent mixtures tested, i.e., chloroform/hexane, dichloromethane/ethyl acetate, ethyl acetate/hexane, and ethyl acetate/methylcyclohexane, did not indicate the occurrence of handedness inversion in the supramolecular assemblies of S- and R-TPE–Ph–PEA.
The helical-assembly induced CPL of S- and R-assemblies, originating from the excited state, was also switchable under different assembly conditions (Fig. 2d and S4d†), indicating that the handedness inversion was an intrinsic rather than a casual phenomenon. The absorption and fluorescence spectra of the assemblies are depicted in Fig. S5 and S6.† In DCM and DCM/HEX (8:2 and 3:7) systems, S-assembly showed a positive Cotton effect in CPL spectra, implying a right-handed helical structure, while in DCM/HEX (2:8 and 1:9) systems, a negative signal was determined, corresponding to a left-handed helical structure. The CPL of R-assembly behaved in the same manner (Fig. S4d†). In addition, both S- and R-assemblies were found to have similar variation of the CPL intensity. Before the Cotton effect inversion, CPL intensity was weakened with increasing the HEX volume ratio.
On the other hand, after inversion CPL intensity enhanced by further increasing the HEX volume ratio, especially when the ratio of DCM/HEX reached 1:9.
Scanning electron microscopy (SEM) was employed to obtain more detailed information on the morphologies of the assemblies. Interestingly, increasing the hexane composition in the DCM solutions of S-TPE–Ph–PEA with a concentration of 1 × 10−4 M resulted in a morphological change from plate to disordered film and to helical fibers (Fig. 3). In DCM, the relatively hydrophobic S- and R-TPE–Ph–PEA molecules would already aggregate due to extended π-conjugation of TPE moieties (Fig. S1†).22 Upon increasing the hexane content in DCM to 20%, right-handed helical assemblies were found as shown in Fig. 3b. Moreover, after further increasing the hexane content to 80% and more, distinct left-handed helical fibers were observed (Fig. 3d and e) which would possibly form from the delicate balance between multiple noncovalent interactions such as hydrogen-bonding interactions, hydrophobic and hydrophilic interactions in DCM/HEX mixtures. Confirmed by SEM, S-assemblies in DCM/HEX (2:8 and 1:9) systems revealed distinct left-handed fibers. These supramolecular helical fibers are highly luminescent and emit circularly polarized light (Fig. 3f). Similar results were found for R-assemblies; left-handed helical assemblies were found in films obtained from a solution containing 20% hexane while right-handed helical fibers were formed in DCM/HEX (2:8 and 1:9) systems (Fig. S7†). For self-assembly from the DCM/HEX 1:9 system, a homogeneous screw pitch was determined to be ca. 140 nm with a width of about 30 nm for S-assembly, and around 190 nm with a width of ca. 40 nm for R-assembly.
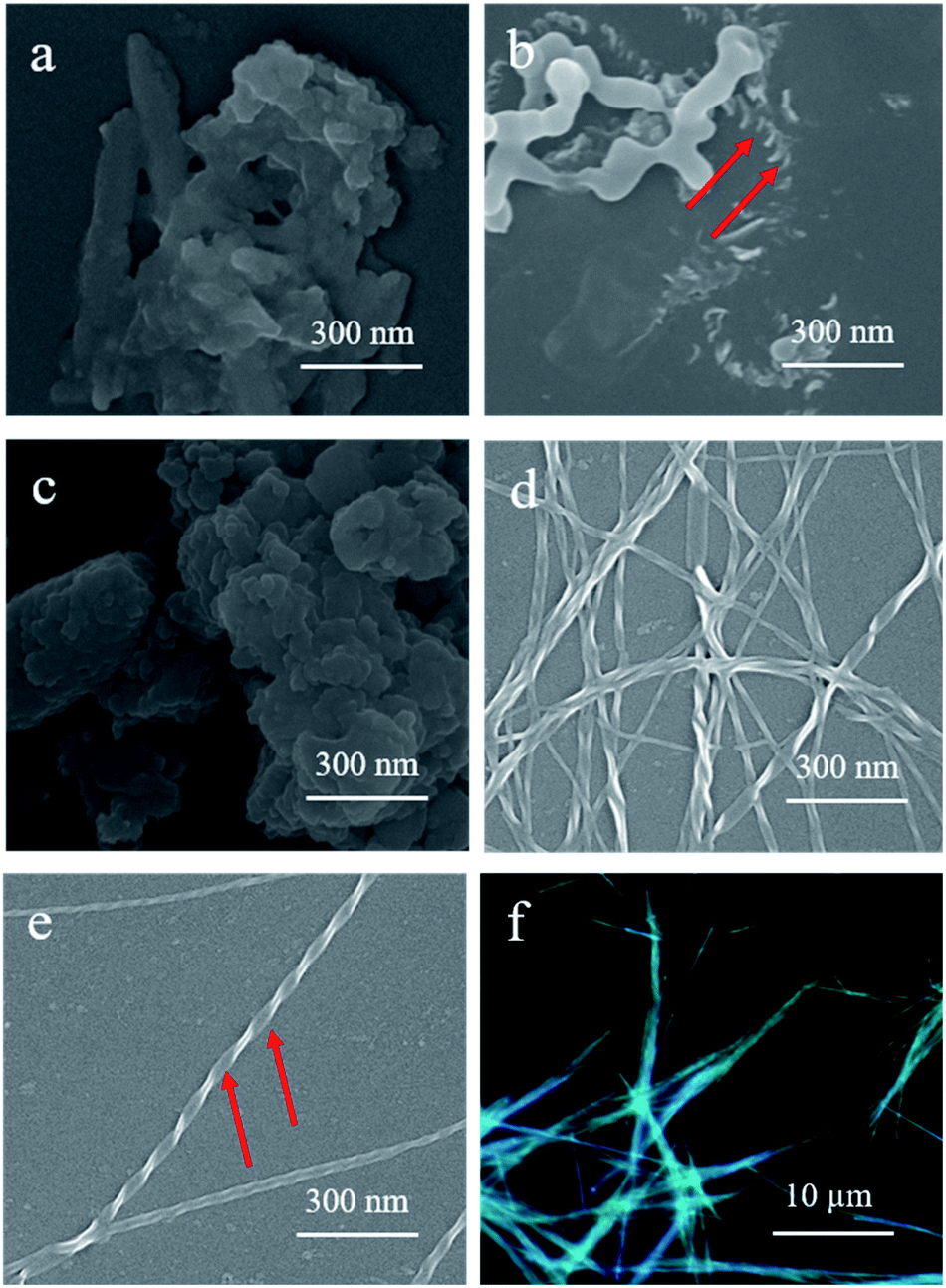 | ||
| Fig. 3 SEM images of S-TPE–Ph–PEA assemblies in DCM/HEX with different ratios: (a) DCM, (b) 8:2, (c) 3:7, (d) 2:8 and (e) 1:9. (f) Confocal fluorescence microscope image of S-TPE–Ph–PEA assemblies in DCM/HEX with a ratio of 1:9. | ||
