
Uranium(VI) complexes with isonicotinic acid: from monomer to 2D polymer with unique U–N bonding†
Yingjie Zhang*a,
Inna Karatchevtsevaa,
Jason R. Priceb,
Igor Aharonovichc,
Fatima Kadia,
Gregory R. Lumpkina and
Feng Lid
aAustralian Nuclear Science and Technology Organisation, Locked Bag 2001, Kirrawee DC, NSW 2232, Australia. E-mail: yingjie.zhang@ansto.gov.au
bAustralian Synchrotron, 800 Blackburn Road, Clayton, VIC 3168, Australia
cSchool of Physics and Advanced Materials, University of Technology Sydney, Ultimo, New South Wales 2007, Australia
dSchool of Science and Health, University of Western Sydney, Locked Bag 1797, Penrith, NSW 2751, Australia
First published on 2nd April 2015
Abstract
Two new uranium(VI) complexes with isonicotinic acid (HINT) have been synthesized and characterized. [(UO2)(NO3)2(HINT)2] (1) has a monomeric structure constructed of a hexagonal bipyramidal uranyl centre, two nitrate anions and two monodentate HINT in trans-positions. [(UO2)(OH)(INT)] (2) has a two-dimensional (2D) polymeric structure constructed of uranyl hydroxyl 1D pillars and μ3-bridging INT anions; the first observation of INT in μ3-bridging mode for U(VI) ion via U–N bonding. Thermal analysis confirmed both complexes lost coordinated INT ligands followed by further decomposition to form U3O8. Raman spectroscopy has confirmed the presence of uranyl ion and INT ligand in both complexes as well as the existence of nitrate vibrations in 1 and hydroxyl vibrations in 2. Their photoluminescence properties have been investigated.
Introduction
Metal–organic complexes and hybrid materials with transition metals and lanthanide ions have been extensively studied in the last fifteen years mainly due to their structural diversity and potential industrial applications.1 Such work on uranium(VI) is also important due to its relevance to the nuclear fuel cycle and the potential impact on the environment. For example, uranyl [U(VI)] complexes are generally soluble and are the major species involved in the migration processes and the formation of U(VI) complexes with environmentally relevant organic ligands such as carboxylates2 is likely to have an impact on the mobility of U(VI) species in the environment.3 In addition, some U(VI) hybrid materials have unique structures and properties making them potential new functional materials.4U(VI) complexes with isonicotinic acid (HINT) have been relatively less studied and the available structures include a 1D polymer with uranyl hydroxyl dimer as a building unit5 and several mixed ligand complexes, e.g. a dimer with CrO4−,6 1D polymers with F− (ref. 7) and a polymer with oxalate.8 In addition, some U(VI) complexes with other monocarboxylate ligands similar to HINT were also documented earlier, e.g. oxypyridine-4-carboxylic acid,9 picolinic acid,10 pyrazine-2-carboxylic acid and pyrimidine-2-carboxylic acid.11 Two conclusions can be drawn from the literature survey on U(VI) complexes with pyridine-based carboxylate ligands. Firstly, monomeric structures are relatively less reported as the carboxylate group tends to form bridging mode linking uranyl centres together. Secondly, U–N bonds are only observed together with the nearby carboxylate group chelating to the same uranyl centre. In fact, no isolated U–N bonding has been reported for U(VI) ion with INT ligand. In this work, we aim to further explore and expand the structural chemistry of U(VI) with HINT and herein report the synthesis, spectroscopic and thermal studies, photoluminescence and crystal structures of two new compounds, [(UO2)(NO3)2(HINT)2] (1) with a monomeric structure and [(UO2)(OH)(INT)] (2) with a 2D layered structure via a unique μ3-coordination mode of INT through bridging carboxylate group and U–N bonding.
Experimental section
Synthesis
[(UO2)(NO3)2(HINT)2] (1). 2.0 mmol of isonicotinic acid (0.246 g) was dissolved in 10 mL of deionised (DI) water. 2.0 mL of uranyl nitrate solution (0.5 M) was then added to the above solution. Yellow crystalline product of 1 (∼0.48 g) was formed after three weeks, with yield of ∼75%. C12H10N4O12U (FW = 640.26): calc. C, 22.51; H, 1.57; N, 8.75; found: C, 22.32; H, 1.65; N, 8.64.[(UO2)(OH)(INT)] (2). 2.0 mL of uranyl nitrate solution (0.5 M), 1.0 mmol of isonicotinic acid (0.123 g), 1.2 mmol of KOH (0.067 g) and 5.0 mL of DI water were added in a 30 mL sealed titanium pressure vessel and left in a 180 °C oven for 48 hours. Yellow crystalline product of 2 (∼0.20 g) was formed after slow cooling (<5 °C h−1) to room temperature in a light yellow solution (pHf ∼ 5.4) with ∼48% yield. C6H5NO5U (FW = 409.14): calc. C, 17.61; H, 1.23; N, 3.42; found: C, 17.54; H, 1.28; N, 3.56.
