
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReview of recent advances in the biodegradability of polyhydroxyalkanoate (PHA) bioplastics and their composites
Kjeld W.
Meereboer
 ab,
Manjusri
Misra
*ab and
Amar K.
Mohanty
*ab
ab,
Manjusri
Misra
*ab and
Amar K.
Mohanty
*ab
aSchool of Engineering, Thornbrough Building, University of Guelph, 50 Stone Road East, Guelph, Ontario, Canada. E-mail: mohanty@uoguelph.ca; mmisra@uoguelph.ca
bBioproducts Discovery and Development Centre, Department of Plant Agriculture, Crop Science Building, University of Guelph, 50 Stone Road East, Guelph, Ontario, Canada
First published on 17th August 2020
Abstract
The detrimental impact of single-use plastics on the environment is daily news across the globe. Single-use plastic packaging materials and other plastic waste originating from petroleum-based sources are continuously building up in landfills and leaching into the environment. Managing plastic waste remains an urgent crisis in the environment and switching to biodegradable plastics can help mitigate some of these issues. This review will summarize recent advances and opportunities to utilize polyhydroxyalkanoates (PHAs) as a biodegradable substitute in some applications where non-biodegradable and petroleum-based plastics are currently used. PHAs are a well-known family of bacteria-based biodegradable plastics and offer an approach to carbon neutrality and support a more sustainable industry. PHAs such as poly(3-hydroxybutyrate) (PHB) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) show biodegradable behaviour in all aerobic and anaerobic environments defined by ASTM standards, and can be used to make completely compostable, and soil and marine biodegradable products – a strong positive compared to the negativity associated with the landfilling of plastics. However, PHAs are relatively expensive compared to petroleum-based alternatives. To reduce the cost, PHAs can be used in biocomposite materials, where bio-based agro-residues are incorporated, while maintaining the performance in certain applications. Organic fillers and fibres composed of cellulosic material can improve the properties of polymers, however, their effect on the marine biodegradable properties of the composite matrix remains an unexplored area. When used in biocomposites with PHAs, they improve biodegradation rates in all environments. In addition to cellulose, other bio-based fillers such as proteins (i.e. distillers dried grains with solubles) and starch have been reported to significantly improve soil and marine biodegradability rates compared to other fibres and fillers. Other components that affect biodegradability are additives (i.e. chain extenders) and compatibilizers (i.e. maleic anhydride etc.) that are added to optimize the service life properties, but are reported to inhibit the biodegradation properties by impacting the hydrophilicity of the polymer and enzyme activity. The multitude of possible combinations of polymers and fillers and fibres, and their effect on the biodegradation of PHA-based biocomposites are a largely unexplored frontier. The potential benefits of PHA-based biocomposites make a strong case for further research into this area.
 Kjeld W. Meereboer | Kjeld has completed his Master of Applied Science (M. A. Sc) degree under the advice of Professor Manjusri Misra and Professor Amar K. Mohanty in Biological Engineering at the University of Guelph (Guelph, Ontario, Canada). Prior to that he received his bachelor's degree in Biological Engineering from the University of Guelph. Mr Meereboer's research interests include biopolymer processing and characterization, sustainable materials development, biodegradation and compostability. He has published two peer reviewed journal papers and 4 conference presentations from his Master's research. |
 Manjusri Misra | Dr Manju Misra is a professor in the School of Engineering and holds a joint appointment in the Dept. of Plant Agriculture at the University of Guelph. Also, she is serving as the Research Program Director for the Ontario Agri-Food Innovation Alliance (Plant – Technology Panel). She is a Fellow of the American Institute of Chemical Engineers (AIChE) and the Royal Society of Chemistry (UK). She has authored more than 700 publications, including 362 peer-reviewed journal papers, 21 book chapters, and 15 granted patents. She was an editor of the CRC Press volume, “Natural Fibers, Biopolymers and Biocomposites”, Taylor & Francis Group, Boca Raton, FL (2005); American Scientific Publishers volume “Packaging Nanotechnology”, Valencia, California (2009); “Polymer Nanocomposites”, Springer (2014) and “Fiber Technology for Fiber-Reinforced Composites”, Woodhead Publishing (2017). She was the chief editor of “Biocomposites: Design and Mechanical Performance” Woodhead Publishing (2015). She was the President of the BioEnvironmental Polymer Society (BEPS) in 2009. She serves in the editorial board of “Journal of Applied Polymer Science”, “Composites Part A: Science and Manufacturing”, “Polymer Testing”, and “Composites Part C: Open Access”. In 2012, Dr Misra received the prestigious “Jim Hammar Memorial Award” from BEPS and the University of Guelph's “Innovation of the year award’ in 2016 for the involvement in developing the “Compostable single-serve coffee pods”. In 2017, Professor Misra has also received the “Andrew Chase Division Award in Chemical Engineering” from the Forest Bioproducts Division of AIChE. In 2019, Professor Misra was awarded with the “Woman of Distinction on Science, Technology, Engineering & Math (STEM)” from the Guelph YWCA-YMCA. Total citations: 32 |
 Amar K. Mohanty | Prof. Amar Mohanty is a Professor and OAC Distinguished Research Chair in Sustainable Biomaterials. He is the Director of the Bioproducts Discovery and Development Centre & University Research Leadership Chair at the University of Guelph, Ontario, Canada. He is the Editor-in-Chief in Sustainable Composites of Composites Part C (An ELSEVIER journal). He is an international leader in the field of bioplastic, biobased materials with a focus on engineering new sustainable materials. He is a highly cited researcher with more than 900 publications to his credit, including 394 peer-reviewed journal papers, and 62 patents awarded/applied (Google Scholar Citations: 34 |
1 Introduction
A seismic shift in economic objectives triggered by the growing and overwhelming evidence from industry suggests that the projected cumulative growth of primary plastic waste produced by 2050 will exceed 25 billion metric tons.1 Combined with the shift towards sustainability using non-petroleum based plastics, the production of bio-based/non-biodegradable and biodegradable plastics projected from 2020 to 20232 is expected to grow 13% per annum. Leading plastic packaging producers are moving towards a goal of 100% recycled, biodegradable or re-useable plastics in their products by 2025.3 This shift towards a sustainable economy has occurred in the recent decade, such that, between 2010 and 2017, bio-based poly(ethylene) (Bio-PE), bio-based poly(ethylene terephthalate) (Bio-PET), poly(lactic acid) (PLA) and poly(hydroxyalkanoate)s (PHAs) have seen production capacity growth of approximately 22%, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000%, 300% and 41% respectively.4 Replacement of petroleum-based plastics with bio-based alternatives is a more sustainable pathway to plastic production due to their lower associated carbon emissions from petroleum extraction and refinement.5
000%, 300% and 41% respectively.4 Replacement of petroleum-based plastics with bio-based alternatives is a more sustainable pathway to plastic production due to their lower associated carbon emissions from petroleum extraction and refinement.5
1.1 Polymer pollution in the environment
Plastic pollution occurs in two fronts, during production (carbon emissions) and their disposal (contaminants and physical hazards) which impacts both the environment and the ecosystem. Replacing petroleum-based polymers with bio-based polymers is a potential solution that produces significantly lower carbon emissions and energy production requirements.6Fig. 1 illustrates the reduced impact of bio-based/biodegradable polymers on the environment relative to some commercial petroleum-based polymers. However, regardless of their production method most plastic waste after their service life ends up being incinerated, landfilled, littered or recycled, resulting in carbon or methane emissions over time. It is the disposal after the service life of biodegradable polymers that further benefits the environment compared to non-biodegradable petroleum-based polymers. Non-biodegradable polymers can leak contaminants or additives into soil and waterways, and physically obstruct animal digestive systems.7 Several industries can thus benefit from this as there are increasing carbon footprint reduction measurements and reporting required due to increased societal pressure on industry to be more environmentally responsible.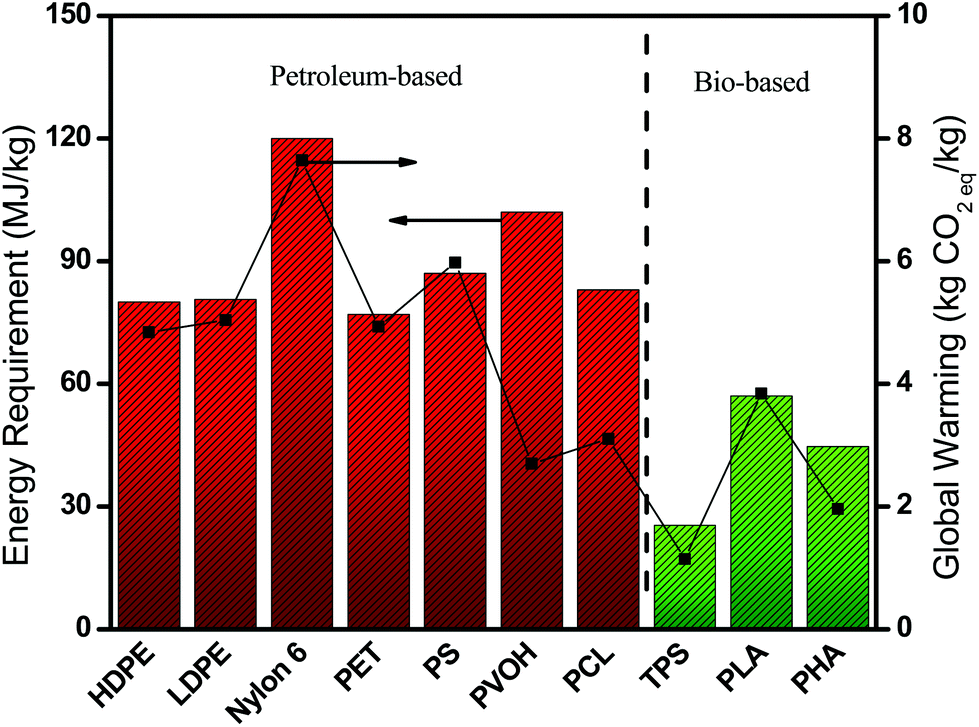 | ||
| Fig. 1 Production energy and CO2 emissions of petroleum-based and bio-based polymers.6,8 | ||
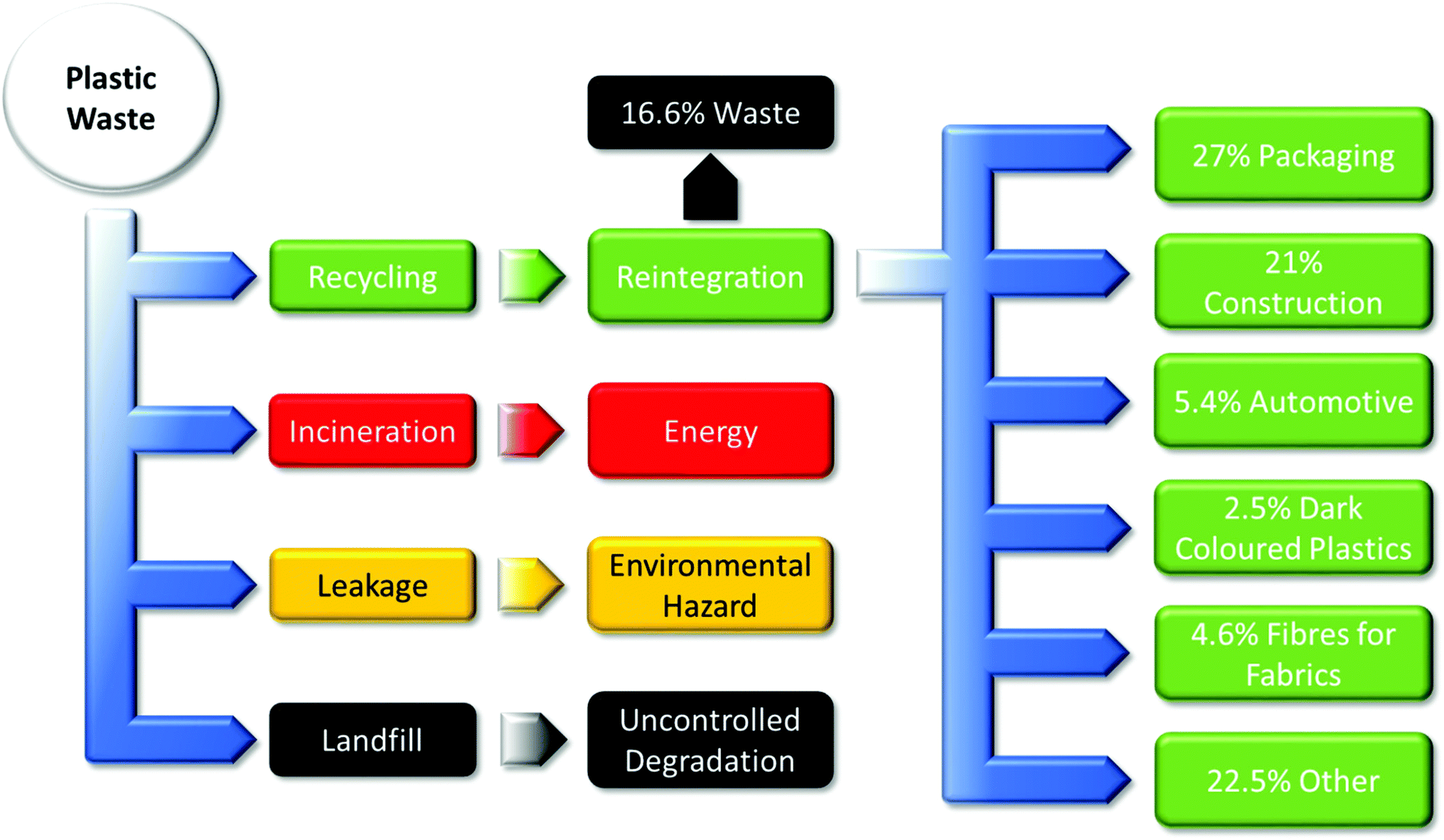 | ||
| Fig. 2 Predominant current polymer waste disposal streams.9 | ||
(i) recycling produces some losses and material degradation;
(ii) incineration produces energy at the cost of material and pollution;
(iii) leakage results in environmental hazards which can harm the surrounding environment;
(iv) and landfills result in uncontrolled degradation that can severely harm the environment from methane production.
A new subset of controlled or managed degradation and disintegration is now being developed with the implementation of compostable polymers in industry, following ASTM standards. The end-of-life is value-added usable compost, which could evolve into a multi-billion dollar industry and generate employment for thousands.
1.1.1.1 Recycling. Recycling plastics is an option to reduce the overall plastic waste produced; however, recycled plastics suffer from reduced mechanical performance. This can be overcome by physically treating (annealing) polymers to increase the modulus and strength, use of chemical stabilizers, blending with other recycled plastics or blending recycled plastics with other polymers as a valorisation method.10 Irrespective of this, recycling generates waste during or at the end of the multi-recycling cycle, with the material being too degraded to use. After many years of implementing recycling, still only a small percentage of plastics in the USA is actually recycled (<10%), compared to non-plastic recycling (25–65%), in 2017.11 Therefore, an alternative method of plastic waste disposal is required when the service life has ended. Possible alternatives include composting, or diversion to alternative energy production (incineration).
1.1.1.2 Landfill. Landfills are suitable for storage of plastic waste, in comparison to other environments, due to the ease of human intervention. However, landfills go through uncontrolled degradation, releasing greenhouse gases into the environment. Landfills can be subdivided into several types, based on the age and the type of material waste. In consideration of municipal solid wastes, there are old landfills and modern landfills. Old landfills have no control of pollution migration and no gas capturing technology in place. Modern landfills are designed to capture the methane produced, for energy generation.12 However, in less developed regions, landfills do not have these measures13 and the off-gases can migrate into the surrounding area.14 Commodity plastics are not generally landfilled, and, unlike organic materials, do not degrade into methane. But this poses another problem because it remains in the landfill indefinitely under anaerobic conditions. Furthermore, if mismanaged waste results in biodegradable polymers being landfilled, the methane generation can make up to 50% of the total gas release.15 Another environmental factor is the proper soil coverage after closing the landfill, to prevent plastic waste from being scattered and dispersed into the environment.16
1.1.1.3 Leakage (litter). Plastic waste in the environment is grouped into two sources, marine-based and land-based, however the relative quantity remains unclear.17 Examples of ocean-based waste that likely remains in the ocean today would be lost cargo and plastic pellets from shipping services,18 or just general shipping waste pollution. Other pathways involve migration of plastics from the land (i.e. litter, landfills) to the ocean by environmental elements.18 Land-based plastic litter can be accidental through environmental elements or intentional, usually by inadequate waste disposal facilities during events or in the public spaces.19 The plastic litter can also originate from landfills, sewage systems and industrial processes which degrade overtime and accumulate in the soil.17 Regardless of the sources, plastics containing additives, such as plasticizers and UV stabilizers, contaminate the soil and marine environment over time and impact animal and cellular organ function.7
With migration of plastics in the environment it is expected that plastic can be found in a number of oceans (surface and sea floor), shorelines and lakes across the globe.17 Plastic ending up in the ocean is of concern due to its movement and the difficulty of human intervention. Plastic debris in the ocean can have concentrations up to 580000 pieces per km2.20 This plastic has been found in the Pacific, Atlantic, and Indian oceans in the past two decades, especially in the North Atlantic gyre and the North Pacific subtropical gyre where garbage patches have seen significant growth,21–23 and it is predicted that 99% of all species of seabirds will ingest plastic by 2050.20 Furthermore, this plastic lingers in the environment due to its durability if not exposed to microorganisms or UV radiation.17 This is exacerbated by the protective coatings applied to polymers to ensure that the properties are not damaged by UV exposure during their service life.24 This plastic residing in the environment can't be recycled due to contamination or poor residual mechanical and thermal properties. Therefore, biodegradable plastics hold significant importance in combatting mismanaged waste which is expected to double by 2025.25
1.1.1.4 Compost. Composting is a subset of biodegradation, such that not all compostable materials are biodegradable in other environments such as marine, soil, landfills etc. Furthermore, only a small subset of plastics can be composted, and do not include the commodity recycled ones. Composting can be divided into home composting and industrial composting, with the main difference being the controlled conditions in the industrial composting (∼58 °C, 50–55% solids etc.). In most climates, home composting is slower than industrial composting, but is suitable for composting organic materials due to their short degradation period.26 Composting of bioplastics is mainly limited to industrial composting operations and not recommended for home composting.27 Industrial composting is designed for large amounts of organic waste, has a high turnover and produces compost suitable for soil remediation.28
Compostable plastics are predominantly thermoplastic starch (TPS), PLA and poly(butylene adipate terephthalate) (PBAT), making up 83% of the biodegradable plastic produced in 2018. Poly(butylene succinate) (PBS), another biodegradable plastic, has also experienced growing global production capacity due to the significantly lower production cost compared to other biodegradable polymers (i.e. PHAs).29 The biodegradability of these plastics varies, with some (i.e. PLA) being less suitable for home composting due to the long duration,30 such that in the virgin form only industrial composting conditions are suitable. PLA for example shows effective biodegradable properties in the soil when hybridized with cellulose using appropriate methods, but is not currently implemented on an industrial scale.31 Furthermore, bio-sourced plastics utilize renewable carbon, compared to petroleum-based biodegradable polymers (PBAT). Given industrial and commercial composting is not always suitable for compostable plastics, an alternative bio-based biodegradable polymer such as PHAs is well posed for commercial adoption use due to better biodegradable properties in many types of environments.32
Therefore, the use of bio-based and biodegradable plastics that can degrade in natural environments (i.e. soil, ocean water etc.) as a global movement is important because it combats both climate change and plastic pollution – both necessary for sustainable growth and lowering the carbon footprint for the positive environmental effect. An important factor to consider is how bio-based biodegradable plastics differ from other plastics, both in terms of their production and at the end of life. Any modifications in the biodegradation of these plastics, including fibre and filler addition, blends and chemical additions must be considered under a comparable standard under all environmental conditions.
2 Bacterial polyesters
Bio-based polymers can be subdivided into three types, plant-based (i.e. starch, cellulose derivatives and natural rubbers), polymerized bio-monomers (i.e. PLA, polyimides, polyurethanes, poly(butylene succinate) (PBS), bio-PE etc.) and extracted bio-polymers (PHAs).33,34 Further subdivision of polymerized bio-monomers exists with partially bio-based polymers such as nylon-6,10, poly(trimethylene terephthalate) (PTT) and poly(p-phenylene) (PPP).35,36 Bacterial polyesters are a unique subset of bio-based polymers polymerized by microorganisms. PHAs for example can be synthesized enzymatically in vivo by microorganisms as a true natural polyester for intracellular storage,37 while PLA is produced through fermentation as lactic acid and chemically polymerized.38 However, despite different production methods, not all bio-based polymers are biodegradable. Among bio-based polymers, only PLA and PHAs are completely bio-based and biodegradable in some form. However, PLA is compostable, but not marine biodegradable like PHAs, making it unsuitable to combat plastic waste leaking into the environment.39 Moving towards bio-based biodegradable polymers allows for a more sustainable option by implementing a cradle-to-cradle approach,40 where the output of biodegradation becomes the production input for the same polymer within a reasonable frame of time (Fig. 3). For example, despite PE and bio-PE being petroleum based and biobased respectively, their main disposal stream is through either recycling or landfill. In addition, petroleum-based polymers such as PBAT are biodegradable,29 but due to their production method, they do not form a circular cradle-to-cradle approach. PHAs, PLA and cellulosic material are all bio-based and biodegradable, forming a complete carbon cycle; however, PLA is only compostable, not marine biodegradable like PHAs, making it unsuitable to combat the leakage of plastic into the environment.39 While biodegradable plastic can potentially be recycled in the general waste stream, the resulting properties of the recycled plastic are significantly worse and they are more suited for short life cycles.41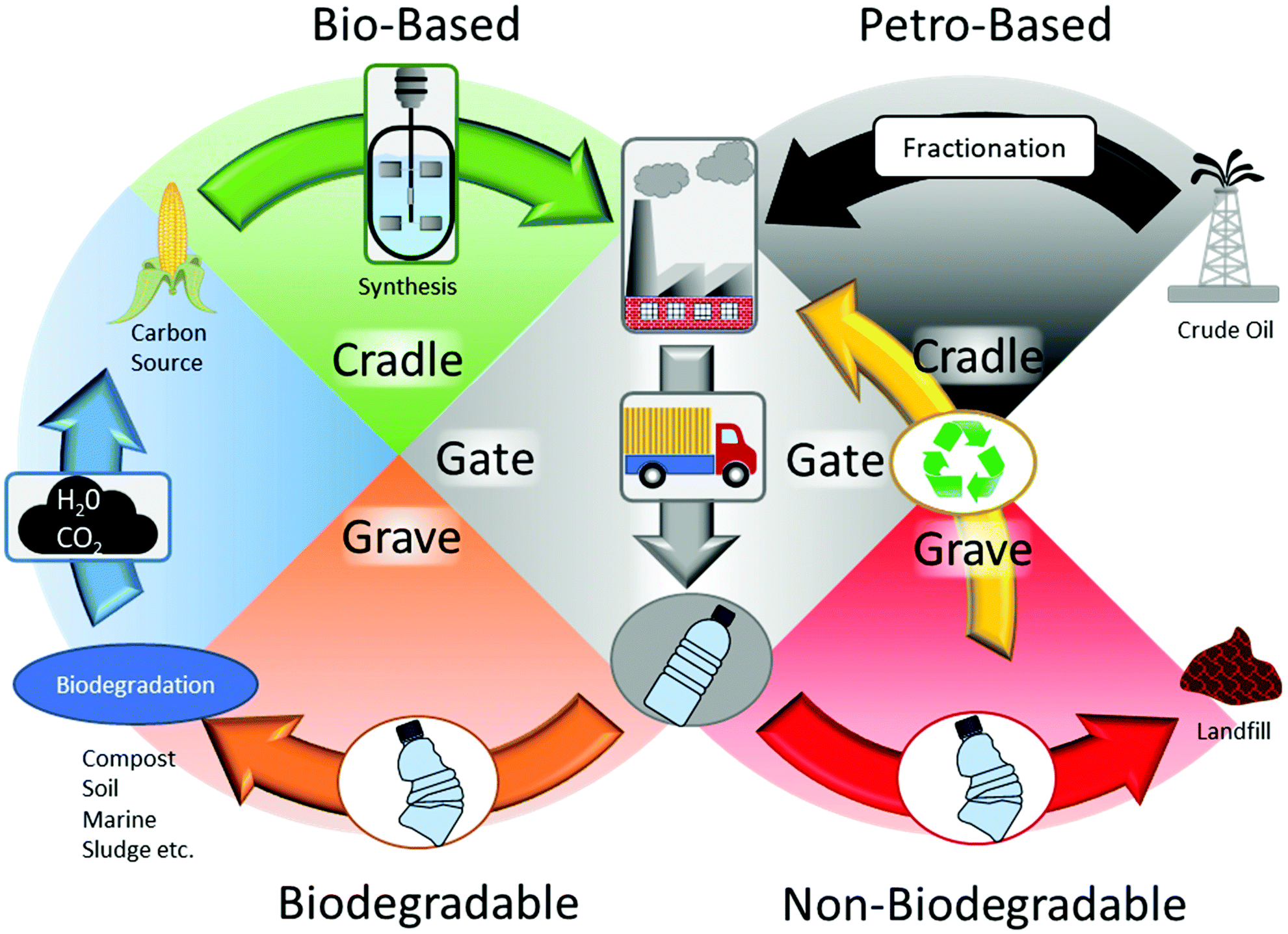 | ||
| Fig. 3 Cradle-to-cradle and cradle-to-grave approach of biodegradable and non-biodegradable plastics. | ||
2.1 PHAs
PHAs are aliphatic polyesters well known for their biodegradable properties and their bacteria based production methods. Over 91 different polyhydroxyalkanoic acid constituents that make up PHAs have been recorded and the number is continuously growing.42 Based on the potential combination of monomer units, an uncountable number of PHA copolymers can be formed. PHAs being biodegradable in various environments are an attractive option to replace current single use plastics11 or plastics that are unsuitable for re-entry into the manufacturing sector due to their poor quality.43 With their biodegradable properties, PHAs form a closed loop cycle from cradle-to-cradle (Fig. 3) that minimizes the impact on the environment.44 However, the functionality and production methods are dependent on the type of PHA.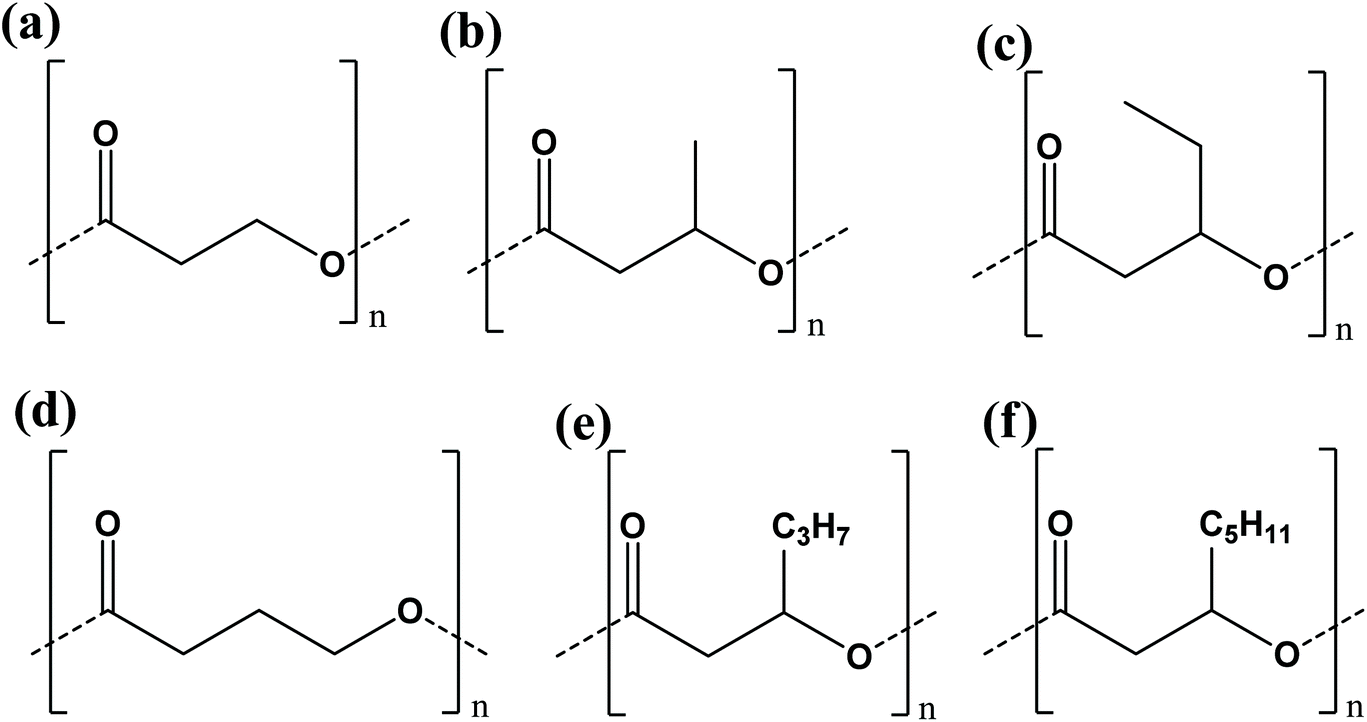 | ||
| Fig. 4 (a) 3-Hydroxypropionate (HP), (b) 3-hydroxybutyrate (HB), (c) 3-hydroxyvalerate (HV), (d) 4-hydroxybutyrate (4HB), (e) 3-hydroxyhexanoate (Hx), and (f) 3-hydroxyoctonaote (HO) chemical structures. | ||
The bacterial production of PHAs begins under growth limiting conditions because they function as an energy storage molecule for microorganisms,49 leading to accumulation in the cell walls of bacteria and archaea.50,51 HB monomers are then bacterially polymerized by PHB synthase of acetoacetyl-CoA,52 however chemical pathways do exist such as ring opening polymerization (ROP) of β-butyrolactone.53 PHBV is manufactured under similar conditions but in the presence of propionic acid. The copolymerization process is initiated enzymatically from acetoacetyl-CoA and 3-ketovaleryl-CoA to form the HB and HV units, respectively.54,55 However, with additional nutrients and energy costs making up more than 75% of the product cost,56 PHAs become a relatively expensive product compared to commercial petro-based polymers of comparable properties.57 For bacterial PHA production, several non-conventional carbon feed sources are used, including mixed carbon sources,58 organic wastes,36 methane,59 peanut oil,60 soybean oil,61,62 palmitate oil,63 waste frying oil,64 margarine waste,65 glycerol etc.66 Despite these advantages, the costs associated with the production of PHAs are still very high (comparative to other bio-polymers) and the properties leave much to be desired. Furthermore, while petroleum-based biodegradable plastics can be incorporated into PHAs, to reduce costs and maintain functionality, the sustainability of production becomes lower.
Applications of PHAs outside of the biomedical industry are mainly in the replacement of single use, disposable plastics, such as plastic tableware, food packaging, plant pots and organic waste collection.72 PHAs also have the potential for substituting poly(ethylene) (PE) in some film applications, e.g. mulch films used to stop weed germination in agricultural farming. PHAs and blends of PHAs with other biodegradable polymers show promise to fulfil the same role as PE with added benefits of degrading during the season to reduce labour costs and farm waste at the end of the season.73,74 In all cases, biodegradation is a critical factor that makes PHAs marketable and defines their applications outside of the biomedical industry. Currently, PHAs’ use in single use plastics for the food industry is limited, despite them showing the potential for bottles, caps, blister packs etc.75 due to odour issues that require additives to remove them.76 However, the biodegradation of the resulting new blends with any additives remains to be fully explored.
2.2 Challenges of biodegradable plastics
Biodegradable plastics usually suffer from limitations, such as poor mechanical properties (Fig. 5), and the inability to blend them with many other polymers without losing their biodegradable functionality. The market value of biodegradable plastics is in their biodegradable performance when disposed and still requires biodegradation testing to ensure that no chemical interactions hinder the overall biodegradation rate. The two main bio-based and biodegradable polymers, PLA and PHB, are both extremely brittle plastics relative to commodity and engineering plastics. Although some PHAs are more ductile as their chain length increases,77–79 the cost of PHAs is significantly higher than that of conventional plastics and other bio-based biodegradable plastics.80 While incorporation of petro-based biodegradable polymers is a potential solution to maintain the biodegradable properties of PHAs, the resulting blends become less environmentally sustainable due to petroleum usage. For example, to make PLA more flexible, it is blended with PBAT and marketed as Ecovio by BASF. Some studies have investigated blending PHAs with other bio-based or biodegradable polymers. PLA was blended with PHB for improved ductility, toughness and thermal stability,81 and poly(ε-caprolactone) (PCL) was blended with PHBV for increased ductility.82 However, the resulting blends suffer from reduced biodegradable performance or costs.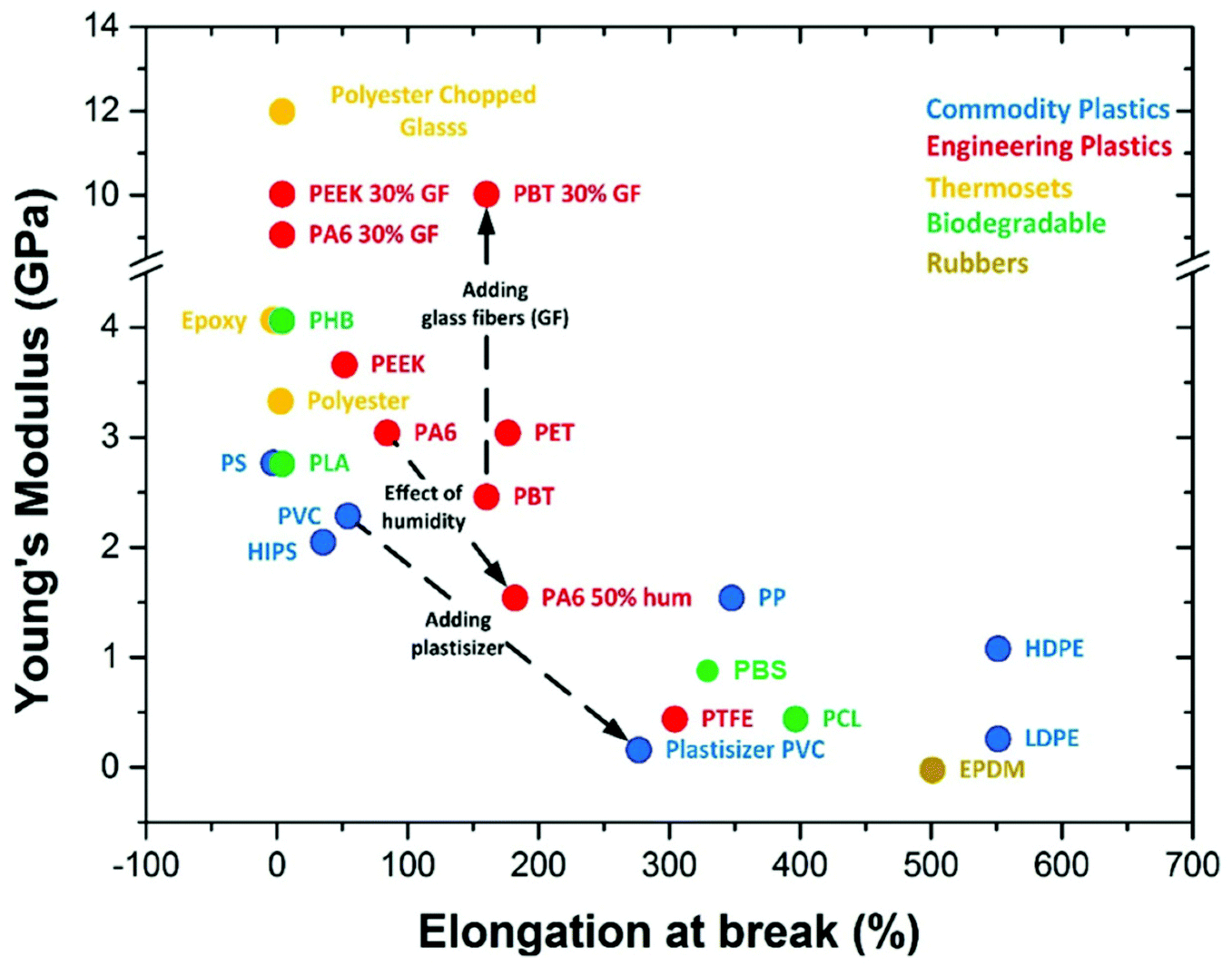 | ||
| Fig. 5 Young's modulus and elongation at break of plastic types. This figure has been reproduced with permission from ref. 87, Coatings, Creative Commons 2015. | ||
The commercial PHB and PHBV have low impact strength, high brittleness and poor flexibility, making them unsuitable for many industrial and commercial applications compared to other biodegradable or commercial polymers.83,84 Much success has been found in varying PHA copolymer compositions, such as increasing the Hx content in PHBHx for improved flexibility marketed as Nodax by Makena,85 or increasing the 4HB content to obtain elastomeric properties.86 However, with increasing complexity of PHA monomer units, the cost of production increases.
Natural fibres are an option to minimize the cost of PHAs while enhancing their best properties such as modulus and biodegradation. However, the effects of natural fibres, composition and compatibilization techniques on biodegradation remain unclear.
3 What is biodegradation?
Biodegradation is the degradation process involving microorganisms, and is widely accepted as selective, and depends on several factors including the physical and chemical properties of biopolymers. The biodegradation process is defined as polymer degradation by biological microorganisms into CO2, H2O, biomass and methane by composting, soil biodegradation, marine biodegradation, or other biodegradation processes.57 It is also termed biotic degradation and can be enhanced or started after some initial abiotic degradation processes occur such as mechanical, oxidative or hydrolytic degradation (Fig. 6) which can increase the surface area of the organism–polymer interface.88,89 Thermal or oxidative degradation is non-selective, occurring to all polymers, and introduces thermal or chemical stressors that scission the chains of polymers into smaller units of oligomers, acids, alcohols, esters, and radicals.90,91 For example, PHAs are less thermally stable than PLA and PCL due to the different thermal degradation mechanisms of random chain scission instead of unzipping depolymerization, and thermal degradation results in a reduced molecular weight along with the esters and alcohol end groups.92 However, thermal energy can also enhance most if not all other forms of degradation, through disproportionally increasing the high energy collisions between reactants or enzymes and reactants. Catalytic degradation of polyolefins results in gas, oil and waxes by clays, acids, zeolites, aluminium oxides, calcium oxides etc. at high temperature.93 High temperatures are not desired for biodegradation of polymers, due to the inactivity of microorganism enzymes.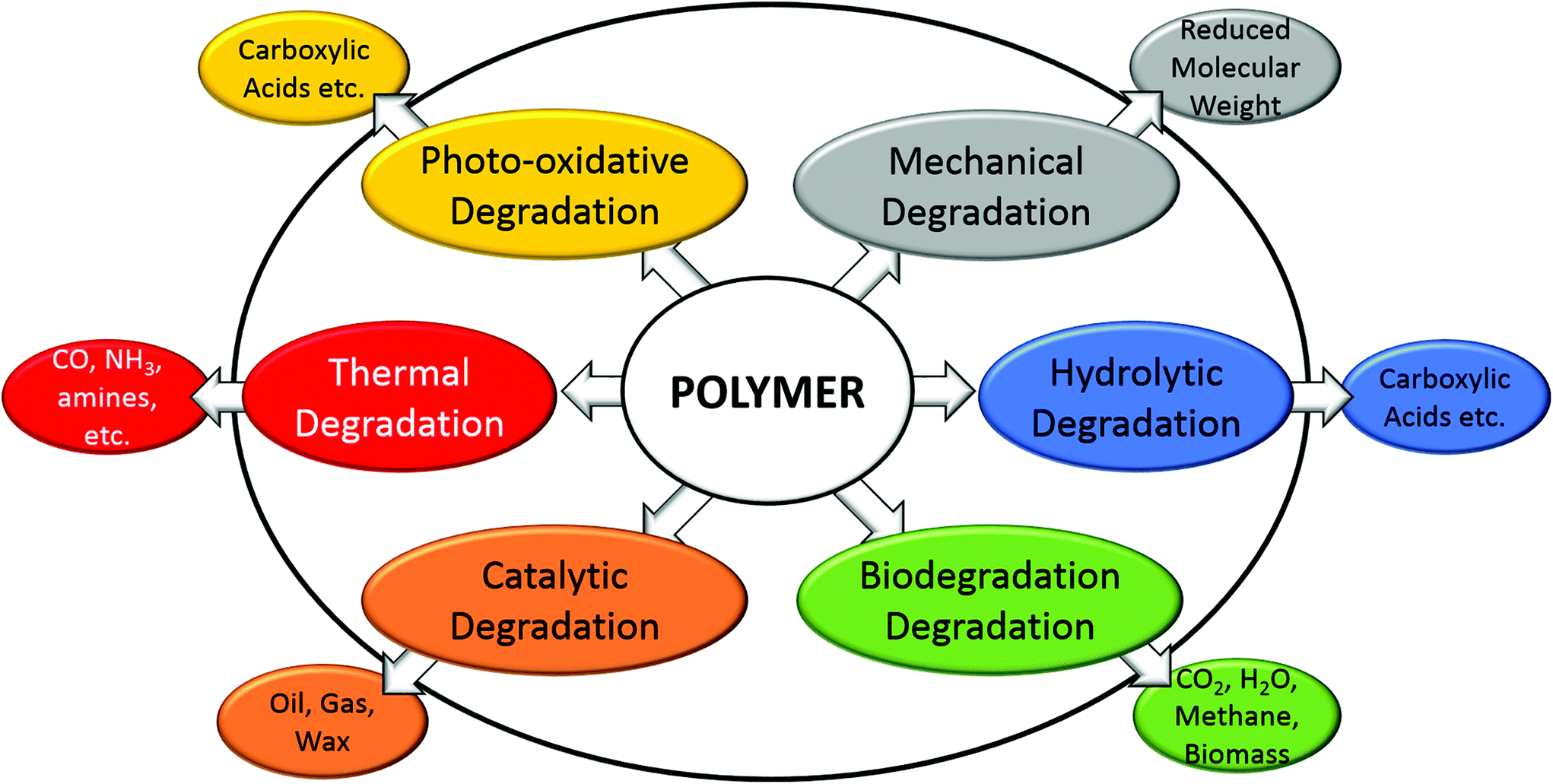 | ||
| Fig. 6 Basic degradation pathways of polymers. | ||
The value of biodegradable polymers lies in rapid degradation under natural conditions (i.e. soil, ocean water etc.), which can become the main marketable attribute, that reduces potential improperly disposed of plastic waste.94 Multiple factors affect biodegradation besides the environment conditions (i.e. temperature, moisture etc.): the polymer composition, molecular weight, crystallinity, composition, chemical structure, reduction potential, hydrophilicity, breakdown products etc.95 but the extent of the effects from some of these factors remains unclear.
When recycling or incineration is unsuitable, biodegradable plastics offer an advantage that has positive effects in environmental waste management. The process can be further enhanced by utilizing bio-based biodegradable polymers, whereby the carbon source input of production biodegrades into CO2, which is then taken up to produce a carbon source, thereby approaching carbon neutrality.
Biodegradation is influenced by the susceptibility of the polymer carbon backbone to microbial attack.96 Degradation of any polymer can be divided into two types: surface erosion and bulk erosion. The biotic (enzymatic) degradation is mainly at the surface. The reason is that enzymes are relatively large particles and are unable to permeate the structure of polymers, in comparison to smaller chemicals, free radicals etc. Abiotic degradation functions as both bulk and surface degradation and is often used as a pre-treatment to biodegradation. In general, bulk erosion is used for breaking the sample apart into smaller pieces (which enhances the rate of surface erosion) and for molecular weight reduction. For example, poly(α-hydroxy-esters) samples must exceed a critical minimum thickness (Lc) of 7.4 cm to undergo surface erosion and not bulk erosion (Fig. 7).97
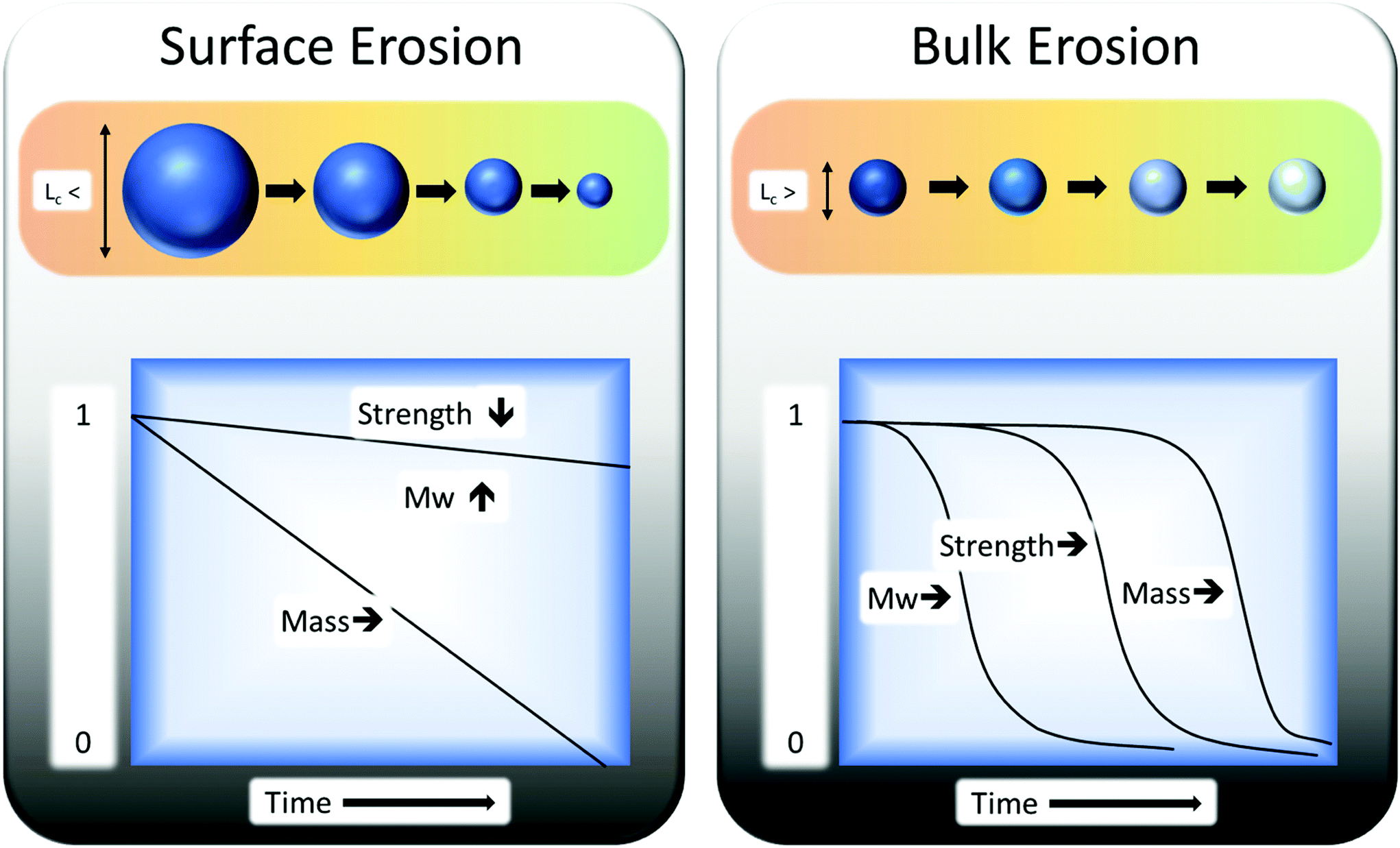 | ||
| Fig. 7 Bulk erosion and surface erosion effects on polymers as a function of time. This figure has been reproduced with permission from ref. 98, Elsevier, Copyright 2008 (License number: 4800311312040). | ||
Enzymatic hydrolysis of polyesters is completed by lipases and esterases to break ester, carbonate, amide and glycosidic bonds.99 Due to this nature, warmer temperatures (15–37 °C) and alkali environments increase the rate of PHA and PLA degradation.100–102 Non-polyesters, considering homochain polymers only, made from entirely carbon backbones are not readily biodegradable. Microbes have difficulty in enzymatically cleaving and degrading aliphatic homochain and heterochain polymers without functional groups (i.e. esters, ethers etc.) such as PE, polycarbonate (PC), polypropylene (PP), polystyrene (PS) and their derivatives with carbon backbones, and any degradation observed is usually minimal.103–106 The degradation of polymers follows the schematic of Fig. 8, where biotic or enzyme catalysed reactions are usually a more efficient method. Following polymer degradation by biotic or abiotic methods, the products can be bio-assimilated by microorganisms and used for other needs such as growth and cellular respiration.
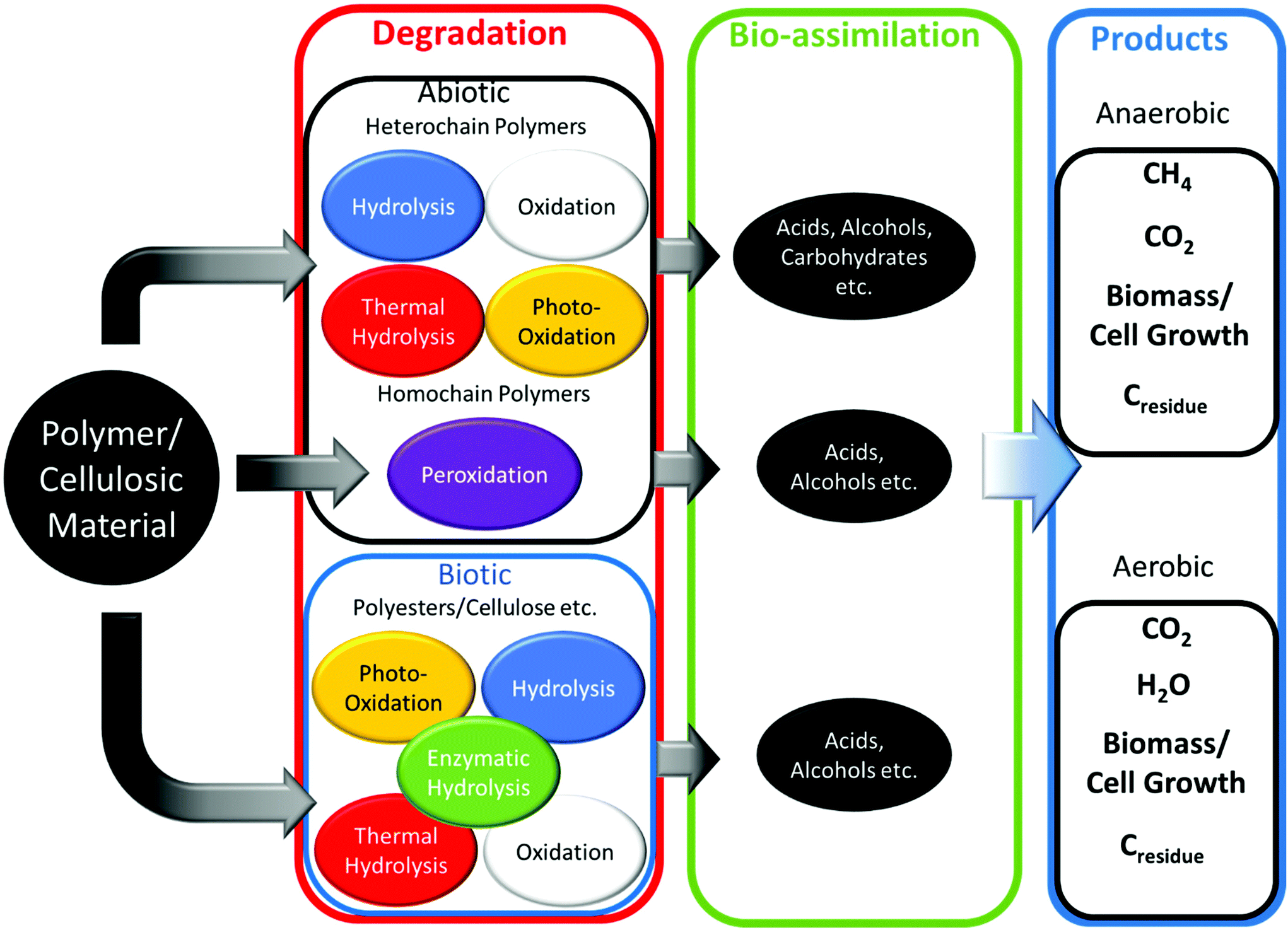 | ||
| Fig. 8 Theoretical biodegradation pathway of polymer materials. | ||
Under environmental conditions, biodegradation of PHA following standards is mainly competed extracellularly by extracellular enzymes that cleave the polymer into small enough units to bio-assimilate.107 During extracellular biodegradation, the PHA is in a highly crystalline form (mainly PHB).36 However, enzyme-catalysed biodegradation can occur in the cells of microorganisms, and is referred to as intracellular biodegradation.108 Intracellular PHAs are present in an amorphous state with disordered conformation covered by protein and phospholipids, and are degraded when no alternative carbon source is available.
3.1 Aerobic vs. anaerobic biodegradation
Biodegradation of polymers can occur under aerobic or anaerobic conditions, leading to varied products. Aerobic degradation (in the presence of oxygen) mainly utilizes oxygen as a final electron acceptor, while microorganisms that perform anaerobic degradation (in the absence of oxygen) use CO2, nitrates, sulphates etc. as an alternate electron acceptor to generate energy for the cell functions.109–111Most biodegradable polymers show evidence of degradation in both aerobic and anaerobic environments.112 For enzymatically degraded polymers, it is the temperature that usually affects whether polymer scission occurs. For example, PLA requires a temperature equal to or greater than its Tg (∼55 °C) to biodegrade effectively.113
The basic chemical equation for aerobic biodegradation is the conversion of organic carbon into CO2 by microorganisms, in the presence of oxygen (eqn (1)). The carbon atom is generally part of a complex structure, and in some cases oxygen can be derived from the polymer itself, such as from polyesters. During cellular respiration of a carbon source (i.e. glucose), the liberated oxygen reacts with free hydrogen ions to produce water (eqn (2)).114
| C + O2 → CO2 | (1) |
| H+ + O2 → H2O | (2) |
Anaerobic biodegradation on an industrial scale following standards is a significantly less studied area compared to aerobic biodegradation, due to the environment control required for the study, and it does not constitute the majority of natural environments where plastic waste could biodegrade (soil and marine) The stoichiometric mass balance of anaerobic biodegradation of plastics and natural fillers is defined by eqn (1), (3) and (4).
| C + 2H2 → CH4 | (3) |
| C6H12O6 → 3CO2 + 3CH4 | (4) |
Under anaerobic conditions, biodegradation results in methane production and some CO2 is produced depending on the residual oxygen in the environment or the type of degraded material. Two types of anaerobic environments exist on a large scale due to commercial actions, biogas facilities and landfills. Biogas facilities deal with anaerobic digestion of organic and plastic materials, capturing the released methane for energy conversion.115 However, landfills are of particular concern, because any uncontrolled biodegradation of organic and plastic materials can result in methane generation into the environment. In 2007, it was estimated that only 10% of the potential methane generated is captured in the United States,116 which has only increased to approximately 20% by 2017.115 Methane is a 25 times more potent greenhouse gas compared to CO2 over a 100-year period. Furthermore, in the waste sector, the largest contributors of methane in the atmosphere are the landfills and solid waste treatment facilities which do not collect biogas, indicating uncontrolled management of these greenhouse gases.117
3.2 Misinterpretation of oxo-degradable plastics
However, another class of biodegradable polymers has been defined as oxo-degradable under ASTM standards. Oxo-degradable plastics undergo abiotic degradation before undergoing either aerobic or anaerobic biodegradation. There is significant concern about defining plastics as oxo-degradable and interpreting them as biodegradable in the plastic community.118 Oxo-degradation is the physical degradation of plastics into smaller units through oxidation, thermal or ultraviolet actions. Through these processes the molecular weight decreases, and potentially these monomer units could be simpler to bio-assimilate. However, this is not always the case if the final units have little to no functionality or a lack of carbon, nor is it a rapid process.Oxo-degradable plastics have previously been termed biodegradable plastics by several literature sources, but discrepancies are found in the pre-treatment which applies unrealistically high or accelerated conditions (i.e. thermal, UV exposure etc.) in temperature and duration respectively. These studies found that 400–600 days are required to obtain at least 30% biodegradation for LDPE films.119,120 ASTM D6954-18 indicates something like an oxo-degradability standard, but it must be used with other biodegradation standards (i.e. soil etc.) in the presence of these abiotic actuators.
There are three tiers to the oxidation and biodegradation standard121 (Fig. 9):
(1) Abiotic degradation to a weight average molecular weight of 5000 or less. Products vary depending on the polymer polarity and the type of initiator. Oxidation of non-polar molecules gives free radicals; UV or heat treatment gives hydrolysed molecules with functional groups.
(2) Biotic degradation in the environment of choice (stated compost, soil or accelerated landfill conditions). The products vary based on the standards followed.
(3) Measure of toxic residue.
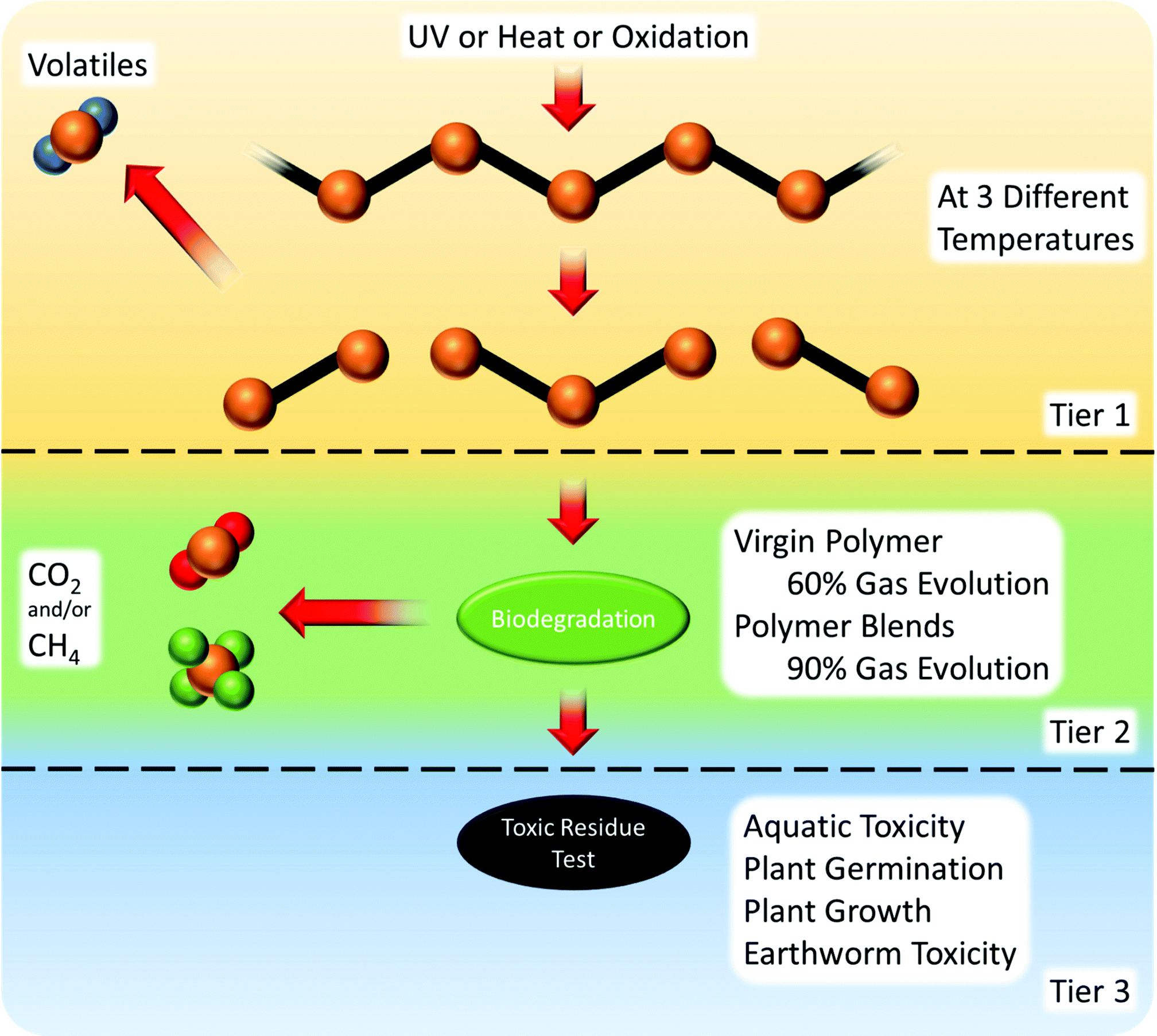 | ||
| Fig. 9 ASTM D6954-18 oxidation and biodegradation standard test procedure.121 | ||
There are some claims of oxo-degradable LDPE plastic packaging and UV-degradable plastics, however they only physically degrade, and do not produce CO2.39 These types of packaging can't be deemed biodegradable in such cases as they physically degrade into microplastics, and biodegradation is not achieved, which is of significant concern for the potential harm chemicals and polymers can cause on the wildlife both physically and chemically when ingested122 and of particular concern is the extremely small particles that can find their way into water systems and cause severe harm to ocean-life.
3.3 Aerobic biodegradation standards for polymers and their limits
This review will consider the American Society of Testing Materials (ASTM) standards that define three main areas of aerobic biodegradation standards: soil biodegradation, composting, and marine biodegradation, and three anaerobic biodegradation standards: sewage sludge biodegradation, anaerobic digestion biodegradation, and accelerated landfill biodegradation.This review does not include standards for unique conditions such as European ISO 14851 and ISO 14852 which are used to certify the OK Biodegradable Water designation in Austria.123 Furthermore, this study does not include aerobic biodegradation standards withdrawn (not updated within the time limit), including the following: ASTM D5209-92 in 2004, ASTM D5271-02 in 2011, and ASTM D6340-07 in 2016, which represent wastewater and sludge aerobic biodegradation, due to them being uncommon processes compared to the available aerobic biodegradation standards. One major limitation of these withdrawn standards is the static test conditions, whereas the natural environment is more susceptible to changes in conditions from climate, weather etc.
Soil biodegradation occurs when biodegradable materials are exposed to soil microbiomes, close enough to the surface to be in an aerobic environment. It does not follow the same level of awareness as composting in society due to there being no implemented collection systems. ASTM D5988-18 and its equivalent ISO 17556 for soil biodegradation only require the initial conditions of the soil to be reported (i.e. pH, moisture content, moisture holding capacity, ash content, carbon to nitrogen, etc.).124 Variations in moisture can alter the degradation rate of hydrolytically driven biological processes, and dry conditions slow down hydrolytic reactions, thus the biodegradation studies may not be comparable. This test can also be complemented with mechanical properties and physical degradation identified in ASTM G160-12, but there is no soil biodegradation ASTM labelling standard that defines whether something can be claimed as soil biodegradable.
The temperature is limited to the range of 20–28 °C, not reflecting a holistic climate approach, however, the soil must be collected from the surface of any natural environment, allowing for some malleability in studies of natural environments. It is also important to note that the efficiency and quantity of cellulose degraders vary based on the type of soil and location,125 which would be further reflected in the polymer degrader diversity. Therefore, climates, region and the temperatures of the soil and the preceding winters all affect the type and the quantity of microorganisms in and around the soil.
Other composting standards available reflect more natural or home composting such as ASTM D5929, which studies the composting of “organic” materials in aerobic conditions under mesophilic conditions (25–45 °C), and has been applied to plastics.32 However, the standard identifies the conditions to be maintained at 40 °C which is beneficial for biodegradation but not a holistic approach to defining the actual reality of composting (ambient temperatures can vary significantly from one geographical jurisdiction to another) and is not specifically designed for plastic materials.
Three phases of composting exist:128
(1) The mesophilic phase: where easily degradable organic matter is broken down. The organic acids produced reduce the pH to 5–5.5. With the rising heat from microorganism activity, the proteins break down and the pH increases to above 8.
(2) The thermophilic phase: where the temperature rises above 40 °C and the degradation of the waste material improves. However, as the temperature continues to increase to 55–65 °C, the microbial activity reduces.
(3) The “maturation phase”: where the compost begins to cool with reduced microbial activity after the readily available carbon sources have been consumed, hence “mature compost”. At this point the mesophilic microorganisms take over and degrade the residual carbon sources over a long slow process.
The three phases of composting are not well reflected in the testing procedures of ASTM D5338-15, which stipulates a stable temperature. Furthermore, education of the end-consumer is needed to impart a better understanding to avoid the improper disposal of the compostable material (which can be detrimental to the environment to the same extent as disposal of non-biodegradable plastics) and allay misperceptions of what compostable means.
ASTM D6400-19 defines that something can be labelled compostable if it meets 3 tiers:
(i) 1st Tier: 90% physically degraded to a particle size <2 mm in 90 days.
(ii) 2nd Tier: 90% converted to CO2 within 180 days.
(iii) 3rd Tier: no toxic residue in compost, and at least 90% germination rate and biomass of plants grown in compost compared to blank.
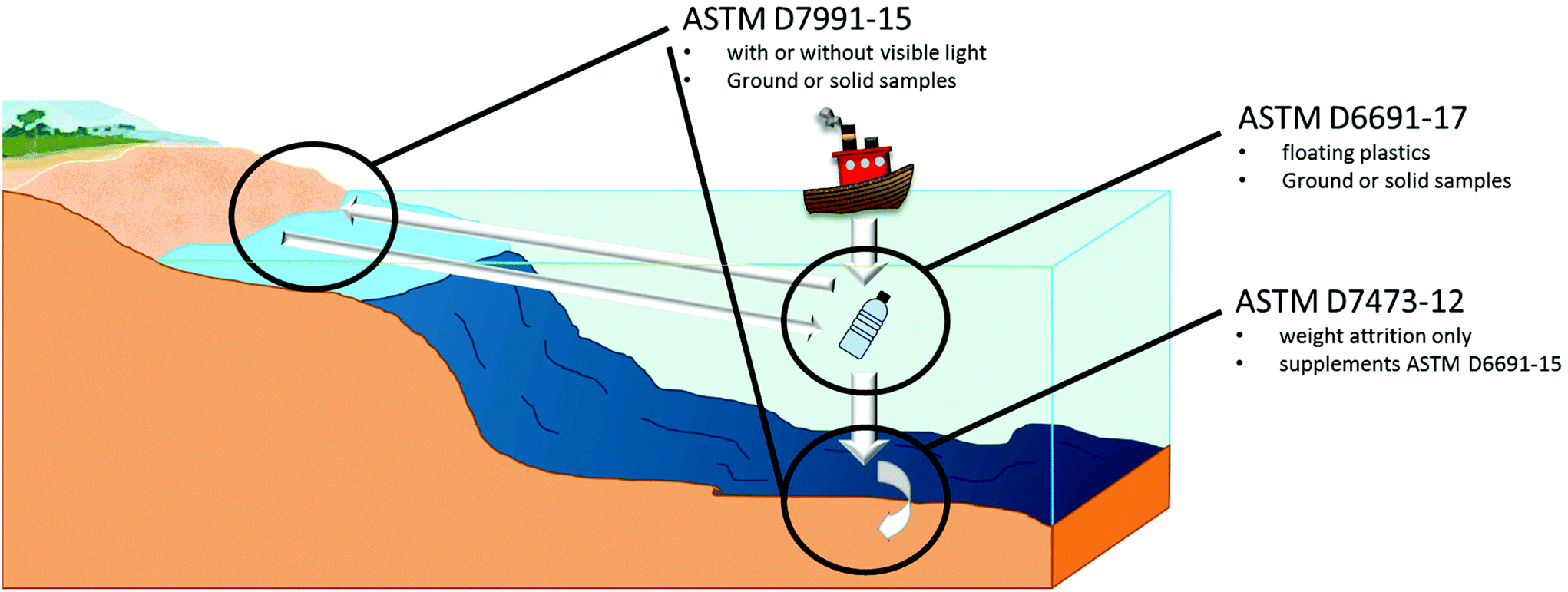 | ||
| Fig. 10 Marine biodegradation and degradation ASTM standards. | ||
A major limitation of marine degradation is no standard defines testing methods for polymers in deep waters where temperatures are well below the optimal growth conditions for bacteria,130 and pressure inhibits the rate of biodegradation and bio-assimilation of PHAs by microorganisms.131 Furthermore, near the ocean floor, water movement is minimal, and anoxic (oxygen absence) conditions can be common near the bottom sediment.132 Although ocean sediment is reported to have a greater microorganism consortium that can degrade PHAs,133 it may not be enough to mitigate these unfavourable conditions. Furthermore, water absorption is positively correlated with temperature,134 which can detrimentally affect biodegradation in cooler climates and conditions. Considering that marine biodegradation will vary across the globe it is understandable that there must be freedom in study. Several limitations have also been identified for biodegradation in aquatic environments, such as differences between lab and real world conditions, benthic environments, toxicity tests etc.135
To evaluate and claim that marine biodegradability is difficult due to the lack of standards identifying a threshold, ASTM D7081-05 did indicate a threshold for biodegradation to identify if polymers were marine biodegradable (30% CO2 theoretical evolution and 70% physically degraded below 2 mm in 180 days). However this could only be applied on polymers tested using ASTM D6691-17, and the standard has since been withdrawn.136 “OK Biodegradable Marine” designation is a valid claim that can be made with 90% biodegradation achieved relative to cellulose or absolute, in addition to physical degradation to a particle size below 2 mm in 180 days, and is supplementary to ASTM D6691.137
3.4 Anaerobic biodegradation standards for polymers and their limits
Three main anaerobic studies exist, defining varying ratios of total solids seen in wastewater treatment, landfills and anaerobic digesters. During anaerobic studies, since biogas is measured, dissolved CO2 in water is not directly measured and must be accounted for afterwards which is not done so in some older studies.138As of the current date of this review, the standard has since been withdrawn for not being updated in a timely manner but is still used in some studies. ISO 13975 is also similar to ASTM D5210-92, for its inoculum type, however, the temperature can be either 35 or 55 °C.135
(1) Biodegradation with household solid wastes of 50% total solids at 30 °C for 4 weeks.
(2) Static digestion with a sludge gestate total solid content of 35, 45 or 60% and a temperature of 35 °C.
Landfills are generally packed densely to conserve space; therefore, biodegradation may be limited due to the lack of water or available CO2 or oxygen to properly degrade over time, in addition to the lack of exposure to other degradation mechanisms due to the depth. The ideal scenario is mismanaged biodegradable plastics end up in the appropriate stream such that it can aerobically degrade instead of anaerobically, resulting in methane release in the environment. Among types of PHA biodegradation, it is the least researched due to the limited studies found at this time.
4 PHA biodegradation
As a biodegradable polymer, PHA has a few significant advantages for applications: (i) slow release of chemicals such as fertilizers or pesticides in agriculture; (ii) photoactivation to induce pollutant oxidation; (iii) leaving no residue behind within a short period of time;140 and (iv) biodegradation pathways are similar to starch (about 90% of microorganisms that degrade starch can also degrade short chain PHAs141). However, starch degraders can usually only bio-assimilate (take in and utilize) biodegradation products and not perform complete biodegradation with large molecular weight polymers or more complex medium to long chain PHAs. Biodegradation has two types of microorganisms, those that physically degrade PHAs, and those that feed off the by-products of the degradation (butyric acid, valeric acid etc.).In this review, biodegradation studies completed in vitro where samples are examined for degradation and analysed based on the surface diameter of the degraded polymer are not critically evaluated. Studies following ASTM standards, modified ASTM standards, natural environments and those in controlled environments (i.e. laboratory environments), with physical samples (i.e. powder, films etc.), that show the extent of degradation and/or bio-assimilation are detailed critically.
4.1 PHA biodegrading microorganisms
Holistically, short and medium length PHAs are biodegraded by a number of bacteria, including members of the genera: Actinomyces, Alcaligenes, Arthrobacter, Aspergillus, Bacillus, Clostridium, Comamonas, Corynebacterium, Enterobacter, Gracilibacillus, Klebsiella, Micrococcus, Mycobacterium, Nocardia, Pimelobacter, Planococcus, Pseudomonas, Pseudoalteromonas, Staphylococcus, Streptomyces, Variovorax.130,142–150 The dominant PHA destructors in aerobic and anaerobic environments have been reported by the members in the bacterial genera of Variovorax, Stenotrophomonas, Acinetobacter, Pseudomonas, Bacillus, Burkholderia, Cupriavidus, Mycobacterium and Streptomyces.144,151 Fungi can also degrade PHA and are reported to be even more effective PHA degraders compared to bacteria.152 Known PHA degrading fungi are in the division Ascomycota, Basidiomycetes, Deuteromycetes, and Zygomycotina in aerobic and anaerobic environments.153,154 Thus, it is reasonable to define PHAs as readily biodegradable in most anaerobic and aerobic environments.10Under favourable conditions, biodegradation follows the ideal bacterial growth curve with a lag, exponential, stationary and death phase, where the biodegradation plateaus between the end of the stationary phase and the start of the death phase.155 However, if conditions are unfavourable, or fluctuate throughout biodegradation, the rates can vary significantly from the theoretical model due to this complex association between biodegradation and bio-assimilation of the products.
4.2 Extracellular PHA biodegradation
The extracellular biodegradation of PHA is mediated by protein lipases and hydrolases. The process follows the general step-wise approach of polymer breakdown into shorter chain polymers by hydrolytic depolymerases in the presence of water, followed by further conversion of PHAs into trimer and dimer units, which are then processed by lipases and hydrolases.156 The extracellular PHA depolymerases are most studied; their protein structure consists of three main domains: (i) a binding domain responsible for surface absorption and disruption of the polymer structure; (ii) a linker domain that links the binding domain to the catalytic domain; and (iii) a catalytic domain that cleaves the PHA and any available dimers/trimers in two (Fig. 11).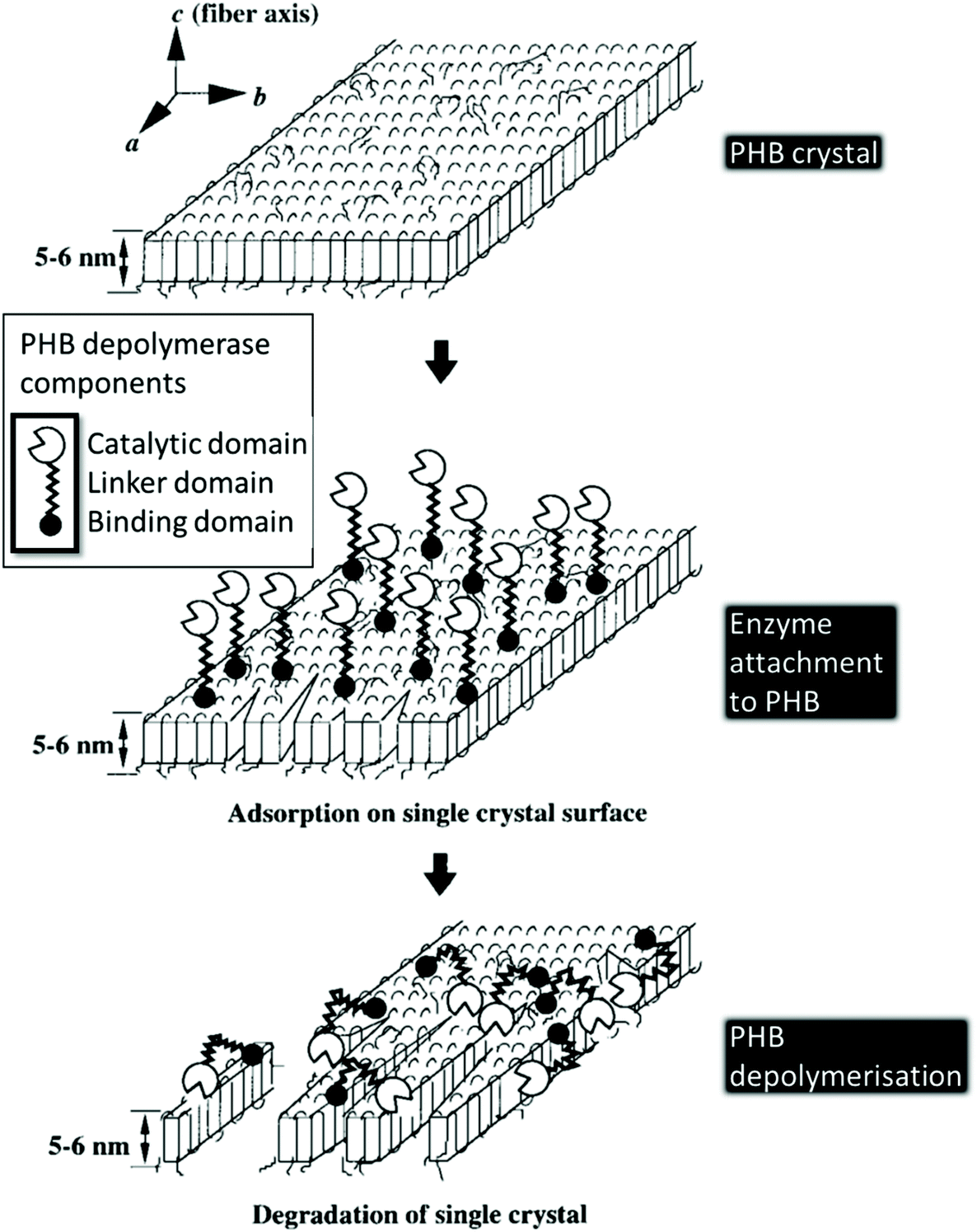 | ||
| Fig. 11 Single PHB crystal enzymatic degradation by PHB depolymerase. This figure has been reproduced with permission from ref. 163, John Wiley and Sons, Copyright 1999 (License number: 4800311035232). | ||
Enzymatic hydrolysis of PHAs is a two-step process;
(1) Adsorption of enzymes upon the surface of the polymer and active sites.
(2) Enzymatic induced hydrolytic cleavage of PHA bonds which is induced by the hydrophobic domain binding site and the catalytic site respectively.143,157
The hydrolysis process is not very specific and releases oligomers of various sizes into the surrounding medium.158,159 There is observed preference of hydrolytic enzymes toward amorphous surface crystals, with less crystalline polymers and co-polymers being targeted more readily due to their less ordered structure being more specially accessible to the enzymatic action.157,160 The degradation products vary depending on the type of PHA. In PHB, 3-hydroxybutyric acid is produced, while PHBV products are 3-hydroxybutyric acid and 3-hydroxyvaleric acid.161 These acids are then taken into the cell to be metabolized into other compounds or more PHAs.52,162 Furthermore, the type of PHA impacts the way biodegradation proceeds effectively.
4.3 PHA attributes that affect biodegradation
The efficiency of PHA biodegradation is closely coupled with the physical and chemical attributes of the polymer type. Degradation rates depend on the: (i) crystallinity; (ii) copolymers; and (iii) copolymeric structure. In PHBV, the introduction of 3-hydroxyvalerate has a greater amorphous region, which is more susceptible to enzymatic attack,53,164 due to eased water penetration,94 absorption,165 and susceptibility of the isodimorphic crystal region to the enzyme catalytic domain.166 Thus, it follows that biodegradation varies based on the crystallinity induced by the processing method.167 Furthermore, based on the copolymer ratio, the amorphous region can be altered and the enzymatic depolymerase activity can be maximized as seen in Fig. 12.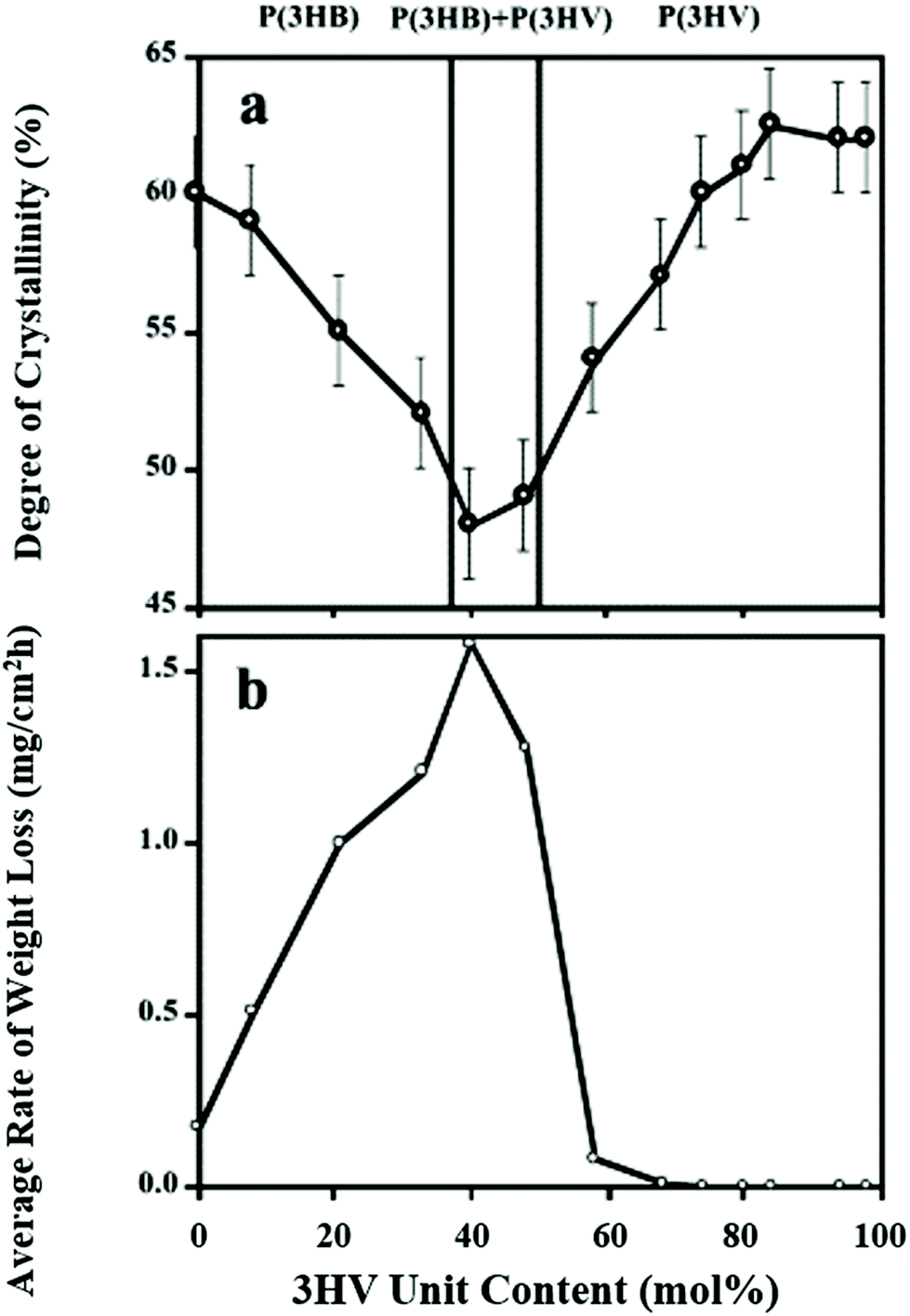 | ||
| Fig. 12 (a) PHB and PHBV crystallinity from WAXD patterns. (b) Rate of weight loss by 2 μg of Ralstonia pickettii type 1 PHA depolymerase due to 3HV ratio. This figure has been reproduced with permission from ref. 168, Elsevier, Copyright 2004 (License number: 4800311458150). | ||
However, other studies indicate that a PHA depolymerase is more effective on PHB than PHBV and most effective on PH4B, although the results are measured in weight loss and the molecular weights vary significantly.143 This indicates that other enzymes or factors play a part in degrading PHAs. For example, excess side chains may inhibit the rate of interaction with the target location, and other actions coupled with the enzymatic hydrolysis can further enhance biodegradation. Water diffusion, promoted by temperature, can induce some hydrolytic degradation which increases active sites.169 Ester bonds of polyesters are also sensitive to hydrolysis and can result in a reduction in molecular weight. The lowering of molecular weight invalidates the Arrhenius model for biodegradation,170 because enzymatic hydrolysis is coupled with ester hydrolysis and overall biodegradation, and not driven by a single actuator. The depolymerisation steps are indicated to be rate limiting steps in PHA biodegradation,171 thus indicating that any single form can reduce the biodegradable product life expectancy after its service life.
As biodegradation proceeds, and mass loss increases, erosion of the interlamellar phase of PHA begins in the initial stages,174 where the disordered chains are targeted first before samples are eroded in the crystalline regions (Fig. 13). The interlamellar phase or amorphous region gives PHAs their flexibility, and with their degradation it is expected that the crystallinity would increase. These characteristics are seen within 30 days of soil biodegradation175 and 60 days within marine biodegradation.176 However, given enough time the crystallinity is expected to be reduced by up to 60%, which occurs in soil biodegradation after 200 days or 30 days in a controlled composting environment due to the intensified conditions.175 The amorphous region is known to allow permeation of moisture and enzymes, and therefore its degradation would increase the surface area of available crystalline regions. The crystalline regions make up the majority of a crystalline PHA such as PHB and their degradation is expected to reduce the overall polymer crystallinity.
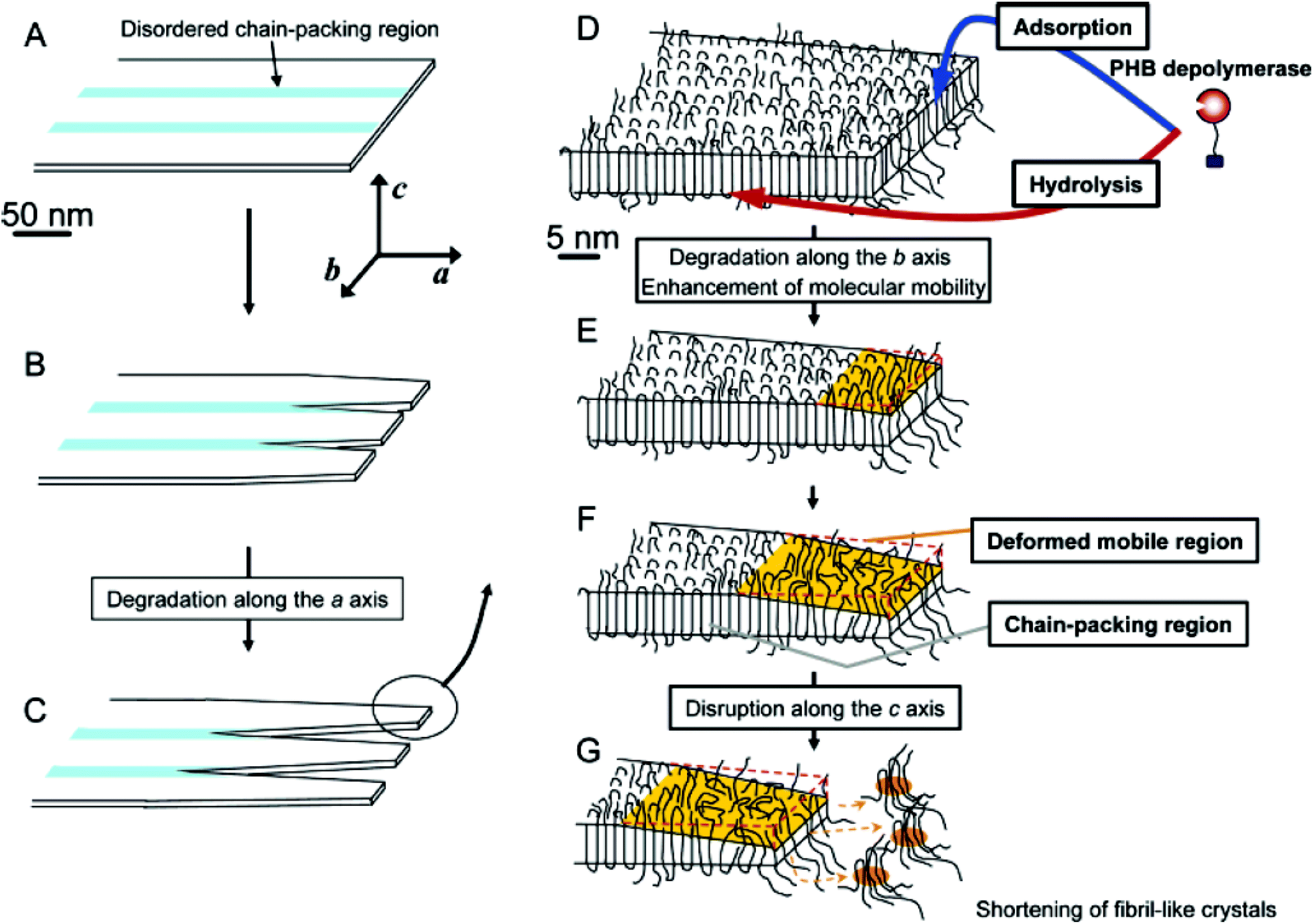 | ||
| Fig. 13 Schematic model of the enzymatic degradation behaviour of lamellar crystals in P(HB) solution-grown single crystals by PHB depolymerase. Reprinted (adapted) with permission from (Numata et al. H. enzymatic degradation processes of poly[(R)-3-hydroxybutyric acid] and poly[(R)-3-hydroxybutyric acid-co-(R)-3-hydroxyvaleric acid] single crystals revealed by atomic force microscopy: effects of molecular weight and second-monomer composition on erosion) on April 1, 2020. Copyright (2005) American Chemical Society.166 | ||
Another aspect to consider is the availability of enzymes to cleave the medium chain length PHAs. Jendrossek identified several microorganisms having short chain length PHA degrading enzymes,183 and several short chain PHAs and other aliphatic polyesters such as poly (ethylene adipate) (PEA), poly (ethylene succinate) (PES) and poly (2,2,6,6-tetramethylpiperidenyloxyl-4-yl methacrylate) (PTMA) can be enzymatically depolymerized by similar enzymes, indicating a significant advantage to short chain PHAs. These enzymes attach the ester bond but have low specificity, not being overly discriminative toward the range of side chains.158 For example, PHB has one carbon long side chain and some PHB depolymerases enzymatically hydrolyze PHBP,66 which can be the result of PHBPs having no side chain. The enzyme affinity is likely determined by the affinity towards the PHA side chain.184 However, these enzymes do not function on medium and long chain PHAs effectively. Research availability of medium chain length depolymerases is minimal with very few species isolated with the particular enzymes,185 which is further reflected by the few poly (3-hydroxyoctanoate-co-3-hydroxydecanoate) degraders seen in Streptomyces.186 This may be why medium chain length polymers such as PHBO with 10% HO content only show between 88–95% biodegradation in anaerobic and aerobic environments.182 Thus, the use of high carbon PHA monomers such as 3-hydroxydecanoate in medium chain length PHAs can impede biodegradability by reducing the number of chemically reactive binding sites in the side chain structure for enzymes to attack.
The observed effect of crystallinity and copolymer ratio does not apply to all biodegradation studies. Lower molecular weight polymers have enhanced motility and significantly reduced size, which should make enzymatic degradation and bio-assimilation faster, but improvements are not always reflected in solvent casting. The method of sample production (i.e. solvent casting) or the sample recovery method can similarly leave behind impurities or toxic residue depending on the solvent187 that may negatively impact the enzymatic activity or microorganism functionality and produce irregular relationships between PHA copolymer compositions and their biodegradation rates.171,188 Furthermore, there is evidence that PHB is a more readily used product than more complex PHA monomers with larger side chains such as HV, 4Hb and Hx148 which is expected to result in faster biodegradation rates, however, this is not always proven, and it has not been determined if its due to crystallinity alone.
Biodegradation of films begins with surface pitting and degradation at the edges (Fig. 14), such that the surface area to volume ratio increases as time goes by. Furthermore, by changing the production method of films from solvent casting to electrospinning, the surface area and water permeability can be increased leading to a significant improvement in biodegradation rate, measured by a mass loss increase from 40% to 100% in a 28 day period.193 This method also increases the surface roughness, further enhancing the surface area of thin films.
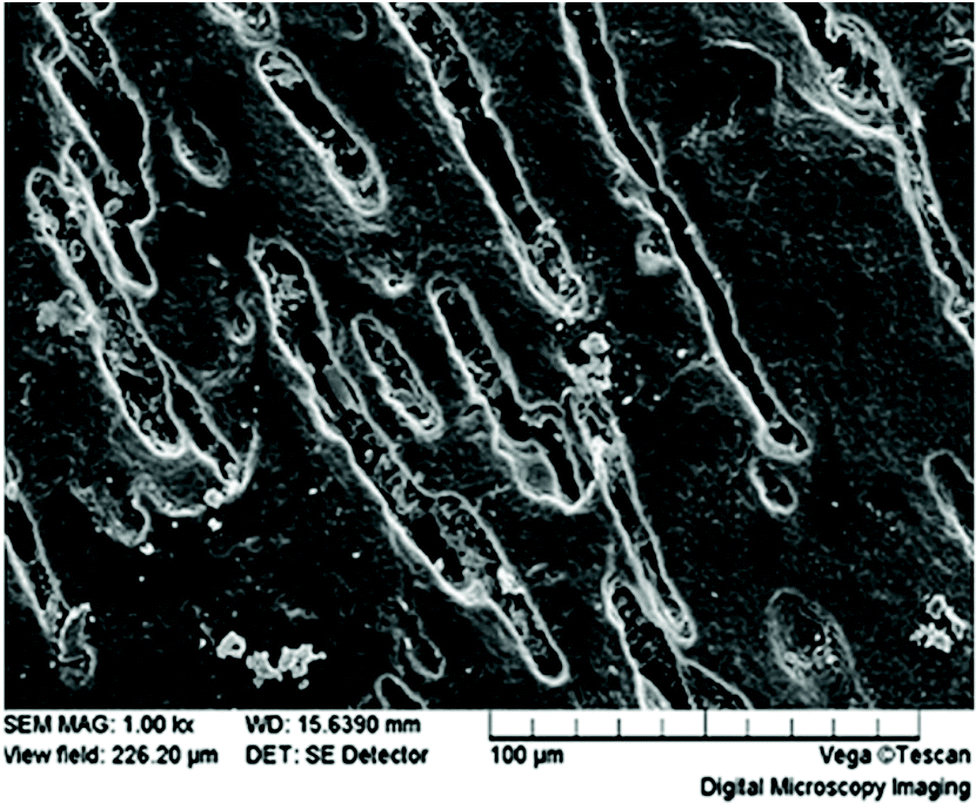 | ||
| Fig. 14 PHBV film surface at 35 days under composting conditions. This figure has been reproduced with permission from ref. 177, Elsevier, Copyright 2004 (License number: 4800311458150). | ||
Biodegradation of PHA pellets is generally slower than that of films to a certain extent depending on the type of PHA. PHB films benefit more from thin film morphology compared to PHBV.151 According to ISO 14855 PHB pellets biodegrade by 54% in 45 days and 92% in 78 days,194 which is comparable to PHB plates of 1.2mm thickness.32 Therefore, when comparing biodegradations studies, it is important to consider the morphology of the samples. However, the comparisons become challenging as only a few completed studies followed ASTM standards.
4.4 Chemical additives and blending effect on PHA biodegradation
The addition of other chemical additives or blends all affects PHA biodegradation on both a chemical and physical scale by impeding enzymatic degradation. Non-biodegradable co-polyesters and chemicals minimize the interactable area between enzymes and biodegradable polymers. This can be further exacerbated if the non-biodegradable polymer makes up the continuous phase with PHA dispersed within,195 or the biodegradable polymer is unsuited for the particular environment (i.e. PLA in marine water). This is mainly reflected in the rate of enzymatic hydrolysis.Plasticization of PHAs with tributyl citrate (TBC) or oxypropylated glycerol reduces the crystallinity, but also slows down the biodegradation rate in all blend ratios in soil, compost and anaerobic sewage sludge.32,189,200 However, this may be due to the preservative effects (inhibits microbial growth) of glycerols as a food additive, its reduced bio-assimilation due to oxypropylation, and the synthetic production method and hydrophobicity of TBC.201,202 PLA has been plasticized with acetyl tributyl citrate which inhibited hydrolysis action due to increased hydrophobicity whereas triethyl citrate based plasticizers are more hydrophilic and can increase PLA degradation,203,204 and these effects likely play a part in reducing PHB degradation. Furthermore, the way plasticizers are closely associated with polymer chains may inhibit the enzyme interaction with binding groups.
PHA/PLA blends similarly reduce the crystallinity and should result in improved biodegradation of PHA components. However, virgin PLA is not readily biodegradable in soil205 and marine water,190 due to the suboptimal temperatures.206 Some instances of 25–30% PLA in PHB, PHBV and PH4B have shown evidence of similar or slower physical degradation or biodegradation rates in soil conditions but no complete biodegradation is reported.175,207,208 However, according to ASTM D5988-12, a PHA/PLA blend was seen to degrade to 99% in 176 days, but the ratio and presence of additives were not identified.209 The addition of PLA, PBAT or PBS into PHBHx has also resulted in a reduced biodegradation rate (3–23%, 10–90% and 20–90% respectively) compared to PHBHx in marine water. The level of reported biodegradation is the result of PLA, PBAT and PBS biodegradation in the blends, despite the virgin polymers not degrading in marine water.190 There is no reported reason behind the unexpected biodegradation of PLA, PBAT and PBS in marine water but it may be the presence of the highly amorphous hydroxyhexanoate,210 which interferes with the crystallization of the other blend components or promoting biofilm development. Some success has been found in PHBV/PBAT/flax composites where soil degradation rate of PHBV and flax in the composite was enhanced with PBAT addition, however this was attributed to phase separation that improved microbial ingress into the samples.211
In higher temperature environments (∼58 °C) such as controlled composting and anaerobic digestion, the biodegradation of PLA is common, and it is expected that there would be benefits of incorporating PHAs into PLA to improve the compostability. However, PLA/PHBV 70/30 blends have shown no biodegradation improvement compared to PLA and PHBV alone (all 90–92% biodegradation in 200 days) under ASTM D5338-15,175 or have even a reduced biodegradation rate in compost,212 and under anaerobic digestion.213 The effects are similarly reflected in PLA/PHB/ATBC and PLA/PHB/PEG based blends where disintegration properties were the same as virgin PLA or the PHB is reported to slightly inhibit PLA degradation under ISO 20200.214 There is also evidence that PHAs biodegrade slower than PLA in anaerobic digestion at 52 °C than PLA alone.213 Considering the cost of PHA production being about double compared to PLA,80 it is not economical to seek compostability or anaerobic digestion improvements with the addition of PHAs. However, for mismanaged waste, industrial composting is not a suitable representation for the natural environment and virgin PLA is not suitable for biodegradation in natural environments.
Overall, it has been established that PHAs fully and rapidly (time frame) biodegrade in the natural environment. Of the available studies few illustrate the biodegradability of PHAs in a comparable manner.
4.5 PHA soil biodegradation
Recent PHA soil biodegradation studies are indicated in Table 1. Studies defined by mass loss did not indicate (%) biodegradation based on CO2 evolution. Soil biodegradation is known to be an incredibly dynamic environment that can vary across the globe. The process is considered relatively slow compared to other aerobic and anaerobic processes, due to the mild conditions. Cellulose for example can take nearly 200 days to degrade by 90%,194 however, if the conditions are favourable, the reference material can take significantly less.215 Therefore, biodegradation of PHAs will vary between studies.| PHA (co-monomer %) | Protocol and source | Conditions | Form | Days | Results | Ref. | |
|---|---|---|---|---|---|---|---|
| 1 | PHBV (12% HV) | ASTM D5988-12, agriculture field soil | 23–25 °C, 20% moisture(w.b.) | Film | 200 | 35% biodegradation | 175 |
| 2 | PHBV (2% HV) | ASTM D5988-03, forest soil | 25 °C | Powder | 350 | 70% biodegradation | 150 |
| 3 | PHB | Non-ASTM, fertile garden soil | 30 °C, 80% relative humidity, 10 cm depth | Nano-fibre Film | 28 | 100% mass loss | 193 |
| 4 | PHB | Non-ASTM, field soil | 21 and 28 °C, 50% moisture(w.b.) | Film | 35 | 60 and 95%, mass loss | 181 |
| 5 | PHBV (12% HV) | 90 and 100% mass loss | |||||
| 6 | PHBHx (12% Hx) | 92 and 100% Mass Loss | |||||
| 7 | PH4B (10% 4HB) | 35 and 28 | 100 & 100% Mass Loss | ||||
| 8 | PHB | ASTM D5988, natural mature soil | 11–30 °C, 17–23% moisture(w.b.) | Film | 112 | 60% mass loss | 196 |
| 9 | PHBV (8% HV) | 60% mass loss | |||||
| 10 | PHB | ASTM D5988-03, commercial soil | 23 °C, 33% moisture(w.b.) | Film | 80 | 82% mass loss | 60 |
| 11 | PHA | 76% mass loss | |||||
| 12 | PHA | ASTM D5988-03, a mixture of topsoil, farm soil and sand | 20 °C, 60% of water holding capacity | Film | 660 | 70% biodegradation | 113 |
| 13 | PHA | Non-ASTM, farmland topsoil | 35% moisture(w.b.) | Film | 140 | 32% mass loss | 191 |
| 14 | PHA | Non-ASTM, farmland topsoil | 35% moisture(w.b.) | Film | 60 | 33% mass loss | 218 |
| 15 | PHA | Non-ASTM, farmland topsoil | 25 °C, 35% moisture(w.b.) | Plate | 120 | 35% mass loss | 219 |
| 16 | PHA | Non-ASTM, farmland topsoil | 30–40% moisture(w.b.) | Film | 60 | 22% mass loss | 192 |
PHAs are considered the most promising soil biodegradable materials, with 100% biodegradation in 90 days, whereas PCL would take up to 270 days according to ASTM D5988.215 In non-ASTM studies, PHBV is degraded by 40% in 120 days (25–30 °C) while no degradation was observed for PBS and PLA.216 However, only 25–30% of microorganisms are PHA degraders, the rest only bioassimilate the products of enzymatic hydrolysis.142 Furthermore, soil biodegradation rates have been reported with several different conditions: (i) soil source; (ii) soil moisture content and pH; and (iii) temperature of soil biodegradation studies. Based on the literature, there is no standard that defines a (%) biodegradation limit for polymers, to indicate whether it is biodegradable or only shows biodegradable behaviour. The existing OK Soil degradable certification states that it must be 90% biodegraded absolute or relative to the reference sample in 2 years under ASTM 5988,217 of which few studies have continued for that long.
Measurements most often reference the location and depth of the sample in the soil, and in rare cases identify the microorganism community. However, the characteristics of the soil not only affect the conditions, but also how the conditions will change over the duration of the experiment. For example, PHBV (8% HV) powder tested in sand (dry silica with low organic material) achieved 80% biodegradation after 600 days,176 which is significantly longer than most soil biodegradation tests. Zaidi et al.211 reported no degradation of PHBV (3% HV) in botany sand in a natural environment within 110 days. Sand has relatively poor moisture holding capacity, substantiating these slow biodegradation rates that take months to years for any evidence and by mixing in organic soil and farm soil into sand, the biodegradation rate of PHAs is not seen to improve113 (Table 1, entry 12). The degradation of PHBV (9.8% HV) film samples indicates minimal physical degradation after 5 months in a mixture of beach sand, horse manure and fertile soil.221 The effect of moisture holding capacity is also reflected in Clarion loam soil 6% PHA biodegradation in 5 months after watering once a week,222 even though clarion loam soil is rated to have very good lytic activity among many other types of soil.125
The limitations of sandy soil can be improved in natural environments near the coast, where frequent water permeation happens continually such that the biodegradation of starch can be 4 times faster than garden soil.215 Other natural soil biodegradation studies include soil sourced from South African agricultural fields, black soil and leaf mould, oil palm cultivation soil, soil from Russia and Vietnam, and soil from California landfills. Field soil and landfill soil appear to have the highest rate of PHA biodegradation activity, being the only samples approaching 100% PHA degradation142,144,151,175,181,205,223,224 (Table 1, entries 1 and 4–7). This may be due to the microbial diversity found in moist soil with consistent environments that maximize microbial population growth and diversity.
The location of soil in a specific area is of significant impact on the potential biodegradation rate. Between subsoil and topsoil, the fungi community is richer in topsoil225 and therefore the biodegradation rate with increased microorganisms will be significantly higher. Variations in the natural environments such as humidity under larch and birch trees, and in harsh winters over a yearly basis were reflected in lower microorganism counts and slower mass loss due to lower humidity.144 The environment fosters the microorganisms; comparing PHA degradation in greenhouse soil and farmland soil it was found that PHA degradation is significantly greater in farm land soil for similar sized films/plates under similar conditions192,218,219 (Table 1, entries 14–16). Celestina et al.225 reported that greenhouse soil treated with a fertilizer has a lower bacterial and fungal community count and diversity compared to untreated soil. Therefore, it is important to note the soil source and conditions being used during the biodegradation study of PHAs.
Soil pH is closely related to moisture and the degradation of PHAs, by maximizing enzyme function. Optimal pH for the most common enzymes used in PHB degradation is between 6–9 with some variation between different types.226 Furthermore, PHB and PHBV ester hydrolysis and physical degradation naturally occur in alkali environments without microorganism presence.101 Few studies consider these points, however, it has been indicated that the optimal soil for PHB degradation is saline soil with a pH of 7–8 compared to clay, sandy, tarine, and laterite soil with a pH lower than 7.227 The effect of soil pH on PHAs is similarly caused by alkali treatment of the PHB to improve the hydrophilicity and initiation time of mass loss at the cost of slightly reduced mechanical properties.228 PHA degradation is maximized with high moisture and optimal pH range, however, this does not capture the soil pH and moisture relationship around the world. 50% of the tested regions in North and South America have an acidic surface soil pH (<6.5) and evidence of a positive correlation between moisture content and acidic pH indicates that ideal conditions for natural PHA degradation are very limited.229
For standard purposes, soil biodegradation should be completed at 20–28 °C, but does perform better at higher temperatures. PHA copolymers have greater thermal sensitivity during biodegradation; PHBV (10% HV) degradation at 15, 28 and 40 °C was 11%, 32% and 100% respectively over approximately 200 days.179 The degradation rates of PH4B (10% 4HB), PHBHx (12% Hx), PHBV (12% HV) and PHB have all seen degradation improvements as the study temperature is increased from 21 to 28 °C, with all copolymers benefitting the greatest with increased temperatures181 (Table 1, entries 4–7). The PHA molecular mass reduces faster at higher temperatures in soil, suggesting that it would benefit more from composting conditions. In most cases <40 °C gives the best improvement in PHA degradation rate (due to microbial action or enzyme activity) while temperatures approaching 60 °C were significantly worse for PHA degradation.230 This can be related to the type of microbial species and proteins present and their respective functions as there is a shift from mesophilic to thermophilic microorganisms at 40 °C,128 in addition to PHB depolymerase activity reducing as temperatures increase from 30 to 70 °C.226 In soil biodegradation low temperature (<20 °C) studies are not regularly completed, limiting the available literature on biodegradation in cooler climates and environments.
Considering that there is no standard that defined whether a polymer can be claimed soil biodegradable after achieving a certain extent of degradation, it is difficult to define whether PHAs undergo soil biodegradation. However, there is enough evidence to claim in the literature that PHAs are soil biodegradable provided the conditions and sample morphology are appropriate, but the duration may vary anywhere from 20 days to several hundred days or more.
4.6 PHA composting
Industrial composting is characterized by the controlled temperature of 58 °C, and can be completed industrially. Most studies are completed under industrial compost conditions, as home composting doesn't consistently achieve this high temperature level. Under ISO 14855 (equivalent to ASTM D5338) composting conditions, cellulose degrades by 92% in 45 days194 (Table 2, entry 15). Comparatively under ASTM 5338, a PHA bag has 94% biodegradation in 180 days39 (Table 2, entry 1), and exceeds the degradation of petroleum based biodegradable polymers like PCL,233 and passed the 2nd tier of the ASTM 6400-19 compost labelling standard which is 90% biodegraded in 180 days. PHA composting studies within the last decade are outlined in Table 2.| PHA (co-monomer %) | Protocol and source | Form | Days | Results | Ref. | |
|---|---|---|---|---|---|---|
| 1 | PHA | ASTM D5338 | Bag | 180 | 94% biodegradation | 39 |
| 2 | PHBV (12% HV) | ASTM D5338-15, mushroom compost | Film | 200 | 90% biodegradation | 175 |
| 3 | PHBV (2% HV) | ASTM D5338 | Film | 45 | 95% biodegradation | 150 |
| 4 | PHB | ASTM D5338-98 | 0.24 mm plate | 112–140 | 99–100% mass loss | 32 |
| 5 | 1.2 mm plate | 84–112 | 98–100% mass loss | |||
| 6 | 5 mm plate | 210 | 45% mass loss | |||
| 7 | PHB | ISO 14855-1, compost factory organic waste | Film | 110 | 80% biodegradation | 178 |
| 8 | PHBV (3% HV) | 80% biodegradation | ||||
| 9 | PHBV (20% HV) | 89% biodegradation | ||||
| 10 | PHBV (40% HV) | 90% biodegradation | ||||
| 11 | PH4B | ISO 14855-1, compost factory organic waste | Film | 110 | 90% biodegradation | 178 |
| 12 | PHB | ISO 14855-1, mature compost | Film | 45 | 80% biodegradation | 177 |
| 13 | PHBV (3% HV) | 81% biodegradation | ||||
| 14 | PHB | Non-ASTM, home composting | Tensile sample | 84 | 50% mass loss | 234 |
| 15 | PHB | ISO 14855, mature organic municipal solid waste | Pellets | 78 | 92% mass loss | 194 |
| 16 | PHB | ASTM D5929-96 | 0.5 mm plate | 182 | 100% mass loss | 32 |
| 17 | 1.2 mm plate | 182 | 100% mass loss | |||
| 18 | 3.5 mm plate | 350 | 94% mass loss |
Although industrial composting is considered very controlled, there is still some variability between samples; PHBV films have been reported to take between 45–200 days (Table 2, entries 2 and 3). This variability may have to do with the inoculum source.
Following ASTM D5338-15 or its equivalent ISO 14855-1, a comparison of the organic waste inoculum and mushroom compost inoculum indicate that organic waste is more effective175,178 (Table 2, entries 2 and 11). Organic compost is characterized by a high moisture content from the organic material improving the conditions for microbial growth and its starting microbial load is significantly higher than the inoculum sourced from the mushroom compost.
Biodegradation of PHAs under home composting or industrial composting follows the pattern of other biodegradable plastics studied under the same conditions, influenced by the microbial profile, temperature and pH during the process. PHAs show minimal if any biodegradation in home composting conditions where the temperature is low, or when the pH becomes low.233 Higher temperatures of industrial composting benefit biodegradation by enhancing non-enzymatic and enzymatic catalysed hydrolysis.237 Because of these variable conditions, PHA biodegradation varies between each study. Mergaert et al.179 reported the biodegradation of PHB, PHBV (10% HV) and PHBV (20% HV) to be 4%, 6–17% and 67% in 152 days where the temperature varied from 8–30 °C. Gilmore et al.145 reports that PHBV (26% HV) had 59% mass loss in 186 days, indicating a slower biodegradation which is slightly slower despite the temperature between 40–63 °C throughout the study. The difference is more likely due to the differences in inoculum and temperature profile which is reflected in PHB which degrades by 50% in 84 days in organic waste home compost with 74–89% humidity and a temperature of 34–66 °C234 (Table 2, entry 14). The unpredictability of PHA home composting makes it difficult to study the effects of the temperature profile throughout the composting process.
Several composting studies (Table 2, entries 1, 3–5, 7–11, 15–17) following ASTM D5338 or ISO 14855 have identified PHB and PHBV to pass the 1st and 2nd tier of ASTM D6400. Samples are 90% physically degraded in 90 days, defined by (%) mass loss, and 90% biodegraded in 180 days, defined by (%) biodegradation.
4.7 PHA marine biodegradation
PHAs are the only class of bio-based polymers that exhibit efficient marine biodegradation following ASTM standards, compared to other polymers such as PLA which does not degrade.238 Among other polymers showing biodegradable behaviour in marine water (i.e. PCL, PES, PEA etc.), PHAs outperform most in all water environments,188 but the research in PHA marine biodegradation following repeatable ASTM standards is still very limited. Recent PHA marine biodegradation studies are outlined in Table 3. Over 70% of the world is covered in water and presents a variety of differing conditions in the natural environments which must be investigated to ensure that PHA waste is biodegraded if improperly disposed.| PHA (co-monomer %) | Protocol and source | Conditions | Form | Days | Result | Ref. | |
|---|---|---|---|---|---|---|---|
| a Biodegradation level after removal of the outlier in data for the sample. | |||||||
| 1 | PHB | Non-ASTM, Eutrophic reservoir | 18–25 °C | Film | 42 | 43.5% mass loss | 100 |
| 2 | PHB | Non-ASTM, South China sea | 27–30 °C | Solid | 160 | 62% mass loss | 146 |
| 3 | Film | 58% mass loss | |||||
| 4 | PHBV (11% HV) | Non-ASTM, South China sea | 27–30 °C | Solid | 160 | 87% mass loss | 146 |
| 5 | Film | 54% mass loss | |||||
| 6 | PHB | ASTM D6691, Woods Hole Harbor water | 30 °C | Film | 100 | 90% mass loss | 164 |
| 7 | PH4B (44% 4HB) | ASTM D6691, Woods Hole Harbor water | 30 °C | Film | 100 | 80% mineralization | 164 |
| 8 | PH4B (47% 4HB) | 82% mineralization | |||||
| 9 | PHBV (8% HV) | ASTM D6691, Woods Hole Harbor water | 30 °C | Film | 100 | 85% mineralization | 164 |
| 10 | PHBV (12% HV) | 100% mineralization | |||||
| 11 | PHBV (8% HV) | Non-ASTM, Lorient Harbour | 25 °C | Film | 180 | 36% mass loss | 176 |
| 12 | PHBV (8% HV) | Non-ASTM, Lorient Harbour water + Foreshore sand | 25 °C | Powder | 210 | 90% biodegradation | 176 |
| 13 | PHA 2200 | ASTM D6691-09 | 30 °C | Film | 365 | 52% biodegradation | 238 |
| 14 | PHA 4100 | 82% biodegradation | |||||
| 15 | PHBV (12% HV) | Non-ASTM, Baltic sea water | 17–20 °C | Film | 42 | 60% mass loss | 235 |
| 16 | PHBHx (6.5% HV) | Non-ASTM, coastal sea water | 23 °C | Film | 148 | 89% biodegradation | 106 |
| 17 | PHBHx (7.1% HV) | 195 | 55% (77%)a biodegradation | ||||
| 18 | PHA | Non-ASTM, tropical river water | 28 °C | Film | 86 | 71% mass loss | 239 |
| 19 | PHBHx (11% HV) | Non-ASTM, sea water | 27 °C | Film | 28 | 35% biodegradation | 190 |
| 20 | PHBV | OECD 301, river water | 25 °C | Film | 90 | 90% biodegradation | 150 |
In certain studies, a combination of marine water and sand has been used to study the effect of the different microorganism's biodegradation performance. PHBV shows biodegradation in both water and sand alone as well as a combination. The presence of marine water is seen to promote biodegradation, which suggests a wide distribution of PHA degraders in water or more effective enzymatic hydrolysis. PHBV is reported to take 210 days to degrade by 90% in a sand and seawater medium, but over 600 days to degrade by 80% in only sand176 (Table 3, entries 11 and 12). During non-ASTM studies under non-laboratory conditions, Sridewi et al.133 reported that the degradation of PHAs is high where tide inundation is higher due to the presence of water and the introduced microbial populations are not found in the soil alone. However, the improvement may be attributed to the presence of water and not the microorganisms in it, because sand based environments are reported to have a greater Shannon-weaver diversity index and overall diversity of phylum compared to water environments.240 Therefore, sediment provides a great microbial diversity while marine water may significantly enhance the enzymatic function.
Other inoculum sources for non-natural marine biodegradation include sewage sludge and anaerobic digester sludge. Anaerobic digester sludge outperforms sewage sludge as an inoculum source for marine aerobic biodegradation of PHAs.241
Natural marine environments can also include fresh water sourced from rivers and lakes, where biodegradation of plastics may occur. Compared to other biodegradable polymers, PHBV biodegrades most efficiently in many aquatic environments. PHB and PHBV biodegrade at a different rate in the aquatic environment, having a 6 times faster rate in seawater than fresh water ponds/canals.179 Under favourable conditions, PHA can be degraded in its entirety, within 28 days in both fresh and saltwater rivers/lakes/oceans. PHB and PH4B degrade slightly slower within the 28-day period. Overall, chemically synthesized polymers tend to degrade at a slower rate and to a lower extent and are more dependent on the origin of water: PCL and PEA have comparable biodegradable behaviour in fresh and salt water to PHAs; PES biodegrades well in fresh water only; and PBS and poly(butylene adipate) (PBA) show some biodegradable behaviour in fresh water and minimal in salt water.188 However, bio-based PCL is still not widely accepted as marine biodegradable and does not always show marine biodegradable behaviour.244 In terms of the scope of ocean saltwater, PHAs are well placed to replace plastics that may end up in the ocean. However, at considerably greater ocean depths, PHAs degrade slower.141
While marine biodegradation does not contain an equivalent labelling standard to ASTM D6400, under ASTM D6691 PHB, PHBV (8–12% HV) and PH4B (47% 4HB) biodegrade beyond 90% in 100 days relative to the glucose reference164 (Table 3, entries 6, 8–10), fulfilling part of the OK marine biodegradable standards.137 Therefore, PHAs are marine biodegradable under ASTM D6691 in appropriate environments but physical degradation must still be assessed.
4.8 PHA sewage sludge biodegradation
Sewage sludge biodegradation is the industrial biodegradation of sewage sludge in municipal waste facilities, and sewage sludge biodegradation studies use sewage sludge as an inoculum. However, there is little rationale why PHAs would be found in sewage sludge. Biodegradation in sewage sludge is usually completed at a mesophilic temperature of 35 °C in a non-industrial scale to stabilize sewage sludge for land application, allowing many types of biodegradable polymers to degrade rapidly. Sewage sludge biodegradation is classified by low total solids required in the inoculum, and is more representative of anaerobic water biodegradation. Under these conditions PHAs are still reported to degrade into 50% CO2 and 50% methane,245 and at least 2 times faster than other biodegradable polymers (PBS, PLA and PCL).182,246 Standards of this kind have been withdrawn as anaerobic digestion with higher solids is a more applicable and representative form of anaerobic biodegradation used in sewage sludge processing on an industrial scale. PHA anaerobic sewage sludge biodegradation studies are outlined in Table 4.| PHA (co-monomer %) | Protocol and source | Form | Days | Results | Ref. | |
|---|---|---|---|---|---|---|
| 1 | PHB | ISO 13975, anaerobic sludge | Powder | 10 | 90% biodegradation | 149 |
| 2 | PHBHx (6.5% Hx) | Non-ASTM, wastewater | Film | 85 | 55% biodegradation | 106 |
| 3 | PHBHx (7.1% Hx) | 77% biodegradation | ||||
| 4 | PHB | ASTM D5210-92, diluted sewage sludge | Powder | 12 | 67% biodegradation | 189 |
| 5 | Plate | 19 | 68% biodegradation | |||
| 6 | 3 PHBs (commercial or research) | Non-ASTM, synthetic sludge with anaerobic biomass | Powder | 40 | 50–79% biodegradation | 247 |
| 7 | PHBO(10% HO) | Non-ASTM, wastewater and septic sludge | Powder | 61 | 88% biodegradation | 182 |
| 8 | 141 | 95% biodegradation | ||||
| 9 | PHBV (8% HV) | ASTM D5210-91, wastewater | Film | 40 | 89% biodegradation | 245 |
| 10 | PHB | Non-ASTM, domestic sewage sludge | Powder | 21 | 90% biodegradation | 138 |
| 11 | PHB | Non-ASTM, sewage sludge | Powder | 16 | 87% biodegradation | 15 |
| 12 | PHBV (13% HV) | 96% biodegradation | ||||
| 13 | PHBV (20% HV) | 83% biodegradation |
Sewage sludge biodegradation varies significantly but approximately 40 days are required for PHB films to biodegrade to 90% under ASTM D5210 (Table 4, entries 5 and 9). Following Non-ASTM standards under laboratory conditions, PHA biodegradation can occur in as little as 16 days.
4.9 PHA anaerobic digestion biodegradation
Anaerobic digestion is a part of the wastewater treatment utilized to reduce the organic matter content present in its effluent. The process can be either thermophilic (55 °C) or mesophilic (35 °C) and can be used to manage waste or produce biomethane. For the disposal of polymers, it is a suitable method to produce methane from food contaminated materials that can't be recycled. Under ASTM D5511 PHA biodegrades entirely in 20 days under mesophilic conditions.39 PLA and PCL both require temperatures above 37 °C to show any form of biodegradation under anaerobic conditions.149,246 While thermophilic anaerobic digestion is faster and more beneficial in methane production, mesophilic anaerobic digestion is more common in industry due to the energy input.248 Under anaerobic conditions, PHAs degrade faster under mesophilic conditions, while at thermophilic temperatures, PLA degradation is faster.213 Exploring mesophilic anaerobic biodegradation for PHAs has significant potential as a waste disposal method if plastic ends up in the wastewater treatment systems.4.10 PHA accelerated landfill biodegradation
Accelerated anaerobic landfill conditions are at mesophilic temperatures (35 °C) with a high ratio of solids, reflecting an environment much like a landfill. Landfill conditions are usually uncontrolled due to the quantity of material flowing into landfills continuously. Under optimal conditions and a high enough moisture content, PHB will degrade completely within 9 days, while the more complex PHBV may take longer to bio-assimilate (29% biodegradation in 42 days) after being degraded.148 The bio-assimilation is mainly based on the microorganism consortium.Moisture content is another factor. Under a high enough moisture content (50–90%), PHAs have been reported to completely degrade within 14 days.148,173 However, when the moisture content is below 50%, the biodegradation extent is halved and biodegradation can exceed 180 days.182
The main limitation of PHA anaerobic landfill biodegradation analysis is the few available studies being completed following either ASTM D5526-18 or ASTM D7475-20. Furthermore, while there is consistent evidence that PHAs show favourable anaerobic biodegradation behaviour, there is no labelling standard to define whether it can be claimed as such and it is not desirable to landfill biodegradable plastics unless the off-gases can be captured.
4.11 Conclusions of biodegradation
Several factors affect the biodegradation of PHAs in the natural environment (non-laboratory) including location, temperature, nutrients, microorganisms present, UV light exposure, dissolved oxygen and salinity. In lab scale studies, optimal temperature, moisture content, pH and higher amorphous content consistently promote enzymatic depolymerization of PHAs in all types of aerobic and anaerobic biodegradation. Combined with abiotic factors, the optimal conditions for PHA degradation can be derived (Fig. 15). Other polymers may include factors such as their Tg (i.e. PLA), due to the transition from a glassy to rubbery state resulting in an increased free volume251 around biodegradation temperatures that may impact hydrolytic and enzymatic degradation. Small chain PHAs also do not suffer from thermal degradation under 60 °C.252 Under composting conditions, PHAs would undergo some thermal degradation; however, the conditions become thermophilic and the thermophilic enzymes used are more stable in this environment, becoming the major driving force for PHA degradation.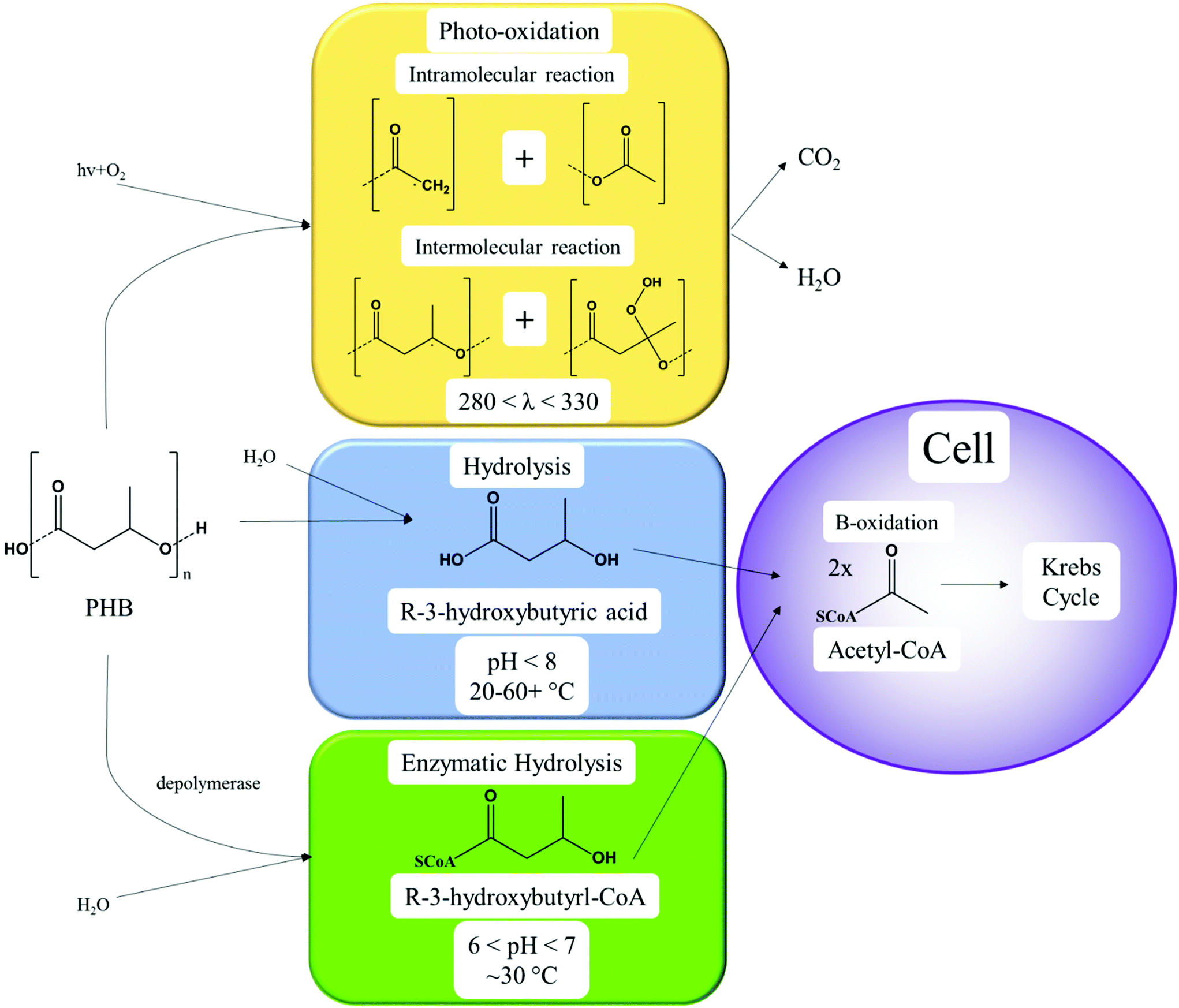 | ||
| Fig. 15 Simplified main pathways and optimal conditions for PHB degradation and bioassimilation in natural environments.91,226,253–255 | ||
The bio-assimilation of PHAs mainly depends on the complexity of the polymer, where less complex polymers are more easily assimilated, but at the same time are limited by their high crystallinity (that limits the degradation rate). Furthermore, the presence of other organic matter can either promote the bio-assimilation of PHA to produce specific products (methane) or can repress the bio-assimilation of PHA in favour of more easily accessible carbon sources (e.g. cellulose, starch, glucose etc.). Mesophilic temperatures are reported to be most favourable for PHAs compared to other biodegradable polymers (i.e. PLA), in both aerobic and anaerobic biodegradation.
The effect of additives and blends of PHAs are important to consider since virgin PHAs are not usually used in industry. Chain extenders, antifouling agents and the synthetically produced plasticizers TBC and glycerol inhibit the biodegradation of PHAs, despite reducing the crystallinity, either by inhibiting the enzymatic action, reducing hydrophilicity or inhibiting the microbial growth. In such cases these can delay the onset of biodegradation for tuneable attributes if so desired.
Inoculum sources provide a significant added diversity of microorganism consortia that is responsible for the variability of PHA biodegradation between studies. In high solid content studies, such as soil biodegradation, marine biodegradation and landfill biodegradation, a high moisture content allows for the microorganisms to have an increased diversity and population that can enhance the biodegradation rate. Therefore, to ensure that biodegradation studies are applicable in research, they should follow ASTM/ISO standards, and also have inoculum sources that are comparable to either a natural environment or comparable inoculum sources that are readily available such as those from waste water treatment plants.
5 PHA-based biocomposites
Biocomposite applications exist mainly in the automotive, packaging, and consumer product industries to reduce material costs and weight. Biocomposites can have improved impact and mechanical modulus compared to the virgin polymer. However, the effects are dependent on the fibre modulus, aspect ratio, morphology and interfacial adhesion of the fibre to the polymer, in addition to the properties of the polymer itself that are being reinforced. The impact and/or modulus are not always improved if the properties of the fibre are lower than the virgin polymer.256 The use of biocomposites introduces a sustainable application for agricultural fibres when their service lives end aside from disposal.10 Cotton for example is still the most produced fibre today,257 and is the most basic natural fibre consisting of predominantly cellulose. Other types of fibres contain cellulosic material such as hemicellulose of lignin depending on the source. In fibres, lignin acts as the cement and cellulose is the rigid structure that gives the biocomposite the increased moduli.258 PHA biocomposites are composites using natural fillers instead of inorganic fillers; it's important to consider the composition of the natural fillers and fibres for biodegradation purposes because they need to be degraded during the process.Natural fibres and fillers, composed of organic materials, are essentially made up of cellulose, hemicellulose and lignin, with some other components depending on the type such as protein, starch, silica and other impurities. Natural fibres are usually composed of the first three, which results in a structure seen in Fig. 16. Variation of these fibre ratios results in different types of fibres (bast, leaf, grass, straw and seeds). Several PHA-based biocomposites have been developed using kenaf,223 abaca,259 flax,260 starch,141 wheat straw,174 sisal,261 hemp,262 cellulose,263 lignin,150 seagrass,264 wood flour,165etc.192,264–267 However, in many cases it is always assumed that the addition of fibre improves the biodegradation of PHAs.
 | ||
| Fig. 16 Basic natural fibre structure. | ||
5.1 PHA-based composite biodegradation
Research on the biodegradation of biocomposites following any ASTM standard is commonly compared to virgin polymers. Several studies of PHA biodegradation have been completed with non-bio-based fillers such as TiO2,193,268 and carbon nanotubes, and organically modified clay claim to slow down the biodegradation of PHAs either by increasing the composite carbon content,269 limiting segmental motion of high molecular chains or limiting diffusion of water molecules into the bulk sample.270 Natural based fillers provide a benefit from a sustainable point of view and the majority are biodegradable in the appropriate environment. Natural rubber and other rubber constituents do show some biodegradation behaviour but unfortunately slow down the biodegradation of PHAs,271,272 as a result of rubber biodegradation being a slow process requiring oxidation.273 PHA-based biocomposites with natural fillers have the potential to reduce the cost and promote the biodegradation of PHAs improving the water diffusion rate and maximizing water absorption.134With the establishment that polysaccharide-based fillers can degrade when in composites, more complex fillers have varying effects that can be considered beneficial for biodegradation. Fibres such as bast when aged in water cause swelling of the cell walls, separating layers which can improve the availability of enzymatically active sites.274 Long sisal fibres 5 mm in length are reported to increase water absorption in PHBV/sisal composites, compared to 0.25 mm fibres. The fibre loading is also positively correlated with water absorption.275 The absorbed moisture not only separates layers, the fibre/matrix can debond, reducing the overall mechanical properties,266 but in turn increase the capacity to absorb water. These factors are attributed to the improved water absorptivity and hydrophilicity of composites working in a similar way that hydrophilic plasticizers improve PHA biodegradation.276 Furthermore, the physical structure of the fibre is important as a larger morphology negatively impacts the biodegradation rate by reducing the surface-to-volume ratio.277
Biodegradation of biocomposites mainly begins around the interface of the matrix and the fibre;205 thus, any fibre treatment may affect the biodegradation rate. While MA grafting is reported to have a minimal effect,134 acetic anhydride and pyradine treatment can slightly inhibit biodegradation but it remains unknown if the end % biodegradation will be affected, because no such study was found at this time that was completed until a plateau.205
The final attribute affecting the biodegradation of composites is the fibre composition. Natural fibres contain a variety of glucose monomer units which can be broken up by cellulases, amylases and cutinases, which are readily produced by microorganisms.99 Dewaxing of jute fibres before composite fabrication can improve the compostability of PHBV/jute composites, by removing the non-polar elements.278 As these natural fibres degrade, they also provide channels that allow water and enzymes into the internal structure of the polymer matrix, thereby enhancing biodegradation through an increased surface area.279 Furthermore, polymers containing proteins and lipids can biodegrade in soil, as the lipid and proteins provide a number of useful nutrients for microorganism growth.280
A major consideration of natural fibres is the limiting effects they have on conventional biodegradation. Lignin is reported to slow down microbial biodegradation and bio-assimilation due to its complex structure and chemical formula, more suited towards fermentation processes.281 Therefore, claiming the biodegradability of biocomposites without an assessment of the individual components as a bare minimum is a fallacy. Furthermore, natural fibres provide the more desirable glucose as a carbon source which can slow down biodegradation by glucose repression. It is still well documented that PHA degradation is repressed in the presence of glucose and cellobiose,282 However, once these specific carbon sources are degraded in a relative area, the PHA availability will be maximized and its biodegradation will resume.
A second misinterpretation is the idea that cellulose derivatives will biodegrade, which is not always correct. Cellulose acetate (CA), a derivative of cellulose, acts like a fibre when not plasticized and can improve the biodegradation of PHA,283 provided its degree of substitution is not high. Increased degree of substitution inhibits the biodegradation of CA, and beyond 2.5, it shows no degradation at all under aerobic composting conditions.284 Cellulose acetate butyrate (CAB) improved PHA elongation at break but reduced degradability285 and lignin-based PHB composites reduced biodegradation performance.286 Thus, research of PHA-based natural fillers and fibre biocomposites can prove to be invaluable in accelerating biodegradation and providing a more sustainable approach in plastic research.
Use of maleic anhydride (MA) is one of the most common fibre-biopolymer grafting methods, although others such as silane treated fibres have been successfully used as well.287 MA grafting does hold other benefits such as reducing odour release during processing.288 However, MA can target the oxygen species in polymers, thus enabling it to functionalize the carboxyl and hydroxyl groups in biopolymers and the hydroxyl groups in cellulose, lignocellulose and hemicellulose.289 This is of particular concern for biodegradation because of the hydrophilicity associated with polar carboxyl and hydroxyl groups.
In PHBV, MA grafting occurs predominantly on the PHB molecule, due to the relative number and chemical/statistical/steric effects. The ethyl group on PHV hinders reaction and reduces acidity, decreasing the availability of hydrogen atoms. Fig. 17 illustrates the reaction scheme between PHBV and MA,290 where the enzyme availability of HB may be inhibited.
 | ||
| Fig. 17 Reaction scheme between PHBV and MA in the presence of an initiator. This figure has been reproduced with permission from ref. 290, Elsevier, Copyright 2007 (License number: 4818250316231). | ||
5.1.1.1 Grafting effect in biodegradation. However, some chemical additives or polymers interfere with enzymatic actions upon the fibre. Of greatest concern is grafting compounds into PHAs, which fundamentally changes the chemical structure and may limit the enzyme function. Several studies of the biodegradation of PHA films grafted to 10 wt(%) acrylic anhydride (AA) or MA have been completed in field soils and greenhouse soils. An increase in the mass loss (%) by 3–5% was observed with the addition of maleic anhydride or acrylic anhydride. The indicated location of grafting for both anhydrides is on the butyrate side chain of PHB, which is expected to reduce the availability of the primary PHA degradation target and inhibit enzyme action. However, the crystallinity of all PHAs is slightly reduced with the grafting and the water absorption increased, as a result of either the increased amorphous region or unreacted anhydride improving the hydrophilicity due to the presence of oxygen species, or a combination thereof and improved the biodegradation rate. With the addition of fibres, the water absorption and mass loss of grafted PHAs perform poorly compared to non-grafted samples,165,191,192,218,219,291 likely due to the available anhydride oxygen species being bonded to fibres.
5.1.1.2 Surface treatment. Surface treatment is an alternative method to improve fibre–matrix interfacial adhesion and the overall properties by targeting the fibre/fillers themselves. In this regard, the polymer does not undergo chain scission. Physical methods include treatments by plasma, electron irradiation and surface roughening which can affect the hydrophilicity by removing hydroxide groups.292,293 Chemical methods such as water washing or NaOH treatment can either wash away undesirable chemicals,294 or functionalize the fibre surface.292 NaOH treatment of fibres is more effective in improving the biocomposite properties when the matrix is more hydrophilic, such as in PHBV compared to PE,84 mainly by substituting the hydrogen for Na+ to increase the polarity.295 Furthermore, NaOH treatment can remove fibre lignin and hemicellulose, roughening the fibre to maximize the surface area of fibres during biodegradation.292
Silane treatment is a form of chemical modification of the fibre surface, which consumes hydroxyl groups on cellulose as new chemical bonds are formed, and is reported to reduce the water absorption in Sisal/PHBV composites.275 The removal of hydroxyl groups can also increase the surface roughness,296 but the method is more commonly used for non-polar polymers due to the reduced fibre hydrophilicity.297
| PHA (co-monomer %)/composite | Protocol and source | Conditions | Form | Days | Results | Ref. | |
|---|---|---|---|---|---|---|---|
| 1 | PHBV (1% HV)/wood flour (i) 80/20, (ii) 50/50 | ASTM G160-12, subtropical field | ∼20 °C, 7cm depth | 1.6 mm plate | 365 | (i) 6.5%, (ii) 12.5% mass loss | 224 |
| 2 | PHBHx (3% Hx)/untreated kenaf 70/30 | Non-ASTM, oil palm cultivation soil | 30 °C, 81% relative humidity | 3 mm plate | 42 | 13% mass loss | 223 |
| 3 | PHBHx (3% Hx)/alkali and acid kenaf 70/30 | Non-ASTM, oil palm cultivation soil | 30 °C, 81% relative humidity | 3 mm plate | 42 | 7.5% mass loss | 223 |
| 4 | PHA/palm fibre (i) 80/20, (ii) 60/40 | Non-ASTM, farmland topsoil | 30–40% moisture(w.b.) | Film | 60 | (i) 72%, (ii) 90% mass loss | 192 |
| 5 | PHA-g-MA/silane treated palm fibre (i) 80/20, (ii) 60/40 | Non-ASTM, farmland topsoil | 30–40% moisture(w.b.) | Film | 60 | (i) 65%, (ii) 82% mass loss | 192 |
| 6 | PHA/marine algae powder (i) 90/10, (ii) 80/20 | Non-ASTM, greenhouse | 25 °C, 35% moisture(w.b.) | Powder | 120 | (i) 70%, (ii) 88% mass loss | 219 |
| 7 | PHA-g-AA/silane marine algae powder (i) 90/10, (ii) 80/20 | Non-ASTM, greenhouse | 25 °C, 35% moisture(w.b.) | Powder | 120 | (i) 61%, (ii) 78% mass loss | 219 |
| 8 | PHB/wood fibre 80/20 | ISO 17556, forest soil with garden soil | 20 °C, 40–60% moisture(w.b.) | Pellet | 195 | 60% biodegradation | 194 |
| 9 | PHA/tea plant fibre (i) 80/20, (ii) 60/40 | Non-ASTM, farmland topsoil | 35% moisture(w.b.) | Film | 140 | (i) 74%, (ii) 89% mass loss | 191 |
| 10 | PHA-g-MA/silane treated tea plant fibre (i) 80/20, (ii) 60/40 | Non-ASTM, farmland topsoil | 35% moisture(w.b.) | Film | 140 | (i) 67%, (ii) 81% mass loss | 191 |
| 11 | PHA/rice husk (i) 80/20, (ii) 60/40 | Non-ASTM, farmland topsoil | 35% moisture(w.b.) | Plate | 60 | (i) 82%, (ii) 95% mass loss | 218 |
| 12 | PHA-g-AA/rice husk (i) 80/20, (ii) 60/40 | Non-ASTM, farmland topsoil | 35% moisture(w.b.) | Plate | 60 | (i) 75%, (ii) 90% mass loss | 218 |
5.1.2.1 Effect of natural fibre/filler type. Soil biodegradation of PHA biocomposites has been completed with several fibres and fillers including starch, soy, lignin, flours, wheat straw etc. Starch based granules are a separate subset among natural fillers due to the alpha repeating units of glucose, providing a direct source of functional carbon in an optimal form with minimal degradation. Increasing the starch content from 10 to 30% increased the % degradation of PHA by 34%, indicating that there are synergistic benefits in the presence of fibres.298 Wei et al.299 reported PHB/potato peel waste composite films where the potato peel waste component degraded in the early stages through analysis of the melt temperature, which is the result of potato peel waste being composed mainly of starch (66%) despite containing some cellulose, hemicellulose and lignin.300
The complex structure of lignin is not easily degraded in soil biodegradation and showed evidence of no biodegradation compared to other natural fillers when in a PHA matrix in an aerobic environment.298 Although lignin is reported to act as a channel for degradation of the polymer matrix in anaerobic environments,301 it severely retarded degradation in anaerobic environments.182 Wheat straw contains 16–25% lignin,302 which is expected to inhibit the degradation of PHAs. Avella et al.174 reported PHBV/wheat straw 70/30 samples degraded by 23% in 180 days in garden soil, but no improvement in the rate of biodegradation was found compared to PHBV. PHA/wood flour 50/50 composites under natural and laboratory conditions are reported to show wood flour degradation improvements although the two studies vary from 12.5% in 365 days to 50% in 84 days respectively, which can be attributed to the higher surface area and moisture in laboratory studies.165,224 Furthermore, depending on the type of wood flour the lignin content can vary from 21–34%.303
The presence of lignin is still not enough to determine if the biodegradation rate will be impacted and to what extent. PHA/rice husk 80/20 biocomposites improved the water absorption of PHA and increased the % mass loss in farmland topsoil from 33 to 75% in 60 days,218 despite rice husk containing 25–30% lignin. However, rice husk also contains 50% cellulose and 20% silica,304 the former being easily degradable and contains only glucose unlike hemicellulose and lignin. Hemicellulose does provide benefits; being the amorphous fraction of the fibre, it is more easily hydrolysed. Peach palm fibre contains a significant amount of hemicellulose which is composed of a number of other hexose and pentose monomers besides glucose,305 and 0–25% weight ratios in PHBV showed a slight improvement in physical disintegration.221 Abaca fibre contains 66% cellulose, 24% hemicellulose and 12% lignin, giving a balance between the cellulose and hemicellulose, with little lignin,306 and 10% abaca fibre improves the degradation of PHBV from 30% to 50% in 180 days in regularly watered gardening soil at 25–30 °C.205 Kenaf fibre has a comparable cellulose and lignin content to that of abaca fibre with 13% hemicellulose,257 and PHBHx/kenaf 70/30 composites improve the degradation from 5.5 to 13% in 42 days223 (Table 5, entry 2). Therefore, it is not only the ratio of cellulose, hemicellulose and lignin that plays a role in the biodegradation improvement of PHA, but also the composition of the hemicellulose monomers (i.e. hexose, pentose glucose etc.).
Natural fillers such as grains include large amounts of protein besides cellulose, hemicellulose and lignin. Protein provides a nitrogen source, which after processing can be more readily available after protein degradation. PHA/soy 67/33 composites degraded by 89% in 168 days, exceeding similar ratios of PHA/starch composites.298 The nitrogen can be utilized by microorganisms for proteins and cell growth. The proteins (with a variety of essential amino acids), oils and cellulosic fibre found in DDGS307 can also provide suitable nutrients to allow growth of microorganisms and are significantly cheaper. The composition of DDGS in PHA/DDGS composites has allowed for comparable degradation to that of PHA/starch composites of similar weight ratios.298 Additionally, Madbouly et al.222 reported that 10% DDGS increased the biodegradation rate of PHA by 6 times. However the study was not completed until a plateau, and also utilized clarion loam soil which is known to have very good cellulolytic activity.125
Other natural PHA-based composites include PHA/marine algae powder, which performed poorly compared to PHA/rice husk under similar weight ratios and study conditions218,219 (Table 5, entries 6 and 11). Marine algae powder can have 15–66% polysaccharides (starch based or similar), making it unknown what was affecting the biodegradation rate.308
5.1.2.2 Effect of fibre treatment. Fibre treatment can be used to improve the surface characteristics for not only the interfacial adhesion, but also the interaction between water/enzymes and the fibres. The most basic fibre treatment studied in soil biodegradation is alkali or acid treatment. Acetic acid and pyradine treated abaca fibre slightly improved the water absorption but failed to improve the biodegradation any further than PHBV/abaca fibre composites.205 Joyyi et al.223 reported that alkali and acid treated kenaf fibres in PHBHx/kenaf 70/30 biocomposites had no effect on water absorption and reduced the degradation (Table 5, entry 3). The alkali treatment is indicated to remove the hemicellulose and lignin,309 which would result in a reduced amorphous fraction that may limit its susceptibility to enzymatic and hydrolytic attack. Hybridization of solubilized PLA and cellulose using N,N-dimethylformamide and 1-butyl-3-methylimidazolium acetate successfully improved the soil biodegradable performance of PLA/cellulose composite films and is attributed to the reduced crystallinity of PLA and cellulose,31 however the effectiveness of this approach has not been explored in PHA composite biodegradation.
Silane treatment of fibre surfaces has been studied on several types of fibre by Wu et al. 20% loading of marine algae powder,219 palm fibre192 or tea plant fibre191 increased the soil biodegradation rate of PHA by 100% (Table 5, entries 5, 7 and 10). However, with treatment of the fibres with tetraethyl orthosilicate or tetraethoxysilane, the biodegradation rate of the composites decreased by approximately 10%. The crosslinking effect upon the fibre can interfere with the enzyme activity, in addition to increasing the molecular weight. Furthermore, the ethyl groups on the silane agent are hydrophobic which can reduce the water absorptivity of the fibres. Acetylated or silane treated fibres similarly reduced the physical degradation in PBS-based composites in soil310 and composting.311
Other forms of fibre treatment completed in composite soil biodegradation are regenerated cellulose (lyocell) and peat (decayed matter) which represent special attributes that make them suitable for low release of fertilizers. PHBV/lyocell 60/40 biocomposites were studied in soil biodegradation with 80% humidity, but showed no improvement in biodegradation compared to PHBV. Shibata et al.216 reported PHBV matrix coated lyocell and did not allow the advantages of the filler to enhance the degradation of the composite. These characteristics are not seen in PBS/lyocell and PLA/lyocell composites and can be attributed to the hydrophobicity of PHAs. In such cases the fertilizer encapsulated by PHA can be released only once degradation of PHA proceeds to a certain extent. Peat is the decayed form of organic matter, and compared to wood flour it has a lower moisture absorption but still improves the biodegradation rate of the composite to a greater degree.312 Peat also has the added benefit of providing beneficial nutrients to plant growth.
The main application of soil biodegradation of PHA biocomposites is modulation of encapsulated nutrients, fertilizers and other compounds. PHA based composites with oil palm fibres have already been studied to create slow release fertilizers in the agricultural industry.313 This solution can be applied to several PHA biocomposites if PHA biodegradation needs to be accelerated and low-cost natural fillers.
| PHA (co-monomer %)/composite | Protocol and source | Form | Days | Results | Ref. | |
|---|---|---|---|---|---|---|
| 1 | PHBV (20% HV)/lignin 80/20 | ASTM D5338, mature compost | Film | 60 | 85% biodegradation | 150 |
| 2 | PHA/soy 67/33 | Non-ASTM, home composting (21–61 °C) | Small pot | 84 | 100% mass loss | 317 |
| 3 | PHA/DDGS 80/20 | 100% mass loss | ||||
| 4 | PHA/starch 90/10 | 57% mass loss | ||||
| 5 | PHA/lignin 80/20 | 21% mass loss | ||||
| 6 | PHB/jute (i) 90/10, (ii) 80/20, (iii) 70/30 | Non-ASTM, home composting (34–66 °C) | Tensile sample | 84 | (i) 65%, (ii) 68%, (iii) 85% mass loss | 234 |
| 7 | PHB/lyocell (i) 90/10, (ii) 80/20, (iii) 70/30 | Non-ASTM, home composting (34–66 °C) | Tensile sample | 84 | (i) 55%, (ii) 58%, (iii) 68% mass loss | 234 |
| 8 | PHB/hemp (i) 90/10, (ii) 80/20, (iii) 70/30 | Non-ASTM, home composting (34–66 °C) | Tensile sample | 84 | (i) 58%, (ii) 62%, (iii) 68% mass loss | 234 |
| 9 | PHBV (3% HV)/cellulose 97/3, 90/10, 75/25, 55/45 | ISO 20200, mature compost | Plate | 47 | 100% mass loss | 314 |
| 10 | PHB/wood fibre 80/20 | ISO 14855, organic municipal solid waste | Pellet | 78 | 95% biodegradation | 194 |
| 11 | PHBV (5% HV)/wheat straw (i) 90/10, (ii) 80/20, (iii) 70/30 | Modified ASTM D5338, mature compost | Film | 48 | (i) 55%, (ii) 60%, (iii) 65% biodegradation | 174 |
Under natural composting conditions, PHBV/starch composites showed a remarkable improvement in degradation rate, increasing PHBV (12% HV) degradation from 7–25% and 49%, with 30 and 50% starch loadings respectively. The increased starch content is also reported to increase the PHBV degradation rate in the composite.316 Starch is well known to have an amylose and an amylopectin fraction, the former being amorphous in nature which can reduce the crystallinity of the composite. Furthermore, starch is hydrophilic in nature, benefitting enzymatic catalysed hydrolytic degradation and is further reflected by its faster rate of soil biodegradation compared to cellulose.171 These attributes are also seen in hemicellulose, but lignin is reported to reduce the crystallinity; however, it does reduce the degradation extent.150 Lignin composites perform poorly compared to starch under composting conditions with both a PHA and PLA matrix.317 Therefore, a crystallinity reduction is not adequate to improve the biodegradation rate and some hydrophilic functions as well as a relative ease of degradation is required. This is further illustrated by PHBV/wheat straw composites which reduced the crystallinity but had a negative impact on the degradation rate in compost.174 The main cause is attributed to the high lignin (16–25%) and low cellulose.302
Aside from starch, neither cellulose nor lignin benefits PHA composting in high ratios. Gunning et al. studied PHB based jute, hemp and lyocell composites. With 30% loadings of the natural filler, jute increased % degradation from 50% to 85% while hemp and lyocell increased it to 68%234 (Table 6, entries 6–8). The mass rate improvement can be attributed to the slightly lower cellulose and higher hemicellulose content in jute fibre.
Another consideration found in composting is the presence of protein which reflects the effect seen in soil biodegradation. PHA Soy 67/33 and PHA/DDGS 80/20 were both reported to have 100% degradation in 84 days, unlike PHA/starch which was at 57%317 (Table 6, entries 2–4). Both natural fillers contain nitrogen sources for microbial growth. The effect of DDGS is also seen in PBAT, which made the composite degradation initial stages comparable to cellulose.318 The DDGS composition is approximately 19% cellulose, 17% hemicellulose, 5% starch and 30% protein,294 making it an amorphous, hydrophilic, nitrogen containing source suitable for biodegradable composites.
| PHA (co-monomer %)/composite | Protocol and source | Conditions | Form | Days | Results | Ref. | |
|---|---|---|---|---|---|---|---|
| 1 | PHBV (12% HV)/starch (i) 70/30, (ii) 50/50 | Non-ASTM, tropical coastal water | 26–32 °C | Tensile sample | 365 | (i) 80%, (ii) 100% mass loss | 141 |
| 2 | PHBV (19% HV)/starch (i) 75/25, (ii) 50/50 | Non-ASTM, anaerobic digester water | 30 °C | Film 150 μm | (i) 21, (ii) 8 | 100% mass loss | 241 |
| 3 | PHBV (19% HV)/starch (i) 75/25, (ii) 50/50 | Non-ASTM, anaerobic digester water | 30 °C | Film 800 μm | (i) 32, (i) 21 | (i) 85%, (ii) 100% mass loss | 241 |
| 4 | PHBV (5% HV) composite/seagrass (i) 90/10, (ii) 80/20 | Modified ASTM D6691, seawater and sediment | 30 °C | Pellets | 216 | (i) 20%, (ii) 27% biodegradation | 264 |
| 5 | PHBV (5% HV) composite/seagrass 80/20 | Non-ASTM, mariculture centre | 12–27 °C | Tensile sample | 365 | 23% mass loss | 264 |
Treatment of fibres with acids, alkalines, or silanes as per the literature found in this review caused no improvement in the water absorption and reduced the degradation in PHAs. The processes either removed the hemicellulose fraction, increasing crystallinity, or crosslinked the fibres, inhibiting the enzymatic functions. Grafting of the PHA with anhydrides did improve the extent of degradation – however, with the addition of fibres, the grafted PHAs performed poorly as crosslinked fibres inhibited enzymatic activity. Other forms of structural treatment such as cellulose regeneration (lyocell) gave no improvement, and the use of decaying organic matter improved the biodegradation of PHAs.
6 Conclusion
For a sustainable circular economy that mitigates the negative effect of conventional plastics on the environment and the eco-system, research and development into bio-based biodegradable polymers is imperative. Not only can bio-based biodegradable polymers reduce greenhouse gas emissions, but they can combat mismanaged waste that leaks into the environment, where human intervention is ineffective. And depending on the end-of-life, there is significant employment generation potential for value-added compost. To implement biodegradable polymers a deep understanding of the environments and their corresponding standards is necessary. Soil and marine environments are representative of a large pool where mismanaged waste ends up, such that studies in natural biodegradation environments can supplement development in the improvement of biodegradable polymers.PHAs are considered the most readily biodegradable polymer among many (i.e. PLA, PBS, PBAT etc.), supported by the literature, in aerobic environments (soil, compost and marine), and show promising biodegradable behaviour in anaerobic (sewage sludge, digesters and landfills) environments. PHAs benefit from biotic degradation by several types of bacterial and fungal enzymes. Furthermore, the surface area can be maximized and the crystallinity lowered based on different processing techniques or longer chain PHAs to optimize the biodegradation performance. The characteristics all improve the permeation of moisture and the enzyme interaction that accelerate the enzymatic catalysed hydrolysis. Furthermore, PHA degradation products are easily assimilated into usable products for microbial growth.
However, the addition of certain additives (chain extenders) and other biodegradable/non-biodegradable polymers that are used to improve its properties can be detrimental to its biodegradable performance. The negative effects of Joncryl, antifouling agents and non-degradable or hydrophobic plasticizers inhibit the onset and/or extent of PHA biodegradation in aerobic and anaerobic environments. Although these attributes are beneficial for biodegradation modulation in the agricultural industry, it is not desirable for single use plastic consumer waste. Blending PHAs with PLA, PBAT etc. all inhibit the biodegradation of PHAs in most environments, except for compost, where PLA does outperform PHAs.
To further improve the biodegradation of PHAs, incorporation of natural fibres and fillers can accelerate the rate of biodegradation based on the composition of fibres, while inorganic fillers either inhibit biodegradation or do not degrade at all. Natural fibres high in hemicellulose and cellulose benefit from improved hydrophilicity and eased biodegradation respectively. Fillers with high lignin and cellulose with low hemicellulose can inhibit the biodegradation of PHAs due to their complexity and low hydrophilicity respectively. Starch based fillers provide high hydrophilicity and eased biodegradation, allowing PHA biodegradation to significantly improve. Furthermore, by incorporating fillers with proteinaceous materials such as DDGS and soy, the biodegradation of PHAs can improve beyond starch and natural fillers without. However, the research in the area of PHA biocomposite biodegradation is severely limited in marine environments, where a significant mismanaged plastic waste is found.
Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
Funding support: The authors would like to thank the following financial supporters to carry out this research: (i) Natural Sciences and Engineering Research Council (NSERC), Canada Discovery Grants (Project # 400320); (ii) the Ontario Ministry of Agriculture, Food and Rural Affairs (OMAFRA) – University of Guelph, the Bioeconomy Industrial Uses Research Program Theme (Project # 030054, 030177, 030255, 030361, 030486); (iii) OMAFRA-University of Guelph Gryphon's Leading to the Accelerated Adoption of Innovative Research (LAAIR) Program (Project # 298635); and (iv) the Ontario Research Fund, Research Excellence Program; Round-9 (ORF-RE09) from the Ontario Ministry of Economic Development, Job Creation and Trade, Canada (Project #053970 and # 054345). This research was also benefited from the facility funding to the Bioproducts Discovery and Development Centre (BDDC) lab by FedDev Ontario; Ontario Ministry of Agriculture, Food, and Rural affairs (OMAFRA); Canada Foundation for Innovation (CFI); Federal Post-Secondary Institutions Strategic Investment Fund (SIF); and matching funds from the province of Ontario, Bank of Montreal (BMO) and numerous University of Guelph's Alumni.Other support: The authors are extremely grateful to Mr Atul Bali, Competitive Green Technologies, Leamington Ontario and Dr Milka Popov, University of Guelph to review, edit and comment on this article during its preparation.
References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 25–29 CrossRef PubMed.
- Nova Institute, Eur. Bioplastics, https://www.european-bioplastics.org/, (accessed 11 March 2020).
- New Plastic Economy, The New Plastic Economy 2019 Progress Report, 2019 Search PubMed.
- H. Storz and K.-D. Vorlop, Landbauforsch. Volk., 2014, 63, 321–332 Search PubMed.
- M. S. Masnadi, H. M. El-Houjeiri, D. Schunack, Y. Li, J. G. Englander, A. Badahdah, J. C. Monfort, J. E. Anderson, T. J. Wallington, J. A. Bergerson, D. Gordon, J. Koomey, S. Przesmitzki, I. L. Azevedo, X. T. Bi, J. E. Duffy, G. A. Heath, G. A. Keoleian, C. McGlade, D. Nathan Meehan, S. Yeh, F. You, M. Wang and A. R. Brandt, Science, 2018, 361, 851–853 CrossRef CAS PubMed.
- F. Gironi and V. Piemonte, Energy Sources, Part A, 2011, 33, 1949–1959 CrossRef CAS.
- F. Gallo, C. Fossi, R. Weber, D. Santillo, J. Sousa, I. Ingram, A. Nadal and D. Romano, Environ. Sci. Eur., 2018, 30(1), 13 CrossRef PubMed.
- K. G. Harding, J. S. Dennis, H. von Blottnitz and S. T. L. Harrison, J. Biotechnol., 2007, 130, 57–66 CrossRef CAS PubMed.
- Deloitte Sustainability, Blueprint for plastics packaging waste: Quality sorting and recycling Final report, 2018 Search PubMed.
- J. D. Badia, O. Gil-Castell and A. Ribes-Greus, Polym. Degrad. Stab., 2017, 137, 35–57 CrossRef CAS.
- ACS, Discovery Report The Future of Plastic, 2020 Search PubMed.
- L. M. Chu, in Encyclopedia of Ecology, Elsevier, 2008, pp. 2099–2103 Search PubMed.
- P. Agamuthu, Waste Manage. Res., 2013, 31, 1–2 CrossRef CAS PubMed.
- Department of Health, Important Things to Know About Landfill Gas, https://www.health.ny.gov/environmental/outdoors/air/landfill_gas.htm, (accessed 13 January 2020).
- K. Budwill, P. M. Fedorak and W. J. Page, Appl. Environ. Microbiol., 1992, 58, 1398–1401 CrossRef CAS PubMed.
- S. Rayne, Afr. J. Environ. Sci. Technol., 2008, 3, 1–3 Search PubMed.
- Reviews of Environmental Contamination and Toxicology, ed. D. M. Whitacre, Springer International Publishing, Cham, 2014, vol. 227 Search PubMed.
- Y. L. Tharpes, Univ. Miami Inter-American Law Rev., 1989, 579–614 Search PubMed.
- A. Cierjacks, F. Behr and I. Kowarik, Ecol. Indic., 2012, 13, 328–337 CrossRef.
- C. Wilcox, E. Van Sebille, B. D. Hardesty and J. A. Estes, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11899–11904 CrossRef CAS PubMed.
- A. Cózar, F. Echevarría, J. I. González-Gordillo, X. Irigoien, B. Úbeda, S. Hernández-León, Á. T. Palma, S. Navarro, J. García-de-Lomas, A. Ruiz, M. L. Fernández-de-Puelles and C. M. Duarte, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10239–10244 CrossRef PubMed.
- K. L. Law, S. Moret-Ferguson, N. A. Maximenko, G. Proskurowski, E. E. Peacock, J. Hafner and C. M. Reddy, Science, 2010, 329, 1185–1188 CrossRef CAS PubMed.
- E. Van Sebille, M. H. England and G. Froyland, Environ. Res. Lett., 2012, 7(4) DOI:10.1088/1748-9326/7/4/044040.
- L. Guedri-Knani, J. L. Gardette, M. Jacquet and A. Rivaton, Surf. Coat. Technol., 2004, 180–181, 71–75 CrossRef CAS.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- EPA, Composting At Home, https://www.epa.gov/recycle/composting-home, (accessed 13 January 2020).
- World Centric, Compostable Plastics, http://www.worldcentric.org/biocompostables/bioplastics, (accessed 25 April 2020).
- Environmental Protection Agency, An Analysis of Composting as an Environmental Remediation Technology, 1998 Search PubMed.
- S. Gibbens, What you need to know about plant-based plastics, 2018, https://www.nationalgeographic.com/environment/2018/11/are-bioplastics-made-from-plants-better-for-environment-ocean-plastic/, (accessed 11 March 2020) Search PubMed.
- J. H. Song, R. J. Murphy, R. Narayan and G. B. H. Davies, Philos. Trans. R. Soc., B, 2009, 364, 2127–2139 CrossRef CAS PubMed.
- A. Xu, Y. Wang, J. Gao and J. Wang, Green Chem., 2019, 21, 4449–4456 RSC.
- M. T. Gutierrez-Wing, B. E. Stevens, C. S. Theegala, I. I. Negulescu and K. A. Rusch, Environ. Eng. Sci., 2011, 28, 477–488 CrossRef CAS.
- A. Rudin and P. Choi, in The Elements of Polymer Science & Engineering, Elsevier, 2013, pp. 521–535 Search PubMed.
- R. P. Babu, K. O'Connor and R. Seeram, Prog. Biomater., 2013, 2, 8 CrossRef PubMed.
- I. Voevodina and A. Kržan, Bio-based Polymers, 2014 Search PubMed.
- G. Q. Chen, Plastics from Bacteria, 2010, vol. 14 Search PubMed.
- Y. J. Kim, S. Y. Choi, J. Kim, K. S. Jin, S. Y. Lee and K. J. Kim, Biotechnol. J., 2017, 12(1), 1600649 CrossRef PubMed.
- C. Lee and S. Hong, Mod. Chem. Appl., 2014, 2(4) DOI:10.4172/2329-6798.1000144.
- J. Greene, Ecol. Pollut. Environ. Sci., 2018, 1, 13–18 Search PubMed.
- M. Braungart, W. McDonough and A. Bollinger, J. Cleaner Prod., 2007, 15, 1337–1348 CrossRef.
- S. Kuciel, P. Kuźniar and M. Nykiel, Polimery/Polymers, 2018, 63, 31–37 CAS.
- A. Steinbüchel and H. E. Valentin, FEMS Microbiol. Lett., 1995, 128, 219–228 CrossRef.
- S. M. Al-Salem, P. Lettieri and J. Baeyens, Waste Manage., 2009, 29, 2625–2643 CrossRef CAS PubMed.
- G. Braunegg, G. Lefebvre and K. F. Genser, J. Biotechnol., 1998, 65, 127–161 CrossRef CAS PubMed.
- P. M. Visakh, RSC Green Chem., 2015, 2015-Janua, 1–17 Search PubMed.
- J. Choi and S. Y. Lee, Biotechnol. Bioeng., 1999, 62, 546–553 CrossRef CAS PubMed.
- F. B. Rebah, D. Prévost, R. D. Tyagi and L. Belbahri, Appl. Biochem. Biotechnol., 2009, 158, 155–163 CrossRef PubMed.
- B. A. Ramsay, I. Saracovan, J. A. Ramsay and R. H. Marchessault, J. Environ. Polym. Degrad., 1994, 2, 1–7 CrossRef CAS.
- J. Juengert, S. Bresan and D. Jendrossek, Bio-Protoc., 2018, 8, 1–15 Search PubMed.
- M. Koller, L. Maršálek, M. M. de Sousa Dias and G. Braunegg, Nat. Biotechnol., 2017, 37, 24–38 CAS.
- C. Kourmentza, J. Plácido, N. Venetsaneas, A. Burniol-Figols, C. Varrone, H. N. Gavala and M. A. M. Reis, Bioengineering, 2017, 4, 1–43 CrossRef PubMed.
- N. Korotkova and M. E. Lidstrom, J. Bacteriol., 2001, 183, 1038–1046 CrossRef CAS PubMed.
- I. Vroman and L. Tighzert, Materials, 2009, 2, 307–344 CrossRef CAS.
- J. Chen, W. Li, Z. Z. Zhang, T. W. Tan and Z. J. Li, Microb. Cell Fact., 2018, 17, 1–12 CrossRef PubMed.
- Q. Chen, Q. Wang, G. Wei, Q. Liang and Q. Qi, Appl. Environ. Microbiol., 2011, 77, 4886–4893 CrossRef CAS PubMed.
- Y. Wang and G.-Q. Chen, in Sustainable Polymers from Biomass, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, pp. 11–33 Search PubMed.
- H. Verhoogt, B. A. Ramsay and B. D. Favis, Polymer, 1994, 35, 5155–5169 CrossRef CAS.
- D. S. Sheu, W. M. Chen, J. Y. Yang and R. C. Chang, Enzyme Microb. Technol., 2009, 44, 289–294 CrossRef CAS.
- P. Strong, B. Laycock, S. Mahamud, P. Jensen, P. Lant, G. Tyson and S. Pratt, Microorganisms, 2016, 4, 11 CrossRef PubMed.
- A. O. Pérez-Arauz, A. E. Aguilar-Rabiela, A. Vargas-Torres, A. I. Rodríguez-Hernández, N. Chavarría-Hernández, B. Vergara-Porras and M. R. López-Cuellar, Food Packag. Shelf Life, 2019, 20, 100297 CrossRef.
- P. Kahar, T. Tsuge, K. Taguchi and Y. Doi, Polym. Degrad. Stab., 2004, 83, 79–86 CrossRef CAS.
- T. Tsuge, T. Yamamoto, K. Yano, H. Abe, Y. Doi and S. Taguchi, Macromol. Biosci., 2009, 9, 71–78 CrossRef CAS PubMed.
- D. Dennis, M. McCoy, A. Stangl, H. E. Valentin and Z. Wu, J. Biotechnol., 1998, 64, 177–186 CrossRef CAS PubMed.
- R. A. J. Verlinden, D. J. Hill, M. A. Kenward, C. D. Williams, Z. Piotrowska-Seget and I. K. Radecka, AMB Express, 2011, 1, 1–8 CrossRef PubMed.
- C. Morais, F. Freitas, M. V. Cruz, A. Paiva, M. Dionísio and M. A. M. Reis, Int. J. Biol. Macromol., 2014, 71, 68–73 CrossRef CAS PubMed.
- B. Andreeßen and A. Steinbüchel, Appl. Environ. Microbiol., 2010, 76, 4919–4925 CrossRef PubMed.
- G. A. Vikhoreva, N. R. Kil'deeva, G. A. Bonartseva, M. B. Fedorov, O. N. Mokhova and L. S. Gal'braikh, Appl. Biochem. Microbiol., 2007, 43, 611–615 CrossRef.
- L. Garrido, I. Jiménez, G. Ellis, P. Cano, J. M. García-Martínez, L. López and E. De La Peña, J. Appl. Polym. Sci., 2011, 119, 3286–3296 CrossRef CAS.
- N. Zhao, K. Wei, J. Chen, S. Zhang, Y. Wang and X. Wang, Mater. Lett., 2006, 61, 1071–1076 Search PubMed.
- E. I. Paşcu, J. Stokes and G. B. McGuinness, Mater. Sci. Eng., C, 2013, 33, 4905–4916 CrossRef PubMed.
- A. Ghebi, K. Khoshnevisan, N. Ketabchi, M. A. Derakhshan and A. A. Babadi, Nanomed. Res. J., 2016, 1, 107–111 Search PubMed.
- Plastics Europe, Biodegradable Plastics, https://www.plasticseurope.org/en/about-plastics/what-are-plastics/large-family/biodegradable-plastics, (accessed 13 January 2020).
- A. Corbin, J. Cowan, C. A. Miles, D. Hayes, D. John and D. Inglis, Using Biodegradable Plastics as Agricultural Mulches, Washington, 2013 Search PubMed.
- S. Dharmalingam, Biodegradation and Photodegradation of Polylactic Acid and Polylactic Acid/Polyhydroxyalkanoate Blends Nonwoven Agricultural Mulches in Ambient Soil Conditions, University of Tennessee, 2014 Search PubMed.
- P. Poltronieri and P. Kumar, in Handbook of Ecomaterials, Springer International Publishing, Cham, 2018, pp. 1–30 Search PubMed.
- L. P. Amaro, M. A. Abdelwahab, A. Morelli, E. Chiellini and M. K. Ed, in Recent Advances in Biotechnology, ed. L. Pérez Amaro, M. A. Abdelwahab, A. Morelli, F. Chiellini and E. Chiellini, Bentham Science Publishers, 2016, vol. 2, pp. 3–74 Search PubMed.
- Z. A. Raza, S. Riaz and I. M. Banat, Biotechnol. Prog., 2018, 34, 29–41 CrossRef CAS PubMed.
- H. M. Chang, Z. H. Wang, H. N. Luo, M. Xu, X. Y. Ren, G. X. Zheng, B. J. Wu, X. H. Zhang, X. Y. Lu, F. Chen, X. H. Jing and L. Wang, Braz. J. Med. Biol. Res., 2014, 47, 533–539 CrossRef CAS PubMed.
- I. Noda, P. R. Green, M. M. Satkowski and L. A. Schechtman, Biomacromolecules, 2005, 6, 580–586 CrossRef CAS PubMed.
- K. Changwichan, T. Silalertruksa and S. H. Gheewala, Sustainability, 2018, 10, 1–15 CrossRef.
- T. Gerard and T. Budtova, Eur. Polym. J., 2012, 48, 1110–1117 CrossRef CAS.
- V. Chiono, G. Ciardelli, G. Vozzi, M. G. Sotgiu, B. Vinci, C. Domenici and P. Giusti, J. Biomed. Mater. Res., Part A, 2008, 85, 938–953 CrossRef PubMed.
- S. S. Ahankari, A. K. Mohanty and M. Misra, Compos. Sci. Technol., 2011, 71, 653–657 CrossRef CAS.
- P. Russo, C. Carfagna, F. Cimino, D. Acierno and P. Persico, Adv. Polym. Technol., 2013, 32(S1), E313–E322 CrossRef CAS.
- E. Rudnik, in Compostable Polymer Materials, Elsevier, 2019, vol. 21, pp. 49–98 Search PubMed.
- G. Q. Chen, Chem. Soc. Rev., 2009, 38, 2434–2446 RSC.
- V. K. Rastogi and P. Samyn, Coatings, 2015, 5, 887–930 CrossRef CAS.
- N. Lucas, C. Bienaime, C. Belloy, M. Queneudec, F. Silvestre and J. E. Nava-Saucedo, Chemosphere, 2008, 73, 429–442 CrossRef CAS PubMed.
- T. Kijchavengkul, R. Auras, M. Rubino, S. Selke, M. Ngouajio and R. T. Fernandez, Polym. Degrad. Stab., 2010, 95, 2641–2647 CrossRef CAS.
- E. Bugnicourt, P. Cinelli, A. Lazzeri and V. Alvarez, Handb. Sustainable Polym., 2015, 899–928 Search PubMed.
- M. Michalak, M. Kwiecień, M. Kawalec and P. Kurcok, RSC Adv., 2016, 6, 12809–12818 RSC.
- Y. Aoyagi, K. Yamashita and Y. Doi, Polym. Degrad. Stab., 2002, 76, 53–59 CrossRef CAS.
- M. Liu, J. K. Zhuo, S. J. Xiong and Q. Yao, Energy Fuels, 2014, 28, 6038–6045 CrossRef CAS.
- Biodegradable Polymers for Industrial Applications, ed. R. Smith, 2005 Search PubMed.
- R. Boopathy, Bioresour. Technol., 2000, 74, 63–67 CrossRef CAS.
- R. Jayasekara, I. Harding, I. Bowater and G. Lonergan, J. Polym. Environ., 2005, 13, 231–251 CrossRef CAS.
- F. von Burkersroda, L. Schedl and A. Göpferich, Biomaterials, 2002, 23, 4221–4231 CrossRef CAS PubMed.
- R. van Dijkhuizen-Radersma, L. Moroni, A. van Apeldoorn, Z. Zhang and D. Grijpma, in Tissue Engineering, Elsevier, 2008, pp. 193–221 Search PubMed.
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 2012, 64, 72–82 CrossRef.
- T. G. Volova, M. I. Gladyshev, M. Y. Trusova and N. O. Zhila, Polym. Degrad. Stab., 2007, 92, 580–586 CrossRef CAS.
- I. I. Muhamad, L. K. Joon and M. A. M. Noor, Malaysian Polym. J., 2006, 1, 39–46 Search PubMed.
- L. Xu, K. Crawford and C. B. Gorman, Macromolecules, 2011, 44, 4777–4782 CrossRef CAS.
- S. K. Ghosh, S. Pal and S. Ray, Environ. Sci. Pollut. Res. Int., 2013, 20, 4339–4355 CrossRef CAS PubMed.
- J. Arutchelvi, M. Sudhakar, A. Arkatkar, M. Doble, S. Bhaduri and P. V. Uppara, Indian J. Biotechnol., 2008, 7, 9–22 CAS.
- J. Jacquin, J. Cheng, C. Odobel, C. Pandin, P. Conan, M. Pujo-Pay, V. Barbe, A. L. Meistertzheim and J. F. Ghiglione, Front. Microbiol., 2019, 10, 1–16 CrossRef PubMed.
- S. Wang, K. A. Lydon, E. M. White, J. B. Grubbs, E. K. Lipp, J. Locklin and J. R. Jambeck, Environ. Sci. Technol., 2018, 52, 5700–5709 CrossRef CAS PubMed.
- J. Mergaert, A. Webb, C. Anderson, A. Wouters and J. Swings, Appl. Environ. Microbiol., 1993, 59, 3233–3238 CrossRef CAS PubMed.
- R. Handrick, S. Reinhardt, D. Schultheiss, T. Reichart, D. Schüler, V. Jendrossek and D. Jendrossek, J. Bacteriol., 2004, 186, 2466–2475 CrossRef CAS PubMed.
- M. B. Pedersen, P. Gaudu, D. Lechardeur, M.-A. Petit and A. Gruss, Annu. Rev. Food Sci. Technol., 2012, 3, 37–58 CrossRef CAS PubMed.
- R. Kleerebezem and M. C. van Loosdrecht, Curr. Opin. Biotechnol., 2007, 18, 207–212 CrossRef CAS PubMed.
- K. Budwill, J. Environ. Polym. Degrad., 1996, 4, 91–102 CrossRef CAS.
- T. Ishigaki, W. Sugano, A. Nakanishi, M. Tateda, M. Ike and M. Fujita, Chemosphere, 2004, 54, 225–233 CrossRef CAS PubMed.
- E. F. Gómez and F. C. Michel, Polym. Degrad. Stab., 2013, 98, 2583–2591 CrossRef.
- D. J. Kelly, N. J. Hughes and R. K. Poole, in Helicobacter pylori, American Society of Microbiology, 2001, pp. 113–124 Search PubMed.
- S. Tanigawa, Biogas: Converting Waste To Energy, 2017 Search PubMed.
- N. J. Themelis and P. A. Ulloa, Renewable Energy, 2007, 32, 1243–1257 CrossRef CAS.
- K. H. Rostkowski, C. S. Criddle and M. D. Lepech, Environ. Sci. Technol., 2012, 46, 9822–9829 CAS.
- New Plastic Economy, Oxo-Degradable Plastic Packaging Is Not a Solution To Plastic Pollution, 2017 Search PubMed.
- I. Jakubowicz, N. Yarahmadi and V. Arthurson, Polym. Degrad. Stab., 2011, 96, 919–928 CrossRef CAS.
- E. Chiellini, A. Corti and G. Swift, Polym. Degrad. Stab., 2003, 81, 341–351 CrossRef CAS.
- ASTM International, Standard Guide for Exposing and Testing Plastics that Degrade in the Environment by a Combination of Oxidation and Biodegradation, 2018 Search PubMed.
- R. C. Thompson, C. J. Moore, F. S. V. Saal and S. H. Swan, Philos. Trans. R. Soc., B, 2009, 364, 2153–2166 CrossRef CAS PubMed.
- Vinçotte, OK biodegradable WATER: Initial acceptance tests, http://www.tuv-at.be/green-marks/doc-center/, (accessed 7 December 2019).
- ASTM International, Standard Test Method for Determining Aerobic Biodegradation of Plastic Materials in Soil, 2018 Search PubMed.
- L. Schaiidt and F. Detrick, Appl. Microbiol., 1956, 6, 108–114 CrossRef.
- ASTM International, Standard test method for determining aerobic biodegradation of plastic materials under controlled composting conditions, Incorporating Thermophilic Temperatures, 2015 Search PubMed.
- ASTM International, Standard Specification for Labeling of Plastics Designed to be Aerobically Composted in Municipal or Industrial Facilities, 2019 Search PubMed.
- M. Tuomela, Degradation of lignin and other 14 C-labelled compounds in compost and soil with an emphasis on white-rot fungi, University of Helsinki, 2002 Search PubMed.
- ASTM International, Standard Test Method for Determining Aerobic Biodegradation of Plastics Buried in Sandy Marine Sediment under Controlled Laboratory Conditions, 2015 Search PubMed.
- T. D. Leathers, N. S. Govind and R. V. Greene, J. Polym. Environ., 2002, 8, 119–124 CrossRef.
- K. E. Gonda, D. Jendrossek and H. P. Molitoris, Hydrobiologia, 2000, 426, 173–183 CrossRef CAS.
- H. Brandl and P. Püchner, Biodegradation, 1991, 2, 237–243 CrossRef CAS.
- N. Sridewi, K. Bhubalan and K. Sudesh, Polym. Degrad. Stab., 2006, 91, 2931–2940 CrossRef CAS.
- J. D. Badia, T. Kittikorn, E. Strömberg, L. Santonja-Blasco, A. Martínez-Felipe, A. Ribes-Greus, M. Ek and S. Karlsson, Polym. Degrad. Stab., 2014, 108, 166–174 CrossRef CAS.
- J. P. Harrison, C. Boardman, K. O’Callaghan, A. M. Delort and J. Song, R. Soc. Open Sci., 2018, 5(5), 171792 CrossRef PubMed.
- ASTM International, Standard Specification for Non-Floating Biodegradable Plastics in the Marine, 2005 Search PubMed.
- Vinçotte, OK biodegradable MARINE: Initial acceptance tests, http://www.tuv-at.be/green-marks/doc-center/, (accessed 7 December 2019).
- P. Puechner, W. R. Mueller and D. Bardtke, J. Environ. Polym. Degrad., 1995, 3, 133–143 CrossRef CAS.
- O. Eriksson and G. Finnveden, Energies, 2017, 10(4), 539 CrossRef.
- A. P. Heitmann, I. Coura Rocha, P. P. de Souza, L. C. A. Oliveira and P. S. Patrícia, Catal. Today, 2018, 0–1 Search PubMed.
- S. H. Imam, S. H. Gordon, R. L. Shogren, T. R. Tosteson, N. S. Govind and R. V. Greene, Appl. Environ. Microbiol., 1999, 65, 431–437 CrossRef CAS PubMed.
- M. Tansengco and I. Dogma, Acta Biotechnol., 1999, 19, 191–203 CrossRef CAS.
- K. Mukai, K. Yamada and Y. Doi, Polym. Degrad. Stab., 1993, 41, 85–91 CrossRef CAS.
- A. N. Boyandin, S. V. Prudnikova, M. L. Filipenko, E. A. Khrapov, A. D. Vasil'ev and T. G. Volova, Appl. Biochem. Microbiol., 2012, 48, 28–36 CrossRef CAS.
- D. F. Gilmore, S. Antoun, R. W. Lenz, S. Goodwin, R. Austin and R. C. Fuller, J. Ind. Microbiol., 1992, 10, 199–206 CrossRef CAS.
- T. G. Volova, A. N. Boyandin, A. D. Vasil'ev, V. A. Karpov, I. V. Kozhevnikov, S. V. Prudnikova, V. P. Rudnev, B. B. Xuån, V. V. Dũng and I. I. Gitel'zon, Microbiology, 2011, 80, 252–260 CAS.
- Y. Doi, Y. Kanesawa, N. Tanahashi and Y. Kumagai, Polym. Degrad. Stab., 1992, 36, 173–177 CrossRef CAS.
- A.-Z. Dunja-Manal, M. Rolf-Joachim and D. Wolf-Dieter, J. Biotechnol., 2001, 86, 113–126 CrossRef.
- H. Yagi, F. Ninomiya, M. Funabashi and M. Kunioka, Polym. Degrad. Stab., 2014, 110, 278–283 CrossRef CAS.
- H. Chen, Assessment of Biodegradation in Different Environmental Compartments of Blends and Composites Based on Microbial Poly(hydroxyalkanoate)s, University of Pisa, 2012 Search PubMed.
- A. N. Boyandin, S. V. Prudnikova, V. A. Karpov, V. N. Ivonin, N. L. Dô˜, T. H. Nguyê˜n, T. M. H. Lê, N. L. Filichev, A. L. Levin, M. L. Filipenko, T. G. Volova and I. I. Gitelson, Int. Biodeterior. Biodegrad., 2013, 83, 77–84 CrossRef CAS.
- B. I. Sang, K. Hori, Y. Tanji and H. Unno, Appl. Microbiol. Biotechnol., 2002, 58, 241–247 CrossRef CAS PubMed.
- D. Y. Kim and Y. H. Rhee, Appl. Microbiol. Biotechnol., 2003, 61, 300–308 CrossRef CAS PubMed.
- T. G. Volova, A. N. Boyandin, A. D. Vasiliev, V. A. Karpov, S. V. Prudnikova, O. V. Mishukova, U. A. Boyarskikh, M. L. Filipenko, V. P. Rudnev, B. Bá Xuân, V. Viêt Dũng and I. I. Gitelson, Polym. Degrad. Stab., 2010, 95, 2350–2359 CrossRef CAS.
- M. H. Zwietering, I. Jongenburger, F. M. Rombouts and K. Van't Riet, Appl. Environ. Microbiol., 1990, 56, 1875–1881 CrossRef CAS PubMed.
- T. Kobayashi, K. Uchino, T. Abe, Y. Yamazaki and T. Saito, J. Bacteriol., 2005, 187, 5129–5135 CrossRef CAS PubMed.
- K. Numata, H. Abe and Y. Doi, Can. J. Chem., 2008, 86, 471–483 CrossRef CAS.
- T. M. Scherer, R. C. Fuller, R. W. Lenz and S. Goodwin, Polym. Degrad. Stab., 1999, 64, 267–275 CrossRef CAS.
- R. A. Slepecky and J. H. Law, J. Bacteriol., 1961, 82, 37–42 CrossRef CAS PubMed.
- L. Shang, Q. Fei, Y. H. Zhang, X. Z. Wang, F. Dai-Di and H. N. Chang, J. Polym. Environ., 2012, 20, 23–28 CrossRef CAS.
- C. Eldsäter, A. C. Albertsson and S. Karlsson, Acta Polym., 1997, 48, 478–483 CrossRef.
- M. Hakkarainen, Adv. Polym. Sci., 2002, 157, 113–138 CrossRef CAS.
- T. Iwata and Y. Doi, Macromol. Chem. Phys., 1999, 200, 2429–2442 CrossRef CAS.
- C. Thellen, M. Coyne, D. Froio, M. Auerbach, C. Wirsen and J. A. Ratto, J. Polym. Environ., 2008, 16, 1–11 CrossRef CAS.
- C. S. Wu, J. Appl. Polym. Sci., 2006, 102, 3565–3574 CrossRef CAS.
- K. Numata, Y. Kikkawa, T. Tsuge, T. Iwata, Y. Doi and H. Abe, Biomacromolecules, 2005, 6, 2008–2016 CrossRef CAS PubMed.
- S. J. Holland, A. M. Jolly, M. Yasin and B. J. Tighe, Biomaterials, 1987, 8, 289–295 CrossRef CAS PubMed.
- L. Feng, Y. Wang, Y. Inagawa, K. Kasuya, T. Saito, Y. Doi and Y. Inoue, Polym. Degrad. Stab., 2004, 84, 95–104 CrossRef CAS.
- Y. Huang, C. Zhang, Y. Pan, Y. Zhou, L. Jiang and Y. Dan, Polym. Degrad. Stab., 2013, 98, 943–950 CrossRef CAS.
- M. Deroiné, A. Le Duigou, Y. M. Corre, P. Y. Le Gac, P. Davies, G. César and S. Bruzaud, Polym. Degrad. Stab., 2014, 105, 237–247 CrossRef.
- M. V. Arcos-Hernandez, B. Laycock, S. Pratt, B. C. Donose, M. A. L. Nikolič, P. Luckman, A. Werker and P. A. Lant, Polym. Degrad. Stab., 2012, 97, 2301–2312 CrossRef CAS.
- I. Keen, L. J. Raggatt, S. M. Cool, V. Nurcombe, P. Fredericks, M. Trau and L. Grøndahl, J. Biomater. Sci., Polym. Ed., 2007, 18, 1101–1123 CrossRef CAS PubMed.
- M. C. Morse, Q. Liao, C. S. Criddle and C. W. Frank, Polymer, 2011, 52, 547–556 CrossRef CAS.
- M. Avella, G. La Rota, E. Martuscelli, M. Raimo, P. Sadocco, G. Elegir and R. Riva, J. Mater. Sci., 2000, 35, 829–836 CrossRef CAS.
- S. Muniyasamy, O. Ofosu, M. J. John and R. D. Anandjiwala, J. Renewable Mater., 2016, 4, 133–145 CrossRef CAS.
- M. Deroiné, G. César, A. Le Duigou, P. Davies and S. Bruzaud, J. Polym. Environ., 2015, 23, 493–505 CrossRef.
- Y. X. Weng, Y. Wang, X. L. Wang and Y. Z. Wang, Polym. Test., 2010, 29, 579–587 CrossRef CAS.
- Y. X. Weng, X. L. Wang and Y. Z. Wang, Polym. Test., 2011, 30, 372–380 CrossRef CAS.
- J. Mergaert, C. Anderson, A. Wouters, J. Swings and K. Kersters, FEMS Microbiol. Lett., 1992, 103, 317–321 CrossRef CAS PubMed.
- T. G. Volova, N. O. Zhila, E. I. Shishatskaya, P. V. Mironov, A. D. Vasil'Ev, A. G. Sukovatyi and A. J. Sinskey, Polym. Sci., Ser. A, 2013, 55, 427–437 CrossRef CAS.
- T. G. Volova, S. V. Prudnikova, O. N. Vinogradova, D. A. Syrvacheva and E. I. Shishatskaya, Microb. Ecol., 2017, 73, 353–367 CrossRef CAS PubMed.
- T. W. Federle, M. A. Barlaz, C. A. Pettigrew, K. M. Kerr, J. J. Kemper, B. A. Nuck and L. A. Schechtman, Biomacromolecules, 2002, 3, 813–822 CrossRef CAS PubMed.
- D. Jendrossek, Appl. Microbiol. Biotechnol., 2007, 74, 1186–1196 CrossRef CAS PubMed.
- S. Miyazaki, K. Takahashi, M. Shiraki, T. Saito, Y. Tezuka and K. I. Kasuya, J. Polym. Environ., 2000, 8, 175–182 CrossRef CAS.
- V. Martínez, P. G. de Santos, J. García-Hidalgo, D. Hormigo, M. A. Prieto, M. Arroyo and I. de la Mata, Appl. Microbiol. Biotechnol., 2015, 99, 9605–9615 CrossRef PubMed.
- J. Mergaert and J. Swings, J. Ind. Microbiol. Biotechnol., 1996, 17, 463–469 CrossRef CAS.
- U. Siemann, in Scattering Methods and the Properties of Polymer Materials, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, vol. 130, pp. 1–14 Search PubMed.
- K. Kasuya, K. Takagi, S. Ishiwatari, Y. Yoshida and Y. Doi, Polym. Degrad. Stab., 2002, 59, 327–332 CrossRef.
- M. T. Gutierrez-Wing, B. E. Stevens, C. S. Theegala, I. I. Negulescu and K. A. Rusch, J. Environ. Eng., 2010, 136, 709–718 CrossRef CAS.
- H. Sashiwa, R. Fukuda, T. Okura, S. Sato and A. Nakayama, Mar. Drugs, 2018, 16, 1–11 CrossRef PubMed.
- C. S. Wu, Polym. Degrad. Stab., 2013, 98, 1473–1480 CrossRef CAS.
- C. S. Wu, H. T. Liao and Y. X. Cai, Polym. Degrad. Stab., 2017, 140, 55–63 CrossRef CAS.
- N. Altaee, G. A. El-Hiti, A. Fahdil, K. Sudesh and E. Yousif, SpringerPlus, 2016, 5(1), 762 CrossRef PubMed.
- M. Seggiani, P. Cinelli, S. Verstichel, M. Puccini, S. Vitolo, I. Anguillesi and A. Lazzeri, Chem. Eng. Trans., 2015, 43, 1813–1818 Search PubMed.
- J. S. Yoon, M. C. Chang, M. N. Kim, E. J. Kang, C. Kim and I. J. Chin, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 2543–2551 CrossRef CAS.
- C. A. Woolnough, L. H. Yee, T. S. Charlton and L. J. R. Foster, PLoS One, 2013, 8, 1–10 CrossRef PubMed.
- S. Duangphet, D. Szegda, J. Song and K. Tarverdi, J. Polym. Environ., 2014, 22, 1–8 CrossRef CAS.
- A. L. P. d. L. Freitas, L. R. Tonini Filho, P. S. Calvão and A. M. C. de Souza, Polym. Test., 2017, 62, 189–195 CrossRef CAS.
- W. Dong, B. Zou, Y. Yan, P. Ma and M. Chen, Int. J. Mol. Sci., 2013, 14, 20189–20203 CrossRef PubMed.
- L. Savenkova, Z. Gercberga, V. Nikolaeva, A. Dzene, I. Bibers and M. Kalnin, Process Biochem., 2000, 35, 573–579 CrossRef CAS.
- J. D. Fonseca, A. M. Latifi, A. Orjuela, I. D. Gil and G. Rodríguez, Dynamic Simulation and Optimisation of an Industrial Process for Tributyl Citrate Production, Elsevier Masson SAS, 2016, vol. 38 Search PubMed.
- M. Râpə, R. N. Darie-Nitə, E. Grosu, E. E. Tənase, A. R. Trifoi, T. Pap and C. Vasile, J. Optoelectron. Adv. Mater., 2015, 17, 1778–1784 Search PubMed.
- A. Höglund, M. Hakkarainen and A. C. Albertsson, Biomacromolecules, 2010, 11, 277–283 CrossRef PubMed.
- H. Kranz, N. Ubrich, P. Maincent and R. Bodmeier, J. Pharm. Sci., 2000, 89, 1558–1566 CrossRef CAS PubMed.
- N. Teramoto, K. Urata, K. Ozawa and M. Shibata, Polym. Degrad. Stab., 2004, 86, 401–409 CrossRef CAS.
- M. A. Abdelwahab, A. Flynn, B. S. Chiou, S. Imam, W. Orts and E. Chiellini, Polym. Degrad. Stab., 2012, 97, 1822–1828 CrossRef CAS.
- Y. X. Weng, L. Wang, M. Zhang, X. L. Wang and Y. Z. Wang, Polym. Test., 2013, 32, 60–70 CrossRef CAS.
- J. Wang and Q. Mao, Adv. Polym. Technol., 2012, 32, 474–485 CrossRef.
- D. Redondo, C. Peñalva, J. Val, F. Braca and M. Pérez, Acta Hortic., 2019, 1252, 69–76 CrossRef.
- H. Sato, R. Murakami, A. Padermshoke, F. Hirose, K. Senda, I. Noda and Y. Ozaki, Macromolecules, 2004, 37, 7203–7213 CrossRef CAS.
- Z. Zaidi, D. Mawad and A. Crosky, Front. Mater., 2019, 6, 1–12 CrossRef.
- R. Y. Tabasi and A. Ajji, Polym. Degrad. Stab., 2015, 120, 435–442 CrossRef CAS.
- S. Hegde, E. Dell, C. Lewis, T. A. Trabold and C. A. Diaz, 21st IAPRI World Conf. Packag. 2018 – Packag. Driv. a Sustain. Futur., 2019, pp. 730–737.
- M. P. Arrieta, J. López, E. Rayón and A. Jiménez, Polym. Degrad. Stab., 2014, 108, 307–318 CrossRef CAS.
- A. Modelli, B. Calcagno and M. Scandola, J. Environ. Polym. Degrad., 1999, 7, 109–116 CrossRef CAS.
- M. Shibata, S. Oyamada, S. Kobayashi and D. Yaginuma, J. Appl. Polym. Sci., 2004, 92, 3857–3863 CrossRef CAS.
- Vinçotte, OK biodegradable SOIL: Initial acceptance tests, 2012 Search PubMed.
- C. S. Wu, J. Polym. Environ., 2014, 22, 384–392 CrossRef CAS.
- C. S. Wu, Polym.-Plast. Technol. Eng., 2016, 55, 391–402 CrossRef.
- W. D. Luzier, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 839–842 CrossRef CAS PubMed.
- K. C. Batista, D. A. K. Silva, L. A. F. Coelho, S. H. Pezzin and A. P. T. Pezzin, J. Polym. Environ., 2010, 18, 346–354 CrossRef CAS.
- S. A. Madbouly, J. A. Schrader, G. Srinivasan, K. Liu, K. G. McCabe, D. Grewell, W. R. Graves and M. R. Kessler, Green Chem., 2014, 16, 1911–1920 RSC.
- L. Joyyi, M. Z. Ahmad Thirmizir, M. S. Salim, L. Han, P. Murugan, K. I. Kasuya, F. H. J. Maurer, M. I. Zainal Arifin and K. Sudesh, Polym. Degrad. Stab., 2017, 137, 100–108 CrossRef CAS.
- C. M. Chan, L. J. Vandi, S. Pratt, P. Halley, D. Richardson, A. Werker and B. Laycock, Sustainable Mater. Technol., 2019, 21, e00099 CrossRef CAS.
- C. Celestina, J. L. Wood, J. B. Manson, X. Wang, P. W. G. Sale, C. Tang and A. E. Franks, Sci. Rep., 2019, 9, 1–12 CrossRef CAS PubMed.
- R. Z. Sayyed, S. J. Wani, A. A. Alyousef, A. Alqasim, A. Syed and H. A. El-Enshasy, PLoS One, 2019, 14, 1–14 CrossRef PubMed.
- A. Manna and A. K. Paul, Biodegradation, 2000, 11, 323–329 CrossRef CAS PubMed.
- H. Tsuji and K. Suzuyoshi, J. Appl. Polym. Sci., 2003, 90, 587–593 CrossRef CAS.
- E. W. Slessarev, Y. Lin, N. L. Bingham, J. E. Johnson, Y. Dai, J. P. Schimel and O. A. Chadwick, Nature, 2016, 540, 567–569 CrossRef CAS PubMed.
- M. Kim, A. Lee, J. Yoon and I. Chin, Eur. Polym. J., 2000, 36, 1677–1685 CrossRef CAS.
- E. Rudnik and D. Briassoulis, J. Polym. Environ., 2011, 19, 18–39 CrossRef CAS.
- R. K. Sadi, G. J. M. Fechine and N. R. Demarquette, Polym. Degrad. Stab., 2010, 95, 2318–2327 CrossRef CAS.
- D. S. Rosa, R. P. Filho, Q. S. H. Chui, M. R. Calil and C. G. F. Guedes, Eur. Polym. J., 2003, 39, 233–237 CrossRef CAS.
- M. A. Gunning, L. M. Geever, J. A. Killion, J. G. Lyons and C. L. Higginbotham, Polym. Test., 2013, 32, 1603–1611 CrossRef CAS.
- M. Rutkowska, K. Krasowska, A. Heimowska, G. Adamus, M. Sobota, M. Musioł, H. Janeczek, W. Sikorska, A. Krzan, E. Žagar and M. Kowalczuk, J. Polym. Environ., 2008, 16, 183–191 CrossRef CAS.
- S. Luo and A. N. Netravali, Polym. Degrad. Stab., 2003, 80, 59–66 CrossRef CAS.
- J. Mergaert, C. Anderson, A. Wouters and J. Swings, J. Environ. Polym. Degrad., 1994, 2, 177–183 CrossRef CAS.
- J. Greene, PLA and PHA Biodegradation in the Marine Environment, Chico, 2012 Search PubMed.
- Y. H. Ho, S. N. Gan and I. K. P. Tan, Appl. Biochem. Biotechnol. – Part A Enzym. Eng. Biotechnol., 2002, 102–103, 337–347 CrossRef CAS.
- E. Halliday, S. L. McLellan, L. A. Amaral-Zettler, M. L. Sogin and R. J. Gast, PLoS One, 2014, 9(3), e90815 CrossRef PubMed.
- B. A. Ramsay, V. Langlade, P. J. Carreau and J. A. Ramsay, Appl. Environ. Microbiol., 1993, 59, 1242–1246 CrossRef CAS PubMed.
- J. Mergaert, A. Wouters, C. Anderson and J. Swings, Can. J. Microbiol., 1995, 41, 154–159 CrossRef CAS PubMed.
- H. Tsuji and K. Suzuyoshi, Polym. Degrad. Stab., 2002, 75, 357–365 CrossRef CAS.
- A. R. Bagheri, C. Laforsch, A. Greiner and S. Agarwal, Global Challenges, 2017, 1, 1700048 CrossRef PubMed.
- P. K. Shin, M. H. Kirn and J. M. Kim, J. Environ. Polym. Degrad., 1997, 5, 33–39 CAS.
- H. Yagi, F. Ninomiya, M. Funabashi and M. Kunioka, Polym. Degrad. Stab., 2013, 98, 1182–1187 CrossRef CAS.
- N. Benn and D. Zitomer, Front. Environ. Sci., 2018, 5, 1–9 Search PubMed.
- R. A. Labatut, L. T. Angenent and N. R. Scott, Water Res., 2014, 53, 249–258 CrossRef CAS PubMed.
- H. M. El-mashad, R. Zhang and J. P. Greene, J. Environ. Sci. Eng. A, 2012, 1, 108–114 CAS.
- J. E. Weaver, Effect of Inoculum Source on the Rate and Extent of Anaerobic Biodegradation, North Carolina State University, 2013 Search PubMed.
- A. Shrivastava, in Introduction to Plastics Engineering, Elsevier, 2018, pp. 1–16 Search PubMed.
- Y. Chen, I.-N. Chou, Y.-H. Tsai and H.-S. Wu, J. Appl. Polym. Sci., 2013, 130, 3659–3667 CrossRef CAS.
- J. Eggers and A. Steinbüchel, J. Bacteriol., 2013, 195, 3213–3223 CrossRef CAS PubMed.
- V. B. Ivanov, E. Y. Khavina, S. I. Voinov and A. A. Ol'khov, Int. Polym. Sci. Technol., 2009, 36, 19–22 CrossRef.
- Y. Cui, J. P. Barford and R. Renneberg, Anal. Lett., 2007, 40, 2915–2924 CrossRef CAS.
- Green Biorenewable Biocomposites, ed. V. K. Thakur and M. R. Kessler, Apple Academic Press, 2016 Search PubMed.
- A. Monti and E. Alexopoulou, in Green Energy and Technology, 2013, vol. 117 Search PubMed.
- M. N. Cazaurang-Martinez, P. J. Herrera-Franco, P. I. Gonzalez-Chi and M. Aguilar-Vega, J. Appl. Polym. Sci., 1991, 43, 749–756 CrossRef CAS.
- M. Shibata, K. I. Takachiyo, K. Ozawa, R. Yosomiya and H. Takeishi, J. Appl. Polym. Sci., 2002, 85, 129–138 CrossRef CAS.
- R. A. Shanks, A. Hodzic and S. Wong, J. Appl. Polym. Sci., 2004, 91, 2114–2121 CrossRef CAS.
- C. Moliner, J. D. Badia, B. Bosio, E. Arato, T. Kittikorn, E. Strömberg, R. Teruel-Juanes, M. Ek, S. Karlsson and A. Ribes-Greus, Chem. Eng. Commun., 2018, 205, 226–237 CrossRef CAS.
- É. B. Hermida and V. I. Mega, Composites, Part A, 2007, 38, 1387–1394 CrossRef.
- P. Gatenholm, J. Kubát and A. Mathiasson, J. Appl. Polym. Sci., 1992, 45, 1667–1677 CrossRef CAS.
- M. Seggiani, P. Cinelli, E. Balestri, N. Mallegni, E. Stefanelli, A. Rossi, C. Lardicci and A. Lazzeri, Materials, 2018, 11(5), 772 CrossRef.
- M. Avella, E. Martuscelli, B. Pacucci, M. Raimo and A. Marzetti, J. Appl. Polym. Sci., 1993, 49, 2091–2103 CrossRef CAS.
- N. M. Barkoula, S. K. Garkhail and T. Peijs, Ind. Crops Prod., 2010, 31, 34–42 CrossRef CAS.
- R. Muthuraj, M. Misra and A. K. Mohanty, J. Appl. Polym. Sci., 2017, 134, 1–10 Search PubMed.
- S. P. Yew, H. Y. Tang and K. Sudesh, Polym. Degrad. Stab., 2006, 91, 1800–1807 CrossRef CAS.
- L. S. Montagna, I. C. Oyama, R. d. C. B. C. Lamparelli, A. P. Silva, T. L. D. A. Montanheiro and A. P. Lemes, J. Renewable Mater., 2019, 7, 117–128 Search PubMed.
- D. Puglia, E. Fortunati, D. A. D'Amico, L. B. Manfredi, V. P. Cyras and J. M. Kenny, Polym. Degrad. Stab., 2014, 99, 127–135 CrossRef CAS.
- R. Bhatt, D. Shah, K. C. Patel and U. Trivedi, Bioresour. Technol., 2008, 99, 4615–4620 CrossRef CAS PubMed.
- K. Kuntanoo, S. Promkotra and P. Kaewkannetra, World Acad. Sci. Eng. Technol., 2013, 7, 1799–1803 Search PubMed.
- K. Rose and A. Steinbuchel, Appl. Environ. Microbiol., 2005, 71, 2803–2812 CrossRef CAS.
- A. Le Duigou, A. Bourmaud and C. Baley, Ind. Crops Prod., 2015, 70, 190–200 CrossRef.
- R. Dangtungee, J. Tengsuthiwat, P. Boonyasopon and S. Siengchin, J. Thermoplast. Compos. Mater., 2015, 28, 879–895 CrossRef CAS.
- T. Freier, C. Kunze, C. Nischan, S. Kramer, K. Sternberg, M. Saß, U. T. Hopt and K. P. Schmitz, Biomaterials, 2002, 23, 2649–2657 CrossRef CAS PubMed.
- T. Ohura, Y. Aoyagi, K. I. Takagi, Y. Yoshida, K. I. Kasuya and Y. Doi, Polym. Degrad. Stab., 1999, 63, 23–29 CrossRef CAS.
- A. K. Mohanty, M. A. Khan and G. Hinrichsen, Compos. Sci. Technol., 2000, 60, 1115–1124 CrossRef CAS.
- T. Bayerl, M. Geith, A. A. Somashekar and D. Bhattacharyya, Int. Biodeterior. Biodegrad., 2014, 96, 18–25 CrossRef.
- P. Faibunchan, Y. Nakaramontri, W. Chueangchayaphan, S. Pichaiyut, C. Kummerlöwe, N. Vennemann and C. Nakason, J. Polym. Environ., 2018, 26, 2867–2880 CrossRef CAS.
- T. K. Kirk and R. L. Farrell, Annu. Rev. Microbiol., 1987, 41, 465–501 CrossRef CAS PubMed.
- C. Phithakrotchanakoon, Y. Rudeekit, S. Tanapongpipat, T. Leejakpai, S. I. Aiba, I. Noda and V. Champreda, Biologia, 2009, 64, 246–251 CAS.
- C. S. Wu, Carbohydr. Polym., 2014, 105, 41–48 CrossRef CAS PubMed.
- R. M. Gardner, C. M. Buchanan, R. Komarek, D. Dorschel, C. Boggs and A. W. White, J. Appl. Polym. Sci., 1994, 52, 1477–1488 CrossRef CAS.
- E. El-Shafee, G. R. Saad and S. M. Fahmy, Eur. Polym. J., 2001, 37, 2091–2104 CrossRef CAS.
- P. Mousavioun, G. A. George and W. O. S. Doherty, Polym. Degrad. Stab., 2012, 97, 1114–1122 CrossRef CAS.
- R. Karnani, M. Krishnan and R. Narayan, Polym. Eng. Sci., 1997, 37, 476–483 CrossRef CAS.
- A. K. Bledzki, A. A. Mamun and O. Faruk, eXPRESS Polym. Lett., 2007, 1, 755–762 CrossRef CAS.
- S. W. Hwang, J. K. Shim, S. Selke, H. Soto-Valdez, M. Rubino and R. Auras, Macromol. Mater. Eng., 2013, 298, 624–633 CrossRef CAS.
- M. Avella, G. Bogoeva-Gaceva, A. Buzõarovska, M. Emanuela Errico, G. Gentile and A. Grozdanov, J. Appl. Polym. Sci., 2007, 104, 3192–3200 CrossRef CAS.
- C. S. Wu and H. T. Liao, Polym. Degrad. Stab., 2014, 99, 274–282 CrossRef CAS.
- H. S. Lee, D. Cho and S. O. Han, Macromol. Res., 2008, 16, 411–417 CrossRef CAS.
- S. G. Lee, S.-S. Choi, W. H. Park and D. Cho, Macromol. Symp., 2003, 197, 089–100 CrossRef CAS.
- N. Zarrinbakhsh, A. K. Mohanty and M. Misra, Biomass Bioenergy, 2013, 55, 251–259 CrossRef CAS.
- L.Y Mwaikambo and M. P. Ansell, Angew. Makromol. Chem., 1999, 272, 108–116 CrossRef CAS.
- A. Kumar Sinha, H. K. Narang and S. Bhattacharya, Mater. Today: Proc., 2017, 4, 8993–8996 CAS.
- Y. Xie, C. A. S. Hill, Z. Xiao, H. Militz and C. Mai, Composites, Part A, 2010, 41, 806–819 CrossRef.
- H. A. Kratsch, J. A. Schrader, K. G. McCabe, G. Srinivasan, D. Grewell and W. R. Graves, HortTechnology, 2015, 25, 119–131 CAS.
- L. Wei, S. Liang and A. G. McDonald, Ind. Crops Prod., 2015, 69, 91–103 CrossRef CAS.
- A. U. Mahmood, J. Greenman and A. H. Scragg, Enzyme Microb. Technol., 1998, 22, 130–137 CrossRef CAS.
- S. Peterson, K. Jayaraman and D. Bhattacharyya, Composites, Part A, 2002, 33, 1123–1134 CrossRef.
- F. Carvalheiro, T. Silva-Fernandes, L. C. Duarte and F. M. Gírio, Appl. Biochem. Biotechnol., 2009, 153, 84–93 CrossRef CAS PubMed.
- C. M. Clemons, in Functional Fillers for Plastics, ed. M. Xanthos, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2010, pp. 249–270 Search PubMed.
- M. S. Ismail and A. M. Waliuddin, Constr. Build. Mater., 1996, 10, 521–526 CrossRef.
- B. C. Bolanho, E. D. G. Danesi and A. D. P. Beléia, Carbohydr. Polym., 2015, 124, 196–200 CrossRef CAS PubMed.
- S. W. Saragih, R. Lubis, B. Wirjosentono and E. Eddyanto, AIP Conf. Proc., 2018, 2049(December) DOI:10.1063/1.5082463.
- K. Liu, J. Agric. Food Chem., 2011, 59, 1508–1526 CrossRef CAS PubMed.
- M. Øverland, L. T. Mydland and A. Skrede, J. Sci. Food Agric., 2019, 99, 13–24 CrossRef PubMed.
- I. Ismojo, E. Yuanita, E. M. Rosa, L. Calvin and M. Chalid, Proc. 5Th Int. Symp. Appl. Chem., 2019, 2175, 020059 Search PubMed.
- H. P. S. Abdul Khalil and H. Ismail, Polym. Test., 2000, 20, 65–75 CrossRef.
- B. P. Calabia, F. Ninomiya, H. Yagi, A. Oishi, K. Taguchi, M. Kunioka and M. Funabashi, Polymers, 2013, 5, 128–141 CrossRef.
- S. Thomas, A. A. Shumilova, E. G. Kiselev, S. V. Baranovsky, A. D. Vasiliev, I. V. Nemtsev, A. P. Kuzmin, A. G. Sukovatyi, R. P. Avinash and T. G. Volova, Int. J. Biol. Macromol., 2020, 155, 1373–1384 CrossRef CAS PubMed.
- A. S. Harmaen, A. Khalina, H. M. Ali and I. N. Azowa, Int. J. Polym. Sci., 2016, 2016 DOI:10.1155/2016/323010.
- E. L. Sanchez-Safont, J. Gonzalez-Ausejo, J. Gamez-Perez, J. M. Lagaron and L. Cabedo, J. Renewable Mater., 2016, 4, 123–132 CrossRef CAS.
- M. P. Arrieta, E. Fortunati, F. Dominici, E. Rayón, J. López and J. M. Kenny, Polym. Degrad. Stab., 2014, 107, 139–149 CrossRef CAS.
- S. H. Imam, L. Chen, S. H. Gorden, R. L. Shogren, D. Weisleder and R. V. Greene, J. Environ. Polym. Degrad., 1998, 6, 91–98 CrossRef CAS.
- J. A. Schrader, K. G. McCabe, D. Grewell and W. R. Graves, Acta Hortic., 2017, 1170, 1101–1108 CrossRef.
- S. Muniyasamy, M. M. Reddy, M. Misra and A. Mohanty, Ind. Crops Prod., 2013, 43, 812–819 CrossRef CAS.
- A. M. Ismail and M. A. B. Gamal, J. Appl. Polym. Sci., 2010, 115, 2813–2819 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |