
Accelerating PLQY and RISC rates in deep-blue TADF materials with the acridin-9(10H)-one acceptor by tuning the peripheral groups on carbazole donors†
Yongqiang
Mei
a,
Di
Liu
 *ab,
Jiuyan
Li
*a and
Jiahui
Wang
a
*ab,
Jiuyan
Li
*a and
Jiahui
Wang
a
aState Key Laboratory of Fine Chemicals, College of Chemical Engineering, Dalian University of Technology, 2 Linggong Road, Dalian, 116024, China. E-mail: liudi@dlut.edu.cn; jiuyanli@dlut.edu.cn
bState Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou, 510640, China
First published on 17th October 2022
Abstract
Blue thermally activated delayed fluorescence (TADF) emitters usually suffer from poor color purity and low efficiencies, especially deep-blue emitters. Here, acridin-9(10H)-one (acridone, AD), featuring an orthogonal and highly rigid conformation, was used as an acceptor to construct a series of deep-blue TADF emitters (3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD), which effectively restricted intramolecular relaxation and produced narrow full widths at half maximum of ∼55 nm. By extending the π-skeleton of the carbazole donor by tuning the peripheral groups on the carbazole ring to slightly increase the donor strength, both the energy splittings between the S1 (1CT) and T1 (3LE) states and the T1 and T2 (3CT) states were gradually reduced, which facilitated the multichannel reverse intersystem crossing (RISC) and realized high kRISC values of 105 s−1 for 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD. At the same time, the extended transition dipole moment along with high molecular rigidity led to an extremely high radiative transition rate constant kR of 108 s−1. 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD exhibited external quantum efficiencies of 17.4% and 17.3% in doped organic light-emitting diodes (OLEDs) with CIE coordinates of (0.15, 0.11) and (0.15, 0.13), respectively. Tuning the peripheral groups on carbazole, even without changing donor distortion, proved to be a practical strategy for enhancing TADF efficiencies while maintaining color purity.
Introduction
Pure organic thermally activated delayed fluorescence (TADF) materials possessing small energy differences (ΔEST) between the singlet and triplet states have drawn increasing attention from both the academic and industrial communities because they can simultaneously take advantage of both singlet and triplet excitons via the reverse intersystem crossing (RISC) process for light emission in organic light-emitting diodes (OLEDs) and thereby achieve an internal quantum efficiency (IQE) of up to 100%.1–3 A small ΔEST is key for TADF emitters to achieve efficient RISC processes and high performances in OLEDs. A traditional way to obtain a small ΔEST is to increase the dihedral angle between the donor (D) and acceptor (A) moieties to facilitate the spatial separation of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).1–6 In some other cases, small groups with steric hindrance have been introduced (on the bridge) between the D and A units to obtain highly twisted structures and reduce the overlap integrals of the frontier molecular orbitals (FMOs), resulting in a small ΔEST as well.7–11 Although TADF materials can show the delocalization of molecular orbitals through these design strategies, the weak overlap between the HOMO and LUMO orbitals at the junction of the D and A units often results in smaller oscillator strength and increased non-radiative decay.11–13 Considering the above points, it is not very difficult to find that a trade-off between ΔEST and radiative decay could be realized and the best compromise is to manipulate the dihedral angles between the D and A moieties under the premise of maintaining overlap integrals of the FMOs to some extent, which is rather tricky. It is even more challenging for deep-blue (CIEy < 0.15) TADF emitters13 as a wider optical band gap and higher triplet energy as well as lower lying intramolecular charge transfer excited states require relatively weak D and/or A units. In most cases, carbazole and its derivatives are selected as the weakest donors for deep-blue emitters; however, their five-membered-ring structure and thus the lower repulsion effect in comparison with other six-membered-ring donors (like acridine and phenoxazine) usually impart small dihedral angles between the D and A moieties.7,13–17 As a result, unfavorable ΔEST values are usually obtained, resulting in relatively inefficient RISC processes and low efficiency in OLEDs. Therefore, the efficiencies of deep-blue TADF emitters can be enhanced by selecting appropriate D and A groups. Recently, to solve this problem, various D and A units have been utilized to develop deep-blue TADF emitters.1,3,7–9,11,13 For example, there have been many reports on highly efficient pure and deep-blue TADF-OLEDs using carbonyl-containing emitters.1,13,17–19 The carbonyl group employs sp2 hybridization, in which the electronic transition from the n orbital to the orthogonally overlapped π* orbital is beneficial for a small ΔEST.13,20 However, because the TADF molecules emit light from the charge transfer (CT) state, TADF molecules based on the widely used benzophenone acceptor usually show a broad emission profile with a relatively large full width at half maximum (FWHM) due to the low rigidity or planarity of benzophenone, having a negative influence on the color purity of the emitters.13,16 Therefore, to increase the rigidity of carbonyl-containing acceptors, oxygen or nitrogen atoms have been applied as bridges to construct novel carbonyl-containing novel acceptors, such as xanthone and acridone, in which the ether or amine bridge fixes the two phenyl rings to prevent their free rotation and reduce vibrational relaxation. As a result, narrow photoluminescence (PL) and high performance can be achieved.21–24Although the less distorted conformations of carbazole (Cz)-based molecules cannot generate a small ΔEST or efficient TADF feature,1,4,7,14,15,25 appropriate HOMO/LUMO overlap is favorable for large radiative decay rates (kR). For example, high kR values on the order of 108 s−1 have been observed in Cz-based TADF compounds.26,27 Therefore, rational molecular design strategies are strongly desired for Cz-containing deep-blue or pure-blue TADF emitters to both boost kR and maintain a low ΔEST. To realize this goal, the introduction of a peripheral group at the 3,6-site or a steric hindrance group at the 1,8-site of the Cz donor was explored to extend the HOMO delocalization or increase D–A dihedral angles, respectively.7,9,16,17,25–33 Among the various peripheral groups, phenyl and tert-butylphenyl groups are frequently used since they not only improve the transition dipole moment to enhance the photoluminescence quantum yields (PLQYs) but also reduce the ΔEST to promote the RISC process.17,30,32,33 Furthermore, they can improve molecular stability.17,32,33
Herein, we have developed a group of donor–acceptor–donor (D–A–D)-type deep-blue TADF emitters (3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD) with highly rigid acridone (AD) as the acceptor and carbazole as the donor. The molecular design focused on adjusting the donor strength and donor conjugation length by varying the number and structure of the peripheral groups at the 3- or 3,6-sites of the Cz donors to tune the energy alignment of the singlet and triplet excited states and enhance the RISC process and TADF performance on the premise of maintaining deep-blue emission. It was observed that by introducing peripheral groups such as phenyl and tert-butylphenyl on the Cz donors, the dihedral angles between the Cz donors and the π-bridge and the optical band gap (Eg) of the molecules hardly changed. Mainly owing to the unique high rigidity of the 10-phenylacridone acceptor that suppressed intramolecular rotation and vibrational motion to reduce the 1CT state relaxation, a high kR of 108 s−1 was achieved for the whole group of TADF emitters. Furthermore, with the incorporation of two peripheral groups that were either phenyls or tert-butylphenyls in 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, the oscillator strengths (f) were enhanced and the PLQYs increased to ∼80%; at the same time, both the ΔEST and ΔETT (T1 − T2) were reduced. As a result, the RISC process was facilitated to reach a high kRISC of 105 s−1 for 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD. The simultaneous possession of a high kR of 108 s−1 and high kRISC of 105 s−1 is a merit of blue TADF emitters. 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD not only exhibited high external quantum efficiencies (EQEs) of 17.4% and 17.3% in their deep-blue OLEDs but also exhibited very high color purity with CIE coordinates of (0.15, 0.11) and (0.15, 0.13) and FWHMs of only 54 and 56 nm, respectively, which are the smallest FWHMs for deep-blue TICT (twisted intramolecular charge transfer)-TADF materials reported so far.
Results and discussion
Synthesis, thermal and electrochemical properties
The chemical structures and synthetic routes of 3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD are shown in Scheme 1. The important intermediate 3,6-DF-AD was synthesized according to a literature method.23,34 The target compounds were then prepared through a nucleophilic substitution reaction between different donor intermediates and the acridone intermediate 3,6-DF-AD in the presence of cesium carbonate in anhydrous N,N-dimethylformamide (DMF) at 165 °C. The products were thoroughly purified via repeated column chromatography and recrystallization to reach high purity for use in spectroscopy and OLED studies. The synthetic and structural characterization details are provided in the ESI.†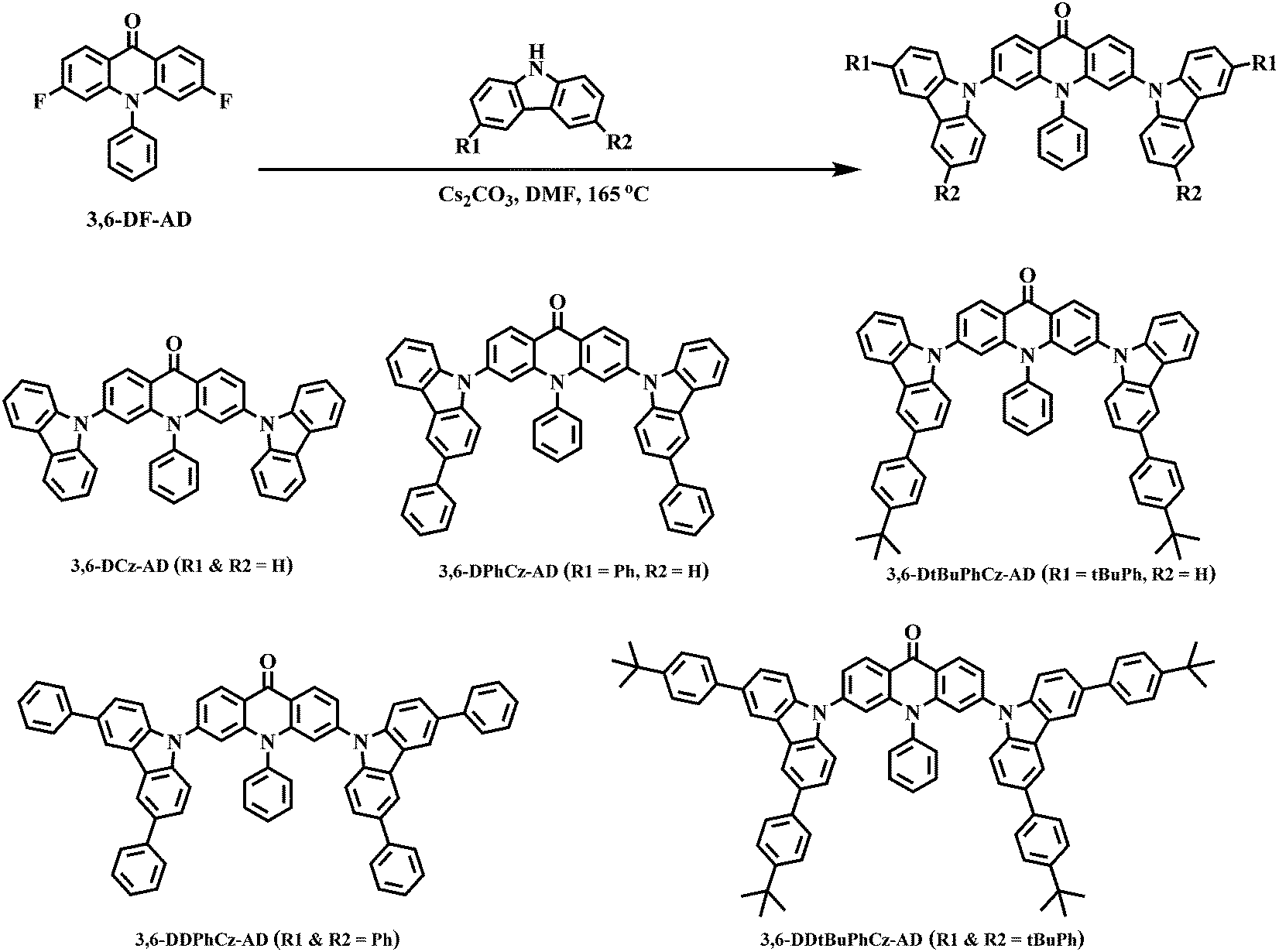 | ||
| Scheme 1 Chemical structures and synthetic routes of the acridone–carbazole TADF emitters. | ||
The thermal properties of all compounds were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), and the TGA traces are shown in Fig. S1(a) (ESI†). The decomposition temperatures (Td) of these compounds were determined by TGA at 5% weight loss to be 452.5 °C (3,6-DCz-AD), 464.5 °C (3,6-DPhCz-AD), 524.3 °C (3,6-DtBuPhCz-AD), 517.2 °C (3,6-DDPhCz-AD) and 544.1 °C (3,6-DDtBuPhCz-AD) (Table 1). Notably, all compounds show good thermal stability with high Td. Additionally, no phase transition was observed for each compound in the DSC measurements throughout the detected temperature range from 30 to 300 °C under the present measuring conditions (Fig. S1(b) in the ESI†), implying that these molecules may have very good amorphous stability with relatively high glass transition temperatures (Tg).2,23 This is reasonable considering their high molecular weights.
 | ||
| Fig. 1 Cyclic voltammograms of the TADF emitters. | ||
| Compound | λ abs [nm] | λ em [nm] | E S [eV] | E T [eV] | ΔESTb [eV] | HOMOc [eV] | LUMOc [eV] | E g [eV] | T d [°C] |
|---|---|---|---|---|---|---|---|---|---|
| a Absorption and fluorescence peak wavelengths in dilute toluene solutions at room temperature. b Estimated from the LT-PL and LT-PH spectra at 77 K in frozen 2-MeTHF solutions, ΔEST: the experimentally determined singlet–triplet energy splitting using ΔEST = ES − ET. c Determined from electrochemical measurements. d Decomposition temperature (Td) at 5 wt% weight loss obtained from TGA measurements. | |||||||||
| 3,6-DCz-AD | 367/390 | 401/422 | 3.08 | 2.68 | 0.40 | −5.63 | −2.85 | 2.78 | 452.5 |
| 3,6-DPhCz-AD | 370/384 | 404/423 | 2.95 | 2.64 | 0.31 | −5.59 | −2.88 | 2.71 | 464.5 |
| 3,6-DtBuPhCz-AD | 370/388 | 406/422 | 2.91 | 2.63 | 0.28 | −5.55 | −2.87 | 2.68 | 524.3 |
| 3,6-DDPhCz-AD | 373/390 | 425 | 2.93 | 2.67 | 0.26 | −5.61 | −2.87 | 2.74 | 517.2 |
| 3,6-DDtBuPhCz-AD | 373/391 | 432 | 2.87 | 2.65 | 0.22 | −5.46 | −2.87 | 2.59 | 544.1 |
The electrochemical redox properties of the compounds were measured by cyclic voltammetry (CV) in dry DCM and DMF solution in the absence of oxygen at room temperature. As shown in Fig. 1, during the cathodic scan in the CV measurement, all compounds revealed a reversible reduction wave with almost identical onset potentials of the first reduction wave (Eonsetred). This was because the reduction of these molecules occurred on the electron-deficient acridone ring. In the anodic scan in the CV measurement, these compounds exhibited quasi-reversible oxidation waves. It is clear that with the incorporation of more phenyl or tert-butylphenyl groups on the Cz donors, the oxidation of these compounds occurred at less positive potentials. These electron-donating peripheral groups make the oxidation of the Cz donors a little easier. However, the Eonsetox of these analogues did not shift severely, leading to only small variations in their band gaps (Eg), implying that this structural modification still retained the emitting color of these TADF emitters in the desired blue region. The HOMO and LUMO energy levels of these compounds were calculated from the onset potentials of the first oxidation (Eonsetox) and reduction (Eonsetred) wave following the equations EHOMO = −(Eonsetox + 4.4) eV and ELOMO = −(Eonsetred + 4.4) eV, respectively. The data are summarized in Table 1.
Theoretical calculations
The HOMO, HOMO−1 and LUMO levels and S1 and T1 state energies were calculated using density functional theory (DFT) and time-dependent DFT (TD-DFT) with B3LYP/6-31G (d), and are shown in Fig. 2. Interestingly, for all molecules, due to the small distorted dihedral angle (θ1 ≈ 50°, Fig. 2(a)) between the A and D units, their LUMOs are primarily localized at the AD moiety, whereas their HOMOs are mainly distributed on the Cz units and partly extended to the AD units (Fig. 2(b)), resulting in appropriate overlap of the HOMO and LUMO while retaining the separation of the HOMO and LUMO to some extent, which should be favorable to large oscillator strength and high kR. Furthermore, the calculated HOMO levels of these compounds show a regular increasing trend from −5.50 eV to −5.24 eV in the order of 3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD. This is consistent with the overall electron-donating ability order of Cz < PhCz < tBuPhCz < DPhCz < DtBuPhCz in these molecules. However, the LUMO of these molecules remains almost unchanged, finally resulting in a slow decrease in the energy gap (Eg) from 3.66 eV for 3,6-DCz-AD to 3.38 eV for 3,6-DDtBuPhCz-AD. At the same time, with a slight increase in the donor strength in the order of Cz < PhCz < tBuPhCz < DPhCz < DtBuPhCz, the S1 state energies exhibit a similar decreasing trend from 3.15 eV for 3,6-DCz-AD to 2.93 eV for 3,6-DDtBuPhCz-AD, but the T1 state energies remain almost constant at ∼2.65 eV for all the compounds. As a result, the ΔEST values gradually reduced in the order of 0.49, 0.40, 0.38, 0.34 and 0.30 eV for 3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, respectively, which is increasingly favorable for efficient RISC and TADF processes. It should be noted that even for 3,6-DDtBuPhCz-AD, which has the lowest S1 state energy due to the presence of the strongest donor among these analogues, its fluorescence is still in the deep-blue region. It is also interesting that by incorporating two peripheral groups in 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, their S1 and S2 states come much closer to each other than in the other analogues (Fig. 2(c)). This is reasonable since two degenerate HOMOs (HOMO and HOMO−1) are located on two Cz donors. Therefore, it can be expected that these two analogues may have good PLQYs as the randomly distributed HOMO and HOMO−1 could lead to multiple transition channels from the singlet excited states to the ground state.35 | ||
| Fig. 2 (a) Optimized geometries in the ground states (S0), (b) spatial distributions and energy levels of the HOMO, HOMO−1, and LUMO, and calculated energies of the S1 and T1 states and the gap Eg, (c) energy alignment of the excited states and schematic supposition of the RISC and luminescence mechanisms for 3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD. | ||
To further study the transition characteristics of the singlet and triplet excited states for these compounds, natural transition orbital (NTO) analysis was performed,36 and the results are shown in Fig. S2 in the ESI.† For the S1 states, the particles are mainly localized on the AD units owing to their same acceptor and similar dihedral angles, while the holes differ significantly. The hole orbitals of 3,6-DCz-AD are distributed on the two donor moieties (Cz), partially extend to the central AD–Ph units and overlap with the particle orbital. As a result, due to a relatively big spatial overlap between the particles and holes,37–39 the S1 state exhibited mixed CT and LE excitation characteristics, featuring both n–π* and π–π* transition properties. With the incorporation of Ph or tBuPh as the peripheral groups of the Cz rings in 3,6-DPhCz-AD and 3,6-DtBuPhCz-AD, the spreading of holes on the central AD ring is reduced. On further increasing the number of Ph or tBuPh groups in 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, the distribution of holes extended from the carbazole ring to all the peripheral Ph or tBuPh groups; at the same time, the overlap of holes and particles on the central AD rings was further reduced. Therefore, on increasing the delocalized degree of holes by incorporating peripheral groups on carbazole rings, the CT characteristics of the S1 states become increasingly dominant and the n–π* transition ratios of S1 states are effectively enhanced. Accordingly, the S0 → S1 transitions of 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD have larger oscillator strengths (f = 0.49 for 3,6-DDPhCz-AD and 0.50 for 3,6-DDtBuPhCz-AD) than 3,6-DCz-AD (f = 0.39), 3,6-DPhCz-AD (f = 0.46) and 3,6-DtBuPhCz-AD (f = 0.46), which should improve the radiative transition efficiency and PLQYs (vide infra). Interestingly, unlike their S1 states, the S0 → T1 transitions for all these emitters show LE characteristics. Such LE characteristics of the triplet states are crucial for enhancing the RISC process by increasing spin–orbit coupling (SOC) when the S1 states are of CT character, according to the El-Sayed rule.2,3,5,10 In general, the RISC process is significantly enhanced when the ΔEST is small and the SOC constant is large.5,9,10,40,41 However, it is obvious that these emitters have different ΔEST values (Fig. 2(b)), implying that there may be different RISC processes under external excitation. Therefore, to further evaluate the RISC process in these emitters, TD-DFT calculations were performed to predict the electronic properties and energy levels of their T2–5 states. As shown in Fig. S2 (ESI†), all the detected triplet states (3LE or 3HLCT), except for the T2 state (3CT), have different excited state characteristics from the S1 states (1CT), which is favorable for efficient SOC between the T2–5 and S1 states. Additionally, along with the gradually reduced ΔEST (S1 and T1), the ΔETT (T1 and T2) is simultaneously reduced from 0.22 eV to 0.17, 0.16, 0.14, and 0.12 eV (Fig. 2(c)) with the increasing incorporation of peripheral groups on the carbazole ring. At the same time, the higher-lying triplet states, including T3, T4 and T5, are pulled down to some extent, causing these triplet states to align closely with very small energy differences between two neighbouring states. Therefore, considering both the gradually improved SOC values and the reduced energy splittings (both ΔEST and ΔETT), the RISC mechanisms for these analogues are proposed in the following ways (Fig. 2(c)). For the parent compound 3,6-DCz-AD with both a large ΔEST (S1 and T1) of 0.49 eV and ΔETT (T1 and T2) of 0.22 eV, the RISC process is supposed to occur through the T1 → T2 → S1 path.5,9,15,25 For 3,6-DPhCz-AD and 3,6-DtBuPhCz-AD, which possess both reduced ΔEST (S1 and T1) and ΔETT (T1 and T2) and lowered higher-lying triplet states, the RISC process may occur through strong T1 → T2 → S1 and weak T1 → T3–5 → S1 paths.15,16,25 The involvement of higher-lying triplet states in 3,6-DPhCz-AD and 3,6-DtBuPhCz-AD must improve the RISC efficiency in comparison with the parent 3,6-DCz-AD. With the further reduced ΔEST (S1 and T1) and more condensed triplet states in 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, the contribution of higher-lying triplet states to the RISC is more important, which must lead to efficient multi-channel T1–T2–5–S1 processes with high RISC rate constants2–5,9,10,16,25,40,41 (vide infra). These results indicate that adding peripheral groups to donors could be an intriguing approach to optimizing the radiative transition and RISC process by adjusting the electronic characteristics and energy alignment of the singlet and triplet states for TADF emitters, so that the triplet excitons can be efficiently harvested in OLEDs.
Photophysical properties
The ultraviolet-visible (UV-Vis) absorption and photoluminescence (PL) spectra for all TADF emitters were recorded at a low concentration (10−5 M) in toluene (TOL) at room temperature, and their low-temperature fluorescence (LT-FL) and phosphorescence (LT-PH) spectra were measured in 2-MeTHF solutions at 77 K; the relevant photophysical data are summarized in Table 1. As shown in Fig. 3(a), all the emitters show similar absorption profiles with two major absorption bands at 280–320 and 325–400 nm with well-resolved fine vibronic structures, which can be assigned to the π–π* and n–π* transitions of the AD (Fig. S3, ESI†) and carbazole fragments.23a It is also noted that with the incorporation of peripheral groups on the carbazole ring, the absorption tails in the long-wavelength direction extend to about 420 nm, which are absent in the intrinsic absorptions of the AD or carbazole moieties.23b In addition, with increasing donor strength in the order of Cz < PhCz < tBuPhCz < DPhCz < DtBuPhCz, the absorption starts at increasingly long wavelengths in the order of 3,6-DCz-AD < 3,6-DPhCz-AD < 3,6-DtBuPhCz-AD < 3,6-DDPhCz-AD < 3,6-DDtBuPhCz-AD (Fig. 3(a)). This indicates that the long-wavelength absorption must be caused by the weak intramolecular CT (ICT) transitions from the D to A units, showing a hybrid CT and LE mixing excitation character.14 The absorption result is consistent with the NTO calculations. | ||
| Fig. 3 (a) Absorbance in dilute toluene solutions at room temperature, (b) PL spectra in dilute toluene solution at room temperature, LT-PL and LT-PH spectra in frozen 2-MeTHF solutions at 77 K, and PL spectra in doped DPEPO films (7 wt%) for all five TADF emitters. | ||
Upon photoexcitation, all these compounds emit bright blue fluorescence, as shown in Fig. 3(b). However, the fluorescence spectral profiles differ with different peripheral groups. For the parent compound 3,6-DCz-AD, a deep-blue fluorescence with a main peak at 401 nm and a fine vibronic structure was found in toluene solution. With the introduction of Ph or tBuPh on carbazole rings, the vibronic structure in the fluorescence spectra of 3,6-DPhCz-AD and 3,6-DtBuPhCz-AD are not as clear as in 3,6-DCz-AD. Upon further increasing the number of peripheral groups in 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, the vibronic structure almost disappeared in their fluorescence spectra. At the same time, the fluorescence spectra in toluene solutions show a regular red-shift with the electron-donating ability of the donors gradually increasing in the order of Cz <PhCz < tBuPhCz < DPhCz < DtBuPhCz. The ICT transition dominates the fluorescence, which typically exhibits broad and structureless emission. As shown in Fig. S4 (ESI†), the regular bathochromic shift and spectral broadening with increasing solvent polarity further confirm the ICT nature of these emitters, especially in polar solvents. Despite the obvious influence of the peripheral groups on the S1 state nature and energy, the fluorescence of all the emitters peaked from 401 to 432 nm (Table 1), all belonging to the deep-blue region. Thin films of these emitters (doped in DPEPO host at 7 wt%) all exhibit broad and red-shifted fluorescence compared to those in solution. The electronic transitions with CT character should dominate in the solid films because the electronic states usually interact with the local environments.15 Generally, for emissions derived from CT-dominated S1 states, the FWHMs tend to be large. However, the five emitters (3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD) exhibit FWHMs of 35 (0.24 eV), 50 (0.35 eV), 56 (0.39 eV), 59 (0.40 eV) and 63 (0.41 eV) nm in toluene solution and 49 (0.30 eV), 66 (0.40 eV), 46 (0.27 eV), 49 (0.29 eV) and 66 (0.39 eV) nm in doped films, respectively, which are among the lowest values for donor–acceptor (D–A) or donor–acceptor–donor (D–A–D)-type blue TADF materials.2,3,7,8,15 This should benefit from the rigid and planar structures of both the AD acceptor and carbazole donors, which definitely help to suppress the non-radiative deactivation processes including the intramolecular rotations and vibrational motions. The short-wavelength emission below 460 nm along with the narrow FWHMs of these TADF emitters may predict deep-blue or pure-blue emission with CIEy coordinates lower than 0.17.14
The low-temperature PL (LT-PL) and phosphorescence (LT-PH) of these emitters were measured in frozen 2-Me-THF glass at 77 K and are shown in Fig. 3(b). Once again, the fine vibronic structure of the fluorescence spectrum of 3,6-DCz-AD (380–450 nm) confirmed the mixed 1CT and 1LE character of the S1 state, and the structureless fluorescence spectra of the other four analogues confirmed the 1CT nature of their S1 states. The phosphorescence of all five emitters showed well-resolved vibronic features, confirming the 3LE nature of their T1 states. The experimentally detected electronic natures of the S1 and T1 states are consistent with the theoretical results (Fig. S2 and Table S1, ESI†). The S1 and T1 state energies were estimated from the highest-energy fluorescence and phosphorescence peaks (Fig. 3(b)) at 77 K in 2-Me-THF to be 3.08/2.68, 2.95/2.64, 2.91/2.63, 2.93/2.67, and 2.87/2.65 eV for 3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, respectively, and accordingly, the ΔEST values were calculated to be 0.40, 0.31, 0.28, 0.26 and 0.22 eV, respectively. Furthermore, the S1 and T1 energies of the TADF emitter films doped in the DPEPO host (7 wt%) were determined by measuring the LT-FL and LT-PH spectra on a Hitachi F-7000 fluorescence spectrometer at 77 K. As shown in Fig. S5 (ESI†), the LT-PH spectra of 3,6-DCz-AD and 3,6-DPhCz-AD exhibited fine vibronic structures, confirming that their T1 states come from 3LE states, which is the same as the case in 2-Me-THF solutions (Fig. 3(b)). For 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, the LT-PH spectra were broad and featureless. This is reasonable because the more polar host DPEPO can stabilize the CT part and make them dominant but the discernible vibronic structures around 500 and 550 nm indicate that their T1 states feature both CT and LE characteristics. The ΔEST values were gradually reduced for these emitters upon introducing more peripheral groups on the carbazole donors. In particular, the ΔEST values of 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD are very small and thus, good TADF properties can be expected from them.
To verify the TADF properties, time-resolved PL (TRPL) experiments were carried out with the doped films of these emitters in the DPEPO host (7 wt%). As shown in Fig. 4(a) and (b), the transient PL decay curves of these doped films displayed biexponential decay features with a prompt component and a delayed component at room temperature. The lifetimes of the prompt fluorescence (τPF) were determined by the TCSPC technique to be 1.1–2.3 ns (Fig. S6 and Table 2). The delayed components have the exact same spectra as the prompt fluorescence (PF) for each compound (Fig. S7, ESI†), confirming their delayed fluorescence (DF). As shown in Fig. 4(a), the parent compound 3,6-DCz-AD showed a long DF lifetime (τDF) of 1368.2 μs. With the incorporation of peripheral groups on carbazole rings, the τDF values of 3,6-DPhCz-AD and 3,6-DtBuPhCz-AD reduced to 685.1 and 695.1 μs, respectively. For 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, the τDF values further reduced to 8.4 and 9.3 μs, respectively. The gradual shortening of τDF could be related to the gradual reduction in ΔEST (between the S1 and T1 states) and the T2–6 states energetically close to the S1 state, both of which are favorable for efficient and fast RISC processes. The short τDF values are favorable for stable TADF emission, especially in OLEDs. In addition, in the temperature-dependent transient PL measurements, the regular enhancement of PL intensity with increasing temperature (Fig. 4(c), (d) and Fig. S8, ESI†), especially in the short time range (e.g. in the first 30 μs for 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD) confirmed the TADF mechanism of the delayed emission for all the emitters.1–5
 | ||
| Fig. 4 Time-resolved transient PL decay curves of the TADF emitters at room temperature (a) and (b) and the temperature-dependent PL decay curves of 3,6-DDPhCz-AD (c) and 3,6-DDtBuPhCz-AD (d). | ||
| Compounds | Φ PL [%] | Φ PF [%] | Φ DF [%] | τ PF [ns] | τ DF [μs] | k R [108 s−1] | k NR [107 s−1] | k ISC [108 s−1] | k RISC [105 s−1] |
|---|---|---|---|---|---|---|---|---|---|
| a k R, kNR, kISC, and kRISC represent the rate constants of radiative transition, non-radiative transition, intersystem crossing, and reverse intersystem crossing, respectively; ΦPL, ΦPF, ΦDF, τPF, and τDF represent the quantum yield of total emission, PF, DF, and average lifetime of PF and DF, respectively. | |||||||||
| 3,6-DCz-AD | 42 | 23 | 19 | 1.1 | 1368 | 2.0 | 27.6 | 4.0 | 0.014 |
| 3,6-DPhCz-AD | 60 | 25 | 35 | 2.3 | 685 | 1.1 | 7.3 | 2.5 | 0.035 |
| 3,6-DtBuPhCz-AD | 72 | 34 | 38 | 1.8 | 695 | 1.9 | 7.5 | 2.9 | 0.031 |
| 3,6-DDPhCz-AD | 83 | 40 | 43 | 1.8 | 8.4 | 2.3 | 4.6 | 2.9 | 2.5 |
| 3,6-DDtBuPhCz-AD | 80 | 36 | 44 | 2.3 | 9.3 | 1.6 | 4.1 | 2.4 | 2.4 |
The PL quantum yields (ΦPL) of the TADF emitters doped in DPEPO films (7 wt%) were found to be 42% (3,6-DCz-AD), 60% (3,6-DPhCz-AD), 72% (3,6-DtBuPhCz-AD), 83% (3,6-DDPhCz-AD) and 80% (3,6-DDtBuPhCz-AD), which in combination with the τPF and τDF allow the calculation of the quantum yield of PF and DF (ΦPF, ΦDF), the rate constants of radiative decay (kR), the intersystem crossing (kISC), and the RISC (kRISC) for each emitter.2–5,14,15,25,35 These photophysical data are summarized in Table 2. Interestingly, the PLQYs of these emitters exhibit a regular increasing trend. This could be because of the moderate overlap between electrons and holes and the gradually increasing delocalized distribution of holes induced by the peripheral groups as well as the increasing oscillator strengths (f, 0.3732, 0.3888, 0.4564, 0.4865, 0.4991), which effectively couple the S1 state with the S0 state. In particular, the PLQYs of 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD are almost double that of the parent 3,6-DCz-AD. Additionally, due to the introduction of the peripheral group on the donor units, their kRISC values gradually improved. Importantly, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD exhibit much larger kRISC values (2.5 × 105 s−1 and 2.4 × 105 s−1) than the other analogues, and are two orders of magnitude higher than that of 3,6-DCz-AD (1.4 × 103 s−1). The superior kRISC values of 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD unambiguously confirm the supposed multi-channel RISC mechanisms, as shown in Fig. 2(c).2–5,7,9–13,40 Similar to the reported cases in the literature,2,9,10,25,40 for the present 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, the 3LE (T1) state can up-convert to the higher or intermediate triplet states 3CT (T2) and 3HLCT/3LE (T3–T5) by efficient VC, which should be efficient and fast due to the tiny energy differences (ΔETT) between the two adjacent triplet states. Some of these triplet states can be efficiently converted into the 1CT state based on their different orbital angular momentums. The energetically close-lying states (1CT) ≈ 3CT (T2) ≈ 3LE (T1, T3–5) together with strong SOC between the 1CT and these triplet states led to multi-channel RISC processes and generated high kRISC (2.5 × 105 s−1 and 2.4 × 105 s−1) for 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD. In contrast, the parent compound 3,6-DCz-AD could not undergo the same RISC process due to its much larger ΔEST (1CT–3LE, 0.40 eV). Instead, its RISC process may involve the 1CT → 3LE (T1) → 3CT (T2) → 1CT cycle.9,10,12–14 However, as illustrated by the NTO result (Fig. S2, ESI†), its T2 state is dominated by CT characteristics with a calculated energy of 2.88 eV by TD-DFT, which has the same electronic state and weak SOC effect as the 1CT state. Therefore, a relatively large ΔEST (1CT–3CT) along with a small SOC matrix element between 1CT and 3CT finally led to an inferior RISC process with a rather low kRISC for 3,6-DCz-AD.
To verify the feasibility of the up-conversion of these intermediate triplet states into the S1 state for these TADF emitters, the SOC matrix element values (SOCMEVs) were calculated using reported methods,42 and the data are listed in Table S1 in the ESI.† Except for the T2 (3CT) state, all the triplet states of these emitters have significant SOCMEVs (〈S1|ĤSOC|Tn〉) with the corresponding S1 (1CT) state, confirming the RISC feasibility of these triplet states. For the parent compound 3,6-DCz-AD, although the SOCMEV between each of its triplet states and S1 is higher than the corresponding values of the other analogues, the lowest kRISC was experimentally obtained (Table 2). This is understandable since both the SOC effect and energy differences (ΔEST) combine to determine the RISC process, while the very large ΔEST and even ΔETT dominate to suppress the RISC efficiency and speed, even if the T1 is first converted to T2 and then to S1.9,16 Upon increasing the peripheral phenyl or tert-butylphenyl groups on the carbazole donor rings, the 〈S1|ĤSOC|T1〉 and 〈S1|ĤSOC|T4〉 values all remarkably increased compared to the corresponding values of their analogues with single peripheral groups. For example, the 〈S1|ĤSOC|T1〉 of 3,6-DDPhCz-AD (0.926) is higher than that of 3,6-DPhCz-AD (0.893), and the 〈S1|ĤSOC|T4〉 of 3,6-DDPhCz-AD (0.371) is higher than that of 3,6-DPhCz-AD (0.166). In addition to the different electronic characteristics of the triplet states and the singlet states in these conversion processes, the closer-aligned T1–5 states and smaller ΔEST values for 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD should be favorable for easier RISC. The ΔEST, VC and SOC between the excited states with different spin multiplicities of TADF emitters can be rationally manipulated by varying the peripheral groups on the donor units, which finally optimize and facilitate the RISC and TADF characteristics.
Electroluminescence properties
To evaluate the electroluminescence (EL) performance of these blue TADF materials, multiple-layer OLEDs were fabricated with the structure of indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene) (PEDOT:PSS) (40 nm)/1,1-bis[(di-4-tolylamino)phenyl]cyclohexane (TAPC) (20 nm)/1,3-bis(N-carbazolyl)benzene (mCP) (5 nm)/7 wt% emitter:DPEPO (20 nm)/DPEPO (5 nm)/1,3,5-tris(3-pyridyl-3-phenyl)benzene (TmPyPB) (40 nm)/LiF (1 nm)/Al (200 nm), (device B1, B2, B4, B5 for 3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, respectively). The device architecture, energy level diagram and chemical structures of the used materials in these devices are shown in Fig. S9 in the ESI.† PEDOT:PSS and LiF were used as the hole- and electron-injecting layers, and TAPC and TmPyPB as the hole- and electron-transporting layers, respectively. mCP and DPEPO with sufficiently high triplet energies were utilized as exciton-blocking layers on both sides of the emitting layer (EML)2,11 to confine the triplet excitons within the EML. In addition, the electron-transporting and higher-triplet-energy DPEPO was selected as the host to prevent reverse energy transfer from the dopant to the host. The doping concentration of the TADF emitter in the DPEPO host was set as 7 wt% after optimization. The current density–voltage–brightness (J–V–B) characteristics, EL spectra and efficiency curves are illustrated in Fig. 5, and the EL data are summarized in Table 3. | ||
| Fig. 5 EL spectra (a), J–V–B characteristics (b) and efficiency curves (c), (d) of TADF-OLEDs B1–B5. | ||
| Devices | V on [V] | η c [cd A−1] | η p [lm W−1] | EQEb [%] | λ EL [nm] | FWHM [nm] | CIE (x, y) |
|---|---|---|---|---|---|---|---|
| a Abbreviations: Von, turn-on voltage at a brightness of 1 cd m−2; EQE, external quantum efficiency; ηc, current efficiency; ηp, power efficiency, λEL, EL peak wavelength; FWHM, full width at half maximum; CIE (x, y), Commission International de I’Eclairage coordinates. b Order of measured values: maximum, then at 10 and 100 cd m−2. | |||||||
| B1 | 4.1 | 5.8 | 4.4 | 5.4/2.3/0.9 | 441 | 71 | 0.17, 0.13 |
| B2 | 4.2 | 13.6 | 10.1 | 13.1/10.1/4.8 | 448 | 56 | 0.16, 0.11 |
| B3 | 3.9 | 18.5 | 14.9 | 15.8/12.6/7.9 | 454 | 58 | 0.16, 0.13 |
| B4 | 3.5 | 16.9 | 15.2 | 17.4/16.3/12.4 | 450 | 54 | 0.15, 0.11 |
| B5 | 4.0 | 16.1 | 12.6 | 17.3/14.8/11.7 | 455 | 56 | 0.15, 0.13 |
All the devices exhibited deep-blue emission with EL peaks at 440–455 nm and CIE coordinates from (0.17, 0.13) to (0.15, 0.13). It is worth noting that devices B2–B5 exhibited very high color purity with FWHMs of 56, 58, 54 and 56 nm, respectively. In particular the FWHMs of 54 and 56 nm for 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD should be the smallest FWHMs for blue TICT-TADF materials reported so far.1,3,7–9,14 On the contrary, the 3,6-DCz-AD-based device B1 exhibited a larger FWHM of 71 nm, which is probably due to an extra peak at 625 nm assigned to the exciplex formation between 3,6-DCz-AD and TmPyPb.43 The CIEy coordinates of devices B1–B5 were 0.10–0.13, satisfying the requirements of blue color for display technologies.
As shown by the J–V–B characteristics in Fig. 5(b), the 3,6-DCz-AD-based device B1 delivered almost the highest current density but the lowest brightness at a given voltage, finally resulting in the lowest efficiency. B1 exhibited a maximum external quantum efficiency (EQEmax) of 5.4%, which exceeded the theoretical upper limit of conventional fluorescent OLEDs (5%, assuming the light out-coupling efficiency is 0.2) with strong efficiency roll-off and inferior CIE coordinates of (0.17, 0.13). Such EQE should not be as high as expected for TADF-OLEDs. This is reasonable considering the rather low kRISC (103 s−1), relatively low PLQY (42%) and low TADF ratio (<50%) of 3,6-DCz-AD, all of which were mainly caused by the absence of the peripheral groups on the carbazole donors. In this situation, the T1 excitons of 3,6-DCz-AD could not effectively up-convert to the S1 state via the RISC process to emit light; instead, most of the T1 excitons may be consumed by certain annihilation processes including triplet–triplet annihilation (TTA) and singlet–triplet annihilation (STA), which eventually enhance the unwanted efficiency roll-off.14
Fortunately, with the incorporation of the peripheral groups on the carbazole donors, the 3,6-DPhCz-AD- and 3,6-DtBuPhCz-AD-based devices B2 and B3 exhibited much improved performances, with CIE coordinates of (0.16, 0.11) and (0.16, 0.13), and EQEmax values of 13.1% and 15.8%, respectively. The greatly improved efficiencies and low efficiency roll-off of devices B2 and B3 relative to the control device B1 could be attributed to the increased PLQY and enhanced TADF feature with higher kRISC values of 3,6-DPhCz-AD and 3,6-DtBuPhCz-AD, which are modified by the peripheral groups on the Cz donor rings. Upon further increasing the number of the peripheral groups in 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD, their devices B4 and B5 realized further improved EQEmax values of 17.4% and 17.3% with relatively low efficiency roll-off, respectively. At the same time, the blue color purities of the two devices were further improved with CIE coordinates of (0.15, 0.11) and (0.15, 0.13) for B4 and B5, respectively. Although the 3,6-DDPhCz-AD- and 3,6-DDtBuPhCz-AD-based devices B4 and B5 have comparable EL peak wavelengths to those of B2 and B3, the blue color purities were greatly improved and approached the deep-blue region. This could be because 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD have remarkably shorter delayed fluorescence lifetimes (below 10 μs, Table 2) than their analogues 3,6-DPhCz-AD (685 μs), 3,6-DtBuPhCz-AD (695 μs) and 3,6-DCz-AD (1368 μs), which suppress the exciton trap and release possibility, intermolecular interactions, and molecular rotation and vibrations, all of which are proved to cause fluorescence spectrum broadening of TADF emitters. Table S2 (ESI†) summarizes the CIE coordinates and EQEs of some typical deep-blue TICT-TADF devices reported in the past decade. In 2016, Lee and coworkers reported a DMTADC-based blue OLED that realized an EQE of 17.5% with CIE coordinates of (0.15, 0.12).44 Cui and coworkers developed a blue TADF emitter Cz-TRZ4 that exhibited an EQE of 18.3% with CIE coordinates of (0.15, 0.10) in a doped OLED.7 Kim reported a high EQE of 20.7% with CIE coordinates of (0.14, 0.18) using TMCz-BO (which contains tetramethylcarbazole as the donor and the famous rigid oxygen-bridged boron–related structure as the acceptor) as a doped emitter.9 As far as we know, these examples represent the highest EQEs for deep-blue TICT-TADF OLEDs reported in recent years. However, it is clear that the high efficiencies in most of these cases were occasionally accompanied by unsatisfactory CIE coordinates. Recently, the Liao group reported a blue TADF emitter PXZN-B that exhibited a maximum EQE of 12.7% with CIE coordinates of (0.13, 0.147).41 Hatakeyama reported a blue MR-TADF-OLED with a pretty good CIE coordinates of (0.13, 0.09) and an EQE of 13.5% using BN-doped nanographene as the key material.45 It is evident that the EQEs and CIE coordinates of the present 3,6-DDPhCz-AD- and 3,6-DDtBuPhCz-AD-based devices B4 and B5 are among the best-performing for deep-blue TICT-TADF materials and devices if both the efficiency and color purity are simultaneously evaluated. We attribute the excellent performance of 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD to their enhanced TADF feature, reflected in the sufficiently low ΔEST, significantly high kRISC (105 s−1) and increased PLQYs, all of which were obtained by increasing the number and electron-donating ability of peripheral groups from one Ph to two tBuPh groups on Cz.
Conclusion
In conclusion, a series of deep-blue TADF emitters 3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD were designed and synthesized using acridone (AD) and carbazole (Cz) as the acceptor and donor, respectively, which have the same D–A–D backbone but different attaching groups on the 3- or 3,6-sites of Cz moieties to fine-tune the singlet and triplet excited state alignment. Upon increasing the electron-donating ability and number of peripheral groups on the Cz ring in the order of 3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD to 3,6-DDtBuPhCz-AD, the 3LE (T1) energy level gradually approached the 1CT (S1) and 3CT (T2) states; at the same time, the higher-lying triplet states (T3–5) were more closely aligned, resulting in reduced ΔEST and ΔETT. As a result, a multi-channel RISC process was achieved in 3,6-DDPhCz-AD to 3,6-DDtBuPhCz-AD to realize high kRISC (105 s−1). Furthermore, the incorporation of these peripheral groups enlarged the HOMO delocalization, which in combination with high molecular rigidity led to gradually increased kR (108 s−1) and PLQYs. The deep-blue OLEDs based on 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD exhibited high EQEs of 17.4% and 17.3% with CIE coordinates of (0.15, 0.11) and (0.15, 0.13), respectively. These two TADF emitters are advantageous not only in that their EQEs are among the highest values ever reported for deep-blue TICT-type TADF materials but also because their FWHMs of 54 and 56 nm, respectively, are the narrowest reported so far for TICT-TADF emitters. For most reported Cz-based TADF emitters, the TADF feature is usually realized by increasing the dihedral angles between the Cz donor and the bridge or acceptor unit to reduce HOMO/LUMO overlap and achieve a tiny ΔEST, which is usually at the expense of kR and PLQY due to limited molecular conjugation. In contrast, the excellent performance of the present blue TADF materials was achieved via modification on the periphery of the Cz ring, which proved to be an effective method to not only guarantee high kRISC and TADF nature but also improve PLQY due to HOMO delocalization.Experimental section
General procedure for the synthesis of 3,6-DCz-AD, 3,6-DPhCz-AD, 3,6-DtBuPhCz-AD, 3,6-DDPhCz-AD and 3,6-DDtBuPhCz-AD
3,6-DF-AD was first synthesized according to literature methods.23,34 A mixture of 3,6-DF-AD (200 mg, 0.65 mmol), carbazole or corresponding carbazole derivative (1.43 mmol), cesium carbonate (847 mg, 2.6 mmol) and 15 mL dry N,N-dimethylformamide (DMF) was stirred at 165 °C under a nitrogen atmosphere. After cooling to room temperature, the mixture was poured into water and the precipitate was filtered under reduced pressure and purified by column chromatography on silica gel using DCM as the eluent to afford a solid, which was then deeply purified by repeated recrystallization from CHCl3/CH3OH to afford the pure product.Author contributions
Yongqiang Mei: conceptualization, investigation, writing-original draft. Di Liu: investigation, project administration, resource, supervision. Jiuyan Li: funding acquisition, investigation, supervision, writing-review and editing. Jiahui Wang: data curation, visualization.Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22078051 and U1801258), Fundamental Research Fundamental Funds for the Central Universities (DUT22LAB610), and Open Fund of the State Key Laboratory of Luminescent Materials and Devices (South China University of Technology, 2021-skllmd-03) for the financial support of this work.References
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402–6406 CrossRef CAS PubMed.
- W. Li, M. Li, W. Li, Z. Xu, L. Gan, K. Liu, N. Zheng, C. Ning, D. Chen, Y. C. Wu and S. J. Su, ACS Appl. Mater. Interfaces, 2021, 13, 5302–5311 CrossRef CAS.
- H. Lim, H. J. Cheon, S. J. Woo, S. K. Kwon, Y. H. Kim and J. J. Kim, Adv. Mater., 2020, 32, e2004083 CrossRef.
- W. Li, B. Li, X. Cai, L. Gan, Z. Xu, W. Li, K. Liu, D. Chen and S. J. Su, Angew. Chem., Int. Ed., 2019, 58, 11301–11305 CrossRef CAS PubMed.
- U. Balijapalli, R. Nagata, N. Yamada, H. Nakanotani, M. Tanaka, A. D'Aleo, V. Placide, M. Mamada, Y. Tsuchiya and C. Adachi, Angew. Chem., Int. Ed., 2021, 60, 8477–8482 CrossRef CAS.
- D. H. Ahn, J. H. Maeng, H. Lee, H. Yoo, R. Lampande, J. Y. Lee and J. H. Kwon, Adv. Opt. Mater., 2020, 8, 2000102 CrossRef CAS.
- L. S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2017, 56, 1571–1575 CrossRef CAS.
- S. J. Woo, Y. Kim, S. K. Kwon, Y. H. Kim and J. J. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 7199–7207 CrossRef CAS.
- J. U. Kim, I. S. Park, C. Y. Chan, M. Tanaka, Y. Tsuchiya, H. Nakanotani and C. Adachi, Nat. Commun., 2020, 11, 1765 CrossRef CAS PubMed.
- J. X. Chen, Y. F. Xiao, K. Wang, D. Sun, X. C. Fan, X. Zhang, M. Zhang, Y. Z. Shi, J. Yu, F. X. Geng, C. S. Lee and X. H. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2478–2484 CrossRef CAS.
- P. Stachelek, J. S. Ward, P. L. Dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, ACS Appl. Mater. Interfaces, 2019, 11, 27125–27133 CrossRef CAS.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- X. Cai, B. Gao, X.-L. Li, Y. Cao and S.-J. Su, Adv. Funct. Mater., 2016, 26, 8042–8052 CrossRef CAS.
- M. Cai, M. Auffray, D. Zhang, Y. Zhang, R. Nagata, Z. Lin, X. Tang, C.-Y. Chan, Y.-T. Lee, T. Huang, X. Song, Y. Tsuchiya, C. Adachi and L. Duan, Chem. Eng. J., 2021, 420, 127591 CrossRef CAS.
- J. Hwang, H. Kang, J.-E. Jeong, H. Y. Woo, M. J. Cho, S. Park and D. H. Choi, Chem. Eng. J., 2021, 416, 129185 CrossRef CAS.
- Y. Luo, S. Li, Y. Zhao, C. Li, Z. Pang, Y. Huang, M. Yang, L. Zhou, X. Zheng, X. Pu and Z. Lu, Adv. Mater., 2020, 32, e2001248 CrossRef.
- J. Liang, C. Li, X. Zhuang, K. Ye, Y. Liu and Y. Wang, Adv. Funct. Mater., 2018, 28, 1707002 CrossRef.
- L. Wang, X. Cai, B. Li, M. Li, Z. Wang, L. Gan, Z. Qiao, W. Xie, Q. Liang, N. Zheng, K. Liu and S. J. Su, ACS Appl. Mater. Interfaces, 2019, 11, 45999–46007 CrossRef CAS PubMed.
- K. Ke, J.-X. Chen, M. Zhang, K. Wang, Y.-Z. Shi, H. Lin, C.-J. Zheng, S.-L. Tao and X.-H. Zhang, Sci. China Mater., 2019, 62, 719–728 CrossRef CAS.
- L. G. Franca, Y. Long, C. Li, A. Danos and A. Monkman, J. Phys. Chem. Lett., 2021, 12, 1490–1500 CrossRef CAS.
- Y. Ran, G. Yang, Y. Liu, W. Han, G. Gao, R. Su, Z. Bin and J. You, Mater. Horiz., 2021, 8, 2025–2031 RSC.
- N. Aizawa, A. Matsumoto and T. Yasuda, Sci. Adv., 2021, 7, eabe5769 CrossRef CAS.
- (a) Y. Mei, D. Liu, J. Li, H. Li and W. Wei, J. Mater. Chem. C, 2021, 9, 5885–5892 RSC; (b) Y. Mei, D. Liu, J. Li, R. Dong, M. Ma, W. Wei and Y. Lan, Mater. Today Chem., 2022, 23, 100645 CrossRef CAS.
- P. Pander, A. Swist, R. Motyka, J. Soloducho, F. B. Dias and P. Data, J. Mater. Chem. C, 2018, 6, 5434–5443 RSC.
- L. Gan, K. Gao, X. Cai, D. Chen and S. J. Su, J. Phys. Chem. Lett., 2018, 9, 4725–4731 CrossRef CAS.
- C. Cheng, Y. Jiang, H. Wang, W. Lou, Y. Zhu, C. Deng, D. Wang, T. Tsuboi, G. Li and Q. Zhang, J. Mater. Chem. C, 2021, 9, 3088–3095 RSC.
- G. Kreiza, D. Berenis, D. Banevičius, S. Juršėnas, T. Javorskis, E. Orentas and K. Kazlauskas, Chem. Eng. J., 2021, 412, 128574 CrossRef CAS.
- C. Y. Chan, L. S. Cui, J. U. Kim, H. Nakanotani and C. Adachi, Adv. Funct. Mater., 2018, 28, 1706023 CrossRef.
- W.-W. Tao, K. Wang, J.-X. Chen, Y.-Z. Shi, W. Liu, C.-J. Zheng, Y.-Q. Li, J. Yu, X.-M. Ou and X.-H. Zhang, J. Mater. Chem. C, 2019, 7, 4475–4483 RSC.
- C.-Y. Chan, M. Tanaka, Y.-T. Lee, Y.-W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Nat. Photonics, 2021, 15, 203–207 CrossRef CAS.
- R. K. Konidena, J. Lim and J. Y. Lee, Chem. Eng. J., 2021, 416, 129097 CrossRef CAS.
- Y. Zhang, D. Zhang, T. Tsuboi, Y. Qiu and L. Duan, Sci. China: Chem., 2019, 62, 393–402 CrossRef CAS.
- F.-M. Xie, Z.-D. An, M. Xie, Y.-Q. Li, G.-H. Zhang, S.-J. Zou, L. Chen, J.-D. Chen, T. Cheng and J.-X. Tang, J. Mater. Chem. C, 2020, 8, 5769–5776 RSC.
- L. A. Andronico, A. Quintavalla, M. Lombardo, M. Mirasoli, M. Guardigli, C. Trombini and A. Roda, Chem, 2016, 22, 18156–18168 CrossRef CAS PubMed.
- X. Cai, D. Chen, K. Gao, L. Gan, Q. Yin, Z. Qiao, Z. Chen, X. Jiang and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1704927 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- H. Liu, Q. Bai, W. Li, Y. Guo, L. Yao, Y. Gao, J. Li, P. Lu, B. Yang and Y. Ma, RSC Adv., 2016, 6, 70085–70090 RSC.
- Y. Liu, H. Liu, Q. Bai, C. Du, A. Shang, D. Jiang, X. Tang and P. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 16715–16725 CrossRef CAS.
- Z. Lin, R. Kabe, K. Wang and C. Adachi, Nat. Commun., 2020, 11, 191 CrossRef CAS.
- A. Khan, X. Tang, C. Zhong, Q. Wang, S. Y. Yang, F. C. Kong, S. Yuan, A. S. D. Sandanayaka, C. Adachi, Z. Q. Jiang and L. S. Liao, Adv. Funct. Mater., 2021, 31, 2009488 CrossRef CAS.
- (a) Y.-K. Chen, J. Jayakumar, C.-M. Hsieh, T.-L. Wu, C.-C. Liao, J. Pandidurai, C.-L. Ko, W.-Y. Hung and C.-H. Cheng, Adv. Mater., 2021, 33, 2008032 CrossRef CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- P. L. dos Santos, J. S. Ward, M. R. Bryce and A. P. Monkman, J. Phys. Chem. Lett., 2016, 7, 3341–3346 CrossRef CAS.
- I. Lee and J. Y. Lee, Org. Electron., 2016, 29, 160–164 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc03448d |
| This journal is © The Royal Society of Chemistry 2022 |