Both complexes were further characterized by elemental analysis, scanning electron microscope-electron disperse spectroscopy (SEM-EDS), thermogravimetric and differential thermal analysis (TG/DTA), Raman spectroscopy, photoluminescence and single crystal X-ray diffraction. SEM-EDS confirmed the presence of C, N, O and U in both complexes (Fig. S1 and S2†).
Characterization
Elemental analyses were carried out using a Perkin-Elmer 2400 CHN elemental analyzer. SEM-EDS was conducted under an accelerating voltage of 20 kV with a Zeiss Ultra Plus SEM (Carl Zeiss NTS GmbH, Oberkochen, Germany). The Raman spectra were recorded on a Perkin Elmer Raman station 400 with Micro300 Microscope and excitation laser 785 nm in the range 2000–100 cm−1. TG/DTA was made on a SEIKO 6300 Thermal Analyzer from room temperature to 1000 °C at a heating rate of 10 °C min−1 and an air flow rate of 300 cm3 min−1. Fluorescence emission spectra were measured using a Cary Eclipse Fluorescence Spectrometer.X-ray diffraction
The single crystal X-ray diffraction measurements were carried out on the MX2 beamline at the Australian Synchrotron. Diffraction data were collected using Si<111> monochromated synchrotron X-ray radiation (λ = 0.72930) at 100 (2) K with BlueIce software12 and were corrected for Lorentz and polarization effects using the XDS software.13 The structures were solved by direct methods and the full-matrix least-squares refinements were carried out using SHELX suite of programmes.14Results and discussion
Structure descriptions and discussion
The crystal data and refinement details for 1 and 2 are summarized in Table 1. The asymmetric unit of 1 contains half a uranyl ion, coordinated by a nitrate anion and a monodentate HINT. The expansion by symmetry suggests that 1 has a monomeric structure with a hexagonal bipyramidal uranyl ion coordinated by two nitrate groups and two HINT, in trans-positions (Fig. 1a and b). The uranyl group is ideally linear [O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UO angle of 180.00(2)°] with the UO bond length of 1.765 (3) Å. The U–O bond lengths in the equatorial plane of the uranyl ion range from 2.543(3) to 2.554(3) Å for nitrate anions and 2.311(3) Å for the carboxylate O atom from HINT ligands. A ball-stick packing view (Fig. 1c) shows how the monomers pack in the crystal lattice. The calculated inter-molecule hydrogen bonds are summarized in Table 2. Apart from strong hydrogen bonds between the terminal carboxylate oxygen atoms (O5) and the nitrogen atoms (N2) of the pyridine rings, uranyl oxygen atoms (O1) and majority of the nitrate oxygen atoms (O2 and O4) are involved in weak hydrogen bonding with the C–H from the pyridine rings leading the monomeric structure into three dimensions. Such neutral uranyl nitrate complexes with monodentate O-donor ligands in trans-arrangement are common15 and the closely related one is the uranyl nitrate complex with pyridine-3-carboxylic acid.16
UO angle of 180.00(2)°] with the UO bond length of 1.765 (3) Å. The U–O bond lengths in the equatorial plane of the uranyl ion range from 2.543(3) to 2.554(3) Å for nitrate anions and 2.311(3) Å for the carboxylate O atom from HINT ligands. A ball-stick packing view (Fig. 1c) shows how the monomers pack in the crystal lattice. The calculated inter-molecule hydrogen bonds are summarized in Table 2. Apart from strong hydrogen bonds between the terminal carboxylate oxygen atoms (O5) and the nitrogen atoms (N2) of the pyridine rings, uranyl oxygen atoms (O1) and majority of the nitrate oxygen atoms (O2 and O4) are involved in weak hydrogen bonding with the C–H from the pyridine rings leading the monomeric structure into three dimensions. Such neutral uranyl nitrate complexes with monodentate O-donor ligands in trans-arrangement are common15 and the closely related one is the uranyl nitrate complex with pyridine-3-carboxylic acid.16
| Complex | 1 | 2 |
|---|---|---|
| a R1 = Σ∥Fo| − |Fc∥/|Fo|.b wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2. | ||
| Formula | C12H10N4O12U | C6H4NO5U |
| Formula weight | 640.27 | 409.14 |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | C2/c | Pbca |
| a (Å) | 14.539(3) | 10.191(2) |
| b (Å) | 9.16999(18) | 8.6030(17) |
| c (Å) | 13.700(3) | 18.665(4) |
| β (°) | 116.31(3) | 90 |
| Volume (Å3) | 1637.2(7) | 1636.4(6) |
| Z | 4 | 8 |
| μ (mm−1) | 4.648 | 9.108 |
| Min./Max. θ [°] | 2.787/28.278 | 2.239/24.993 |
| dcalcd (g cm−3) | 2.597 | 3.321 |
