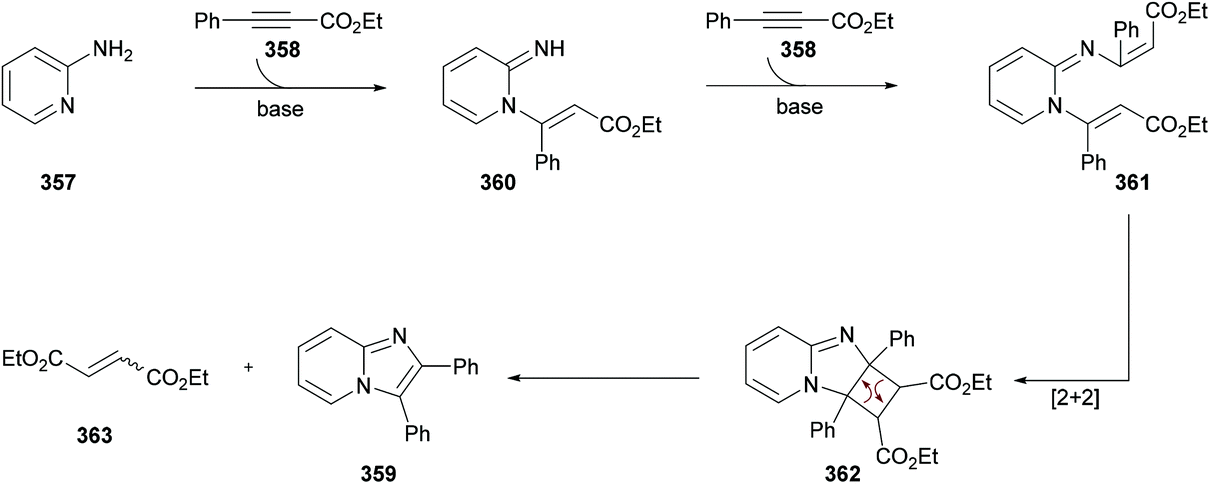
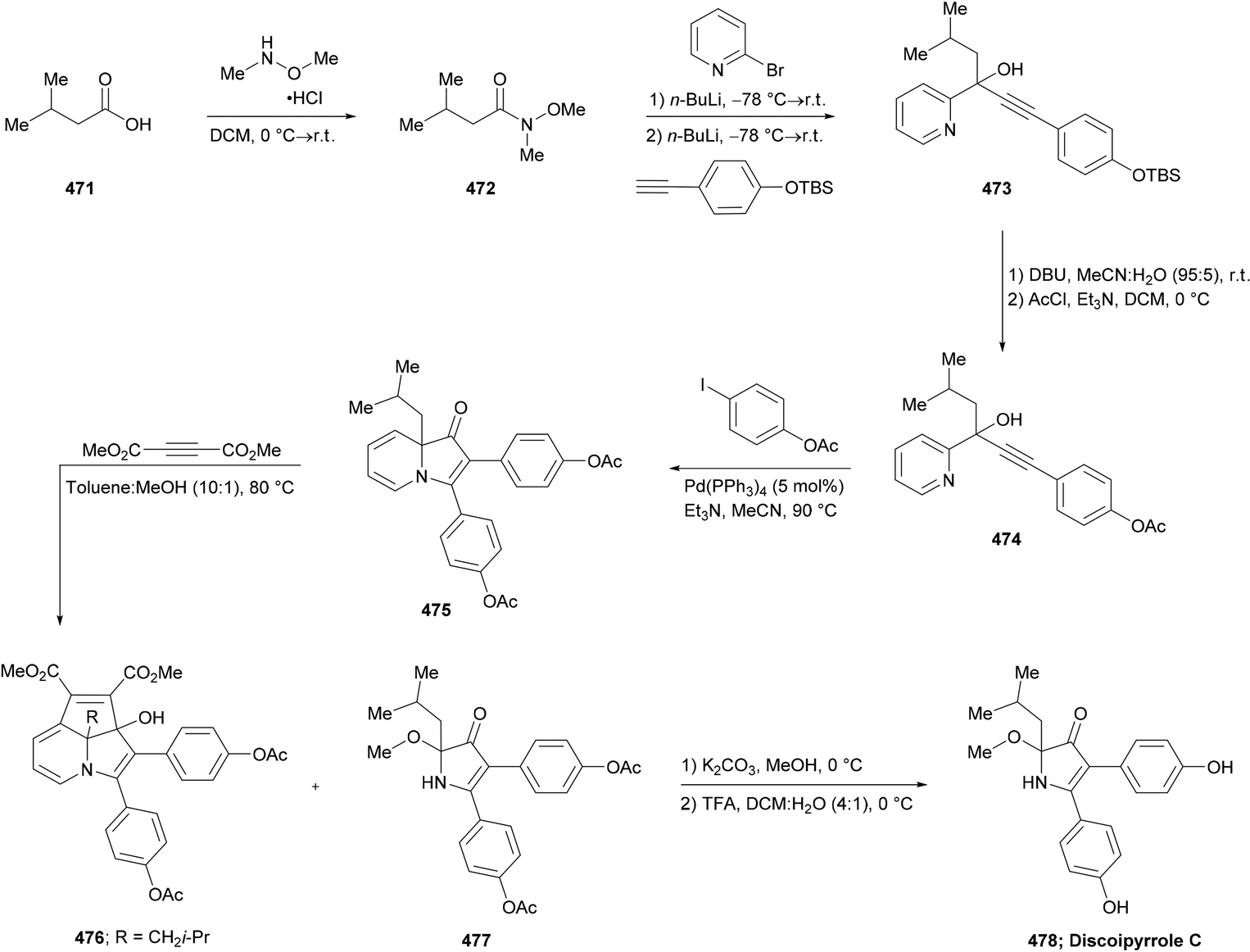
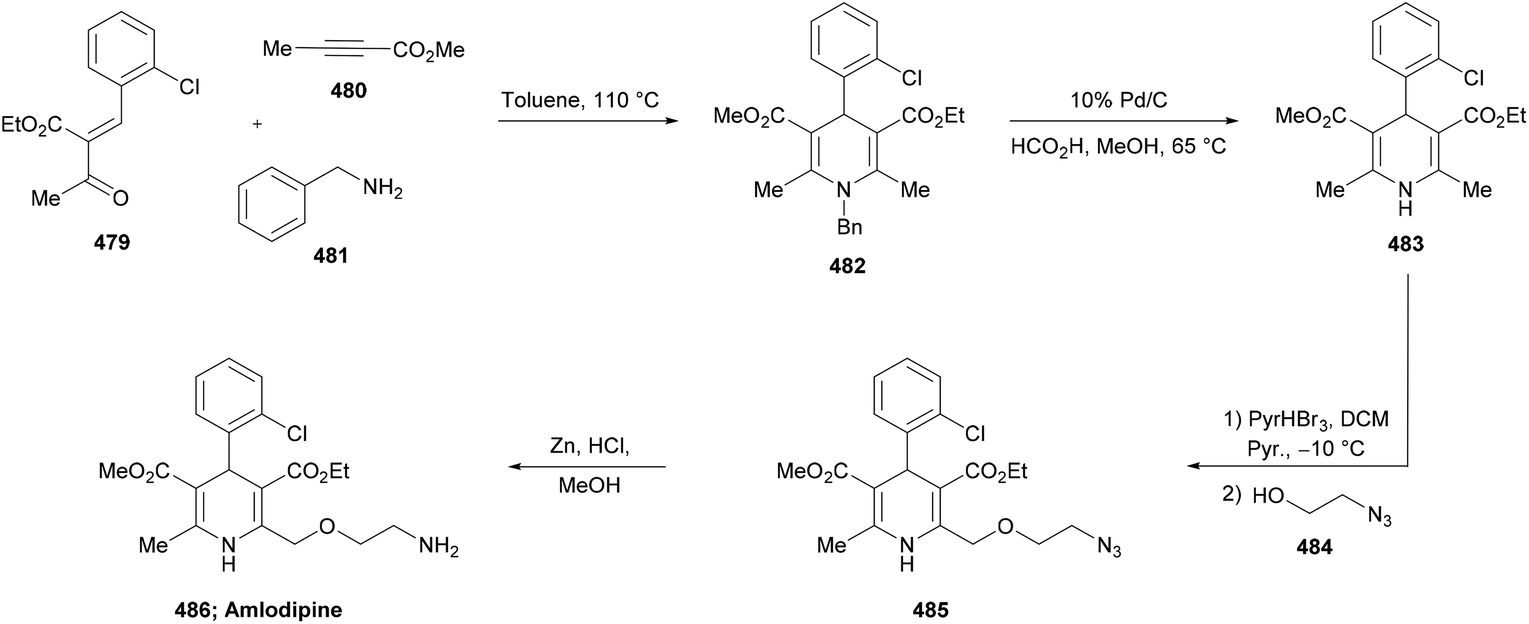
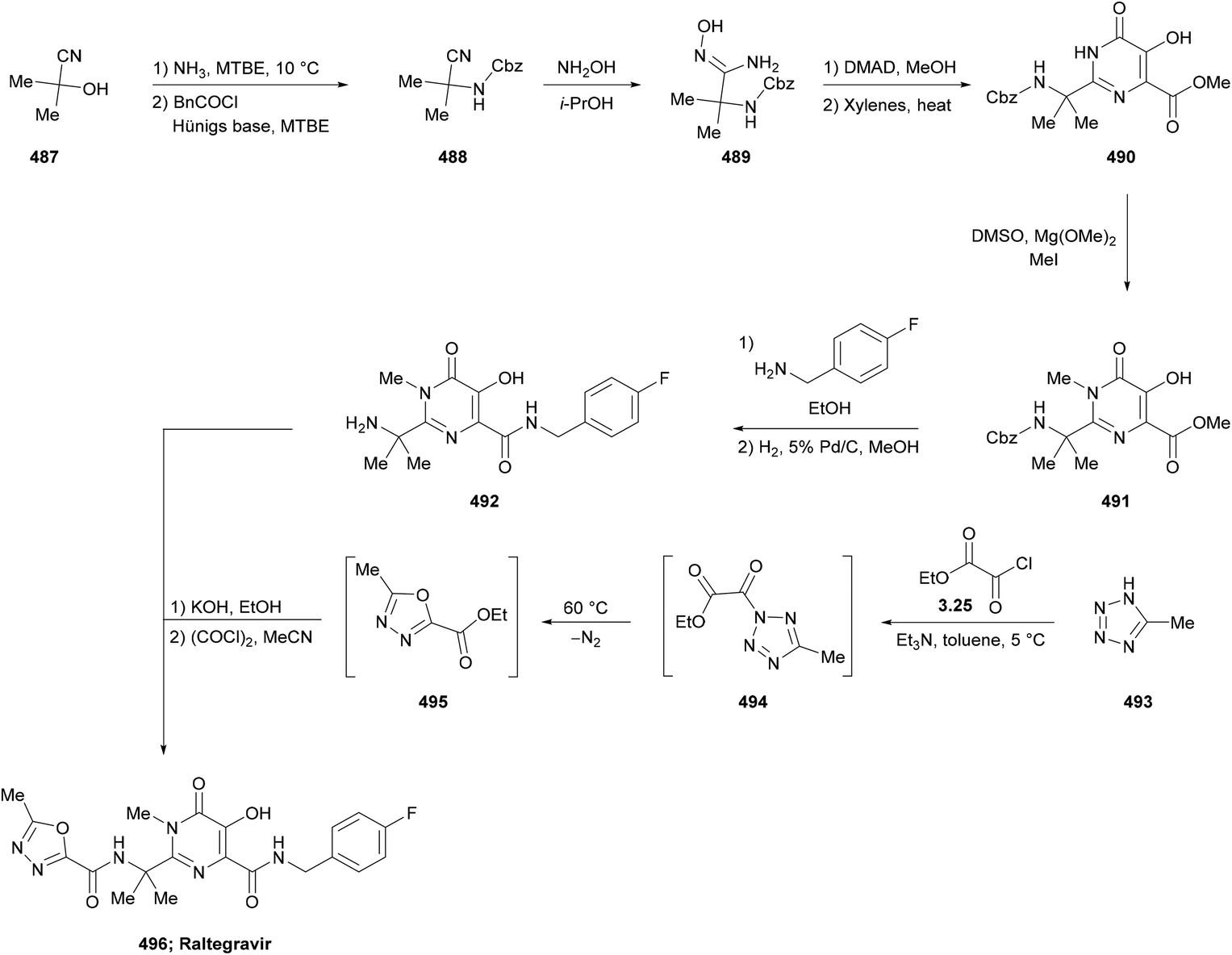
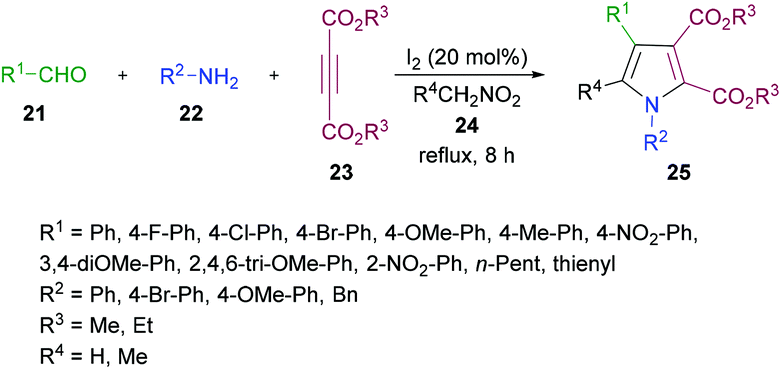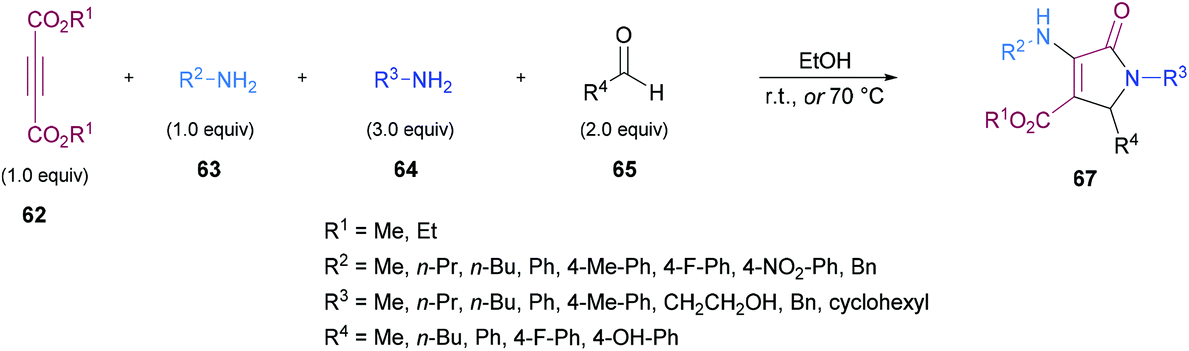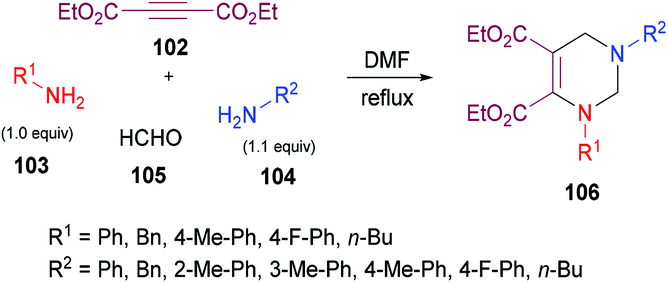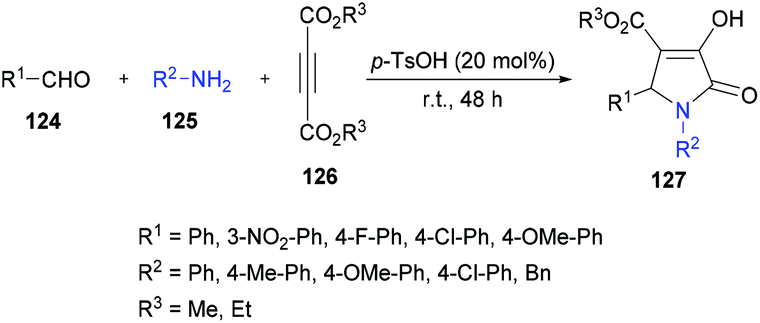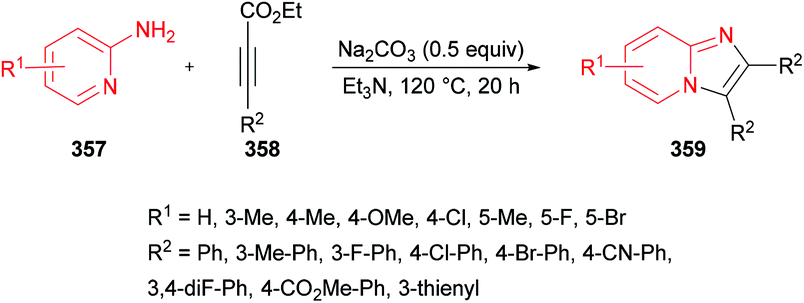DOI:
10.1039/D0QO00698J
(Review Article)
Org. Chem. Front., 2020,
7, 3734-3791
New frontiers in the transition-metal-free synthesis of heterocycles from alkynoates: an overview and current status
Received
13th June 2020
, Accepted 27th August 2020
First published on 22nd September 2020
Abstract
Heterocycles are among the well-established classes of compounds, constituting a wide range of organic molecules. These structural motifs not only have a dominant presence in a wide variety of drugs but are also equally present as ubiquitous fragments of numerous vitamins, biologically active natural products, biomolecules, and synthetic drug candidates. Their key role in medicinal chemistry in the design of safe, effective, and efficacious drug molecules gives an impetus to the scientific community to access heterocyclic entities from robust chemical reactions. In this review, we summarize a diverse range of synthetic methods employing alkynoates as key starting materials to furnish useful bioactive heterocyclic frameworks (nitrogen, oxygen, sulfur, fused- and hetero-spirocycles), thus offering new opportunities and expanding the toolbox of synthetic chemistry reactions under transition-metal-free conditions. Several reaction parameters such as the use of easily available starting materials, commercially accessible reagents, and ease of synthesis (moderate temperatures, short reaction times, high yields, limited by-products) are included in the discussion alongside the reaction scope and limitations as well as mechanistic insights. The application of these methodologies in the synthesis of natural products and pharmaceuticals has also been discussed. We hope that the diverse utilities of alkynoates in the delivery of various heterocycles will fulfil the objectives of medicinal chemists in overcoming the formidable challenges associated with drug discovery and development.
 Imtiaz Khan | Imtiaz Khan received his PhD degree from The University of Nottingham, United Kingdom under the direction of Professor Hon Wai Lam where he developed new synthetic methodologies involving C–H functionalization of inert bonds for the synthesis of carbo- and heterocycles. Later, he worked as a postdoctoral research associate in the same group for one year. He then spent two years (2016–2018) as a Leverhulme Trust research fellow at Cardiff University, before joining The University of Manchester, United Kingdom in 2018. He has a good contribution towards the scientific and academic community with numerous research publications in highly prestigious international journals and various presentations in both national and international conferences. He has published over 60 peer-reviewed articles and reviews with an impact factor of >250 that have been cited over 1800 times. He is serving as an editorial board member of several journals including Current Organic Chemistry, Current Organocatalysis, Mini-Reviews in Medicinal Chemistry, and Anti-Cancer Agents in Medicinal Chemistry. His research interests include the development of novel synthetic methodologies for the efficient synthesis of biologically active heterocycles and carbocycles, frustrated Lewis pair (FLP) chemistry and integrated chemo- and biocatalysis. |
 Sumera Zaib | Sumera Zaib received her PhD degree in biochemistry from Hazara University, Mansehra, Pakistan. She has also been a visiting DAAD research fellow at Bonn University, Germany. She is currently serving as an assistant professor in the Department of Biochemistry at the University of Central Punjab, Lahore, Pakistan. She has produced >80 research publications and reviews in international journals of high impact with an h-index of 20 and an impact factor of >250. She is also the recipient of the research productivity award from the Pakistan Council of Science & Technology (PCST) in recognition of her contribution to the scientific field. Her research interests include the identification of potent structural leads of biological importance. |
 Aliya Ibrar | Aliya Ibrar received her PhD degree in organic chemistry from Quaid-i-Azam University, Islamabad, Pakistan under the supervision of Prof. Dr Aamer Saeed, working on the design and synthesis of coumarinyl-based bioactive heterocycles. She is the recipient of the Indigenous 5000 Ph.D. Fellowship from the Higher Education Commission of Pakistan. She has also been a visiting research fellow at the University of Oxford (UK), working in the group of Prof. S. G. Davies. She has also served as an assistant professor of organic chemistry at the COMSATS University, Islamabad, Abbottabad campus, Pakistan, before joining the University of Haripur, Pakistan. Her current research interests include heterocyclic chemistry, focused mainly on the development of new synthetic methodologies, and medicinal chemistry with a particular interest in the discovery and development of bioactive five- and six-membered heterocycles. |
1. Introduction
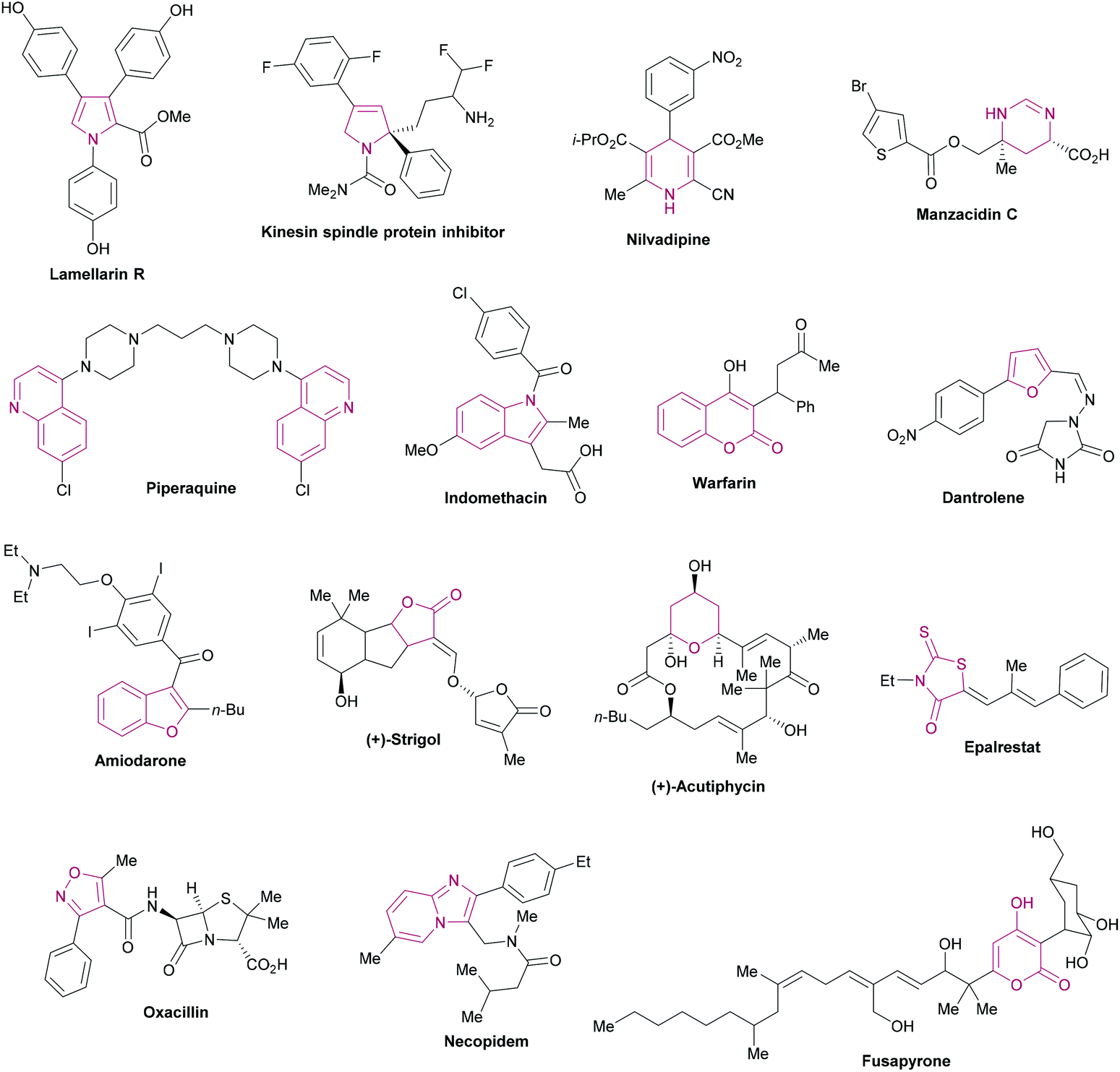
Heterocycles are ubiquitous fragments of numerous natural products, pharmaceuticals, designed bioactive drug candidates and agrochemicals.1,2 Their significant prevalence in drug candidates improves the solubility and reduces the lipophilic character of the drug molecules.3,4 In the drug discovery field, heterocyclic entities demonstrate a wide range of biological activities such as anti-inflammatory, antitumor, antidepressant, antibiotic, anti-HIV, antimalarial, antimicrobial, antiviral, and anticancer activities. In addition, these heterocycles also inhibit various enzymes (aldehyde/aldose reductase, α-glucosidase, α-amylase, cholinesterase, monoamine oxidase, urease, β-glucoronidase, alkaline phosphatase) for effective lead identification and the treatment of several chronic disorders.5–26 These heteroarenes are also well-recognized core structural units in organic chemistry2 and materials sciences.27Fig. 1 shows the representative examples of heterocycles reported in the literature and included in the current review/discussion.
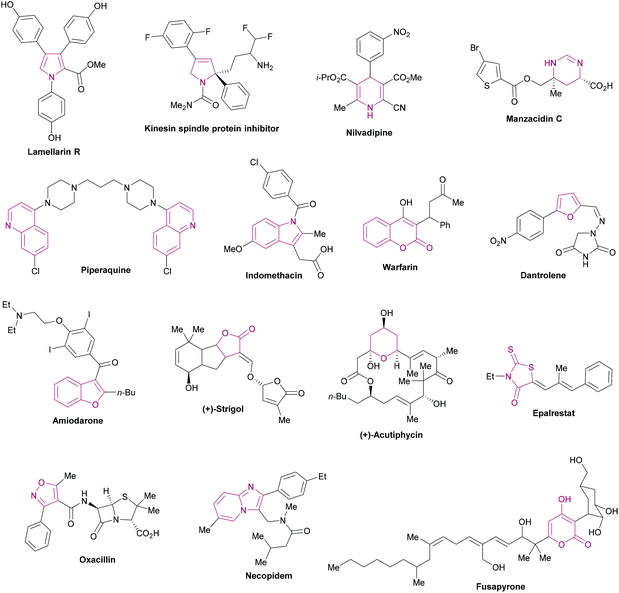 |
| | Fig. 1 Selected examples of heterocycles reported in the literature and included in the current review. | |
Recognizing the well-established significance of heterocycles in synthetic and medicinal chemistry as well as in the drug discovery arena, their easy, facile and practical synthesis remains a continuous challenge. In this pursuit, several efficient protocols have been established using transition-metal catalysts such as silver,28,29 gold,30–33 copper,34–36 cobalt,37 iron,38–40 palladium,41,42 ruthenium,43–45 mercury,46 and rare-earth metals.47 Several specific transformations such as cycloaddition reactions,48–52 cycloisomerization reactions,53,54 Sonogashira reaction,55,56 C–H bond functionalization processes,57–59 metathesis reactions60–62 and multi-component reactions63–65 have also been reported to produce heterocyclic structures. In addition, various starting materials such as azides,66–68 α-oxoesters,69 isocyanides,70–73 arylglyoxals,74 and diazo compounds75,76 have also been employed to deliver a range of heterocycles. Despite these fascinating developments, the generation of molecular/modular complexity in the pharmaceutical industry with good atom- and step-economy and broad functional group tolerance from readily available starting materials and commercial feedstock continues to be a driving force for synthetic chemists to develop sustainable methods. In addition, transition-metal-free reactions offer several advantages over transformations involving transition-metal catalysts. For instance, transition-metal-catalyzed protocols are often very sensitive to air and moisture and require very toxic and expensive catalysts and non-commercial supporting ligands. The removal of trace amounts of transition-metal residues from the desired products is quite costly and challenging while crucial, especially in the pharmaceutical industry. In many cases, special additives and cocatalysts are also critical to promote the efficiency and selectivity of transformations. Finally, the large consumption of transition-metal catalysts does not indeed meet the requirement of sustainable synthesis.77–80 Therefore, alternative approaches to form various bonds particularly in heterocyclic synthesis under transition-metal-free and generally practical and sustainable conditions are highly desirable obviating the need for metal catalysts, ligands, additives and cocatalysts.
In the present review, we summarize and showcase the utility of transition-metal-free approaches for the construction of a variety of heterocycles. These easy, practical and efficient synthetic methods involve the use of alkynoates as cheap, readily accessible and simple feedstock. Alkynoates are significantly important, diverse and powerful building blocks in organic chemistry due to their unique and inherent properties such as the electronic bias on the carbon–carbon triple bond posed by electron-withdrawing groups. A range of chemical reactions could be exploited involving the reactivity of zwitterions derived from activated acetylenic esters with nucleophilic trivalent phosphines such as Wittig reactions,81 annulations,82 and cycloadditions.83 In addition, the reaction between trivalent phosphorus compounds and acetylenic esters in the presence of organic acids such as CH acids, NH acids, OH acids, or SH acids to give a vinyl trivalent phosphonium cation as the important intermediate has also been studied extensively.84 Other carbon-, nitrogen-, or sulfur-based nucleophiles could be employed as Michael donors while alkynoates act as Michael acceptors. Alkynoates can also undergo isomerization into the corresponding allenes under basic conditions which could be utilized in conjugate addition. Meanwhile, the unusual reactivity of the isocyanide functionality with alkynoates (DMAD) offers opportunities for the generation of complex and diverse drug-like small heterocyclic compounds using the multi-component reaction (MCR) methodology.85 Thus, this review will consider the use of the triple bond directly attached to an electron-withdrawing ester group and its transformation into double or single bonds. The mechanistic details of the selective transformations as well as the post-synthetic manipulations (Suzuki–Miyaura cross-coupling, Buchwald–Hartwig amination, oxidation and hydrogenolysis reactions among many others) have also been included. The robust use of these methods in the synthesis of natural products, pharmaceuticals and bioactive drug candidates has also been discussed. Selected examples of heterocycles accessed by using DMAD and DEAD as coupling units have been reviewed previously;86–92 thus they have not been discussed exhaustively here and are excluded from the scope of this review except a few representative examples demonstrating the diversity of transformations.
2. Synthetic methods
2.1. Synthesis of nitrogen-containing heterocycles
Nitrogen-containing heteroaromatic skeletons are well-recognized structural motifs of numerous pharmaceuticals, natural products and bioactive drug molecules. These heterocyclic entities also find several useful applications in organic dyes for solar cells and as important building units in organic chemistry.93 Therefore, environmentally benign and green synthetic approaches to access these N-heterocycles need unlocking under mild reaction conditions.
2.1.1. Synthesis of pyrroles.
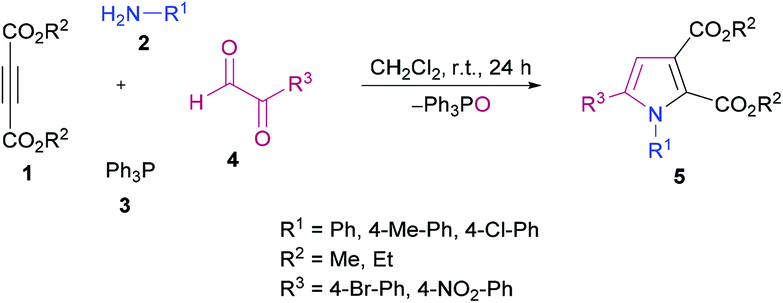
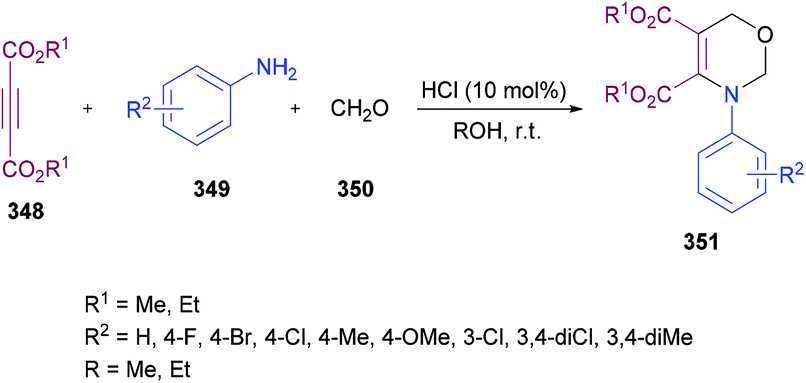
Anary-Abbasinejad and co-workers94 developed a facile one-pot multi-component approach to access highly functionalized pyrroles 5 (Scheme 1). The reaction was performed using dialkyl acetylenedicarboxylates 1, aromatic amines 2, triphenylphosphine 3, and arylglyoxals 4 in dichloromethane at room temperature. Various amines and arylglyoxals were tested to broaden the reaction scope and the anticipated products were furnished in good yields.
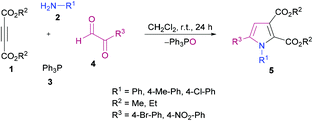 |
| | Scheme 1 Synthesis of highly functionalized pyrroles via a multi-component approach. | |
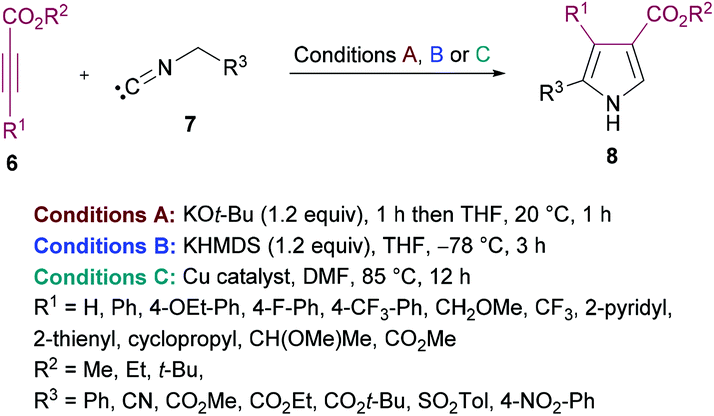
de Meijere and co-workers95 reported a formal cycloaddition of α-metallated methyl isocyanides 7 and electron-deficient acetylenes 6 to access a range of oligosubstituted pyrroles 8. The corresponding products were isolated in 25–97% yields (Scheme 2). Three different procedures have been developed. Initially, potassium tert-butoxide was employed (method A) which was replaced with potassium bis(trimethylsilyl)amide (KHMDS) added simultaneously with the corresponding acetylene (method B). Lastly, the reactions were performed using a copper catalyst. Several copper sources were employed; however, CuSPh remained the optimal. In all the copper catalysts, the σ-donating character of the counterion was the common property. Using all the three methods, various isocyanides and electron-deficient alkynes were successfully cyclized into the corresponding products.
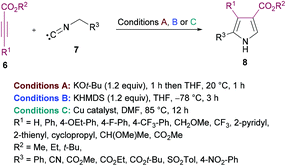 |
| | Scheme 2 Synthesis of oligosubstituted pyrroles from substituted methyl isocyanides and acetylenes. | |
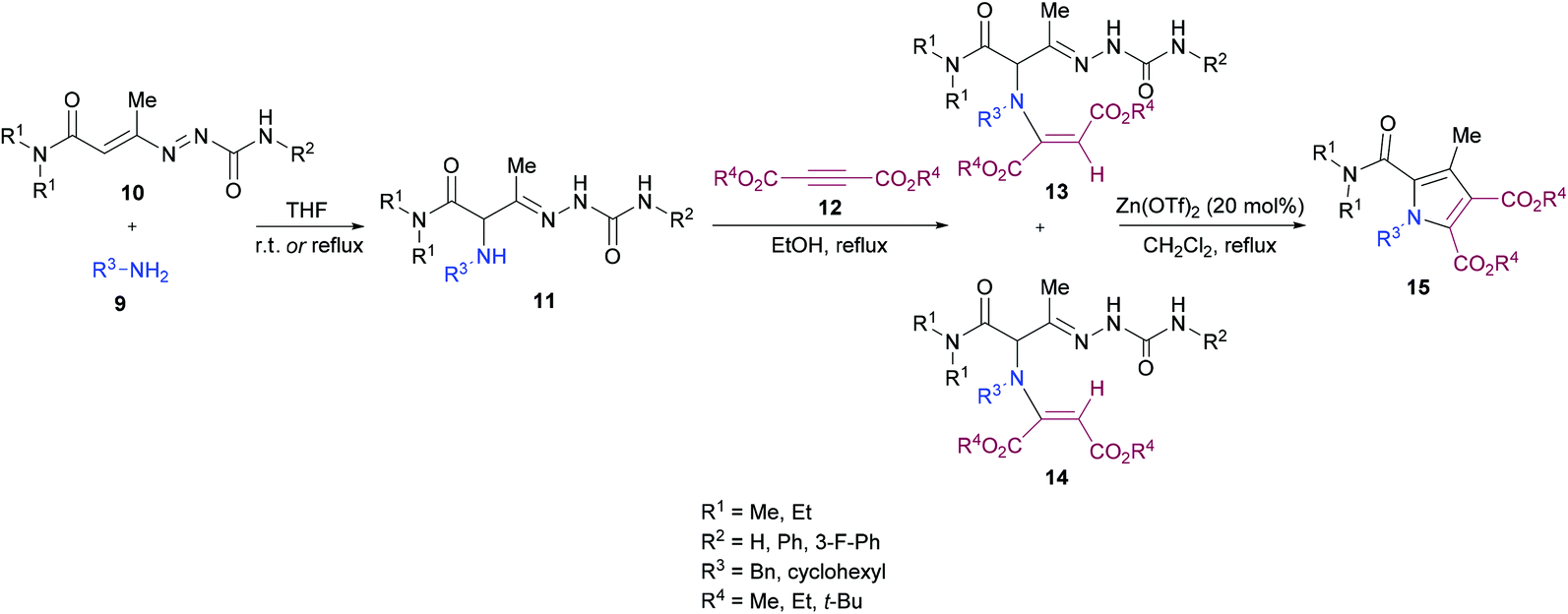
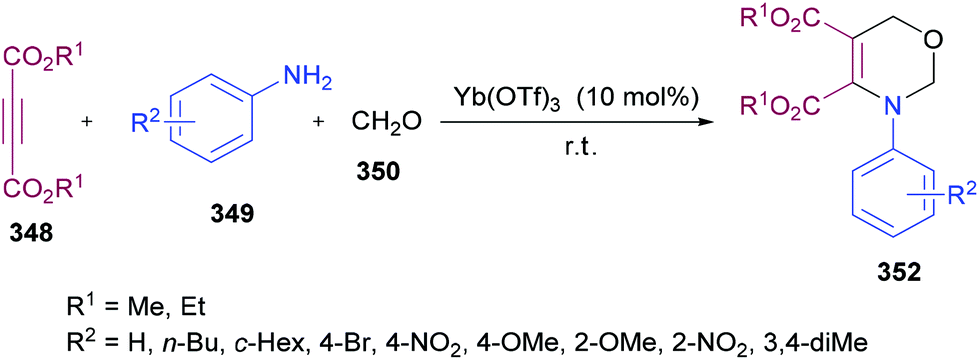
Mantellini and co-workers96 established a facile Lewis acid-catalyzed approach for the construction of highly functionalized pyrroles 15 from α-aminohydrazones 11 and dialkyl acetylenedicarboxylates 12 (Scheme 3). α-Aminohydrazones were in turn synthesized from primary amines 9 and 1,2-diaza-1,3-butadienes 10via Michael addition. Initially, various Lewis/Brønsted acid catalysts were screened and the results revealed that Zn(OTf)2 alongside ZnCl2, Zn(TfO)2, In(TfO)3, Yb(TfO)3, InCl3 and InBr3 produces the best conversions; however, in the case of Zn(OTf)2, the reaction time was 0.5 h. Using this catalyst, various pyrrole derivatives were synthesized. This methodology was expedient due to the use of simple and easily accessible starting materials like 1,2-diaza-1,3-butadienes, used as building units in the construction of aza-heterocycles, as well as the mild experimental conditions.
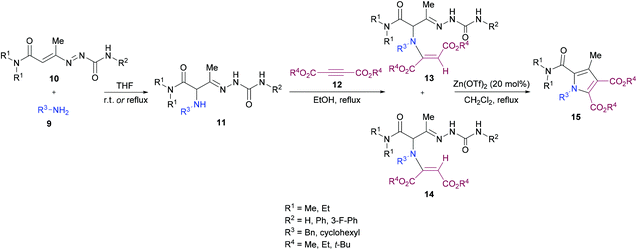 |
| | Scheme 3 Lewis acid-catalyzed synthesis of functionalized pyrroles. | |
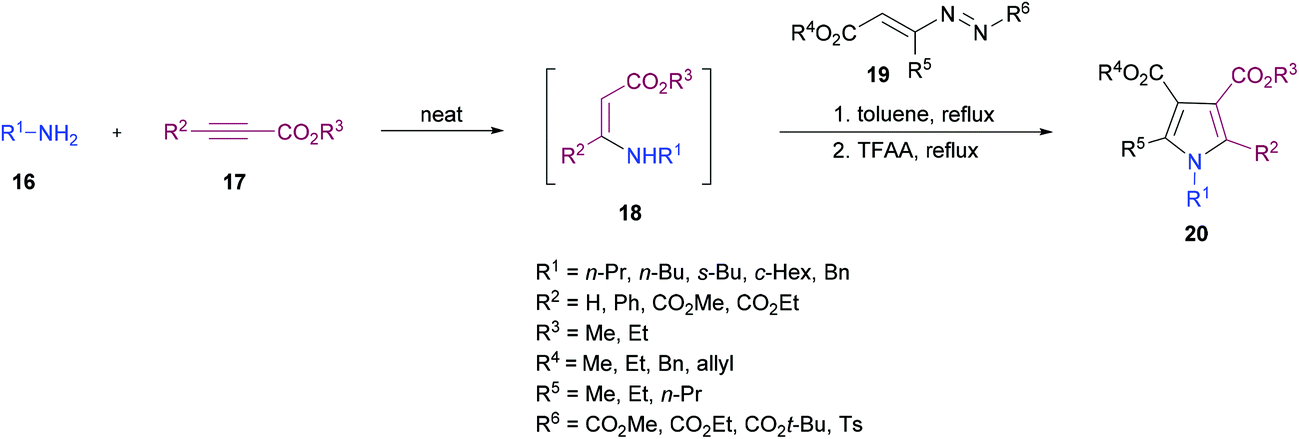
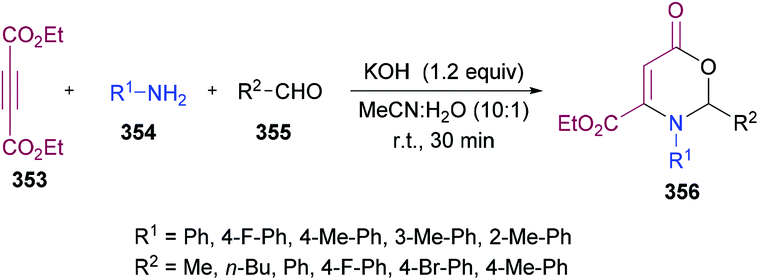
Favi and co-workers97 introduced a new acid-catalyzed sequential three-component protocol for the rapid assembly of functionalized pyrroles 20 from amines 16, alkynoates 17 and 1,2-diaza-1,3-dienes 19 (Scheme 4). The reaction proceeds in a regioselective manner with the formation of an enamino ester intermediate followed by Michael addition and subsequent azaheterocyclization. After extensive screening of reaction parameters, suitable conditions were identified. The substrate scope was examined using a model system. Various amines, alkynoates and 1,2-diaza-1,3-dienes were tolerated to afford the desired pyrrole derivatives. In the case of aliphatic amines, the acid-promoted enamine–azoene cyclization failed due to the reduced nucleophilicity of the amine nitrogen atom. Moreover, DMAD, DEAD, ethyl propiolate and ethyl 3-phenylprop-2-ynoate were successfully coupled delivering the anticipated products in moderate to good yields. The reaction conditions also tolerated various 1,2-diaza-1,3-dienes with a variety of different functional groups.
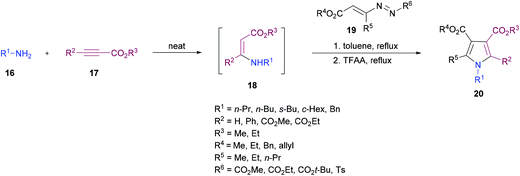 |
| | Scheme 4 Acid-catalyzed sequential three-component synthesis of substituted pyrroles from amines, alkynoates and 1,2-diaza-1,3-dienes. | |
In another study, Das and co-workers98 demonstrated the synthesis of polysubstituted pyrrole derivatives 25 from an iodine-catalyzed one-pot four-component process involving aldehydes 21, amines 22, dialkyl acetylenedicarboxylates 23 and nitromethane 24. The corresponding tetrasubstituted pyrroles were isolated in good yields (Scheme 5). Using 20 mol% of molecular iodine, the scope of pyrrole formation was examined. A range of aldehydes, amines, and dialkyl acetylenedicarboxylates were tested. Both aromatic and heteroaromatic aldehydes with various functional groups (electron-rich, electron-deficient) participated well in the reaction. However, aliphatic aldehydes remained reluctant to produce the corresponding pyrroles.
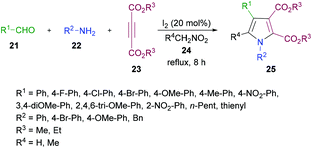 |
| | Scheme 5 Iodine-catalyzed four-component synthesis of tetrasubstituted pyrroles from aldehydes, amines, alkynoates and nitromethane. | |
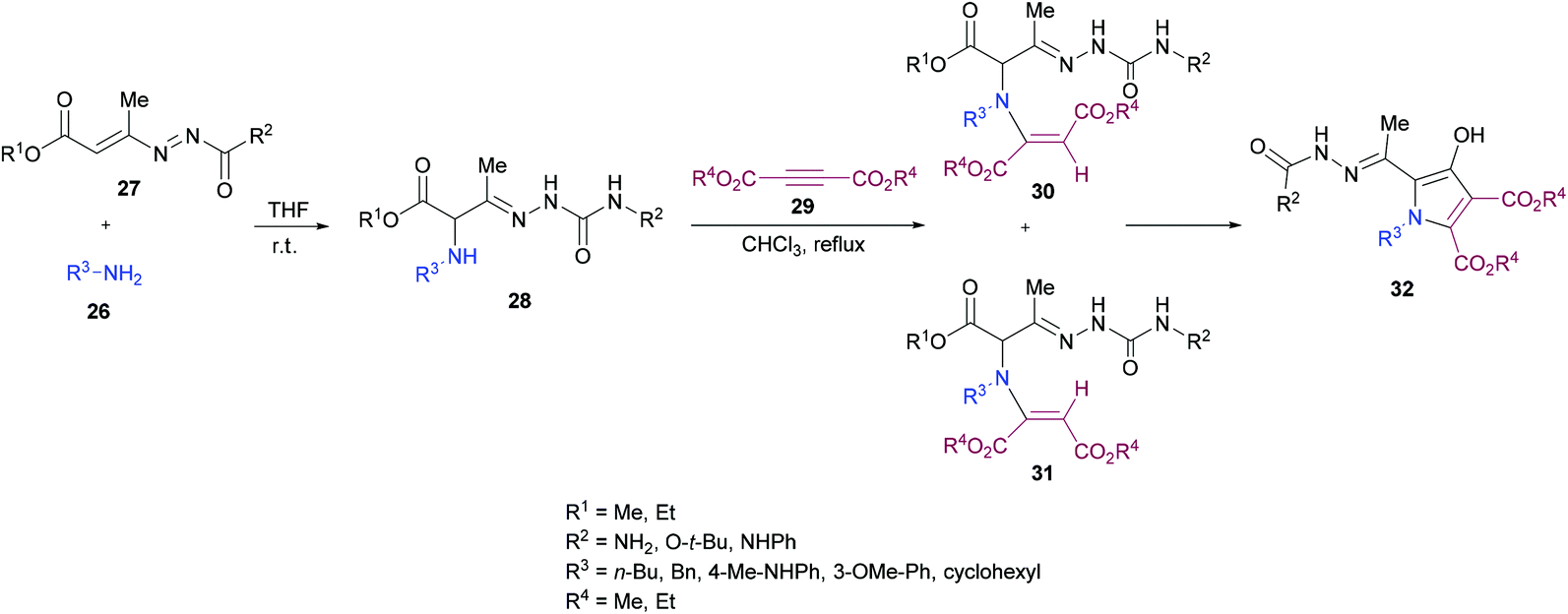
Building on their previous work,96 Mantellini and co-workers99 demonstrated the synthesis of pyrrole derivatives 32 (Scheme 6). The reaction sequence involves the generation of α-aminohydrazones 28 by Michael addition of primary amines 26 to 1,2-diaza-1,3-dienes 27 followed by the coupling with dialkyl acetylenedicarboxylates 29 to produce α-(N-enamino)-hydrazones 30, 31 which were cyclized to the corresponding pyrroles 32. α-Amino ketones were replaced with α-aminohydrazones in this methodology. Among the different Lewis acids screened for this transformation, Zn(OTf)2 (20 mol%) produced the best results in dichloromethane under reflux. In general, this method was widely applicable for the construction of more complex systems using mild conditions from stable and easily accessible building blocks.
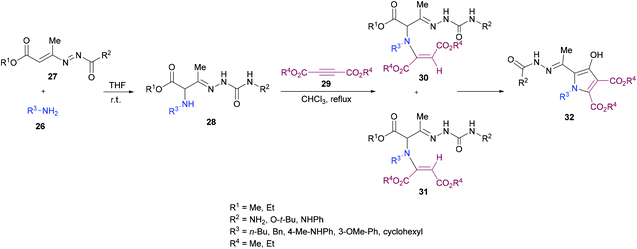 |
| | Scheme 6 Zinc(II) triflate-catalyzed divergent synthesis of polyfunctionalized pyrroles. | |
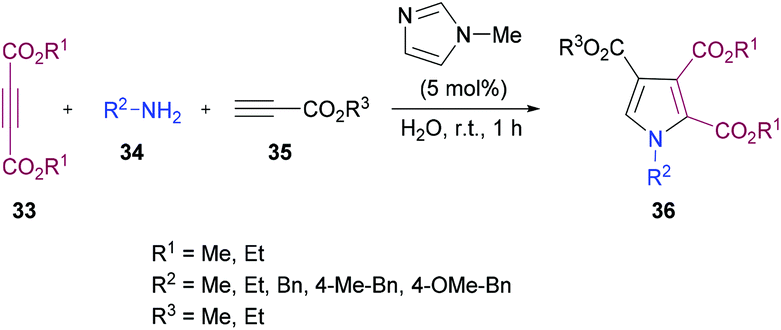
Sayyed-Alangi and co-workers100 synthesized a range of functionalized pyrroles 36 from a one-pot multi-component methodology using dialkyl acetylenedicarboxylates 33, primary amines 34 and propiolates 35 in the presence of N-methylimidazole in water at room temperature (Scheme 7). The products were furnished in good yields. The easy availability of the starting materials and their cyclization to pyrrole compounds in water were the key features of this procedure.
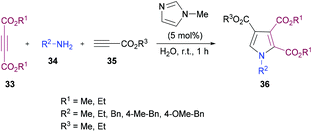 |
| | Scheme 7 Synthesis of highly functionalized pyrroles from primary amines and activated acetylenes in water. | |
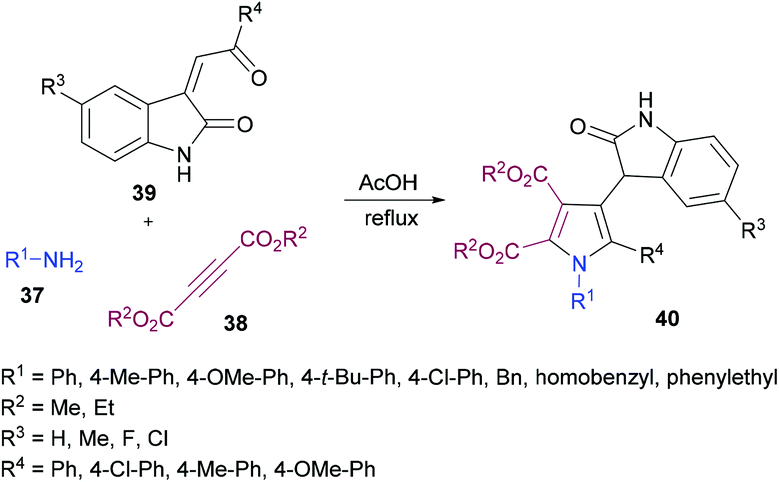
Yan and co-workers101 also utilized a one-pot domino methodology to prepare pentasubstituted pyrroles 40 from arylamines 37, acetylenedicarboxylates 38, and 3-phenacylideneoxindoles 39 (Scheme 8). Initially, p-methylaniline was treated with dimethyl acetylenedicarboxylate to furnish the β-enamino ester which was coupled with 3-p-chlorophenacylideneoxindole in refluxing acetic acid to afford the corresponding pentasubstituted pyrrole in 74% yield. Later, the scope and versatility of the domino reaction were explored with differently substituted arylamines. Benzylamine, α-phenylethylamine and β-phenylamines were used. In addition, 3-phenacylideneoxindoles incorporating both electron-donating (methyl, methoxy) and inductively electron-withdrawing (chloro) substituents participated efficiently under the optimized set of reaction conditions. All the reactions provided smooth cyclization to nitrogen heterocycles in good yields. It is interesting to note that isatinyl substituted products obtained using this method are unusual because under normal circumstances the reaction of isatin and 3-isatinylidene compounds usually provides spirocyclic oxindoles.102
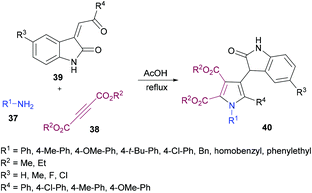 |
| | Scheme 8 One-pot synthesis of tetrasubstituted pyrroles from arylamines, acetylenedicarboxylates and 3-phenacylideneoxindoles. | |
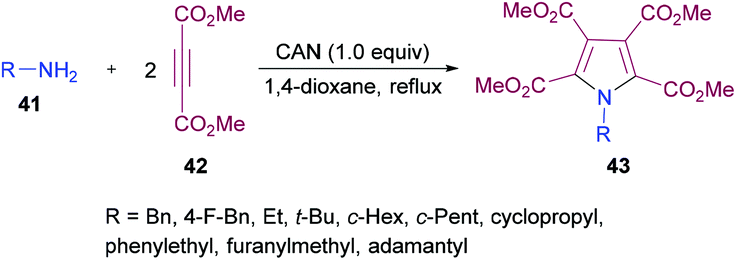
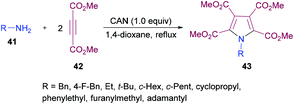
Madabhushi and co-workers103 reported a facile approach for the preparation of fully functionalized pyrrole derivatives 43 from amine 41 and dimethyl acetylenedicarboxylate 42 using ceric(IV) ammonium nitrate (Scheme 9). The cyclocondensation of two units of DMAD and one unit of primary amine resulted in the formation of a symmetrical pentasubstituted pyrrole. The best results were obtained in 1,4-dioxane using 1.0 equivalent of CAN. A diverse variety of primary amines (benzylamine, α-methyl benzylamine, ethylamine, t-butylamine, cyclopropylamine, cyclopentylamine, cyclohexylamine, adamantylamine) were successfully coupled to form the corresponding N-alkyl pyrrole compounds in generally good yields.
 |
| | Scheme 9 CAN-mediated synthesis of pentasubstituted pyrroles from dimethyl acetylenedicarboxylates and amines. | |
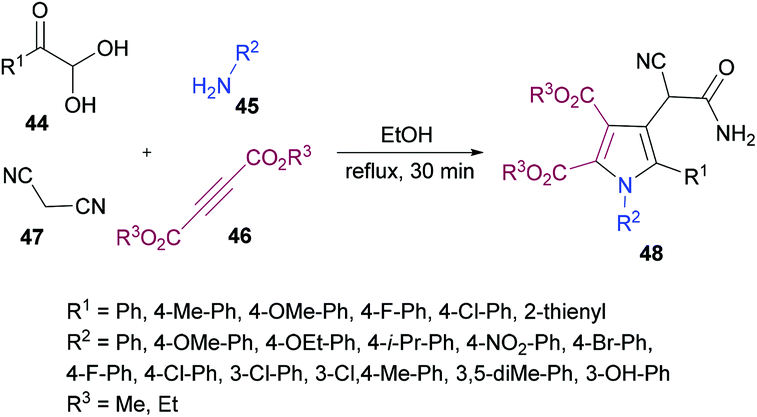
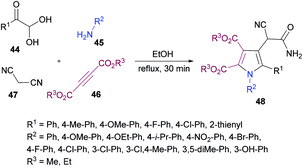
Huang, Shi and co-workers104 described a highly efficient metal-free multi-component domino procedure for the construction of multisubstituted pyrroles 48 from the reaction of arylglyoxal monohydrate 44, aniline 45, alkynoate 46, and malononitrile 47 (Scheme 10). Various reaction parameters were screened to obtain the optimal conditions. The results indicated that using equimolar quantities of all the starting materials, the desired product was isolated in 81% yield in ethanol under reflux conditions. Under the optimal conditions, the substrate scope was investigated. Various substituents (methoxy, methyl, fluoro, chloro) on the aryl ring of glyoxal as well as heteroarylglyoxal worked well alongside aliphatic substituents. Similarly, anilines containing electron-rich and electron-deficient substituents were also well endured and the corresponding pyrrole products were obtained in good yields. However, the reaction showed no progress when methyl (or ethyl) 2-cyanoacetate was employed with phenylglyoxal monohydrate, 4-methylaniline, and dimethyl but-2-ynedioate under the optimized reaction conditions.
 |
| | Scheme 10 Four-component domino synthesis of polysubstituted pyrroles. | |
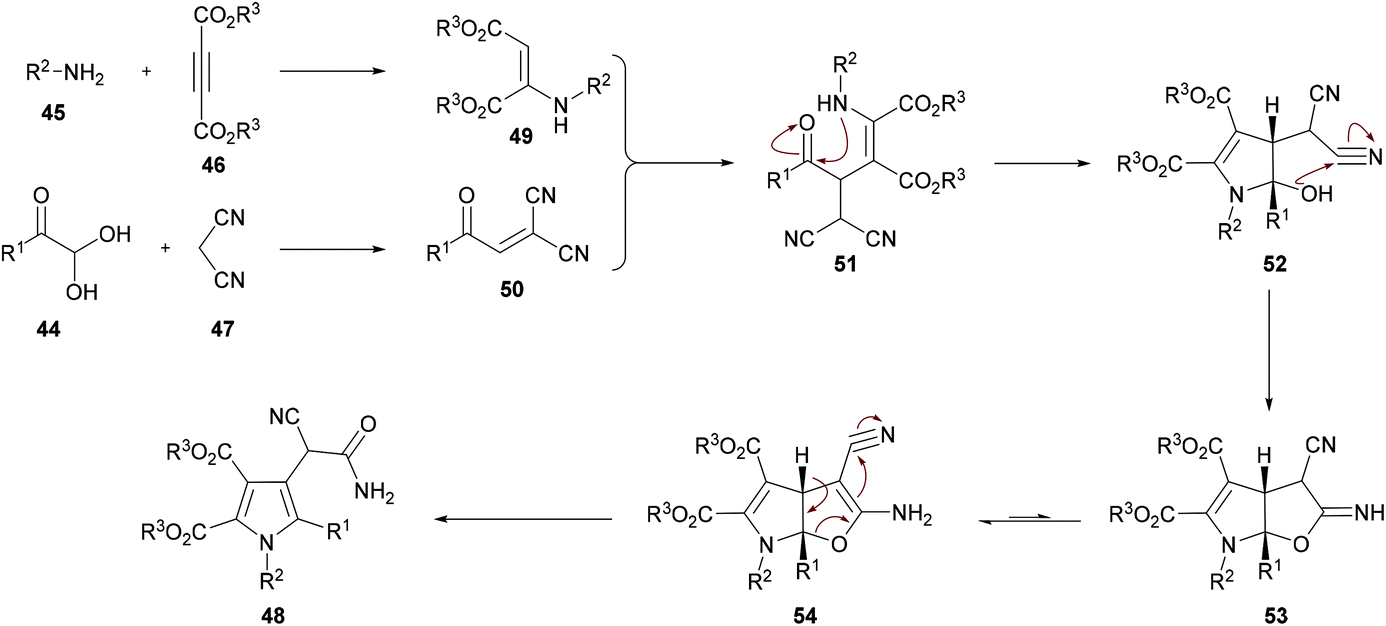
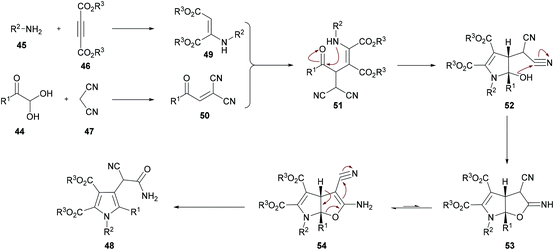
Based on the literature reports (Shu et al., 2013)105 and the results obtained from this domino process, the proposed mechanism is illustrated in Scheme 11 which starts with the addition of aniline 45 to alkyne 46 to generate intermediate 49. Simultaneously, Knoevenagel condensation of arylglyoxal monohydrate 44 and malononitrile 45 produced intermediate 50. Michael addition of β-enamino ester 49 to intermediate 50 afforded 51, which was cyclized via intramolecular nucleophilic addition to deliver intermediate 52. Another intramolecular cyclization of 52 produced 53, which tautomerized to the more stable intermediate 54. Finally, a ring-opening reaction of intermediate 54 furnished the polysubstituted pyrrole 48. Furthermore, density functional theory (DFT) calculations were performed at the B3LYP/6-31G level of theory for intermediates 53 and 54 and the results suggested that intermediate 54 could be easily transformed into the more stable pyrrole product 48.
 |
| | Scheme 11 Proposed mechanism for the formation of highly substituted pyrroles 48. | |
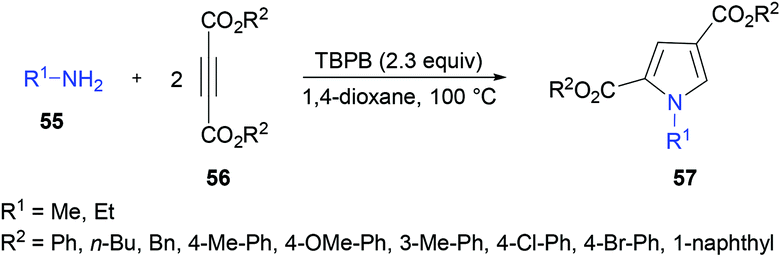
Liu and co-workers106 described a tandem cyclization for the construction of heterocycles through the formation of new carbon–carbon and carbon–nitrogen bonds (Scheme 12). 1,2,4-Trisubstituted 1H-pyrroles 57 were synthesized from amines 55 and alkynoates 56 in the presence of tert-butyl perbenzoate (TBPB). A model reaction was used to develop the optimized reaction parameters using dimethyl but-2-ynedioate and p-toluidine in dioxane at 100 °C in the presence of TBHP. Dimethyl 1-p-tolyl-1H-pyrrole-2,4-dicarboxylate was obtained in 92% yield when 2.3 equivalents of TBHP was employed in dioxane. Using these conditions, the substrate scope was investigated. A diverse range of alkynoates and amines were coupled to generate heterocyclic products. The presence of electronically different substituents on the amine ring imparted a significant effect on the yield of the corresponding product. Substrates bearing electron-rich groups provided superior results whereas those bearing strongly electron-deficient (NO2) groups did not show reactivity. Aliphatic amines also successfully delivered the corresponding pyrroles in excellent yields. On the other hand, diethyl but-2-ynedioate produced the desired pyrrole in 79% isolated yield. However, other alkynoates such as ethyl but-2-ynoate, ethyl 3-phenylpropiolate, and methyl oct-2-ynoate did not produce any product under the optimized reaction conditions.
 |
| | Scheme 12 TBPB-mediated synthesis of trisubstituted pyrroles from alkynoates and amines. | |
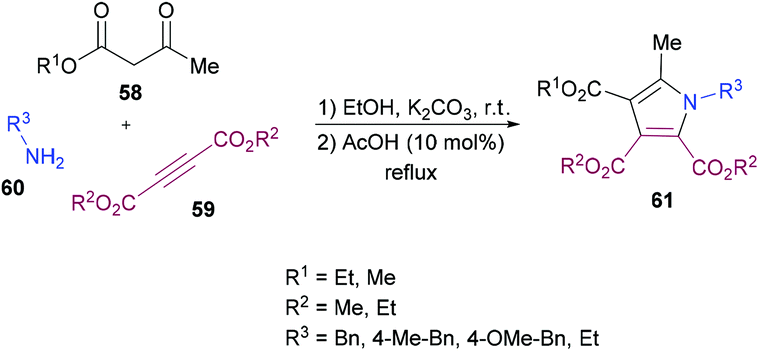
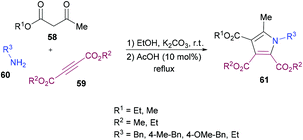
Mehrabi and co-workers107 developed a metal-free one-pot two-step synthetic protocol involving alkyl acetoacetates 58, dialkyl acetylenedicarboxylates 59 and amines 60 to access a range of pentasubstituted pyrroles 61 (Scheme 13). The reaction sequence produces 4-hydroxypenta-1,3-diene-tricarboxylates in the first step followed by cyclization with amines. The heterocyclic products were isolated in excellent yields. The reaction parameters were mainly optimized for the second step where the solvent, temperature, time, and amount of acid were screened. Although 4-hydroxypenta-1,3-diene-tricarboxylates underwent cyclization successfully at room temperature, a slight increase in the yield was noticed under reflux conditions. With an optimized set of reaction conditions, the possibility of using various starting materials to produce functionalized pyrroles was explored. The results indicated that the reactions proceeded smoothly to furnish the pyrrole product in good to excellent yields. Aromatic amines bearing different substituents as well as alkyl amines were successfully coupled to 4-hydroxypenta-1,3-diene-tricarboxylates which were generated from various alkyl acetoacetates and dialkyl acetylenedicarboxylates.
 |
| | Scheme 13 Synthesis of pentasubstituted pyrroles from alkyl acetoacetates, dialkyl acetylenedicarboxylates and amines. | |
2.1.2. Synthesis of dihydropyrroles.
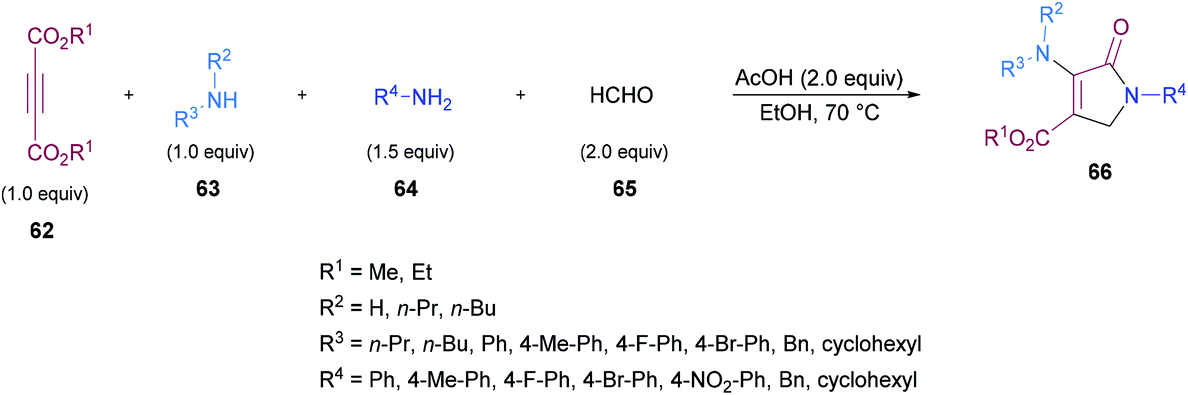
Jiang and co-workers108 demonstrated a multi-component methodology to construct tetra- and pentasubstituted dihydropyrroles 66, 67 in good yields (Scheme 14). This process involves but-2-ynedioates 62, amines 63, 64, and aldehydes 65 as starting materials. Various parameters including solvents, additives, reactant ratios, and temperature were investigated for the identification of the best reaction conditions. Using different starting materials, a range of tetrasubstituted dihydropyrroles were synthesized, no matter whether the same or different primary or secondary amines of aliphatic or aromatic nature were employed. Different substituents (methyl, fluoro, bromo) at the aryl ring were successfully tolerated.
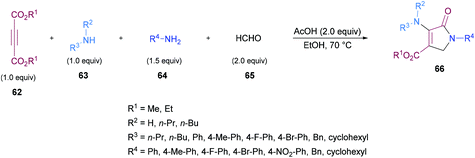 |
| | Scheme 14 Multi-component synthesis of polysubstituted dihydropyrroles. | |
Similarly, some efforts were made to optimize the reaction conditions to produce pentasubstituted dihydropyrroles 67 using alkynoates 62, amines 63, 64 and aldehydes 65; however, the order of the addition of reactants was changed. Again, the compatibility of different functional groups was tested and the corresponding dihydropyrrole products were isolated in good to excellent yields (Scheme 15).
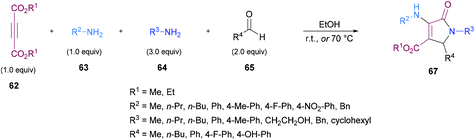 |
| | Scheme 15 Synthesis of multisubstituted dihydropyrroles from alkynoates, amines and aldehydes. | |
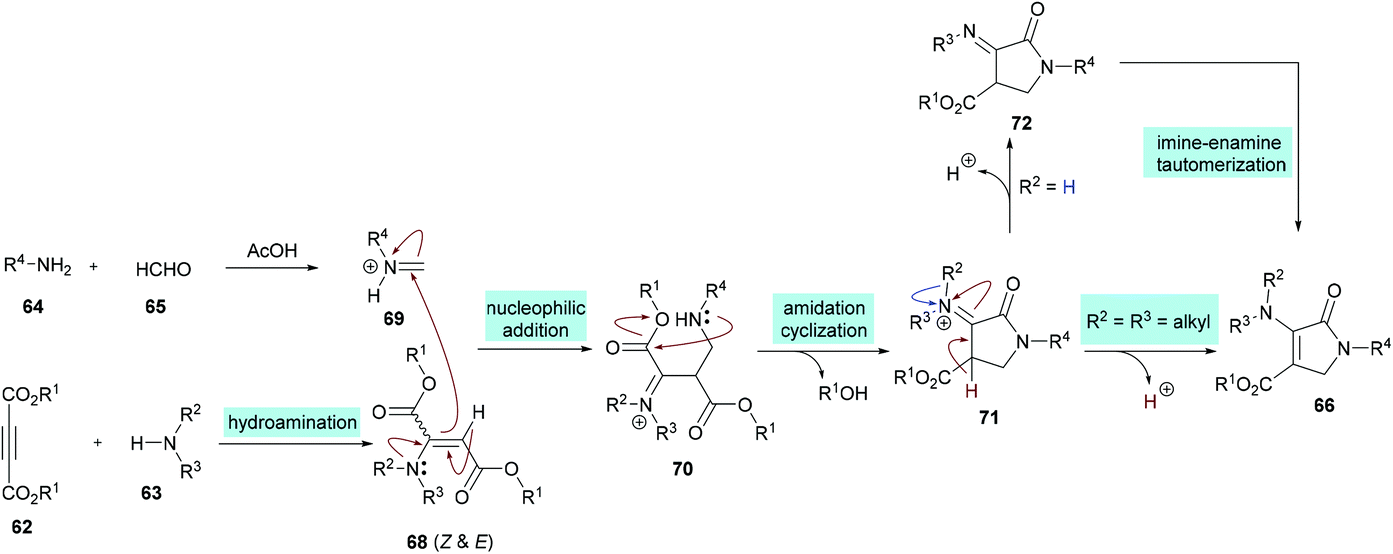
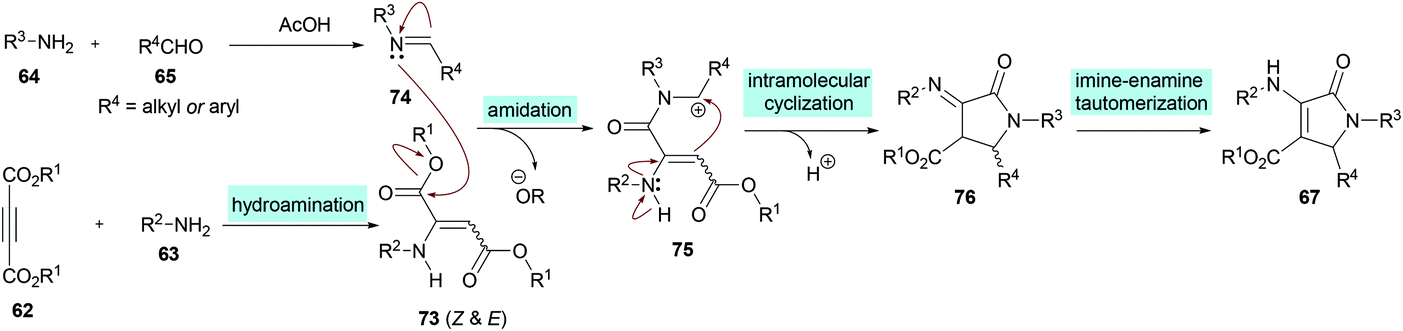
Two different mechanistic approaches were considered for the formation of tetrasubstituted and polysubstituted dihydropyrroles. The first one comprises the domino hydroamination/nucleophilic addition/amidation–cyclization sequence (Scheme 16), whereas the second one involves the hydroamination/amidation/intramolecular cyclization/imineenamine tautomerization sequence (Scheme 17).
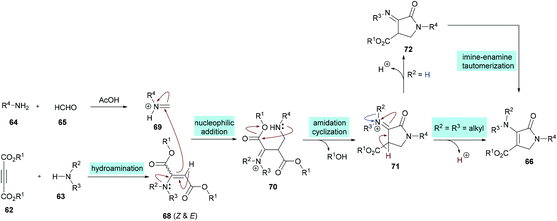 |
| | Scheme 16 Proposed mechanism for the formation of polysubstituted dihydropyrroles 66. | |
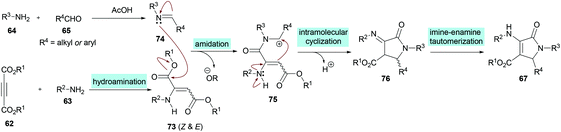 |
| | Scheme 17 Proposed mechanism for the formation of polysubstituted dihydropyrroles 67. | |
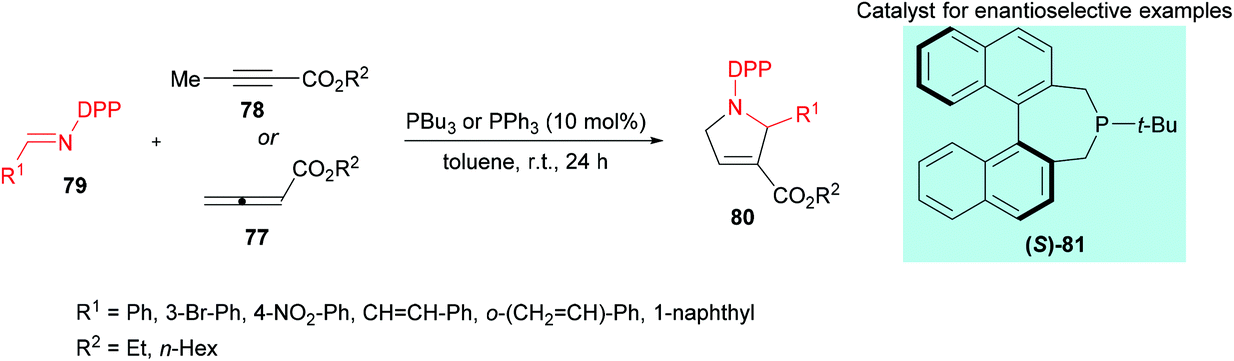
Marinetti and co-workers109 investigated a [3 + 2] cyclization reaction between allenoates 77 or 2-butynoates 78 and imines 79 using a phosphine catalyst to deliver pyrrolines 80 (Scheme 18). The effect of imine protecting groups on the enantioselectivity was explored by comparing N-Ts- and N-DPP-imines. Under the optimized reaction conditions, good conversions were obtained when N-DPP-imines were used. In addition, the use of a DPP protecting group remains advantageous over the Ts group due to its easy deprotection. In the asymmetric version, good yields and enantioselectivities were observed using the (S)-81 catalyst.
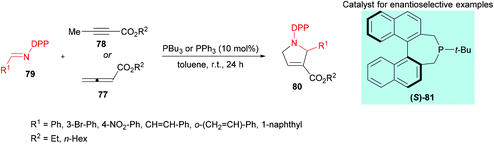 |
| | Scheme 18 Phosphine-catalyzed synthesis of pyrrolines from N-DPP-imines and allenoates/2-butynoates. | |
2.1.3. Synthesis of dihydropyridines.
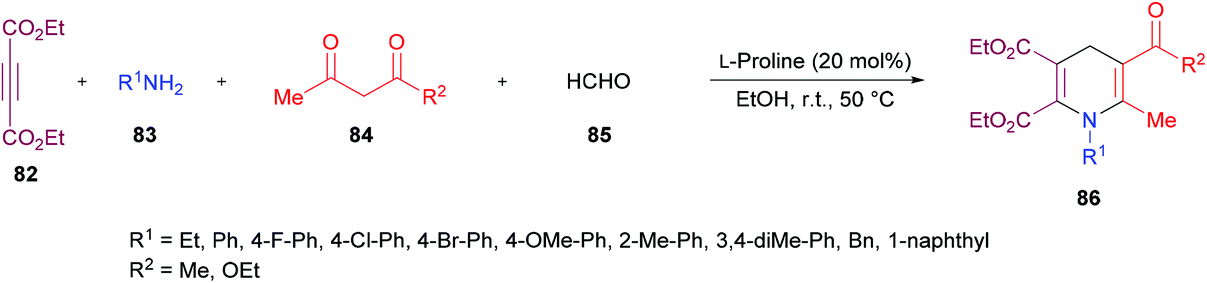
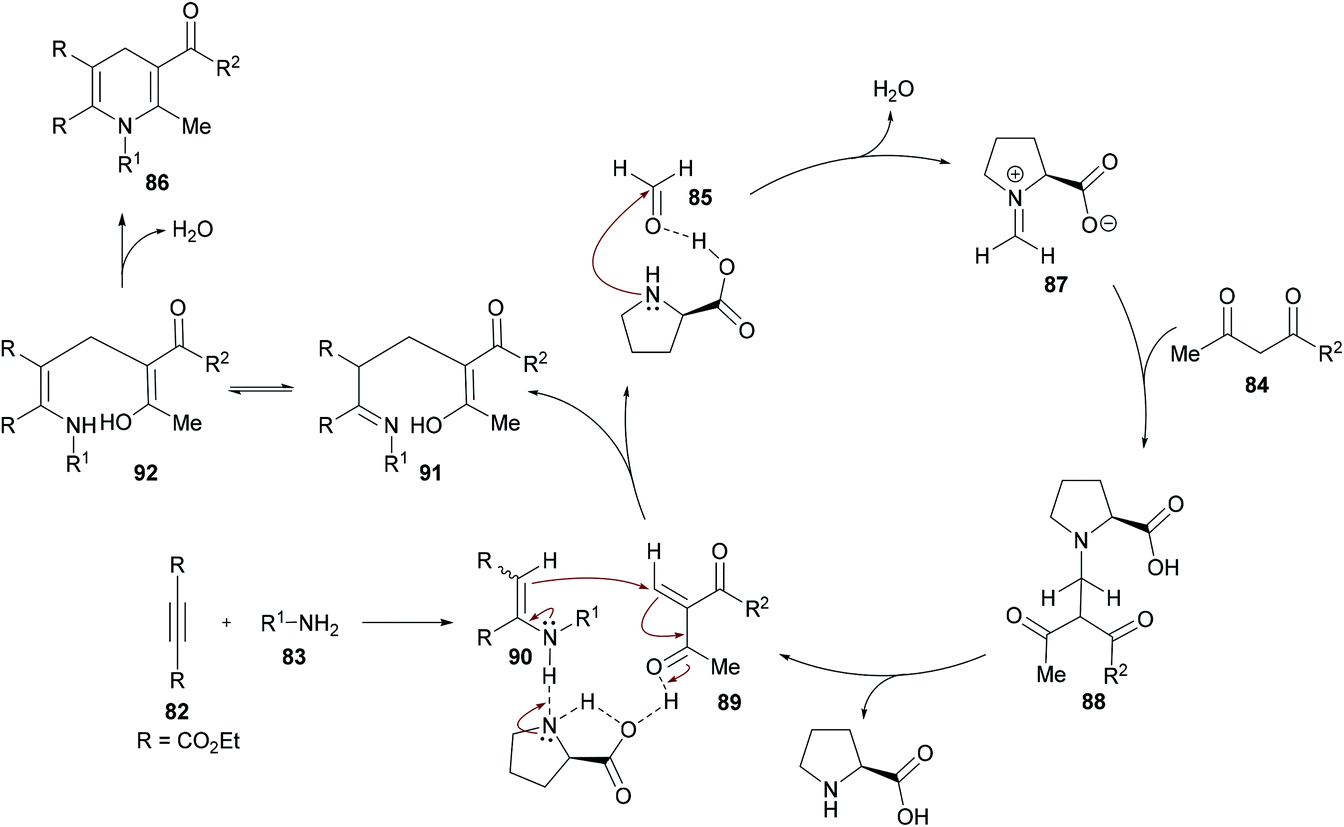
Jiang and co-workers110 developed a new L-proline-catalyzed one-pot four-component reaction methodology for the facile synthesis of densely functionalized 1,4-dihydropyridines 86 from alkynoates 82, amines 83, 1,3-dicarbonyl compounds 84 and aldehydes 85 (Scheme 19). The process operates under environmentally benign conditions involving various reactions such as hydroamination/Knoevenagel condensation/Michael-type addition/intramolecular cyclization and delivers 1,4-dihydropyridines in moderate to good yields. After screening various catalysts, L-proline was identified as the optimal. Other reaction parameters including solvents, reactant ratios, and temperature were also considered to obtain the optimized set of conditions. The scope and diversity of 1,4-dihydropyridines were realized where the different electronic nature of the functional groups on the aryl ring of the amines showed a minor impact on the reactivity. As such, electron-rich substituents showed preference over electron-deficient systems producing the anticipated products in higher yields. Sterically hindered amines were also effective coupling partners alongside aliphatic primary amines that showed higher reactivity than the aromatic ones. A limited diversification around the other coupling partners was also investigated.
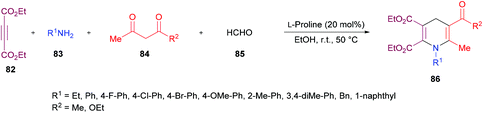 |
| | Scheme 19
L-Proline-catalyzed synthesis of highly functionalized 1,4-dihydropyridines. | |
The authors proposed the reaction mechanism which starts with the activation of aldehyde carbonyl by hydrogen bonding from L-proline and subsequent attack of the proline nitrogen on the activated carbonyl to generate intermediate 87 which on Knoevenagel condensation with 1,3-dicarbonyl compound 84 furnished 88. Knoevenagel product 89 was obtained with the regeneration of L-proline.111–113 Meanwhile intermediate 90 was formed by the addition of amines to alkynes which on Michael-type addition to 89 furnished intermediate 91. The tautomerization of 91 to 92 followed by an intramolecular cyclization provided 1,4-dihydropyridine 86 (Scheme 20).
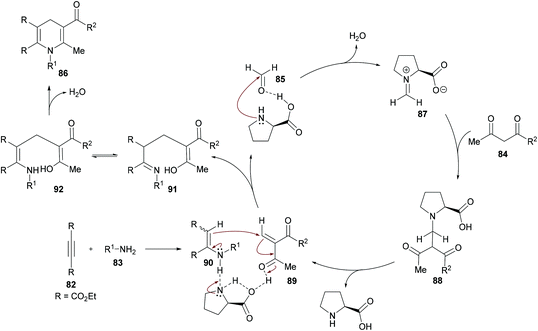 |
| | Scheme 20 Proposed mechanism for the formation of 1,4-dihydropyridines 86. | |
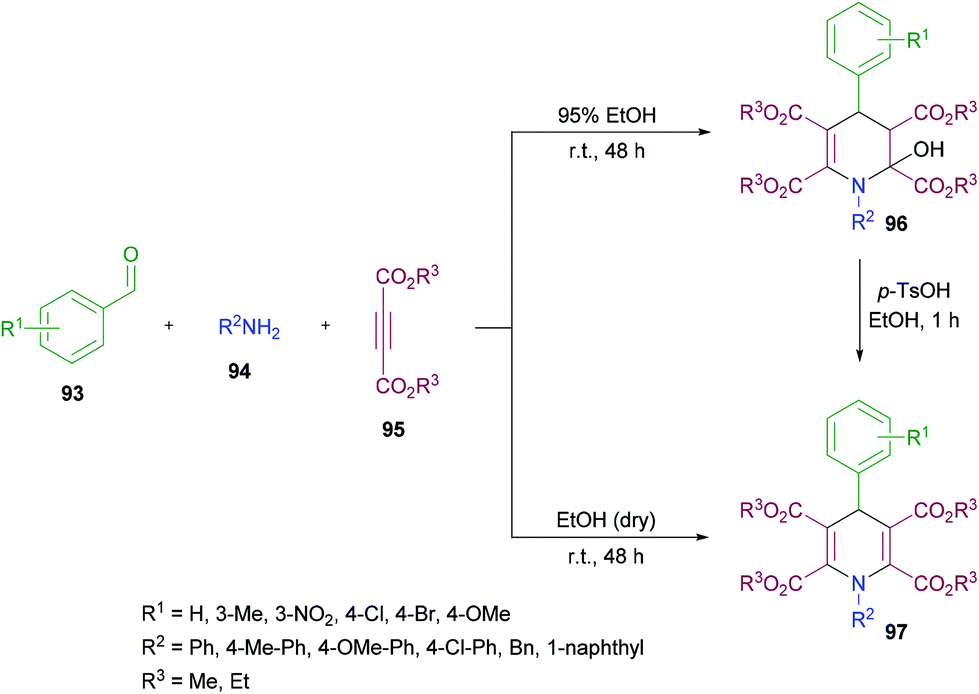
Yan and co-workers114 established a one-pot three-component methodology incorporating aldehydes 93, aromatic amines 94 and acetylenedicarboxylates 95 to afford 2-hydroxyhydropyridines 96 (Scheme 21). Based on the literature reports (Ziyaei-Halimehjani & Saidi, 2008; Glotova et al., 2009; Li et al., 2009; Yavari et al., 2010; Jiang et al., 2010)115–119 that conjugate addition of aniline to acetylenedicarboxylates smoothly provides 2-(phenylamino)but-2-enedioate, the authors believed that double conjugate addition is possible if another molecule of acetylenedicarboxylates is present in the reaction system. With this idea in mind, they started investigating a range of conditions to identify the optimal parameters for this methodology. The required product was formed in 71% yield in ethanol at room temperature with the presence of an unexpected hydroxyl group at the C-2 position presumably resulting from a hydration step in the reaction process. Other solvents including tetrahydrofuran, acetonitrile, dichloromethane and chloroform retarded the reaction. Similarly, elevated temperatures and the addition of a base to the reaction pot provided a complex mixture of unidentified compounds. The reaction scope was assessed with several aromatic aldehydes as well as amines and the desired dihydropyridine products were isolated in 70–87% yields. Both electron-donating and electron-withdrawing substituents were equally tolerated in the reaction. α-Naphthylamine and benzylamine also provided the corresponding 2-hydroxyhydropyridines in good isolated yields. In addition, the reaction was also performed under inert conditions using absolute ethanol to avoid the formation of 1,4-dihydropyridines 97 bearing a hydroxyl group at the C-2 position and the required products were isolated in 68–81% yields. Similar results were obtained when p-toluenesulfonic acid (TsOH) was employed as a catalyst to convert 2-hydroxyhydropyridines 96 to 1,4-dihydropyridines 97 in ethanol under reflux.
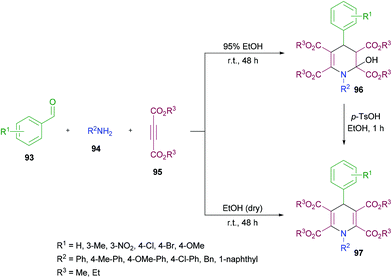 |
| | Scheme 21 Three-component synthesis of 2-hydroxyhydropyridines and 1,4-dihydropyridines from aromatic aldehydes, arylamines and acetylenedicarboxylates. | |
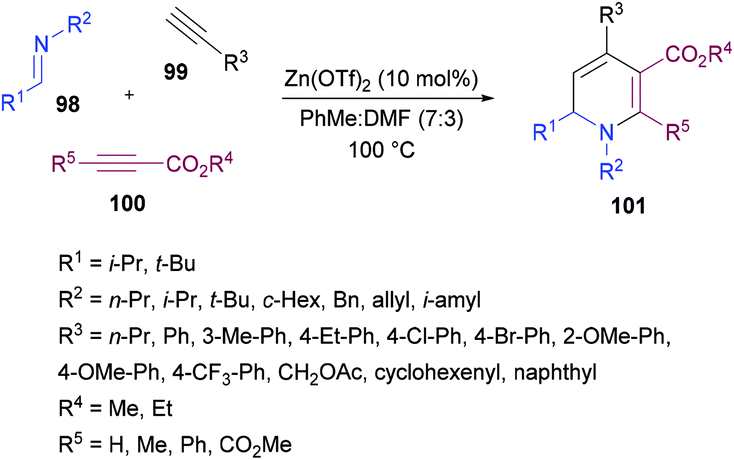
Tehrani and co-workers120 developed a Zn(OTf)2-catalyzed three-component cascade approach to produce less explored 1,6-dihydropyridines 101 from aldimines 98, terminal alkynes 99 and electron-deficient alkynes 100. The heterocyclic products were achieved in good to excellent chemical yields (Scheme 22). In a mixture of solvents (toluene and DMF), various reaction parameters such as the catalyst and temperature were screened. Other catalysts including ZnBr2, ZnI2, Zn(OAc)2, Cu(OTf)2 and CuBr2 were less effective. The reaction scope was explored by testing a range of aldimines and ketimines in the first instance. The anticipated products were isolated in good yields. The variation of the alkyne component was also examined. Both aromatic and aliphatic alkynes were successfully tolerated. The presence of electron-rich and electron-deficient substituents on the aryl ring of alkynes had no influence on the reactivity of this cascade process. The use of electron-deficient propiolates instead of DMAD produced the 1,6-dihydropyridine heterocycle with the regiospecific incorporation of the ester moiety.
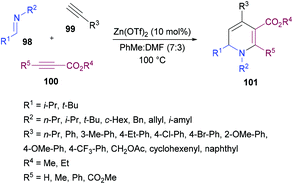 |
| | Scheme 22 Zn(OTf)2-catalyzed three-component synthesis of highly functionalized 1,6-dihydropyridines from aldimines and two alkynes. | |
2.1.4. Synthesis of tetrahydropyrimidines.
Jiang and co-workers121 unearthed a facile and convenient one-pot multi-component approach involving alkynoates 102, amines 103, 104 and formaldehyde 105 to access a range of diversely functionalized tetrahydropyrimidines 106 (Scheme 23). This domino reaction works smoothly in the absence of a catalyst with the production of nitrogenous heterocycles in high yields. The relay process comprised of hydroamination, Mannich-type reaction and amine-aldehyde dehydration–cyclization sequence. After extensive screening of reaction parameters, including solvent, time and reactant ratios, the best results were furnished when but-2-ynedioic acid diethyl ester 102/aniline 103/formaldehyde 105/benzylamine 104 are used in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:4:1.1–1.2 in DMF. Although, both aliphatic and aromatic amines were effective coupling partners, the reaction proceeded much faster with aliphatic primary amines. The reaction was also insensitive to electron-donating and electron-withdrawing substituents present on the aromatic amines. All the tetrahydropyrimidine products were formed in good to excellent yields except the ortho-aryl-substituted product which was obtained in a lower yield (62%).
:1:4:1.1–1.2 in DMF. Although, both aliphatic and aromatic amines were effective coupling partners, the reaction proceeded much faster with aliphatic primary amines. The reaction was also insensitive to electron-donating and electron-withdrawing substituents present on the aromatic amines. All the tetrahydropyrimidine products were formed in good to excellent yields except the ortho-aryl-substituted product which was obtained in a lower yield (62%).
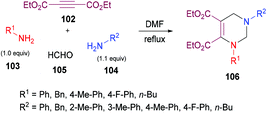 |
| | Scheme 23 Catalyst-free one-pot multi-component synthesis of polysubstituted tetrahydropyrimidines. | |
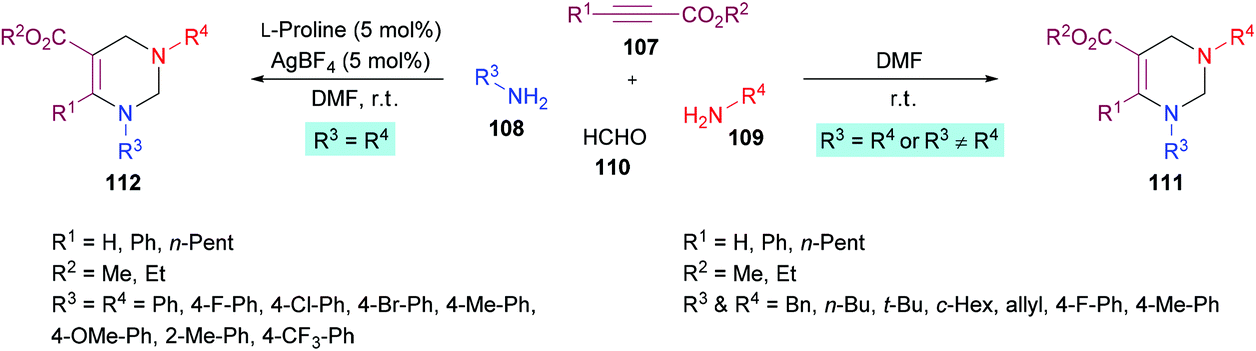
In a subsequent study, Jiang and co-workers122 developed two multi-component methodologies towards polysubstituted pyrimidine derivatives 111, 112. In the metal-free approach, electron-deficient alkynes 107, amines 108, 109 and formaldehyde 110 were coupled to obtain the heterocyclic product in an efficient yield (Scheme 24). The product was furnished with high regioselectivity with Markovnikov addition only. Upon the addition of two different amines, the reactivity was in favour of aliphatic amine while no product was formed when aromatic amine was added first. Various aliphatic amines were employed in this method to deliver aza-heterocycles with high yields and regioselectivities. In the second approach where aromatic amines served as potential substrates, extensive catalyst screening was performed to identify the optimized conditions favouring the hydroamination–cyclization reaction sequence. In this pursuit, AgBF4/L-proline was identified as the most effective catalyst. The best working conditions include AgBF4 (5 mol%) and L-proline (5 mol%) in DMF at room temperature, furnishing the desired product in 80% yield in 6 h. Using this set of conditions, various anilines bearing electron-rich (Me, OMe) and electron-deficient (F, Cl, Br, CF3) substituents were illustrated as suitable coupling partners and the corresponding products were formed in good yields.
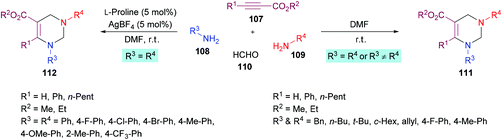 |
| | Scheme 24 One-pot multi-component divergent synthesis of polysubstituted tetrahydropyrimidines. | |
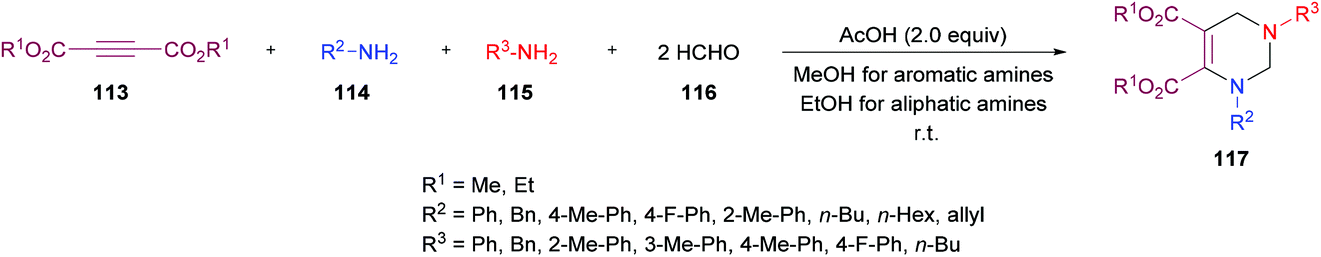
Next, the same group123 investigated a multi-component methodology to construct functionalized tetrahydropyrimidines 117 from but-2-ynedioates 113, amines 114, 115, and formaldehyde 116 (Scheme 25). After screening of the initial reaction conditions, more ambient conditions were developed, and the results were compared with the heat-promoted MCRs.124 The products were obtained at much lower temperature and in much shorter time with higher yields.
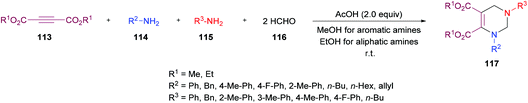 |
| | Scheme 25 Multi-component synthesis of polysubstituted tetrahydropyrimidines from alkynoates, amines and formaldehyde. | |
2.1.5. Synthesis of quinolines.
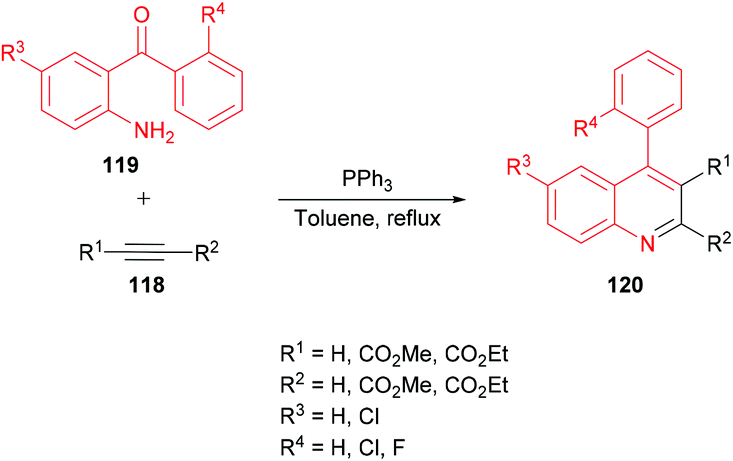
Asghari and co-workers125 established the synthesis of quinoline derivatives 120 by the addition of acetylenic esters 118 to aromatic amines 119 in the presence of triphenylphosphine (Scheme 26). The initial addition of triphenylphosphine to the acetylenic ester led to functionalized phosphoranes which upon the intramolecular Wittig reaction produced the desired quinoline compounds.
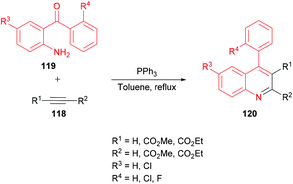 |
| | Scheme 26 Phosphine-catalyzed synthesis of 4-arylquinolines from aromatic aminoketones and alkynoates. | |
2.1.6. Synthesis of pyrrolidines and indolines.
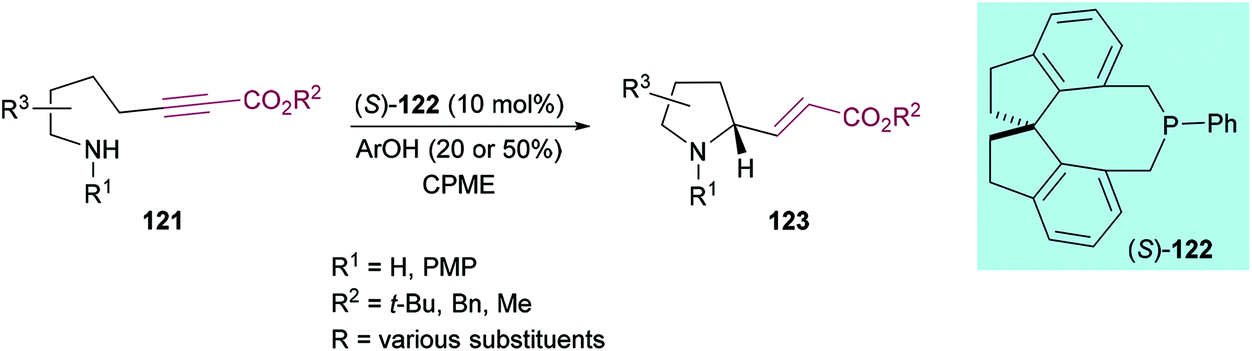
Fu and co-workers126 demonstrated a straightforward phosphine-catalyzed intramolecular approach to access highly functionalized enantioenriched nitrogenous heterocycles (pyrrolidines and indolines) 123via γ C–N bond formation using alkynoate substrates 121 (Scheme 27). This enantioselective strategy also successfully tolerated various electron-withdrawing and electron-donating functional groups on the aromatic ring delivering indoline products with good enantioselectivity but with insignificant reduction in the yield. The presence of a methyl group ortho to nitrogen did not impede the reaction and smooth cyclization was observed towards the corresponding product in 67% yield and 88% ee.
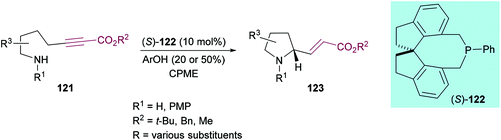 |
| | Scheme 27 Phosphine-catalyzed synthesis of pyrrolidines and indolines. | |
2.1.7. Synthesis of pyrrolidinones.
Based on the results obtained from the three-component methodology for the formation of 2-hydroxyhydropyridines or 1,4-dihydropyridines, Yan and co-workers127 combined the three-component methodology with an acid-catalyzed dehydration step to afford 1,4-dihydropyridines in a convenient fashion. However, the expected product was not furnished; rather 2-hydroxytetrahydroxypyridine was formed and an unexpected pyrrolidinone derivative was generated as the major product (Scheme 28). The result was interesting because there was no literature report to construct pyrrolidinones 127 from electron-deficient alkynes in this manner. The reaction conditions were then optimized and it was revealed that stirring the aldehyde with amine in the presence of 20 mol% of p-toluenesulfonic acid for 30 min followed by the introduction of acetylenedicarboxylate provided the desired polysubstituted pyrrolidinone in 62% yield after 24 h. Various amines and aldehydes with dimethyl or diethyl acetylenedicarboxylates were successfully coupled with the generation of the desired pyrrolidinone products in good yields.
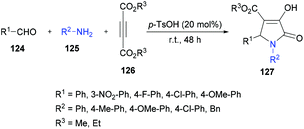 |
| | Scheme 28 Three-component synthesis of pyrrolidinones from aromatic aldehydes, arylamines and acetylenedicarboxylates. | |
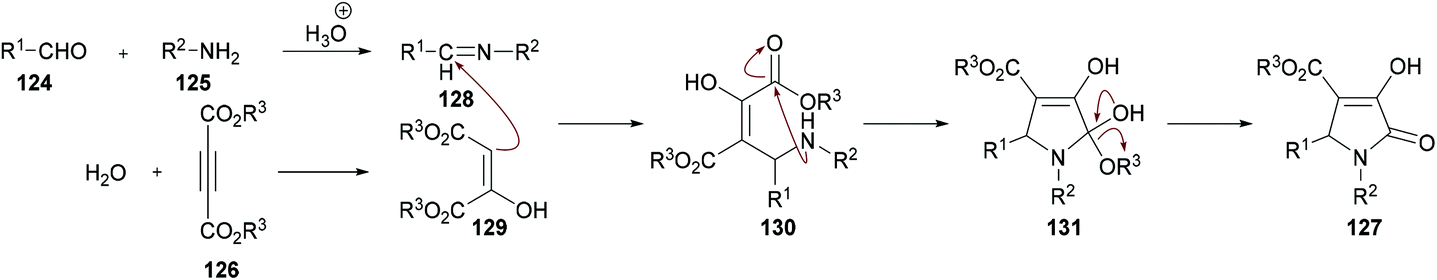
The proposed mechanism for this transformation suggests that the acid-catalyzed condensation of the aromatic aldehyde 124 with arylamine 125 produced imine intermediate 128. Upon the addition of acetylenedicarboxylate 126 in the presence of water, intermediate 129 was generated by the nucleophilic addition of water to an electron-deficient alkyne. Then, addition of 129 to imine 128 provided intermediate 130 which was cyclized via an intramolecular attack of the amine nitrogen on the ester carbonyl resulting in the formation of five-membered ring 131. Finally, the desired product 127 was delivered by the elimination of protonated alcohol (Scheme 29).
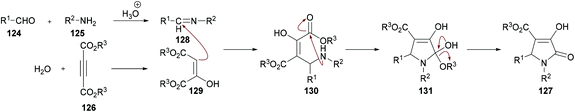 |
| | Scheme 29 Proposed mechanism for the formation of pyrrolidinones 127. | |
2.1.8. Synthesis of indoles.
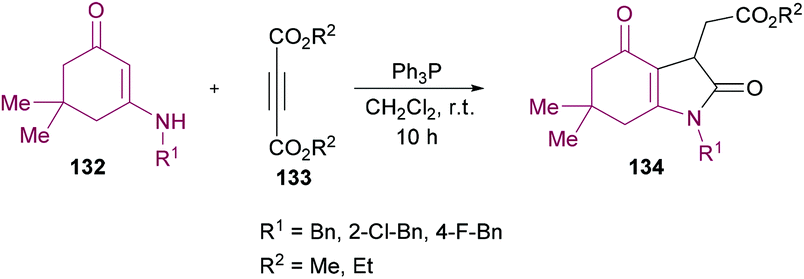
Nasiri and co-workers128 synthesized a variety of alkyl 2(1-benzyl-2,4-dioxo-2,3,4,5,6,7-hexahydro-1H-indol-3-yl)acetates 134 using enaminones 132 and acetylenic esters 133 as starting materials (Scheme 30). The reactions were catalyzed by triphenylphosphine and the corresponding products were isolated in moderate yields (61–65%).
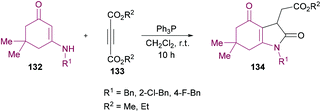 |
| | Scheme 30 Phosphine-catalyzed synthesis of alkyl 2(1-benzyl-2,4-dioxo-2,3,4,5,6,7-hexahydro-1H-indol-3-yl)acetates from enaminones and acetylenic esters. | |
2.1.9. Synthesis of lactams.
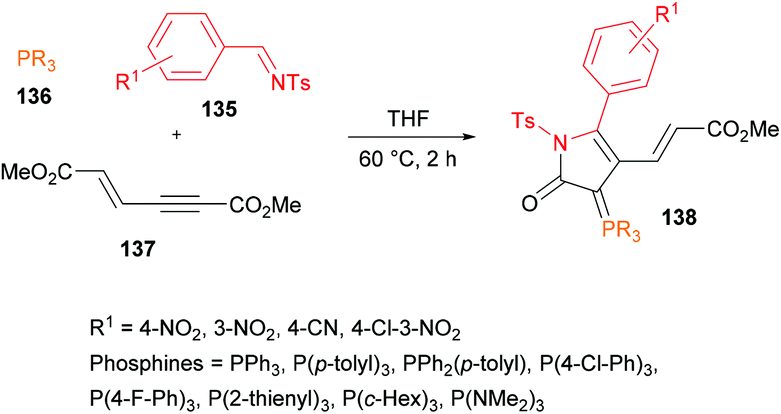
Based on the previous efforts to use alkynoates for lactone synthesis (Deng & Chuang, 2011),129 Chuang and co-workers130 elaborated the approach while using N-tosyl aldimines 135, phosphines 136 and alkynoates 137 to deliver fluorescent γ-lactams 138 (Scheme 31). The highest reactivity was achieved in an aprotic etherate solvent (THF) at 60 °C. The reaction conditions were comparable to those previously developed for lactone synthesis.129 The scope and generality of the three-component methodology revealed the use of various triarylphosphines and electron-deficient aldimines. A range of γ-lactam products were accessed in 49–79% yields while using different triarylphosphines containing electron-donating and electron-withdrawing substituents and 4-nitrobenzaldimine. The coupling of more nucleophilic tricyclohexylphosphine with 4-cyano aldimine furnished the desired product in a poor yield (22%). Consistent with the previous report (Deng et al., 2012),131 electron-withdrawing aldimines were only compatible in the multicomponent reaction.
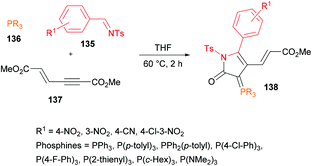 |
| | Scheme 31 One-pot multi-component synthesis of γ-lactams containing an α-phosphorus ylide moiety. | |
2.1.10. Synthesis of succinimides.
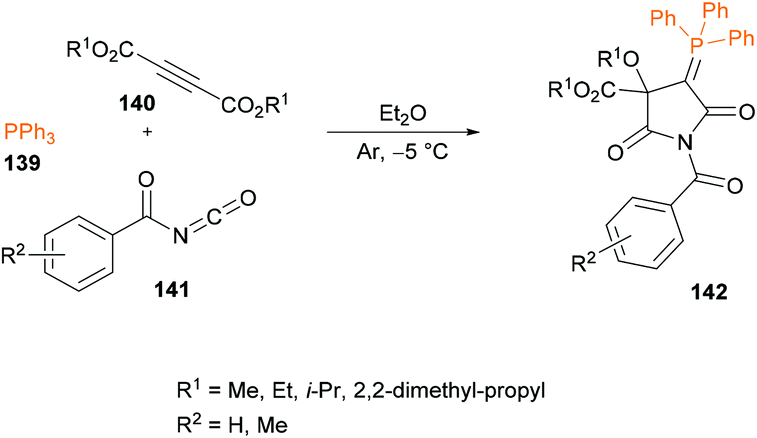
Esmaeili and co-workers132 described a one-pot three-component reaction for the generation of N-benzoyl-2-triphenylphosphoranylidene succinimide derivatives 142 from triphenylphosphine 139, acetylenic esters 140 and aroyl isocyanates 141 (Scheme 32). The reaction performs well under metal-free conditions and the anticipated products were formed in good to high yields. Several properties such as structural, electronic, energetic and mechanistic properties were explored with density functional theory (DFT) calculations using the Gaussian 09 computational package.133–135
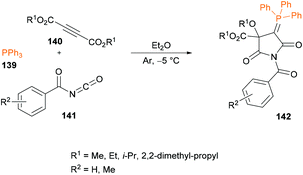 |
| | Scheme 32 Three-component synthesis of highly functionalized triphenylphosphoranylidene succinimides. | |
2.2. Synthesis of oxygen-containing heterocycles
Oxygen-containing heterocycles constitute a privileged class of heterocyclic compounds that contribute enormously to the development of new medicinal and bioactive drug molecules.136 Therefore, the development of new synthetic methods using mild reaction conditions remains an attractive field.
2.2.1. Synthesis of coumarins.
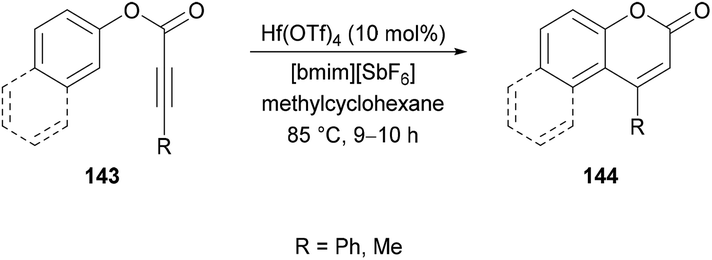
Song and co-workers137 successfully developed an intramolecular hydroarylation method for the regioselective synthesis of coumarins 144 using aryl phenylpropiolates 143 (Scheme 33). The intramolecular cyclization was catalyzed by Hf(OTf)4 in a mixture of an ionic liquid and methylcyclohexane. The reaction was high yielding at 85 °C for 9–10 h. Aryl 2-butynoate was also tested as a promising substrate and the corresponding coumarin was obtained in 89% yield. However, a lower stabilizing effect of the alkyl group compared to the phenyl group promoted self-oligomerization of the alkynes featuring alkyl substituents. In general, the explored methodology employing an ionic liquid provided an alternative approach to access a diverse variety of products where conventional methods showed a hindered scope.
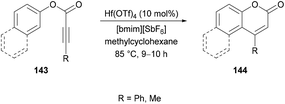 |
| | Scheme 33 Hf(OTf)4-catalyzed regioselective synthesis of coumarins. | |
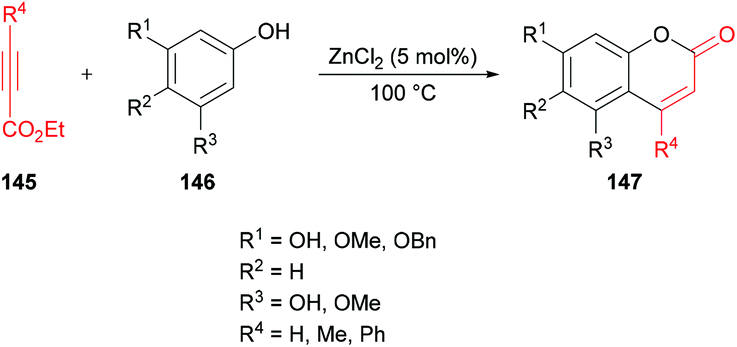
Costa and co-workers138 made use of a Lewis acid (zinc chloride) as an efficient catalyst for the construction of coumarins 147 by the hydroarylation of acetylenic esters 145 with oxygenated phenols 146 under solvent-free conditions (Scheme 34). The products were formed in moderate to good yields. The initial reaction of benzene-1,3,5-triol and ethyl propiolate provided the required coumarin in 88% yield. It was worth noting that similar yields were reported by Kaufman and Kelly;139 however, a stoichiometric amount of the catalyst (ZnCl2) was required, and the reaction time was 12 h. This procedure was improved, delivering the desired coumarins in pleasing yields after a reaction time of 5–60 minutes in most of the cases.
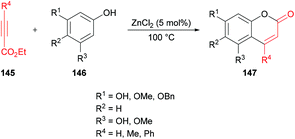 |
| | Scheme 34 Synthesis of coumarins through zinc chloride catalyzed hydroarylation of acetylenic esters with phenols. | |
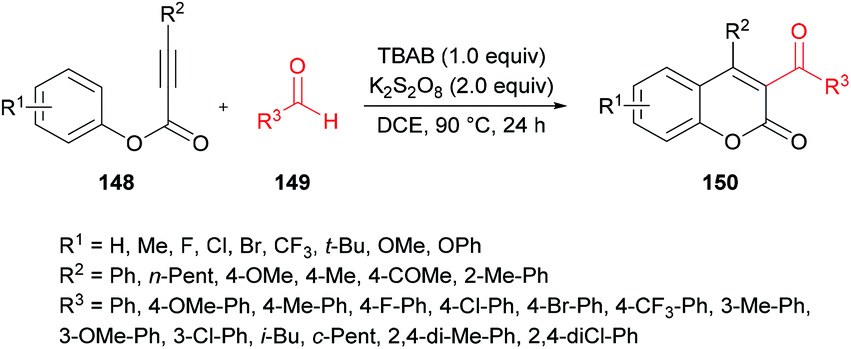
Mi et al.140 developed a metal-free tandem acylation/cyclization method using alkynoates 148 and aldehydes 149 for the generation of substituted coumarins 150 (Scheme 35). This method offers the instantaneous formation of two new C–C bonds via C–H functionalization. Regarding the literature methods,141–148 which suffer several drawbacks including a hindered substrate scope, low atom-economy and toxic reagents, the present protocol realizes a general and direct approach to 3-acylcoumarins. The reaction scope was fairly general, and several functional groups showed great compatibility under the optimal conditions. However, due to a strongly electron-deficient group (CF3) on the phenoxy ring of the alkynoate, the desired product was furnished in 38% yield. In addition, the steric effect was also quite distinct on the phenoxy ring and in the case of an ortho-substituted system, no reactivity was observed. Alkynoates bearing alkyl substituents were also unreactive.
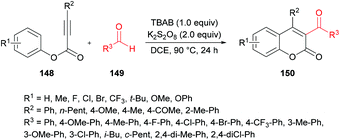 |
| | Scheme 35 Synthesis of 3-acyl-4-arylcoumarins via metal-free tandem oxidative acylation/cyclization of alkynoates and aldehydes. | |
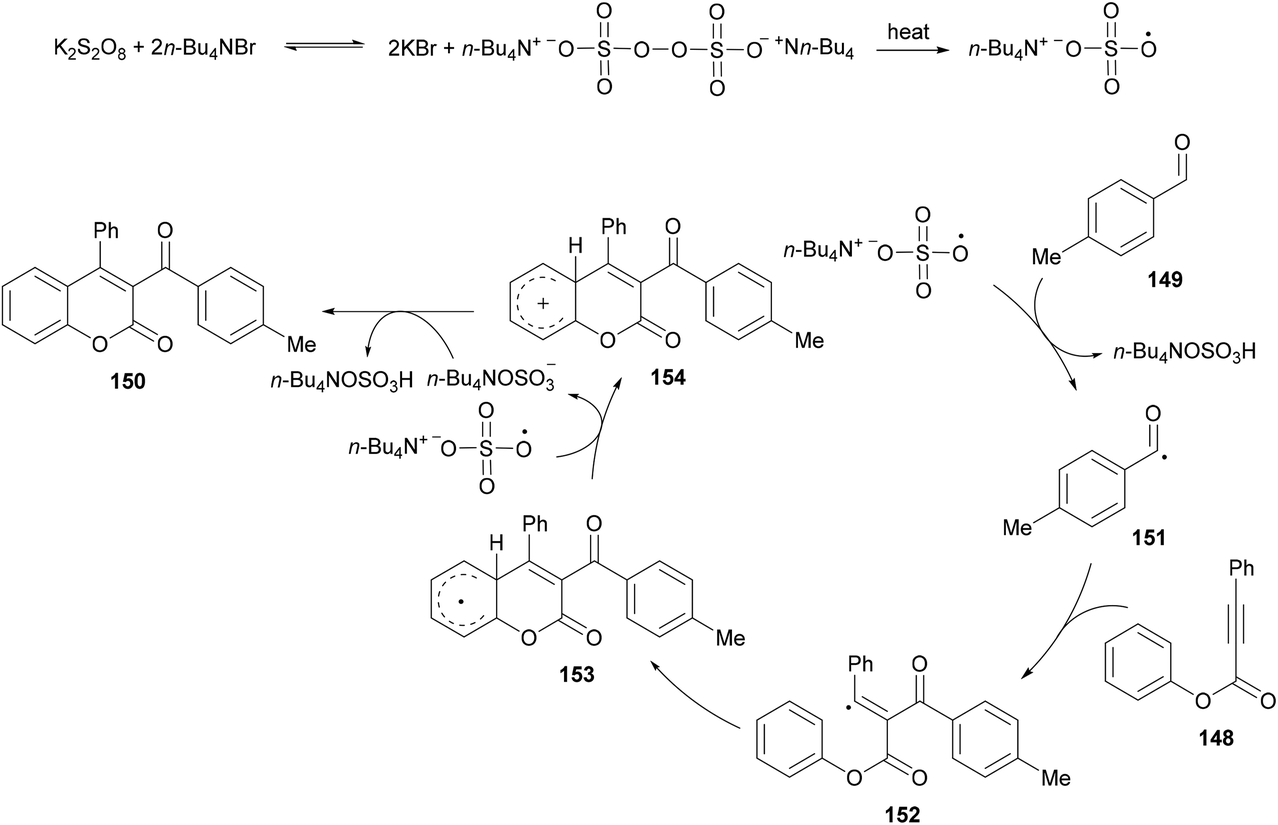
Based on the previous reports (Liu et al., 2011; Shi & Glorius, 2013; Luo et al., 2014),149–151 the authors proposed a radical mechanism for tandem oxidative acylation/cyclization (Scheme 36). Peroxydisulfate was converted to bis(tetrabutylammonium) peroxydisulfate upon reaction with TBAB which was transformed into tetrabutylammonium sulfate radical anions on heating.152,153 The reaction of the tetrabutylammonium sulfate radical with 149 produces the acyl radical 151 which adds selectively to the α-position of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in 148 producing vinyl radical 152. Cyclization of 152 then afforded the radical intermediate 153 which subsequently converted into cation 154via single electron transfer (SET). The intermediate 154 was deprotonated to give the desired product 150.
O bond in 148 producing vinyl radical 152. Cyclization of 152 then afforded the radical intermediate 153 which subsequently converted into cation 154via single electron transfer (SET). The intermediate 154 was deprotonated to give the desired product 150.
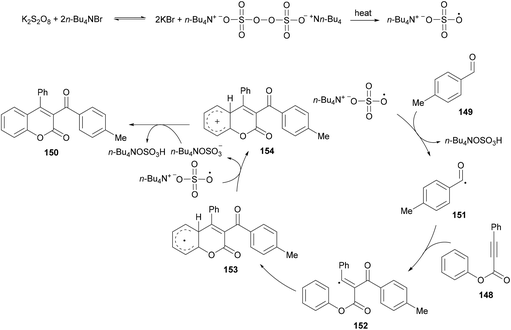 |
| | Scheme 36 Proposed mechanism for the formation of 3-acyl-4-arylcoumarins 150. | |
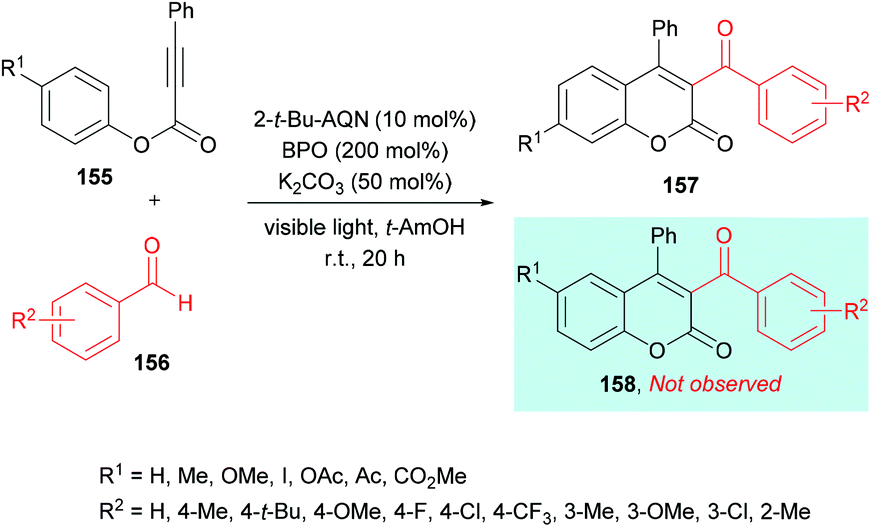
Similarly, Yamaguchi, Itoh and co-workers154 developed the synthesis of 3-acyl-4-arylcoumarins 157 using alkynoates 155 and simple aldehydes 156 under radical cyclization conditions (Scheme 37). A photocatalyzed generation of acyl radicals was achieved using aldehydes under metal-free and mild reaction conditions. Based on previous reports (Tada et al., 2011; Tada et al., 2012; Shimada et al., 2013),155–157 anthraquinone derivatives (AQNs) were employed where an O-centered radical of 3AQN* can trap a hydrogen atom. Phenyl-2-propynate and p-tolualdehyde were chosen as model substrates to investigate the reactivity profile of various reaction parameters in the radical addition/cyclization strategy. Photocatalysts including eosin Y, Ru(bpy)3Cl2 and 2-tert-butylanthraquinone (2-t-Bu-AQN) were employed alongside a range of oxidants (BPO, K2S2O8, H2O2, DTBP) under visible light irradiation. The role of bases (K2CO3, Na2CO3, KOH, Et3N) was also investigated. The screening results indicated that acylcoumarins could be furnished in the best isolated yields when the reaction mixture containing 2-t-Bu-AQN (10 mol%), BPO (200 mol%), and K2CO3 (50 mol%) in t-amyl alcohol was irradiated with four 23 W lamps under an argon atmosphere. Benzaldehydes bearing various substituents such as methyl, tert-butyl, fluoro, chloro, trifluoromethyl, and methoxy at the 3- and 4-positions of the phenyl ring produced the corresponding 3-acyl-4-arylcoumarins in 47–77% yields. Next, the scope of the reaction was examined with various alkynoates representing structural diversification at the para-position of the phenoxy ring. Both electron-donating (methyl, methoxy) and electron-withdrawing (iodo, ester, acetyl, acetoxy) groups were compatible and furnished the desired products in 58–79% isolated yields. With meta-substituted alkynoates, two regioisomers were obtained except in the case of methoxy-substituted substrates; however, ortho-substituted substrates failed to produce the desired coumarin.
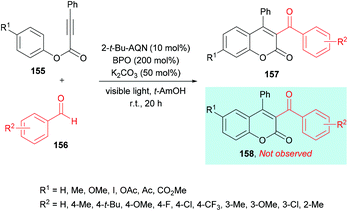 |
| | Scheme 37 Synthesis of 3-acyl-4-arylcoumarins from alkynoates and aldehydes under radical cyclization conditions. | |
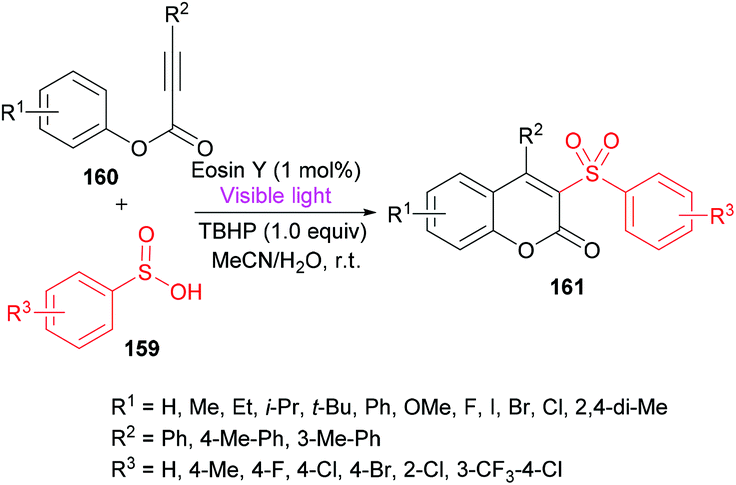
Wang and co-workers158 utilized sulfinic acids 159, versatile and readily available intermediates,159,160 for the formation of coumarin derivatives 161 (Scheme 38). The oxidative cyclization of phenyl propiolates 160 was achieved at room temperature under metal-free conditions. This visible-light induced transformation offered several advantages including broad functional group acceptance, good yields and high regioselectivity except alkyl substituents which were found to be unreactive.
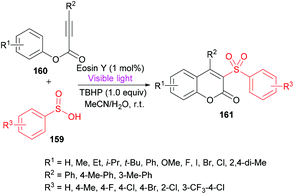 |
| | Scheme 38 Synthesis of coumarins through visible light initiated oxidative cyclization of aryl propiolates with sulfinic acids. | |
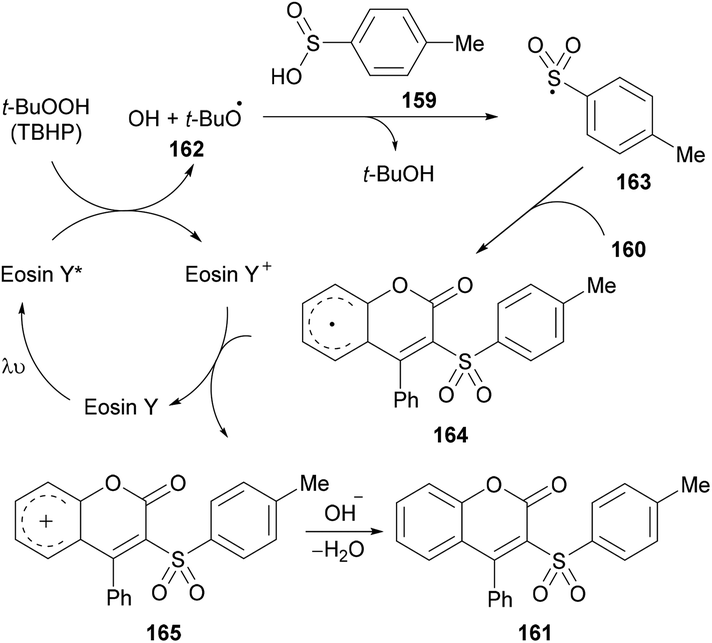
After several control experiments and based on the literature reports (Lu et al., 2013; Luo et al., 2014; Wei et al., 2014; Ouyang et al., 2014),150,161–163 the authors proposed a mechanism for this photoreaction (Scheme 39). The initial radical formation (tert-butoxyl radical 162) took place by a SET process by the reaction of the excited state of eosin Y* with TBHP. Next, the corresponding sulfonyl radical 163 was produced by the abstraction of a hydrogen radical from 4-methylbenzenesulfinic acid 159 which was transformed into carbocation intermediate 165via radical intermediate 164. Finally, intermediate 166 was deprotonated to deliver the desired 3-sulfonated coumarins 161.
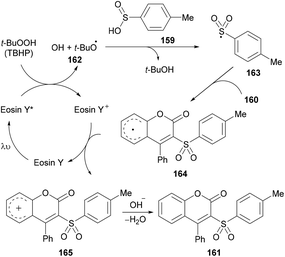 |
| | Scheme 39 Proposed mechanism for the formation of 3-sulfonated coumarins 161. | |
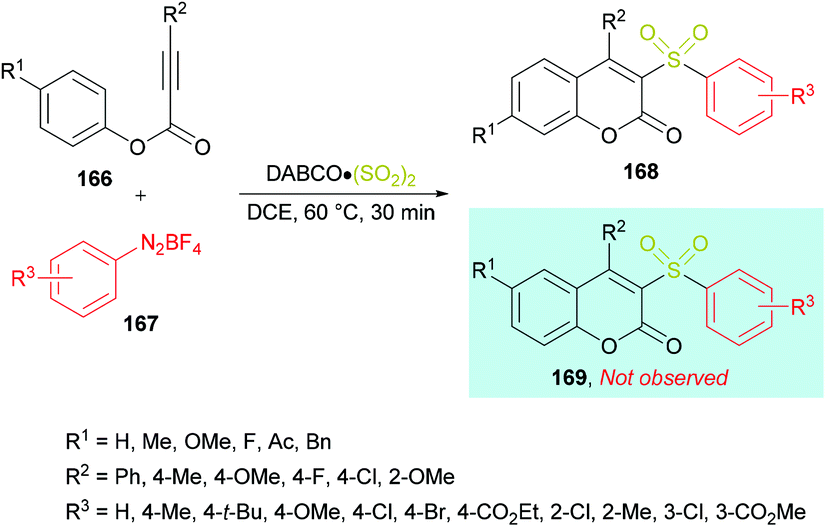
Wu and co-workers164 reported a catalyst-free approach for the efficient synthesis of 3-sulfonated coumarins 168 from aryl propiolates 166 (Scheme 40). Sulfonyl radicals were produced from aryldiazonium tetrafluoroborates 167 in the presence of DABCO·(SO2)2. This tandem process operates through various reaction sequences such as radical addition, spirocyclization, and 1,2-migration of esters. Phenyl propiolate and phenyldiazonium tetrafluoroborate served as the model substrates whereas DABSO was employed as a sulfonyl radical precursor. Various reaction parameters including solvents and temperature were studied to obtain the optimal conditions (aryl propiolate (0.2 mmol), DABCO·(SO2)2 (0.4 mmol), phenyldiazonium tetrafluoroborate (0.24 mmol), DCE (1.5 mL), under an argon atmosphere, 60 °C, 30 min). Under this set of conditions, aryldiazonium tetrafluoroborates and alkynoates were smoothly cyclized to the desired 3-sulfonated coumarins in good to excellent yields. Several electronically different substituents (methyl, methoxy, tert-butyl, chloro, bromo, ester) on the aromatic ring of aryldiazonium tetrafluoroborates showed remarkable tolerance and produced the coumarin products in 60–95% isolated yields. Quinolinyl aryldiazonium tetrafluoroborate was also tolerated, albeit generating the sulfonated coumarin in 31% yield. On the other hand, phenyl propiolates incorporating an aryl group attached to the alkyne component showed similar reactivity. Both electron-rich (methyl, methoxy) and electron-deficient (fluoro, chloro) substituents engendered the sulfonylated heterocycle in 85–92% yields; however, the presence of an alkyl group (tert-butyl) completely hindered the reaction. Similarly, substrates containing substituents such as benzyl, methoxy, fluoro, and acetyl at the para-position of the phenoxy ring were also examined.
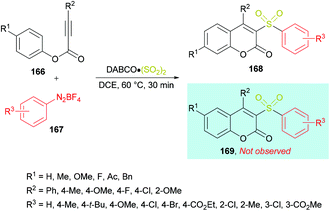 |
| | Scheme 40 Synthesis of 3-sulfonated coumarins from aryl propiolates and aryldiazonium tetrafluoroborates in the presence of DABSO. | |
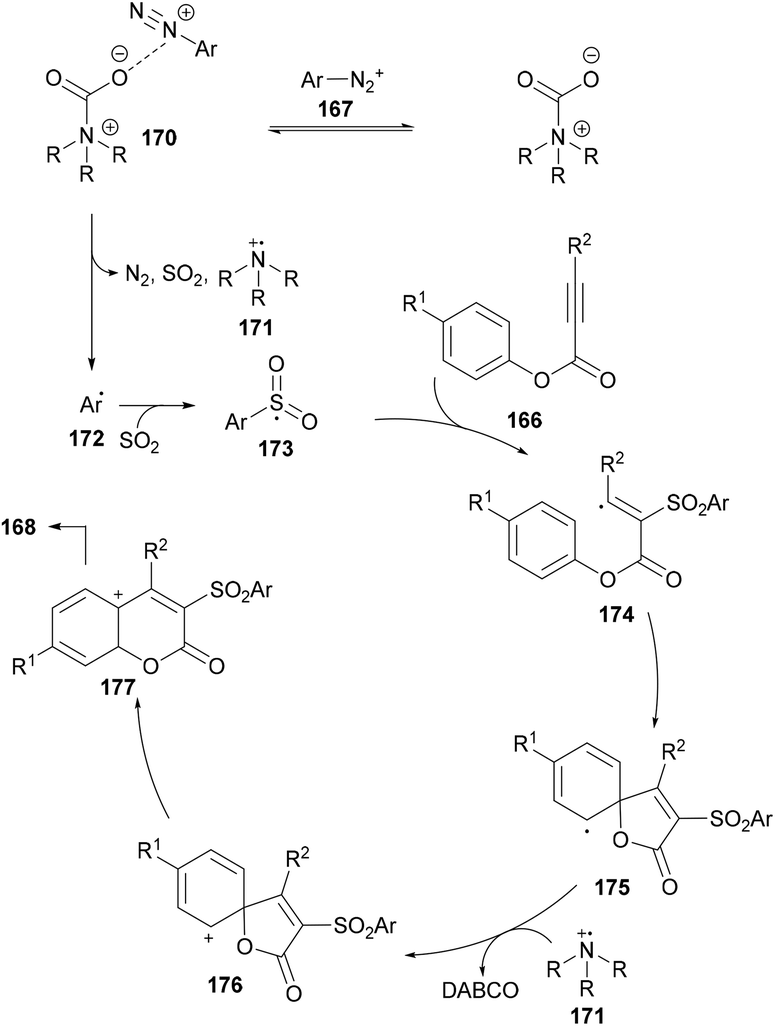
After performing several mechanistic experiments and in line with the literature reports, the authors proposed a possible reaction mechanism which starts with the coordination of arydiazonium cation 167 with DABSO to form the complex 170via electrostatic interaction.165 Homolytic cleavage of the N–S bond166 in 170 then furnished the radical cation intermediate 171, SO2 and aryl radical 172 which combines with sulfur dioxide to produce the sulfonyl radical 173. The attack of the radical 173 on the triple bond of the alkynoate generates intermediate 174 which on subsequent spirocyclization provides intermediate 175. Oxidation by radical cation 171 then affords 176167 followed by 1,2-ester migration to intermediate 177. The desired sulfonylated product 168 was obtained upon aromatization of intermediate 177 (Scheme 41).
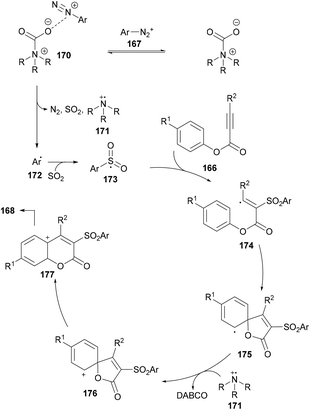 |
| | Scheme 41 Proposed mechanism for the formation of 3-sulfonated coumarins 168. | |
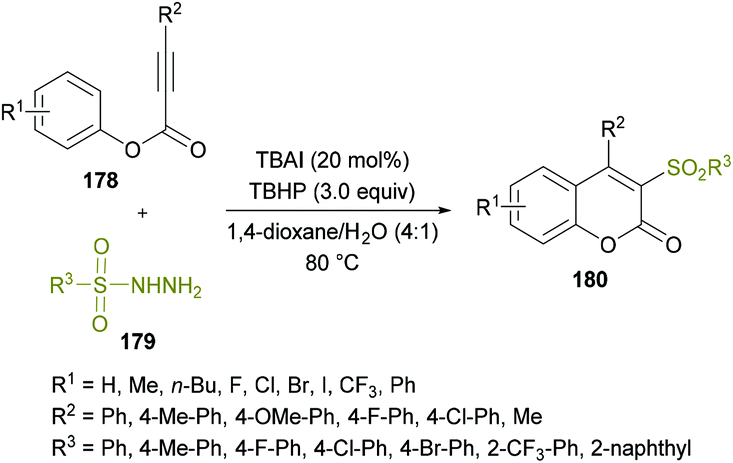
Due to the widespread applications of sulfone functionalities in organic and medicinal chemistry,168–177 the synthesis of organic molecules with these groups remained an attractive pursuit for many research groups. Recently, Wang and co-workers178 developed a new metal-free approach to construct structurally diverse 3-sulfonated coumarins 180 (Scheme 42). The easily available starting materials including alkynoates 178 and sulfonylhydrazides 179 were coupled via the direct difunctionalization strategy in the presence of TBAI and TBHP as the catalyst and oxidant, respectively. This procedure is generally convenient for the synthesis of 3-sulfonated coumarins as compared to the previously developed methods,179–183 which possess several limitations including harsh reaction conditions, low chemical yields and tedious work-up procedures. The reaction scope was reasonably broader, tolerating several electron-poor and electron-rich substrates effectively; however, the reactivity was stalled by steric factors leading to trace amounts of the desired compounds. Similar results were afforded when alkyl sulfonylhydrazides, such as methyl sulfonylhydrazide, were used as the substrates. Mechanistically, radical-promoted intramolecular cyclization/oxidation was among the key steps for the formation of the title product.
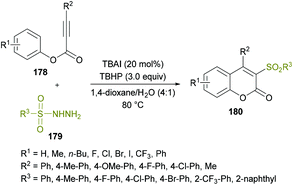 |
| | Scheme 42 Synthesis of 3-sulfonated coumarins via metal-free arylsulfonylation of alkynoates with sulfonylhydrazides. | |
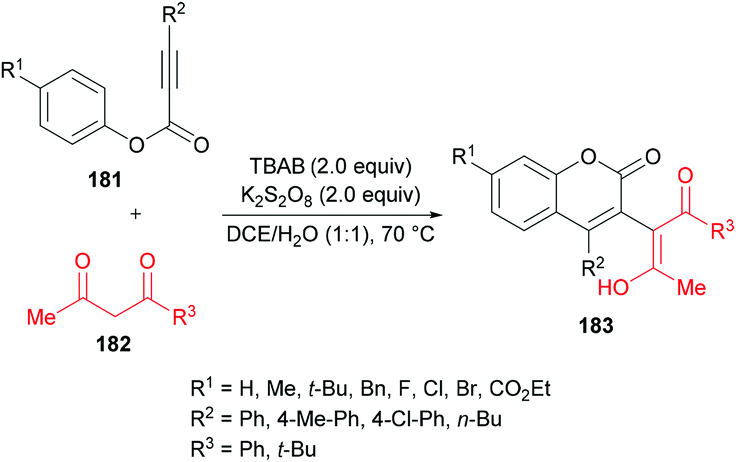
Liu et al.184 extended the scope of 3-functionalized coumarins 183 by using a range of alkynoates 181 and acetylacetone 182. The 3-acetonylcoumarins were accessed in moderate yields (Scheme 43). Again, the key step in the mechanistic process was an oxidative tandem 5-exo dearomative spirocyclization and ester migration.
