
N-Heterocyclic carbene-catalyzed radical ring-opening acylation of oxime esters with aldehydes†
Zhihao
Zhang
a,
Xin
Zou
a,
Zhenjiang
Li
 *ab,
Yu
Gao
a,
Yuanyuan
Qu
a,
Yusheng
Quan
a,
Yi
Zhou
a,
Jinlan
Li
a,
Jie
Sun
c and
Kai
Guo
*ab
*ab,
Yu
Gao
a,
Yuanyuan
Qu
a,
Yusheng
Quan
a,
Yi
Zhou
a,
Jinlan
Li
a,
Jie
Sun
c and
Kai
Guo
*ab
aCollege of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, 30 Puzhu Road South, Nanjing 211816, P. R. China. E-mail: zjli@njtech.edu.cn
bState Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, 30 Puzhu Road South, Nanjing 211816, P. R. China. E-mail: guok@njtech.edu.cn
cCollege of Food Science and Light Industry, Nanjing Tech University, 30 Puzhu Road South, Nanjing 211816, P. R. China
First published on 1st September 2021
Abstract
A cross-coupling of cycloketone oxime esters with aldehydes catalyzed by N-heterocyclic carbenes via a radical pathway was established. This modular protocol features easy operation, no external oxidants or reductants, and a broad functional group compatibility. The merit of this method was showcased by late-stage modification of complicated aldehydes derived from the medical intermediate pregnenolone and natural product diacetone-D-glucose.
Introduction
Owing to their unique nucleophilicity and Lewis basicity, N-heterocyclic carbenes (NHCs) have arisen as prominent organocatalysts with wide applications in chemical synthesis.1 Commonly, NHC-catalyzed transformations proceed by the generation of Breslow,2 π-extended Breslow3 or acyl azolium intermediates4 in electron-pair-transfer pathways. In contrast, the emerging realm of NHC catalyzed single-electron-transfer (SET) reactions still remains underdeveloped,5 but has opened new avenues for developing novel catalytic modes and promising synthetic strategies.6 Thus, the development of novel reaction types via radical NHC catalysis is highly desirable.Since the pioneering disclosure of the reducing ability of the enolate form of the Breslow intermediate by the Fukuzumi group,7 elegant radical transformations mediated by NHCs have been reported by Scheidt,8 Studer,9 Rovis,10 Chi,11 Ye,12 Ohmiya,13 and other groups.14 Recently, we also developed a regioselective, intermolecular 1,2-cyanoalkylacylation of alkenes with oxime esters15 and aldehydes by NHC organocatalysis (Scheme 1A).16 A SET from the enolate form of the Breslow intermediate to a cyclobutanone oxime ester occurs to give a persistent aldehyde-derived ketyl radical and a transient primary cyanoalkyl radical after radical transposition via C–C bond cleavage. Radical addition of the primary cyanoalkyl radical to styrene occurs preferentially to lead to the more stable benzyl radical, which then engages in radical–radical cross-coupling with the persistent ketyl radical to afford the three-component coupling product.
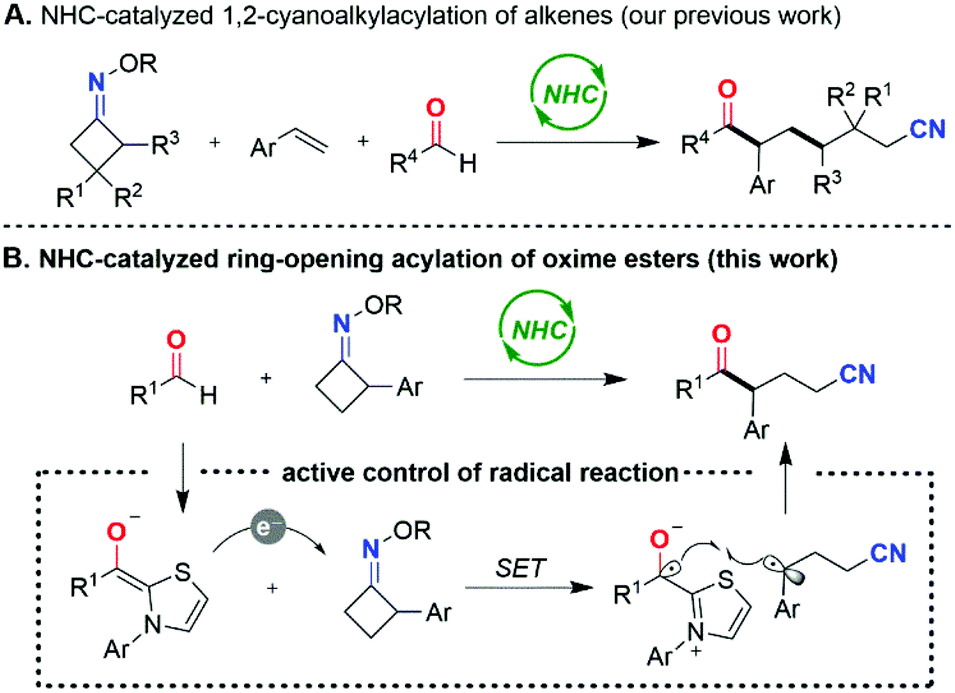 | ||
| Scheme 1 Synthetic strategy. | ||
Enlightened by this finding, we surmised that if the cycloketone oxime esters themselves can form the more stable benzyl radicals, the direct radical cross-coupling of aldehydes with such cycloketone oxime esters might be realized. Herein, we report an NHC-catalyzed radical ring-opening acylation of oxime esters with aldehydes under transition-metal-free and redox-neutral conditions, granting a straightforward, flexible access to ketonitrile structures (Scheme 1B). It is noteworthy that the products are a class of versatile building blocks.
Results and discussion
At the outset, 4-methylbenzaldehyde 1a and O-(tert-butoxycarbonyl) oxime 2a were chosen as model substrates to investigate the ring-opening acylation of oxime esters in the presence of the carbene precursor. Indeed, in our most recent work,16 the two-component cross-coupling of an oxime and an aldehyde was observed;17 thus a brief optimization of the carbene catalyst, solvent, and base led to the identification of optimal conditions, which involved the use of the N-2,6-diisopropylphenyl-substituted cycloheptane-fused thiazolium precatalyst C1 (20 mol%), dichloromethane (DCM), and cesium carbonate to furnish the two-component coupling product ketonitrile 3aa in 95% yield (Table 1, entry 1).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (3aa, %) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.12 mmol), C1 (20 mol%), and Cs2CO3 (0.15 mmol) in DCM (1.0 mL) at 60 °C for 12 h under Ar. b Yield determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. c Using 10 mol% C. Boc = tert-butyloxycarbonyl, DBU = 1,8-diazabicyclo[5.4.0]-7-undecene, and TMG = 1,1,3,3-tetramethylguanidine. | ||
| 1 | None | 95 |
| 2 | Using 10 mol% C1 | 66 |
| 3c | C2 instead of C1 | 53 |
| 4c | C3 instead of C1 | 40 |
| 5c | C4 instead of C1 | 30 |
| 6c | C5 instead of C1 | 1 |
| 7c | C6 instead of C1 | 0 |
| 8c | C7 instead of C1 | 0 |
| 9c | C8 instead of C1 | 0 |
| 10 | DCE instead of DCM | 12 |
| 11 | MeCN instead of DCM | 1 |
| 12 | THF instead of DCM | 3 |
| 13 | DMSO instead of DCM | 0 |
| 14 | Na2CO3 instead of Cs2CO3 | 6 |
| 15 | K2CO3 instead of Cs2CO3 | 93 |
| 16 | DBU instead of Cs2CO3 | 10 |
| 17 | TMG instead of Cs2CO3 | 43 |
| 18 | No NHC catalyst | 0 |
| 19 | No base | 0 |
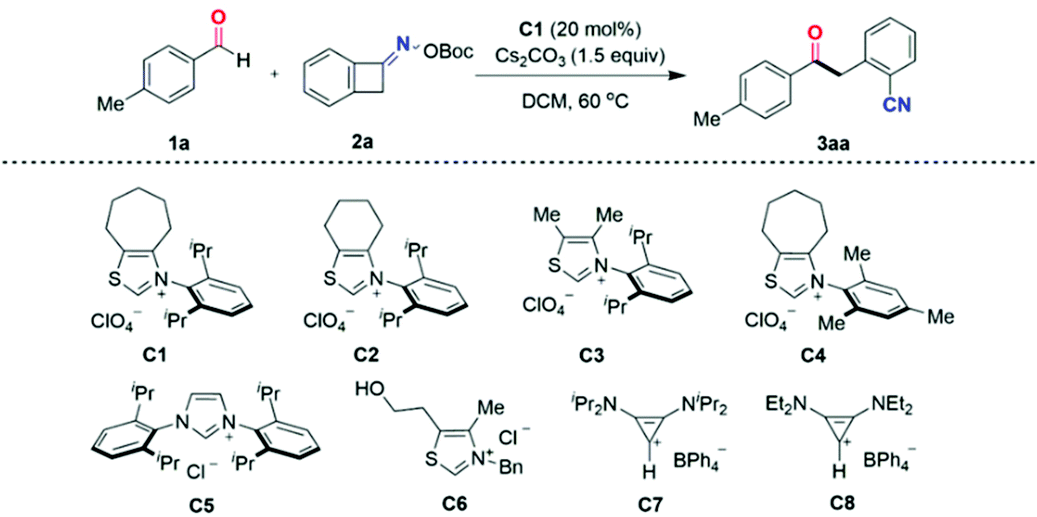
It was found that both the backbone architecture and the N-aryl substituent of the thiazolium salts played a significant role in reaction efficiency (Table 1, entries 2–5). Employing 10 mol% carbene precursor C, such as the six-membered-ring thiazolium salt bearing N-2,6-diisopropylphenyl-substituent C2 (entry 3), or the thiazolium salt with an acyclic backbone C3 (entry 4), or the seven-membered-ring thiazolium salt bearing an N-mesityl-substituent C4 (entry 5) resulted in reduced yields. The sterically demanding N-2,6-diisopropylphenyl group and a cycloheptane backbone were identified as the optimum structure (entry 2). Increasing the precatalyst C1 loading to 20 mol% further boosted the product yield (entry 1). On the other hand, the commercially available imidazolium- and thiazolium-type NHC precatalysts C5 and C6 were quite ineffective in this transformation (entries 6 and 7), as were non-heterocyclic-based carbenes derived from bis(amino)cyclopropenium salts18C7 and C8 (entries 8 and 9).
