
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrends in activity for the oxygen evolution reaction on transition metal (M = Fe, Co, Ni) phosphide pre-catalysts†
Junyuan
Xu
 a,
Junjie
Li
a,
Dehua
Xiong
a,
Bingsen
Zhang
b,
Yuefeng
Liu
b,
Kuang-Hsu
Wu‡
b,
Isilda
Amorim
a,
Wei
Li
a and
Lifeng
Liu
*a
a,
Junjie
Li
a,
Dehua
Xiong
a,
Bingsen
Zhang
b,
Yuefeng
Liu
b,
Kuang-Hsu
Wu‡
b,
Isilda
Amorim
a,
Wei
Li
a and
Lifeng
Liu
*a
aInternational Iberian Nanotechnology Laboratory, Av. Mestre Jose Veiga, 4715-330 Braga, Portugal. E-mail: lifeng.liu@inl.int
bShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang, Liaoning 110016, China
First published on 5th March 2018
Abstract
Transition metal phosphides (TMPs) have recently emerged as a new class of pre-catalysts that can efficiently catalyze the oxygen evolution reaction (OER). However, how the OER activity of TMPs varies with the catalyst composition has not been systematically explored. Here, we report the alkaline OER electrolysis of a series of nanoparticulate phosphides containing different equimolar metal (M = Fe, Co, Ni) components. Notable trends in OER activity are observed, following the order of FeP < NiP < CoP < FeNiP < FeCoP < CoNiP < FeCoNiP, which indicate that the introduction of a secondary metal(s) to a mono-metallic TMP substantially boosts the OER performance. We ascribe the promotional effect to the enhanced oxidizing power of bi- and tri-metallic TMPs that can facilitate the formation of MOH and chemical adsorption of OH− groups, which are the rate-limiting steps for these catalysts according to our Tafel analysis. Remarkably, the tri-metallic FeCoNiP pre-catalyst exhibits exceptionally high apparent and intrinsic OER activities, requiring only 200 mV to deliver 10 mA cm−2 and showing a high turnover frequency (TOF) of ≥0.94 s−1 at the overpotential of 350 mV.
Introduction
The oxygen evolution reaction (OER) is a half-cell reaction that is essential for a number of electrochemical devices such as water electrolyzers and metal–air batteries.1,2 The OER involves four concerted proton-coupled electron transfer steps, and is both thermodynamically and kinetically demanding. Without a catalyst, the OER usually takes place at a high overpotential leading to a large energy loss. Presently, ruthenium (Ru)/iridium (Ir)-based materials are the state-of-the-art OER catalysts, which show excellent catalytic activity in both acidic and alkaline solutions.3,4 However, Ru and Ir are precious and among the rarest metals in the Earth's crust, and hence practically they cannot be employed on a large scale. Moreover, recent studies have pointed out that both RuO2 and IrO2 suffer from dissolution under high anodic potentials and exhibit poor long-term operational stability.5–7 To enable widespread deployment of electrochemical energy conversion devices, in particular water electrolyzers, developing efficient and durable OER catalysts comprising low-cost earth-abundant elements is becoming a pressing need and has drawn considerable attention in recent years.Lately, metal non-oxides including carbide,8,9 chalcogenide,10–13 nitride,14,15 and phosphide16–21 have been extensively investigated as alternatives to the conventional metal oxide OER catalysts. Although an in-depth molecular-level understanding of the OER process taking place on TMP catalysts currently still remains out of reach, it is generally believed that the metalloid nature of TMP together with the metal-oxo/hydroxo species in situ formed on TMP under OER conditions synergistically contribute to the enhanced oxygen evolution. In this sense, TMP is a good pre-catalyst for OER, and it has been proven to be more active, in many cases, than the corresponding oxide/hydroxide containing the same TM species, likely due to the fact that more catalytically active sites can be exposed during the electrochemical coldworking of TMP during the OER.22 Using TMP as OER pre-catalysts, a benchmark anodic current density of 10 mA cm−2 has been achieved at an overpotential (η) as low as 200–300 mV in concentrated alkaline solution (e.g. 1.0 M KOH),21,23–25 and this outperforms many non-precious OER catalysts reported recently and favorably compares to RuO2/IrO2 noble metal catalysts. At present, many researches on TMP pre-catalysts are focused on nanostructure engineering with an aim to expose more catalytically active sites to improve the catalytic activity.21,26–29 Besides, doping TMP with a secondary transition metal (TM) is also an effective approach to enhancing the catalytic performance. Bi-metallic TM(A+B)P pre-catalysts with an optimal atomic ratio of metal A over metal B (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in most previous reports), e.g. nano-porous (Co0.52Fe0.48)2P,18 Ni0.51Co0.49P films,19 NiCoP nano-plates,20 FeCoP nanoarrays,23 and NiCoP/GO nanoparticles,24 have demonstrated OER performance much better than that of the mono-metallic counterparts (i.e. TMAP and TMBP). Furthermore, η10 of tri-metallic Fe10Co40Ni40P was reported to be small as 250 mV in 1.0 M KOH, substantially outperforming other mono- or bi-metallic TMP catalysts tested under the same conditions.30
:1 in most previous reports), e.g. nano-porous (Co0.52Fe0.48)2P,18 Ni0.51Co0.49P films,19 NiCoP nano-plates,20 FeCoP nanoarrays,23 and NiCoP/GO nanoparticles,24 have demonstrated OER performance much better than that of the mono-metallic counterparts (i.e. TMAP and TMBP). Furthermore, η10 of tri-metallic Fe10Co40Ni40P was reported to be small as 250 mV in 1.0 M KOH, substantially outperforming other mono- or bi-metallic TMP catalysts tested under the same conditions.30
Notwithstanding remarkable progress, the intrinsic electrocatalytic activity of TMP pre-catalysts, reflected by the turnover frequency (TOF), remains low. For instance, assuming all metal cations are catalytically active, the TOF value calculated for hierarchically porous urchin-like Ni2P – one of the most active TMP OER catalysts reported by far (with a η10 as low as 200 mV), is only 1.5 × 10−2 s−1 at η = 350 mV.21 Moreover, how the number of TM components (i.e. mono-metallic, bi-metallic, or tri-metallic) and their specific combinations (i.e. FeCo, FeNi, CoNi, or FeCoNi) affect the OER activity of TMP pre-catalysts has not been systematically explored yet. Herein, we report the fabrication and electrocatalytic OER performance of a series of TM (TM = Fe, Co, and Ni) monophosphide nanoparticles (NPs) supported on carbon nanofibers (CNFs) for use as pre-catalysts. We have studied the influence of catalyst composition on both the apparent and intrinsic OER activities, which shows a clear ascending trend as the number of TM species in TMPs increases. Remarkably, the tri-metallic phosphide FeCoNiP exhibits the best OER performance among others, requiring only an overpotential of 200 mV to deliver 10 mA cm−2 (with a TM loading of merely 0.06 mg cm−2) and showing a high TOF value of ≥0.94 s−1 at η = 350 mV. This is by far the best OER activity achieved by TMP based pre-catalysts.
Results and discussion
The TMP pre-catalysts were prepared by chemical reduction of the corresponding TM cations in the presence of sodium borohydride (NaBH4) and CNFs in ethylene glycol (EG) solution, followed by a post-phosphorization treatment at 300 °C using sodium hypophosphite (NaH2PO2) as the source of phosphorus. To ensure the deposition of TM NPs on CNFs, the CNFs were first pre-treated in acid to make the surface hydrophilic. Seven phosphides with different TM components were obtained, which are denoted as FeP, NiP, CoP, FeNiP, FeCoP, CoNiP, and FeCoNiP, respectively. For bi- and tri-metallic catalysts, only TMPs having equimolar TM components (i.e. with an atomic ratio of 1:1 or 1:1:1) were investigated, given that TMPs with such a composition turned out to be most active for OER as demonstrated by recent theoretical and experimental studies about TMP-based catalysts.18–20,23,31 Our inductively coupled plasma mass spectroscopy (ICP-MS) analyses confirmed that the atomic ratios of TM components in bi- and tri-metallic phosphide pre-catalysts are close to 1:1 and 1:1:1, respectively, and the total loading of TM species in each TMP sample is ca. 20 wt% (Fig. S1, ESI†).
The morphology and microstructure of all TMP pre-catalysts were examined by transmission electron microscopy (TEM). After acid-treatment, most CNFs retained the hollow tubular structure, though the graphitic layers of the fiber walls became distorted and some CNFs were partially unzipped (Fig. S2, ESI†). The TMP NPs were found to load on both the inner and outer walls of CNFs with a high density. These NPs are highly crystallized and have a typical diameter of 3–6 nm (Fig. 1 and S3–S8†). Owing to the small crystallite size, the X-ray diffraction (XRD) patterns of all TMP pre-catalysts either display broad humps or are featureless without any resolvable diffraction peaks (Fig. S9, ESI†). Fig. 1a shows a representative low-magnification TEM image of FeCoNiP, where dense distribution of NPs on CNF are clearly distinguished, in stark contrast to the bare CNF support (Fig. S2, ESI†). Although the standard powder diffraction crystallographic data of Fe–Co–Ni–P compounds are not available, our extensive high-resolution TEM (HRTEM) imaging and fast Fourier-transform electron diffraction (FFT-ED) analyses indicated that these FeCoNiP NPs may adopt an orthorhombic structure with lattice constants of a = 5.6046 Å, b = 3.4899 Å, and c = 6.4018 Å (Fig. 1b), similar to that of Fe1.4Co0.6P (ICDD no. 04-014-4290, a = 5.7716 Å, b = 3.5431 Å, and c = 6.6145 Å). Examinations were also carried out in the high-angle annual dark-field scanning transmission electron microscopy (HAADF-STEM) mode, where the nanoparticulate feature of FeCoNiP and its distribution on CNF can be seen more clearly (Fig. 1c). The elemental mapping result manifests that Fe, Co, Ni, and P elements all cover the CNF surface uniformly, as evidenced in Fig. 1d–h. TEM investigations of mono- and bi-metallic TMP pre-catalysts were performed, and the results are shown in Fig. S3–S8 (ESI†).
 | ||
| Fig. 1 Morphology and microstructure of FeCoNiP pre-catalysts. (a) TEM image, (b) HRTEM image. Insets: particle size distribution, zoomed view of a single FeCoNiP NP and the corresponding FFT-ED pattern. (c) STEM-HAADF image. (d–h) Elemental maps of C, Fe, Co, Ni and P. | ||
The electrocatalytic activity of the TMP pre-catalysts towards the OER was investigated in 1.0 M O2-saturated KOH using cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). The catalyst ink was prepared by dispersing 5 mg of pre-catalyst powders in a mixture of 1 mL ethanol and 50 μL Nafion solution. 50 μL of ink was then cast on a glassy carbon (GC) electrode, leading to a loading of ca. 0.3 mg cm−2 for each pre-catalyst (TM loading: ca. 0.06 mg cm−2). Prior to the catalytic test, pre-activation was carried out by repetitive CV scans at 5 mV s−1 in the potential range of 1.0–1.6 V vs. reversible hydrogen electrode (RHE) until a steady state CV curve was obtained. Fig. 2a shows the cathodic branches of iR-corrected CV curves of all samples after pre-activation (Fig. S10, ESI†). The η value needed at a specific anodic current density has been broadly used as an extrinsic performance indicator of OER catalysts.32,33 The η values needed for all TMP pre-catalysts to deliver 10, 20, 50, and 100 mA cm−2 (i.e. η10, η20, η50, and η100) are compared in Fig. 2b. From the polarization curves, several notable trends have been observed: (i) η value decreases as the number of TM species in TMPs increases, namely, η (mono-metallic TMP) > η (bi-metallic TMP) > η (tri-metallic TMP); (ii) in the low η region (e.g. η < 350 mV), the apparent OER activity follows the order: FeP < NiP < CoP < FeNiP < FeCoP < CoNiP < FeCoNiP (Fig. 2b). To reach the benchmark current density of 10 mA cm−2, FeP needs a η value of 325 mV, while FeCoNiP merely demands a η value of 200 mV, showing a significant improvement in OER activity. Even to afford a high current density of 100 mA cm−2, η100 of FeCoNiP is as low as 270 mV. More significantly, a superior mass activity of 5000 mA mgFeCoNi−1 can be obtained at η = 330 mV (Fig. S11, ESI†); (iii) in the high η region (e.g. η > 400 mV), the trend (i) is still valid, but the order in the apparent OER activity has changed to FeP < CoP < NiP < FeCoP < FeNiP < CoNiP < FeCoNiP. From the above observations, we conclude that for the phosphide pre-catalysts with the same arity of TM species, the TMPs containing Fe are the least active for OER, e.g. for mono-metallic TMPs, FeP is not as active as NiP and CoP, and for bi-metallic TMPs, the activity of FeNiP and FeCoP is lower than that of CoNiP; having Co in TMPs helps reduce η in the low overpotential region, as evidenced by the activity trend of FeP < NiP < CoP for mono-metallic TMPs, and of FeNiP < FeCoP < CoNiP for bi-metallic TMPs; while having Ni in TMPs is beneficial for boosting the anodic current in the high overpotential region, as illustrated by the change in activity trend shown above, i.e. FeP < CoP < NiP for mono-metallic TMPs and FeCoP < FeNiP < CoNiP for bi-metallic TMPs. In the whole overpotential range under investigation, tri-metallic FeCoNiP showed the best OER performance, outperforming most non-precious OER electrocatalysts reported recently (Table S1, ESI†) as well as the state-of-the-art commercial RuO2 NPs (η10 = 276 mV, Fig. S12, ESI†).
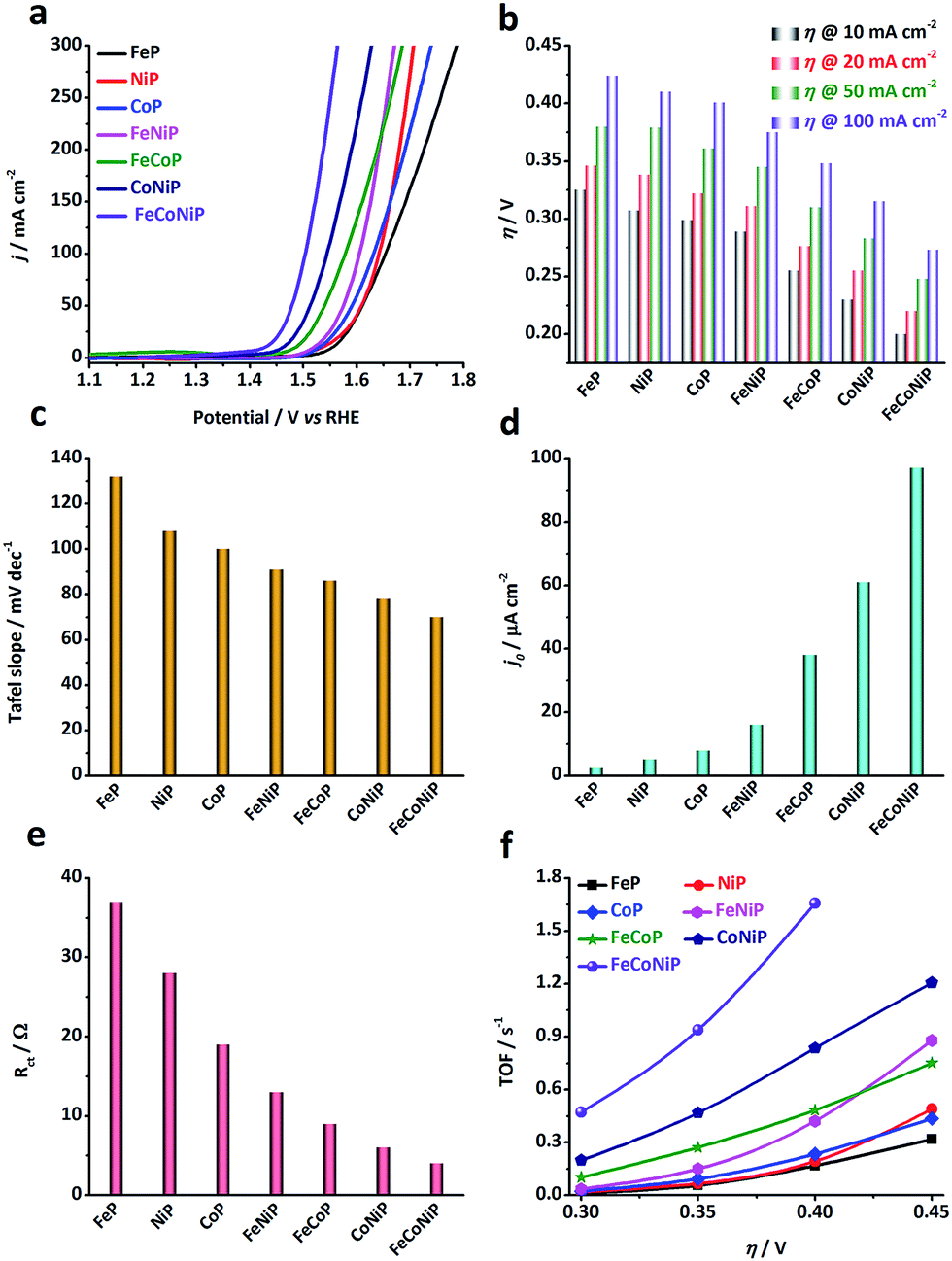 | ||
| Fig. 2 OER performance of different TMP pre-catalysts measured in 1.0 M O2-saturated KOH electrolyte. (a) iR-corrected polarization curves (i.e. the cathodic branches of the CV curves shown in Fig. S10, ESI†) of the TMP pre-catalysts, recorded at a scan rate of 5 mV s−1 in the potential range of 1.0 to 1.8 V vs. RHE. (b) The overpotentials needed to deliver anodic current densities of 10, 20, 50 and 100 mA cm−2 for all TMP pre-catalysts. (c) Tafel slopes, (d) exchange current densities, and (e) charge transfer resistance (Rct) values measured at 1.45 V vs. RHE. (f) TOF values calculated at η = 300, 350, 400 and 450 mV. | ||
The OER kinetics of TMP pre-catalysts was studied by the Tafel analysis. Interestingly, the Tafel slope reduces as the number of TM species in the TMPs increases, following the same order as observed in the low η region, i.e. FeP > NiP > CoP > FeNiP > FeCoP > CoNiP > FeCoNiP (Fig. 2c and S13†). The Tafel slope of FeP is 132 mV dec−1, indicating that the discharge of OH− was the rate-limiting step for FeP; while the Tafel slope of the other TMP pre-catalysts falls in the range of 60–120 mV dec−1, which implies that chemical adsorption of OH− on the pre-catalyst might limit the reaction rate.34 The exchange current density (j0) is a measure of the intrinsic kinetic rate of a reaction, and a high j0 is usually an indication of a good OER electrocatalyst.5,34 In consistence with the OER activity trends observed, j0 of bi- and tri-metallic TMP pre-catalysts is significantly higher than that of mono-metallic TMP pre-catalysts (Fig. 2d), suggesting that the OER kinetics is substantially promoted in mixed TMPs. This was further corroborated by EIS analyses performed at 1.45 V vs. RHE (Fig. 2e and S14†), where the charge transfer resistance (Rct) values of all TMP pre-catalysts follow the same order as that of η, the Tafel slope and j0, confirming that the bi- and tri-metallic TMP pre-catalysts show remarkably improved charge transfer kinetics during the OER, compared to the mono-metallic counterparts. In addition, the equivalent series resistance (Rs), which to a certain extent reflects the ohmic resistance of TMPs, of each pre-catalyst was also extracted from the EIS Nyquist plot and compared in Fig. S14.† The Rs values of all TMPs show the following trend in a narrow range of 0.6–0.7 Ω: Rs (mono-metallic TMP) > Rs (bi-metallic TMP) > Rs (tri-metallic TMP). This indicates that the introduction of secondary TM species may enhance the electrical conductivity of TMP materials.
The electrochemical double-layer capacitance (Cdl) of each TMP pre-catalyst was calculated using CV in the non-faradaic region (Fig. S15 and S16, ESI†).32 Although it is challenging to precisely estimate the number of catalytically active sites, Cdl provides the information about the electrocatalytically accessible surface area which correlates with the number of active sites.32 As shown in Fig. S16,† the introduction of a secondary TM(s) helps to improve Cdl. For instance, Cdl of FeNiP is higher than that of both FeP and NiP, and Cdl of FeCoNiP is superior to that of all bi-metallic TMPs.
However, since the Cdl value of a smooth surface for each TMP catalyst is unknown, it is impossible to calculate the specific activity of TMPs to appraise their intrinsic activity. Therefore, we assess the intrinsic OER activity of TMPs using TOF, assuming all TM species in TMPs are catalytically active (i.e. the lower limit). Fig. 2f shows the TOF values of all the TMP pre-catalysts at η = 300, 350, 400 and 450 mV, respectively. The trend in TOF for all TMP pre-catalysts is found to be the same as that observed for the apparent OER activity (Fig. 2b), indicating that the promotional effect of bi- and tri-metallic phosphides on OER is an intrinsic property. Remarkably, the TOF value of FeCoNiP can be as high as 0.47 and 0.94 s−1 at η = 300 and 350 mV, respectively, significantly higher than that of ultrathin CoNiP nanosheet (0.028 s−1),16 NiCo–porphyrin (0.12 s−1),35 NiFeOx/CFP nanoparticle (0.012 s−1),36 nano-porous Ni0.75V0.25-LDH nanosheet (0.0216 s−1)37 and NiCo-UMOFNs nanosheet (0.18 s−1)38 at η = 300 mV, making it rank among the most active non-precious OER electrocatalysts reported in the literature (Table S1, ESI†).
In order to elucidate the impact of CNF supports on the OER activity, supportless FeCoNiP NPs were synthesized using the same solution-based chemical reduction method in the absence of CNFs, followed by a phosphorization treatment under the same conditions as those used to synthesize other TMP pre-catalysts. These supportless FeCoNiP NPs are crystallized, but have much larger particle size (20–40 nm) than that of CNF-supported FeCoNiP (Fig. S17a–c†). The OER performance of the supportless FeCoNiP as well as bare CNF powders (CNF-P, i.e. CNFs subjected to acid-treatment and phosphorization) was compared to that of supported FeCoNiP pre-catalysts. The CNF-P only generated negligible anodic current up to 1.8 V vs. RHE, indicating that it is catalytically inactive for OER and that the performance observed in FeCoNiP pre-catalysts should primarily originate from the tri-metallic phosphide (Fig. S17d†). This was confirmed by the electrocatalytic measurement of supportless FeCoNiP, which only required 268 mV to deliver 10 mA cm−2, showing high OER activity. EIS study showed that the CNF-supported FeCoNiP has much smaller Rct (4 Ω), compared to the supportless FeCoNiP (Rct = 11 Ω), indicating that the CNF supports can help improve charge transport leading to fast charge transfer kinetics (Fig. S17e†). Furthermore, Cdl of the CNF-supported FeCoNiP was calculated to be 30 mF cm−2 (Fig. S17f and g†), substantially higher than that of supportless FeCoNiP (17 mF cm−2). This indicates that the CNF supports can offer many nucleation sites during the wet chemical reduction of metal cations, allowing for the formation of FeCoNiP NPs with very small particle sizes (few nm) and thereby being able to provide more catalytically active sites. Similar roles of carbon-based supports were also reported previously in carbon nanotube supported NiFe layered double hydroxide OER catalysts.39
In the past few decades, considerable efforts have been devoted to rationalizing the OER activity of different catalytic materials based on a given “descriptor”. The “d-band” model and the orbital occupancy theory have successfully accounted for the trends in OER activity for transition metals/alloys and perovskite transition metal oxides, respectively. Recent studies on the correlation between the outer electrons and the OER activity of metal oxides further suggest that the number of valence electrons is likely the hidden parameter behind all successful OER descriptor models proposed by far.34,40 However, all these approaches are subject to the limitation of the scaling relations, predicting a minimum η value of 370 ± 20 mV to drive the OER.5,34
It was recently demonstrated that developing mixed TM oxides or doping a secondary TM element into a metal oxide is an effective way of breaking the thermodynamic limitation of the scaling relations,41 which may lead to a substantial enhancement in OER activity. The introduction of a secondary TM(s) species into a metal oxide would alter its electronic structure that essentially determines its catalytic properties.34 Having this in mind and considering that the electrocatalytic activity usually only occurs in the near-surface region of catalysts, we have investigated all TMP pre-catalysts using X-ray photoelectron spectroscopy (XPS). Fig. 3a–c present the core level XPS spectra of Fe 2p3/2, Co 2p3/2 and Ni 2p3/2. A binding energy (BE) shift towards a higher BE value has been observed for FeCoNiP relative to bi-metallic TMPs as well as for bi-metallic TMPs relative to mono-metallic TMPs. More interestingly, according to the P 2p spectrum (Fig. 3d), the TM–P peaks located at ca. 130 eV show a regular red shift in BE values for all TMP pre-catalysts, and the BE values decrease following the order FeP > NiP > CoP > FeNiP > FeCoP > CoNiP > FeCoNiP, in consistence with that observed for OER activity. This suggests that partial electron transfer from TM cations to P is enhanced as a secondary metal(s) is introduced into the TMP, and thereby gives rise to the formation of high-valence-state TM species with more oxidizing power. As indicated by our Tafel analysis, the rate-limiting step for all TMP pre-catalysts except FeP may be a chemical step after the first electron transfer step, described as follows (Krasil'shchikov's path 2):34
| MOH + OH− → MO− + H2O | (1) |
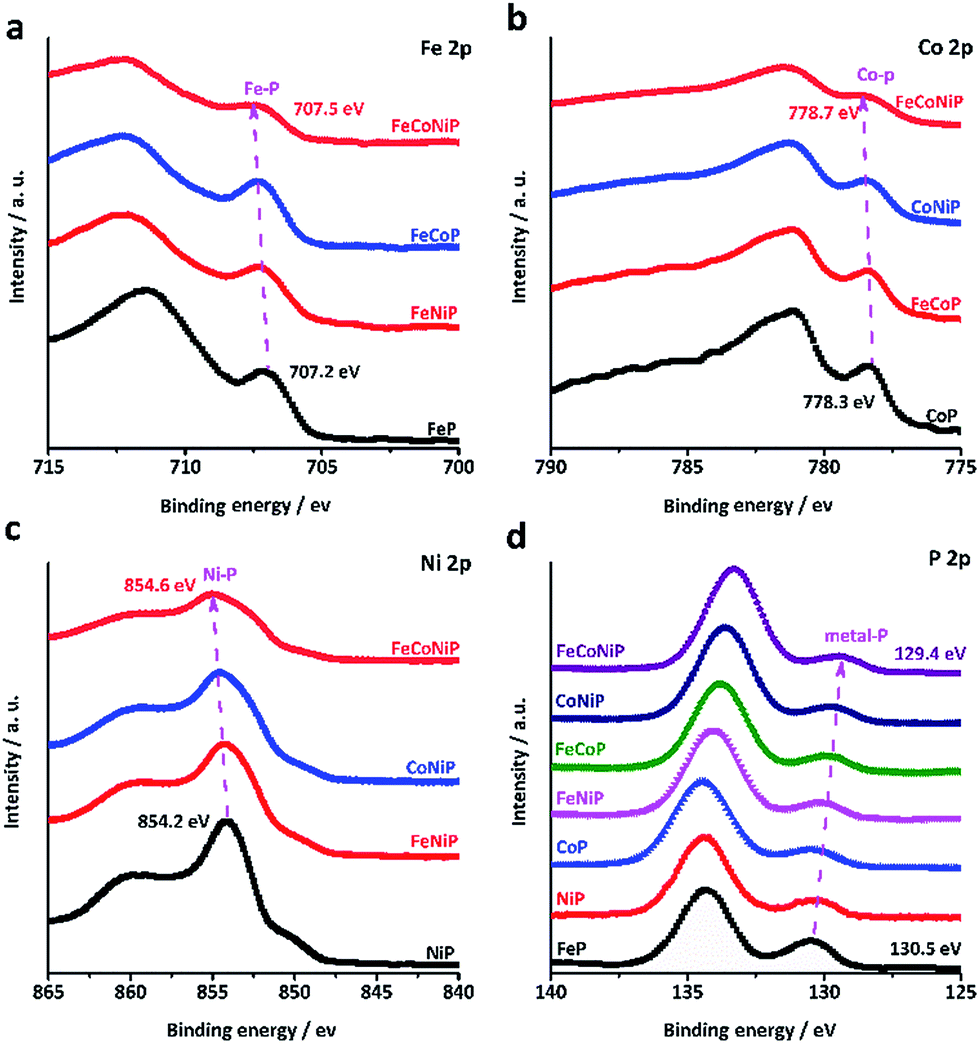 | ||
| Fig. 3 High resolution core level XPS spectra of (a) Fe 2p3/2, (b) Co 2p3/2, (c) Ni 2p3/2 and (d) P 2p of the TMP pre-catalysts. | ||
This chemical adsorption process can be facilitated by the presence of high-valence-state TM species due to the enhanced local electrostatic interaction between metal cations and OH− groups, leading to fast OER kinetics, as evidenced by the trend in the Tafel slope (Fig. 2c). This reasonably explains why the OER activity and the TM–P BE shift of TMP pre-catalysts consistently follow the same trend. In other words, the observed trend in OER activity to a large extent reflects the electronic structure change in TMP as the number of TM components changes. The unique electronic structure effect of multi-metallic TMPs on the OER activity was further verified by comparing the OER performance of tri-metallic FeCoNiP to that of physical mixtures of CoNiP + FeP, FeCoP + NiP and FeNiP + CoP (Fig. S18, ESI†). Both the apparent OER activity and TOF value of FeCoNiP pre-catalysts are remarkably higher than those of the physically mixed TMPs, suggesting that the intrinsic synergy among different TM species in FeCoNiP indeed plays a determinative role in the enhanced OER activity.
The stability of all TMP pre-catalysts was evaluated using chronopotentiometry (CP) at a constant current density of 10 mA cm−2, as shown in Fig. 4. The η value needed to maintain 10 mA cm−2 decreases gradually in the first 2–3 h and then gets stabilized up to at least 24 h, exhibiting excellent long-term stability. The initial potential drop indicates that an activation process exists for all TMP pre-catalysts. In fact, electrochemical in situ de-phosphorization/oxidation has recently been reported in many non-oxide based OER catalysts, and it is believed to help promote the OER performance, as more electrocatalytic active sites would likely be exposed during the electrochemical coldworking.12,13,18,20,27,42,43 We have examined the microstructure and composition of the FeCoNiP pre-catalysts after the activation (Fig. S19†), i.e. after repetitive CV scans at 5 mV s−1 in the potential range of 1.0–1.6 V vs. RHE for 30 cycles. The results showed that densely distributed NPs still retained on the surface of CNFs after activation, but the P content was remarkably decreased, indicating that the initial phosphide was largely converted to (oxy)hydroxide during the activation. Notwithstanding the composition change, the OER performance remained excellent during the whole long-term stability test. Furthermore, we also investigated the chemical states of FeCoNiP pre-catalysts after the OER electrolysis for 2 and 24 h, respectively, using XPS (Fig. S20†). A blue shift in binding energy was observed in the Fe, Co and Ni XPS spectra for both post-OER samples, implying that the valence states of the catalysts became higher upon OER,44 compared to those of the as-prepared FeCoNiP pre-catalysts. Besides, the faradaic efficiency of FeCoNiP pre-catalysts was monitored as a function of OER electrolysis time at a constant current density of 50 mA cm−2, and an efficiency close to 100% was obtained (Fig. S21, ESI†).
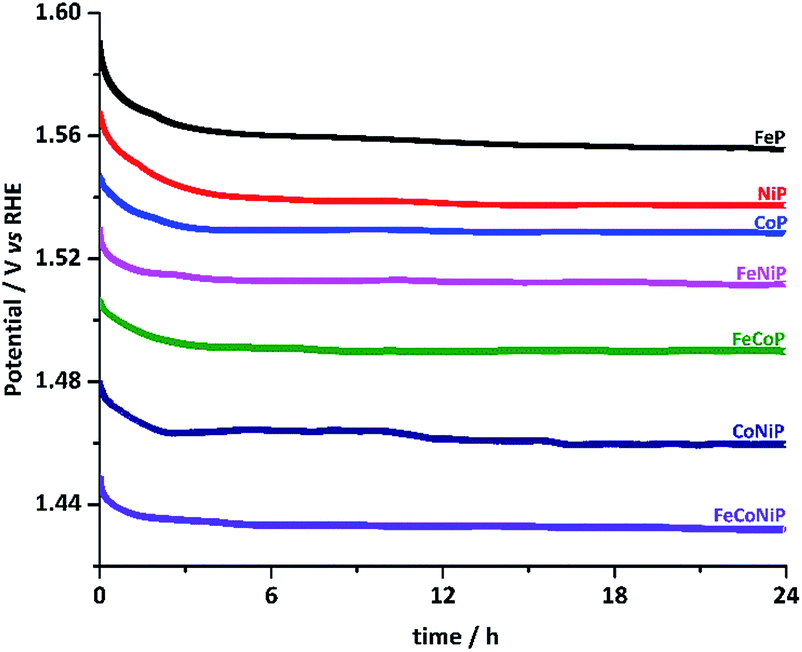 | ||
| Fig. 4 Chronopotentiometric curves for TMP pre-catalysts recorded at a constant current density of 10 mA cm−2. | ||
Conclusions
In summary, we have synthesized a series of transition metal phosphide pre-catalysts with different compositions on carbon nanofiber supports using a simple and cost-effective solution-based chemical reduction method followed by a post-phosphorization treatment. We have systematically investigated the influence of the pre-catalyst composition on the oxygen evolution activity, and observed prominent promotional effect in bi- and tri-metallic TMPs. The trend of TMP pre-catalysts in OER activity is in good agreement with that in chemical shift of TM–P bonds, indicating that the electronegativity plays a determinative role in the OER performance of metal monophosphide pre-catalysts. Remarkably, the tri-metallic FeCoNiP pre-catalysts exhibit both high apparent and intrinsic OER activities, requiring merely 200 mV to afford a benchmark current density of 10 mA cm−2 with a pretty low mass loading and showing a high TOF value of ≥0.94 s−1 at the overpotential of 350 mV. Moreover, they also exhibit excellent long-term stability and can sustain galvanostatic OER electrolysis for 24 hours without degradation. Our research provides important insight into the activity trend of metal phosphide pre-catalysts and offers useful design guidelines for this specific class of earth-abundant OER pre-catalysts. It is worth mentioning that the simple fabrication procedures and the carbon supported structure compatible with industrial electrocatalysts will enable the TMP pre-catalysts we report here to find practical applications in electrochemical energy conversion devices where the OER is involved.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the European Horizon 2020 project “CritCat” under the grant agreement number 686053. L. F. L. acknowledges the financial support from the Portuguese Foundation of Science and Technology (FCT) under the projects “IF/01595/2014” and “PTDC/CTM-ENE/2349/2014” (grant agreement No. 016660).Notes and references
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and S. H. Yang, Energy Environ. Sci., 2015, 8, 1404–1427 CAS.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- M. Huynh, T. Ozel, C. Liu, E. C. Lau and D. G. Nocera, Chem. Sci., 2017, 8, 4779–4794 RSC.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- N. Hodnik, P. Jovanovic, A. Pavlisic, B. Jozinovic, M. Zorko, M. Bele, V. S. Selih, M. Sala, S. Hocevar and M. Gaberscek, J. Phys. Chem. C, 2015, 119, 10140–10147 CAS.
- E. Antolini, ACS Catal., 2014, 4, 1426–1440 CrossRef CAS.
- T. Y. Ma, J. L. Cao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 1138–1142 CrossRef CAS PubMed.
- K. Xu, H. Ding, H. F. Lv, P. Z. Chen, X. L. Lu, H. Cheng, T. P. Zhou, S. Liu, X. J. Wu, C. Z. Wu and Y. Xie, Adv. Mater., 2016, 28, 3326–3332 CrossRef CAS PubMed.
- H. F. Wang, C. Tang, B. Wang, B. Q. Li and Q. Zhang, Adv. Mater., 2017, 29, 1702327 CrossRef PubMed.
- L. L. Feng, G. T. Yu, Y. Y. Wu, G. D. Li, H. Li, Y. H. Sun, T. Asefa, W. Chen and X. M. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS PubMed.
- A. T. Swesi, J. Masud and M. Nath, Energy Environ. Sci., 2016, 9, 1771–1782 CAS.
- C. Tang, N. Y. Cheng, Z. H. Pu, W. Xing and X. P. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351–9355 CrossRef CAS PubMed.
- K. Xu, P. Z. Chen, X. L. Li, Y. Tong, H. Ding, X. J. Wu, W. S. Chu, Z. M. Peng, C. Z. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed.
- F. Yu, H. Q. Zhou, Z. Zhu, J. Y. Sun, R. He, J. M. Bao, S. Chen and Z. F. Ren, ACS Catal., 2017, 7, 2052–2057 CrossRef CAS.
- X. F. Xiao, C. T. He, S. L. Zhao, J. Li, W. S. Lin, Z. K. Yuan, Q. Zhang, S. Y. Wang, L. M. Dai and D. S. Yu, Energy Environ. Sci., 2017, 10, 893–899 CAS.
- G. Zhang, G. C. Wang, Y. Liu, H. J. Liu, J. H. Qu and J. H. Li, J. Am. Chem. Soc., 2016, 138, 14686–14693 CrossRef CAS PubMed.
- Y. W. Tan, H. Wang, P. Liu, Y. H. Shen, C. Cheng, A. Hirata, T. Fujita, Z. Tang and M. W. Chen, Energy Environ. Sci., 2016, 9, 2257–2261 CAS.
- J. Yu, Q. Q. Li, Y. Li, C. Y. Xu, L. Zhen, V. P. David and J. S. Wu, Adv. Funct. Mater., 2016, 26, 7644–7651 CrossRef CAS.
- H. F. Liang, A. N. Gandi, D. H. Anjum, X. B. Wang, U. Schwingen-schlögl and H. N. Alshareef, Nano Lett., 2016, 16, 7718–7725 CrossRef CAS PubMed.
- B. You, N. Jiang, M. L. Sheng, M. W. Bhushan and Y. J. Sun, ACS Catal., 2016, 6, 714–721 CrossRef CAS.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef.
- C. Tang, R. Zhang, W. B. Lu, L. B. He, X. Jiang, A. M. Asiri and X. P. Sun, Adv. Mater., 2017, 29, 1602441 CrossRef PubMed.
- J. Y. Li, M. Yan, X. M. Zhou, Z. Q. Huang, Z. M. Xia, C. R. Chang, Y. Y. Ma and Y. Q. Qu, Adv. Funct. Mater., 2016, 26, 6785–6796 CrossRef CAS.
- H. W. Huang, C. Yu, C. T. Zhao, X. T. Han, J. Yang, Z. B. Liu, S. F. Li, M. D. Zhang and J. S. Qiu, Nano Energy, 2017, 34, 472–480 CrossRef CAS.
- Y. P. Zhu, Y. P. Liu, T. Z. Ren and Z. Y. Yuan, Adv. Funct. Mater., 2015, 25, 7337–7347 CrossRef CAS.
- L. A. Stern, L. G. Feng, F. Song and X. L. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 CAS.
- M. Ledendecker, S. K. Calderon, C. Papp, H. P. Steinruck, N. Anto-nietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- Y. Yan, B. Y. Xia, X. M. Ge, Z. L. Liu, A. Fisher and X. Wang, Chem.–Eur. J., 2015, 21, 18062–18067 CrossRef CAS PubMed.
- Z. Zhang, J. H. Hao, W. S. Yang and J. L. Tang, RSC Adv., 2016, 6, 9647–9655 RSC.
- D. Li, H. Baydoun, C. N. Verani and S. L. Brock, J. Am. Chem. Soc., 2016, 138, 4006–4009 CrossRef CAS PubMed.
- C. C. L. McCrory, S. H. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- J. S. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- R. L. Doyle and M. E. G. Lyons, Photoelectrochemical Solar Fuel Production, Springer, 2016, ch. 2, pp. 41–104 Search PubMed.
- J. Q. Sun, H. j. Yin, P. R. Liu, Y. Wang, X. D. Yao, Z. Y. Tang and H. J. Zhao, Chem. Sci., 2016, 7, 5640–5646 RSC.
- H. T. Wang, H. W. Lee, Y. Deng, Z. Y. Lu, P. C. Hsu, Y. Y. Liu, D. C. Lin and Y. Cui, Nat. Commun., 2015, 6, 7261 CAS.
- K. Fan, H. Chen, Y. F. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. S. Li, Y. Luo and L. C. Sun, Nat. Commun., 2016, 7, 11981 CrossRef CAS PubMed.
- S. L. Zhao, Y. Wang, J. C. Dong, C. T. He, H. J. Yin, P. F. An, K. Zhao, X. F. Zhang, C. Gao, L. J. Zhang, J. W. Lv, J. Q. Liu, A. M. Khattak, N. A. Khan, Z. W. Wei, J. Zhang, S. Q. Liu, H. J. Zhao and Z. Y. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- M. Gong, Y. G. Li, H. L. Wang, Y. Y. Liang, J. Z. Wu, J. G. Zhou, J. Wang, T. Regier, F. Wei and H. J. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- F. Calle-Vallejo, N. G. Inoglu, H. Y. Su, J. I. Martinez, I. C. Man, M. I. M. Koper, J. R. Kitchin and J. Rossmeisl, Chem. Sci., 2013, 4, 1245–1249 RSC.
- N. B. Halck, V. Petrykin, P. Krtil and J. Rossmeisl, Phys. Chem. Chem. Phys., 2014, 16, 13682–13688 RSC.
- J. Y. Xu, X. K. Wei, J. D. Costa, J. L. Lado, B. Owens-Bair, L. P. L. Goncalves, S. P. S. Fernandes, M. Heggen, D. Y. Petrovykh, R. E. Dunin-Borkowski, K. Kovnir and Y. V. Kolen'ko, ACS Catal., 2017, 7, 5450–5455 CrossRef CAS.
- K. Xu, H. Cheng, L. Q. Liu, H. F. Lv, X. J. Wu, C. Z. Wu and Y. Xie, Nano Lett., 2017, 17, 578–583 CrossRef CAS PubMed.
- M. Favaro, W. S. Drisdell, M. A. Marcus, J. M. Gregoire, E. J. Crumlin, J. A. Haber and J. Yano, ACS Catal., 2017, 7, 1248–1258 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05033j |
| ‡ Present address: School of Chemical Engineering, University of New South Wales, Sydney NSW 2052, Australia. |
| This journal is © The Royal Society of Chemistry 2018 |