The transmission electron microscope (TEM) studies presented sufficient evidence for the self-assembly process and the growth of helical fibers with inversed helicity in DCM/HEX (1:9) for both S-assemblies (Fig. 4) and R-assemblies (Fig. S6†). At the beginning (ca. 1 min), only slender fibers without a helical structure were observed. With the ongoing assembly process, obvious helical fibers with left-handed chirality were formed as shown in Fig. 4a (2 min). It was worth noting that smaller helical fibers assembled into larger helical fibers through entanglement with each other (Fig. 4b). Helical fibers for R-assemblies observed by TEM are shown in Fig. S8.† At the end of the assembly process, larger nano-helical fibers were dramatically generated with a homogeneous width of about 27 nm and pitches to be ca. 130 nm for S-assembly, and about 35 nm and pitches around 160 nm for R-assembly. It was noted that the obvious helix structure was found only in fibers large enough because the surface tension interaction of solvent restricted the surface morphology.
 | ||
| Fig. 4 TEM images of S-TPE–Ph–PEA assemblies from the DCM/HEX 1:9 system after self-assembling for 2 min (a) and 4 min (b), illustrating the formation and growth of helical fibers of S-TPE–Ph–PEA assemblies from solutions with a concentration of 1 × 10−4 M. | ||
Preliminary computational studies were carried out to understand the assembly of the compounds under different polarity conditions. As shown in Fig. S11–S21,† the rotation of two dihedral angles ϕ1 and ϕ2 would bring different conformations to both S- and R-TPE–Ph–PEA molecules. In view of this, a computational study was carried out to determine the most stable conformers in different solvents. We carried out a potential energy scan (PES) on the TPE–Ph–PEA molecules by varying the degree of ϕ1 and ϕ2 (Fig. S12 and S13†). Considering all possibilities, four possible conformations for each enantiomer were found. These conformers were then optimized in solvation models with hexane and DCM and are summarized in Fig. S14 and S15.† From frequency analysis, the relative Gibbs free energies of each conformer were calculated and are summarized in Table S1.† When using DCM as a solvation model, S3 and R3 conformers were found to have the lowest energy for S-TPE–Ph–PEA and R-TPE–Ph–PEA, respectively, in which the dihedral angle ϕ1 was found to twist by about 35°, while ϕ2 twisted by about 90°. On the other hand, in the hexane solvation model, one conformer with the lowest energy was located, S4 for S-TPE–Ph–PEA and R4 for R-TPE–Ph–PEA, in which the dihedral angle ϕ1 was found to twist by about −140°, while ϕ2 twisted by about 90°. Since ϕ1, resulting from the C–C rotation between two phenyl rings, can lead to the formation of both cis-like and trans-like conformations of the chiral building blocks (Fig. S14†), it is believed to play a significant role in molecular packing in the helical assemblies during solvent drying which could induce the handedness inversion in such self-assembly systems. The non-covalent bonding interactions like intermolecular hydrogen bonding between the amide groups, π–π interaction, and C–H–π interaction could provide additional stability to these handedness invertible helical luminescent aggregates.
We envisioned that we could predict the possible packing models of S- and R-TPE–Ph–PEA molecules according to the experimental PXRD patterns, and thereby gain insight into the molecular packing of the assemblies. Computational models for the most stable conformer of S-TPE–Ph–PEA in hexane, S4, were minimized using the COMPASSII force field in different space groups, and the corresponding simulated XRD patterns were generated (Fig. S16†). It is shown in Fig. S17† that the simulated packing model for S4 has a head-to-tail arrangement in a unit cell (b), and the molecules are packed to form a chain-like structure with a left-handed helix (c). Similar simulations were performed to predict the possible packing models for compounds in DCM based on their experimental powder XRD patterns. The simulated XRD pattern is shown in Fig. S19.† It corresponds to the predicted packing model as shown in Fig. S20,† where the most stable conformer S3 in DCM assembles into a right-handed form structure. Combining X-ray diffraction experiments of S-TPE–Ph–PEA assemblies (Fig. S9†) and a computational study in DCM and hexane, the possible mechanisms of molecular packing models of S-TPE–Ph–PEA were proposed to illustrate handness inversion (Fig. 5) during assembly processes in different solutions. Interestingly, the lowest energy conformers S3 and R3 in DCM solution have cis-like conformation, while the lowest energy conformers S4 and R4 in hexane have trans-like conformation. Both S3 and S4 or R3 and R4 showed the same chirality as their corresponding molecular chirality in solution; however, the corresponding assemblies show handedness inversion after properly assembling during solvent evaporation due to different spatial structures of conformers. This exciting result gives a broad view to fabricate helical inversion systems that simply design one configuration chiral molecule with cis-like and trans-like conformations just by regulating their packing structures.
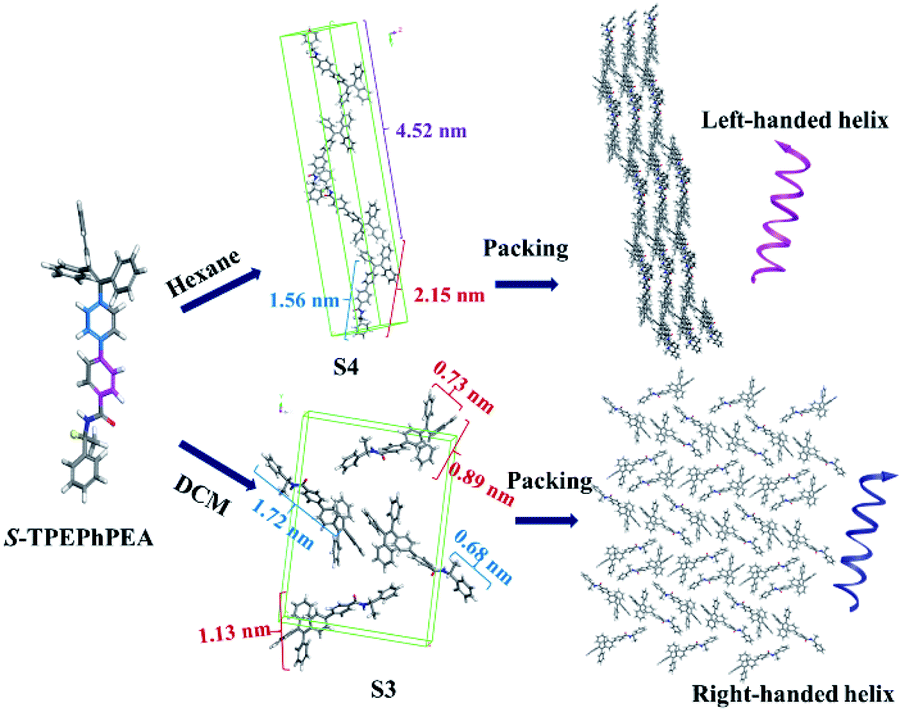 | ||
| Fig. 5 Possible molecular assemblies computationally determined for S-TPE–Ph–PEA in DCM and hexane. | ||
Conclusions
In summary, the realization of both left- and right-handed fluorescent helical assemblies from each enantiomer of a chiral aggregation induced emission active tetraphenylethylene derivative has been demonstrated by modulating the polarity of the processing solution without changing the molecular chirality. The S-enantiomer forms right-handed helical assemblies in more polar solution while left-handed helical assemblies are obtained from less polar solution for this enantiomer. Thus, the inversion of supramolecular chirality has been achieved in the system. The R-enantiomer exhibits a similar behavior. The driving force for such supramolecular assembly has been attributed to the rotation of the C–C bond between two phenyl rings and different types of molecular packing induced by different non-covalent interactions in assemblies. Moreover, the switching and enhancement of CPL emission were achieved by helicity inversion of the assemblies which is a challenging task but is highly desirable for device applications involving chirality switchable CPL. These results would provide new insights into preparing both left- and right-handed helical structures through building block conformation-controlled cis-like and trans-like components from the same chiral molecules, thus presenting broad potential applications in chemistry, materials science and life science.