DOI:
10.1039/D4RA01387E
(Review Article)
RSC Adv., 2024,
14, 19752-19779
Recent advances on anticancer and antimicrobial activities of directly-fluorinated five-membered heterocycles and their benzo-fused systems
Received
23rd February 2024
, Accepted 5th June 2024
First published on 19th June 2024
Abstract
Due to the importance of the fluorinated heterocycles as main components of marketed drugs where 20% of the anticancer and antibiotic drugs contain fluorine atoms, this review describes the reported five-membered heterocycles and their benzo-fused systems having directly connected fluorine atom(s). The in vivo and in vitro anticancer and antimicrobial activities of these fluorinated heterocycles are well reported. Some fluorinated heterocycles were found to be lead structures for drug design developments where their activities were almost equal to or exceeded the potency of the reference drugs. In most cases, the fluorine-containing heterocycles showed promising safety index via their reduced cytotoxicity in non-cancerous cell lines. SAR study assigned that fluorinated heterocycles having various electron-donating or electron-withdrawing substituents significantly affected the anticancer and antimicrobial activities.
 Ashraf A. Abbas | Professor Ashraf A. Abbas was born in Egypt in September 1968 and he is presently a Professor of Organic Chemistry, Department of Chemistry, Faculty of Science, Cairo University, Giza, Egypt, since 2009. He graduated from Cairo University in 1990. He received his M.Sc. and Ph.D. degrees in 1994 and 1997, respectively, from Cairo University. He spent six months in the Fakultat fur Chemie, Universitat of Konstanz (Germany) on a DAAD fellowship (1996) to finish his Ph.D. thesis and one year at Tokyo Institute of Technology (TIT, Japan) as a UNESCO fellow (postdoctoral fellowship) from Oct 2001 to Sept 2002. He received several research prizes in chemistry: (1) he was awarded the prize of Prof. Dr. Mohamed Abdel Salam in Chemistry (2001) for young scientists provided by the Academy of Scientific Research and Technology (Egypt). (2) He was awarded the Cairo University encouragement prize in Chemistry in 2004. (3) He was awarded the Third World Academy of Science (TWAS) prize in chemistry for young scientists in 2004, provided by ICTP-Strada, Triesta-Italy. (4) He was awarded the State Award in Chemistry, Egypt, 2005. He has published many papers in the field of synthesis and applications of macrocycles and bis-heterocycles chemistry. |
 Thoraya A. Farghaly | Thoraya Abd Elreheem Farghaly was born in Cairo, Egypt, in 1974. She received her B.Sc. (1996), M.Sc. (2002) and Ph.D. (2005) degrees from the University of Cairo. She is a Professor of organic chemistry in the Chemistry Department, Faculty of Science, University of Cairo. She has worked at Umm Al-Qura University since 2014. She joined the scientific school of Prof. A. S. Shawali in 1997 and published more than 220 papers in the area of the chemistry of hydrazonoyl halides, heterocyclic chemistry and bioactive heterocyclic compounds. |
 Kamal M. Dawood | Kamal M. Dawood graduated from Cairo University, Egypt, in 1987 and received his PhD in 1995 from Cairo University. In 1997, he was awarded the UNESCO Fellowship at TIT for one year, and in 1999, he was awarded the JSPS Fellowship for two years and in both fellowships, he worked with Professor T. Fuchigami at the Tokyo Institute of Technology (TIT) in the field of “Anodic Selective Fluorination of Heterocycles'. Further, he was awarded the Alexander von Humboldt (AvH) Fellowship at Hanover University in 2004–2005 with Prof. A. Kirschning (in the area of polymer supported palladium catalyzed cross coupling reactions) and as AvH three short visits in 2007, 2008 and 2012 with Prof. P. Metz at TU-Dresden (in the field of Metathesis Reactions in Domino Processes). Since May 2007 – to date, he has been appointed as a full Professor of Organic Chemistry, Faculty of Science, Cairo University. He worked as a Professor of Organic Chemistry at the Chemistry Department, Kuwait University from Sept. 2013 till Aug 2017. He received a number of National Awards: Cairo University Award in Chemistry (2002), the State Award in Chemistry (2007), Cairo University Award for Academic Excellence (2012) and the Cairo University Merit Award (2017). He has published about 160 scientific papers, reviews and book chapters in distinguished international journals. There are more than 3700 citations of his work (Scopus h-index 33). |
1. Introduction
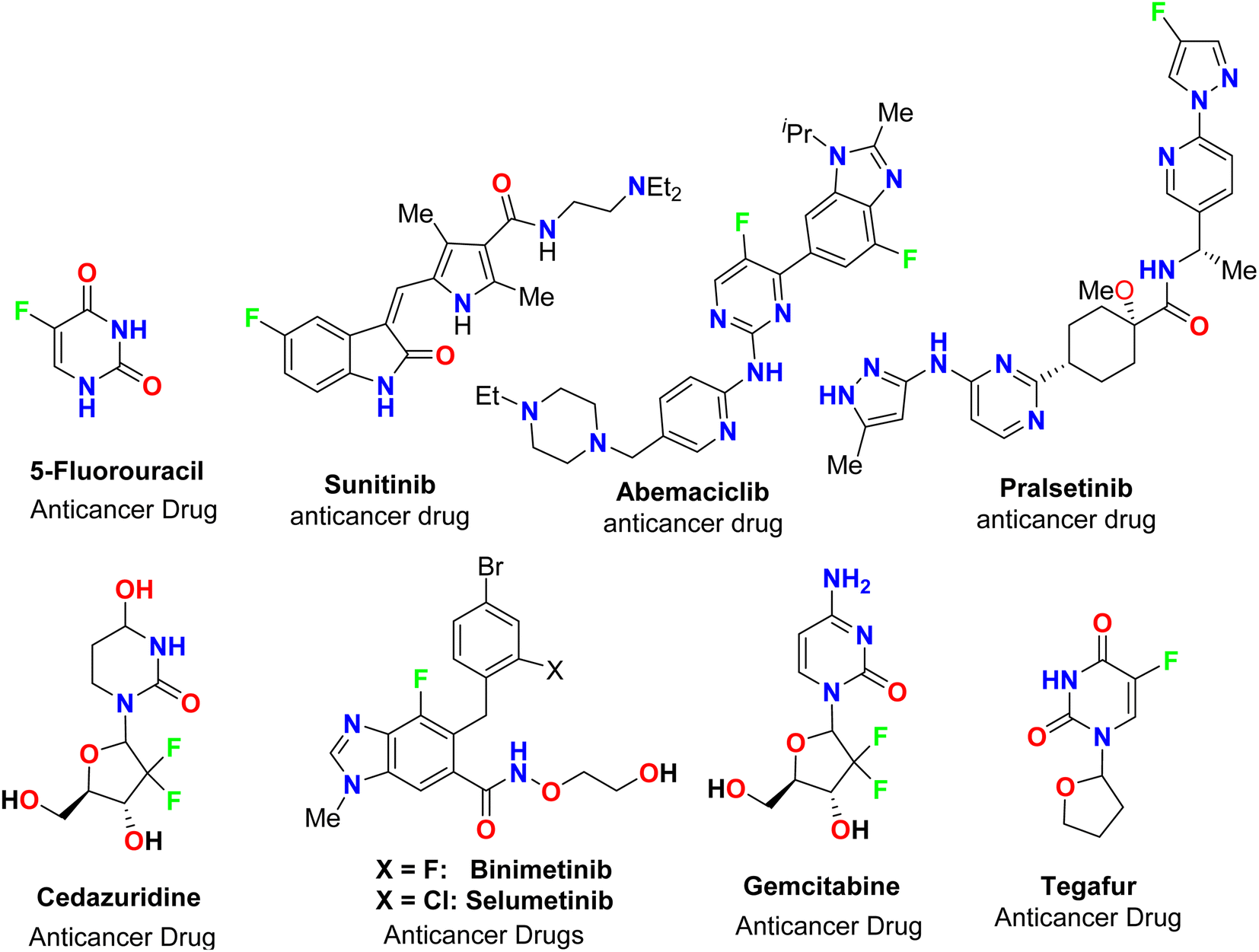
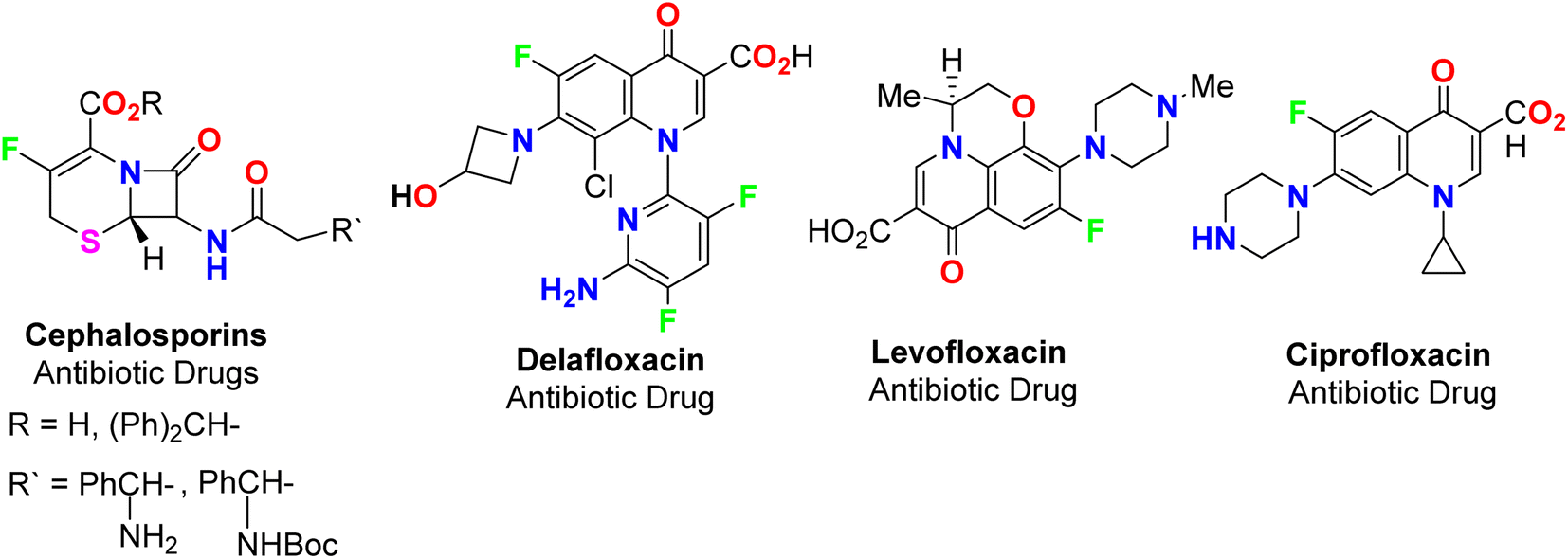
The spread of various types of cancers and microbes worldwide, which frequently leads to death, is regarded one of the world's greatest challenges in eliminating them. Multidrug resistance (MDR) is one of the primary causes of the failure of cancer chemotherapy, where cancer cells can survive several chemotherapy treatments with various structures and mechanisms of action. Multidrug-resistant bacteria (MDR bacteria) are bacteria that resist some antimicrobial drugs.1–3 Researchers are devoted to finding effective drugs with minimum toxicity to avoid serious side effects of cancer drugs. One of the research methods is to introduce a new active moiety or atoms on the heterocyclic compounds that enhance their effectiveness. Researchers have discovered that ring-fluorinated five- and six-membered nitrogen heterocyclic compounds have promising antibacterial and anticancer potencies.4,5 Due to the high electronegativity and small size of the fluorine atom, the introduction of fluorine atom(s) into biologically active heterocyclic compounds can remarkably improve chemical and physical properties of entire molecules. These properties are acidity, basicity, solubility, lipophilicity, stability and hydrogen bonding interactions and may greatly enhance their biological potencies.6–9 In addition, direct fluorination via replacing hydrogen with fluorine on heteroaromatic rings was very effective for the metabolic stability of the new fluorinated heteroaromatics.10 About 20% of the marketed pharmaceutical drugs contain fluorine atom(s).11,12 One of the oldest fluorinated anticancer drugs was 5-fluorouracil (Fig. 1), which was approved in 1957.13 Some directly fluorinated heterocycles such as sunitinib (Fig. 1) was the first anticancer drug approved by the FDA in 2006 for the treatment of renal cell carcinoma (RCC).14 Sunitinib was able to inhibit VEGF(R)-2 (vascular endothelial growth factor receptor), a tyrosine receptor kinase, which is important for the direct regulation of tumor angiogenesis.15–17 Abemaciclib was approved in 2017 for the treatment of advanced or metastatic breast cancers18 and pralsetinib (Fig. 1) was approved in 2020 for the treatment of metastatic fusion-positive non-small-cell lung cancer.19 In addition, cedazuridine,20 binimetinib,21 selumetinib22 and gemcitabine23 (Fig. 1) were also approved as anticancer drugs in the market. Moreover, fluorinated heterocycles were reported as the core component of some antibiotic drugs that were approved in the market, such as cephalosporins (β-lactam-based fluorinated thiazine derivatives), delafloxacin, levofloxacin and ciprofloxacin, as shown in Fig. 2.24–26 The importance of fluorine-based small molecules in drug discovery forced chemists and drug developers to overcome challenges associated with the insertion of fluorine atom(s) into small organic molecules. Several synthetic routes to fluorinated heterocycles were described in the literature, particularly chemical and electrochemical tools or employing readily available fluorinated reagents. Chemical fluorination of heterocyclic systems was achieved using either electrophilic or nucleophilic fluorinating agents such as F2, SF4, XeF2, Et2NSF3 (DAST), or N-fluoropyridinium triflates, or using the fluorinated building blocks such as CF3COCH3, CF3CO2Et, or CF2Br2.27–31 Electrochemical fluorination protocol employed the use of stable fluoride ion sources such as alkyl ammonium fluoride ionic liquids (Et3N·nHF, or Et4NF·nHF (n = 3, 4, 5)) under constant current or constant potential conditions.32–37 The late-stage fluorination was reported as exceptional, effective access for the synthesis of complex small molecules involving metal-catalysed procedures to facilitate the C–F bond formation of such molecules with potential industrial applications.38 The above facts attracted our attention to collect all reports about the anticancer and antibacterial activities of the directly fluorinated five-membered and their fused heterocyclic compounds during the last two decades, from 2003 till the end of 2023. We highly expect that this review article will help a wide range of researchers interested in pharmaceutical drugs to discover effective derivatives for the treatment of various cancers and widespread bacterial diseases.
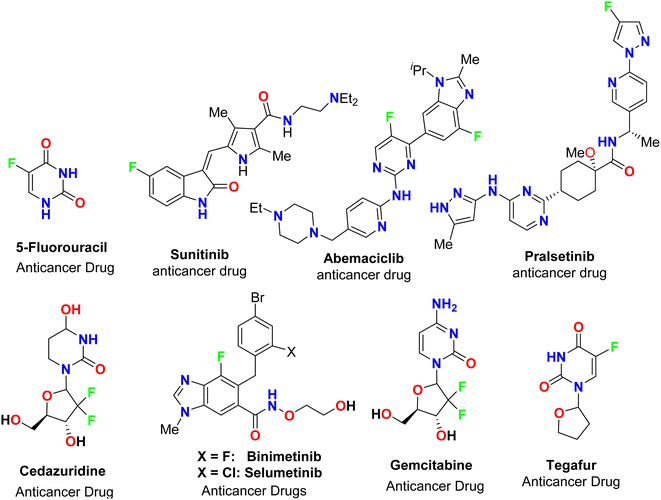 |
| | Fig. 1 Ring-fluorinated heterocycles as commercial anticancer drugs. | |
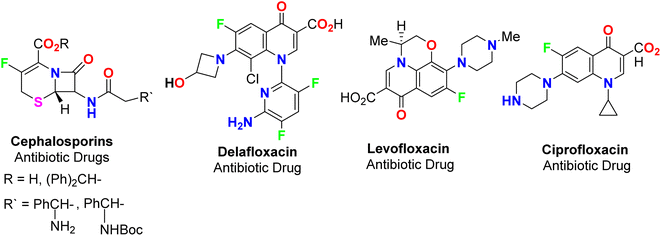 |
| | Fig. 2 Ring-fluorinated heterocycles as commercial antibiotic drugs. | |
2. Anticancer activity of fluorinated heterocycles
2.1. Anticancer activity of fluorobenzofuran derivatives
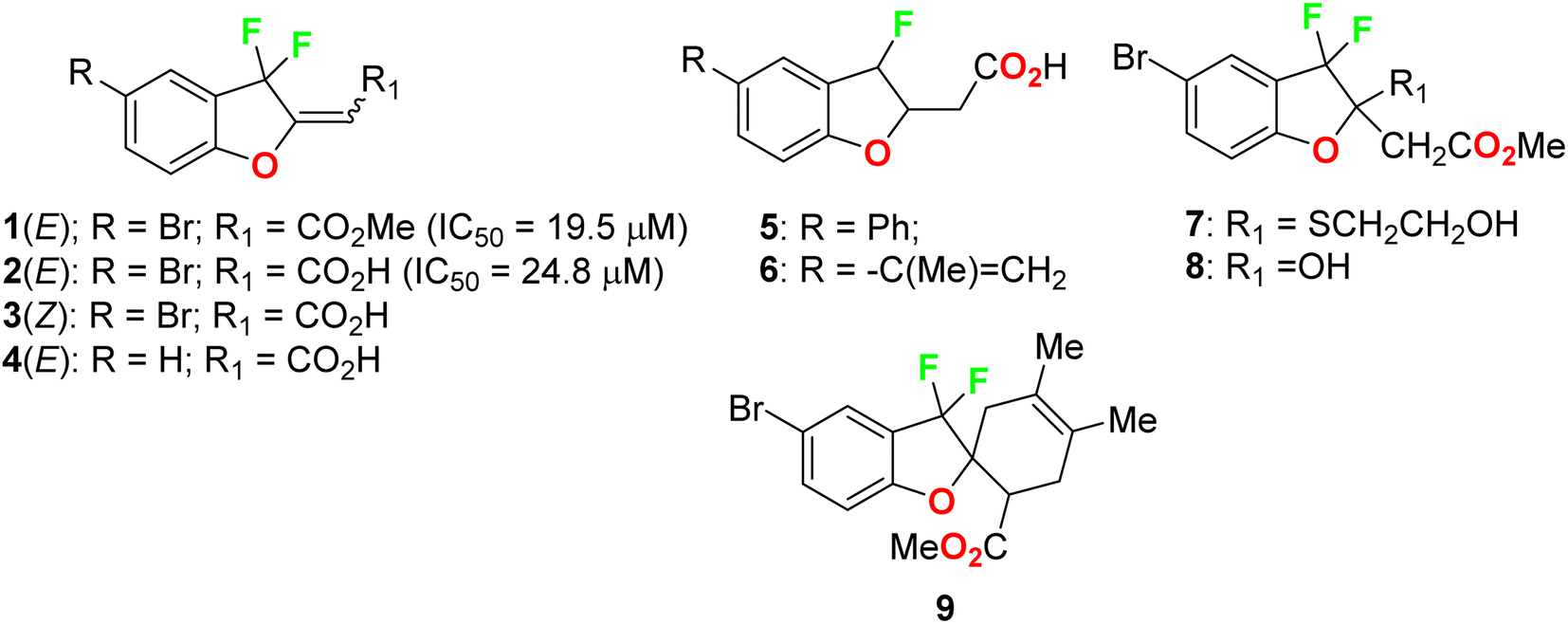
Ayoub et al. screened the anticancer activity of the fluorinated benzofuran derivatives 1–9 (Fig. 3) against the human colorectal adenocarcinoma cell line HCT116 using a WST-1 assay.39 Only compounds 1 and 2 inhibited the proliferation by approximately 70% with IC50 values of 19.5 and 24.8 μM. Both compounds 1 and 2 were reported to exhibit inhibition of the expression of the antiapoptotic protein Bcl-2 and concentration-dependent cleavage of PARP-1, as well as DNA fragmentation by approximately 80%.
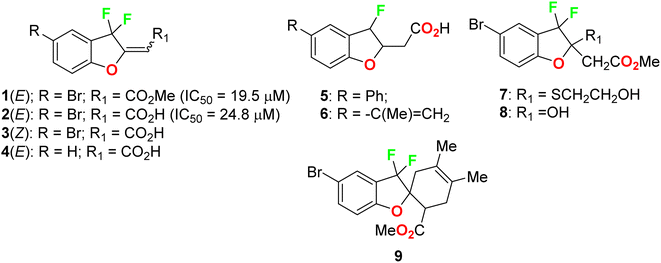 |
| | Fig. 3 Structures of fluorinated benzofurans 1–9. | |
2.2. Anticancer activity of fluoroindole derivatives
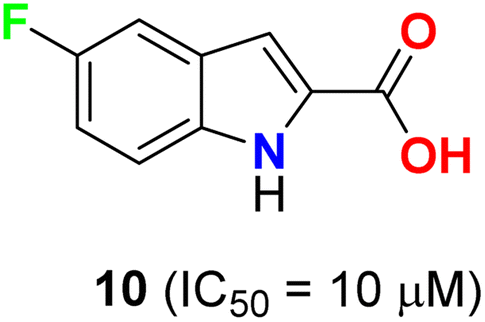
Pidugu et al. tested indole-2-carboxylic acids 10 (Fig. 4) as inhibitors of the human apurinic/apyrimidinic endonuclease 1 (APE1).40 The result indicated that compound 10 formed aggregates and was a weak inhibitor of APE1 under conditions that disrupted compound aggregation. 5-Fluoroindole-2-carboxylic acid (10) was found to inhibit APE1 with an IC50 of 10 μM. APE1 was elevated in cancers and was correlated with increased tumor progression, decreased survival and reduced sensitivity to chemotherapy.
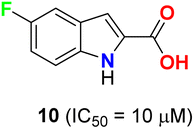 |
| | Fig. 4 Structure of 5-fluoroindole-2-carboxylic acid 10. | |
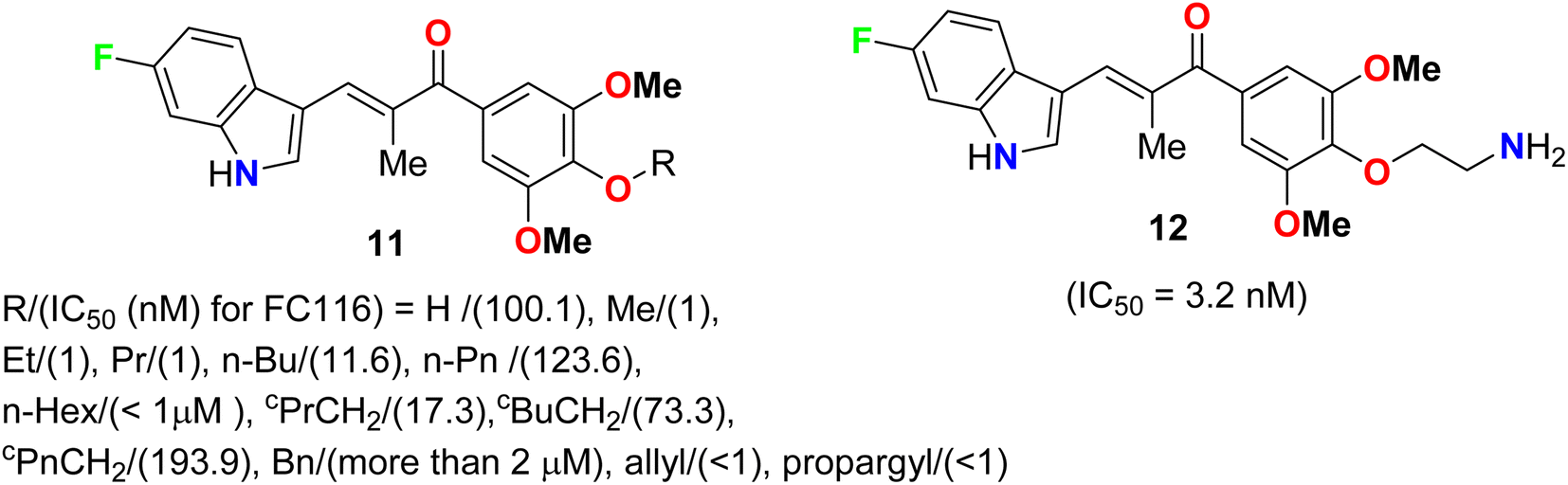
Two series of 6-fluoroindole derivatives 11 and 12 (Fig. 5) were synthesized and screened for their in vivo cytotoxicity against HCT-116 CRC in a low nanomolar range.41 Among them, compound 12 could induce G2/M phase arrest via regulating cyclin B1 expression, produce excess reactive oxygen species (ROS), and target tubulin in CRC cells. Compound 12 significantly increased the G2/M phase population in HCT-116 cells, indicating an anti-microtubule mechanism. Cyclin B1, a G2/M marker protein, was up- and down-regulated after 12 and 48 hours of treatment, indicating its high cytotoxicity and cell death after 48 hours. Compound 12 treatment significantly reduced β-tubulin proteolysis at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :300 pronase ratio. Molecular docking confirmed the binding mode and activity of 12 with tubulin, with the 5-methoxyl and 1-amino functional groups interacting through hydrogen bonds. The additional aminoethyl group formed hydrogen bonds with Y202, confirming the direct binding target of 12. In vivo, compound 12 significantly suppressed tumor growth, achieving 65.3% and 73.4% at doses of 5 and 10 mg kg−1 d−1 (i.v. 21 d), much better than 54.1% of Taxol at 7 mg kg−1. In addition, compound 12 showed better in vivo tolerance compared to that of 11 (R = Me) (only 3 mg kg−1 tolerance, intraperitoneal, i.p.) and no major organ-related toxicity. 4-Aminoethoxyphenyl indole-chalcone 12 represents the lead compound in the chemotherapy of CRC for further drug development.
:300 pronase ratio. Molecular docking confirmed the binding mode and activity of 12 with tubulin, with the 5-methoxyl and 1-amino functional groups interacting through hydrogen bonds. The additional aminoethyl group formed hydrogen bonds with Y202, confirming the direct binding target of 12. In vivo, compound 12 significantly suppressed tumor growth, achieving 65.3% and 73.4% at doses of 5 and 10 mg kg−1 d−1 (i.v. 21 d), much better than 54.1% of Taxol at 7 mg kg−1. In addition, compound 12 showed better in vivo tolerance compared to that of 11 (R = Me) (only 3 mg kg−1 tolerance, intraperitoneal, i.p.) and no major organ-related toxicity. 4-Aminoethoxyphenyl indole-chalcone 12 represents the lead compound in the chemotherapy of CRC for further drug development.
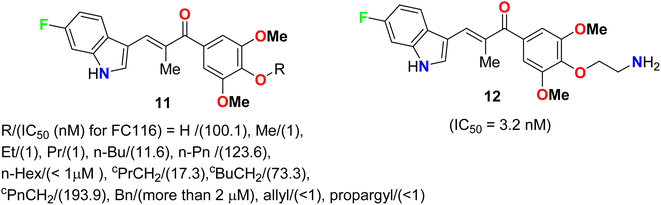 |
| | Fig. 5 Structure of 6-fluoroindole derivatives 11 and 12. | |
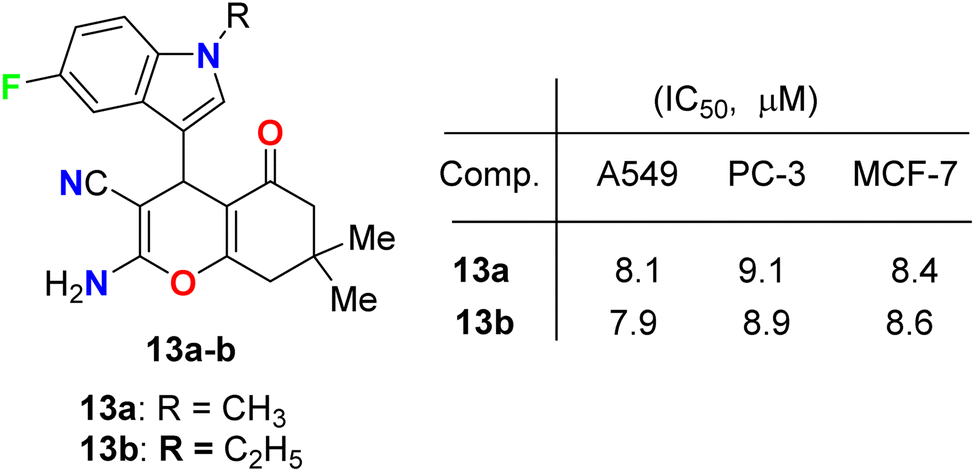
Fluoroindole-tethered chromene derivatives 13a,b (Fig. 6) were synthesized and evaluated for anticancer properties against A549, PC-3, and MCF-7 cancer cell lines. The derivatives 13a and 13b displayed promising cytotoxic activity with IC50 values ranging between 7.9–9.1 μM. Molecular docking revealed that 13a and 13b had a good binding affinity towards tubulin protein, better than the positive control (crolibulin), and the molecular dynamic simulations further demonstrated the stability of ligand–receptor interactions.42
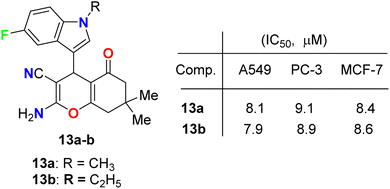 |
| | Fig. 6 Structure of fluoroindole-tethered chromene derivatives 13a,b. | |
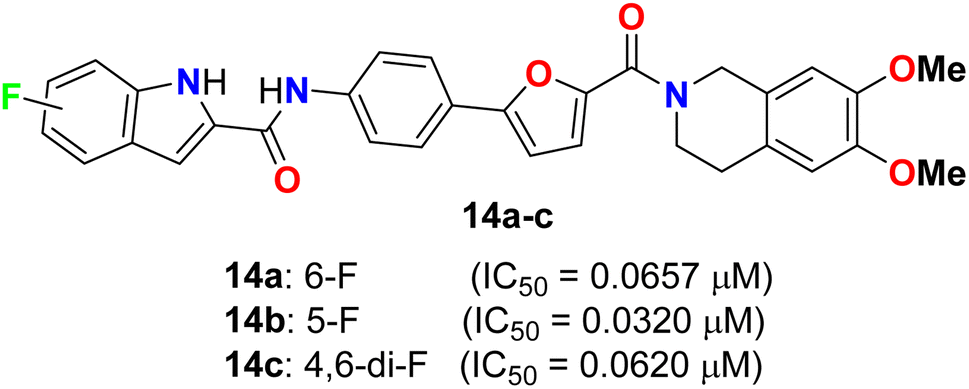
Yang and his colleagues designed and synthesized a series of phenylfuran-bisamide derivatives 14a–c (Fig. 7) as P-glycoprotein (P-gp) inhibitors based on target-based drug design and screened on MCF-7/ADR cells. The results revealed that the introduction of electron-withdrawing fluorine substituent (6-F, 5-F, 4,6-di-F) at position 2 of indole was beneficial to the reversal activity. The reversal activity could be ordered as: 14b (5-F) > 14c (4,6-di-F) > 14a (6-F), which demonstrated that fluorine with stronger electron-drawing ability at 5-position of indole might be more favourable to the reversal activity of target skeleton.43 The fluorinated derivative 14b exhibited low cytotoxicity and promising MDR reversal activity (IC50 = 0.0320 μM, reversal fold = 1163.0), 3.64-fold better than third-generation P-gp inhibitor tariquidar (IC50 = 0.1165 μM, reversal fold = 319.3). The results of western blot and rhodamine 123 accumulation verified that compound 14b exhibited excellent MDR (multi-drug reversal) activity by inhibiting the efflux function of P-gp but not expression. Furthermore, molecular docking showed that compound 14b bound to target P-gp by forming the double H-bond interactions with residue Gln 725. These results suggest that compound 14b might be a potential MDR reveal agent acting as a P-gp inhibitor in clinical therapeutics and provide insight into design strategy and skeleton optimization for the development of P-gp inhibitors.43
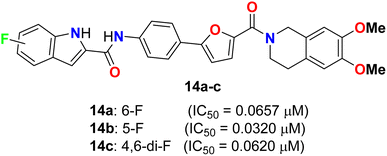 |
| | Fig. 7 Structure of phenylfuran-bisamide derivatives 14a–c. | |
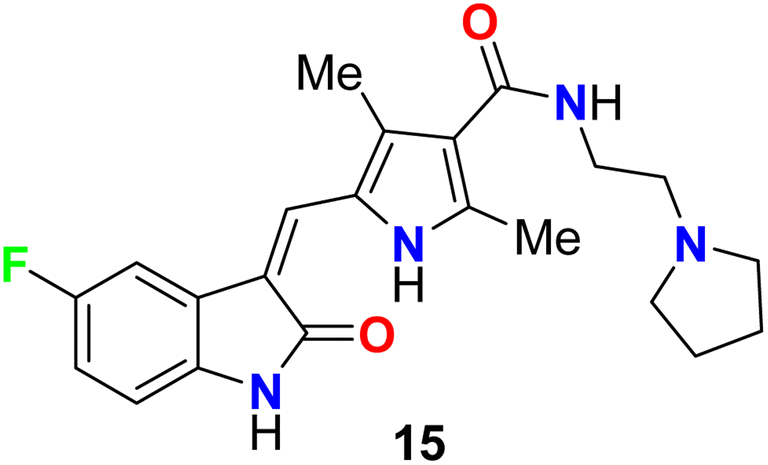
The fluorinated indolone derivative 15 (Fig. 8) was reported by London et al. as a good candidate for the treatment of canine mast cell tumors (MCT) in dogs. Compound 15 had significant biological activity against canine MCTs and was administered on a continuous schedule. The objective response rate (ORR) for 145 dogs that received 15 was 42.8% (21 complete responses, 41 partial responses), and the duration of tumor progression was 18.1 weeks. Clinical trials of this compound showed that spontaneous tumors in dogs were good for the evaluation of therapeutics in a clinical stage.44
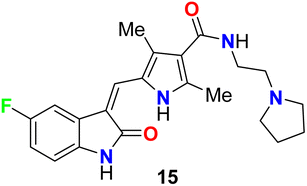 |
| | Fig. 8 Structure of fluorinated indolone derivative 15. | |
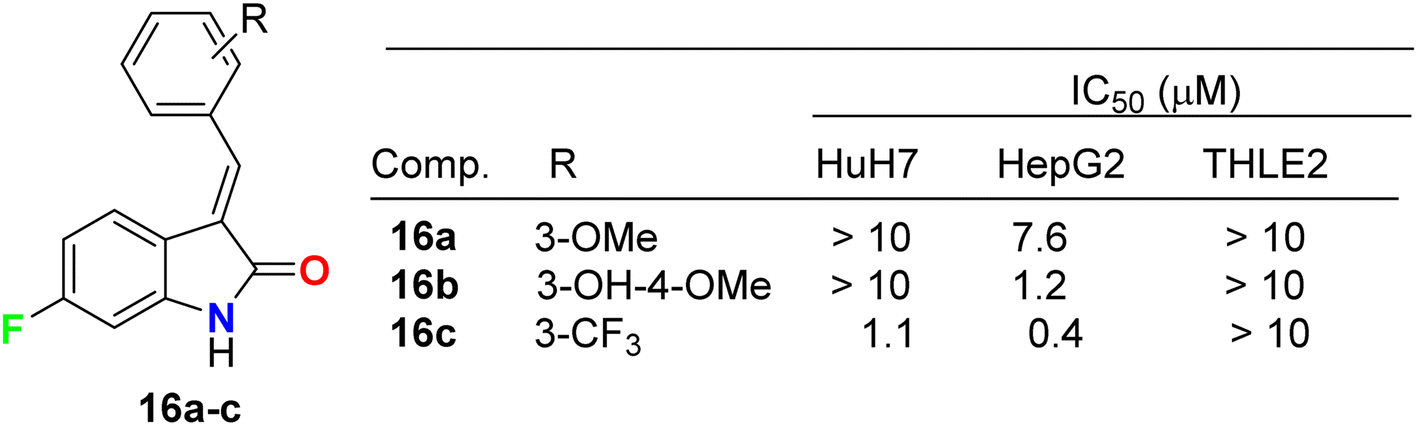
Three fluorinated indolinone derivatives 16a–c (Fig. 9) were synthesized and examined as multi-targeted kinase inhibitors against the hepatocellular carcinoma (HCC) cell lines HuH7 and HepG2. Compound 16c showed the best activity against both cell lines HuH7 and HepG2 with IC50 values 1.1 and 0.4 μM more potent than the sunitinib reference drug with IC50 values 4.7 and 4.5 μM in HuH7 and HepG2, respectively. Compound 16c demonstrated a good safety profile via its reduced cytotoxicity in the non-cancerous THLE2 liver cell line (IC50 > 10 μM) compared with sunitinib IC50 = 4.5 μM. The selectivity ratios (SR) of the tested compound 16c against HuH7/THLE2 and HepG2/THLE2 were SR = 6.4 and 17.2, respectively, indicating increased anti-proliferative activity in tumor versus normal cells. Thus, compound 16c showed a promising safety index with comparable efficacy with sunitinib (SR = 10.8 and 20.4).45 The study investigated the biochemical mechanism of anti-proliferative effects of the target indolinone derivatives. It showed a dose-dependent inhibition of Erk and Akt phosphorylation in HuH7 cells, while HepG2 cells showed reduced Akt phosphorylation but not Erk phosphorylation. The study also demonstrated a reduction in cell cycle marker cyclin-D1.
 |
| | Fig. 9 Structure of fluorinated indolinone derivatives 16a–c and its IC50 values. | |
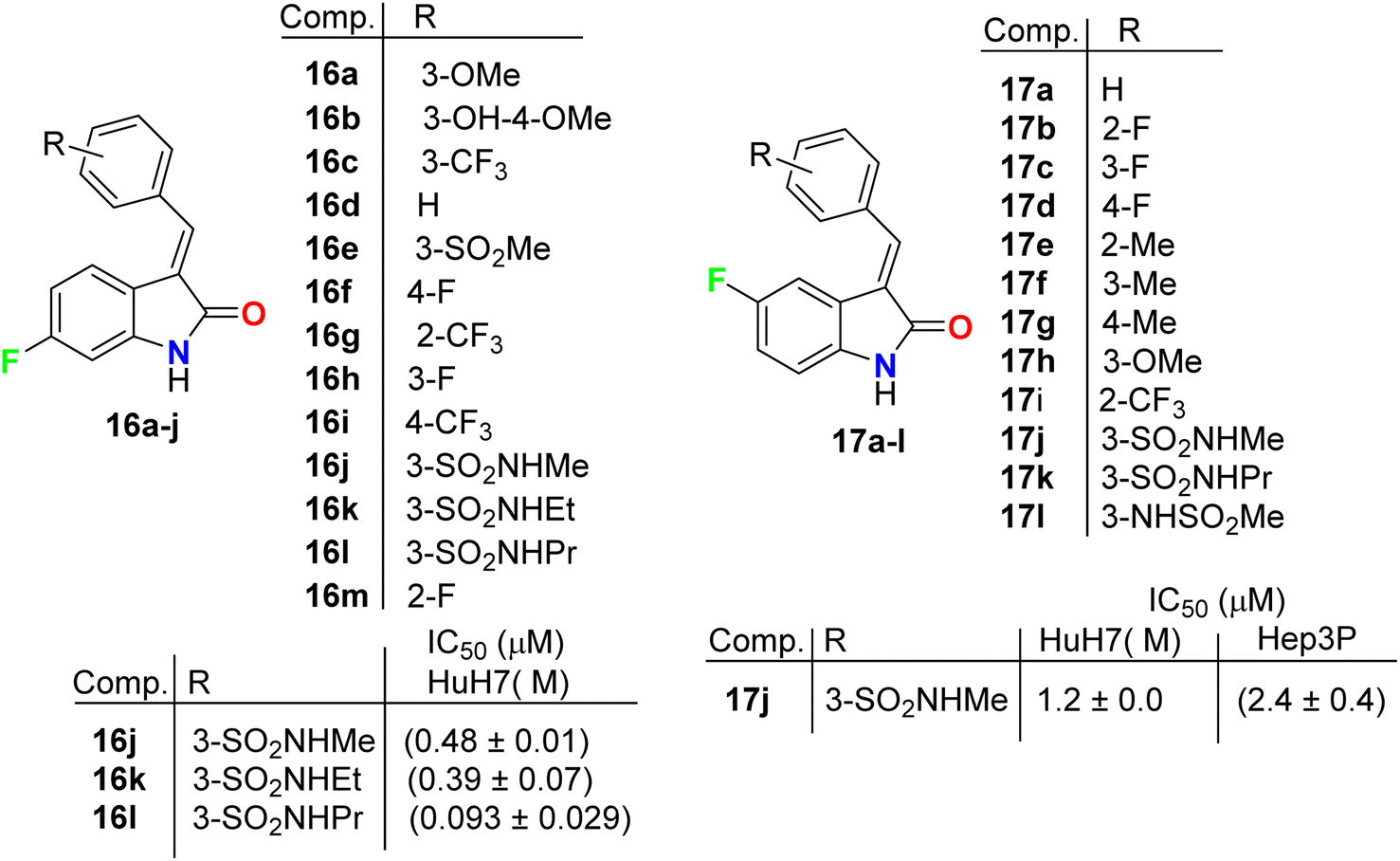
Chen et al. described the synthesis of the 5(6)-monofluorinated indolin-2-one, series 16 and 17 bearing the aminosulfonyl moiety, and screened their growth inhibitory activities on HCC (HuH7 and Hep3B) cells (Fig. 10). It was reported that they showed large variations of activities with IC50 values 0.09 μM to >30 μM (HuH7) and 0.36–13.6 μM (Hep3B). The 6-monofluorinated indolin-2-one 16l was found to be the most potent compound in this series, where IC50 values were 0.09 μM (HuH7) and 0.36 μM (Hep3B), superior to both sunitinib and sorafenib. The IC50 values of sunitinib and sorafenib were 5.6 and 5.4 (HuH7) and 5.2 and 5.4 (Hep3B). Thus, structure 16l was considered as a lead structure for drug design investigations.46 All fluorinated indolin-2-one derivatives inhibited the phosphorylation of RTKs in HuH7, notably FGFR4 and HER3, and decreased tumor load in a mouse model.
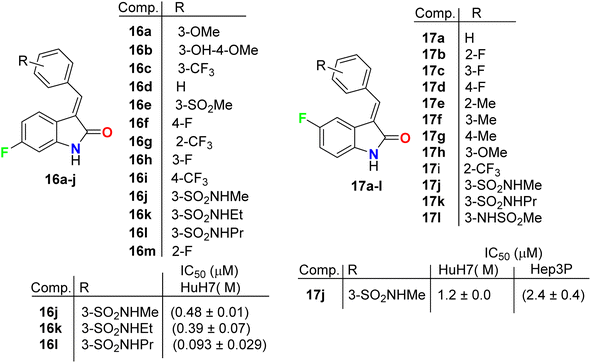 |
| | Fig. 10 Structure of fluorinated indolinone derivatives 16a–k and 17a–l. | |
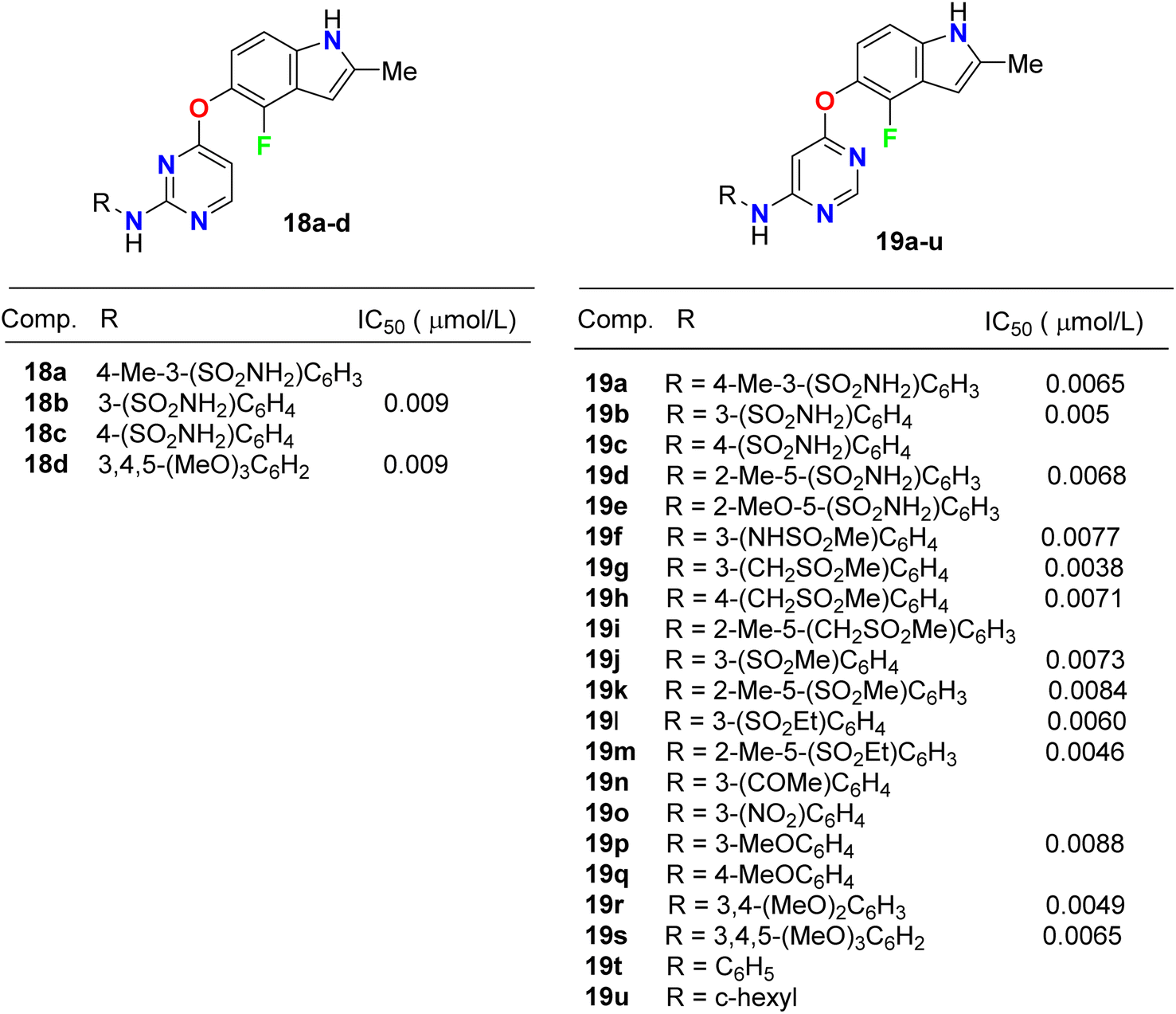
Inhibition of the VEGFR-2 signaling pathway has already become one of the most promising approaches for the treatment of cancer. Gao et al. designed and synthesized a series of 4-fluoroindole derivatives 18 and 19 and evaluated their inhibitory activity of VEGF(R)-2 kinase inhibition (Fig. 11). Most of these compounds demonstrated high anti-angiogenesis activities via VEGFR-2 in enzymatic proliferation assays on a nanomolar scale. In particular, compound 19g showed significant activity with an IC50 value of 3.8 nM. SAR was reported to study the effect of substitutions at the aniline ring linked to the pyridine motif on the activity. The SAR result declared that the removal of the p-methyl group led to a higher potency against VEGFR-2. The presence of the p-sulfonamide group dramatically decreased the potency by 3.4-fold (IC50 = 31 nM) than the m-sulfonamide group (IC50 = 9 nM). Thus, the effect of the position of substituent was crucial. Compound 18c showed similar potency with compound 18b (IC50 = 31 nM). Furthermore, 4,6-disubstituted compounds showed an improved VEGFR-2 inhibitory potency than those having 3,4-disubstituted or trimethoxy groups on the ring. SAR was also reported to investigate the effect of substitutions at the aniline ring linked to the pyrimidine motif on the activity. Compounds (19f–19o) with an electron-withdrawing group at the aniline ring exhibited high VEGFR-2 inhibitory activities; particularly, compound 19g displayed a potent inhibitory activity with an IC50 value of 3.8 nM. The selectivity of derivative 19g was evaluated on a panel of tyrosine kinases, where it showed good potency against VEGFR-1, PDGFR-α, and PDGFR-β, with high selectivity over VEGFR-3 and excellent selectivity over ErbB2, ErbB4, EGFR, ABL, EPH-A2, FGFR-1, FGFR-2, and IGF-1R. The described results confirmed that such compounds could act as lead structures for the development of more selective anticancer medication.47
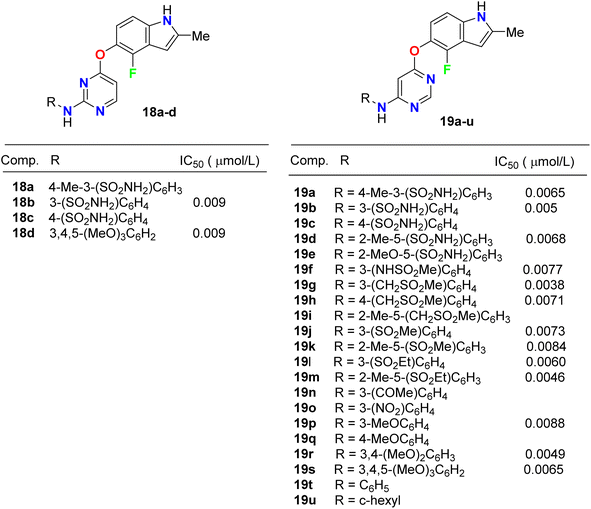 |
| | Fig. 11 Structure of 4-fluoroindole derivatives 18 and 19. | |
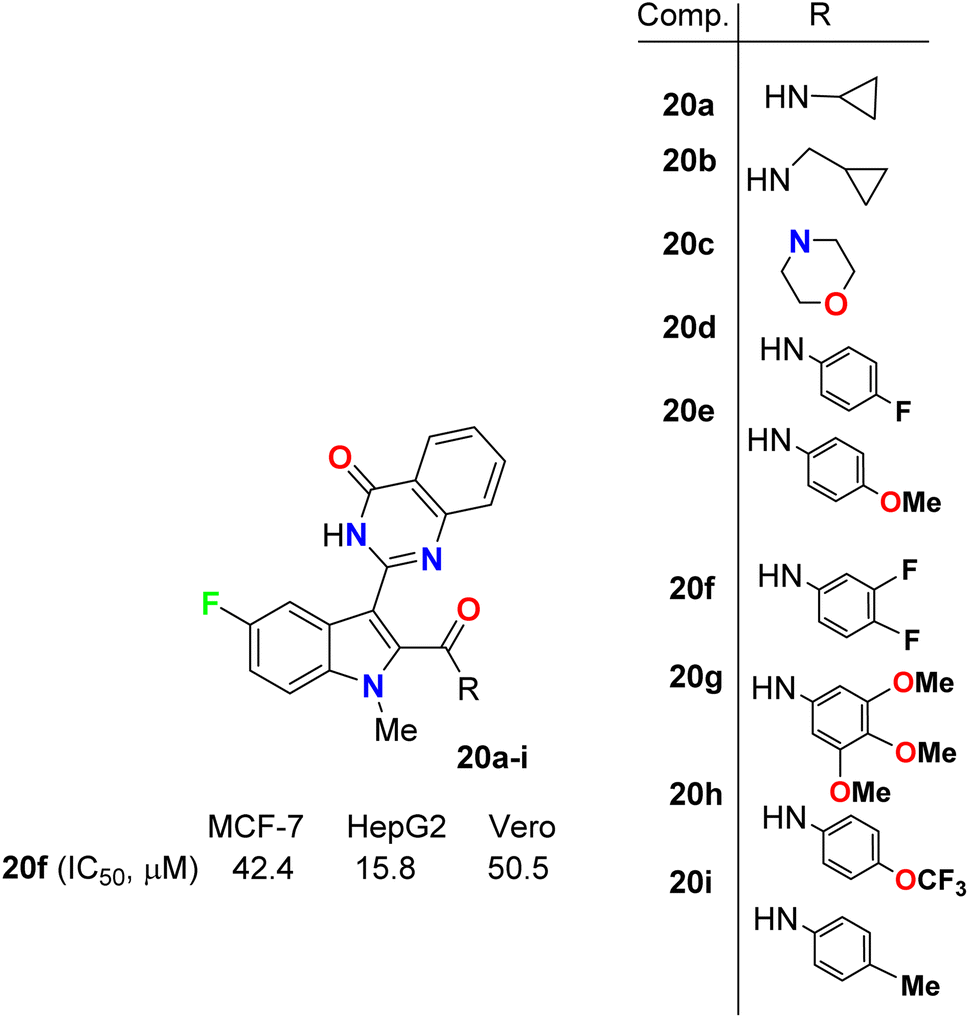
Gokhale et al. described the synthesis of a series of quinazolinone-based 5-fluoroindole hybrids 20a–i (Fig. 12) and studied their in vitro cytotoxic activity using a sulphorhodamine B (SRB) bioassay method. The bioassay was carried out against MCF-7 (human breast adenocarcinoma), HepG2 (human liver hepatocellular) and the non-tumorigenic Vero cell lines. Among the synthesized compounds, 20a, 20f, and 20i provided significant activities against the tested cell lines. It was also reported that HepG2 cells were more sensitive to all the synthesized hybrids than MCF-7 cells. Compound 20f presented the highest potency against MCF-7, HepG2 and Vero cell lines with IC50 values of 42.4, 15.8 and 50.5 μM compared with 23.7, 18.8 and 45 μM for the standard reference 5-fluorouracil. The presence of three fluorine atoms in 20f might be responsible for its higher activity in all the cell lines. Thus, compound 20f showed substantial anticancer activity and could be used as a lead for further investigations where it had very low toxicity against the non-cancerous Vero cell line.48
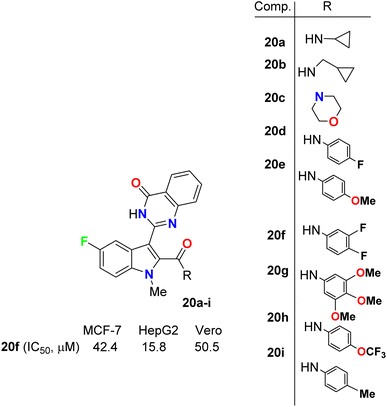 |
| | Fig. 12 Structure of 5-fluoroindole derivatives 20a–i. | |
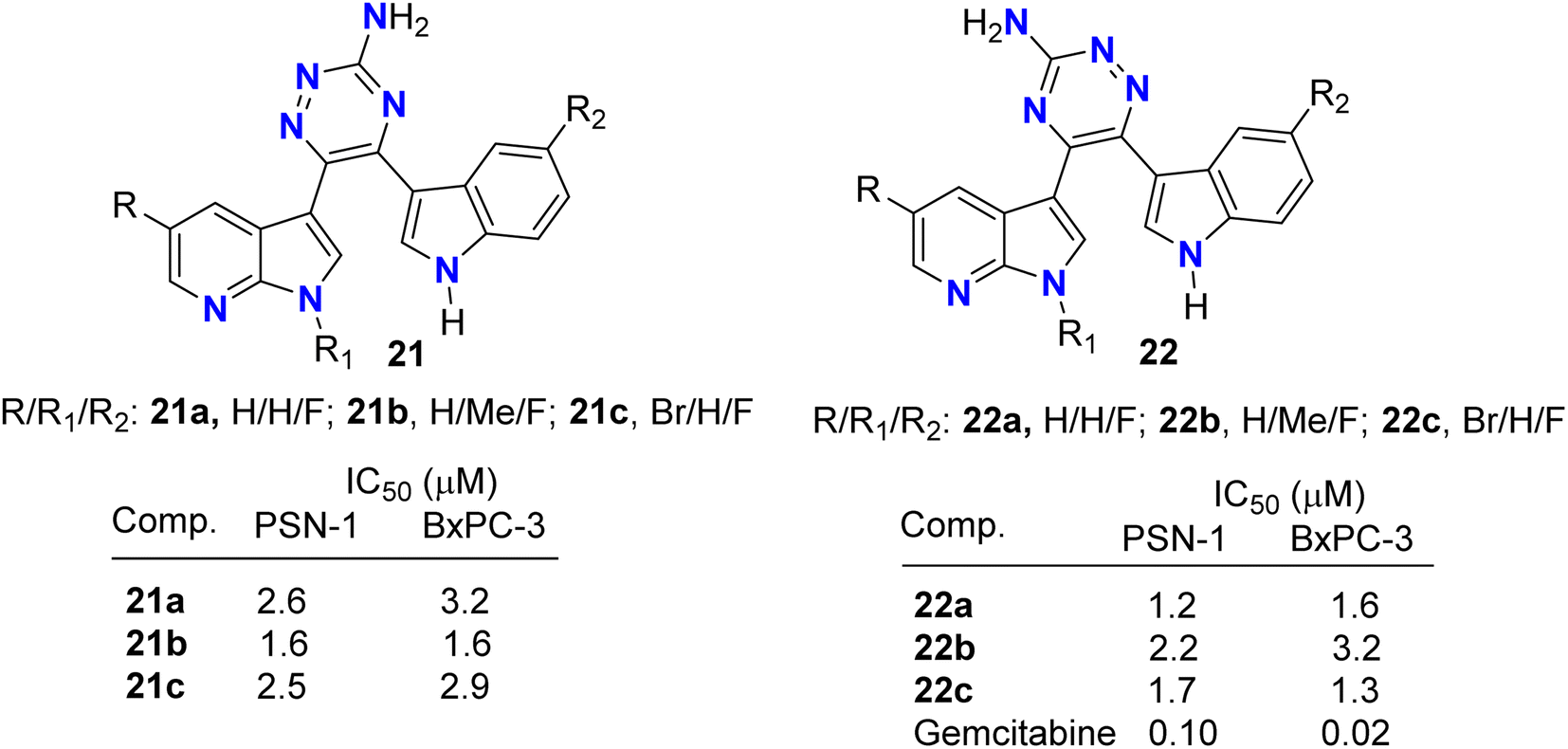
Pecoraro and co-workers49 synthesized two series of the amino-triazine derivatives 21 and 22 (Fig. 13) to test their ability to lock into the nucleotide-binding pocket, occupying the space of the ATP and hampering the kinase enzymatic activity. The modulatory effect of the amino-triazine derivatives on the pyruvate dehydrogenase kinases (PDKs) was tested, where many derivatives were found to modulate the PDKs enzymatic activity with a marked selectivity against the PDK1 and PDK4 isoforms. Considering the high degree of similarity between PDK1 and HSP90 (heat shock protein 90), compounds 21c and 22c were tested for their ability to inhibit its enzymatic activity (Fig. 13). Interestingly, the two fluorinated derivatives, 21c and 22c, were extremely active against HSP90. These results were also validated by molecular modelling, which demonstrated the ability of the newly synthesized triazine derivatives to fit into the nucleotide-binding pocket of PDK1. The newly developed molecules of types 21 and 22 also exhibited very promising anti-proliferative activities against KRAS (mutated pancreatic cancer) wild-type and mutant PDAC cells, namely BxPC-3 and PSN-1, respectively, with IC50 values ranging from low-micromolar to sub-nanomolar level. These findings supported further development of new classes of PDK inhibitors relevant to PDAC treatment.
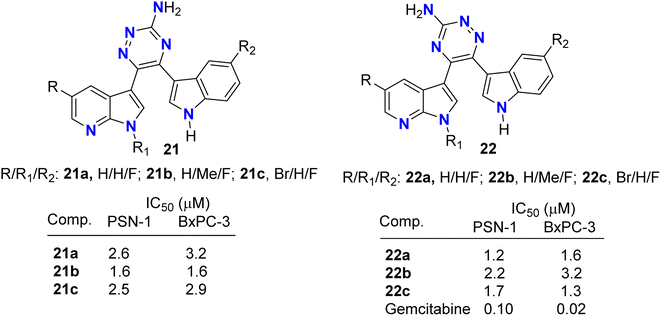 |
| | Fig. 13 Structure of aminotriazine derivatives 21 and 22. | |
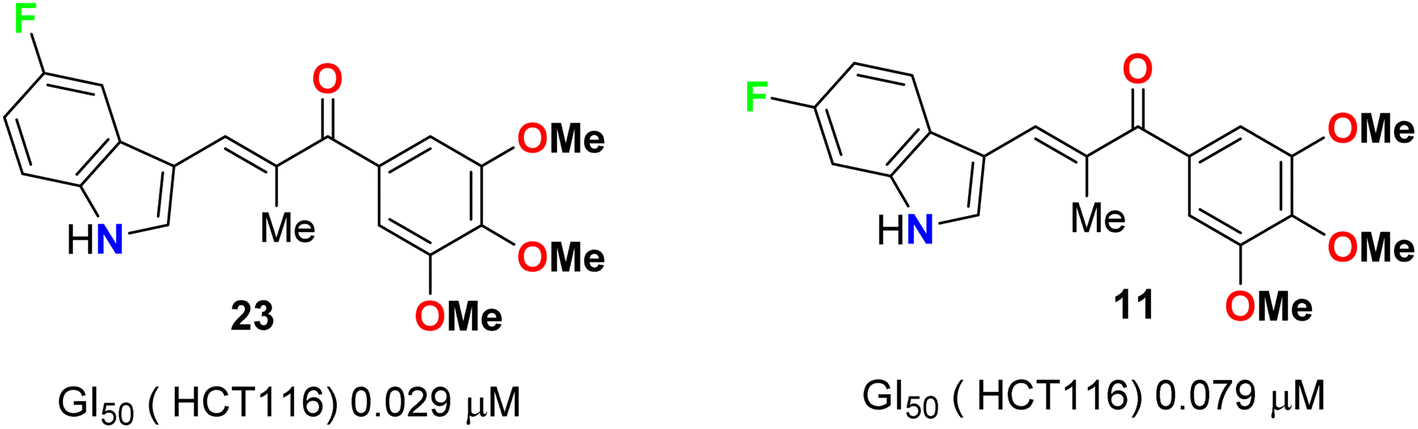
A series of chalcone-based 5(6)-fluoroindole derivatives 23 and 11 (Fig. 14) were designed, and their inhibitory activity against colorectal cancer (CRC) was explored. Compound 11 exhibited significant inhibitory activity toward HCT116 cells (IC50 = 4.52 nM), CT26 cells (IC50 = 18.69 nM), CRC organoids (IC50 = 1.8–2.5 nM), HCT116-xenograft mice (TGI value of 65.96% at the dose of 3 mg kg−1), and APCmin/+ mice (adenoma number inhibition rate of 76.25% at the dose of 3 mg kg−1). Meanwhile, the related mechanisms mediated by 11 for CRC were also studied in detail. This study supported that indole-chalcone compounds were a class of promising lead microtubule-targeting drugs (MTAs) and highlighted the potential of 11 to combat CRC.50
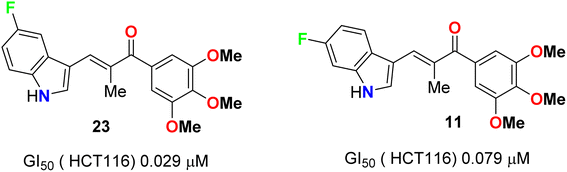 |
| | Fig. 14 Structure of 5(6)-fluoroindole derivatives 23 and 11. | |
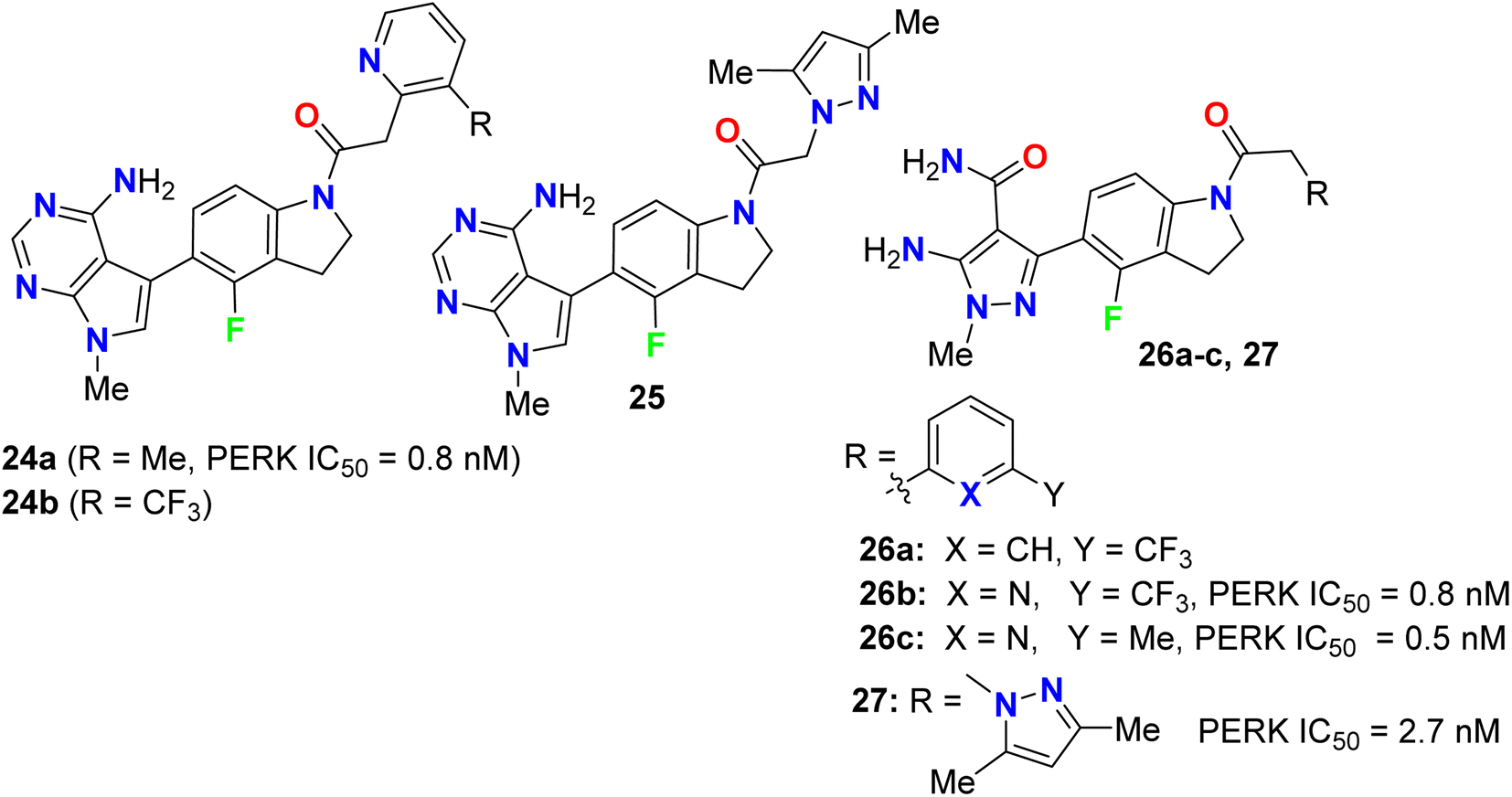
Synthesis and anticancer activity of the 4-fluoroindoline derivatives 24a,b and 25 were studied (Fig. 15). The anticancer activity was measured via selective inhibition of endoplasmic reticulum kinase (PERK) enzyme assay. All the synthesized 4-fluoroindoline derivatives displayed PERK inhibitory activity with IC50 ranging between 0.5 and 2.5 nM. The derivative 24a showed a 3-fold increase in the PERK inhibitory activity (IC50 = 0.8 nM) compared with its non-fluorinated analogue (IC50 = 2.5 nM). Thus, the effect of fluorine substitution was substantial for the inhibition of PERK. Owing to the potent activity of 24a, it was selected for advancement to preclinical development. In addition, all the fluorinated compounds 26a–c and 27 (Fig. 15) were tested for activity against Endoplasmic Reticulum Kinase (PERK) enzyme and exhibited a pIC50 value > 6.9 against PERK [where pIC50 = −log(IC50)].51,52
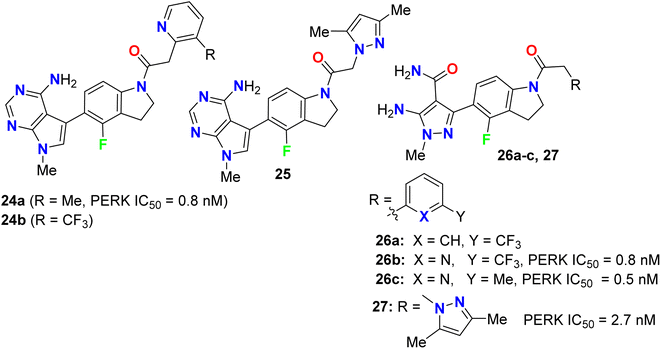 |
| | Fig. 15 Structure of 4-fluoroindoline derivatives 24a,b, 25, 26a–c and 27. | |
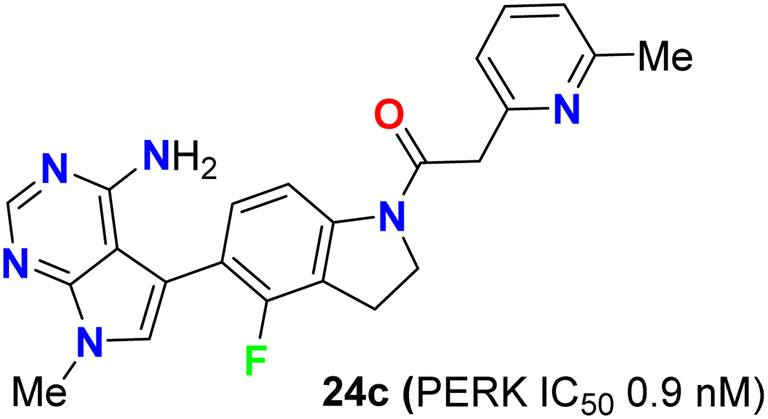
The biological study of compound 24c (Fig. 16) was extensively reported.53 It was found that treatment of mice with compound 24c led to inhibition of tumor growth in multiple human tumor xenografts. This compound was chosen as a candidate for preclinical studies. It was found to be an ATP-competitive inhibitor of PERK enzyme activity with an IC50 of 0.9 nM. It was very selective for PERK against a panel of 300 kinases with IC50 in the range of 10–30 nM. When compound 24c was orally administered to mice, it showed a time and dose-dependent pharmacodynamic response in the pancreas as measured by PERK autophosphorylation. Dose-dependent inhibition of multiple human tumor xenograft growth in mice was reported for twice daily dosing of compound 24c. Thus, compound 24c was assigned as a lead for the development of any PERK inhibitor in human subjects.53
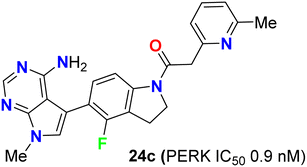 |
| | Fig. 16 Structure of 4-fluoroindoline derivative 24c. | |
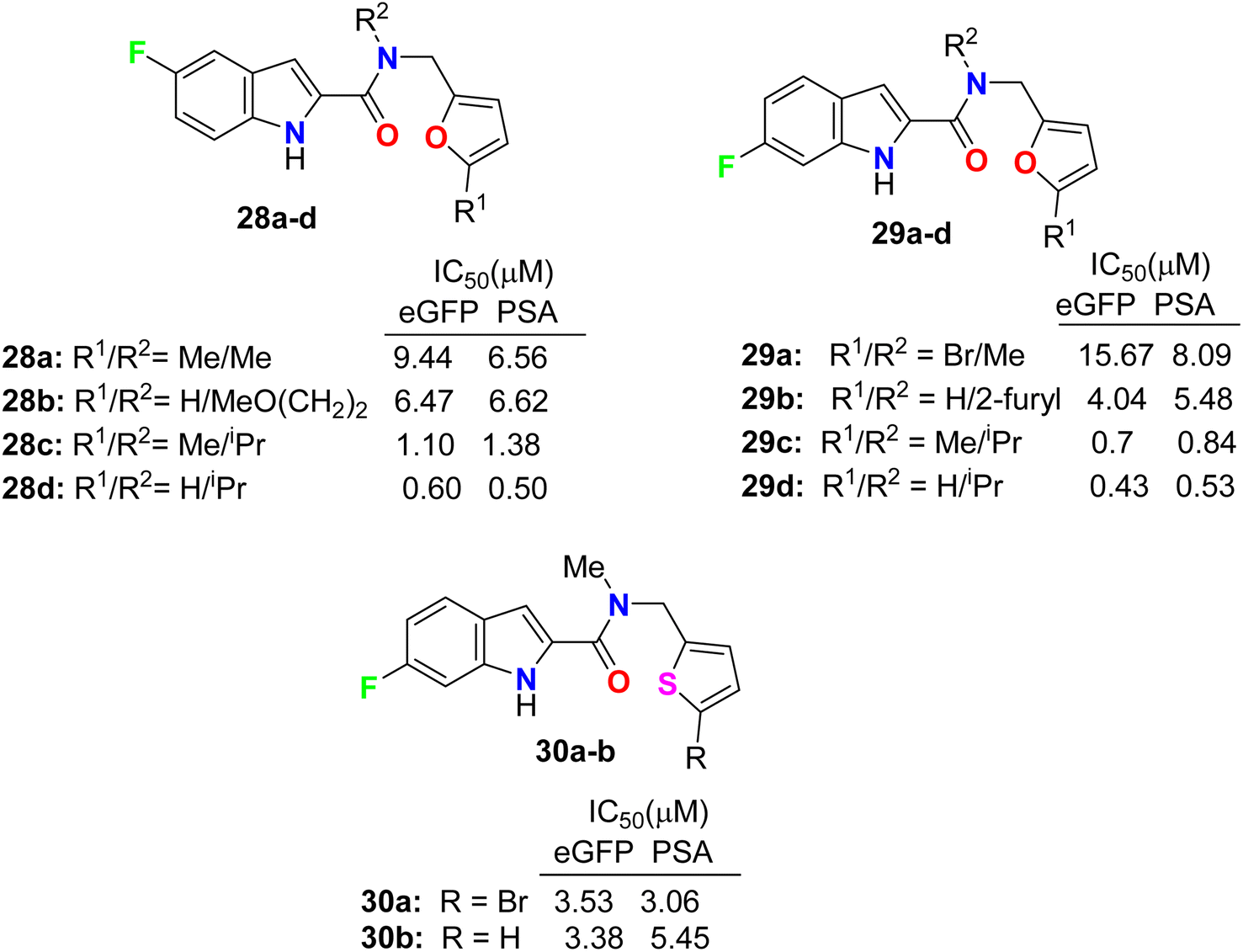
Some 5(6)-fluoroindole-carboxamide derivatives, 28, 29 and 30 (Fig. 17), were designed and described as inhibitors of androgen receptor binding function-3 (AR-BF3) using an enhanced green fluorescent protein (eGFP) AR transcriptional bioassay. Most of the synthesized compounds were noticed as promising selective AR-BF3 inhibitors and exhibited substantial activity against wild-type and drug-resistant prostate cancer cells. From the tested series, three compounds, 29c, 28d, and 29d, were the most potent BF3 inhibitors with eGFP IC50 values of 0.7, 0.6, and 0.43 μM, respectively. Compounds 29c, 28d, and 29d also had the ability to reduce the levels of prostate-specific antigen (PSA), which is a serine protease, and showed IC50 values of 0.84, 0.5, and 0.53 μM, respectively. Compound 29c had more solubility than compounds 28d and 29d at high concentrations; thus, compound 29c, having IC50 values of 0.70 and 0.84 μM in eGFP and PSA, respectively, was assigned as a possible candidate for future clinical applications.54,55
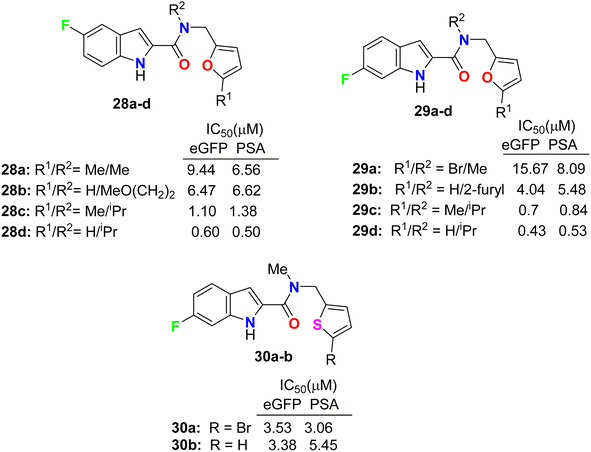 |
| | Fig. 17 Structure of 5(6)-fluoroindole-carboxamide derivatives 28, 29 and 30. | |
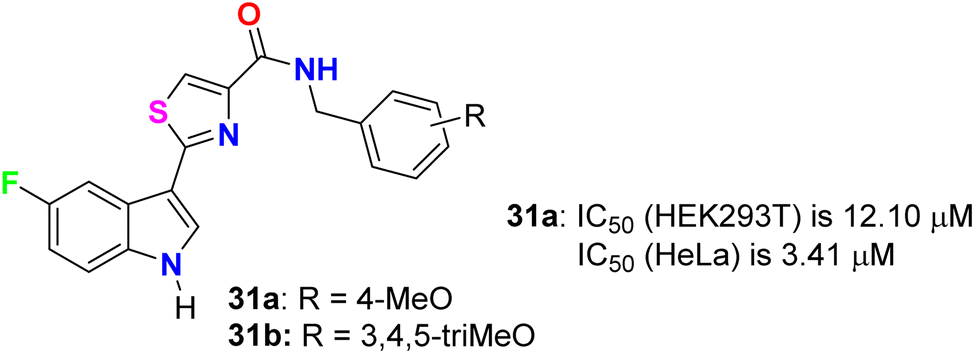
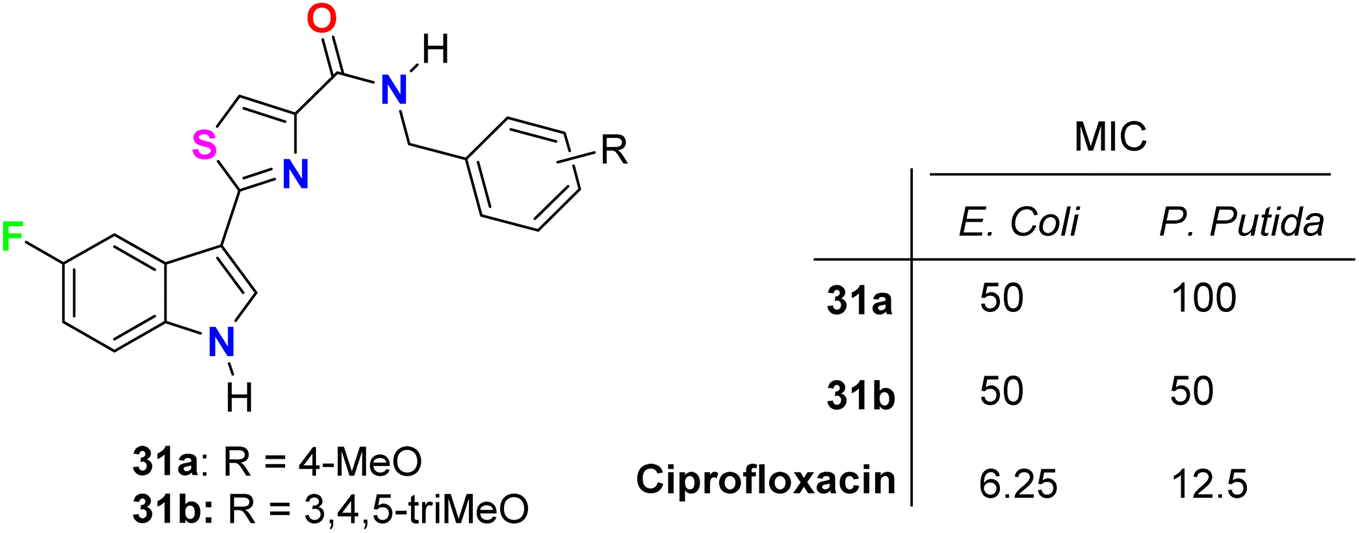
Tantak et al. described the synthesis of the thiazole-based 5-fluoroindole derivatives 31a,b (Fig. 18) and investigated their in vitro anticancer activities in human cervical (HeLa), breast (MDA-MB- 231), embryonic kidney 293 (HEK293T), prostate (PC-3, LNCaP and castration-resistant prostate cancer cell line C4-2) cancer cell lines. The MTT bioassay was conducted in the presence and absence of FBS (fetal bovine serum) using doxorubicin as a reference drug. Compound 31a exhibited selective cytotoxicity towards HEK293T and HeLa cells with IC50 values of 12.10 and 3.41 μM, compared with doxorubicin IC50 = 0.84 and 0.45 μM, respectively, without FBS; however, the analogue 31b was inactive. In addition, compound 31a showed lower potency against HeLa cell lines with IC50 = 32.48 μM and was inactive towards HEK293T, in the presence of FBS. The mechanism of action disclosed that the cytotoxicity against HeLa cells employed the induction of cell death by apoptosis.56
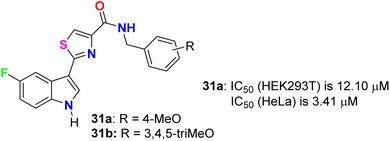 |
| | Fig. 18 Structure of thiazole-based 5-fluoroindole derivatives 31a,b. | |
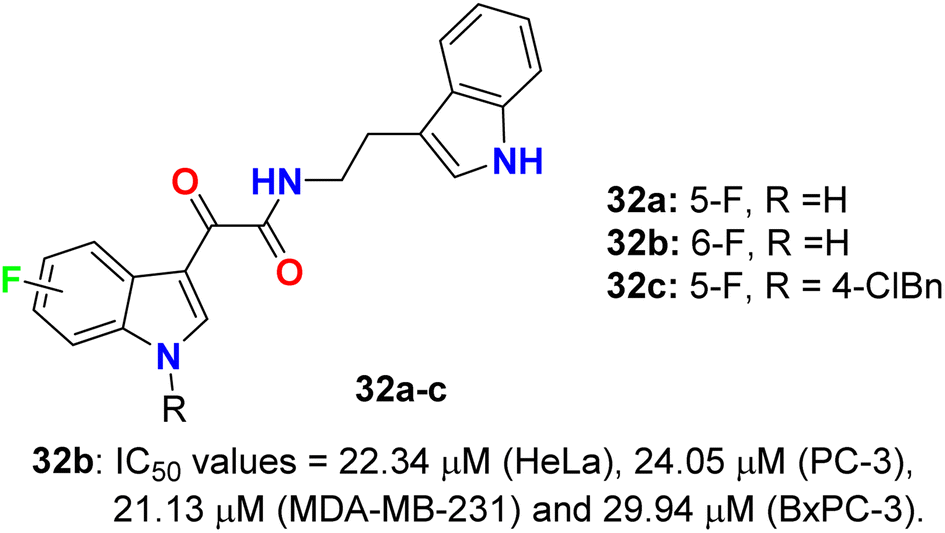
The glyoxylamide-based 5(6)-fluoroindole derivatives 32a–c (Fig. 19) were synthesized and assigned for their in vitro cytotoxicity against a panel of human cancer cell lines; HEK293T, MDA-MB-231, HeLa, PC-3, C4-2, and pancreatic BxPC-3 using doxorubicin as a reference drug. The bioassay experiments disclosed that 6-fluoroindole 32b demonstrated the best cytotoxicity with IC50 values 22.34 μM (HeLa), 24.05 μM (PC-3), 21.13 μM (MDA-MB-231) and 29.94 μM (BxPC-3). Treatment of 32b with PC-3 cells led to enhancing the levels of cleaved PARP1, indicating that compound 32b induced apoptosis in PC-3 cells. Interestingly, compound 32b displayed no toxicity in mammalian cells. Compounds 32a, and 32c were completely inactive towards all the tested cell lines.57
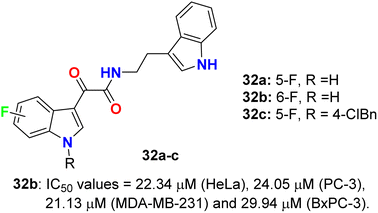 |
| | Fig. 19 Structure of 5(6)-fluoroindole derivatives 32a–c. | |
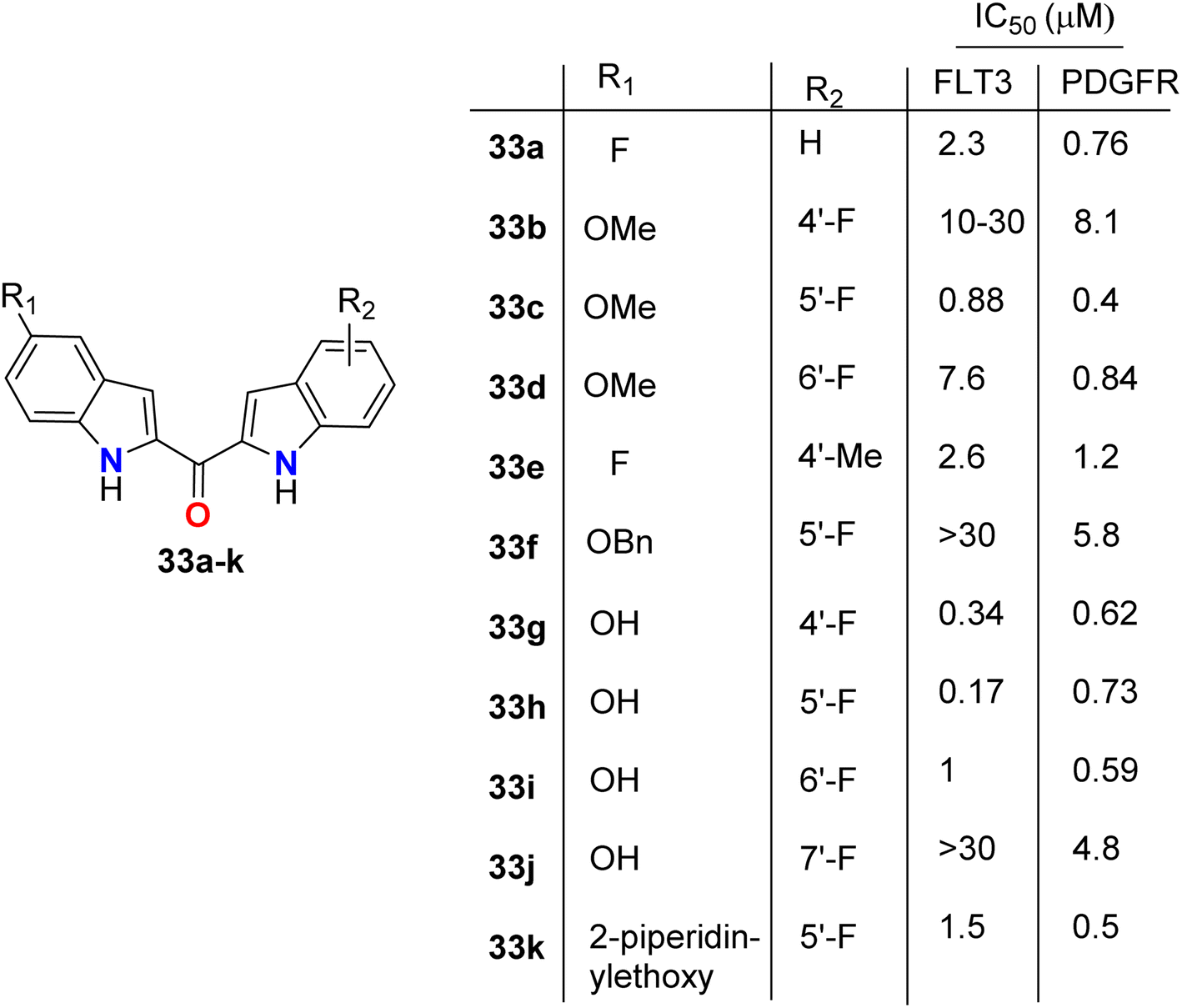
The directly fluorinated bis-indole derivatives 33a–k (Fig. 20) were synthesized by Mahboobi et al. and examined their inhibitory activity against Fms-like tyrosine kinase 3 (FLT3), which is active in many cases of acute myeloid leukemia (AML), and the platelet-derived growth factor receptor tyrosine kinase (PDGFR). Most of the derivatives exhibited selectivity towards both FLT3 and PDGFR kinases; in particular, compounds 33g and 33h were the best active FLT3 inhibitors with IC50 values of 0.34 and 0.17 μM, respectively. Compounds 33c and 33k showed the highest PDGFR inhibitory activity with IC50 = 0.4 and 0.5 μM, respectively. SAR study showed that the 5-F substitution (33a) reduces activity at both kinases, where fluorine atom might interact with the inner pocket as indicated by the results of the 5-methoxy, 5-benzyloxy, and 5-piperidinylethoxy derivatives 33c, 33f, and 33k, respectively. The larger substituents with polar termini were superior to the more hydrophobic groups, where a 10-fold decrease of PDGFR inhibition from 5-piperidinylethoxy 33k to 5-benzyloxy 33f compounds was reported. It was mentioned that the more lipophilic group was preferentially transferred into the interior via hydrophobic patches.58
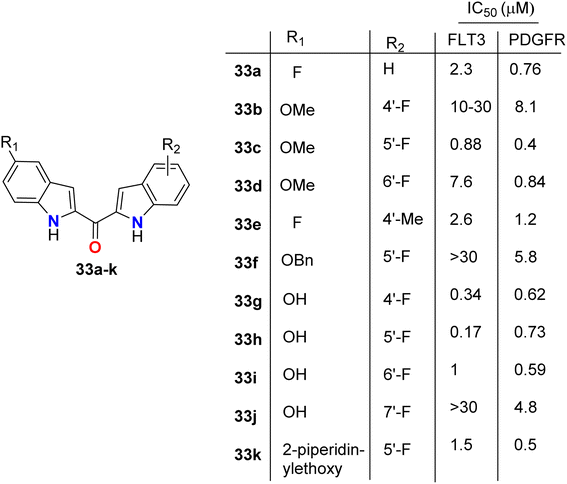 |
| | Fig. 20 Structure of fluorinated bis-indole derivatives 33a–k. | |
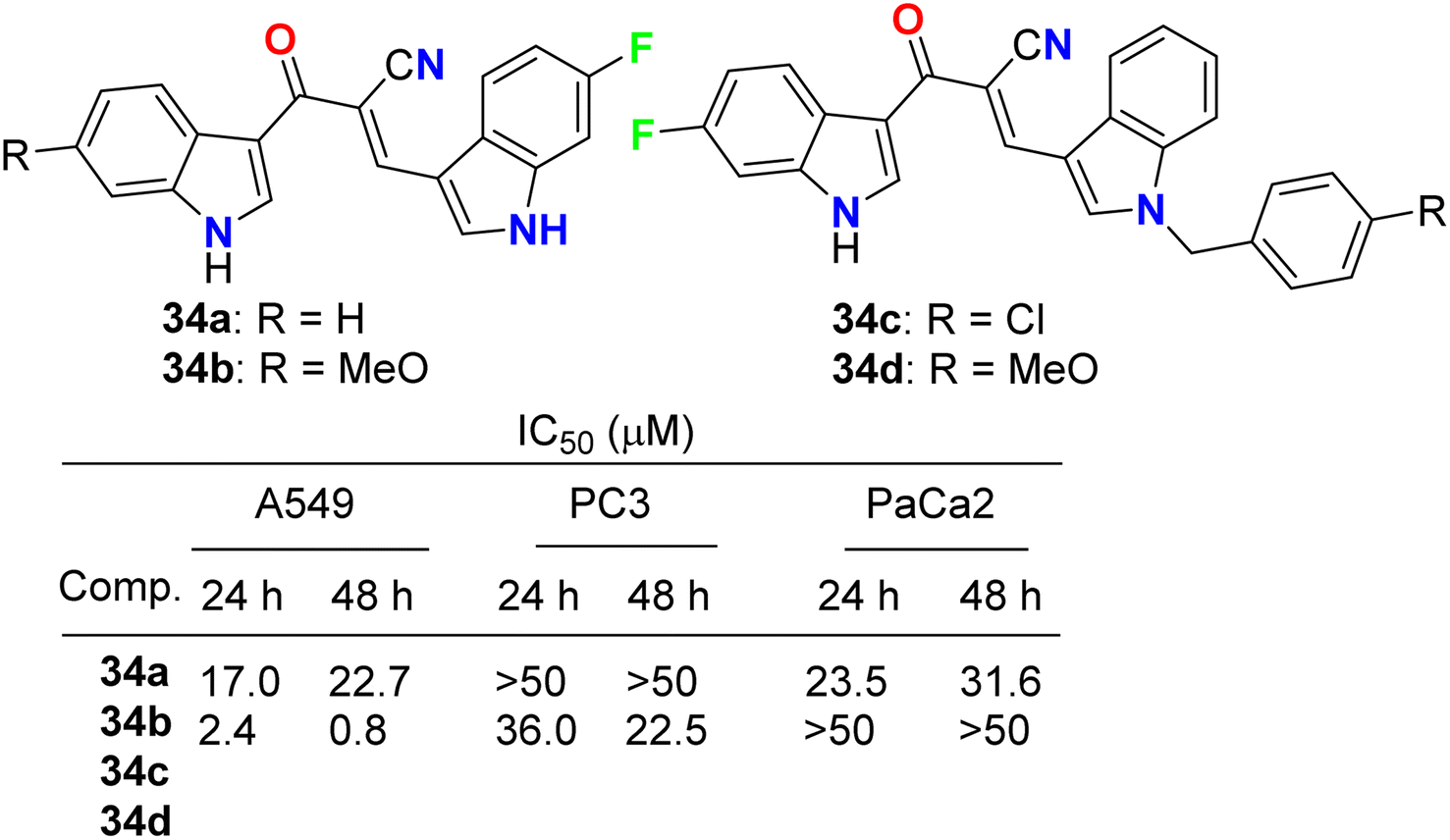
Kumar et al. reported the synthesis and anticancer activity of the fluorinated-indole derivatives 34a–d (Fig. 21). The bioassay experiment was carried out against three human cancer cell lines: prostate (PC3), lung (A549), and pancreas (PaCa2), following the WST-8 protocol. The synthesized compounds showed moderate to high anticancer activities, and in particular, compound 34b was found to be the most potent inhibitor and selective against A549 cells with an IC50 value of 0.8 μM. The designed hybrids were found to increase tubulin polymerization, giving these hybrids a chance to act as microtubule-stabilizing agents. SAR study assigned that compound 34b, having fluoro and methoxy groups at the C-6 position of the two indole rings, exhibited the best anticancer activity against A549 cells.59
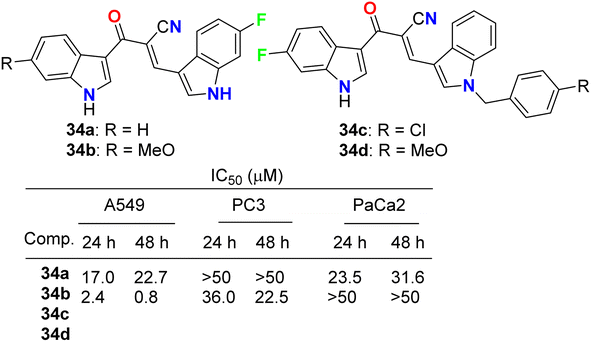 |
| | Fig. 21 Structure of fluorinated-indole derivatives 34a–d. | |
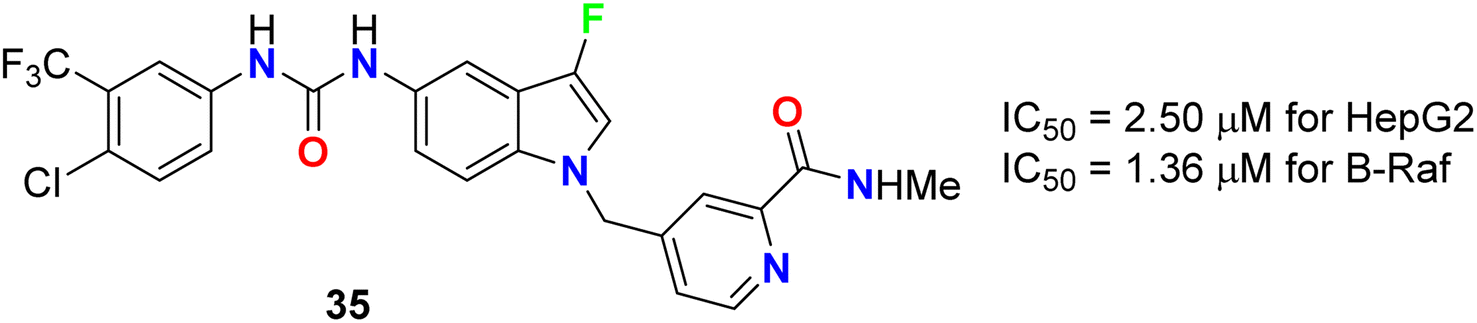
The 3-fluoroindole derivative 35 was designed and synthesized by Wu et al. and evaluated for its inhibitory activity against the HepG2 cell line and B-Raf kinase (Fig. 22). Compounds 35 displayed substantial inhibitory potency in HepG2 cells and B-Raf with an IC50 value of 2.50 and 1.36 μM, compared with the reference drug sorafenib (IC50 values of 14.95 and 0.032 μM), respectively. Compound 35, which showed better inhibitory activity in HepG2 with 6-fold improvement than sorafenib, provided the potential for further research as a lead compound.60
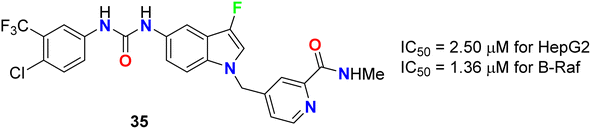 |
| | Fig. 22 Structure of 3-fluoroindole derivative 35. | |
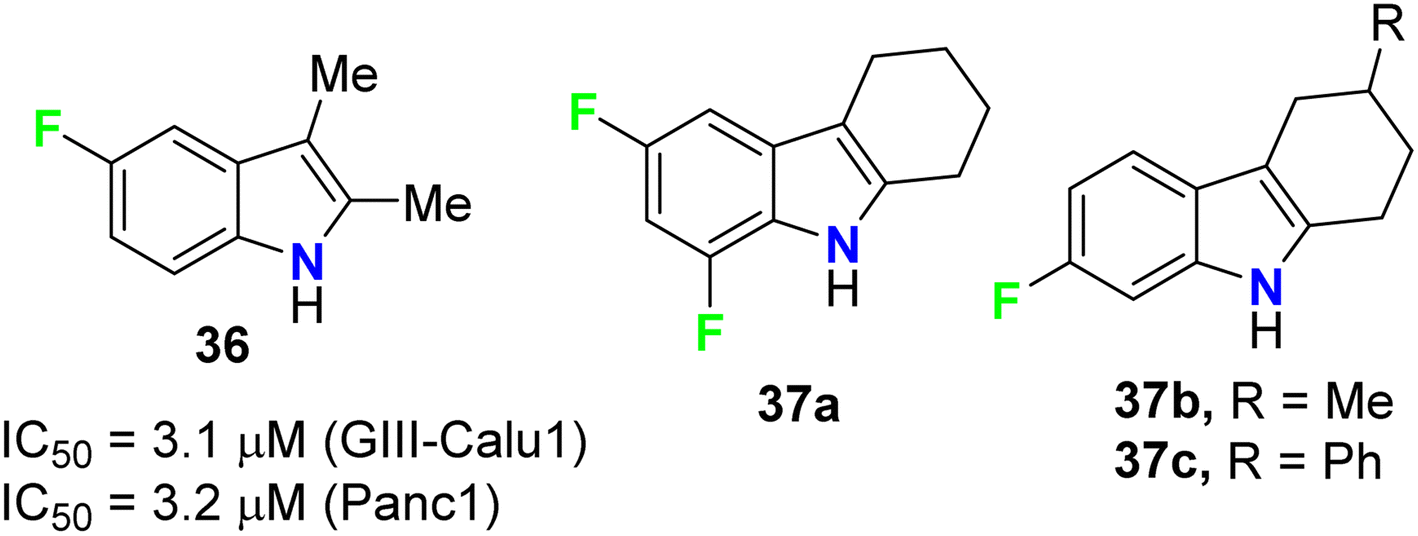
