
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric defluorinative allylation of silyl enol ethers†
Jordi
Duran
 ,
Javier
Mateos
,
Albert
Moyano
and
Xavier
Companyó
*
,
Javier
Mateos
,
Albert
Moyano
and
Xavier
Companyó
*
Section of Organic Chemistry, Department of Inorganic and Organic Chemistry, University of Barcelona, Carrer Martí i Franquès 1, 08028 Barcelona, Spain. E-mail: x.companyo@ub.edu; Web: https://companyolab.com
First published on 24th May 2023
Abstract
The stereocontrolled installation of alkyl fragments at the alpha position of ketones is a fundamental yet unresolved transformation in organic chemistry. Herein we report a new catalytic methodology able to construct α-allyl ketones via defluorinative allylation of silyl enol ethers in a regio-, diastereo- and enantioselective manner. The protocol leverages the unique features of the fluorine atom to simultaneously act as a leaving group and to activate the fluorophilic nucleophile via a Si–F interaction. A series of spectroscopic, electroanalytic and kinetic experiments demonstrate the crucial interplay of the Si–F interaction for successful reactivity and selectivity. The generality of the transformation is demonstrated by synthesising a wide set of structurally diverse α-allylated ketones bearing two contiguous stereocenters. Remarkably, the catalytic protocol is amenable for the allylation of biologically significant natural products.
Introduction
α-Substituted carbonyl compounds are ubiquitous in natural products, agrochemicals, and pharmaceuticals. Therefore, synthetic methodologies able to construct such privileged motifs in a selective and catalytic manner are amongst the most fundamental transformations in organic chemistry.1 Unlike other parent carbonyl compounds, such as aldehydes and carboxylic acids,2 the asymmetric α-allylation of ketones still represents an unmet challenge in organic synthesis.3 In this regard, asymmetric allylic alkylation (AAA) reactions represent a powerful tool for the stereoselective insertion of allyl groups.4 Several catalytic protocols are known under transition-metal5 and organic Lewis-base6 catalysis, encompassing a wide array of nucleophiles.7 Indeed, the implementation of non-stabilised ketone enolates into transition-metal-catalysed AAA schemes embody an alternative approach for stereoselective α-allylation of ketones.8 Specifically, transformations employing silyl enol ethers (SEEs) as ketone surrogates are mainly catalysed by chiral iridium complexes and are restricted to linear, non-substituted substrates (Fig. 1A).9 As a result, the corresponding allylated ketones feature a single asymmetric carbon formed at the β position, while the challenging carbonyl α position does not exhibit stereogenicity (Fig. 1A).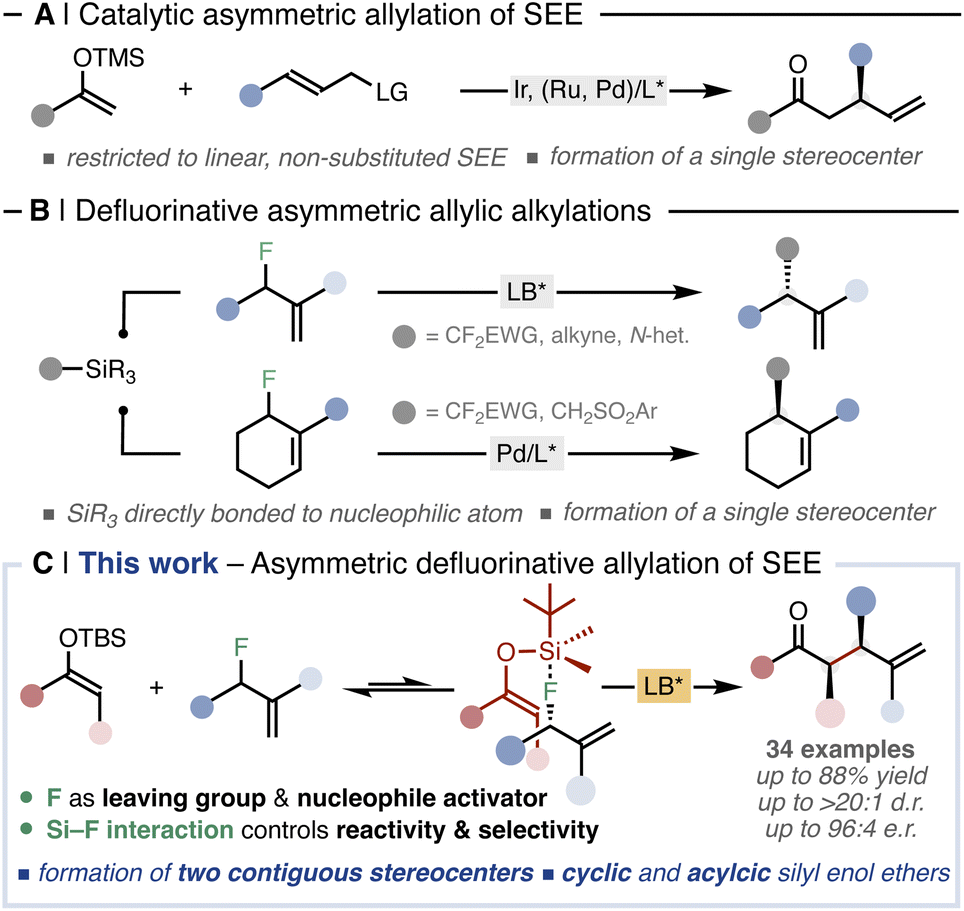 | ||
| Fig. 1 (A) Precedents in the allylation of SEE and (B) in defluorinative AAAs. (C) Catalytic defluorinative construction of α-allyl ketones. | ||
The activation and cleavage of strong C–F bonds is currently attracting significant attention, providing novel alternative electrophiles.10 As the C–F bond is the strongest single bond that carbon forms (BDE(Csp3–F) = up to 112 kcal mol−1), defluorinative reactions need to overcome high thermodynamic barriers.11 Thus, leveraging the ability of these highly polarized bonds to act as electrophilic counterparts in mild, controllable and selective catalytic transformations is becoming a central topic in modern organic chemistry.12 Shibata pioneered the use of allyl fluorides as unconventional electrophiles in AAA reactions with different silylated C-nucleophiles under Lewis-base catalysis (Fig. 1B, up).13 Subsequently, Vilotijevic expanded this strategy using N-silylated heterocycles as latent nucleophiles.14 Recently, Trost and Hartwig have implemented allyl fluorides in transition-metal catalysed defluorinative AAAs (Fig. 1B, bottom).15
The common feature of all these defluorinative transformations13–15 is that the silylated nucleophilic moieties do not comprise a prochiral center. Consequently, the corresponding allylated products bear only one stereogenic carbon (Fig. 1B). As a result, the simultaneous, catalytic formation of two or more asymmetric carbons via defluorinative allylation has not been addressed yet.
Herein, we report the first asymmetric allylation of SEEs able to form α-allylated ketones bearing two contiguous stereocenters in a single catalytic event (Fig. 1C). We demonstrate that a defluorinative strategy using allyl fluorides and Si-derived fluorophiles represents an effective approach to unlock the α-allylation of ketones in a regio-, diastereo- and enantiocontrolled manner, without the need of external, stoichiometric fluoride sources. The methodology leverages the unique features of fluorine to act in a concerted way both as a leaving group and as an activator of the fluorophilic SEE through Si–F interaction, overcoming thus the high thermodynamic demand for C–F bond cleavage. The exploitation of such interaction allows the formation of a highly organised transition state which is crucial for the regio- and the diastereochemical outcome. The reaction proceeds through a dynamic kinetic resolution of the racemic allyl fluorides where both the racemisation of the starting material and the enantioselectivity of the transformation are controlled by the chiral catalyst. Altogether this methodology provides a general catalytic protocol enabling the allylation of a wide set of cyclic and acyclic ketones, including complex natural products, in good yields and with excellent regio- and stereoselectivities.
Results and discussion
Reaction design and optimisation
We focused our initial studies by testing the direct allylation of cyclohexanone 1a with the classical allyl carbonate 2a catalysed by the achiral Lewis base DABCO (5a).6 Although the ionisation of the leaving group generates tert-butoxide anion that should be able to enolise 1a, no product was observed after 18 h (Fig. 2A, entry 1). Next, we decided to pre-form the SEE 1b of cyclohexanone to avoid the enolisation step, but no reactivity was observed either (Fig. 2B, entry 2). The use of an external fluoride source to activate 1b yielded only cyclohexanone with unreacted 2a (Fig. 2C, entry 3). We then hypothesised that the combination of a fluorinated allylating agent (2b) with SEE 1b could be a convenient alternative (Fig. 2D, entry 4). Gratifyingly, the use of allyl fluoride 2b enabled the formation of the α-allylated product 3a with 82% yield and with excellent regio- and diastereocontrol. This result points out that the simultaneous presence of the silicon moiety on the nucleophile and the fluorine atom on the electrophile is crucial for reactivity and selectivity. Next, we surveyed a wide set of chiral Lewis bases in different reaction conditions.16 This screening revealed (DHQD)2PHAL (5b) as the best catalyst in a THF/CH2Cl2 mixture, affording product 3a in 75% yield, good regiocontrol (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 branched vs. linear), complete syn diastereoselectivity (>20:1) and 95:5 e.r. after 72 h (Fig. 2D, entry 5). In an attempt to accelerate the asymmetric catalytic process, we tested the reaction in the presence of (nBu)4NF as additive. Despite full conversion of 2b was reached in 18 h, only the linear allyl ketone 4a was formed in 33% yield (Fig. 2C, entry 6). Hence, while the activation of 1b is crucial for reactivity, the uncontrolled formation of its enolate is detrimental for the desired selectivity.17
:1 branched vs. linear), complete syn diastereoselectivity (>20:1) and 95:5 e.r. after 72 h (Fig. 2D, entry 5). In an attempt to accelerate the asymmetric catalytic process, we tested the reaction in the presence of (nBu)4NF as additive. Despite full conversion of 2b was reached in 18 h, only the linear allyl ketone 4a was formed in 33% yield (Fig. 2C, entry 6). Hence, while the activation of 1b is crucial for reactivity, the uncontrolled formation of its enolate is detrimental for the desired selectivity.17
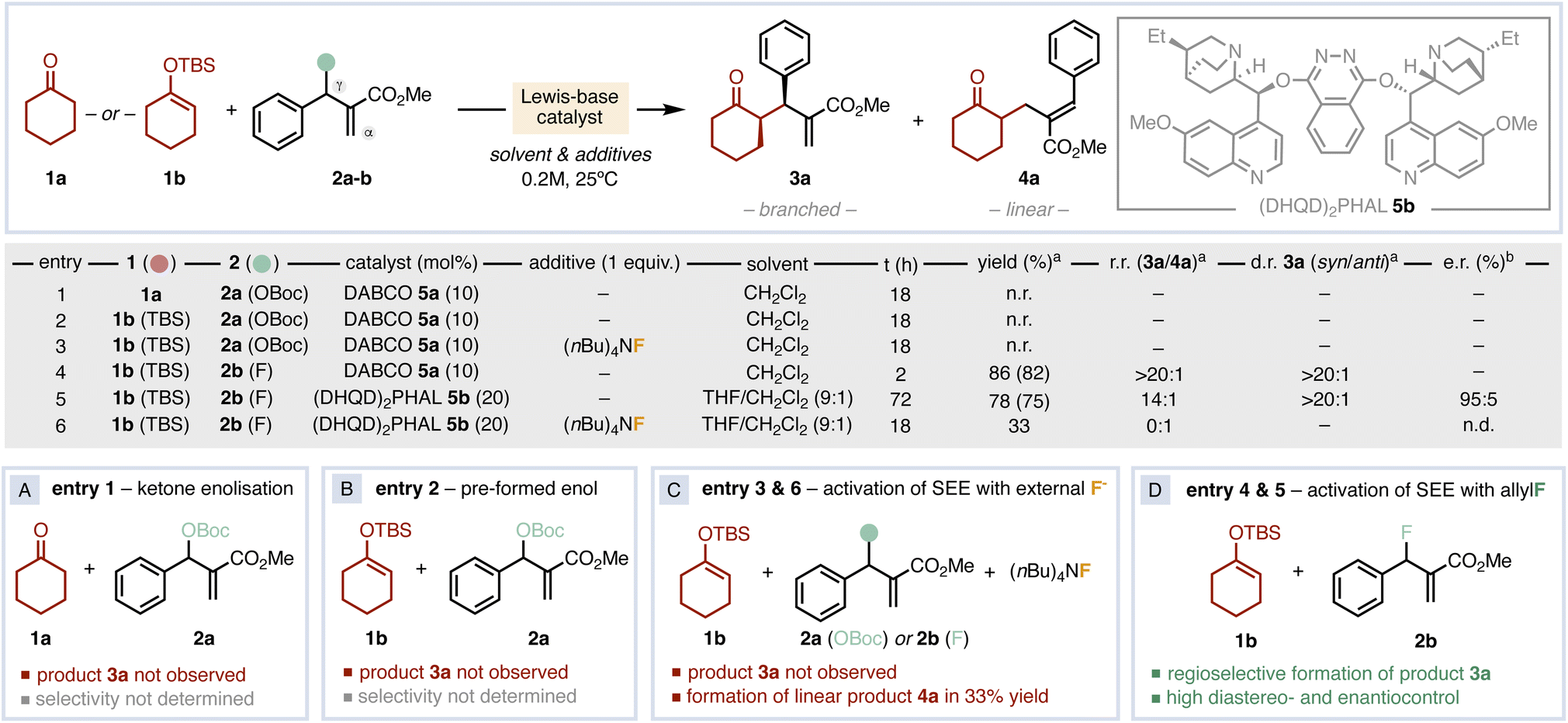 | ||
| Fig. 2 Reaction design and optimization. Selected results determined by 1H-NMRa (in parenthesis the isolated yield) and chiral HPLC.b n.d.: not determined. | ||
Mechanistic studies
Compelled by the excellent regio- and diastereoselectivity of the reaction, we studied its mechanism. We initially focused our attention on the exquisite diastereocontrol, as none of the catalytic defluorinative AAA reported to date13–15 generate more than one stereogenic carbon. First, we questioned whether the ammonium intermediate A could be detected (Fig. 3A), as it is commonly invoked as the competent electrophilic catalytic species in Lewis-base catalysed AAAs.6 Upon mixing 2b with 20 mol% of DABCO in CD2Cl2, A was quantitatively formed as a 75:25 E/Z mixture in 4 h. The release of fluoride anion was confirmed by 19F-NMR.16 Conversely, A was not formed after 24 h under the effect of (DHQD)2PHAL (Fig. 3A). Upon monitoring the reaction course by in situ NMR, the free catalyst 5b was identified as the catalyst resting state while no traces of the corresponding ammonium species A could be detected.16
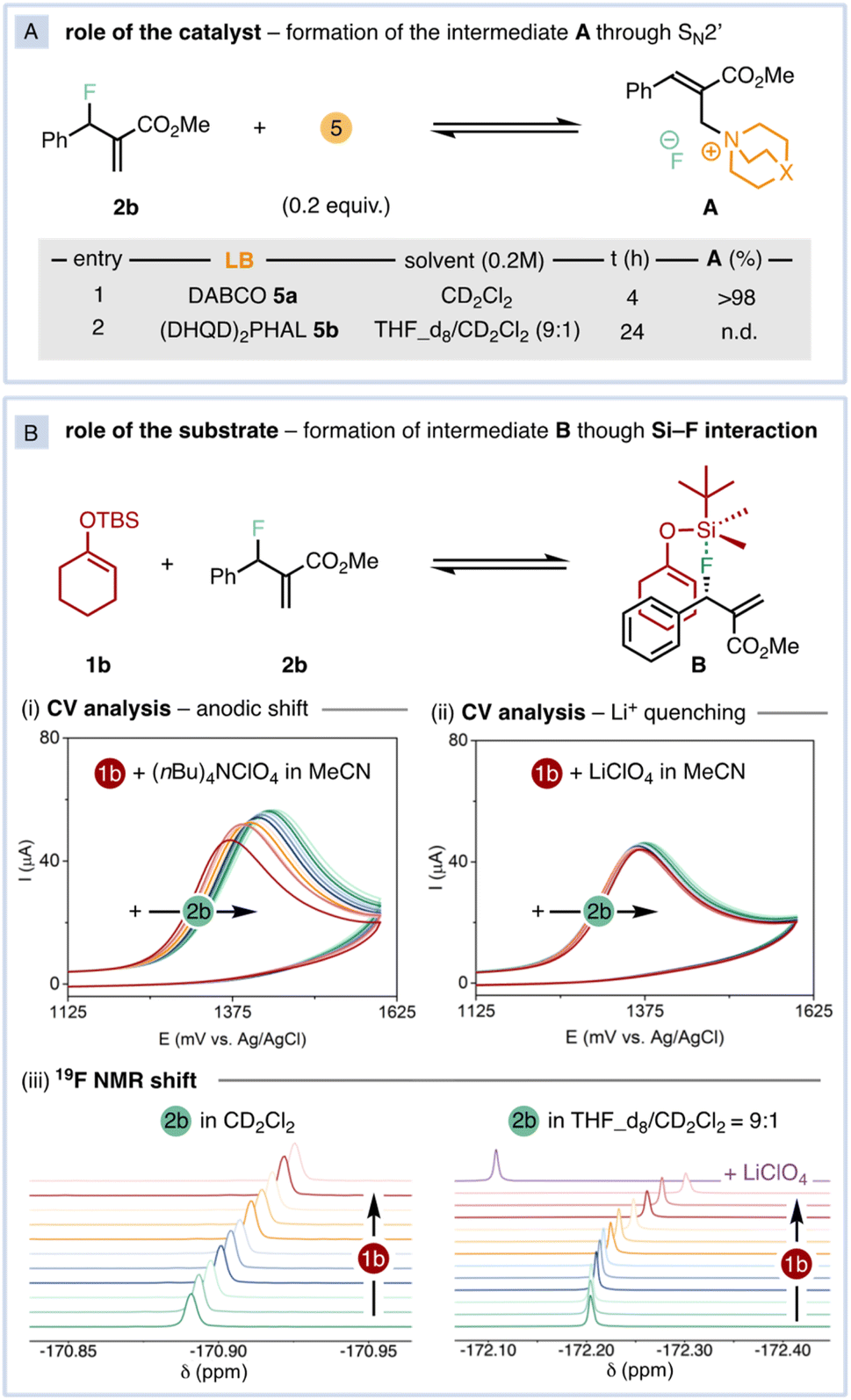 | ||
| Fig. 3 (A) Role of the catalyst. In situ formation of the ammonium intermediate A. (B) Role of the substrate: formation of the hypervalent intermediate B through Si–F interaction. (i and ii) CV titration of 1b (5 mM) adding aliquots of 2b (0–50 mM) using either (nBu)4NClO4 (0.1 M) or LiClO4 (0.1 M) as supporting electrolyte in MeCN. (iii) 19F-NMR titration of 2b (0.2 M) adding aliquots of 1b (0–1.4 M). n.d.: not detected. | ||
Next, we turned our attention to the silicon-assisted activation of the fluorine atom in 2b.10 By using electroanalytical and spectroscopic techniques, we uncovered the existence of a Si–F interaction (Fig. 3B).18 The oxidation potential of SEE 1b undergoes an anodic shift upon sequential additions of 2b (Fig. 3B(i)) in CV measurements using (nBu)4NClO4 as supporting electrolyte. This observation is consistent with the formation of the pentacoordinated, trigonal bipyramid intermediate B, whose central silicon atom becomes inherently electron-deficient due to hypervalent bonding.19 In such configuration, the most electronegative fluorine atom occupies an apical position while the enol ether is located in an equatorial position that facilitates back π-bonding with the electron-depleted silicon atom.16 When the CV titration was performed with LiClO4 as supporting electrolyte, the oxidation potential of 1b remained constant upon increasing concentrations of 2b (Fig. 3B(ii)). Considering the ability of lithium ions to activate C–F bonds towards nucleophilic displacements15c,20 with its strong Lewis acidity,21 the presence of lithium in solution prevents the formation of intermediate B. These trends are also observed by 19F-NMR in the actual reaction conditions (Fig. 3B(iii)). Successive addition of controlled amounts of 1b to a solution of 2b shifts the fluorine resonance towards higher fields, coherently with the formation of the pentacoordinated intermediate B with concomitant increase of electron density on the apical ligands. However, the 2b fluorine signal is deshielded upon addition of LiClO4 in THF_d8/CD2Cl2, indicating a net charge transfer from F to Li atoms (Fig. 3B(iii), right).16 Indeed, the reactivity is completely suppressed in the presence of 1 equiv. of LiClO4. According to the acquired spectroscopic information, we propose that the high regio- and diastereoselectivity of the reaction is governed by the aforementioned Si–F interaction.
Next, we monitored the kinetic and stereochemical reaction profiles to prove the feasibility of our proposed model (Fig. 4). The reaction exhibits an initial fast regime where only (S)-2b is consumed (Fig. 4A(i), black traces). Around 50% of conversion, the remaining allyl fluoride 2b is essentially in its (R) form (Fig. 4A(i), green traces). At this point, the reaction kinetics shifts to a second, slower regime. Indeed, an enantiopure sample of (S)-2b was completely racemised in the presence of 20 mol% of 5b after 72 h.16 These observations indicate that the asymmetric reaction proceeds through dynamic kinetic resolution of the allyl fluoride 2b, where the attack of the catalyst to B is likely the rate-determining step in the first regime whereas the catalyst-promoted racemisation of 2b determines the reaction rate during the second regime.
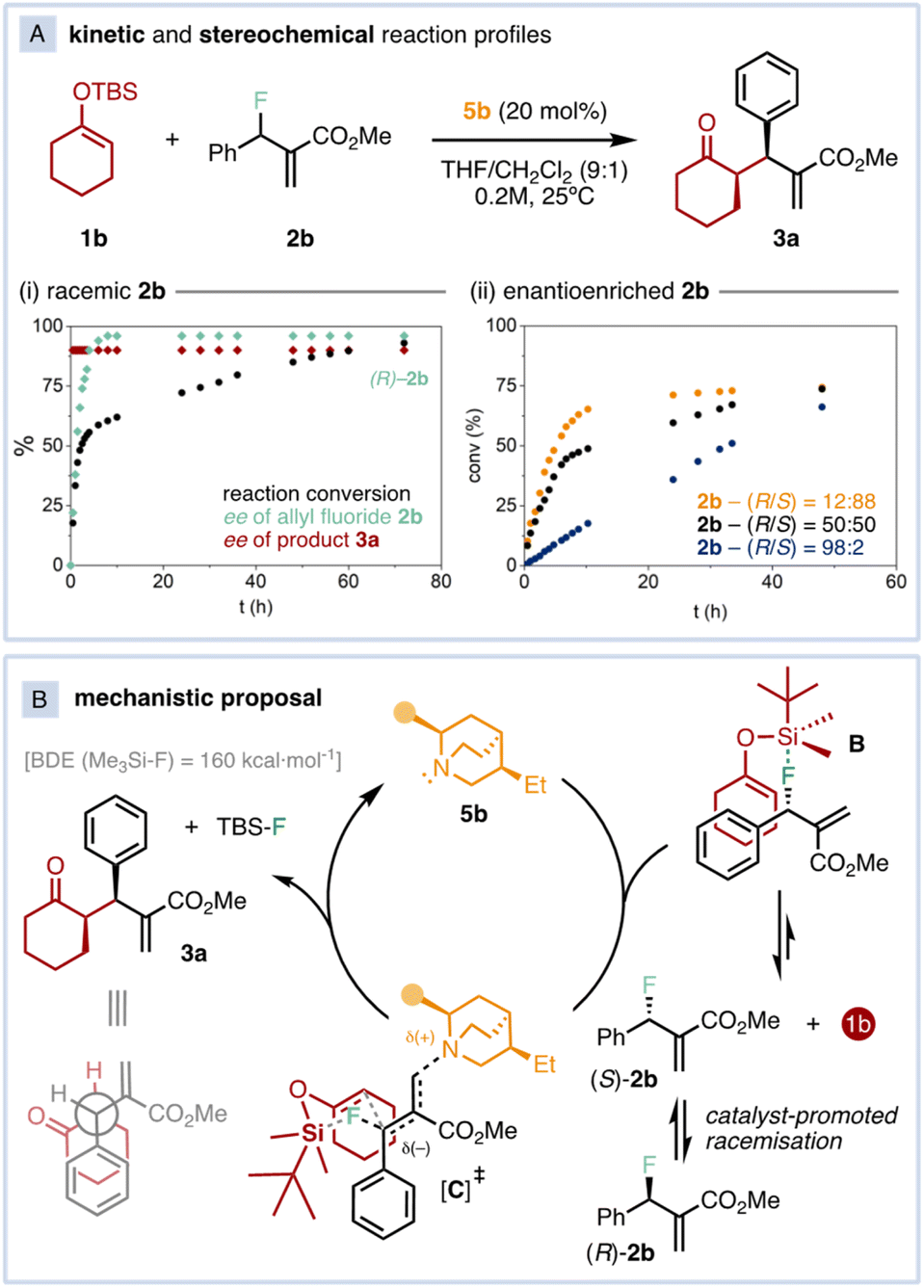 | ||
| Fig. 4 (A) Kinetic and stereochemical reaction profiles (i) starting from a rac-2b sample, and (ii) starting from enantioenriched 2b samples. (B) Mechanistic proposal. | ||
The kinetic profiles using enantioenriched allyl fluorides further confirm this hypothesis (Fig. 4A(ii)). While the reaction using highly-enantioenriched (R)-2b (R/S = 98:2) proceeds through a single, slow regime controlled by substrate racemisation (Fig. 4A(ii), blue traces), the kinetic profile employing a scalemic mixture of 2b (R/S = 12:88) features a faster initial regime up to 70% conversion (Fig. 4A(ii), yellow traces). Based on the observed Si–F interaction, the impossibility to detect any charged species, the acquired mechanistic information together with crucial control experiments, as well as key literature precedents,13c,d the stereochemical outcome of the reaction can be rationalised through transition state Cvia SNi substitution (Fig. 4B).22 In this scenario, the C–F bond activation takes place by simultaneous Si/F and Lewis-base/allyl moiety interactions, leading to the chair-like, six-membered transition state without formation of fully charged species. The bulkiest acrylate/catalyst residue, located in equatorial position, shields the upper face of the allyl fluoride. Thus, the nucleophilic methine of the SEE attacks the electrophile by the lower face in C, substituting the fluorine with retention of configuration and affording product 3a with syn diastereoselectivity.
Generality of the catalytic methodology
Having identified the optimal catalytic conditions and disclosed a coherent reaction mechanism, we next evaluated the generality and limitations of the asymmetric defluorinative allylation of SEE (Fig. 5). A series of six-membered SEEs reacted smoothly under these conditions. The corresponding allylated ketones adorned with different functional groups, such as a gem-dimethyl (3b), an acetal (3c), an ether (3d), and a tertiary amine (3e), as well as fused aromatic rings (3i and 3k), were isolated in good yields and excellent stereoselectivities. Cyclic SEEs with other ring sizes were also successfully allylated (3f–h). Next, we studied the reactivity with acyclic nucleophiles. The SEE derived from acetophenone afforded the allylated products (3n–3p) in excellent yields end enantioselectivities. Acyclic substituted SEEs could also be allylated under the optimised conditions. Despite products 3l and 3m are formed in good yields (66% and 78% respectively), the stereoselectivity is substantially eroded. Subsequently, we investigated how the electronic nature of the allyl fluoride influenced the reaction outcome. Allyl fluorides decorated with halogens at the para position of the aryl ring are well tolerated (3q, 3r, 3w). Strong electron-withdrawing groups, including trifluoromethyl (3s), cyano (3t) and methyl ester (3u) groups, are also competent substrates. Strong electron-donating groups, such as methoxy in 3v, hampered the reactivity, affording the allylated ketone in a low 34% yield but with good stereocontrol (>20:1 d.r. and 92:8 e.r.). Substitution can also be allocated in other positions of the aryl ring. While the meta-substituted derivative 3y was formed in 43% yield, the corresponding para and ortho derivatives 3x and 3z were isolated in 75% and 60% yield respectively, with e.r. ranging from 88:12 to 96:4 and >20:1 d.r. Allyl fluorides bearing fused aromatic (3aa and 3ab) and heteroaromatic (3ac) rings also engage in the catalytic asymmetric allylation. Finally, we investigated the impact of the ester group on the allyl fluoride. We observed that the bulkier is the ester alkyl group, the slower is the catalytic process. While ethyl and n-hexyl-derived substrates furnished ketones 3ad and 3ae in good results, the tert-butyl derivative 3af could only be obtained in trace amounts. Remarkably, the catalytic methodology can be successfully scaled up to the gram scale without compromising neither the yields nor the selectivity (3a and 3w). The absolute configuration was ascertained by anomalous X-ray diffraction of the brominated ketone 3w. Attempts to expand the generality of the catalytic transformation to other reaction partners, such as α,α-disubstituted SEE to construct a quaternary stereocenter or aliphatic allyl fluorides, were unsuccessful.16 Under the optimised asymmetric conditions, the former mainly afforded the linear regioisomer 4, while the latter provided sluggish reactivity and complex reaction mixtures. Finally, we tested the catalytic defluorinative transformation in the challenging late-stage allylation of chiral biorelevant compounds. The α-allylated ketones derived from (+)-camphor (3ag), pregnenolone (3ah) and estrone (3ai) were isolated in 6–52% of yield and 2:1–10:1:0:0 d.r. The three allylated natural products encompass structurally distinct ketone functionalities, including a six- and a five-membered as well as a linear ketone, thus demonstrating the generality and versatility of the defluorinative allylation. It is worth to note that an increase of the steric encumbrance on the nucleophilic atom of the SEE favours the formation of the linear regioisomer, as in the case of substrates 3l and 3ag.16
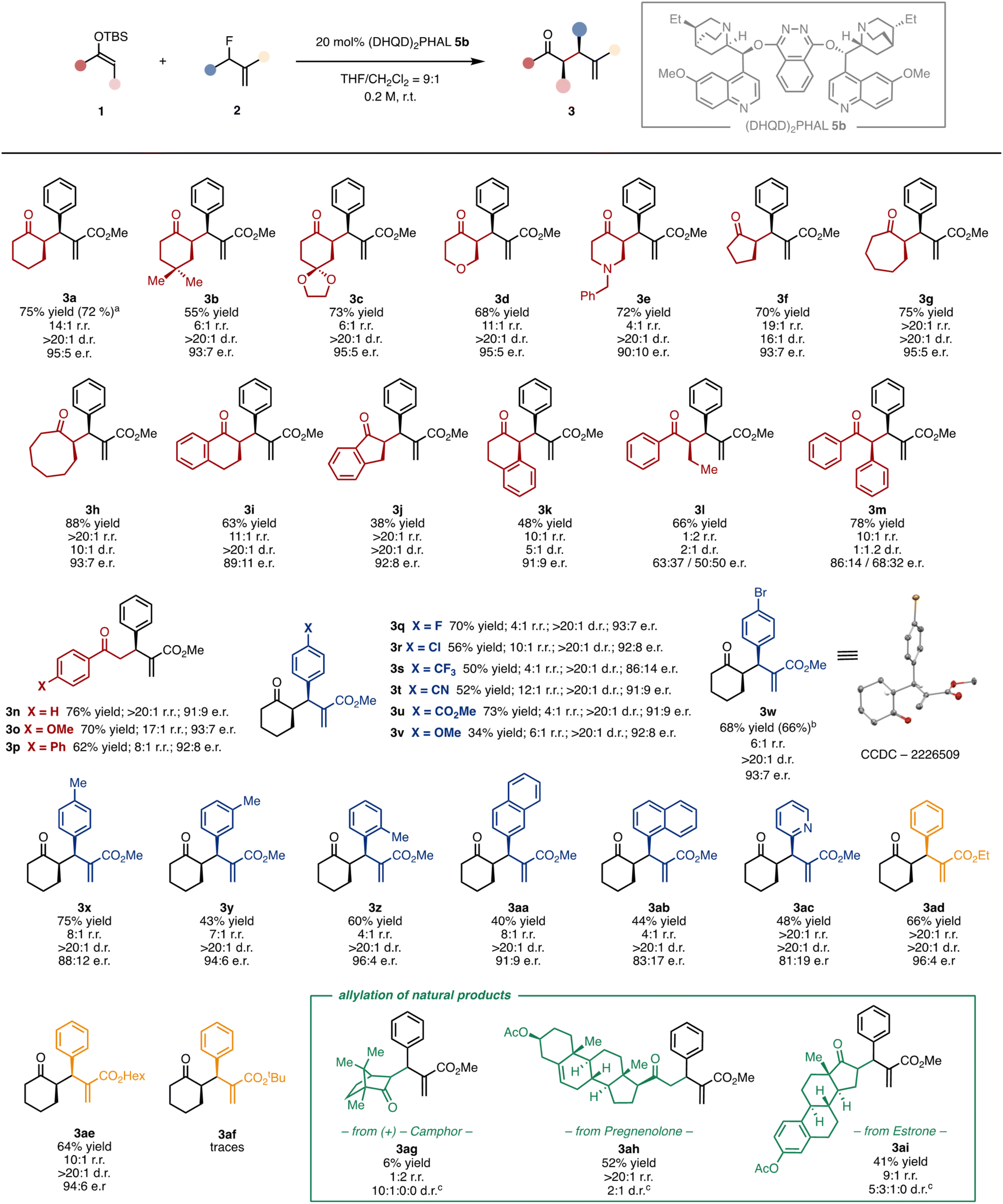 | ||
| Fig. 5 Reaction scope. General conditions: an ordinary vial equipped with a magnetic stirring bar is charged with 0.2 mmol of 2, 0.6 mmol of 1, 1 mL of THF/CH2Cl2 (9:1) and 0.04 mmol of catalyst 5b, and the resulting solution is stirred at room temperature until full conversion. Reaction performed at 5 mmola and 1 mmolb scale. Relative configuration not determined.c | ||
To further validate the synthetic utility of the method, we developed a series of diastereoselective manipulations of 3a for the construction of complex relevant products bearing multiple contiguous stereocenters (Fig. 6). While alkene hydrogenation of 3a catalysed by Pd/C afforded product (R,R,R)-6a in quantitative yield in favour of the compound with anti-configuration of the newly formed stereocenter (2:1 d.r.), the hydrogenation mediated by the Crabtree catalyst selectively furnished the all-syn ketone (R,R,S)-6b with high diastereocontrol (1:10 d.r.) and excellent yield (92% yield, Fig. 6 red). On the other hand, Luche-type reduction of the ketone moiety of 3a and subsequent acid-catalysed cyclisation afforded the α-methylene-δ-lactone 7 in 65% yield over the two steps and complete diastereocontrol (>20:1 d.r., Fig. 6 blue). The α-methylenelactone motif is present in several natural products with broad spectrum of biologically relevant activities as well as in anti-cancer drug candidates.23
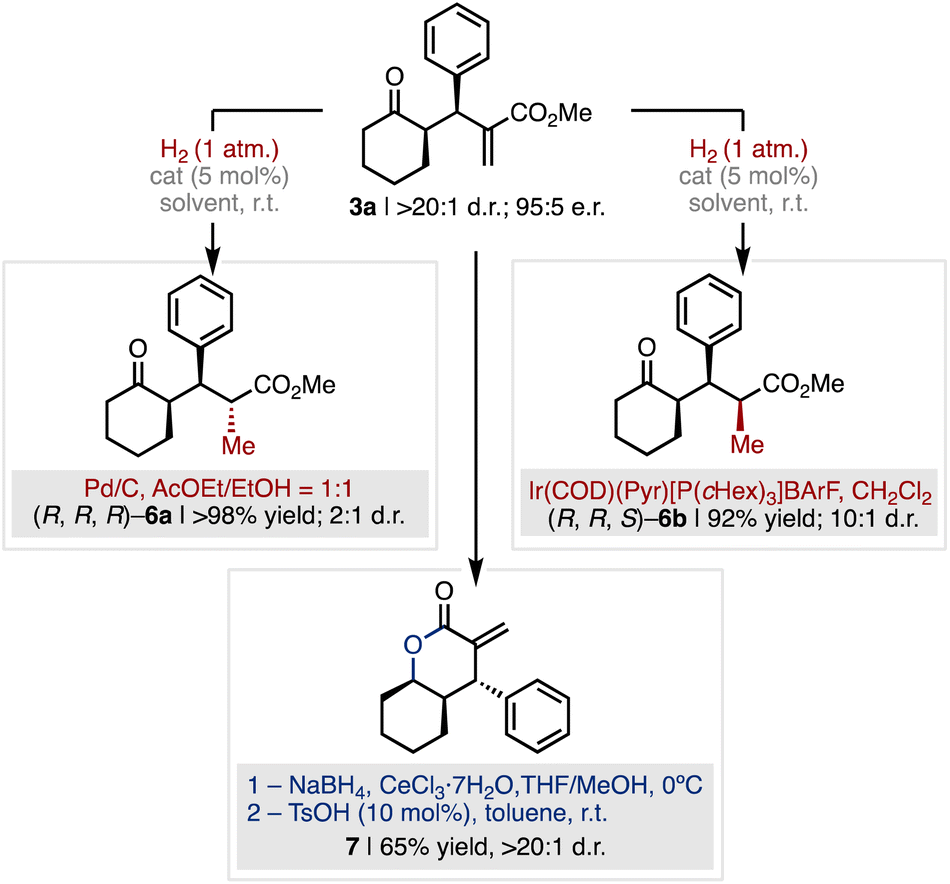 | ||
| Fig. 6 The diastereoselective manipulation of product 3a enables to increase chemical complexity for the synthesis of relevant scaffolds. | ||
Conclusions
In conclusion, we have developed a general, robust, and versatile methodology for the construction of α-allyl ketones in highly regio-, diastereo- and enantioselective fashion. The formation of a supramolecular intermediate through Si–F interaction between the two reaction partners is essential to accomplish the challenging C–F bond-cleavage with concomitant activation of the SEE, as well as to master the selectivity of the whole process. We envisage that the exploitation of such interactions will overcome common pitfalls in AAA reactions while unlocking novel reactivity in asymmetric catalysis.24Data availability
All the supplementary data associated with this article is already in the ESI.†Author contributions
J. D. and J. M. performed the experiments. J. D., J. M., A. M. and X. C. discussed the results. X. C. directed the project. X. C. wrote the manuscript with assistance from all the authors. All authors contributed to the manuscript and have approved its final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the projects PID2020-116859GA-I00 (X. C.) and PID2020-116846GB-C21 (A. M.) funded by MCIN/AEI/10.13039/501100011033. Lucija Ružaj and Federico Mascetti are acknowledged for the synthesis of starting materials. J. D. thanks Santander Bank and the Faculty of Chemistry of UB for post-bachelor fellowships. Dr B. Limburg and Dr A. Vega-Peñaloza are acknowledged for proofreading the manuscript. The authors also thank the NMR, mass spectroscopy and X-ray diffraction units from the Scientific and Technological Centers (CCiTUB) of UB for their support and advice.Notes and references
-
(a)
F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry Part B: Reactions and Synthesis, Springer, 2001, pp. 1–56 Search PubMed
; (b) V. Andrushko and N. Andrushko, Stereoselective Synthesis of Drugs and Natural Products, John Wiley & Sons, 2013 Search PubMed
- For selected examples, see:
(a)
M. Remes and J. Veselý, Stereoselective Organocatalysis Bond Formation Methodologies and Activation Modes, Wiley, 2013, pp. 267–312 Search PubMed
-
(a) A. R. Cano, A. Zakarian and G. P. McGlacken, Angew. Chem., Int. Ed., 2017, 56, 9278 CrossRef PubMed
-
(a) B. M. Trost and D. L. van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed
-
(a) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS PubMed
-
(a) R. Rios, Catal. Sci. Technol., 2012, 2, 267 RSC
- For selected recent examples with unconventional nucleophiles, see:
(a) H. You, E. Rideau, M. Sidera and S. P. Fletcher, Nature, 2015, 517, 351 CrossRef CAS PubMed
-
(a) A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 2745 CrossRef CAS PubMed
-
(a) T. Graening and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 17192 CrossRef CAS PubMed
-
(a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed
-
(a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC
-
(a) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390 CrossRef CAS PubMed
-
(a) T. Nishimine, K. Fukushi, N. Shibata, H. Taira, E. Tokunaga, A. Yamano, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2014, 53, 517 CrossRef CAS PubMed
-
(a) Y. Zi, M. Lange, C. Schultz and I. Vilotijevic, Angew. Chem., Int. Ed., 2019, 58, 10727 CrossRef CAS PubMed
-
(a) B. M. Trost, H. Gholami and D. Zell, J. Am. Chem. Soc., 2019, 141, 11446 CrossRef CAS PubMed
- For further information, see ESI.†.
- Other reaction parameters, including concentration, temperature, additives, the ratio of reagents, and the protecting group of the enol form did not lead to any substantial improvement. While TMS- and TES-derived SEE provided product 3a in higher yields and lower enantioselectivities, the reactivity was virtually precluded when the bulkiest TIPS-derived SEE was used as nucleophilic counterpart.
-
(a) C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404 RSC
-
(a) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS PubMed
-
(a) M. Bergeron, T. Johnson and J. F. Paquin, Angew. Chem., Int. Ed., 2011, 50, 11112 CrossRef CAS PubMed
-
(a) M. A. Forman and W. P. Dailey, J. Am. Chem. Soc., 1991, 113, 2761 CrossRef CAS
- For a detailed discussion of the reaction mechanism, see ESI† sections G-J.
-
(a) A. Janecka, A. Wyrębska, K. Gach, J. Fichna and T. Janecki, Drug Discovery Today, 2012, 17, 561 CrossRef CAS PubMed
- J. Duran, J. Mateos, A. Moyano and X. Companyó, A preliminary version of this manuscript was deposited in ChemRxiv repository, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-wxmzv.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, mechanistic investigations. CCDC 2226509. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01498c |
| This journal is © The Royal Society of Chemistry 2023 |