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (201704086 and 51733007), Foundation of Hunan Educational Committee (18K030), China Postdoctoral Science Foundation (2017M622592), and China Scholarship Council (201908430092). We acknowledge the South African Centre for High Performance Computing (CHPC) for donating its cluster facility, which was used to perform the computational work presented in this article.Notes and references
- Y. J. Zhang, R. Suzuki, T. J. Ye and Y. Iwasa, Science, 2014, 344, 725 CrossRef CAS
; Photoactive Functional Soft Materials: Preparation, Properties, and Applications, ed. Q. Li, Wiley-VCH, Weinheim, 2019 Search PubMed
- M. Bartók, Chem. Rev., 2010, 110, 1663 CrossRef CAS
- L. Wang, K. G. Gutierrez-Cuevas, A. Urbas and Q. Li, Adv. Opt. Mater., 2016, 4, 247 CrossRef CAS
- S. I. Sakurai, K. Okoshi, J. Kumaki and E. Yashima, J. Am. Chem. Soc., 2006, 128, 5650 CrossRef CAS
- S. Zahn and J. W. Canary, Science, 2000, 288, 1404 CrossRef CAS
- E. Ohta, H. Sato, S. Ando, A. Kosaka, T. Fukushima, D. Hashizume, M. Yamasaki, K. Hasegawa, A. Muraoka, H. Ushiyama, K. Yamashita and T. Aida, Nat. Chem., 2011, 3, 68 CrossRef CAS
- S. Akine, S. Sairenji, T. Taniguchi and T. Nabeshima, J. Am. Chem. Soc., 2013, 135, 12948 CrossRef CAS
- G. Liu, L. Zhu, W. Ji, C. Feng and Z. Wei, Angew. Chem., Int. Ed., 2016, 55, 2411 CrossRef CAS
- M. Mathews, R. Zola, S. Hurley, D. Yang, T. J. White, T. J. Bunning and Q. Li, J. Am. Chem. Soc., 2010, 132, 18361 CrossRef CAS
- Y. Li, C. Xue, M. Wang, A. Urbas and Q. Li, Angew. Chem., Int. Ed., 2013, 52, 13703 CrossRef CAS
- R. E. Kohman, A. M. Kunjapur, E. Hysolli, Y. Wang and G. M. Church, Angew. Chem., Int. Ed., 2018, 57, 4313 CrossRef CAS
- D. Yang, P. Duan, L. Zhang and M. Liu, Nat. Commun., 2017, 8, 15727 CrossRef CAS
- S. Zhang, L. Kang, X. Wang, L. Tong, L. Yang, Z. Wang, K. Qi, S. Deng, Q. Li and X. Bai, Nature, 2017, 543, 234 CrossRef CAS
- J. Han, J. You, X. Li, P. Duan and M. Liu, Adv. Mater., 2017, 29, 1606503 CrossRef
- H. Hayasaka, T. Miyashita, M. Nakayama, K. Kuwada and K. J. Akagi, J. Am. Chem. Soc., 2012, 134, 3758 CrossRef CAS
- A. Kousar, J. Liu, N. Mehwish, F. Wang, A. Dang-i and C. Feng, Mater. Today Chem., 2019, 11, 217 CrossRef CAS
- K. Toyofuku, M. A. Alam, A. Tsuda, N. Fujita, S. Sakamoto, K. Yamaguchi and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 6476 CrossRef CAS
- G. Liu, J. Sheng, H. Wu, C. Yang, G. Yang, Y. Li, R. Ganguly, L. Zhu and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 6467 CrossRef CAS
- P. Xing and Y. Zhao, Acc. Chem. Res., 2018, 51, 2324 CrossRef CAS
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS
- K. Watanabe, H. Iida and K. Akagi, Adv. Mater., 2012, 24, 6451 CrossRef CAS
- M. H. Y. Chan, S. Y. L. Leung and V. W. W. Yam, J. Am. Chem. Soc., 2018, 140, 7637 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04179c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |