
Unveiling the size effect of Pt-on-Au nanostructures on CO and methanol electrooxidation by in situ electrochemical SERS†
Xing
Chen
ab,
Miao-Miao
Liang
b,
Juan
Xu
b,
Han-Lei
Sun
b,
Chen
Wang
b,
Jie
Wei
b,
Hua
Zhang
*b,
Wei-Min
Yang
b,
Zhi-Lin
Yang
 b,
Jian-Jun
Sun
*a,
Zhong-Qun
Tian
b and
Jian-Feng
Li
*b
b,
Jian-Jun
Sun
*a,
Zhong-Qun
Tian
b and
Jian-Feng
Li
*b
aMinistry of Education Key Laboratory for Analytical Science of Food Safety and Biology, Fujian Provincial Key Laboratory of Analysis and Detection Technology for Food Safety, College of Chemistry, Fuzhou University, Fuzhou, Fujian 350108, China. E-mail: jjsun@fzu.edu.cn
bState Key Laboratory of Physical Chemistry of Solid Surfaces, Fujian Key Laboratory of Advanced Materials, iChEM, College of Chemistry and Chemical Engineering, College of Materials, Department of Physics, Xiamen University, Xiamen 361005, China. E-mail: Li@xmu.edu.cn; zhanghua@xmu.edu.cn
First published on 4th February 2020
Abstract
In situ monitoring of electrocatalytic processes at solid–liquid interfaces is essential for the fundamental understanding of reaction mechanisms, yet quite challenging. Herein, Pt-on-Au nanocatalysts with a Au-core Pt-satellite superstructure have been fabricated. In such Pt-on-Au nanocatalysts, the Au cores can greatly amplify the Raman signals of the species adsorbed on Pt, allowing the in situ surface-enhanced Raman spectroscopy (SERS) study of the electrocatalytic reactions on Pt. Using the combination of an electrochemical method and in situ SERS, size effects of Pt on the catalytic performance of the core–satellite nanocomposites towards CO and methanol electrooxidation are revealed. It is found that such Pt-on-Au nanocomposites show improved activity and long-term stability for the electrooxidation of CO and methanol with a decrease in the Pt size. This work demonstrates an effective strategy to achieve the in situ monitoring of electrocatalytic processes and to simultaneously boost their catalytic performance towards electrooxidation.
Direct methanol fuel cells (DMFCs) have been regarded as one of the most promising fuel cells for commercial applications and have become a highly hot topic in the electrochemical field due to their excellent advantages.1–5 Although great progress has been achieved for the development of DMFCs, there are still many problems that seriously hinder their commercialization. For example, the electrocatalytic oxidation of methanol on Pt catalysts is a self-poisoning process.6,7 CO generated by the methanol dehydrogenation reaction adsorbs on the surface of Pt and occupies the active sites, which prevents the further oxidation of methanol and reduces the activity of the catalyst. Therefore, it is urgent to search for more efficient and poison-resistant catalysts.
It has been demonstrated that the particle size of the catalyst could significantly affect its electrocatalytic performance. However, debates still exist for the size effect on the activity towards the electrooxidation of methanol. Bergamaski et al.8 found that the optimum Pt nanoparticle size for the electrooxidation of methanol is 3–10 nm. For Pt nanocatalysts with a size smaller than 5 nm, Tang et al.9 reported that the mass specific activity of the catalyst increased with the size of Pt in the range of 2.2–3.8 nm, but it began to decrease with further increase in the particle size. In contrast, the results obtained by Zeng et al.10 show that the mass specific activity of the Pt nanocatalyst decreases with an increase in the Pt nanoparticle size in the range of 2.2–4.0 nm. One of the most important reasons accounting for such contradictory results is that catalysts with different particle sizes are usually prepared by different methods, which would greatly affect their catalytic performance.
On the other hand, the in situ study of the electrochemical process is also of significant importance for the development of efficient electrode materials.11 Surface-enhanced Raman spectroscopy (SERS) is molecular vibrational spectroscopy with extremely high surface sensitivity even down to the single molecule level.12–17 It can provide rich structural information of molecules and has thus been extensively applied in the study of the adsorption and reaction of molecules at surfaces or interfaces.18–21 Moreover, combined with electrochemical methods, SERS can be directly used to in situ monitor the electrocatalytic reactions occurring at solid/liquid interfaces.22–24 However, only a few metals like Au, Ag, and Cu with a rough surface can generate strong SERS effects. The SERS effect for other transition metals including Pt, Pd, Ru, etc. that are frequently used in DMFCs is too weak to realize the in situ detection of the surface species on them. Such disadvantages significantly restrict the application of electrochemical SERS in electrocatalysis.
To solve the aforementioned limitation of SERS, a “borrowing” strategy has been developed via the fabrication of Au-transition metal core–shell or core–satellite nanostructures.25,26 Using the “borrowing” strategy, in situ monitoring of reactions carried out on transition metal nanocatalysts becomes possible.27–30 However, most of these studies only focus on some probe surface reactions, such as the conversion of 4-nitrothiophenol (4-NTP) to 4-aminothiophenol (4-ATP), instead of the practical catalytic reactions that are of great importance in the chemical industry. Furthermore, such nanostructures are usually prepared by one-pot methods, making it hard to fine-tune the structure of the catalysts. Thus, it is still a great challenge to study the structure–activity relationships, such as the size effect or the composition effect, of the catalysts by SERS.
Herein, we prepared a bifunctional nanocatalyst with a core–satellite superstructure (Pt-on-Au) via a self-assembly method, where Au nanoparticles are surrounded by Pt nanoparticles. The Au core significantly improves the CO-tolerance and long-term stability of the Pt nanosatellites. At the same time, the Au core also works as a model amplifier to enhance the Raman signals of the molecules adsorbed on the Pt surface. Using such a self-assembly method, Pt-on-Au core–satellite nanocomposites consisting of Pt nanocatalysts with different diameters are fabricated. Thus, the size effect of Pt on the electrooxidation of CO and methanol using the Pt-on-Au bifunctional nanocatalysts was further studied by in situ electrochemical SERS. The electrochemical SERS results correlate well with the electrochemical data and show that in the range of 2–7 nm, the activity towards the electrooxidation of CO and methanol increases with a decrease in the size of Pt.
Pt nanoparticles with different sizes ranging from 2.1–7.6 nm were prepared via a seed-mediated method (see the ESI†). Pt nanoparticles with a size of around 2.1 nm were first prepared using a borane tert-butylamine complex as the reducing agent and oleylamine as the protective agent (Fig. 1a and e).31,32 The as-prepared Pt nanoparticles were then used as the seeds for the further growth of Pt nanoparticles with larger sizes. Using such a method, highly uniform Pt nanoparticles are obtained, and their diameters can be tailored from 2.1–7.6 nm (Fig. 1a–h). As all the Pt nanoparticles with different sizes were prepared via a similar method, the influence of the preparation method on the catalytic performance had been excluded. Therefore, a more accurate and general size effect can be obtained using these Pt nanoparticles as the catalyst.
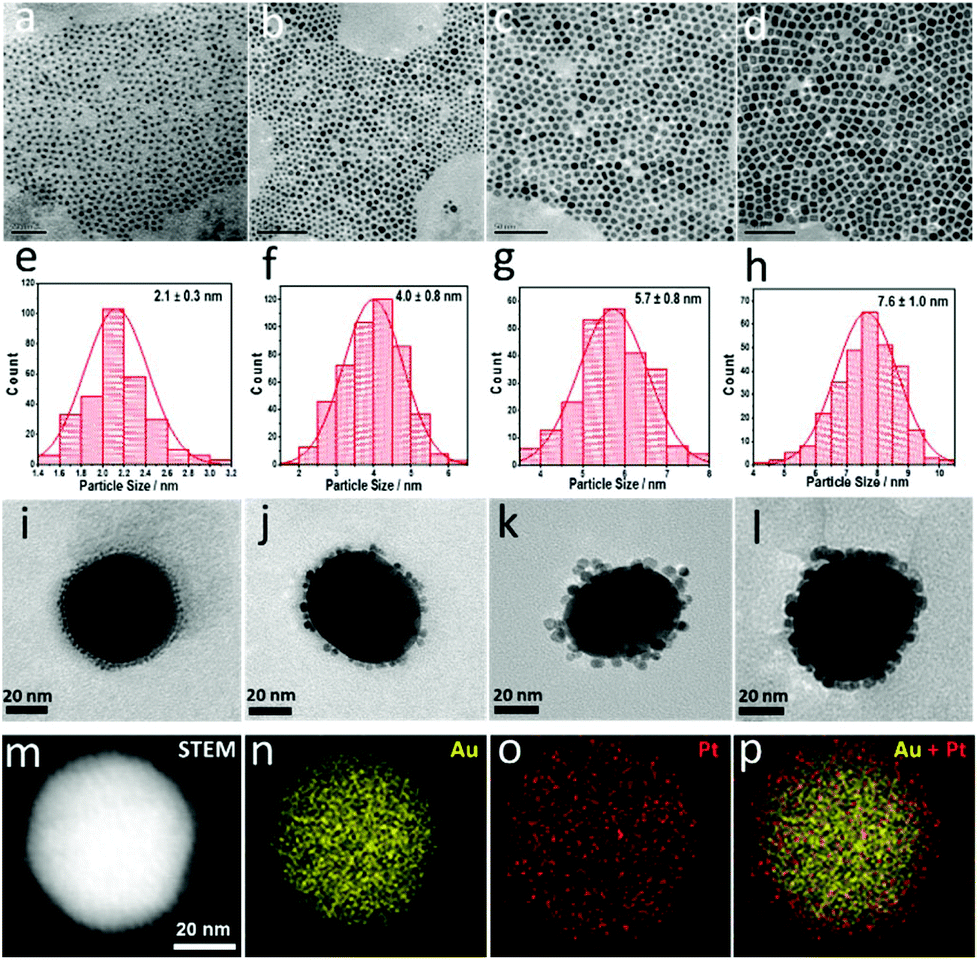 | ||
| Fig. 1 (a)–(d) TEM images of Pt nanoparticles with different sizes. Their size distributions are shown in (e)–(h), respectively. (i)–(l) TEM images of the Pt-on-Au satellite nanostructure catalysts. 55 nm Au NPs (larger particles) are surrounded by Pt NPs (smaller ones) shown in (a)–(d) via electrostatic self-assembly. (m)–(p) High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image and elemental maps of Pt (2.1 nm)-on-Au. | ||
The obtained Pt nanoparticles were then assembled on Au nanoparticles (∼55 nm) to form the Pt-on-Au core–satellite nanocomposites via electrostatic self-assembly. As shown in Fig. 1i–l and Fig. S1,† the Pt nanoparticles, independent of the size, are homogeneously deposited on the Au nanoparticles. Such a core–satellite nanostructure has also been demonstrated by the HAADF-STEM images and elemental maps (Fig. 1m–p), which clearly show that the Pt nanoparticles are distributed around the Au cores.
The as-prepared Pt-on-Au core–satellite nanocomposites are then used in the electrooxidation of CO. To remove the oleylamine on the surface of the Pt nanoparticles, electrochemical activation was conducted before electrochemical tests by performing cyclic voltammetry (CV) in an acid solution, which is widely applied in previous studies.33 As the surface structure of Pt would greatly affect its electrocatalytic performance,34 all the catalysts were activated using the same procedure to exclude the influence of the surface reconstruction during the cleaning process on further electrochemical measurements. As shown in Fig. S2 in the ESI,† the electrochemically active surface areas (ECSAs) increase with an increase in the number of CV cycles and become stable after 50 cycles. This result indicates that oleylamine has been removed from the surface of Pt and the catalyst has been fully activated.
Fig. 2a displays the electrochemical results of the electrooxidation of CO on Pt and Pt-on–Au. The onset potential of the electrooxidation of CO on the pure Pt nanocatalyst is about 0.44 V, but it shifts lower to about 0.41 V for Pt-on-Au (Fig. 2a). The additional peak for Pt-on-Au is attributed to the oxidation of CO adsorbed on the Au surface, as demonstrated by the CV of CO electrooxidation on pure Au nanoparticles (Fig. S3†). Such a result demonstrates that Pt-on-Au shows higher activity towards the electrooxidation of CO compared to pure Pt. XPS characterization of the Pt and Pt-on-Au samples indicates that electronic interactions between Pt and Au exist in Pt-on-Au (Fig. S4†), which is also consistent with previous reports.35 Thus, we believe that the improved performance of Pt-on-Au results from the electronic interactions between Pt and Au.
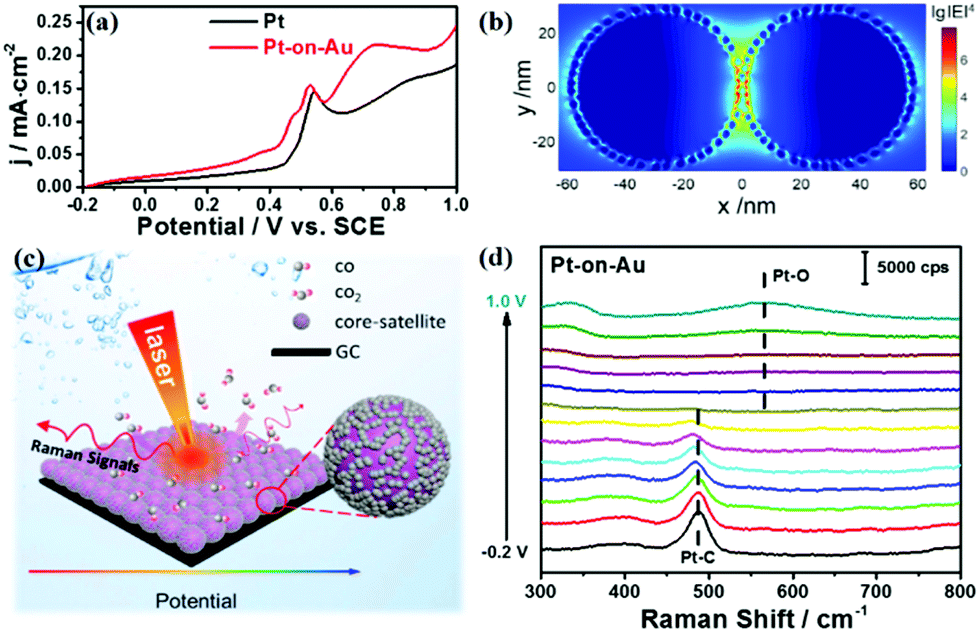 | ||
| Fig. 2 (a) CO stripping voltammograms for Pt and Pt-on-Au core–satellite nanocatalysts in 0.1 M HClO4 at a scan rate of 20 mV s−1. (b) 3D-FDTD simulation for a pair of Pt-on-Au nanocomposites. (c) A schematic diagram of the in situ electrochemical SERS study of CO electrooxidation. (d) The electrochemical SERS spectra for CO electrooxidation in 0.1 M HClO4 on Pt (2.1 nm)-on-Au core–satellite nanocatalysts. | ||
At the same time, the Pt-on-Au core–satellite nanocomposites can also be used as a bifunctional SERS substrate to in situ monitor the catalytic reactions occurring on their surfaces. As shown in Fig. S5,† no Raman signals can be observed for the pure Pt nanoparticles during the CO electrooxidation as a result of the weak plasmonic properties of Pt. However, according to three-dimensional finite-difference time-domain (3D-FDTD) simulations, highly localized and strong electromagnetic (EM) fields are generated around the Au cores, due to the continuum of the delocalized propagation surface plasmon at the Au surface (Fig. 2b). The EM field could significantly enhance the Raman signals from the species adsorbed on the surface of the Pt catalyst by 7 orders of magnitude. Such a high enhancement enables the in situ SERS tracking of the reaction processes and intermediates on the Pt nanocatalysts. Therefore, we then employ electrochemical SERS to in situ monitor the electrooxidation of CO on Pt-on-Au (Fig. 2c). As displayed in Fig. 2d, two peaks located at about 390 and 485 cm−1 are observed, which can be assigned to the stretch modes of Pt–CO with bridge and on-top configurations, respectively.36 As the potential increases, these Pt–CO peaks start to decline, with a simultaneous redshift of the peaks, and completely disappear at about 0.6 V. Another peak located at about 570 cm−1 arises when further increasing the potential to 0.8 V, which is attributed to the Pt–O bending mode of surface platinum oxide.37 This trend correlates well with the electrochemical data and directly illustrates the reaction process of CO electrooxidation.
The size effect of Pt on the electrooxidation of CO on the Pt-on-Au nanocomposite was further studied, and the results are shown in Fig. 3a. The current density of the CO stripping peak decreases with an increase in the size of Pt, and Pt-on-Au with 2.1 nm Pt satellites shows the maximum current density. At the same time, the onset potential for CO electrooxidation increases with an increase in the Pt particle size, showing a minimum value at 2.1 nm (Fig. 3b). Similar trends are also observed for the pure Pt nanoparticles with different sizes (Fig. S6†). As is well known, a smaller onset potential and a larger current density indicate a better activity. Therefore, it demonstrates that decreasing the diameter of the Pt nanoparticles in the Pt-on-Au core–satellite nanocomposites will promote their activity towards the CO electrooxidation, and Pt-on-Au with 2.1 nm Pt shows the best catalytic performance. Such a trend can be explained by the fact that Pt nanoparticles with a smaller size have more coordinatively unsaturated sites and thus boost the formation of the key intermediates like OH during CO electrooxidation.38 Another reason may be that the electronic interactions would become stronger with a decrease in the the Pt size, as there are more Au–Pt interfaces existing in Au–Pt core–satellite nanocomposites with smaller Pt nanoparticles.
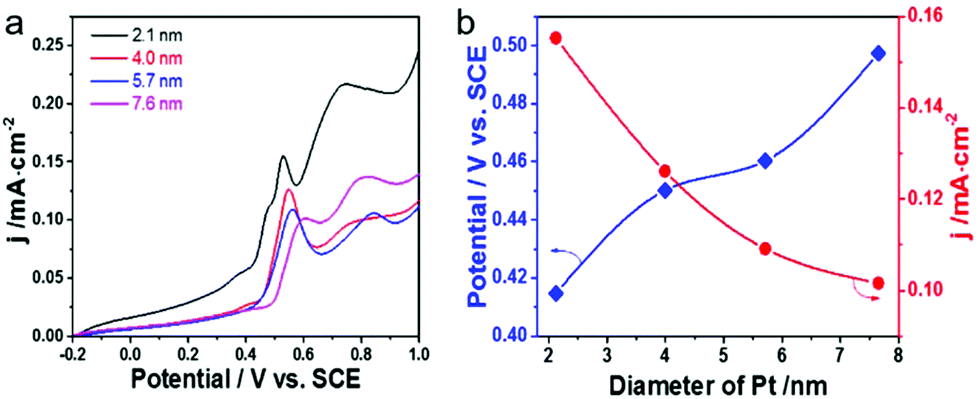 | ||
| Fig. 3 (a) CO stripping voltammograms for Pt-on-Au core–satellite nanocatalysts with different Pt nanoparticle sizes in 0.1 M HClO4 at a scan rate of 20 mV s−1. (b) The corresponding current density of the CO stripping peak and onset potentials of CO electrooxidation as a function of the Pt particle size. | ||
Electrochemical SERS has also been used for the in situ study of the Pt size effect on CO electrooxidation. As displayed in Fig. 4a–c, the in situ SERS spectra during CO electrooxidation for all the Pt-on-Au nanocomposites, independent of the Pt diameter, are similar to those for Pt-on-Au with 2.1 nm Pt nanoparticles (Fig. 2d). The Raman peaks for the stretch modes of Pt–CO decrease with the increase of the potential and completely disappear at about 0.6 V. At the same time, the Raman peak for the Pt–O bending mode appears at higher potentials (∼0.8 V). In order to quantitatively compare the influence of the Pt size on the in situ SERS spectra during CO electrooxidation, the Raman intensity of the Pt–CO bands for different Pt-on-Au nanocomposites is plotted as a function of the potential (Fig. 4d). Though they have very similar trends, it can still be observed that the normalized Raman intensity of Pt–CO declines faster as the diameter of the Pt nanocatalysts decreases. Such results are consistent with the electrochemical data (Fig. 3) and further demonstrate that reducing the Pt diameter could greatly boost the CO electrooxidation activity of the Pt-on-Au nanocomposite.
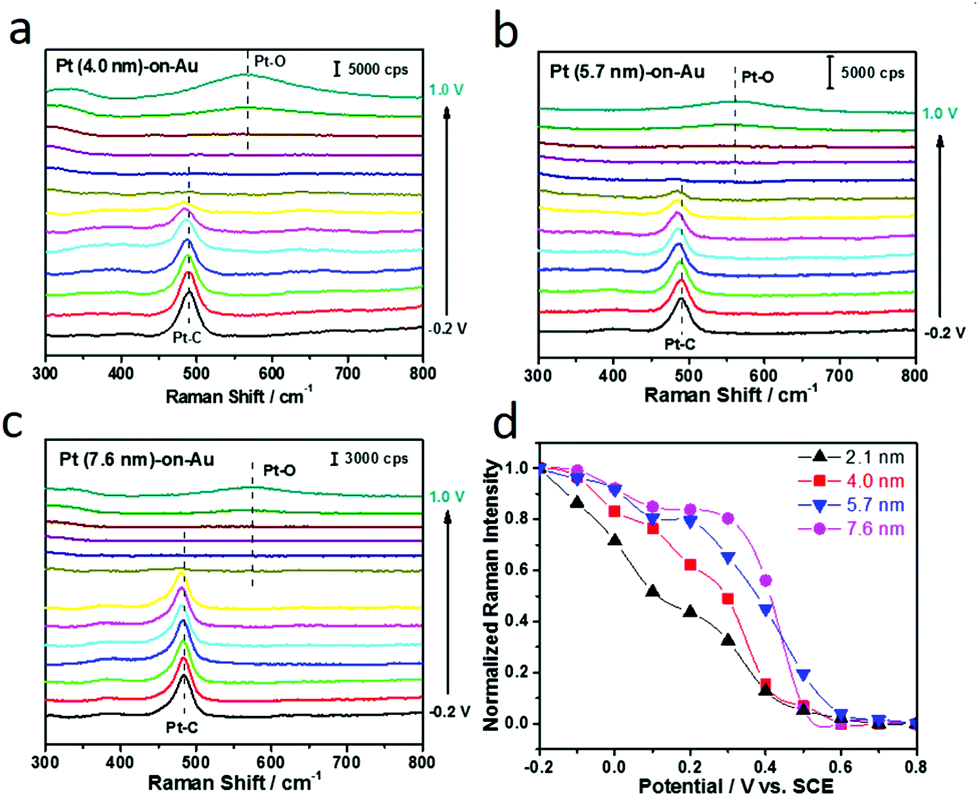 | ||
| Fig. 4 (a)–(c) are the electrochemical SERS spectra for CO electrooxidation in 0.1 M HClO4 on Pt-on-Au with a Pt particle size of 4.0 nm, 5.7 nm, and 7.6 nm, respectively. (d) Normalized Raman intensity of Pt–CO bands as a function of the potential. | ||
We also studied the electrooxidation of methanol on such Pt-on-Au nanocomposites by in situ SERS. As shown in Fig. S7,† the Raman signals for CO adsorbed on Pt can be clearly observed during the electrooxidation of methanol, and their intensity will decrease rapidly with the increase of the potential. Such results directly demonstrate that CO is an intermediate during the methanol electrooxidation on Pt-based nanocatalysts, which may block the Pt surface, leading to the rapid deactivation of the catalysts.
Given the fact that the Pt-on-Au nanocomposites with smaller Pt nanoparticles show much better performance towards CO electrooxidation, we then studied the size effect of Pt on the methanol electrooxidation on the Pt-on-Au nanocomposites. Fig. 5a shows the CV diagrams of methanol electrooxidation on the nanocomposites consisting of Pt nanoparticles with the particle size ranging from 2.1–7.6 nm. Similar to CO electrooxidation, the peak current density of methanol electrooxidation decreases while the onset potential increases with the increase of the Pt size (Fig. 5b), indicating that Pt-on-Au core–satellite nanocomposites with smaller Pt nanoparticles have higher activity for methanol electrooxidation.
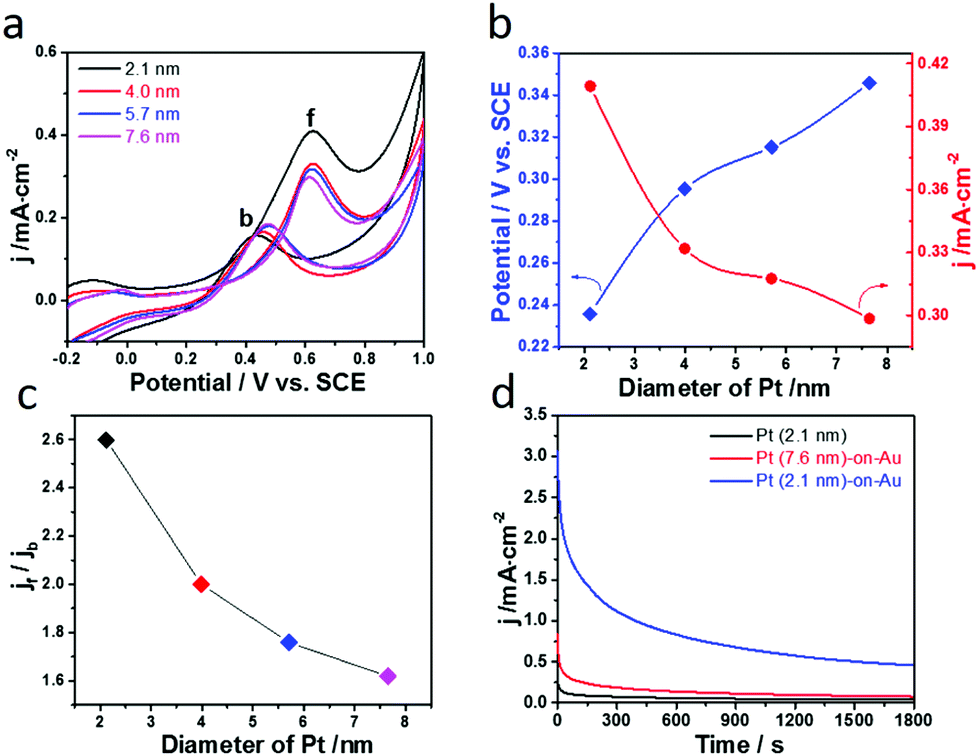 | ||
| Fig. 5 (a) Cyclic voltammograms of methanol electrooxidation catalyzed by the Pt-on-Au core–satellite nanocomposites with different Pt particle sizes in 2 M CH3OH + 0.5 M H2SO4 at a scan rate of 20 mV s−1. (b) The corresponding onset potentials and peak current density (jf) of methanol electrooxidation as a function of Pt particles size. (c) Effect of the Pt size on the jf/jb ratio of methanol electrooxidation. (d) Amperometric i–t curve at a potential of 0.6 V vs. SCE for methanol electrooxidation on Pt and Pt-on-Au satellite nanostructure catalysts. | ||
Furthermore, the CO-tolerance and long-term stability of the Pt-on-Au nanocomposites towards the methanol electrooxidation have also been studied. As demonstrated by the in situ SERS results (Fig. S7†) and previous in situ IR results,39,40 during the electrooxidation of methanol in the forward scan, CO is first generated and would poison the catalyst. Therefore, the ratio between the peak current density in the forward scan (jf) and that in the backward scan (jb) can reflect the CO-tolerance of the catalyst. The larger jf/jb value indicates better CO-tolerance. As shown in Fig. S8a,† the jf/jb value for the Pt-on-Au core–satellite nanocomposites is much higher than that for the pure Pt nanoparticles. This result indicates that the core–satellite nanocomposite is much more resistant to CO poisoning compared with Pt, leading to its improved long-term stability (Fig. S8b†).
The influence of the Pt size on the CO-tolerance of the Pt-on-Au nanocomposite is also investigated. As displayed in Fig. 5c, the jf/jb value decreases with the increase of the Pt size, meaning that the Pt-on-Au nanocomposites with smaller Pt nanoparticles show better CO-tolerance. Such results can be explained by the fact that smaller Pt nanoparticles can promote the electrooxidation of CO, which is a poisoning intermediate generated during methanol electrooxidation (Fig. 3 and 4). As a result, the Pt-on-Au nanocomposites with smaller Pt nanoparticles show much better long-term stability than the nanocomposites with larger Pt nanoparticles due to their improved CO-tolerance (Fig. 5d). Furthermore, it is also observed that the initial activity and long-term stability of Pt-on-Au nanocomposites are much higher than those of pure Pt (Fig. 5d), as CO generated on Pt-on-Au during the electrooxidation of methanol could be more easily removed.
In summary, bifunctional Pt-on-Au core–satellite nanocomposites have been fabricated by assembling of Pt nanoparticles on Au cores. Assembly of Pt on Au could greatly promote the electrooxidation of CO, leading to the improved activity for the Pt-on-Au nanocomposites as compared to the monometallic Pt nanocatalyst. At the same time, the Au core can generate strong electromagnetic fields, which would significantly enhance the Raman signals of the species adsorbed on the Pt satellites. Therefore, the electrocatalytic reactions carried out on the surface of the nanocomposites can be in situ monitored by SERS. Combining the electrochemical method with SERS, we then further studied the size effects of the Pt nanoparticles on the electrooxidation of CO. It has been found that the activity of the Pt-on-Au nanocomposites towards CO electrooxidation increases with the decrease of the Pt size in the range of 2.1–7.6 nm. Furthermore, such Pt-on-Au nanocomposites have also been used in the electrooxidation of methanol, and the effect of the Pt size on the catalytic performance is also revealed. With the decrease of the Pt size, the activity and CO-tolerance of the catalysts increase rapidly, leading to the best activity and long-term stability for the core–satellite nanocomposites with 2.1 nm Pt nanoparticles. This work provides a new strategy to fabricate highly efficient nanocatalysts for DMFCs and to in situ study their structure–activity relationships by SERS.
Author contributions
J.F. Li, J.J. Sun, and H. Zhang designed the experiments. X. Chen, M.M. Liang, H. L. Sun, C. Wang, and J. Wei conducted the experiments. W.M. Yang and Z.L. Yang performed the 3D-FDTD simulations. X. Chen, J. Xu, J.J. Sun, H. Zhang, J.F. Li, and Z.Q. Tian analysed the data. X. Chen, J.J. Sun, H. Zhang and J.F. Li wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21775127, 21703181, 21972117, and 21475023), the Fundamental Research Funds for the Central Universities (20720190044 and 20720190018), the Natural Science Foundation of Fujian Province (2019J01030), Foundation of State Key Laboratory of Coal Clean Utilization and Ecological Chemical Engineering (Grant No. 2016-06), and Program for Changjiang Scholars and Innovative Research Team in University (No. IRT_15R11).References
- S. Sharma and B. G. Pollet, J. Power Sources, 2012, 208, 96–119 CrossRef CAS.
- J. G. Liu, T. S. Zhao, R. Chen and C. W. Wong, Electrochem. Commun., 2005, 7, 288–294 CrossRef CAS.
- W. Chen, J. Cai, J. Yang, M. M. Sartin and Y. X. Chen, J. Electroanal. Chem., 2017, 800, 89–98 CrossRef CAS.
- S. Chumillas, C. Busó-Rogero, J. Solla-Gullón, F. J. Vidal-Iglesias, E. Herrero and J. M. Feliu, Electrochem. Commun., 2011, 13, 1194–1197 CrossRef CAS.
- H. J. Yan, M. C. Meng, L. Wang, A. P. Wu, C. G. Tian, L. Zhao and H. G. Fu, Nano Res., 2016, 9, 329–343 CrossRef CAS.
- S. Wasmus and A. Küver, J. Electroanal. Chem., 1999, 461, 14–31 CrossRef CAS.
- T. Iwasita, Electrochim. Acta, 2002, 7, 3663–3674 CrossRef.
- K. Bergamaski, A. L. Pinheiro, E. Teixeira-Neto and F. C. Nart, J. Phys. Chem. B, 2006, 110, 19271–19279 CrossRef CAS PubMed.
- Y. W. Tang, L. L. Zhang, Y. N. Wang, Y. M. Zhou, Y. Gao, C. P. Liu, W. Xing and T. H. Lu, J. Power Sources, 2006, 162, 124–131 CrossRef CAS.
- J. Zeng and W. Zhou, Appl. Catal., A, 2006, 308, 99–104 CrossRef CAS.
- A. J. Cowan and L. J. Hardwick, Annu. Rev. Anal. Chem., 2019, 12, 323–346 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman and I. Itzkan, Phys. Rev. Lett., 1997, 78, 1667 CrossRef.
- M. Moskovits, L. L. Tay, J. Yang and T. Haslett, Top. Appl. Phys., 2002, 82, 215–227 CrossRef CAS.
- X. Y. Zhang, N. C. Shah and R. P. V. Duyne, Vib. Spectrosc, 2006, 42, 2–8 CrossRef CAS.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, Z. Y. Zhou, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392 CrossRef CAS PubMed.
- S. L. Guan, O. Donovan-Sheppard, C. Reece, D. J. Willock, A. J. Wain and G. A. Attard, ACS Catal., 2016, 6, 1822–1832 CrossRef CAS.
- Y. X. Zou, L. Chen, Z. L. Song, D. Ding, Y. Q. Chen, Y. T. Xu, S. S. Wang, X. F. Lai, Y. Zhang, Y. Sun, Z. Chen and W. H. Tan, Nano Res., 2016, 9, 1418–1425 CrossRef CAS.
- Y. Y. Dong, Y. L. Su, L. L. Du, R. F. Wang, L. Zhang, D. B. Zhao and W. Xie, ACS Nano, 2019, 13, 10754–10760 CrossRef CAS PubMed.
- Y. H. Wang, Y. J. Zhang, M. M. Liang, S. Chen, P. Radjenovic, H. Zhang, Z. L. Yang, X. S. Zhou, Z. Q. Tian and J. F. Li, Angew. Chem., Int. Ed., 2018, 57, 11257–11261 CrossRef CAS PubMed.
- W. B. Cai, B. Ren, X. Q. Li, C. X. She, F. M. Liu, X. W. Cai and Z. Q. Tian, Surf. Sci., 1998, 406, 9–22 CrossRef CAS.
- R. Liu, L. Q. Zhang, C. Yu, M. T. Sun, J. F. Liu and G. B. Jiang, Adv. Mater., 2017, 29, 1604571 CrossRef PubMed.
- Y. L. Li, Y. F. Hu, F. X. Shi, H. X. Li, W. Xie and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 9049–9053 CrossRef CAS PubMed.
- Y. H. Wang, J. B. Le, W. Q. Li, J. Wei, P. M. Radjenovic, H. Zhang, X. S. Zhou, J. Cheng, Z. Q. Tian and J. F. Li, Angew. Chem., Int. Ed., 2019, 58, 16062–16066 CrossRef CAS PubMed.
- C. Y. Li, J. B. Le, Y. H. Wang, S. Chen, Z. L. Yang, J. F. Li, J. Cheng and Z. Q. Tian, Nat. Mater., 2019, 18, 697–701 CrossRef CAS PubMed.
- S. Park, P. Yang, P. Corredor and M. J. Weaver, J. Am. Chem. Soc., 2002, 124, 2428–2429 CrossRef CAS PubMed.
- V. Joseph, C. Engelbrekt, J. Zhang, U. Gernert, J. Ulstrup and J. Kneipp, Angew. Chem., Int. Ed., 2012, 51, 7592–7596 CrossRef CAS PubMed.
- W. Xie, B. Walkenfort and S. Schlücker, J. Am. Chem. Soc., 2013, 135, 1657–1660 CrossRef CAS PubMed.
- M. Ahn and J. Kim, J. Phys. Chem. C, 2016, 117, 24438–24445 CrossRef.
- H. Jeong and J. Kim, Electrochim. Acta, 2018, 283, 11–17 CrossRef CAS.
- C. Wang, X. Chen, T. M. Chen, J. Wei, S. N. Qin, J. F. Zheng, H. Zhang, Z. Q. Tian and J. F. Li, ChemCatChem, 2020, 12, 75–79 CrossRef CAS.
- H. Zhang, C. Wang, H. L. Sun, G. Fu, S. Chen, Y. J. Zhang, B. H. Chen, J. R. Anema, Z. L. Yang, J. F. Li and Z. Q. Tian, Nat. Commun., 2017, 8, 15447 CrossRef CAS PubMed.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS PubMed.
- M. F. Li, Z. P. Zhao, T. Cheng, A. Fortunelli, C. Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov, Z. Y. Lin, E. Zhu, T. Yu, Q. Y. Jia, J. H. Guo, L. Zhang, W. A. Goddard III, Y. Huang and X. F. Duan, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed.
- Q. S. Chen, F. J. Vidal-Iglesias, J. Solla-Gullón, S. G. Sun and J. M. Feliu, Chem. Sci., 2012, 3, 136–147 RSC.
- H. Zhang, X. G. Zhang, J. Wei, C. Wang, S. Chen, H. L. Sun, Y. H. Wang, B. H. Chen, Z. L. Yang, D. Y. Wu, J. F. Li and Z. Q. Tian, J. Am. Chem. Soc., 2017, 139, 10339–10346 CrossRef CAS PubMed.
- P. P. Fang, S. Duan, X. D. Lin, J. R. Anema, J. F. Li, O. Buriez, Y. Ding, F. R. Fan, D. Y. Wu and B. Ren, Chem. Sci., 2011, 2, 531–539 RSC.
- Y. F. Huang, P. J. Kooyman and M. T. Koper, Nat. Commun., 2016, 7, 12440 CrossRef CAS PubMed.
- F. Calle-Vallejo, J. I. Martínez, J. M. García-Lastra, P. Sautet and D. Loffreda, Angew. Chem., Int. Ed., 2014, 53, 8316–8319 CrossRef CAS PubMed.
- J. X. Tang, Q. S. Chen, L. X. You, H. G. Liao, S. G. Sun, S. G. Zhou, Z. N. Xu, Y. M. Chen and G. C. Guo, J. Mater. Chem. A, 2018, 6, 2327–2336 RSC.
- Q. S. Chen, S. G. Sun, Z. Y. Zhou, Y. X. Chen and S. B. Deng, Phys. Chem. Chem. Phys., 2008, 10, 3645–3654 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and calculation details, and Fig. S1–S7. See DOI: 10.1039/c9nr10304j |
| This journal is © The Royal Society of Chemistry 2020 |