| GOF | 1.0543 | 1.107 |
| Final R1a [I > 2σ(I)] | 0.0200 | 0.0305 |
| Final wR2b [I > 2σ(I)] | 0.0573 | 0.0772 |
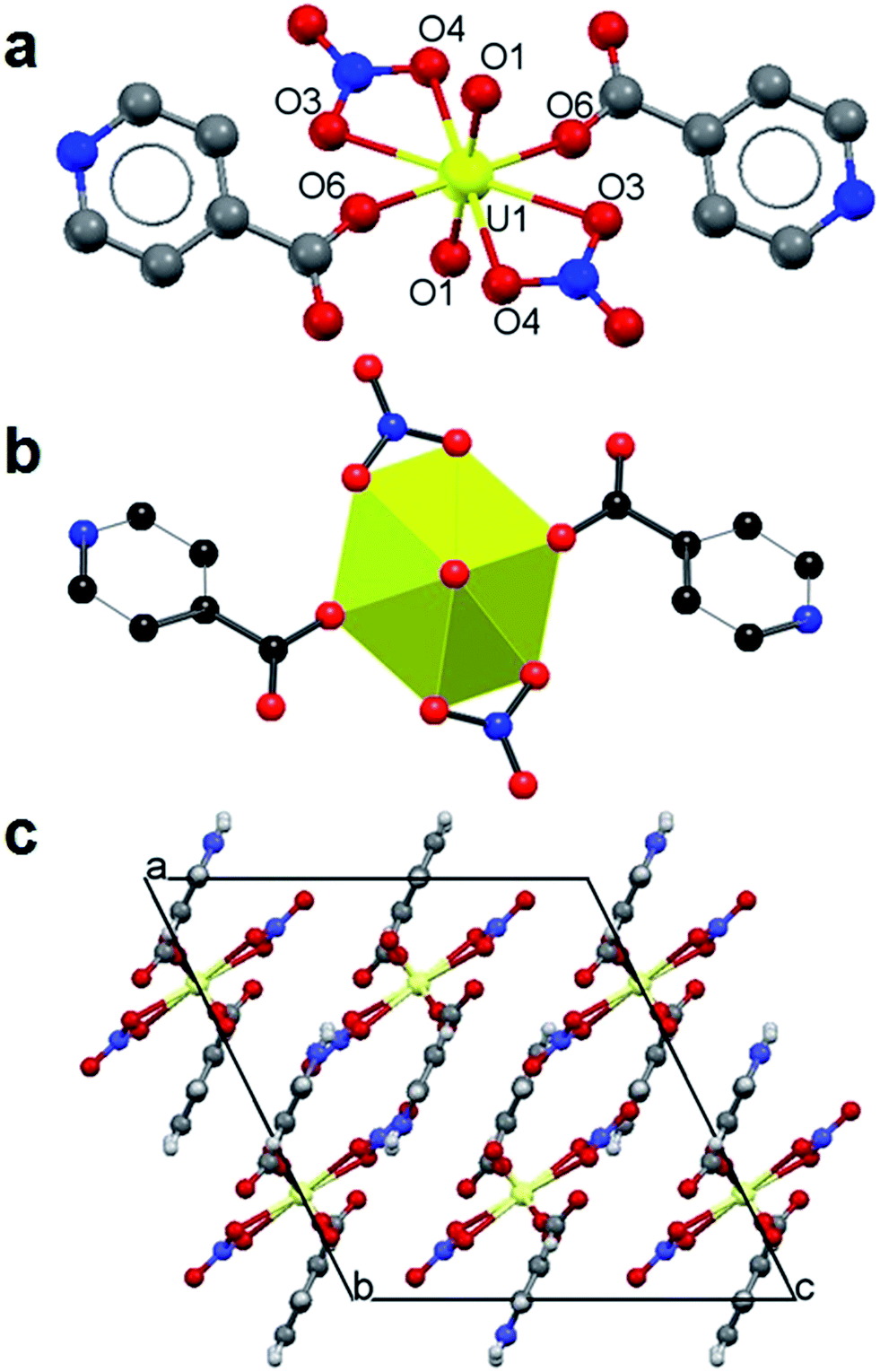 | ||
| Fig. 1 Structure of 1: ball-stick (a) and polyhedral (b) views of the monomeric structure and a packing view along the crystallographic b-axis (c). | ||
| Donor | H | Acceptor | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|---|---|
| Complex 1 | ||||||
| N2 | H2 | O5 | 0.71 | 2.11 | 2.71 | 144 |
| C3 | H3 | O1 | 0.93 | 2.56 | 3.20 | 127 |
| C4 | H4 | O2 | 0.93 | 2.34 | 3.25 | 167 |
| C5 | H5 | O4 | 0.93 | 2.59 | 3.23 | 126 |
| C6 | H6 | O2 | 0.93 | 2.42 | 3.32 | 161 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Complex 2 | ||||||
| O3 | H3 | O2 | 0.85 | 2.11 | 2.94 | 165 |
| C4 | H4 | O1 | 0.93 | 2.38 | 3.13 | 138 |
| C5 | H5 | O1 | 0.93 | 2.46 | 3.30 | 150 |
Complex 2 has a 2D polymeric structure built with pentagonal bipyramidal uranyl centres (Fig. 2a and b) via corner- sharing hydroxyl groups forming 1D uranyl hydroxyl pillars (Fig. 2c) which are further linked through μ3-bridging INT anions to form the 2D layered structure (Fig. 2d) with layers closely packed in the crystal lattice (Fig. 2e). The uranyl unit is normal with OUO angle of 178.57(2)° and UO bond lengths of 1.782(2) and 1.787(4) Å. The U–O bond lengths in the equatorial plane range from 2.316(4) to 2.379(4) Å for hydroxyl oxygen atoms (O–OH) and 2.360(4) to 2.436(4) Å for O–COO atoms. The N atom of the pyridine-ring occupies the fifth coordination position with U–N bond length of 2.574(5) Å. Inter-molecule hydrogen bonds (Table 2) include strong interactions between O-yl (O2) and O–OH (O3) groups and weak interactions between O-yl (O1) and C–H (C4 and C5) leading the 2D structure into three dimensions.
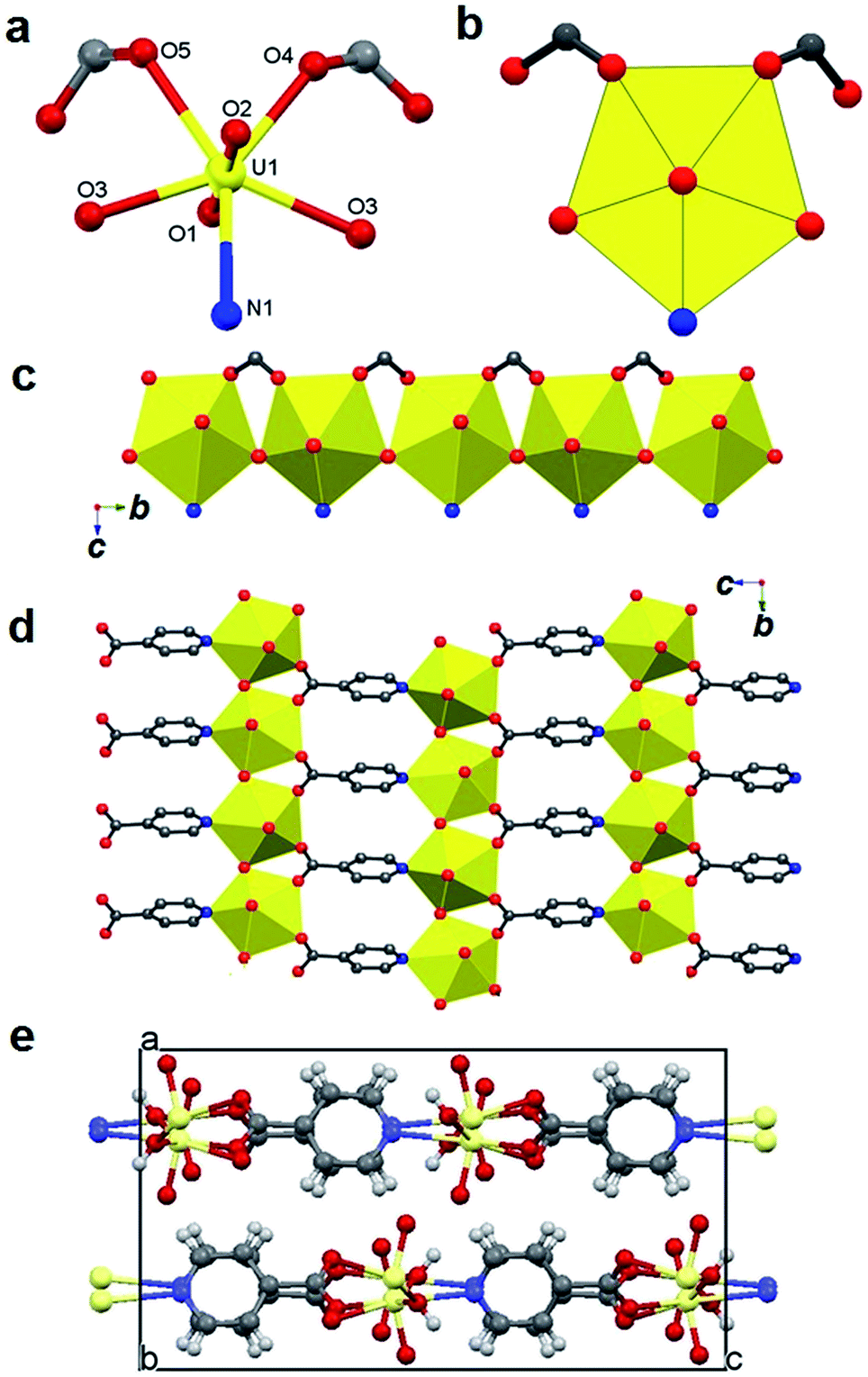 | ||
| Fig. 2 Structure of 2: ball-stick (a) and polyhedral (b) views of the pentagonal bipyramidal uranyl unit, a 1D uranyl hydroxyl pillar (c) formed by corner-sharing hydroxyl groups, a 2D layer (d) formed via μ3-bridging INT ligands and a packing view along the crystallographic b-axis (e). | ||