The solvent had a profound effect on the reaction, as other solvents including 1,2-dichloroethane (DCE), acetonitrile (MeCN), tetrahydrofuran (THF), and dimethyl sulfoxide (DMSO) were found to be unsuitable for this ring-opening acylation transformation (Table 1, entries 10–13). The choice of the base also proved to be the key. K2CO3 gave 3aa in a comparable yield to Cs2CO3, whereas the use of other bases such as Na2CO3, DBU, or TMG instead of Cs2CO3 all led to a stark decrease in yield (entries 14–17). Control experiments revealed that the NHC catalyst and base were necessary for the success of this ring-opening acylation of oxime esters.
With the optimal reaction conditions in hand, the scope and limitations of this ring-opening acylation of oxime esters were investigated (Schemes 2 and 3). The initial focus was on assessing aldehyde diversity. As shown in Scheme 2, the C–C bond cleavage of benzocyclobutenone-derived oxime ester occurred with complete regioselectivity to form the more stable benzyl radical to engage in the reaction. When benzaldehyde 1b was subjected to the standard conditions, the ketonitrile 3ba was obtained in 86% yield. Benzaldehydes 1c–1h bearing a gamut of pendant functionalities at the para position, such as ether (3ca), halides (3da–3fa), cyano group (3ga), and ester (3ha), were well accommodated, giving access to the corresponding coupling products 3ca–3ha with yields ranging from 65% to 91%. Relatively lower reactivity was observed for 4-methoxybenzaldehyde 1c, indicating that the introduction of a strong electron-donating group onto the aryl ring of the aldehyde might be detrimental for the aldehyde-derived Breslow radical intermediate to undergo radical–radical coupling with the benzyl radical. Although a substituent at the para and meta positions of the aryl aldehyde was tolerated (3ea and 3ja), o-substituted benzaldehyde completely inhibited the reaction (data not shown). 2-Naphthaldehyde proved to be a viable coupling partner in this reaction manifold, affording the expected product 3ka in 83% yield. Furthermore, aldehydes containing electron-rich and electron-deficient heteroaromatic rings such as thiophene and pyridine can also be used, as exemplified by 3la and 3ma. As a current limitation, aliphatic aldehydes are not competent as acyl donors in this reaction. This may be attributed to the instability (short lifetime) of the aliphatic aldehyde-derived Breslow radical intermediate. Although cinnamaldehyde was also attempted, no products were observed. Finally, this organocatalytic approach was further applied to the late-stage modification of complex molecules. For example, complex aldehyde derivatives of pregnenolone and diacetone-D-glucose were apt to give serviceable yields of the coupling products 3na and 3oa. It is noteworthy that the thus obtained ketonitrile products 3 could be further converted to biologically active CWJ-a-5 analogues according to a reported procedure.19
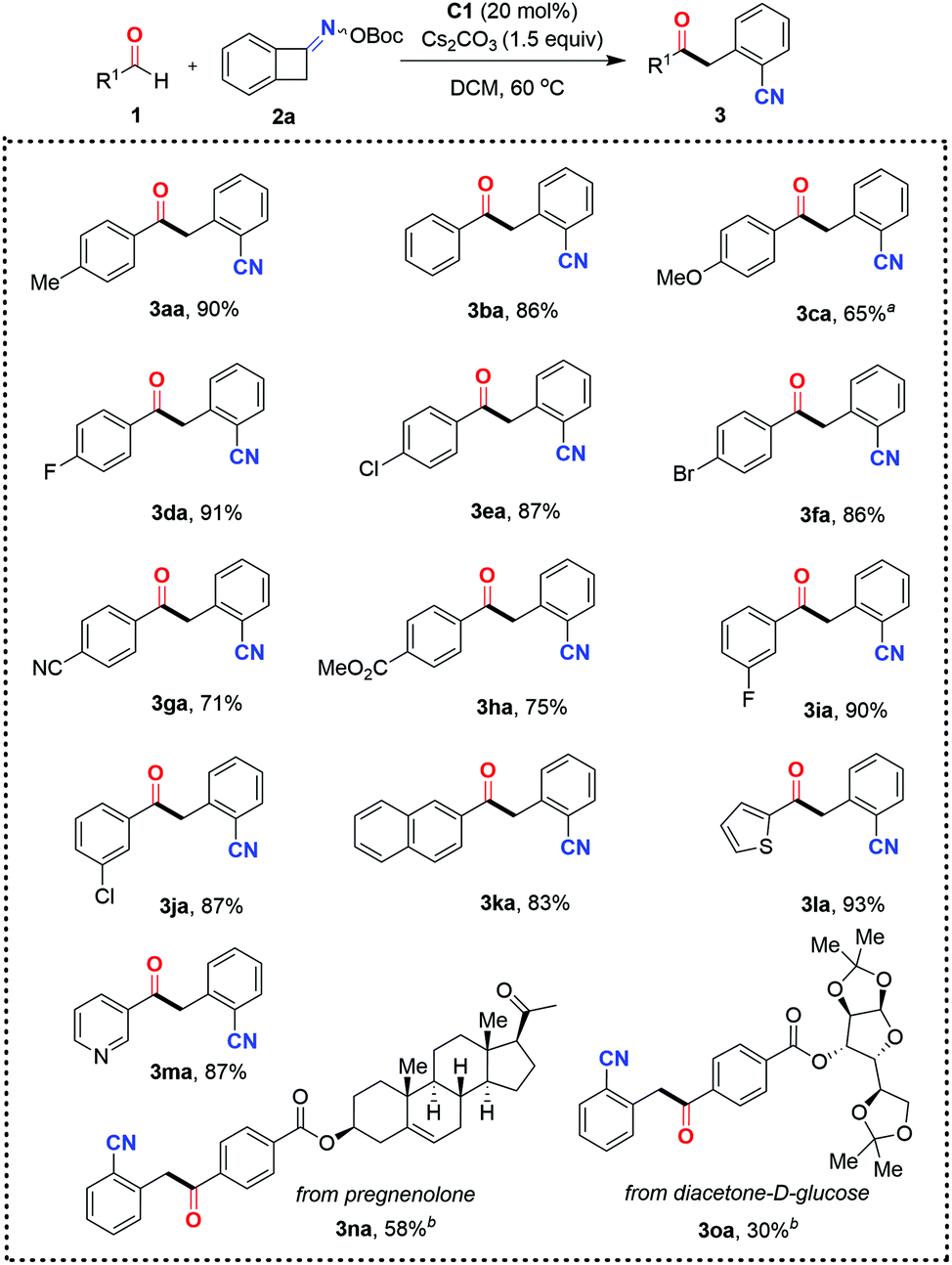 | ||
Scheme 2 Scope with respect to aldehydes. Standard reaction conditions: 1 (0.1 mmol), 2a (0.12 mmol), C1 (20 mol%), and Cs2CO3 (0.15 mmol) in DCM (1.0 mL) at 60 °C for 12 h under Ar; isolated yields based on 1 are given. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Using 40 mol% C1. bUsing 30 mol% C1. Using 40 mol% C1. bUsing 30 mol% C1. | ||
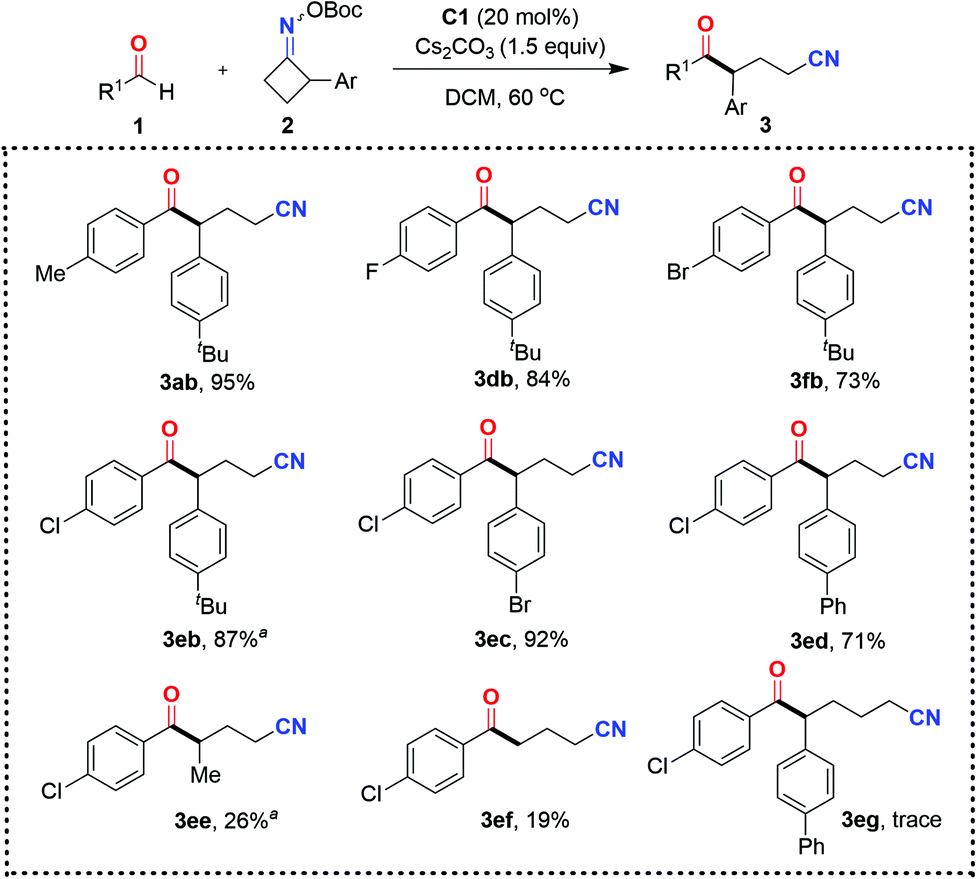 | ||
| Scheme 3 Scope of the NHC-catalyzed ring-opening acylation of oxime esters. Reaction conditions: 1 (0.1 mmol), 2 (0.11 mmol), C1 (20 mol%), and Cs2CO3 (0.15 mmol) in DCM (1.0 mL) at 60 °C for 12 h under Ar; isolated yields based on 1 are given. a0.2 mmol scale. | ||
Our attention then turned to evaluating the scope of cycloketone oxime esters. Gratifyingly, the range of cycloketone oxime esters was not restricted to the benzocyclobutenone-derived oxime ester 2a, and could also encompass α-aryl-substituted cyclobutanone-derived oxime esters (Scheme 3). The cyclobutanone oxime esters 2 with substituents such as tertiary butyl (2b), bromide (2c), and phenyl (2d) groups at the para-position of the aromatic ring reacted smoothly with 4-chlorobenzaldehyde 3e, affording the corresponding products δ-keto nitriles (3eb, 3ec, and 3ed) with yields ranging from 71% to 92%. On the downside, the reaction performed poorly with the α-methyl-substituted cyclobutanone-derived oxime ester 2e and non-substituted cyclobutanone-derived oxime ester 2f, presumably due to the instability of the generated secondary or primary radical in comparison with the benzyl radical. The less-strained, five-membered cycloketone oxime ester 2g was nonproductive under the present organocatalytic conditions. Only a trace amount of the target product 3eg was detected by HRMS.
To shed light on the tentative reaction mechanism, a preliminary mechanistic investigation was performed (Scheme 4). In the presence of 2.0 equivalents of the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), the standard reaction of 4-methylbenzaldehyde 1a and O-(tert-butoxycarbonyl) oxime 2a was totally inhibited. Furthermore, 4-methylbenzaldehyde 1a was reacted with O-(tert-butoxycarbonyl) oxime 2a under standard conditions in the presence of 2.0 equivalents of the radical scavenger 2,6-di-tert-butyl-4-methylphenol (BHT), affording the two-component coupling product 3aa in only 28% yield. This result indicated the radical nature of this transformation.
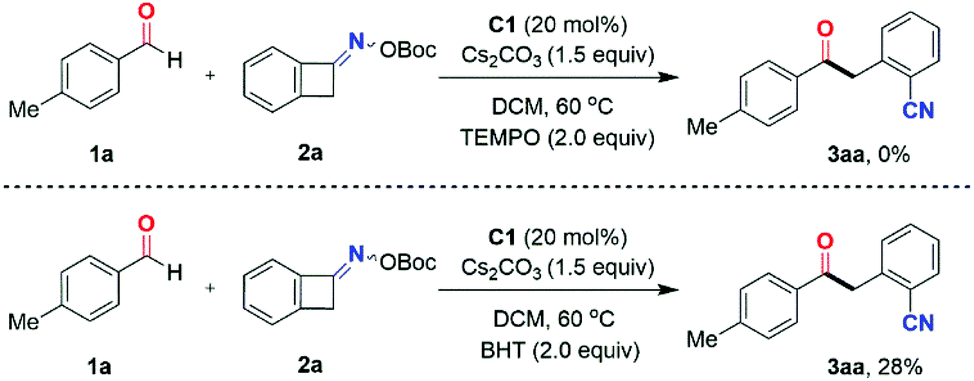 | ||
| Scheme 4 Mechanistic investigations. | ||
On the basis of the above experimental results and the literature,13a,16 a tentative mechanism for the NHC-catalyzed ring-opening acylation of oxime esters is illustrated in Scheme 5. First, NHC reacts with the aldehyde 1 to give the corresponding Breslow intermediate I. Next, the enolate form of the Breslow intermediate II would be generated by deprotonation in the presence of a base and induce the SET event between the enolate II and cyclobutanone oxime ester 2, affording a persistent ketyl radical III and an iminyl radical A, respectively. Subsequent regioselective ring-opening of Avia C–C bond β-cleavage forms the benzyl radical B. Intermediates III and B would undergo radical–radical cross-coupling to furnish the intermediate IV (path a).20 Alternatively, another addition of the benzyl radical B to the enolate form of the Breslow intermediate II might occur, giving a plausible NHC-bound radical intermediate V. SET from the intermediate V to oxime ester 2 would ensue to form the intermediate IV and a new iminyl radical A, respectively (path b). Currently, we cannot exclude either pathway. Ultimately, the species IV would undergo facile elimination to deliver the target product ketonitrile 3 and release the NHC, thereby closing this catalytic redox-neutral cycle.
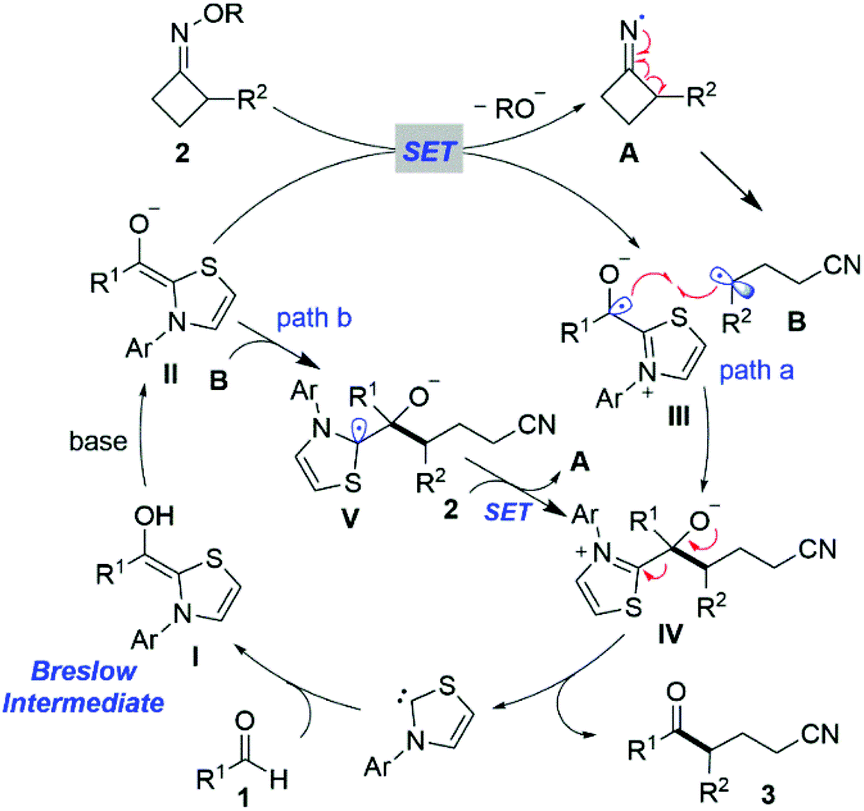 | ||
| Scheme 5 Proposed mechanism for the NHC-catalyzed ring-opening acylation of oxime esters. | ||
Experimental
General procedure for ring-opening acylation of oxime esters
A flame-dried 10 mL resealable screw-cap Schlenk tube containing a magnetic stirring bar was charged with the aldehyde 1 (0.1 mmol, 1.0 equiv.), oxime ester 2 (0.12 mmol, 1.2 equiv.), NHC precursor C1 (8.3 mg, 0.02 mmol, 0.2 equiv.), Cs2CO3 (48.9 mg, 0.15 mmol, 1.5 equiv.), and anhydrous DCM (1.0 mL) under an argon atmosphere. The reaction mixture was stirred in a preheated 60 °C oil bath for 12 h. After that, the mixture was concentrated under reduced pressure. The resulting crude product was purified by silica gel flash column chromatography to give the corresponding products 3aa–3ef.Conclusions
In summary, we have developed an emerging NHC-catalyzed radical cross-coupling of cycloketone oxime esters with readily available aldehydes. By using an N-2,6-diisopropylphenyl-substituted cycloheptane-fused thiazolium salt as the NHC precatalyst, the cyanoalkyl or 2-cyanobenzyl from the cycloketone oxime esters and the acyl from the aldehydes were docked efficiently, giving access to ketonitriles in moderate to good yields. The aromatic nitriles can be used to construct a compound library of topoisomerase I inhibitor CWJ-a-5 analogues. This protocol features easy operation, with no need for external oxidants, reductants or transition-metal catalysts, and a good functional group tolerance. The compatibility of substrate scope covers benzocyclobutenone oxime esters, α-aryl-substituted cyclobutenone oxime esters, and aromatic aldehydes. Complicated aldehydes derived from the medical intermediate pregnenolone and natural product diacetone-D-glucose also proved to be viable coupling partners, affording the corresponding coupling products in synthetically useful yields. Mechanistic studies indicated that a radical pathway was involved. This work further enriches the types of radical reactions catalyzed by NHC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support to this work by the National Natural Science Foundation of China (22078150), the Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Top-Notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP) is gratefully acknowledged.Notes and references
- For recent reviews, see: (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (b) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- R. Breslow, On the Mechanism of Thiamine Action. IV.1 Evidence from Studies on Model Systems, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS.
- R. S. Menon, A. T. Biju and V. Nair, Recent advances in employing homoenolates generated by N-heterocyclic carbene (NHC) catalysis in carbon–carbon bond-forming reactions, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC.
- C. Zhang, J. F. Hooper and D. W. Lupton, N-Heterocyclic Carbene Catalysis via the α,β-Unsaturated Acyl Azolium, ACS Catal., 2017, 7, 2583–2596 CrossRef CAS.
- R. Song and Y. R. Chi, N-Heterocyclic Carbene Catalyzed Radical Coupling of Aldehydes with Redox-Active Esters, Angew. Chem., Int. Ed., 2019, 58, 8628–8630 CrossRef CAS PubMed.
- For selected reviews, see: (a) T. Ishii, K. Nagao and H. Ohmiya, Recent advances in N-heterocyclic carbene-based radical catalysis, Chem. Sci., 2020, 11, 5630–5636 RSC; (b) J. Liu, X.-N. Xing, J.-H. Huang, L.-Q. Lu and W.-J. Xiao, Light opens a new window for N-heterocyclic carbene catalysis, Chem. Sci., 2020, 11, 10605–10613 RSC; (c) L. Dai and S. Ye, Recent advances in N-heterocyclic carbene-catalyzed radical reactions, Chin. Chem. Lett., 2021, 32, 660–667 CrossRef CAS; (d) K.-Q. Chen, H. Sheng, Q. Liu, P.-L. Shao and X.-Y. Chen, N-heterocyclic carbene-catalyzed radical reactions, Sci. China: Chem., 2021, 64, 7–16 CrossRef CAS; (e) Q.-Z. Li, R. Zeng, B. Han and J.-L. Li, Single-Electron Transfer Reactions Enabled by N-Heterocyclic Carbene Organocatalysis, Chem. – Eur. J., 2021, 27, 3238–3250 CrossRef CAS PubMed; (f) H. Ohmiya, N-Heterocyclic Carbene-Based Catalysis Enabling Cross-Coupling Reactions, ACS Catal., 2020, 10, 6862–6869 CrossRef CAS.
- (a) N. Ikuo, I. Shinobu, S. Tomoyoshi, I. Hiroo and F. Shunichi, Redox Behavior of Active Aldehydes Derived from Thiamin Coenzyme Analogs, Chem. Lett., 1997, 26, 707–708 CrossRef; (b) I. Nakanishi and S. Itoh, Electron transfer properties of active aldehydes derived from thiamin coenzyme analogues, Chem. Commun., 1997, 1927–1928, 10.1039/A704559J; (c) I. Nakanishi, S. Itoh, T. Suenobu and S. Fukuzumi, Direct Observation of Radical Intermediates While Investigating the Redox Behavior of Thiamin Coenzyme Models, Angew. Chem., Int. Ed., 1998, 37, 992–994 CrossRef CAS; (d) I. Nakanishi, S. Itoh and S. Fukuzumi, Electron-Transfer Properties of Active Aldehydes of Thiamin Coenzyme Models, and Mechanism of Formation of the Reactive Intermediates, Chem. – Eur. J., 1999, 5, 2810–2818 CrossRef CAS.
- B. E. Maki, A. Chan, E. M. Phillips and K. A. Scheidt, Tandem Oxidation of Allylic and Benzylic Alcohols to Esters Catalyzed by N-Heterocyclic Carbenes, Org. Lett., 2007, 9, 371–374 CrossRef CAS PubMed.
- (a) J. Guin, S. De Sarkar, S. Grimme and A. Studer, Biomimetic Carbene-Catalyzed Oxidations of Aldehydes Using TEMPO, Angew. Chem., Int. Ed., 2008, 47, 8727–8730 CrossRef CAS PubMed; (b) S. De Sarkar, S. Grimme and A. Studer, NHC Catalyzed Oxidations of Aldehydes to Esters: Chemoselective Acylation of Alcohols in Presence of Amines, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef CAS PubMed.
- (a) N. A. White and T. Rovis, Enantioselective N-Heterocyclic Carbene-Catalyzed β-Hydroxylation of Enals Using Nitroarenes: An Atom Transfer Reaction That Proceeds via Single Electron Transfer, J. Am. Chem. Soc., 2014, 136, 14674–14677 CrossRef CAS PubMed; (b) N. A. White and T. Rovis, Oxidatively Initiated NHC-Catalyzed Enantioselective Synthesis of 3,4-Disubstituted Cyclopentanones from Enals, J. Am. Chem. Soc., 2015, 137, 10112–10115 CrossRef CAS PubMed.
- (a) Y. Zhang, Y. Du, Z. Huang, J. Xu, X. Wu, Y. Wang, M. Wang, S. Yang, R. D. Webster and Y. R. Chi, N-Heterocyclic Carbene-Catalyzed Radical Reactions for Highly Enantioselective β-Hydroxylation of Enals, J. Am. Chem. Soc., 2015, 137, 2416–2419 CrossRef CAS PubMed; (b) B.-S. Li, Y. Wang, R. S. J. Proctor, Y. Zhang, R. D. Webster, S. Yang, B. Song and Y. R. Chi, Carbene-catalysed reductive coupling of nitrobenzyl bromides and activated ketones or imines via single-electron-transfer process, Nat. Commun., 2016, 7, 12933 CrossRef PubMed; (c) X. Wu, Y. Zhang, Y. Wang, J. Ke, M. Jeret, R. N. Reddi, S. Yang, B.-A. Song and Y. R. Chi, Polyhalides as Efficient and Mild Oxidants for Oxidative Carbene Organocatalysis by Radical Processes, Angew. Chem., Int. Ed., 2017, 56, 2942–2946 CrossRef CAS PubMed.
- (a) X.-Y. Chen, K.-Q. Chen, D.-Q. Sun and S. Ye, N-Heterocyclic carbene-catalyzed oxidative [3+2] annulation of dioxindoles and enals: cross coupling of homoenolate and enolate, Chem. Sci., 2017, 8, 1936–1941 RSC; (b) L. Dai, Z.-H. Xia, Y.-Y. Gao, Z.-H. Gao and S. Ye, Visible-Light-Driven N-Heterocyclic Carbene Catalyzed γ- and ε-Alkylation with Alkyl Radicals, Angew. Chem., Int. Ed., 2019, 58, 18124–18130 CrossRef CAS PubMed; (c) L. Dai and S. Ye, Photo/N-Heterocyclic Carbene Co-catalyzed Ring Opening and γ-Alkylation of Cyclopropane Enal, Org. Lett., 2020, 22, 986–990 CrossRef CAS PubMed; (d) L. Dai, Y.-Y. Xu, Z.-H. Xia and S. Ye, γ-Difluoroalkylation: Synthesis of γ-Difluoroalkyl-α,β-Unsaturated Esters via Photoredox NHC-Catalyzed Radical Reaction, Org. Lett., 2020, 10, 12960–12966 Search PubMed.
- (a) T. Ishii, Y. Kakeno, K. Nagao and H. Ohmiya, N-Heterocyclic Carbene-Catalyzed Decarboxylative Alkylation of Aldehydes, J. Am. Chem. Soc., 2019, 141, 3854–3858 CrossRef CAS PubMed; (b) T. Ishii, K. Ota, K. Nagao and H. Ohmiya, N-Heterocyclic Carbene-Catalyzed Radical Relay Enabling Vicinal Alkylacylation of Alkenes, J. Am. Chem. Soc., 2019, 141, 14073–14077 CrossRef CAS PubMed; (c) K. Ota, K. Nagao and H. Ohmiya, N-Heterocyclic Carbene-Catalyzed Radical Relay Enabling Synthesis of δ-Ketocarbonyls, Org. Lett., 2020, 22, 3922–3925 CrossRef CAS PubMed; (d) Y. Kakeno, M. Kusakabe, K. Nagao and H. Ohmiya, Direct Synthesis of Dialkyl Ketones from Aliphatic Aldehydes through Radical N-Heterocyclic Carbene Catalysis, ACS Catal., 2020, 10, 8524–8529 CrossRef CAS; (e) T. Ishii, K. Nagao and H. Ohmiya, Radical N-heterocyclic carbene catalysis for β-ketocarbonyl synthesis, Tetrahedron, 2021, 132212 CrossRef CAS; (f) Y. Matsuki, N. Ohnishi, Y. Kakeno, S. Takemoto, T. Ishii, K. Nagao and H. Ohmiya, Aryl radical-mediated N-heterocyclic carbene catalysis, Nat. Commun., 2021, 12, 3848 CrossRef CAS PubMed.
- (a) W. Yang, W. Hu, X. Dong, X. Li and J. Sun, N-Heterocyclic Carbene Catalyzed γ-Dihalomethylenation of Enals by Single-Electron Transfer, Angew. Chem., Int. Ed., 2016, 55, 15783–15786 CrossRef CAS PubMed; (b) J.-L. Li, Y.-Q. Liu, W.-L. Zou, R. Zeng, X. Zhang, Y. Liu, B. Han, Y. He, H.-J. Leng and Q.-Z. Li, Radical Acylfluoroalkylation of Olefins through N-Heterocyclic Carbene Organocatalysis, Angew. Chem., 2020, 59, 1863–1870 CrossRef CAS PubMed; (c) H.-B. Yang, Z.-H. Wang, J.-M. Li and C. Wu, Modular synthesis of α-aryl β-perfluoroalkyl ketones via N-heterocyclic carbene catalysis, Chem. Commun., 2020, 56, 3801–3804 RSC; (d) B. Zhang, Q. Peng, D. Guo and J. Wang, NHC-Catalyzed Radical Trifluoromethylation Enabled by Togni Reagent, Org. Lett., 2020, 22, 443–447 CrossRef CAS PubMed; (e) I. Kim, H. Im, H. Lee and S. Hong, N-Heterocyclic carbene-catalyzed deaminative cross-coupling of aldehydes with Katritzky pyridinium salts, Chem. Sci., 2020, 11, 3192–3197 RSC; (f) M.-S. Liu and W. Shu, Catalytic, Metal-Free Amide Synthesis from Aldehydes and Imines Enabled by a Dual-Catalyzed Umpolung Strategy under Redox-Neutral Conditions, ACS Catal., 2020, 10, 12960–12966 CrossRef CAS; (g) L. Chen, S. Jin, J. Gao, T. Liu, Y. Shao, J. Feng, K. Wang, T. Lu and D. Du, N-Heterocyclic Carbene/Magnesium Cocatalyzed Radical Relay Assembly of Aliphatic Keto Nitriles, Org. Lett., 2021, 23, 394–399 CrossRef CAS PubMed.
- For selected reviews, see: (a) S. Z. Zard, Recent progress in the generation and use of nitrogen-centred radicals, Chem. Soc. Rev., 2008, 37, 1603–1618 RSC; (b) W. Yin and X. Wang, Recent advances in iminyl radical-mediated catalytic cyclizations and ring-opening reactions, New J. Chem., 2019, 43, 3254–3264 RSC; (c) T. Xiao, H. Huang, D. Anand and L. Zhou, Iminyl-Radical-Triggered C–C Bond Cleavage of Cycloketone Oxime Derivatives: Generation of Distal Cyano-Substituted Alkyl Radicals and Their Functionalization, Synthesis, 2020, 52, 1585–1601 CrossRef CAS; (d) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Visible Light-Driven Radical-Mediated C–C Bond Cleavage/Functionalization in Organic Synthesis, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (e) W. Xiao and J. Wu, Recent advances for the photoinduced CC bond cleavage of cycloketone oximes, Chin. Chem. Lett., 2020, 31, 3083–3094 CrossRef CAS.
- Y. Gao, Y. Quan, Z. Li, L. Gao, Z. Zhang, X. Zou, R. Yan, Y. Qu and K. Guo, Organocatalytic Three-Component 1,2-Cyanoalkylacylation of Alkenes via Radical Relay, Org. Lett., 2021, 23, 183–189 CrossRef CAS PubMed.
- In our previous work (Org. Lett., 2021, 23, 183–189), we recorded two-component coupling reactions between a cycloalkane oxime and an aldehyde, and during preparation of this manuscript, an independent work employing cyclopentanone oxime as the substrate was disclosed. H.-B. Yang and D.-H. Wan, C–C Bond Acylation of Oxime Ethers via NHC Catalysis, Org. Lett., 2021, 23, 1049–1053 CrossRef CAS PubMed.
- M. M. D. Wilde and M. Gravel, Bis(amino)cyclopropenylidenes as Organocatalysts for Acyl Anion and Extended Umpolung Reactions, Angew. Chem., Int. Ed., 2013, 52, 12651–12654 CrossRef CAS PubMed.
- K. Yashiro, K. Sakata, I. Hachiya and M. Shimizu, Titanium tetraiodide induced cyclization of cyanoketones into 3-Aryl-1-Iodoisoquinolines, Heterocycles, 2016, 92, 2032–2046 CrossRef CAS.
- D. Leifert and A. Studer, The Persistent Radical Effect in Organic Synthesis, Angew. Chem., 2020, 59, 74–108 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qo01015h |
| This journal is © the Partner Organisations 2021 |