The anticancer activities of the fluorinated 2,3-dimethylindole 36 and tetrahydrocarbazole 37a–c derivatives (Fig. 23) were performed against human pancreas carcinoma (Panc1), lung carcinoma (GIII) (Calu1), kidney adenocarcinoma (ACHN), colon cancer cell (HCT116), non-small cell lung carcinoma (H460), and normal breast epithelium (MCF10A) cell lines. The bioassay results, sing propidium iodide (PI) staining method, confirmed that compound 36 exhibited significant activity against both GIII-Calu1 and Panc1 cell lines with IC50 values 3.1 and 3.2 μM, respectively. The fluorinated tetrahydrocarbazoles 37a–c presented good activity against all the tested cell lines with IC50 ranging between 4.9–7.4 μM. Noteworthy, all the derivatives 36 and 37a–c were inactive against the normal cell line MCF10A.61
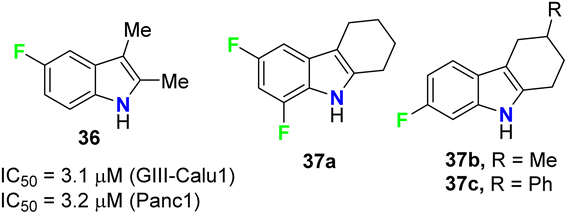 |
| | Fig. 23 Structures of fluorinated 2,3-dimethylindoles 36 and tetrahydrocarbazoles 37a–c. | |
2.3. Anticancer activity of fluorinated pyrazoles and indazoles
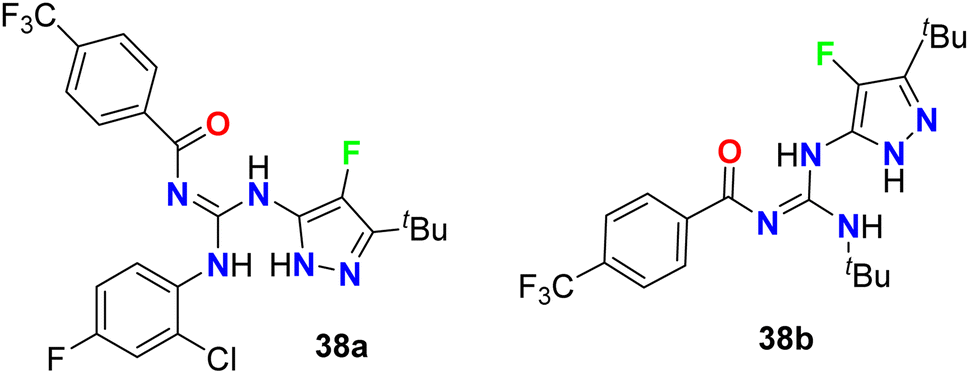
The 4-fluoropyrazole derivatives, 38a and 38b, were patented by Glick et al. as inhibitory agents for hydrolytic activity of mitochondrial F1F0-ATP-ase (Fig. 24). It is known that transport ATP-ases carry out the hydrolysis of ATP, leading to the transport of ions for the treatment of tumors. The inhibitory activity of 4-fluoropyrazole derivatives 38a and 38b against F1F0-ATPase was conducted by testing their ability to inhibit ATP synthesis, and the IC50 values were <10 μM. Furthermore, assessment of compounds 38a and 38b for their cytotoxicity in human Ramos cells was reported, and the bioassay results declared the EC50 values < 10 μM.62
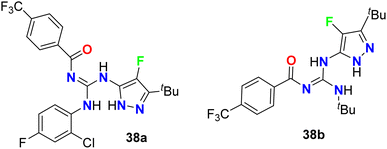 |
| | Fig. 24 The structure of 4-fluoropyrazole derivatives 38a and 38b. | |
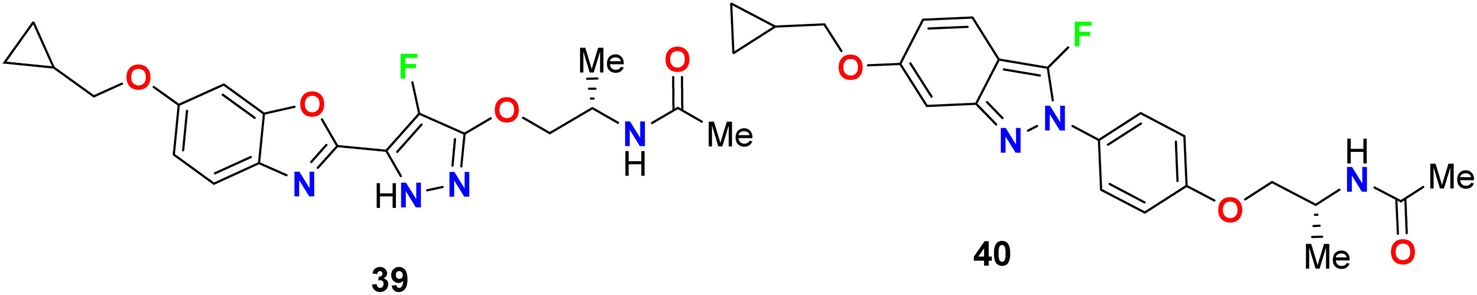
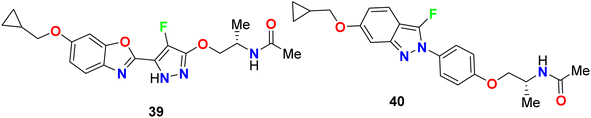
Yasuma et al. patented the synthesis and the acetyl-CoA carboxylase 2 (ACC2) inhibitory activity of fluoropyrazole derivatives 39 and 40 (Fig. 25).63 ACC2 inhibitory action is useful for the treatment of obesity, diabetes and cancer. The inhibitory rates (%) against ACC2 of compounds 39 and 40 were 89% and 99%, respectively, at 10 μM.
 |
| | Fig. 25 Structure of fluoropyrazole derivatives 39 and 40. | |
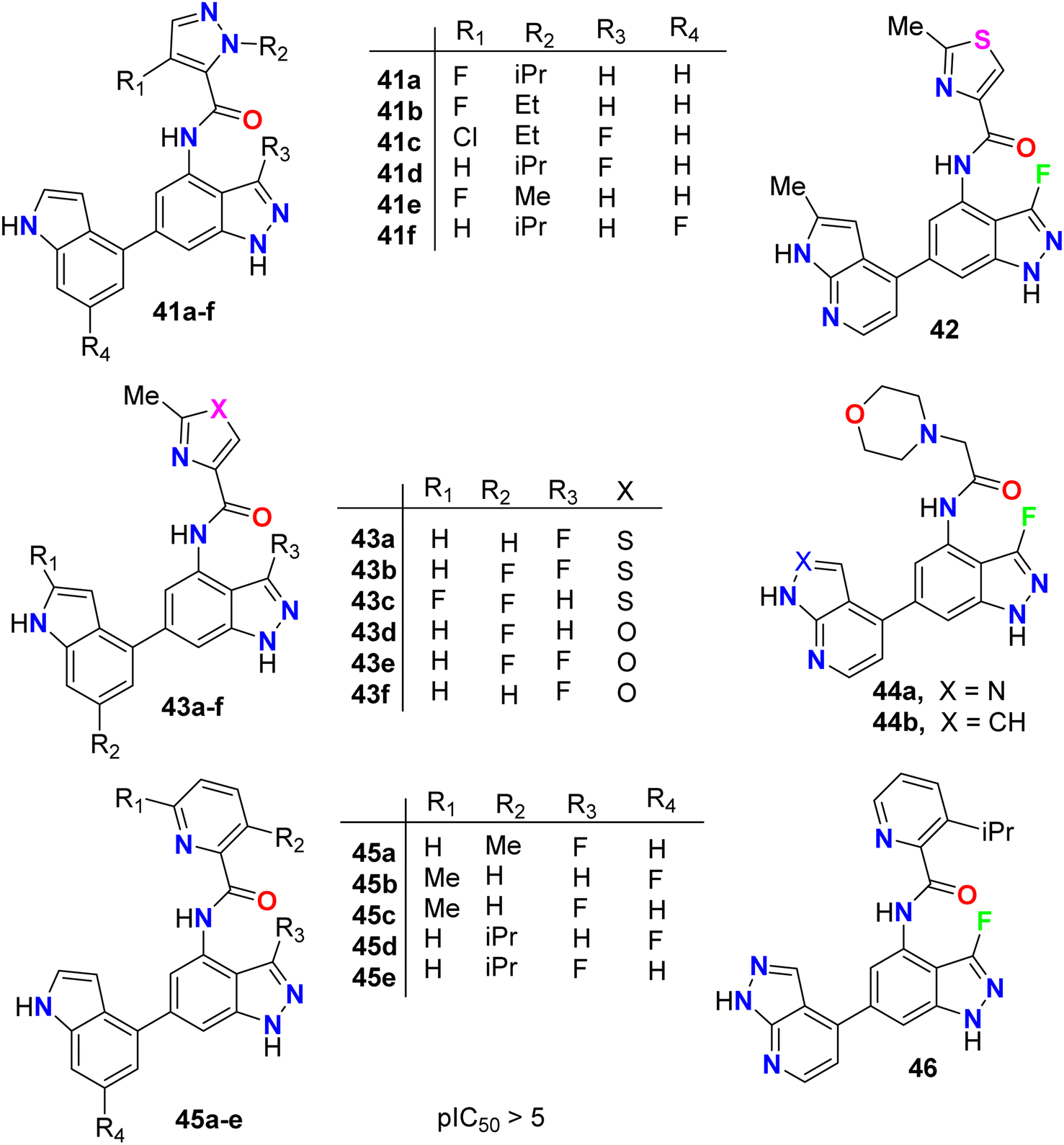
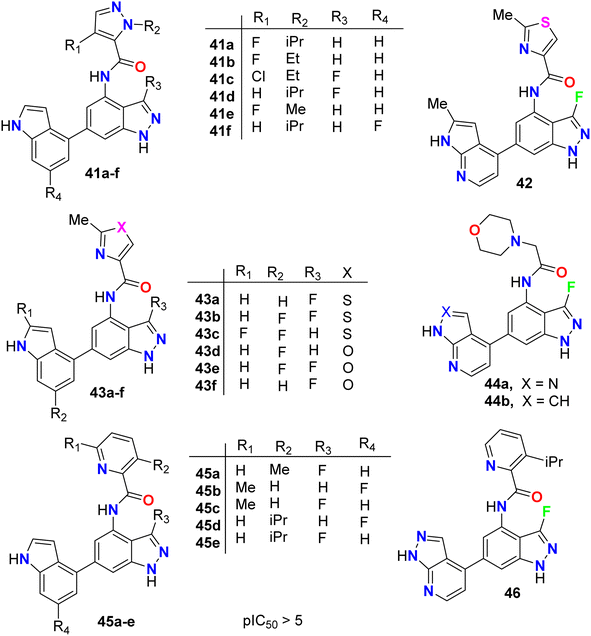
The anticancer activity of the fluorinated indazole derivatives 41–46 (Fig. 26) via inhibition of phosphoinositide-3′-OH kinase (PI3-kinases) was investigated by Baldwin et al.64–67 The prepared examples 41–46 were tested in one or more of the PI3Kδ, PI3Kα, PI3Kβ and/or PI3Kγ assays and were found to have mean pIC50 values ≥5. Certain compounds were also tested in the T-cells using flow cytometry and were reported to have mean pIC50 values ≥5.
 |
| | Fig. 26 Structure of fluorinated indazole derivatives 41–46. | |
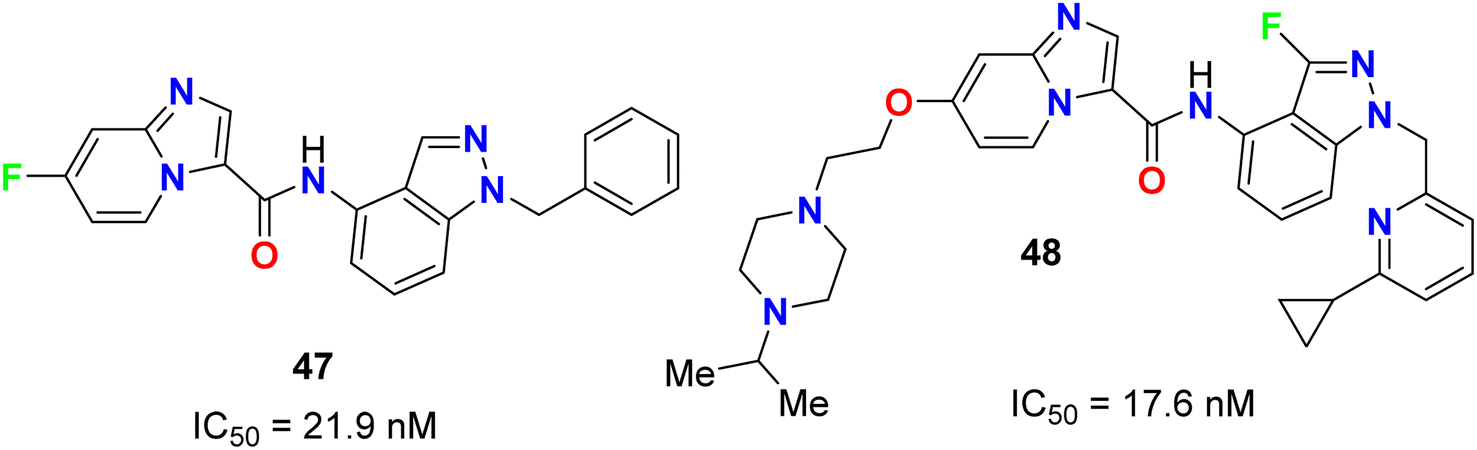
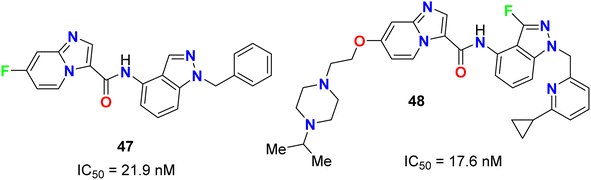
The fluoroindazole derivatives 47 and 48 were estimated by Boys et al. as inhibitors of PDGFR, a type III receptor tyrosine kinase, for the treatment of cancer (Fig. 27). The synthesized compounds 47 and 48 were evaluated for their inhibitory action of PDGFR-β phosphorylation in the human fibroblast cell line (HS27) with IC50 values of 21.9 and 17.6 nM, respectively.68
 |
| | Fig. 27 Structure of fluoroindazole derivatives 47 and 48. | |
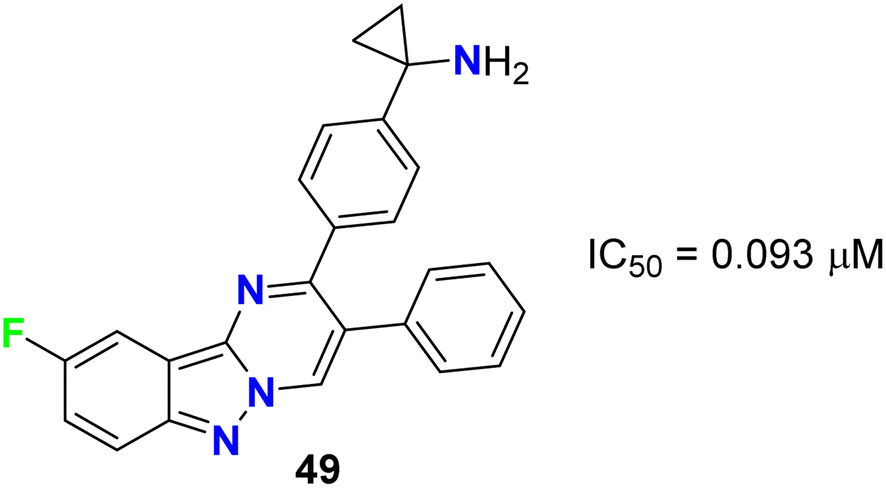
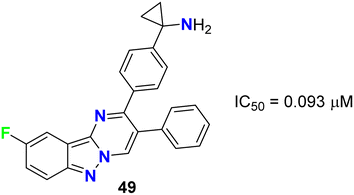
Rehwinkel et al. patented the anticancer activity of the fluorinated pyrimido[1,2-b]indazole 49 (Fig. 28) via its inhibition of PI3 K/Akt. Compound 49 exhibited potent PI3 K/Akt inhibitory activity with an IC50 value of 0.093 μM.69
 |
| | Fig. 28 Structure of fluorinated pyrimido[1,2-b]indazole 49. | |
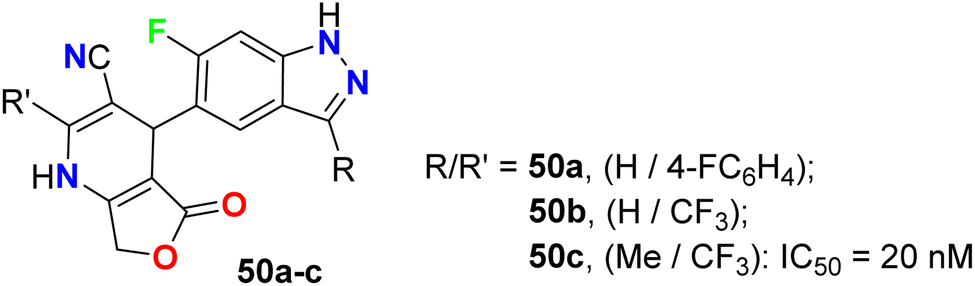
6-Fluoroindazole derivatives 50a–c (Fig. 29), having dihydropyridine moiety in position 5, were synthesized by Michels et al. and evaluated their c-Met tyrosine kinase inhibitory activity through in vitro, ex vivo, and in vivo assays for the treatment of cancer diseases. These compounds were found to have a potent in vitro c-Met tyrosine kinase inhibitory activity with IC50 values of 14–20 nM.70,71
 |
| | Fig. 29 Structure of 6-fluoroindazole derivatives 50a–c. | |
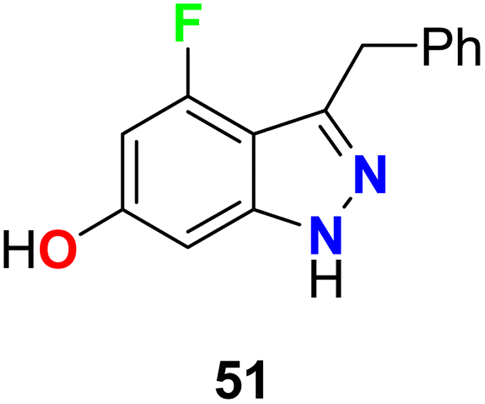
4-Fluoroindazole derivative 51 was tested for its inhibition activity against heat-shock protein (Hsp90) (Fig. 30). This compound showed potent Hsp90 inhibition and represented a good candidate for the treatment of proliferation diseases.72
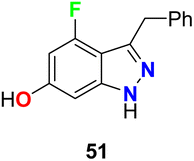 |
| | Fig. 30 Structure of 4-fluoroindazole derivative 51. | |
2.4. Anticancer activity of fluorinated pyrrolo[2,1-c][1,4]benzodiazepines
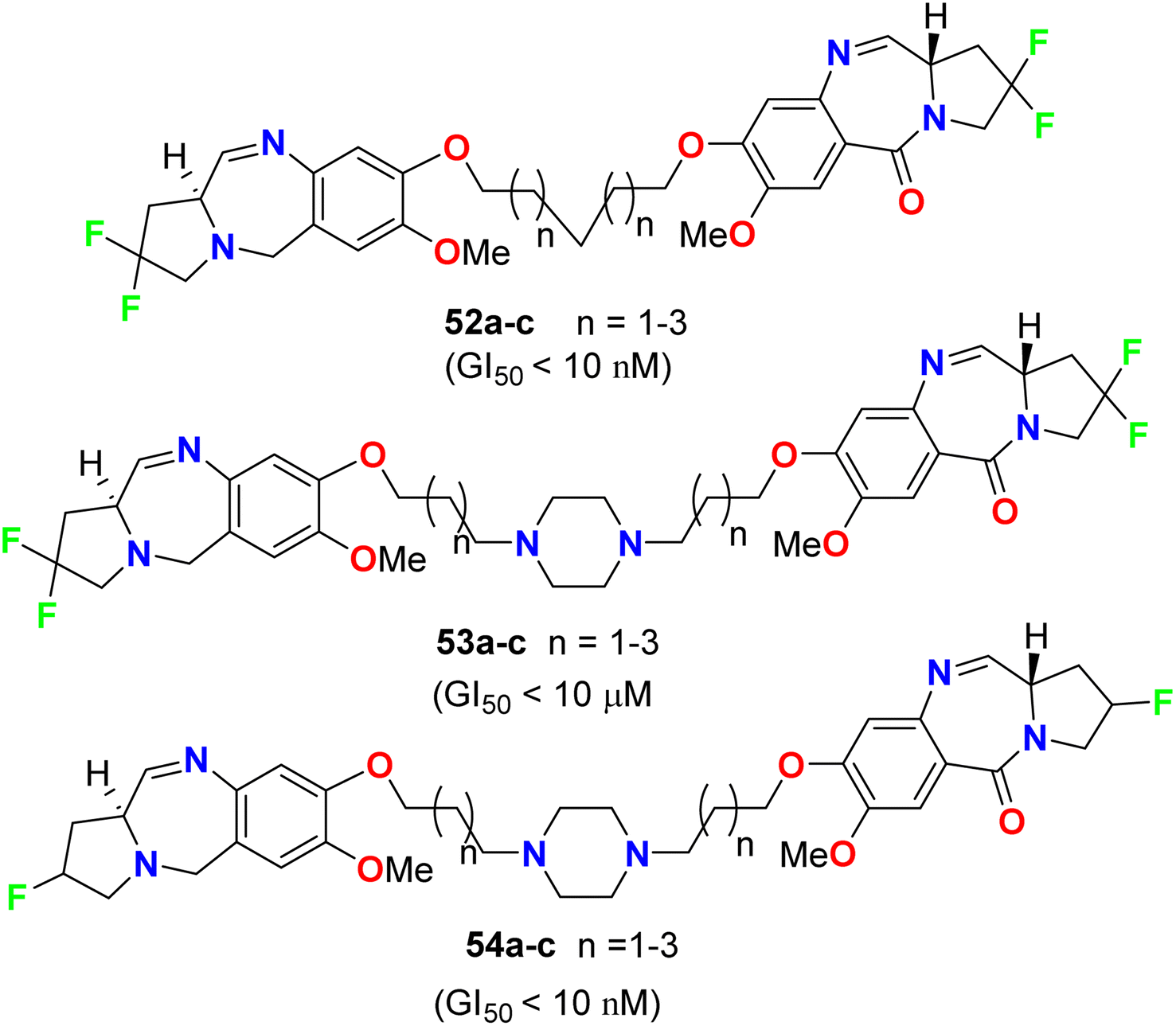
Kamal et al. described the synthesis of some fluorinated-pyrrolo[2,1-c][1,4]benzodiazepines 52–54 (Fig. 31) and studied their anticancer activity derived from nine cancer types (leukemia, non-small cell lung, colon, CNS, melanoma, ovarian, renal, prostate, and breast cancers) using SBR (sulforhodamine B) protein assay to estimate cell viability or growth. Most of the tested substrates demonstrated promising anticancer activity with GI50 values in the nanomolar to micromolar concentration range. The GI50 values (concentration causing 50% inhibition of cell growth) of compounds 52a and 54a, particularly, exhibited excellent inhibition against most of the cancer cell lines with GI50 < 10 nM, but compound 53a showed moderate cytotoxic activity with GI50 < 10 μM. Therefore, the presence of a piperazine ring in the alkylene spacer was responsible for increasing the cytotoxic activity.73
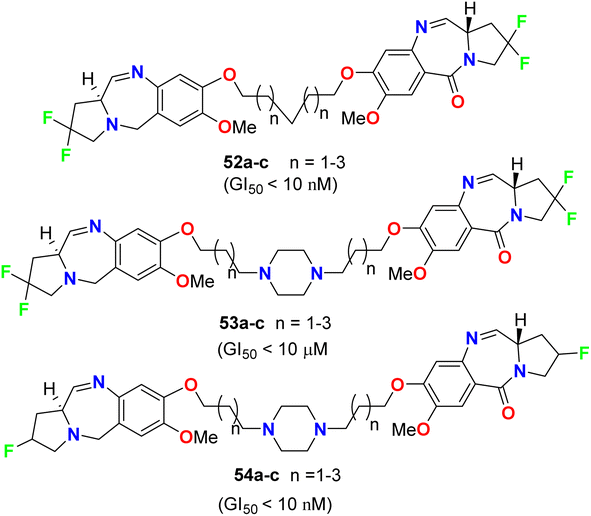 |
| | Fig. 31 Structure of fluorinated-pyrrolo[2,1-c][1,4]benzodiazepines 52–54. | |
2.5. Anticancer activity of fluorinated benzimidazoles
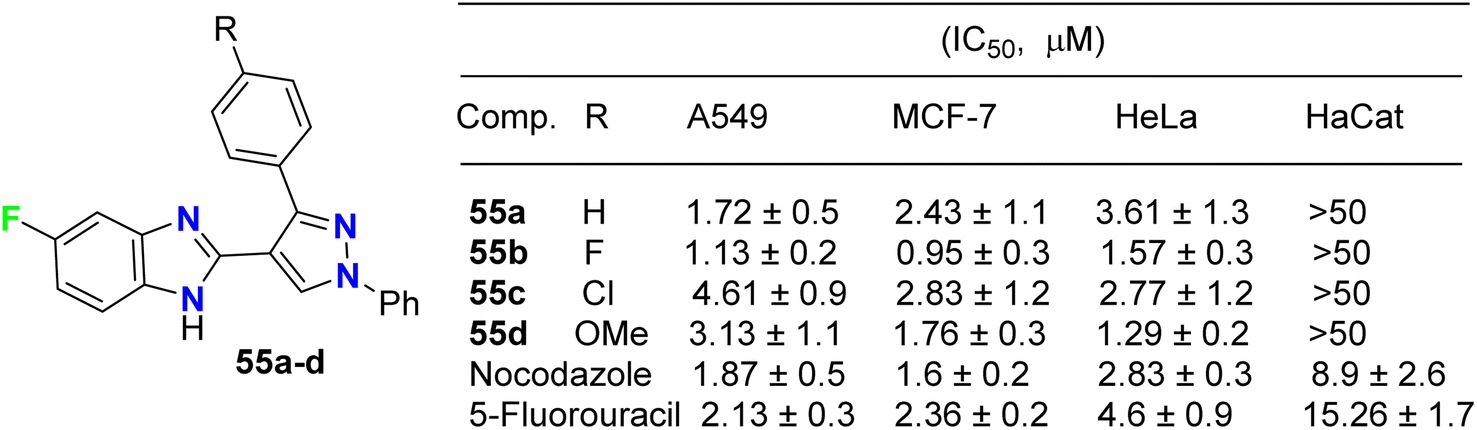
The fluorinated pyrazolylbenzimidazole hybrid molecules 55a–d (Fig. 32) were reported by Reddy et al. and were tested for their anticancer activity against three human tumor cell lines: lung (A549), breast (MCF-7), cervical (HeLa) and against normal keratinocyte (HaCaT) cells using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) growth inhibition assay protocol. All compounds showed potent growth inhibition against all the cell lines A549, MCF-7, and HeLa; particularly, compound 55b was the most potent with IC50 values in the range of 0.95–1.57 μM. Flow-cytometry revealed that all compounds arrested MCF-7 cells in the G1 phase of the cell cycle via the down-regulation of cyclin D2 and CDK2. Fluorescent staining and DNA fragmentation studies showed that cell proliferation was inhibited by induction of apoptosis. Moreover, the compounds led to the collapse of mitochondrial membrane potential (DJm), and increased levels of reactive oxygen species (ROS) were noted. The remarkable biological activity made compound 55b a promising new candidate for the development of cancer therapeutics.74
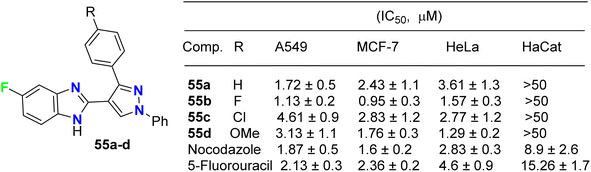 |
| | Fig. 32 Structure of fluorinated pyrazolylbenzimidazole hybrids 55a–d. | |
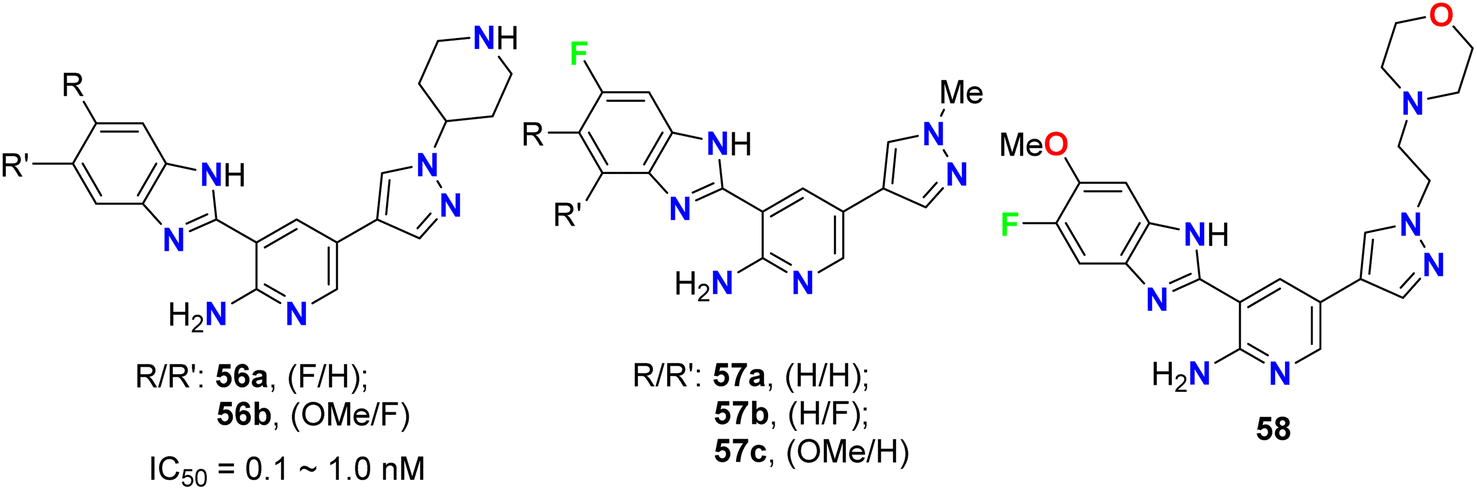
The pyrazolyl-based fluorobenzimidazole hybrids 56–58 (Fig. 33) were assigned as inhibitors of PDK1 and cell proliferation/cell vitality. The inhibitions of PDK1 (Pyruvate Dehydrogenase Kinase 1), which is a protein-coding gene, and the interleukin-1 receptor-associated IRAK-1 and IRAK-4 were evaluated. Compounds 56b, 57a and 57c demonstrated the highest inhibition of PDK1 with IC50 values ranging between 0.1 μM to 1.0 nM; in particular, compound 56b had a powerful inhibition of all PDK1, IRAK-1 and IRAK-4 with IC50 values of 0.1 μM to 1.0 nM.75
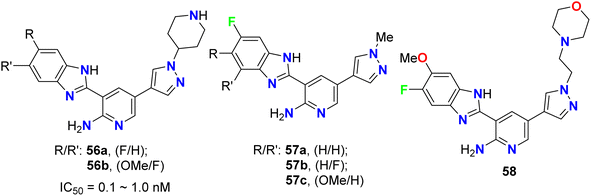 |
| | Fig. 33 Structure of pyrazolyl-based fluorobenzimidazole hybrids 56–58. | |
2.6. Anticancer activity of fluorinated benzothiazoles and benzoxazoles
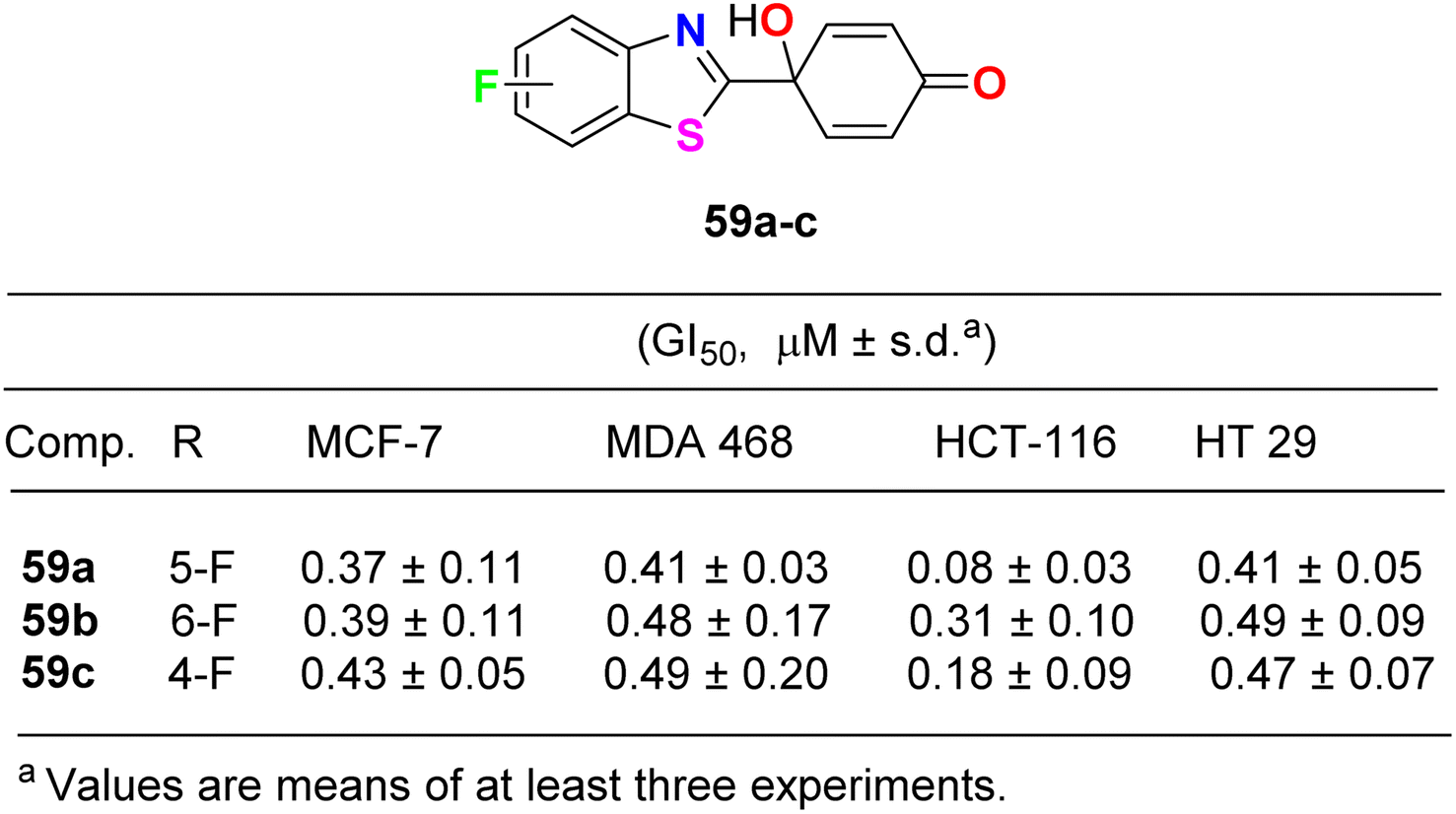
Lion et al. examined the in vitro anticancer activity of the fluorinated benzothiazole derivatives 59a–c (Fig. 34) against the human breast cancer cell lines: MCF-7 (oestrogen receptor positive) and MDA MB 468 (oestrogen receptor negative), and the human colon carcinoma cell lines HCT-116 and HT-29. Interestingly, the 5-fluoro derivative 59a displayed the most potent antiproliferative activity against all the tested cell lines: MCF-7, MDA MB 468, HCT-116 and HT 29 with GI50 values 0.37, 0.41, 0.08 and 0.41 μM, respectively. In addition, the ability of the fluorinated benzothiazoles 59a,b to inhibit human thioredoxin signalling was measured using a modified protocol of the insulin reduction assay. The fluorinated derivatives 59a,b inhibited thioredoxin signalling at micromolar concentrations.76
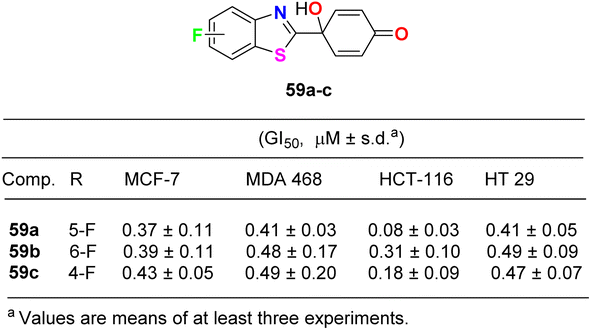 |
| | Fig. 34 Structure of fluorinated benzothiazole derivatives 59a–c. | |
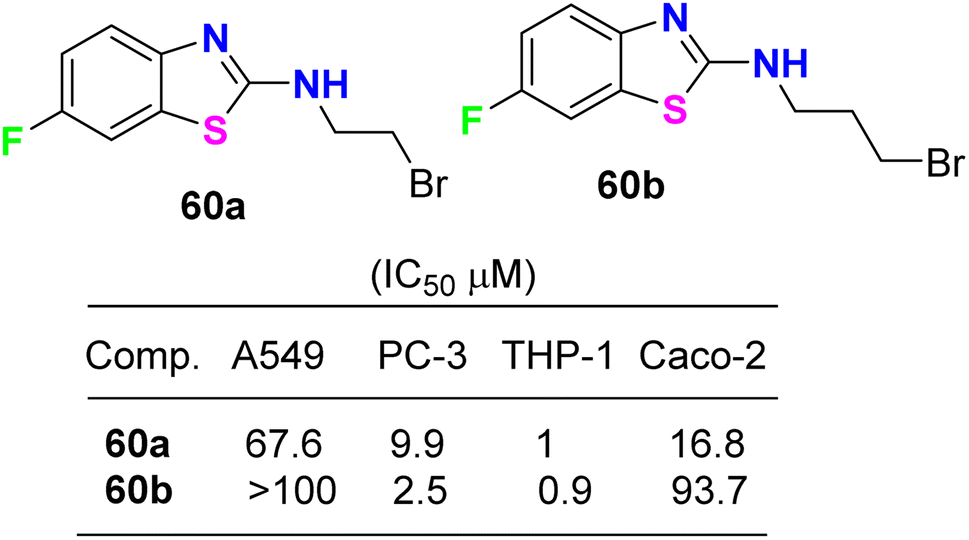
The cytotoxicity of the 6-fluorobenzothiazole derivatives 60a,b (Fig. 35) was evaluated in vitro against four human cancer cell lines: prostate (PC-3), lung (A-549), leukemia (THP-1), and colon (Caco-2). Compounds 60a,b were highly active against leukemia (THP-1) cancer cells and exhibited IC50 values of 1 and 0.9 μM, respectively. A docking study of the synthesized ligands was done on epidermal growth factor receptors using Argus Lab flexible docking to determine their observed activity. Compounds 60a,b served as a useful ‘‘lead’’ for further anticancer drug development.77
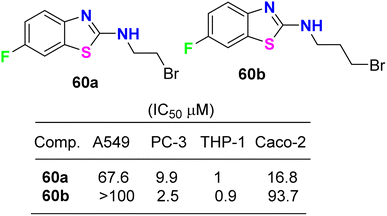 |
| | Fig. 35 Structure of 6-fluorobenzothiazole derivatives 60a,b. | |
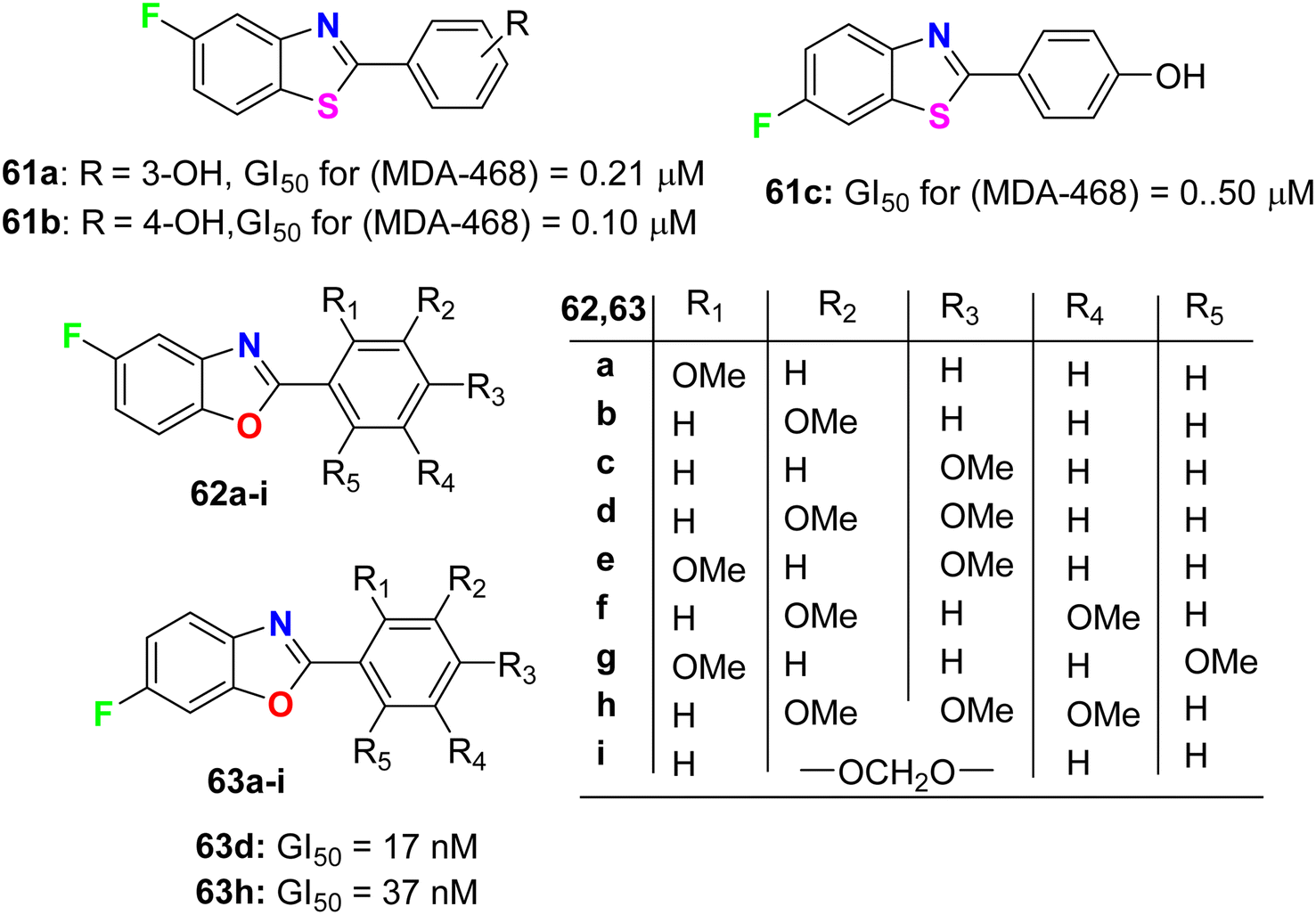
Aiello et al.78 synthesized two series of fluorinated heterocycles, namely the fluorinated benzothiazole 61a–c and benzoxazole 62–63a–i derivatives, and evaluated their anticancer potency against MCF-7 and MDA 468 breast cancer cell lines. The fluorinated benzothiazoles 61a–c were more active against the ER −ve human breast cancer cell line MDA 468, giving GI50 values 0.20–0.5 μM. In the MCF-7 (ER +ve) human breast cancer cell line, the most active compounds were found to be the fluorinated benzothiazole derivatives 61a and 61b (GI50 of 0.57 and 0.40 μM, respectively) (Fig. 36). In addition, the 5-fluorobenzoxazole 62a exhibited high potency against both cell lines MCF-7 and MDA-468 with GI50 values of 0.36 and 0.27 μM, respectively. Overall, the fluorinated benzoxazole series 62a–j (Fig. 36) was more active against the MDA 468 cell line with IC50 varying between the range of 0.017–98.6 μM. Against the generally less sensitive MCF-7 cell line, benzoxazoles displayed activity with GI50 values ranging between 0.36 and 90.7 μM. The 6-fluorobenzoxazole compounds 63d and 63h (bearing 3,4-dimethoxy and 3,4,5-trimethoxy substituents on the phenyl ring) were found to be potently active on the MDA 468 cell lines, giving GI50 values of 17 and 37 nM, respectively.
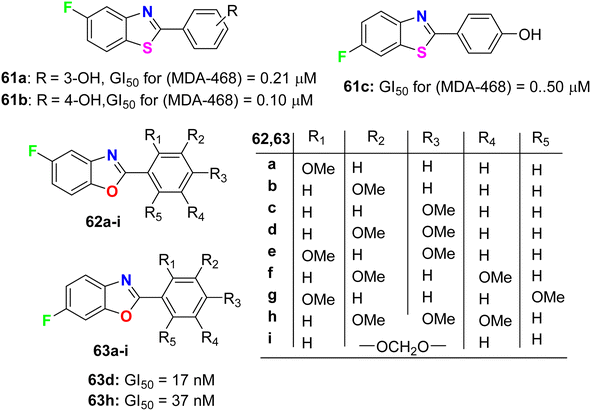 |
| | Fig. 36 Structures of the fluorinated derivatives 61a–c, 62a–i and 63a–i. | |
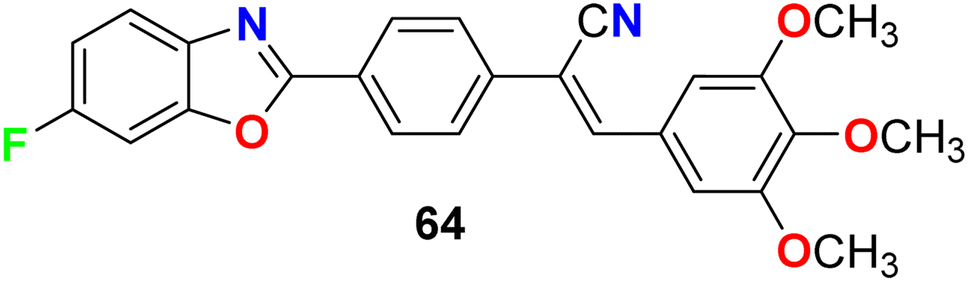
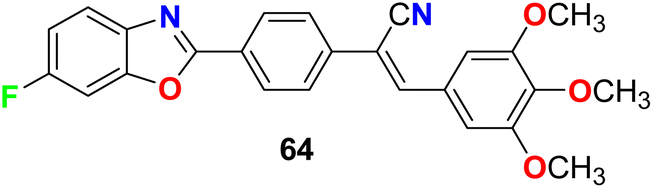
Jauhari et al. synthesized 6-fluorobenzoxazole 64 and examined its anticancer activity against four human cancer cell lines: HEPG-2, HeLa, MCF-7 and the human colon carcinoma (WiDr). Interestingly, compound 64 showed promising inhibitory activity against all the tested cell lines with IC50 values of 11.40, 11.70, 11.24, and 11.42 μM, compared with the control (culture medium only) IC50 values of 15.26, 14.72, 20.12, and 10.22 μM, respectively (Fig. 37).79
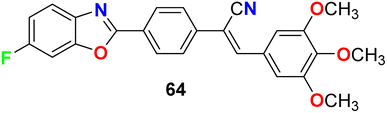 |
| | Fig. 37 Structure of 6-fluorobenzoxazole 64. | |
2.7. Anticancer activity of fluorinated thiazoles and isoxazoles
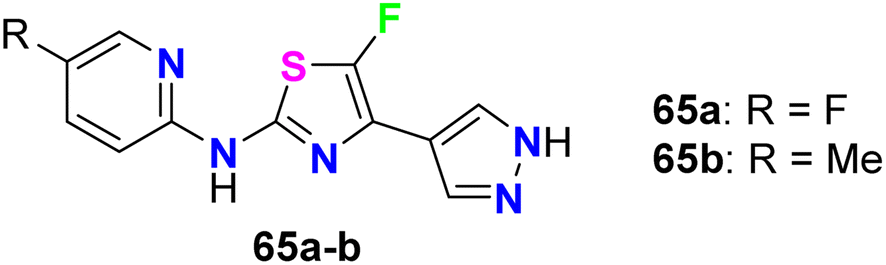
The fluorinated pyrazolylthiazoles 65a,b were profoundly reported as positive allosteric modulators of metabotropic glutamate receptors (mGluRA) (Fig. 38). The activity was tested on recombinant human mGluR4a receptors by disclosing the alteration in intracellular Ca2+ concentration, employing Fluo4-(AM) (the fluorescent Ca2+-sensitive dye) and FLIPR (Fluorometric Imaging Plate Reader). The mean EC50 resulted from at least three separate experiments of the fluorinated compounds in duplicate. The bioassay results confirmed that the reported compounds were positive allosteric modulators of human mGluR4 receptors with EC50 values <100 nM.80
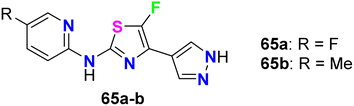 |
| | Fig. 38 Structure of fluorinated pyrazolylthiazoles 65a,b. | |
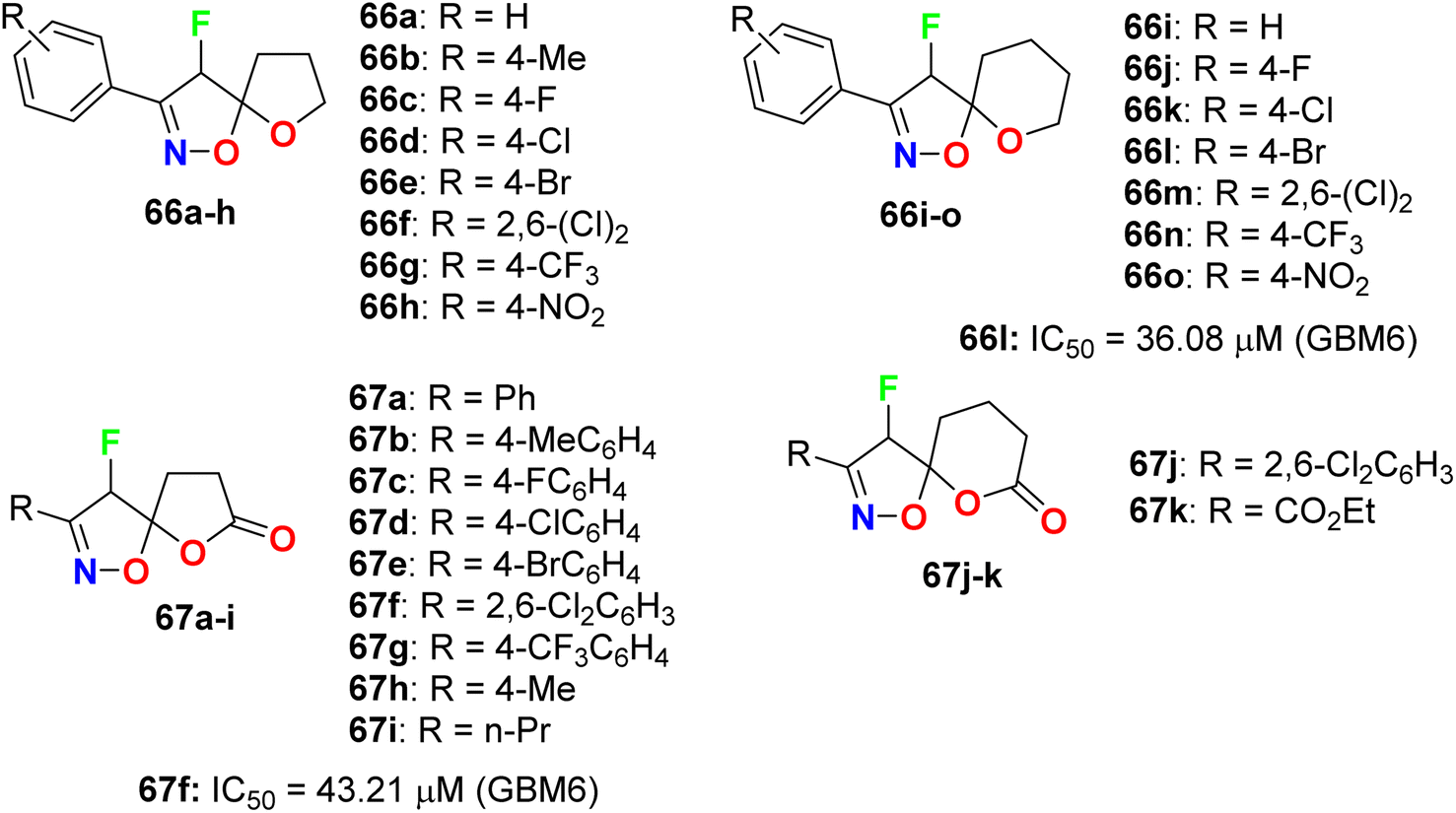
Das et al. synthesized a series of fluorinated spiro-isoxazoline hybrids 66 and 67 (Fig. 39) and probed their cytotoxicity against human glioblastomas (GBM6) and triple-negative breast cancer (MDA MB 231). The bioassay protocol was carried out using the MTT method with concentrations ranging between 0.8 μM to 100 μM for 72 hours. Two fluorinated derivatives, 66l and 67f, exhibited robust anti-proliferative effects against both GBM6 and MDA MB 231 cells with IC50 values 36.08 and 43.21 μM (GBM6) and 68.18 and 79.80 μM (MDA MB 231), respectively. Thus, compound 66l displayed better inhibition than 67f against both GBM6 and MDA MB 231 cells, giving a superior effect for the spiro-ether than the corresponding spiro-lactone in cytotoxicity.81
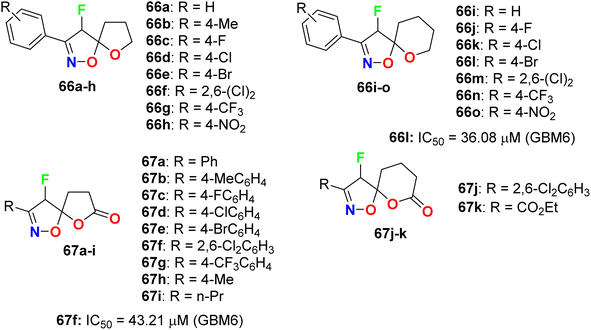 |
| | Fig. 39 Structure of fluorinated spiro-isoxazoline hybrids 66 and 67. | |
2.8. Anticancer activity of fluorinated furans
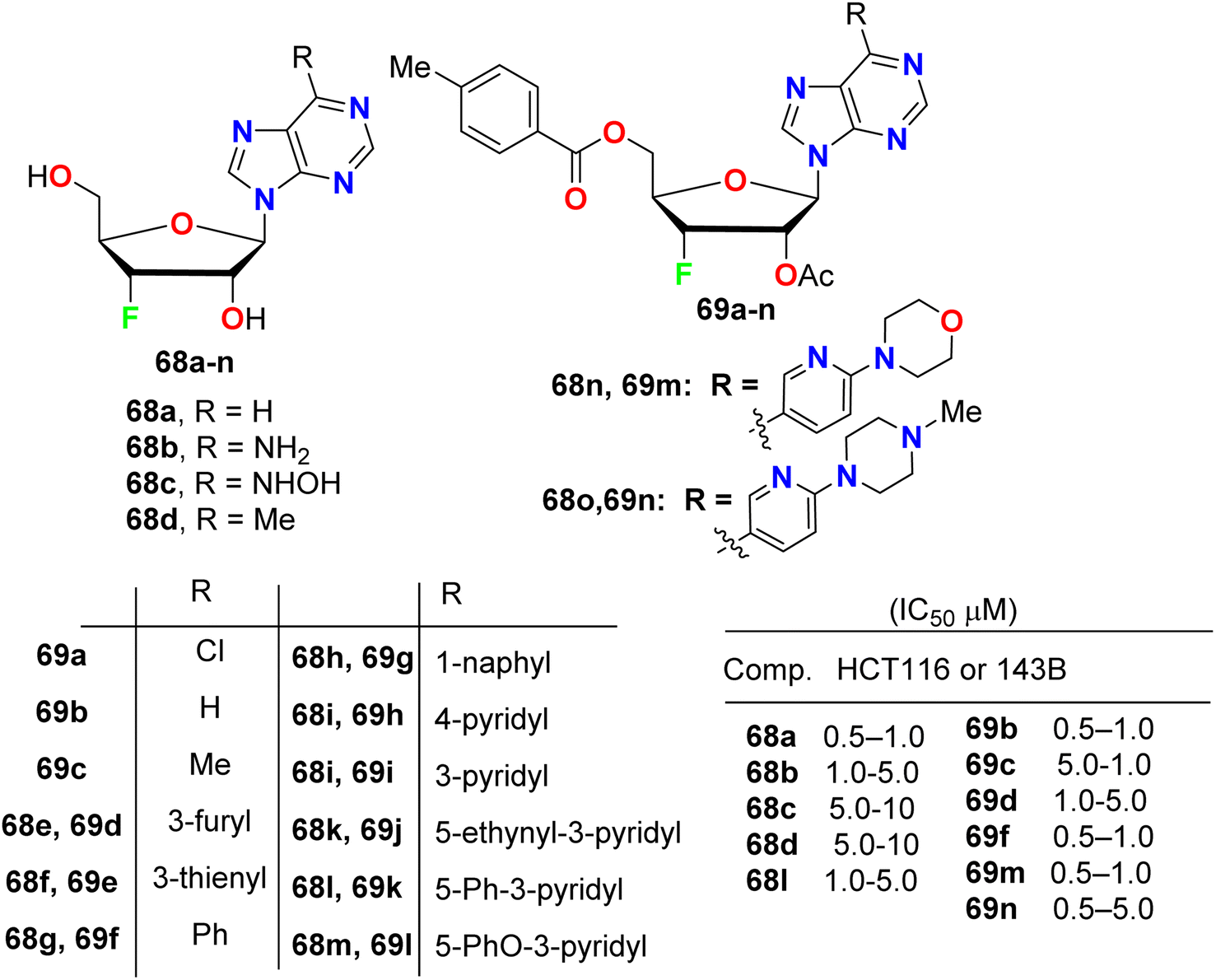
A series of purine nucleosides fluorinated at the 3′-position of the sugar moiety 68 and 69 (Fig. 40) were synthesized by Ren et al. and their anticancer activity was probed. The assigned fluorinated nucleosides were examined against HT116 (colon cancer) and 143B (osteosarcoma cancer) cell lines. Most of these compounds demonstrated inhibition of the growth activity of HT116 and 143B cancer cells at sub- or low-micromolar concentration. The fluorinated sugars 68a, 69b, 69c, 69f, 69m and 69n showed the highest inhibition of cancer cell growth with IC50 values 0.5–1.0 μM against both HT116 and 143B. From the SAR study, it was concluded that the protected fluorinated purine riboside 69c and 69d demonstrated 10-fold higher anticancer activity than their deprotected analogues 68d and 68e. In addition, the 3′-fluorinated purine nucleosides 68b, 68c, 68d, 68l and 69d showed moderate potency, but the other derivatives did not show detectable activity against the evaluated cancer cell lines.82
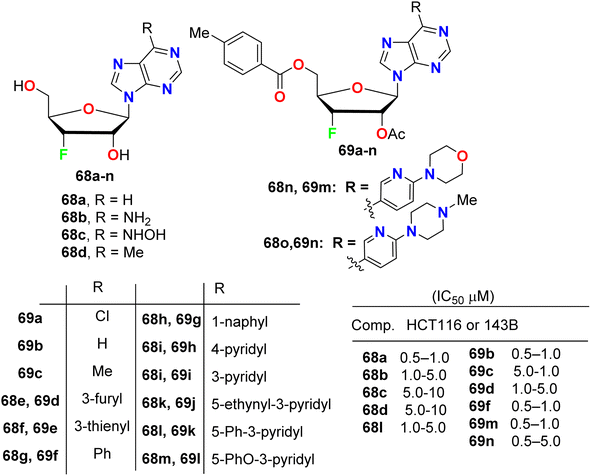 |
| | Fig. 40 Structure of fluorinated purine nucleosides 68 and 69. | |
2.9. Anticancer activity of fluorinated pyrazolopyrimidines
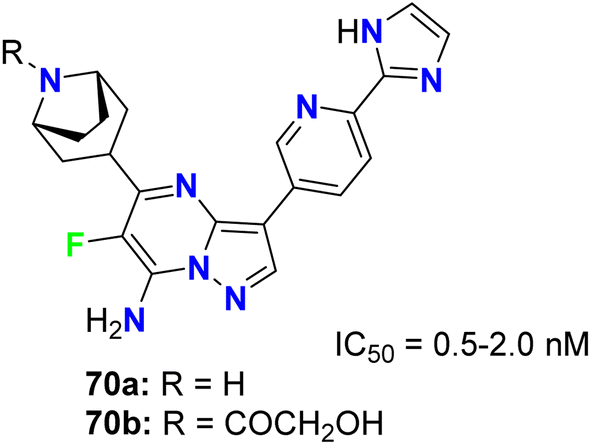
The fluorinated pyrazolopyrimidines 70a,b (Fig. 41) were reported to be useful for the treatment of cancer via the regulation of mTOR (mammalian target of rapamycin), which regulates cell proliferation, autophagy and apoptosis. The IC50 values were determined by dose–response curves in duplicate, and both fluorinated compounds showed mTOR with IC50 values on a nanomolar scale between 0.5–2 nM.83
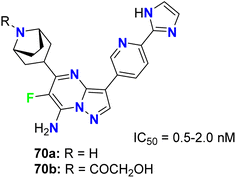 |
| | Fig. 41 Structure of fluorinated pyrazolopyrimidines 70a,b. | |
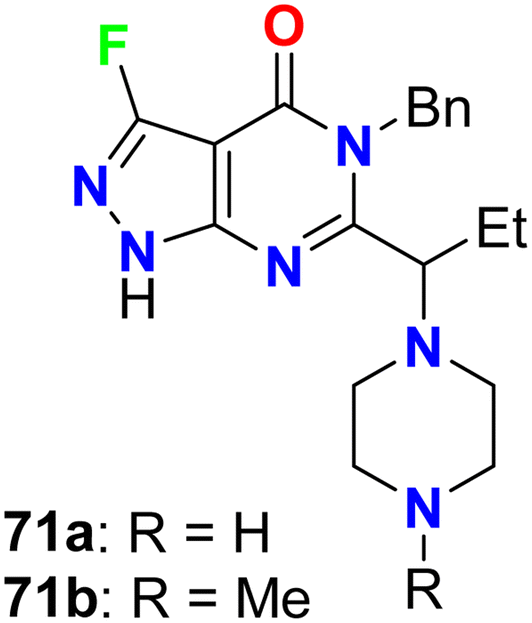
Fraley et al. designed the fluorinated pyrazolo[3,4-d]pyrimidinone derivatives 71a,b (Fig. 42) and assigned them as inhibitors of the mitotic kinesin KSP for the treatment of cellular proliferative diseases, such as breast cancer. Measuring the mitotic arrest and apoptosis was done using FACS (Fluorescence-Activated Cell Sorting) analysis by measuring DNA content in a treated population of cells. The tested compounds 71a,b were reported to inhibit KSP with IC50 values ≤50 μM. The EC50 for mitotic arrest and apoptosis was derived by plotting compound concentration on the x-axis and the percentage of cells in the G2/M phase of the cell cycle. The cytotoxic EC50 was determined by plotting % inhibition of cell growth for each titration point on the y-axis and compound concentration on the x-axis.84
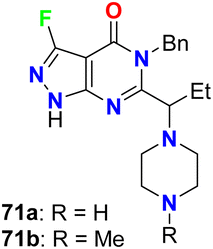 |
| | Fig. 42 Structure of fluorinated pyrazolo[3,4-d]pyrimidinone derivatives 71a,b. | |
3. Antimicrobial activity of fluorinated heterocycles
3.1. Antimicrobial activity of fluorinated indoles
The in vitro antibacterial activity of the thiazole-based 5-fluoroindole molecular hybrids 31a,b (Fig. 18 above) was described by Tantak and his group against two Gram-positive Bacillus subtilis (B. subtilis), and Staphylococcus aureus (S. aureus) and two Gram-negative Escherichia coli (E. coli) and Pseudomonas putida (P. putida) bacterial strains using ciprofloxacin as a reference drug. The tested compounds showed moderate activity towards the bacterial strains, wherein compound 31b showed selective inhibition of the Gram-negative bacteria (E. coli and P. putida), better than the Gram-positive ones (S. aureus and B. subtilis).56
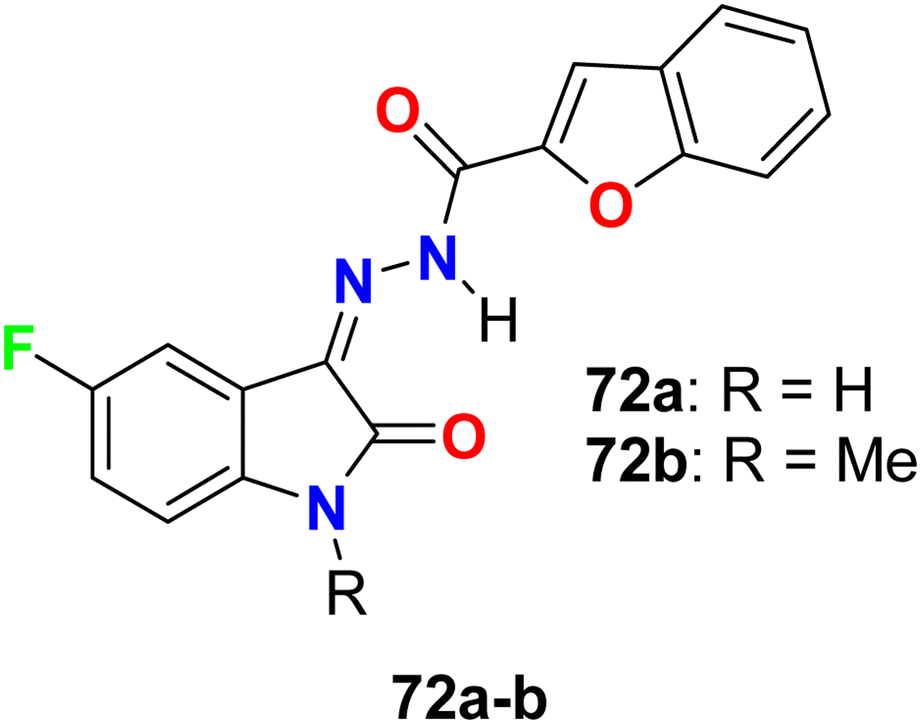
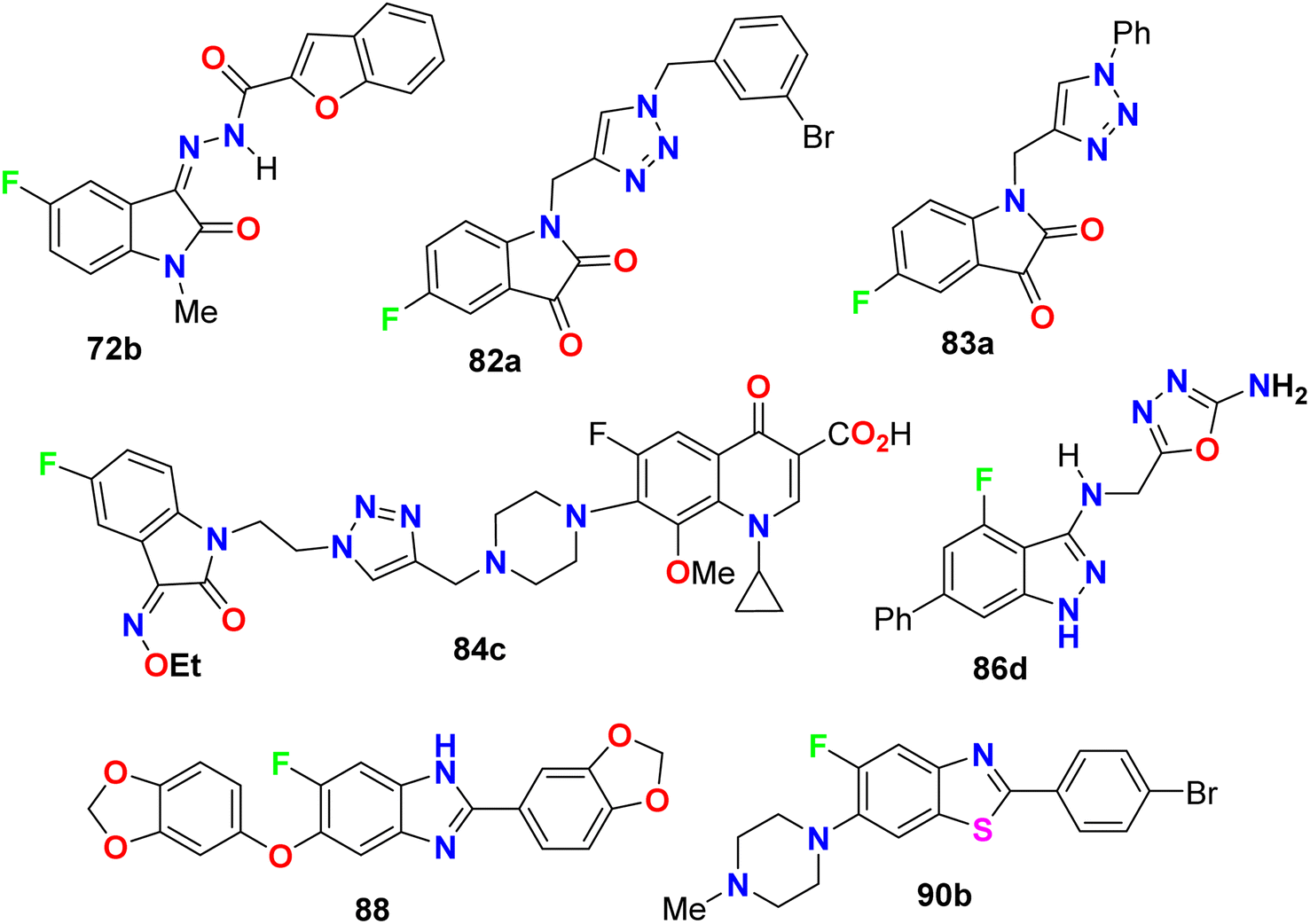
Two hybrids of benzofuran-based 5-fluoroindole 72a,b (Fig. 43) were synthesized and tested for their in vitro antimicrobial activities against three Gram-positive bacteria (B. subtilis, S. aureus, and Staphylococcus epidermis (S. epidermis)), four Gram-negative bacteria (Salmonella typhi (S. typhi), E. coli, Klebsiella pneumoniae (K. pneumoniae) and Pseudomonas aeruginosa (P. aeruginosa)) as well as four fungi (Candida albicans (C. albicans), Aspergillus niger (A. niger), Aspergillus flavus (A. flavus), and Aspergillus fumigates (A. fumigates)) strains using Cup plate method at 100 μg mL−1 using ciprofloxacin and miconazole as positive controls. Both the tested compounds showed significant activity towards the bacterial strains but low activity against the antifungal agents. Compound 72b showed promising antibacterial activity against S. aureus, P. aeruginosa and S. typhi, almost similar or better than the standard ciprofloxacin. Compound 71a showed good activity against S. aureus and B. subtilis and compound 72b possessed good activity against S. epidermis.85
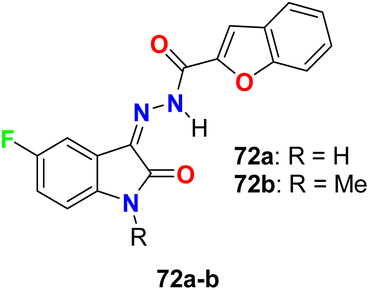 |
| | Fig. 43 Structure of benzofuran-based 5-fluoroindole 72a–b. | |
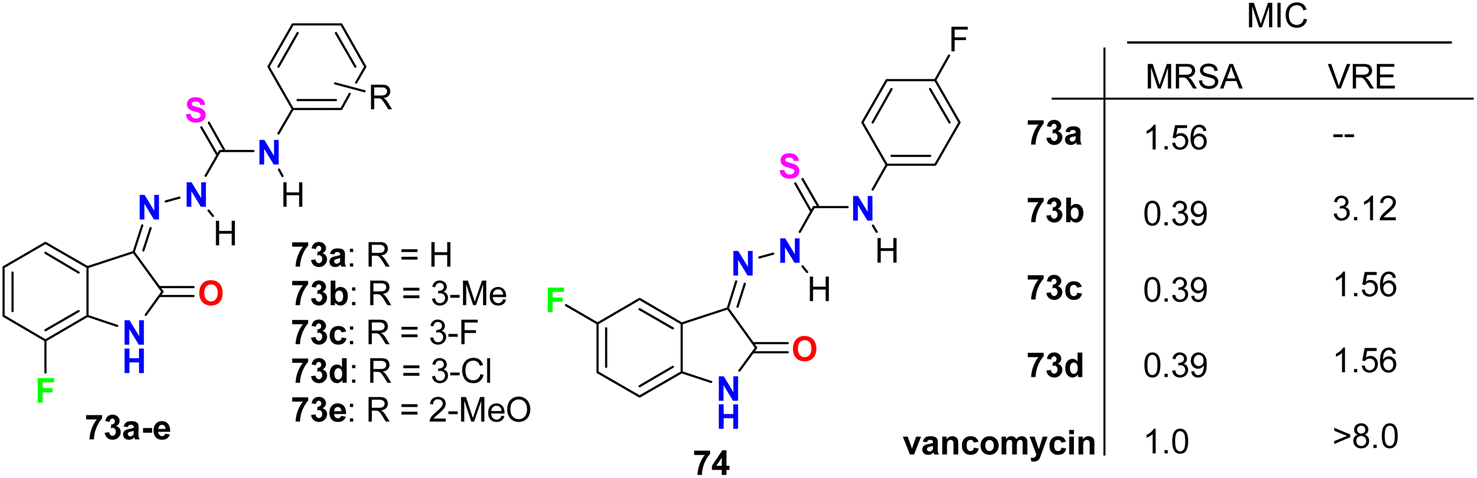
Zhang et al. designed and synthesized some fluorinated indol-2-one molecular hybrids 73a–e and 74 (Fig. 44) and evaluated their in vitro inhibitory activity against the growth of clinically isolated MRSA strain (MRSA = methicillin-resistant S. aureus) and VRE (vancomycin-resistant Enterococcus). The prepared molecular hybrids showed promising activity against the Gram-positive pathogen. Interestingly, compound 73a significantly inhibited the MRSA (Chaoyang) strain, with a MIC value of 1.56 mg L−1, comparable to vancomycin (MIC = 1.0 mg L−1). The MIC values of compounds 73b, 73c, and 73d were 0.39 mg L−1 against the MRSA (Chaoyang) strain. Compounds 73c and 73d presented anti-MRSA activities much better than their activities against S. aureus, where their MIC value was 0.78 mg L−1. Compounds 73c and 73d also showed promising inhibition of VRE with a MIC value of 1.56 mg L−1. Thus, compounds 73c and 73d displayed distinguished inhibition of MRSA and VRE as Gram-positive bacteria. The presence of fluorine atom at the 7-position led to enhancement of the anti-MRSA activity. Such compounds were considered potential leads in discovering antibacterial inhibitors to combat drug resistance.86
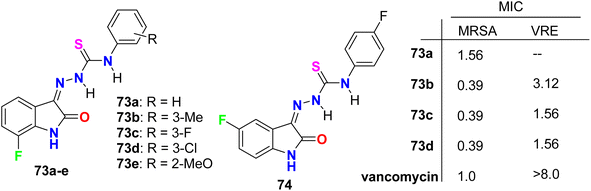 |
| | Fig. 44 Structure of fluorinated indol-2-one molecular hybrids 73a–e and 74. | |
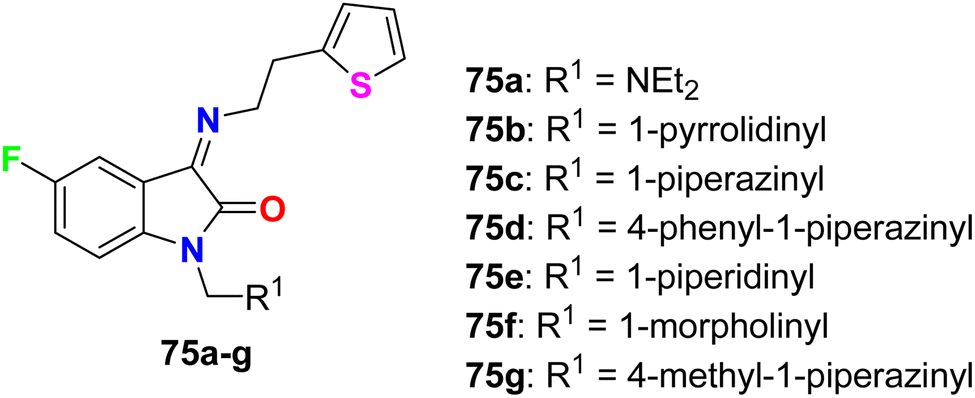
Synthesis of 5-fluoroisatin derivatives 75 was reported and their anti-tubercular activity was evaluated (Fig. 45). The anti-tubercular activity was measured using the Almar blue assay against the strain of Mycobacterium tuberculosis (MTB H37Rv). All compounds showed varied (from weak to good) inhibitory activity against MTB H37Rv, particularly compound 75d, having N-phenylpiperazine group, was the most active one against MTB H37Rv (ATCC-27294) with MIC = 6.25 μg mL−1 as compared to the standard drug isoniazid (with MIC = 1.6 μg mL−1).87
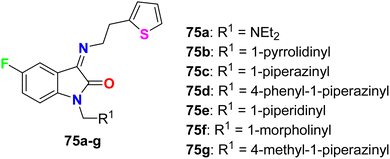 |
| | Fig. 45 Structure of 5-fluoroisatin derivatives 75a–g. | |
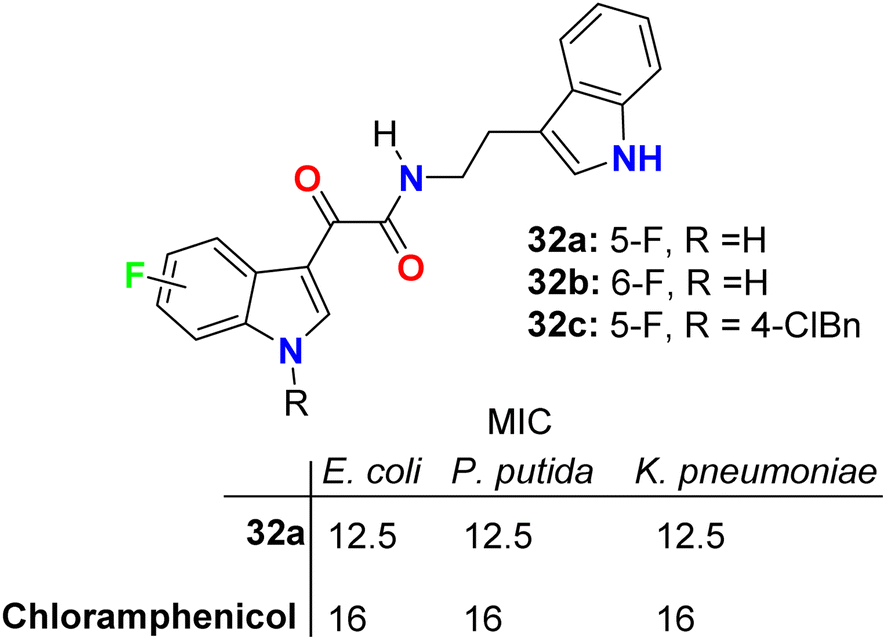
The glyoxylamide-based 5(6)-fluoroindole derivatives 32a–c (Fig. 19 above) were synthesized and evaluated for their in vitro antibacterial activity against two Gram-positive (B. subtilis and S. aureus) and three Gram-negative (E. coli, P. putida, and K. pneumoniae) bacterial strains. Compound 32a displayed the best antibacterial activity against the Gram-negative strain. Compound 32a displayed high antibacterial activity (MIC = 12.5 μg mL−1) against all the tested three Gram-negative bacterial strains, and MIC = 25 μg mL−1 for the tested Gram-positive ones, compared with chloramphenicol reference drug (MIC = 16 μg mL−1) for all bacterial strains. Moving the fluorine atom from position C-5 to C-6 in the indole moiety or arylation of indole N-1 led to the inactive derivatives 32b and 32c. The killing kinetics of compound 32a showed 95% bactericidal activity against the Gram-negative bacteria within the first 2 h, indicating its fast-bactericidal action through quick inhibition of bacterial proliferation. Compound 32a also showed good bactericidal activity against the Gram-positive bacteria, B. subtilis (>80%) and S. aureus (>65%), within the initial 2 h of incubation. In addition, compound 32a showed no toxicity in any of the human cell lines; therefore, this compound appeared to target bacterial-specific pathways and was identified as a promising antibacterial agent.57
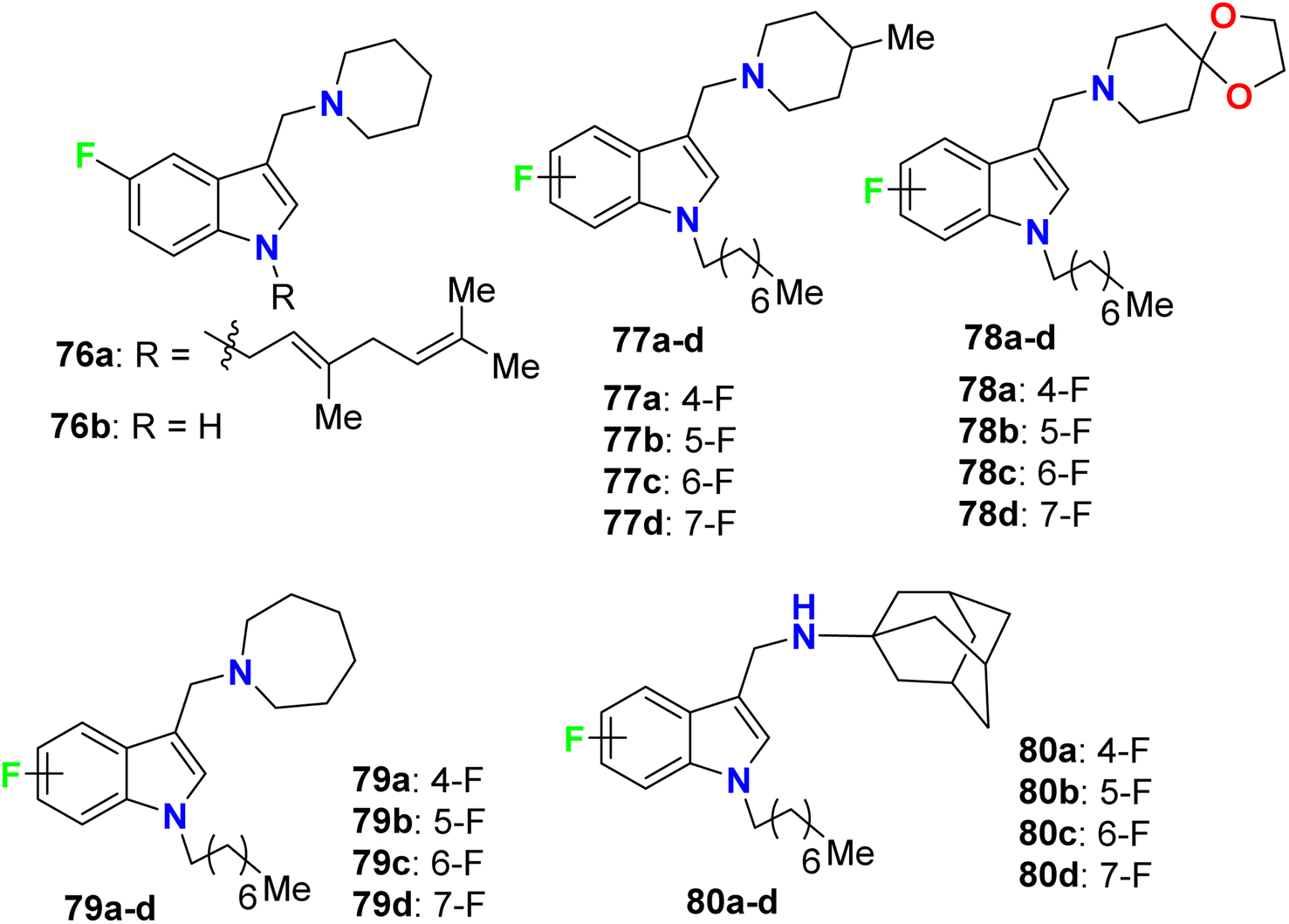
Yang and his research group designed some 4-fluoroindole derivatives 76–80 (Fig. 46) containing geranyl or n-octyl moieties at N-1 and determined their antimycobacterial activity against Mycobacterium bovis (M. bovis) BCG and M. tuberculosis H37Rv using the broth dilution method. The reported fluorinated derivatives showed variable potencies against M. bovis BCG and M. tuberculosis H37Rv. The turbidity method was employed to determine the MIC values required to minimize the bacterial growth by 50% (MIC50). The mycobacterial activity of 76a demonstrated good potencies against M. bovis BCG (MIC50 = 5 μM) and M. tuberculosis H37Rv (MIC50 = 3.5 μM), and compound 76a was found to be soluble at 25 μM, 25 °C and pH 7.4. The SAR study revealed that the 4-fluoro analogues of 77a, 79a and 80a (all had MIC50 = 2 μM) were twice more potent than the other fluorinated regioisomers 77b–d, 79b–d and 80b–d (all had MIC50 = 4 μM) against M. bovis BCG. However, compounds 77a, 78a, 78c, 79a, and 80a were found to be highly effective against M. tuberculosis H37Rv, where all of them presented potent activities with MIC50 values 2–3 μM on the virulent tubercle bacillus.88
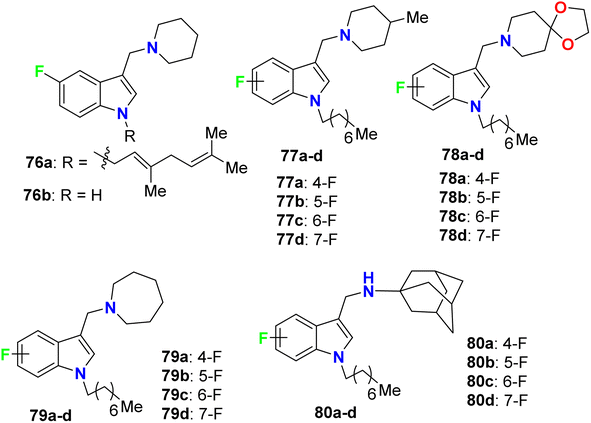 |
| | Fig. 46 Structure of 4-fluoroindole derivatives 76–80. | |
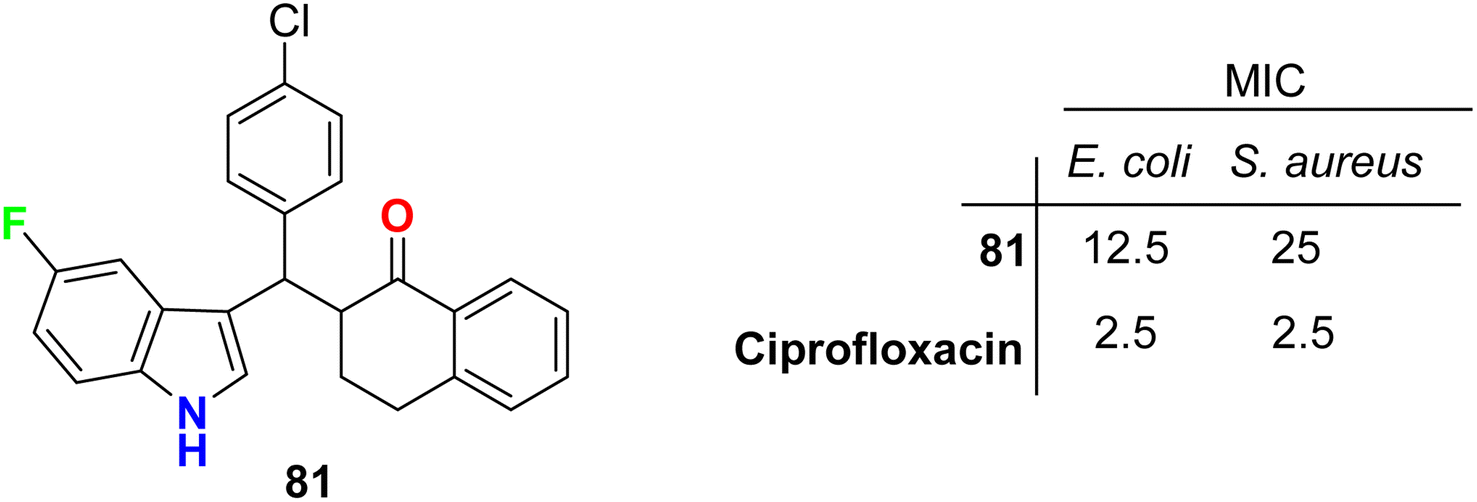
The tetralone-based 5-fluoroindole molecular hybrid 81 (Fig. 47) was synthesized and its antibacterial and antitubercular activities were screened. Compound 81 displayed reasonable inhibitory activity against E. coli and S. aureus with MIC values of 12.5 and 25 μg mL−1, respectively, compared with the ciprofloxacin drug (MIC = 2.5 μg mL−1). It also showed the same potency against both S. typhi and M. tuberculosis with MIC = 50 μg mL−1, compared with the ciprofloxacin drug with MIC = 2.5 and 5.0 μg mL−1, respectively.89
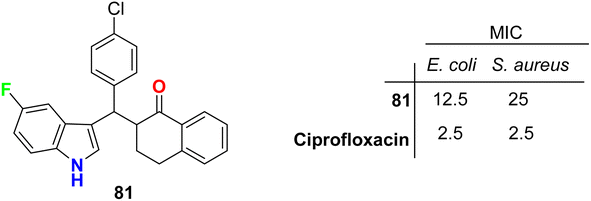 |
| | Fig. 47 Structure of tetralone-based 5-fluoroindole derivative 81. | |
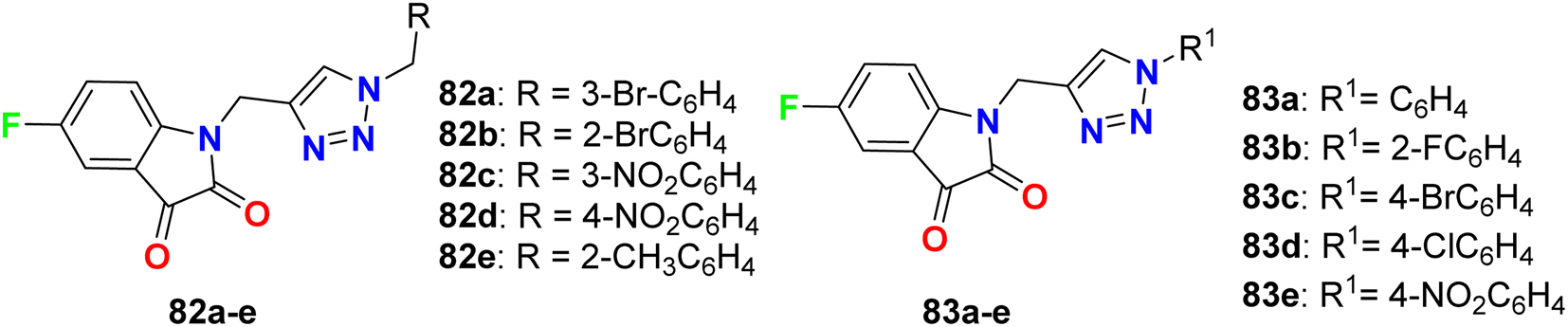
Deswal and co-workers reported the synthesis of fluorinated indolin-2,3-dione derivatives 82 and 83 (Fig. 48) and examined their in vitro antibacterial efficiency against two Gram-positive: B. subtilis, and Staphylococcus epidermidis (S. epidermidis) and two Gram-negative: P. aeruginosa and E. coli bacterial strains. Using ciprofloxacin as reference drug, the serial dilution method was employed. The minimum inhibitory concentration (MIC) results revealed that most of the fluorinated hybrids (82a–e, 83a–e) provided variable activity against tested bacterial strains with MIC ranging between 0.016–0.038 μmol mL−1. Compound 82b displayed high antibacterial activity against S. epidermidis and B. subtilis and both had MIC of 0.0075 μmol mL−1, respectively, and 82c also showed high activity against S. epidermidis with a MIC of 0.0082 μmol mL−1 compared to ciprofloxacin (MIC = 0.0047 mmol mL−1). The other fluorinated hybrids 82 and 83 showed variable activities against the tested bacterial strains with MIC ranging between 0.015–0.038 μmol mL−1. It was found that compounds with electron-withdrawing groups had better antibacterial activity than those having electron-donating groups. The in vitro antifungal activity of 82a–e and 83a–e against two fungal strains (A. niger and C. albicans) revealed moderate to excellent activity; specifically, hybrids 82a, 82d and 83c displayed promising activity for A. niger with MIC values of 0.0075, 0.0082 and 0.0092 μmol mL−1, respectively, better than the antifungal drug fluconazole (MIC, 0.0102 μmol mL−1). For C. albicans, however, the hybrids 82a, 82d and 82e showed good activity (MIC = 0.0090–0.0075 μmol mL−1) comparable to fluconazole (MIC, 0.0051 μmol mL−1).90
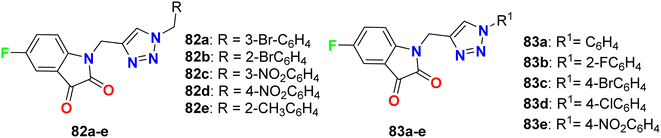 |
| | Fig. 48 Structure of fluorinated indolin-2,3-dione derivatives 82, 83. | |
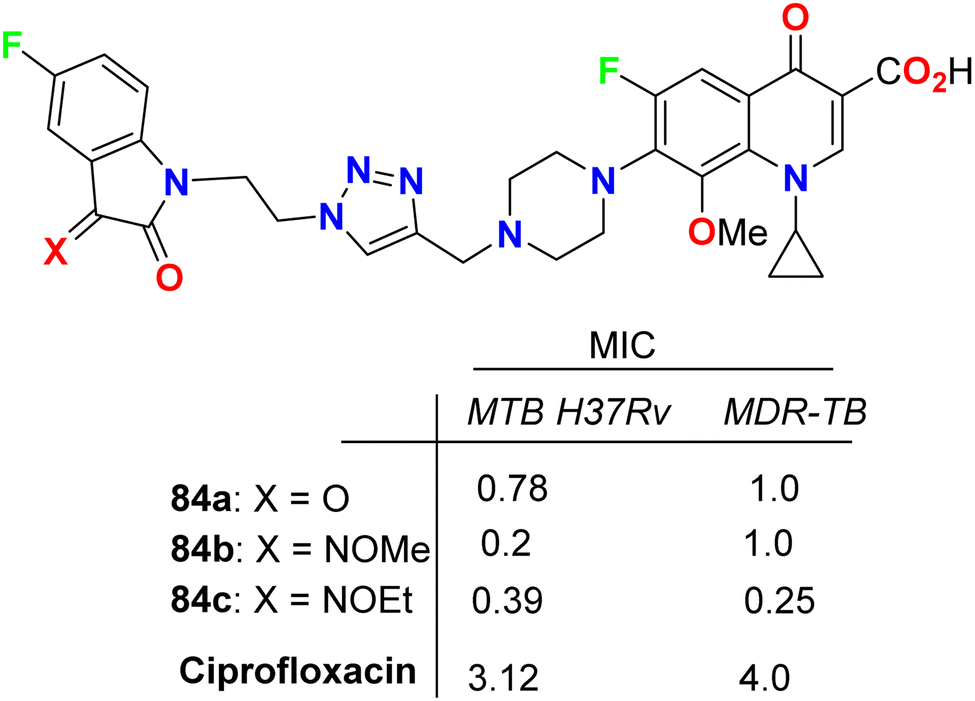
Some 1H-1,2,3-triazole-based 5-fluoroisatin hybrids 84a–c were synthesized and examined for their in vitro anti-mycobacterial activities against multi-drug-resistant tuberculosis (MDR-TB) and M. tuberculosis H37Rv. The tested compounds showed significant inhibitory activity against MDR-TB and MTB H37Rv with MIC values ranging between 0.25–1 μg mL−1 and 0.20–0.78 μg mL−1, respectively, better than the ciprofloxacin drug with MIC = 4.0 and 3.12 μg mL−1, respectively. The hybrid structure 84b (MIC: 0.20 μg mL−1) displayed the best activity with 16-fold inhibitory action against MTB H37Rv better than the reference ciprofloxacin drug. Compound 84c showed extraordinary potency against MDR-TB (MIC = 0.25 μg mL−1), 16 times more active than the reference drug ciprofloxacin. Thus, the reported 5-fluoroisatins 84 were worthy of further development for possible anti-tuberculous drug therapy (Fig. 49).91
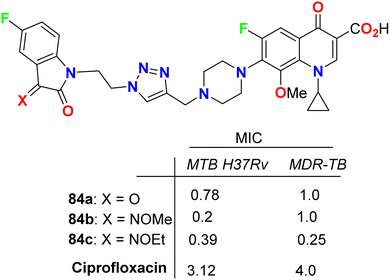 |
| | Fig. 49 Structure of 1H-1,2,3-triazole-based 5-fluoroisatin hybrids 84a–c. | |
3.2. Antimicrobial activity of fluorinated pyrazoles and indazoles
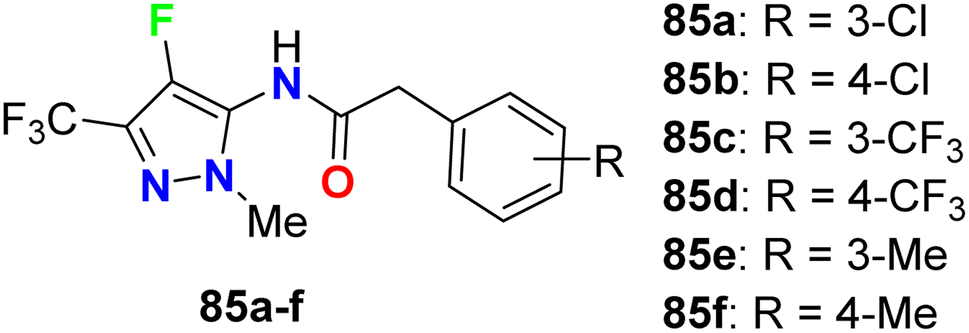
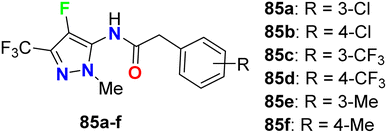
The carboxamide-based 4-fluoropyrazole derivatives 85a–f (Fig. 50) were invented and patented as antifungal agents via measuring their inhibition activity on Sclerotinia sclerotiorum (S. sclerotiorum), Rhizoctonia cerealis (R. cerealis), Gaeumannomyces graminis (G. graminis), and Valsa mali (V. mali). The in vitro inhibition activity of the invented 4-fluoropyrazoles was examined on plant pathogenic fungi at a concentration of 50 mg L−1 using a hypha linear growth rate method. All compounds showed varied antifungal activity ranging between moderate to excellent activity. In particular, compound 85b exhibited excellent inhibitory activity against G. graminis and V. mali with inhibition rates of 100% and 86.9%, respectively, and compound 85e showed an inhibition rate of 92.8% against Rhizoctonia solani.92
 |
| | Fig. 50 Structure of 4-fluoropyrazole derivatives 85a–f. | |
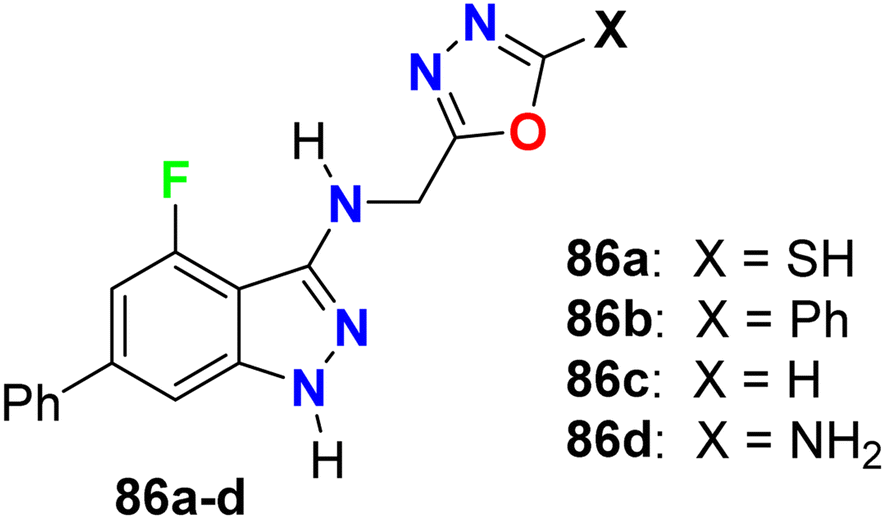
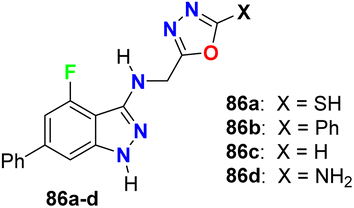
Some oxadiazole containing 4-fluoroindazoles 86a–d (Fig. 51) were tested for their antibacterial activity against B. subtilis and E. coli and antifungal activity against A. niger employing cup-plate bioassay at a concentration of 1000 μg mL−1. Compound 86a presented inhibition with 72.2% against both B. subtilis and E. coli and 50% against A. niger. The unsubstituted oxadiazole compound 86c presented the most potent inhibitory activity (94.4%) against B. subtilis and E. coli with almost equal potency as ciprofloxacin and showed 50% inhibition against A. niger. Moreover, the amino derivative 86d showed 94.4% inhibition against A. niger compared with griseofulvin 93.
 |
| | Fig. 51 Structure of oxadiazole containing 4-fluoroindazoles 86a–d. | |
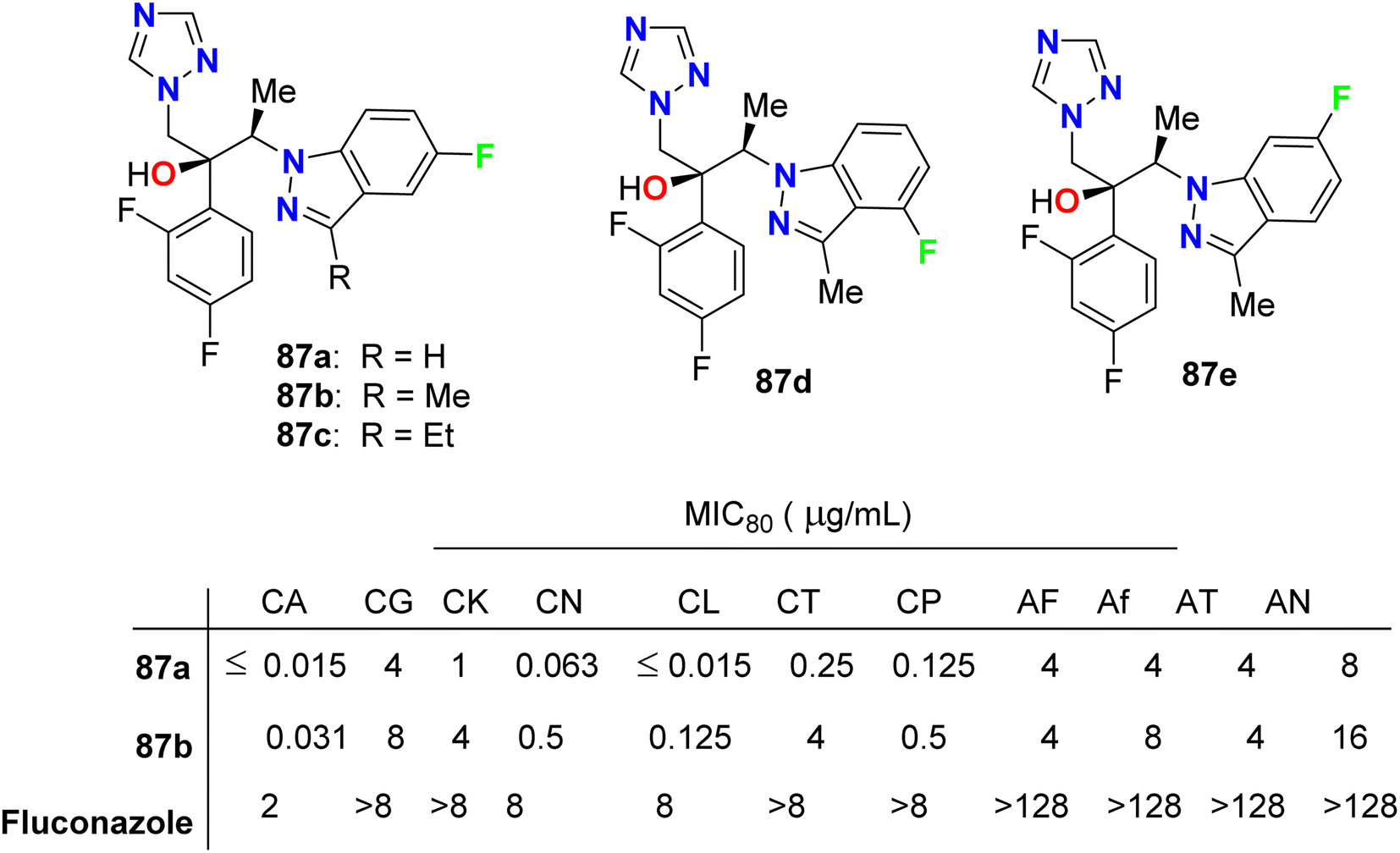
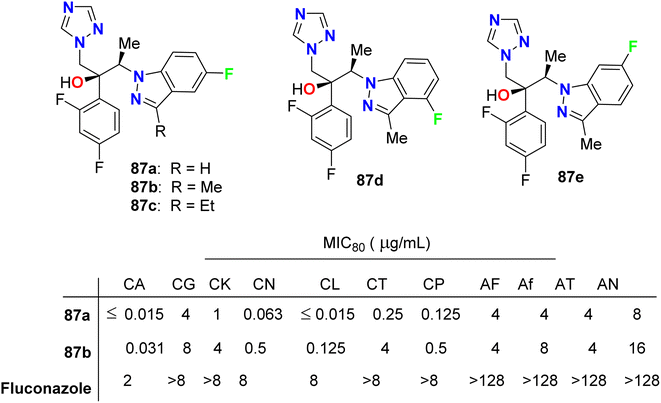
Park et al. described the in vitro antifungal activities of some chiral fluorinated indazole hybrids 87a–e (Fig. 52) against eleven fungi (Candida spp. and Aspergillus spp.); C. albicans, Candida krusei, Candida glabrata, Candida lusitaniae, Cryptococcus neoformans (C. neoformans), Candida tropicalis (C. tropicalis), Candida parapsilosis, Aspergillus fumigatus, A. niger, A. flavus and Aspergillus terreus. The antifungal potencies were determined by in vitro broth microdilution assay. The synthesized compounds displayed variable inhibition potencies against most of the tested fungal pathogens and their MIC80's were determined. The 5-fluoroindazole derivatives 87a and 87b were reported as promising lead candidates for antifungal therapy, where they showed the most potent antifungal activity with a broad spectrum.94
 |
| | Fig. 52 Structure of fluorinated indazole derivatives 87a–e. | |
3.3. Antimicrobial activity of fluorinated benzazoles
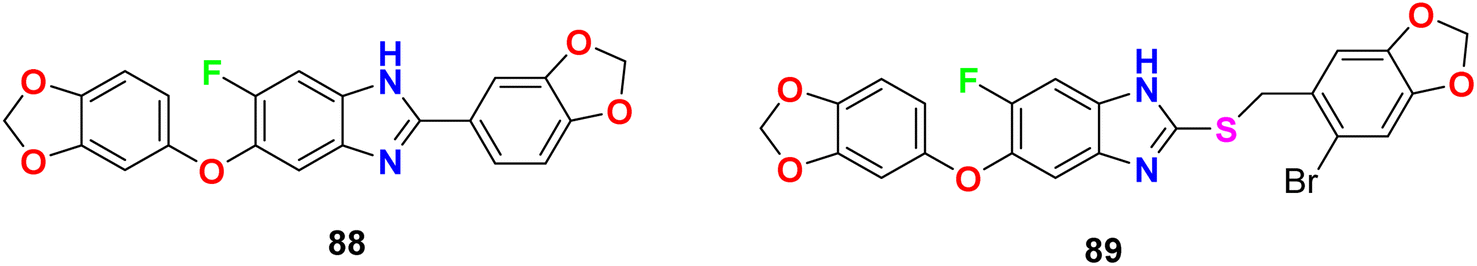
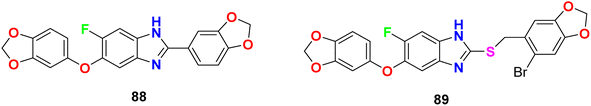
The in vitro anti-mycobacterial activities of the fluorobenzimidazole derivatives 88 and 89 (Fig. 53) against M. tuberculosis H37Rv strain by the MABA method were reported. The tested compounds presented moderate activity. Compounds 88 and 89 displayed antitubercular activity with MIC lower than that determined for the naturally occurring sesamin against the pathogenic MTB H37Rv strain. Compounds 88 and 89 presented better activity against MTB H37Rv both with MIC = 25 μg mL−1, compared to sesamin, the antitubercular natural product (MIC = 50 μg mL−1).95
 |
| | Fig. 53 Structure of fluorobenzimidazole derivatives 88 and 89. | |
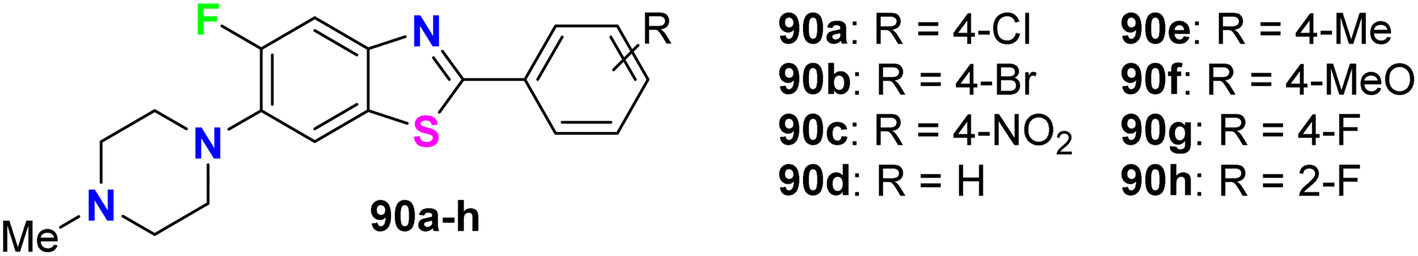
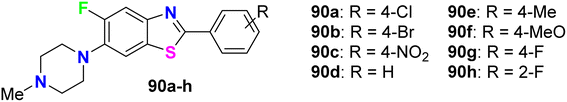
Al-Harthy et al. prepared a series of 5-fluoro-benzothiazole derivatives 90a–h (Fig. 54) and screened their antimicrobial activity against a number of bacterial and fungi strains: E. coli, K. pneumoniae, Acinetobacter baumannii (A. baumannii), P. aeruginosa, and S. aureus. The same compounds were screened against two fungal strains, C. albicans and C. neoformans, at a concentration of 32 μg mL−1. Overall, all compounds presented antibacterial activity against all tested strains, especially S. aureus, and also showed more activity towards Gram-positive bacteria than Gram-negative ones. Compounds 90a and 90b, particularly, displayed the best inhibitory potency against S. aureus, with 92.34% and 81.42% growth inhibition, respectively. Both compounds 90a and 90b had MIC 32 μg mL−1 compared to the tamoxifen reference standard with MIC 10 μg mL−1. Moreover, only compound 90b showed the best inhibitory potency against the fungal strain C. neoformans with 103.06% growth inhibition.96
 |
| | Fig. 54 Structure of 5-fluoro-benzothiazole derivatives 90a–h. | |
Jauhari et al. described the 6-fluorobenzoxazole derivative 64 (Fig. 37 above) as an antimicrobial agent using the disc diffusion method at a concentration of 10 μg mL−1. Interestingly, compound 64 exhibited significant antifungal activity against both A. flavus and A. niger with 90% and 95% growth inhibition, respectively. On the other hand, the antibacterial screening against P. aeruginosa, S. aureus, and K. pneumoniae, showed remarkable activity with inhibition zone diameters of 20–25 mm.80
3.4. Antimicrobial activity of fluorinated benzosiloxaboroles
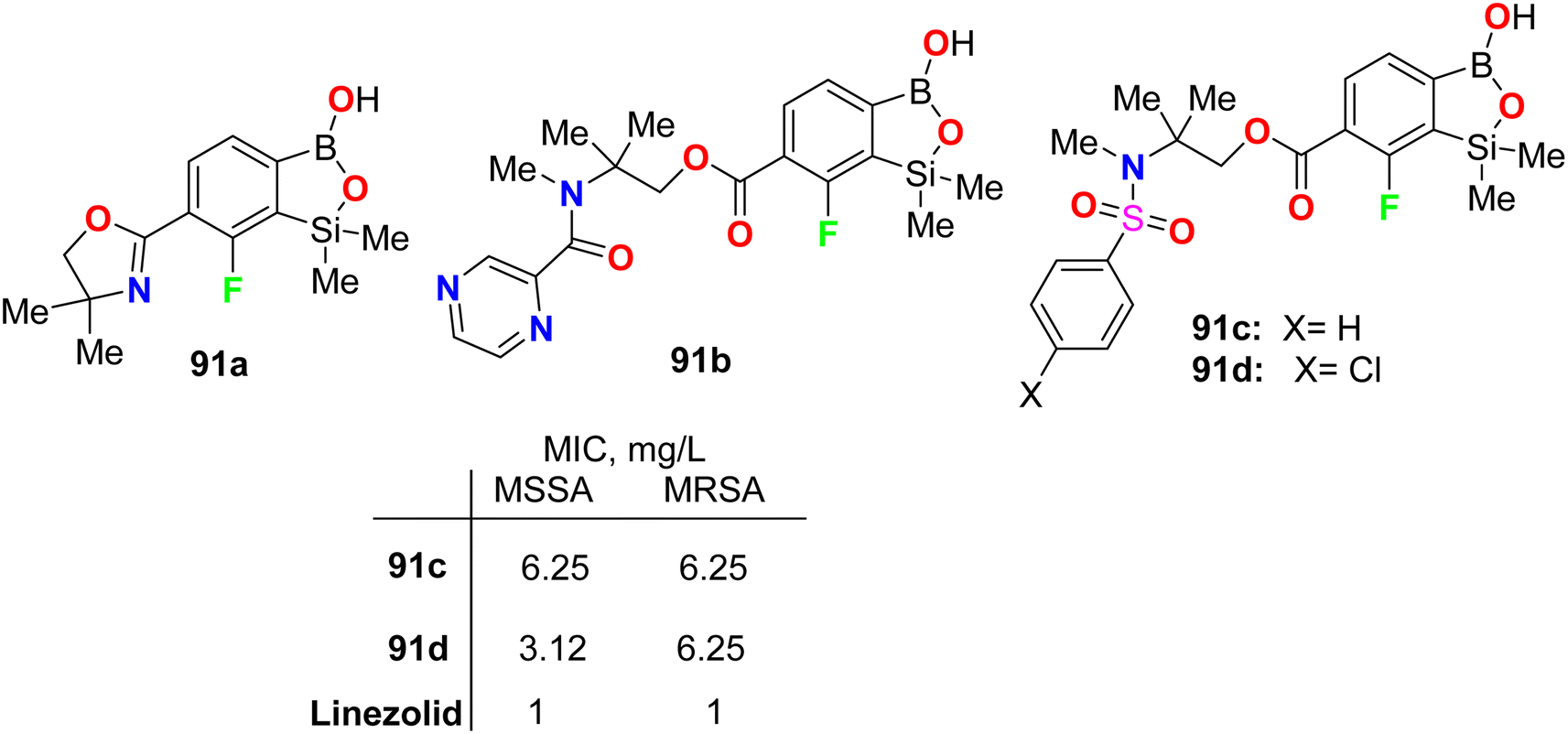
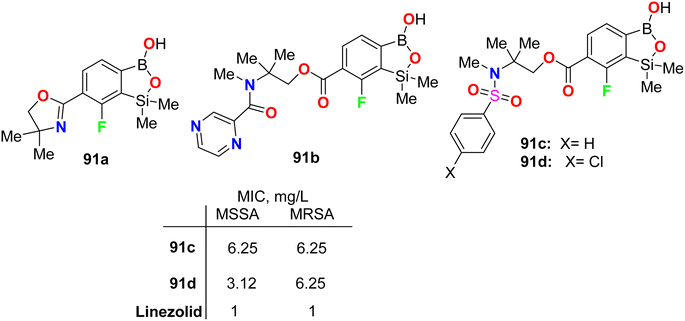
Krajewska et al. synthesized the benzosiloxaborole derivatives 91a–d (Fig. 55) and investigated their antimicrobial activity against the Gram-positive cocci, methicillin-sensitive S. aureus (MSSA) and methicillin-resistant S. aureus (MRSA) as well as the MRSA clinical strains. The tested compounds 91a–d exhibited high activity with MIC ranging between 3.12–6.25 mg L−1. These compounds also presented moderate activity against Enterococcus faecalis and Enterococcus faecium (with MIC values 25–50 mg L−1). The studies confirmed that compound 91d, having sulfonamide moiety, demonstrated the best inhibitory activity (MIC 3.12 mg L−1) and was not cytotoxic at the concentration close to its MIC value for S. aureus and was considered a potential antibacterial agent against S. aureus MRSA.97
 |
| | Fig. 55 Structure of fluorinated benzosiloxaborole derivatives 91a–d. | |
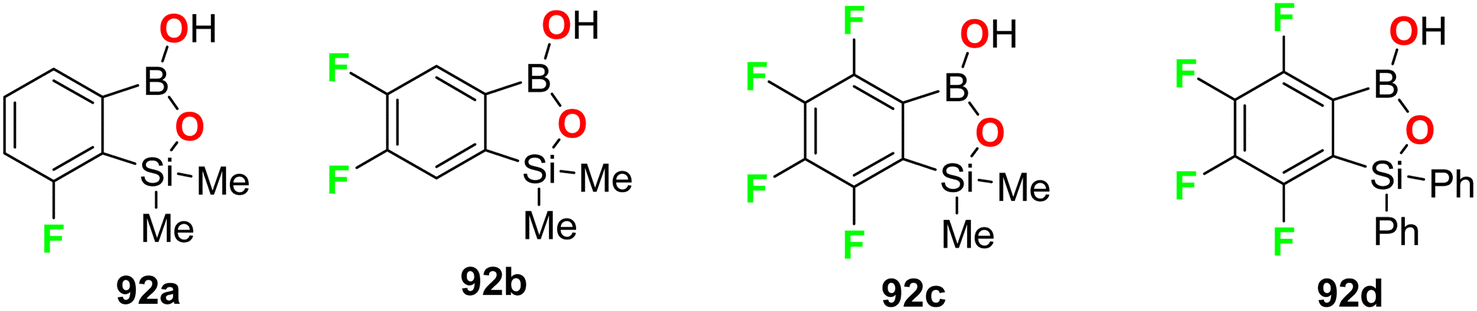
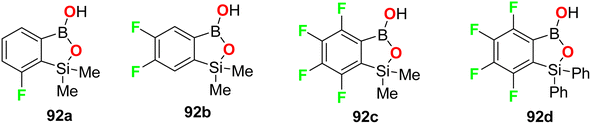
The fluorine-containing benzosiloxaborole derivatives 92a–f (Fig. 56) were designed by Brzozowska et al. and screened for their antimicrobial potency. The bioassay data was based on using 14 bacterial and 7 yeast standard strains in the study, and the MIC and MBC (minimal bactericidal concentration) were calculated following CLSI (Clinical and Laboratory Standards Institute) and EUCAST (European Committee for Antimicrobial Susceptibility Testing) methodology. The fluorinated compounds were more active against yeast strains than against bacteria. Compounds 92b and 92c were the most active, and both demonstrated promising antifungal activity against C. tropicalis with MIC values of 0.78 and 1.56 mg L−1, respectively. Compound 92b showed significant antifungal activity against Candida guilliermondii IBA-155, C. tropicalis IBA-171, Saccharomyces cerevisiae ATCC-9763, Saccharomyces cerevisiae IBA-198 with MIC values ranging between 0.78–6.25 mg L−1, respectively.98
 |
| | Fig. 56 Structure of the fluorine-containing benzosiloxaborole derivatives 92a–d. | |
4. Conclusions
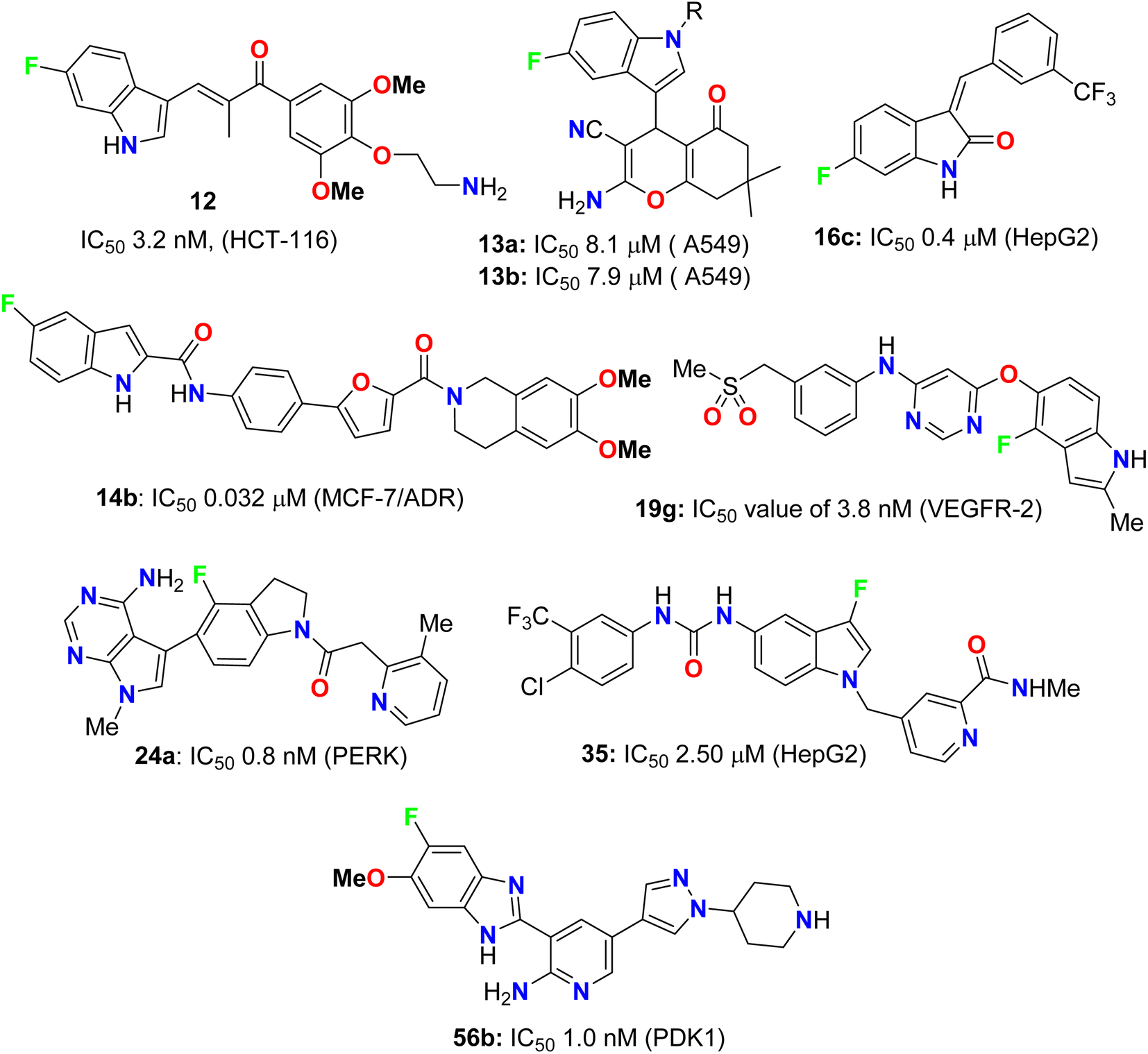
It was reported that 20% of the anticancer and antibiotic drugs approved by the FDA contain fluorine atom(s). The current review article outlines numerous directly ring-fluorinated heterocycles (five-membered and their benzo-fused systems) of potent in vivo and in vitro anticancer and antimicrobial activities. Thereby, many compounds were considered as lead structures for drug design developments. Some fluorinated heterocycles were found to be promising inhibitors and selective against a wide array of human cancer cell lines on a nanomolar scale. Some fluorinated heterocycles were also established as highly potent bactericidal and fungicidal activities via strong inhibition of various bacterial and fungal strains with almost equal or better potency with the appropriate reference drugs. In most cases, the reported fluorine-containing heterocycles demonstrated promising safety index via their reduced cytotoxicity in non-cancerous cell lines, and such derivatives are interesting candidates for anticancer or antibiotic drug discovery. In addition, several fluorinated heterocycles were found to be useful for the regulation of cell proliferation, autophagy and apoptosis. SAR study assigned that the position of a fluorine atom at the ring fluorinated heterocycles greatly affected the anticancer activity to be much better than some of the corresponding reference drugs. Furthermore, fluorinated heterocycles having various electron-donating or electron-withdrawing substituents significantly influenced the anticancer and antimicrobial activities. The mechanism of action disclosed that the cytotoxicity against the cancer cells employed the induction of cell death by apoptosis. This review is expected to open a new avenue for the development of anticancer and antimicrobial therapeutics. Charts 1 and 2 demonstrate the leading structures that showed higher inhibitory activities than reference drugs.
 |
| | Chart 1 Examples of compounds having anticancer activity better than reference drugs. | |
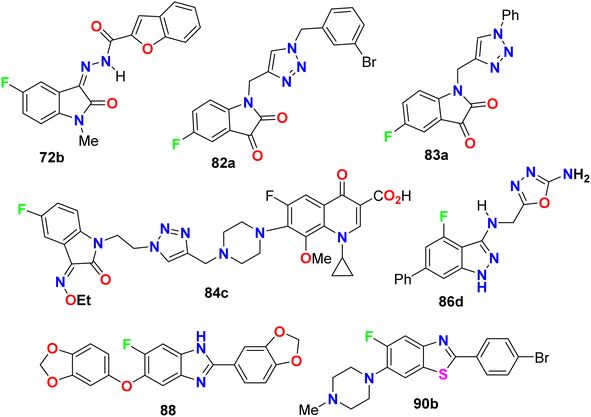 |
| | Chart 2 Examples of compounds having antimicrobial activity better than reference drugs. | |
Data availability
This is a review article, and there is no available data.
Author contributions
All authors, A. A. A., T. A. F. and K. M. D., have equal sharing in all preparation steps for the review in its current final form.
Conflicts of interest
There are no conflicts of interest to declare.
References
- P. Kumar, D.-M. Zhang, K. Degenhardt and Z.-S. Chen, Cells, 2012, 1, 558–575 CrossRef.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS.
- G. Kalin, E. Alp, A. Chouaikhi and C. Roger, Microorganisms, 2023, 11, 2575 CrossRef.
- L.-Y. Zhang, B.-L. Wang, Y.-Z. Zhan, Y. Zhang, X. Zhang and Z.-M. Li, Chin. Chem. Lett., 2016, 27, 163–167 CrossRef CAS.
- S. B. Lagu, R. P. Yejella, S. Nissankararao, R. R. Bhandare, V. S. Golla, B. V. S. Lokesh, M. M. Rahman and A. B. Shaik, PLoS One, 2022, 17, e0265068 CrossRef CAS.
- Fluorine in Heterocyclic Chemistry, ed. V. Nenajdenko, Springer, 2014, vol. 1, issue 2 Search PubMed.
- Fluorinated Heterocyclic Compounds: Synthesis, Chemistry and Applications, ed. V. A. Petrov, Wiley, New York, 2009 Search PubMed.
- Fluorinated Heterocycles, ed. A. Gakh and K. L. Kirk, ACS, Washington, 2009 Search PubMed.
- P. V. Reddy, Organofluorine Compounds in Biology and Medicine, Chapter 9, Organofluorine Compounds as Anticancer Agents, Elsevier, 2015, pp. 265–300 Search PubMed.
- J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS.
- I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Blackwell, Chichester, 2009 Search PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef.
- C. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren, L. Griesbach, R. Duschinsky, R. J. Schnitzer, E. Pleven and J. Scheiner, Nature, 1957, 179, 663–666 CrossRef CAS.
- D. B. Mendel, A. D. Laird, X. Xin, S. G. Louie, J. G. Christensen, G. Li, R. E. Schreck, T. J. Abrams, T. J. Ngai, L. B. Lee, L. J. Murray, J. Carver, E. Chan, K. G. Moss, J. O. Haznedar, J. Sukbuntherng, R. A. Blake, L. Sun, C. Tang, T. Miller, S. Shirazian, G. McMahon and J. M. Cherrington, Clin. Cancer Res., 2003, 9, 327 CAS.
- H. K. Gan, B. Seruqa and J. J. Knox, Expert Opin. Invest. Drugs, 2009, 18, 821–834 CrossRef CAS.
- R. J. Motzer, B. Escudier, A. Gannon and R. A. Figlin, Oncologist, 2017, 22, 41–52 CrossRef CAS.
- L. DeVorkin, M. Hattersley, P. Kim, J. Ries, J. Spowart, M. S. Anglesio, S. M. Levi, D. G. Huntsman, R. K. Amaravadi and J. D. Winkler, Mol. Cancer Res., 2017, 15, 250–258 CrossRef CAS.
- M. Martin and J. A. Garcia-Saenz, Future Oncol., 2020, 16, 2763–2778 CrossRef CAS.
- V. Subbiah, T. Shen, S. S. Terzyan, X. Liu, X. Hu, K. P. Patel, M. Hu, M. Cabanillas, A. Behrang and F. Meric-Bernstam, Ann. Oncol. Off. J. Eur. Soc. Med. Oncol., 2021, 32, 261–268 CrossRef CAS.
- S. Dhillon, Drugs, 2020, 80, 1373–1378 CrossRef CAS.
- P. Koelblinger, J. Dornbierer and R. Dummer, Future Oncol., 2017, 13, 1755–1766 CrossRef CAS.
- X. Zheng, J. Qiu, W. Pan, Y. Gong, W. Zhang, T. Jiang, L. Chen, W. Chen and Z. Hong, Front. Pharmacol, 2022, 13, 938133 CrossRef CAS.
- J. Rieger, S. Durka, J. Streffer, J. Dichgans and M. Weller, Eur. J. Pharmacol., 1999, 365, 301–308 CrossRef CAS.
- J. Qian, Y. Han, J. Li, J. Zhang and C. Hu, Toxicol. in Vitro, 2018, 46, 137–147 CrossRef CAS.
- A. Markham, Delafloxacin: First Global Approval, Drugs, 2017, 77, 1481–1486 CrossRef CAS.
- A. Grillon, F. Schramm, M. Kleinberg and F. Jehl, PLoS One, 2016, 11, e0156690 CrossRef.
- P. L. Kalar, S. Agrawal, S. Kushwaha, S. Gayen and K. Das, Curr. Org. Chem., 2023, 27, 190–205 CrossRef CAS.
- R. Chatterjee and R. Dandela, J. Fluorine Chem., 2023, 268, 110133 CrossRef CAS.
- R. Szpera, D. F. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew Chem. Int. Ed. Engl., 2019, 58, 14824–14848 CrossRef CAS.
- E. V. Nosova, G. N. Lipunova, V. N. Charushin and O. N. Chupakhin, J. Fluorine Chem., 2018, 212, 51–106 CrossRef CAS.
- T. Fujita and J. Ichikawa, Heterocycles, 2017, 95, 694–714 CrossRef CAS.
- M. D. Markus, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- K. M. Dawood, Tetrahedron, 2004, 60, 1435–1451 CrossRef CAS.
- K. M. Dawood and T. Fuchigami, J. Org. Chem., 2004, 69, 5302–5306 CrossRef CAS.
- K. M. Dawood and T. Fuchigami, J. Org. Chem., 2001, 66, 7691–7695 CrossRef CAS.
- K. M. Dawood, H. Ishii and T. Fuchigami, J. Org. Chem., 2001, 66, 7030–7034 CrossRef CAS.
- T. Fuchigami, S. Higashiya, Y. Hou and K. M. Dawood, Rev. Heteroat. Chem., 1999, 19, 67–78 Search PubMed.
- M. G. Campbell and T. Ritter, Org. Process Res. Dev., 2014, 18, 474–480 CrossRef CAS.
- A. J. Ayoub, G. A. El-Achkar, S. E. Ghayad, L. Hariss, R. H. Haidar, L. M. Antar, Z. I. Mallah, B. Badran, R. Grée, A. Hachem and E. Hamade, Int. J. Mol. Sci., 2023, 24, 10399 CrossRef CAS.
- L. S. Pidugu, H. W. Servius, S. E. Sevdalis, M. E. Cook, K. M. Varney, E. Pozharski and A. C. Drohat, PLoS One, 2023, 18, e0280526 CrossRef CAS.
- B. Du, X. Liu, X. Luan, W. Zhang and C. Zhuang, Bioorg. Chem., 2023, 135, 106531 CrossRef CAS.
- M. S. Malik, H. Ather, S. M. A. Ansari, A. Siddiqua, Q. M. S. Jamal, A. H. Alharbi, M. M. Al-Rooqi, R. S. Jassas, E. M. Hussein, Z. Moussa, R. J. Obaid and S. A. Ahmed, Pharmaceuticals, 2023, 16, 333 CrossRef CAS.
- Z. Yang, X. Yang, Y. Li, Y. Cai, Y. Yu, W. Zhuang, X. Sun, Q. Li, X. Bao, X. Ye, J. Tian, B. Wei, J. Chen, Q. Wud, H. Zhang, X. Mou and H. Wang, Eur. J. Med. Chem., 2023, 248, 115092 CrossRef CAS.
- C. A. London, P. B. Malpas, S. L. Wood-Follis, J. F. Boucher, A. W. Rusk, M. P. Rosenberg, C. J. Henry, K. L. Mitchener, M. K. Klein, J. G. Hintermeister, P. J. Bergman, G. C. Couto, G. N. Mauldin and G. M. Michels, Cancer Res., 2009, 15, 3856–3865 CAS.
- H. K. Ho, B. T. Chua, W. Wong, K. S. Lim, V. Teo, H. T. Ong, X. Chen, W. Zhang, K. M. Hui, M. L. Go and A. Ullrich, Mol. Oncol., 2014, 8, 1266–1277 CrossRef CAS.
- X. Chen, T. Yang, A. Deivasigamani, M. K. Shanmugam, K. M. Hui, G. Sethi and M. L. Go, ChemMedChem, 2015, 19, 1548–1558 CrossRef.
- G. R. Gao, M. Y. Li, L. J. Tong, L. X. Wei, J. Ding, H. Xie and W. H. Duan, Chin. Chem. Lett., 2015, 26, 1165–1168 CrossRef CAS.
- N. Gokhale, N. Panathur, U. Dalimba, P. G. Nayak and K. S. R. Pai, J. Heterocycl. Chem., 2016, 53, 513–524 CrossRef CAS.
- C. Pecoraro, M. De Franco, D. Carbone, D. Bassani, M. Pavan, S. Cascioferro, B. Parrino, G. Cirrincione, S. Dall'Acqua, S. Moro, V. Gandin and P. Diana, Eur. J. Med. Chem., 2023, 249, 115134 CrossRef CAS.
- X. Liu, J. Jin, Y. Wu, B. Du, L. Zhang, D. Lu, Y. Liu, X. Chen, J. Lin, H. Chen, W. Zhang, C. Zhuang and X. Luan, Eur. J. Med. Chem., 2023, 257, 115540 CrossRef CAS.
- J. M. Axten, S. P. Romeril, A. Shu, J. Ralph, J. R. Medina, Y. Feng, W. H. H. Li, S. W. Grant, D. A. Heerding, E. Minthorn and T. Mencken, ACS Med. Chem. Lett., 2013, 4, 964–968 CrossRef CAS.
- J. R. Medina, A. K. Charnley and S. P. Romeril, PCT WO2015056180A1, 2015.
- C. Atkins, Q. Liu, E. Minthorn, S. Y. Zhang, D. J. Figueroa, K. Moss, T. B. Stanley, B. Sanders, A. Goetz, N. Gaul and A. E. Choudhry, Cancer Res., 2013, 73, 1993–2002 CrossRef CAS.
- F. Ban, E. Leblanc, H. Li, R. S. Munuganti, K. Frewin, P. S. Rennie and A. Cherkasov, J. Med. Chem., 2014, 57, 6867–6872 CrossRef CAS.
- P. Rennie, A. Tcherkassov, R. N. Young and C. M. Andre, PCT WO2015154169A1, 2015.
- M. P. Tantak, J. Wang, R. P. Singh, A. Kumar, K. Shah and D. Kumar, Bioorg. Med. Chem. Lett., 2015, 25, 4225–4231 CrossRef CAS.
- M. P. Tantak, V. Gupta, K. Nikhil, V. Arun, R. P. Singh, P. N. Jha, K. Shah and D. Kumar, Bioorg. Med. Chem. Lett., 2016, 26, 3167–3171 CrossRef CAS.
- S. Mahboobi, A. Uecker, A. Sellmer, C. Cénac, H. Höcher, H. Pongratz, E. Eichhorn, H. Hufsky, A. Trümpler, M. Sicker and F. Heidel, J. Med. Chem., 2006, 49, 3101–3115 CrossRef CAS.
- D. Kumar, N. M. Kumar, M. P. Tantak, M. Ogura, E. Kusaka and T. Ito, Bioorg. Med. Chem. Lett., 2014, 24, 5170–5174 CrossRef CAS.
- Z. Wu, M. Yan, S. H. Hu, Z. C. Yu, Y. Zhu, Y. D. Cheng, H. C. Liu, Y. M. Zhang, S. H. Yao, W. F. Tang and T. Lu, Chin. Chem. Lett., 2014, 25, 351–354 CrossRef CAS.
- T. O. S. Kumar, K. M. Mahadevan and M. N. Kumara, Int. J. Pharm. Pharm. Sci., 2014, 6, 137–140 Search PubMed.
- G. D. Glick, A. R. Hurd, C. B. Taylor and C. A. Vanhuis, PCT WO2012078874A1, 2012.
- T. Yasuma, M. Kamato, T. Yamashita, H. Hirose, M. Murakami, A. Kina, K. Yonemori, R. Mizojiri, I. Fujimori, T. Fujimoto and Z. Ikeda, PCT WO2012074126A1, 2012.
- I. R. Baldwin, K. D. Down, P. Faulder, S. Gaines, J. N. Hamblin, J. Le, C. J. Lunniss, N. J. Parr, T. J. Ritchie, J. E. Robinson, J. K. Simpson and C. A. P. Smethurst, US Pat., US2011178063A1, 2011 Search PubMed.
- I. R. Baldwin, K. D. Down, P. Faulder, S. Gaines, J. N. Hamblin, J. Le, C. J. Lunniss, N. J. Parr, T. J. Ritchie, J. K. Simpson and C. A. P. Smethurst, PCT WO2009147190A1, 2009.
- I. R. Baldwin, K. D. Down, P. Faulder, S. Gaines, J. N. Hamblin, J. Le, C. J. Lunniss, N. J. Parr, T. J. Ritchie, J. E. Robinson, J. K. Simpson and C. A. P. Smethurst, PCT WO2011067364A1, 2011.
- I. R. Baldwin, K. D. Down, P. Faulder, S. Gaines, J. N. Hamblin, K. L. Jones, P. S. Jones, J. Le, C. J. Lunniss, N. J. Parr, T. J. Ritchie, J. E. Robinson, J. K. Simpson and C. A. P. Smethurst, US Pat., US2012245171A1, 2012 Search PubMed.
- M. L. Boys, R. K. Delisle, E. J. Hicken, A. L. Kennedy, D. A. Mareska, F. P. Marmsater, M. C. Munson, B. Newhouse, B. Rast, J. P. Rizzi, M. E. Rodriguez, G. T. Topalov and Q. Zhao, PTC WO2012082689A1, 2012.
- H. Rehwinkel, A. Haegebarth, O. Politz, R. Neuhaus and U. Boemer, PCT WO2012080237A1, 2012.
- M. Michels, M. Follmann, A. Vakalopoulos, K. Zimmermann, M. Lobell, N. Teusch and S. Yuan, PCT WO2010094405A1, 2010.
- M. Michels, M. Follmann, A. Vakalopoulos, K. Zimmermann, N. Teusch and M. Lobell, PCT WO2011003604A1, 2011.
- P. Chene, A. Floersheimer, P. Furet and J. Schoepfe, PCT WO2006010595A1, 2006.
- A. Kamal, D. R. Reddy, M. K. Reddy, G. Balakishan, T. B. Shaik, M. Chourasia and G. N. Sastry, Bioorg. Med. Chem., 2009, 17, 1557–1572 CrossRef CAS.
- T. S. Reddy, H. Kulhari, V. G. Reddy, V. Bansal, A. Kamal and R. Shukla, Eur. J. Med. Chem., 2015, 101, 790–805 CrossRef CAS.
- M. Calderini, M. Wucherer-Plietker, U. Graedler and C. Esdar, PCT WO2011006567A1, 2011.
- C. J. Lion, C. S. Matthews, G. Wells, T. D. Bradshaw, M. F. Stevens and A. D. Westwell, Bioorg. Med. Chem. Lett., 2006, 16, 5005–5008 CrossRef CAS.
- R. K. Gill, G. Singh, A. Sharma, P. M. S. Bedi and A. K. Saxena, Med. Chem. Res., 2013, 22, 4211–4222 CrossRef CAS.
- S. Aiello, G. Wells, E. L. Stone, H. Kadri, R. Bazzi, D. R. Bell, M. F. Stevens, C. S. Matthews, T. D. Bradshaw and A. D. Westwell, J. Med. Chem., 2008, 51, 5135–5139 CrossRef CAS.
- P. K. Jauhari, A. Bhavani, S. Varalwar, K. Singhal and P. Raj, Med. Chem. Res., 2008, 17, 412–424 CrossRef CAS.
- C. Bolea, PCT WO2010079239A1, 2010.
- P. Das, S. Boone, D. Mitra, L. Turner, R. Tandon, D. Raucher and A. T. Hamme, RSC Adv., 2020, 10, 30223–30237 RSC.
- H. Ren, H. An, P. J. Hatala, W. C. Stevens Jr, J. Tao and B. He, Beilstein J. Org. Chem., 2015, 11, 2509–2520 CrossRef CAS.
- M. A. Siddiqui, Y. Nan, M. F. Patel, P. A. P. Reddy, U. F. Mansoor, Z. Meng, L. D. Vitharana, L. Zhao, A. K. Mandal, D. Liu, S. Tang, A. McRiner, D. B. Belanger, P. J. Curran, C. Dai, A. R. Angeles, L. Yang and M. H. Daniels, PCT WO2011090935A1, 2011.
- M. E. Fraley, G. D. Hartman and W. F. William, PCT WO2003049527A2, 2003.
- B. Bhardwaj and S. C. Jain, Pharma Chem., 2014, 6, 272–278 Search PubMed.
- X. M. Zhang, H. Guo, Z. S. Li, F. H. Song, W. M. Wang, H. Q. Dai, L. X. Zhang and J. G. Wang, Eur. J. Med. Chem., 2015, 101, 419–430 CrossRef CAS.
- B. D. Varpe and S. B. JadhavZ, Int. J. Pharm. Invest., 2021, 11, 189–194 CrossRef CAS.
- T. Yang, W. Moreira, S. A. Nyantakyi, H. Chen, D. B. Aziz, M. L. Go and T. Dick, J. Med. Chem., 2017, 60, 2745–2763 CrossRef CAS.
- K. V. Praveen, J. Renjitha, C. T. F. Salfeena, K. T. Ashitha, S. K. Rangappa, V. Sunil and B. S. Sasidhar, Chem. Biol. Drug Des., 2017, 90, 703–708 CrossRef.
- S. Deswal, R. K. Tittal, D. G. Vikas, K. Lal and A. Kumar, J. Mol. Struct., 2020, 1209, 127982 CrossRef CAS.
- Z. Xu, X. F. Song, M. Qiang and Z. S. Lv, J. Heterocycl. Chem., 2017, 54, 3735–3741 CrossRef CAS.
- X. Dan, W. Wei, H. Jiachuan, L. Zhengyi, Z. Siying and X. Gong, CN Pat., CN116003323A, 2023 Search PubMed.
- R. A. Kusanur and R. Mahesh, Int. J. Life Sci. Pharma Res., 2013, 3, 6–10 CAS.
- J. S. Park, A. Y. Kyung, T. H. Kang, S. Kim and Y. G. Suh, Bioorg. Med. Chem. Lett., 2007, 17, 3486–3490 CrossRef CAS.
- B. Nandha, L. G. Nargund, S. L. Nargund and K. Bhat, Iran. J. Pharm. Res., 2017, 16, 929 CAS.
- T. Al-Harthy, W. M. Zoghaib, R. Stoll and R. Abdel-Jalil, Monatsh. Chem., 2018, 149, 645–651 CrossRef CAS.
- J. Krajewska, K. Nowicki, K. Durka, P. H. Marek-Urban, P. Wińska, T. Stępniewski, K. Woźniak, A. E. Laudy and S. Luliński, RSC Adv., 2022, 12, 23099–23117 RSC.
- A. Brzozowska, P. Ćwik, K. Durka, T. Kliś, A. E. Laudy, S. Luliński, J. Serwatowski, S. Tyski, M. Urban and W. Wróblewski, Organometallics, 2015, 34, 2924–2932 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Thoraya A. Farghaly
a,
Thoraya A. Farghaly