A uranyl hydroxyl complex with INT anion was previously reported.5 It has a 1D polymeric structure constructed with the uranyl hydroxyl dimer as the building unit and μ2-bridging INT anions. In contrast, complex 2 has a 2D layered structure constructed with uranyl hydroxyl 1D pillars linked through μ3-bridging INT anions. It is believed that the relatively higher final solution pH (∼5.4) and consequently further hydrolysis of uranyl species lead to the formation of complex 2 with 1D uranyl hydrolysis pillars. In addition, it is rare to observe INT in μ3-bridging mode for actinide ions and such coordination mode of INT was only found in a Np(V) complex before.17
Thermal stability
The DTA curve of 1 (Fig. S3†) has a small endotherm at ∼275 °C and a large exotherm at ∼450 °C with obvious three continuous decomposition steps. The first two steps from ∼250 to ∼420 °C with a weight loss of ∼38% correspond to the decomposition of HINT ligands (calc. 38.4%) and the last step at ∼450 °C is due to the decomposition of nitrate anions with the final product of U3O8 (residue: calc. 43.8%; found 44.7%). Similarly, the large exotherm at ∼410 °C for 2 (Fig. S3†) corresponds to the decomposition of INT ligand with the final product of U3O8 (residue: calc. 68.6%; found 68.1%). Note uranium oxide phase transitions at high temperature in air have been well documented.18 β-UO3 is first formed at temperatures around 425 to 450 °C, which transforms to γ-UO3 at ∼500 °C. Further phase transition from γ-UO3 to U3O8 occurs at 650 to 710 °C. Consequently, U3O8 is the main oxide phase present after heating to over 710 °C, which has been confirmed previously by both DSC/TG and powder X-ray diffraction studies.18Raman spectroscopy
Raman spectroscopic characterization of both 1 and 2 (Fig. 3) confirmed: (1) the presence of uranyl groups in 1 and 2 with νs (UO2)2+ at 828 and 833 cm−1, corresponding to the calculated UO bond lengths of 1.788(7) Å and 1.778(5) Å,19 respectively, consistent with the values determined by single crystal X-ray diffraction studies; bending vibrations of (UO2)2+ and (U−Oligand) at 286 and 279 cm−1; 2) the presence of INT in 1 and 2 including νas (COO−) and νs (COO−) at 1629–1524 cm−1 and 1403–1372 cm−1 for 1, and 1525 cm−1 and 1409 cm−1 for 2; carboxylate δ (COO−) bending vibrations at 684, 467 and 235 cm−1 for 1, and 686, 442 and 234 cm−1 for 2.
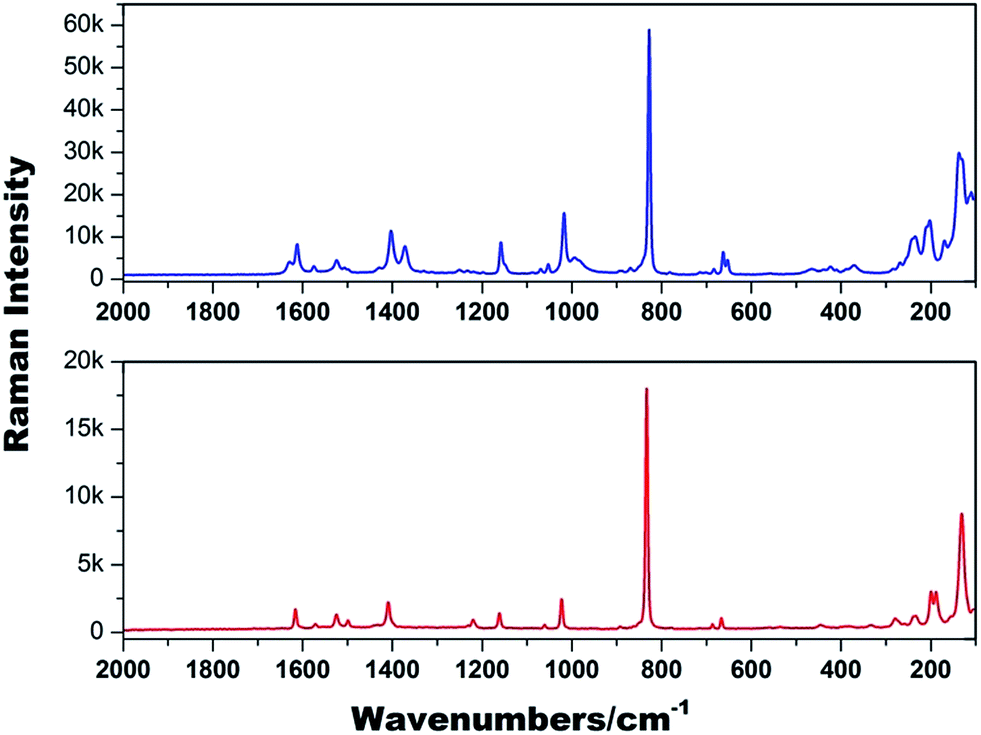 | ||
| Fig. 3 Raman spectra (2000–100 cm−1) of 1 (top) and 2 (bottom). | ||
Two outstanding differences between the two Raman spectra are also apparent. Firstly, the presence of nitrate anions in 1 is evident with the corresponding nitrate vibration modes at 1372, 1070 and 653 cm−1.20 Secondly, the presence of hydroxyl groups in 2 gives δ (U–O–H) bending vibrations at 1220 cm−1. The detailed Raman assignments for the two complexes are summarized in Table S1.†
Photoluminescence
The fluorescence emission spectra for 1, 2 and HINT ligand in fine powders were collected at ambient temperature using their maximum excitation wavelengths. The emission spectrum of HINT has a broad band with a maximum at around 455 nm, similar to the earlier observation.21 The fluorescence emission spectrum of 1 (Fig. 4) has six emission bands at 470, 485, 505, 528, 555 and 579 nm, quite similar to that of uranyl nitrate hexahydrates with typical bands corresponding to the electronic transitions S11 → S00 and S10 → S0ν (ν = 0–4) of the uranyl ion.22 The most intense band is located at 505.0 nm (blue-shifted) for 1 compared to 510.0 nm observed for UNH.22,23 The blue-shift effect is related to the presence of hexagonal bipyramidal uranium centres and has been discussed before.24 The effect of INT in monodentate coordination mode has not been observed. The emission spectrum of 2 (Fig. 4) has a broad feature with the most intense band at 538 nm (red-shifted), together with shoulders at 530 and 553 nm. The slightly red-shift effect for some uranyl carboxylate compounds have been discussed and attributed to the presence of pentagonal bipyramidal uranium centres.24 However, further red-shift and broad nature in the case of 2 could be the combined result of the uranium local coordination environment and the ligand effect via U–N bonding. All in all, the fluorescence emission spectrum of 1 is dominated by the presence of hexagonal bipyramidal uranium centre with no obvious ligand effect. However, the complexation of U(VI) ion by INT anions through μ3-coordination mode via both bridging carboxylate group and U–N bonding has apparently enhanced fluorescence emission of 2 with significant red shift being witnessed. Note the fluorescence lifetimes for both compounds are fairly long, around 0.5 μs (Fig. S4†).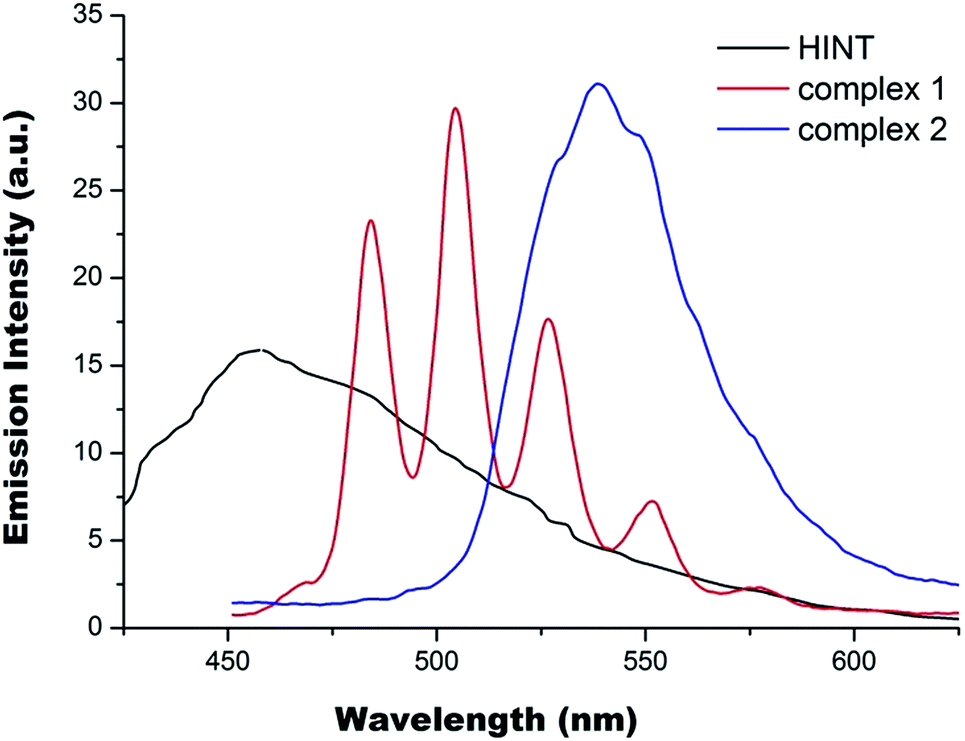 | ||
| Fig. 4 The fluorescence emission spectra of 1, 2 and HINT ligand. | ||
Conclusions
In summary, the reaction of uranyl nitrate and HINT at room temperature affords the formation of a monomeric complex 1 with both coordinated nitrate anions and HINT molecules in trans-positions. In addition, a new uranyl hydroxyl complex 2 with INT was synthesized under hydrothermal conditions. It has a 2D layered structure built with uranyl hydroxyl pillars linked through μ3-bridging INT anions and is the first reported U(VI) compound with INT in μ3-bridging mode via bridging carboxylate group and U–N bonding. The relatively higher final solution pH (∼5.4) and consequently further hydrolysis of uranyl species are believed to be the reason favoring the formation of complex 2. Thermal analysis confirmed both complexes lost organic ligand first, followed by further decomposition to form U3O8. Raman spectroscopy has been successfully used not only to confirm the presence of uranyl ion and INT anion in both complexes but also reveal the existence of nitrate vibrations in 1 and hydroxyl vibrations in 2. Complex 1 has fluorescent emission spectrum similar to uranyl nitrate hexahydrates but slightly blue-shifted due to the presence of hexagonal bipyramidal uranium coordination environment. The significant red shift observed for 2 is thought to be due to the combined result of both pentagonal bipyramidal uranium local coordination environment and the ligand effect via both bridging carboxylate group and U–N bonding.Acknowledgements
The crystallographic data collections were undertaken on the MX2 beamline at the Australian Synchrotron, Victoria, Australia.References
- (a) O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474 CrossRef CAS; (b) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed; (c) C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466 CrossRef CAS PubMed; (d) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC; (e) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- (a) P. Thuéry, Inorg. Chem. Commun., 2008, 11, 616 CrossRef PubMed; (b) P. Thuéry, CrystEngComm, 2008, 10, 79 RSC; (c) P. Thuéry, CrystEngComm, 2009, 11, 1081 RSC; (d) P. Thuéry, Cryst. Growth Des., 2011, 11, 347 CrossRef; (e) J. Lhoste, N. Henry, P. Roussel, T. Loiseau and F. Abraham, Dalton Trans., 2011, 2422 RSC; (f) I. Mihalcea, N. Henry and T. Loiseau, Cryst. Growth Des., 2011, 11, 1940 CrossRef CAS; (g) I. Mihalcea, N. Henry, N. Clavier, N. Dacheux and T. Loiseau, Inorg. Chem., 2011, 50, 6243 CrossRef CAS PubMed; (h) T. Loiseau, I. Mihalcea, N. Henry and C. Volkringer, Coord. Chem. Rev., 2014, 266–267, 69 CrossRef CAS PubMed; (i) F. Abraham, B. Arab-Chapelet, M. Rivenet, C. Tamain and S. Grandjean, Coord. Chem. Rev., 2014, 266–267, 26 Search PubMed.
- P. Crançon and J. Van der Lee, Radiochim. Acta, 2003, 91, 673 CrossRef.
- M. B. Andrews and C. L. Cahill, Chem. Rev., 2013, 113, 1121 CrossRef CAS PubMed.
- E. V. Grechishnikova, Y. N. Mikhailov, A. S. Kanishcheva, L. B. Serezhkina and V. N. Serezhkin, Russ. J. Inorg. Chem., 2005, 50, 1436 Search PubMed.
- L. B. Serezhkina, A. V. Vologzhanina, S. A. Novikov, A. A. Korlyukov and V. N. Serezhkin, Crystallogr. Rep., 2011, 56, 258 Search PubMed.
- J.-Y. Kim, A. J. Norquist and D. O'Hare, Chem. Mater., 2003, 15, 1970 CrossRef CAS.
- D.-S. Liu, T.-Q. Shi, S.-F. Liu, S.-M. Ying and X.-F. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m385 CAS.
- (a) Y.-R. Xie, H. Zhao, X.-S. Wang, Z.-R. Qu, R.-G. Xiong, X. G. Xue, Z. Xue and X.-Z. You, Eur. J. Inorg. Chem., 2003, 3712 CrossRef CAS; (b) S. Lis, Z. Glatty, G. Meinrath and M. Kubicki, J. Chem. Crystallogr., 2010, 40, 646 CrossRef CAS.
- (a) E. V. Grechishnikova, E. V. Peresypkina, A. V. Virovets, Y. N. Mikhailov and L. B. Serezhkina, Koord. Khim., 2007, 33, 468 CrossRef; (b) V. I. Mishkevich, M. S. Grigoriev, A. M. Fedosseev and P. Moisy, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m1243 CAS; (c) W. Aas and M. H. Johansson, Acta Chem. Scand., 1999, 53, 581 CrossRef CAS PubMed; (d) P. R. Silverwood, D. Collison, F. R. Livens, R. L. Beddoes and R. J. Taylor, J. Alloys Compd., 1998, 271, 180 CrossRef; (e) P. Thuéry, Inorg. Chem. Commun., 2009, 12, 800 CrossRef PubMed; (f) R. C. Severance, S. A. Vaughn, M. D. Smith and H.-C. Zur Loye, Solid State Sci., 2011, 13, 1344 CrossRef CAS PubMed.
- M. B. Andrews and C. L. Cahill, CrystEngComm, 2011, 13, 7068 RSC.
- T. M. McPhillips, S. E. McPhillips, H. J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter, R. P. Phizackerley, S. M. Soltis and P. Kuhn, J. Synchrotron Radiat., 2002, 9, 401 CrossRef CAS PubMed.
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- S. Kannan, S. B. Deb, J. S. Gamare and M. G. B. Drew, Polyhedron, 2008, 27, 2557 CrossRef CAS PubMed.
- E. V. Mit'kovskaya, Y. N. Mikhailov, Y. E. Gorbunova, L. B. Serezhkina and V. N. Serezhkin, Russ. J. Inorg. Chem., 2004, 49, 1538 Search PubMed.
- N. A. Budantseva, G. B. Andreev, A. M. Fedoseev, M. Yu. Antipin and J.-C. Krupa, Radiochim. Acta, 2006, 94, 69 CrossRef CAS.
- (a) P. C. Debets, Acta Crystallogr., 1966, 21, 589 CrossRef CAS; (b) E. Gómez-Rebollo, P. Herrero and R. M. Rojas, J. Nucl. Mater., 1997, 245, 161 CrossRef; (c) J. Čejka, J. Sejkora, Z. Mrázek, Z. Urbanec and T. Jarchovský, Neues Jahrbuch für Mineralogie Abhandlungen, 1996, 170, 155 Search PubMed; (d) K. Sudo and A. Okawa, Bull. Res. Inst. Miner. Dressing Metall., Tohoku Univ., 1960, 16(1), 85 Search PubMed.
- J. R. Bartlett and R. P. Cooney, J. Mol. Struct., 1989, 193, 295 CrossRef CAS.
- L. M. Toth and G. M. Begun, J. Phys. Chem., 1981, 85(5) CrossRef CAS.
- W.-T. Chen, Bull. Chem. Soc. Ethiop., 2011, 25(2), 233 CrossRef CAS.
- (a) C. Jacopin, M. Sawicki, G. Plancque, D. Dozi, F. Taran, E. Ansoborlo, B. Amekraz and C. Moulin, Inorg. Chem., 2003, 42, 5015 CrossRef CAS PubMed; (b) A. F. Leung, L. Hayashibara and J. Spadaro, J. Phys. Chem. Solids, 1999, 60, 429 CrossRef.
- (a) Y.-N. Hou, Y.-H. Xing, F.-Y. Bai, Q.-L. Guan, X. Wang, R. Zhang and Z. Shi, Spectrochim. Acta, Part A, 2014, 123, 267 CrossRef CAS PubMed; (b) Y. Zhang, M. Bhadbhade, I. Karatchevtseva, J. R. Price, H. Liu, Z. Zhang, L. Kong, J. Čejka, K. Lu and G. R. Lumpkin, J. Solid State Chem., 2015, 226, 42 CrossRef CAS PubMed.
- (a) I. Mihalcea, N. Henry, C. Volkringer and T. Loiseau, Cryst. Growth Des., 2012, 12, 526 CrossRef CAS; (b) T. Loiseau, I. Mihalcea, N. Henry and C. Volkringer, Coord. Chem. Rev., 2013, 266–267, 69 Search PubMed; (c) I. Mihalcea, N. Henry and T. Loiseau, Eur. J. Inorg. Chem., 2014, 1322 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM-EDS, TG/DTA, Raman assignments. CCDC 1035178 and 1035179. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra01272d |
| This journal is © The Royal Society of Chemistry 2015 |