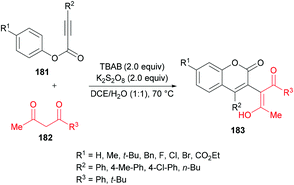 |
| | Scheme 43 Synthesis of 3-functionalized coumarins from alkynoates and acetylacetone. | |
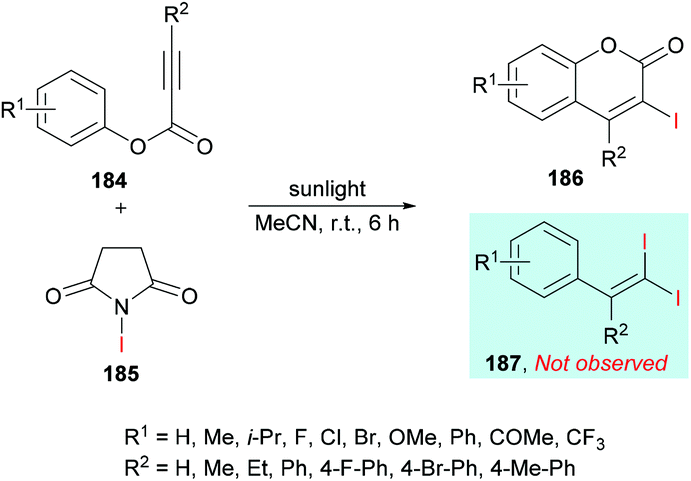
In an effort to develop green organic methodologies using photoredox catalysis, Ni et al.185 used alkynoates 184 for the synthesis of 3-iodocoumarins 186 driven by sunlight as a catalyst (Scheme 44). The process operates under catalyst-free conditions and cascade radical iodination and cyclization using N-iodosuccinimide (NIS)186–189 afforded the coumarin products at room temperature. This methodology was different from their previously developed procedure where the iodination triggered cascade radical decarboxylative migration reaction of alkynoates afforded 1,1-diiodoalkenes at elevated temperatures.190 After extensive screening of reaction parameters, suitable conditions were obtained which were utilized for the exploration of the general applicability of the method. Several sensitive functional groups worked well and delivered iodo-coumarins in pleasing yields. It is also notable that the substrates bearing aliphatic substituents such as methyl and ethyl also worked efficiently affording the corresponding coumarins in excellent yields (83–89%). However, no desired product was realized when phenyl propiolate was employed as the substrate. Also, with sterically hindered substrates, only trace amounts of the desired coumarins were achieved.
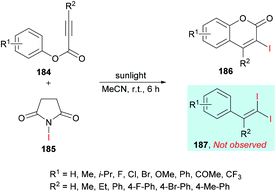 |
| | Scheme 44 Synthesis of 3-iodocoumarins via sunlight-promoted cyclization of alkynoates with N-iodosuccinimide. | |
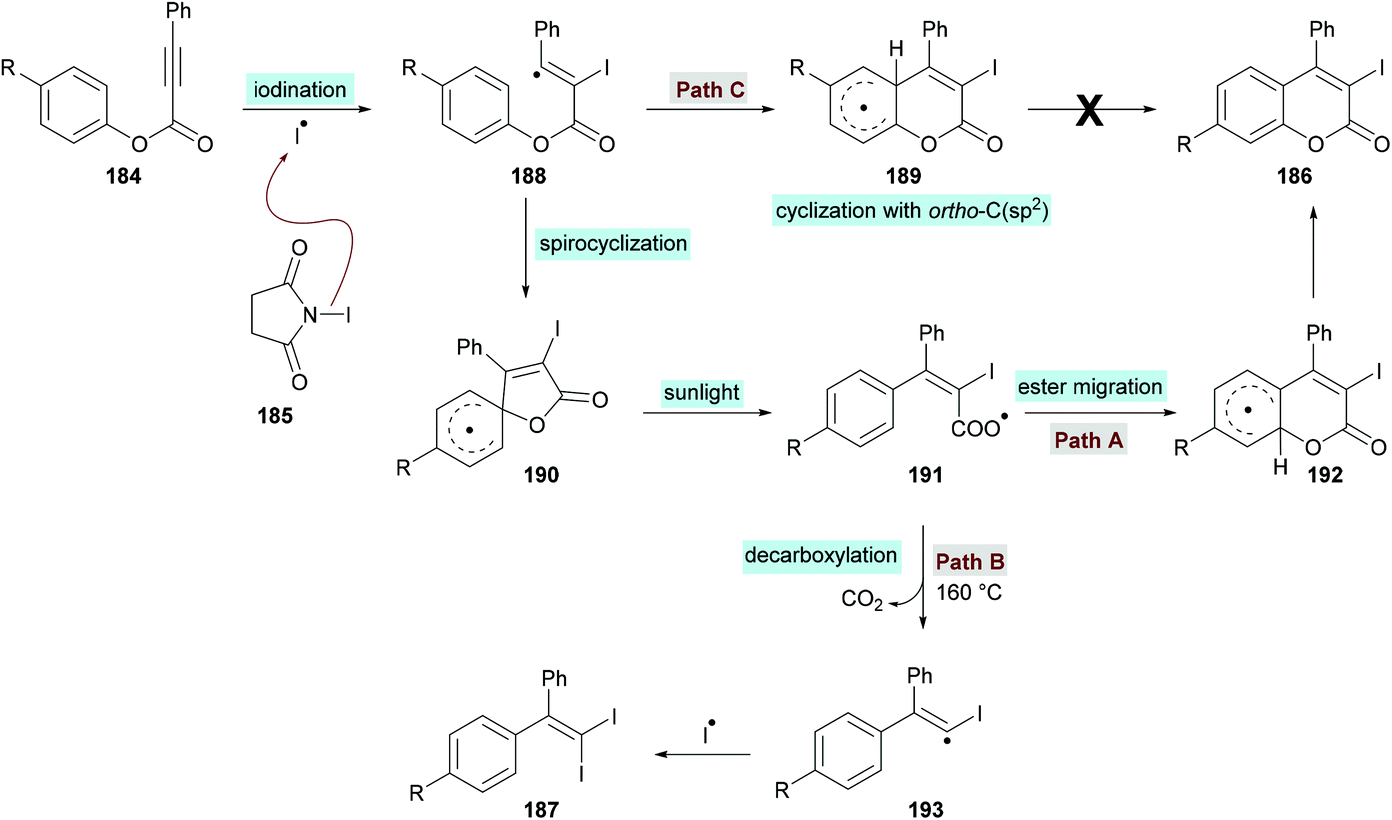
The proposed catalytic mechanism is illustrated in Scheme 45. The addition of the iodine radical (produced from NIS 185 under sunlight irradiation) to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of the alkynoate provided intermediate 188 which on subsequent intramolecular spirocyclization delivered intermediate 190. The intermediate 190 provided carboxyl radical 191 by the cleavage of the C–O bond which on cyclization and deprotonation by NIS gave the desired product 186 (path A). However, according to the previous reports (Mendis & Tunge, 2015; Ni et al., 2016),190,191 intermediate 191 could possibly generate intermediate 193 with the release of a CO2 molecule. Finally, intermediate 193 reacts with NIS to afford the final 1,1-diiodoalkene product 187. It is worth noting that 187 was not observed under the current sunlight irradiation experimental conditions.
C bond of the alkynoate provided intermediate 188 which on subsequent intramolecular spirocyclization delivered intermediate 190. The intermediate 190 provided carboxyl radical 191 by the cleavage of the C–O bond which on cyclization and deprotonation by NIS gave the desired product 186 (path A). However, according to the previous reports (Mendis & Tunge, 2015; Ni et al., 2016),190,191 intermediate 191 could possibly generate intermediate 193 with the release of a CO2 molecule. Finally, intermediate 193 reacts with NIS to afford the final 1,1-diiodoalkene product 187. It is worth noting that 187 was not observed under the current sunlight irradiation experimental conditions.
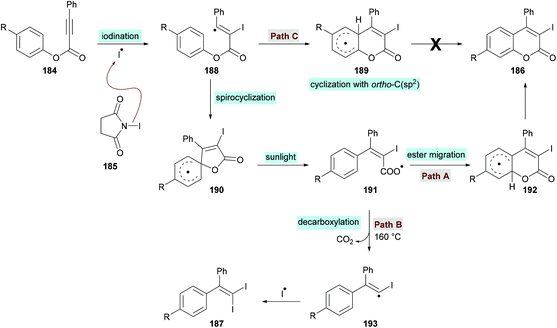 |
| | Scheme 45 Proposed reaction mechanism for the formation of 3-iodocoumarins 186. | |
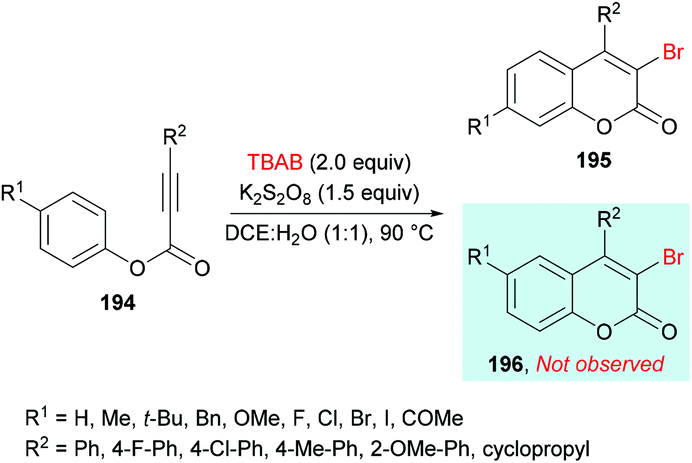
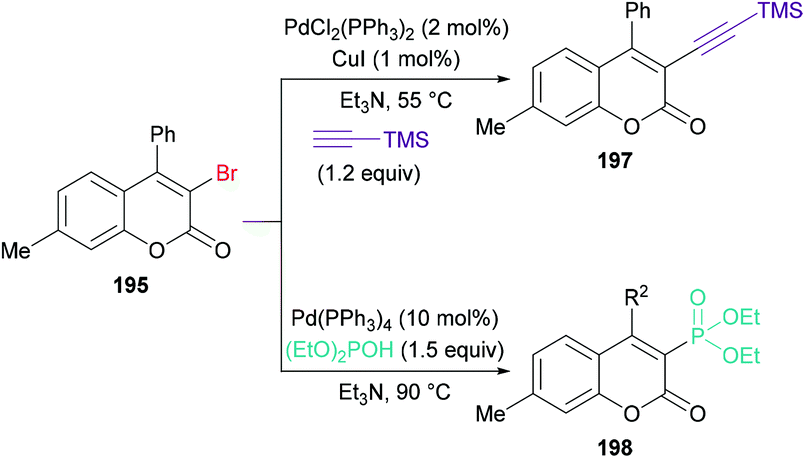
Qiu, Ding and co-workers192 developed a tandem radical bromination of alkynoates 194 to furnish a range of 3-bromocoumarins 195 catalyzed by K2S2O8 (Scheme 46). This methodology differs from the previous reports where liquid bromine was employed as a bromo source193–197 using TBAB and K2S2O8; thus the bromo radical would undergo addition to the CC triple bond of the alkynoate, followed by oxidative spirocyclization and 1,2-migration of the ester group to provide 3-bromocoumarin. Using tolyl alkynoate as a standard substrate with TBAB to develop the optimal conditions, various reaction parameters were investigated. The desired 3-bromo-7-methylcoumarin 195 was isolated, whereas 3-bromo-6-methylcoumarin 196 was not observed as evidenced by Wu and Wang.198–203 Further optimization of the relevant reaction parameters (oxidant, solvent, temperature) was then performed. A mixture of DCE and water (v/v = 1:1) was identified as the best reaction media. The addition of water was critical to hinder the formation of liquid bromine via homocoupling. With the identification of the optimized conditions (2.0 equiv. TBAB, 2.0 equiv. K2S2O8, in DCE/H2O (v/v 1:1) at 90 °C), the versatility of various alkynoates was explored. The presence of various substituents (R1) such as methyl, tert-butyl, benzyl, methoxy, fluoro, chloro, bromo, iodo, and acetyl at the phenyl ring of the alkynoate was productive in the delivery of the desired products in 63–78% yields. In addition, the scope and versatility of the reaction were further explored using various substituents (R2). Aryl groups bearing electron-rich (methyl, methoxy) and electron-deficient (halogens) substituents were compatible and produced 3-bromocoumarins in 64–80% yields. However, phenyl 3-cyclopropylpropiolate showed some hindrance in cyclization and gave the corresponding 3-bromo-4-cyclopropylcoumarin in 42% yield. Other halogen sources such as tetrabutylammonium fluoride (TBAF), tetrabutylammonium chloride (TBAC) and tetrabutylammonium iodide (TBAI) were unsuitable to furnish the desired halogenated product under the optimized reaction conditions. Moreover, keeping in view the synthetic utility of bromo compounds to serve as useful building blocks, synthetic functionalization was performed with the successful installation of alkynyl (197) and phosphite (198) groups onto the coumarin skeleton under palladium catalysis (Scheme 47).
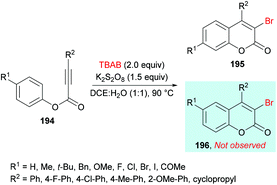 |
| | Scheme 46 K2S2O8-catalyzed synthesis of 3-bromocoumarins from alkynoates and TBAB. | |
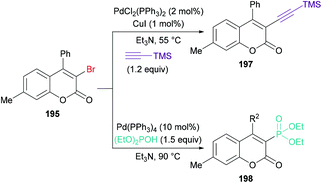 |
| | Scheme 47 Palladium-catalyzed synthetic functionalization of 3-bromocoumarin 195. | |
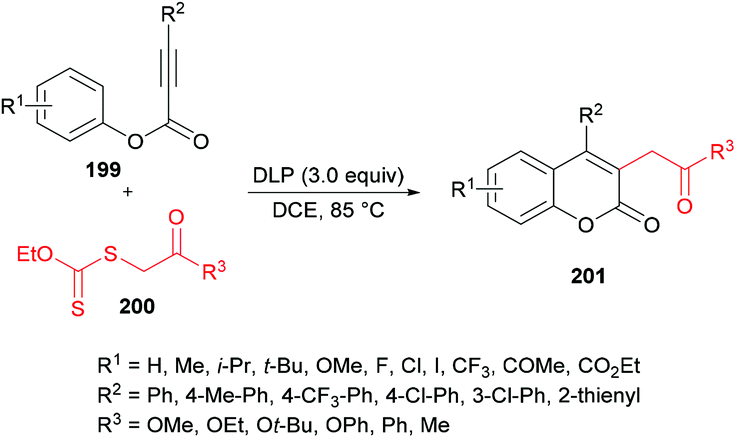
Pan, Yu and co-workers204 demonstrated the synthesis of substituted coumarins 201 by carboannulation of alkynoates 199 with xanthates 200. This dilauroyl peroxide (DLP)-promoted process involves radical addition/cyclization using metal-free conditions (Scheme 48). The effect of different oxidants in a variety of solvents was examined. Phenyl 3-phenylpropiolate and methyl 2-((ethoxycarbonothioyl)thio)acetate were employed in the presence of BPO as an oxidant to develop the optimal conditions. Several other oxidants including DTBP, TBHP, DLP, K2S2O8, PhI(OAc)2 and H2O2 were tested; however, DLP showed superior results and produced methyl 2-(2-oxo-4-phenyl-2H-chromen-3-yl)acetate in 74% yield in dichloroethane solvent. The reaction performed in various other solvents (DCM, MeCN, THF, PhCl, acetone) did not demonstrate any increase in the product yield. The scope and generality of this radical cyclization were expanded using different alkynoate substrates bearing both electron-rich (methyl, isopropyl, tert-butyl, methoxy) and electron-deficient (trifluoromethyl, halogens) substituents on the aryl ring. The corresponding 4-aryl-3-(2-methoxy-2-oxoethyl) coumarins were isolated in moderate to good yields. However, alkynoates incorporating strongly electron-deficient substituents (ester, acetyl) produced the corresponding products in lower yields. 4-Nitrophenyl 3-phenylpropiolate remained unreactive. Heterocyclic substituents at the alkynoate were also compatible, albeit producing the coumarin in 42% yield. The structural diversity was also realized when different xanthates featuring alkyl ester, ketone, and cyano were employed and the corresponding products were isolated in moderate to good yields.
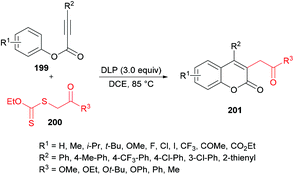 |
| | Scheme 48 Dilauroyl peroxide (DLP)-promoted synthesis of 3-(β-carbonyl)coumarins via radical addition/cyclization of alkynoates with xanthates. | |
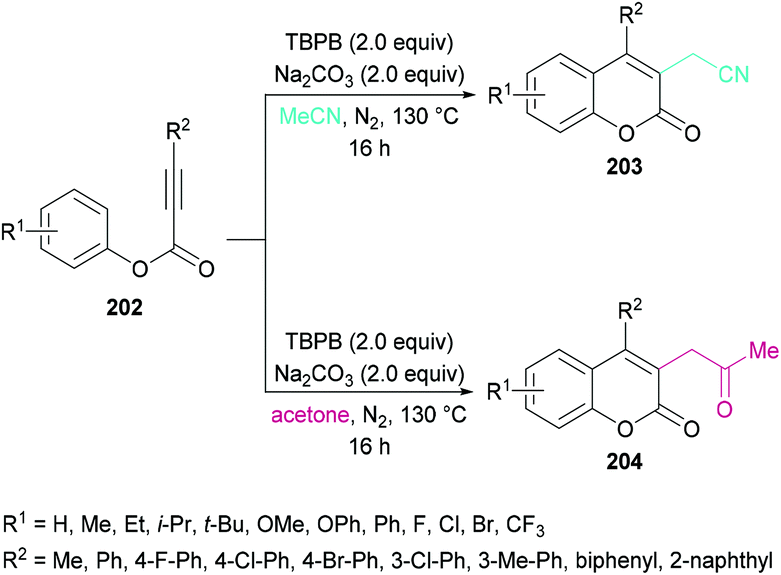
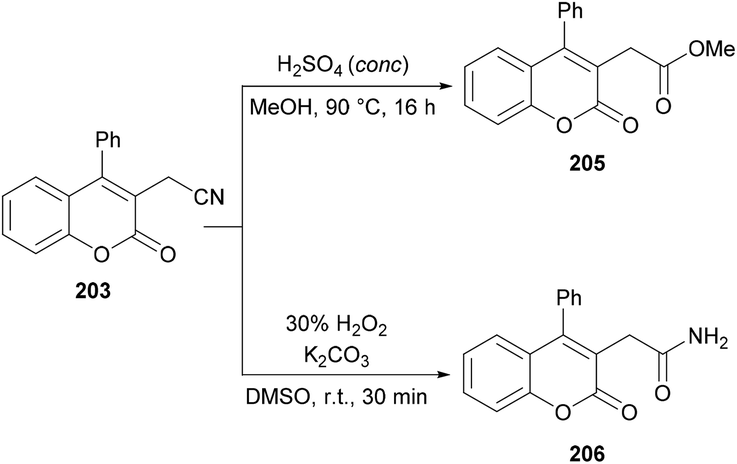
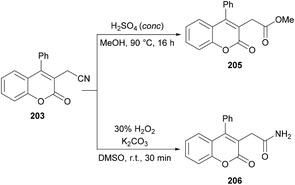
Sun and co-workers205 reported a facile route for the synthesis of cyanomethylated coumarins 203via cyanomethylation and cyclization of aryl alkynoates 202 (Scheme 49). Acetonitrile was used as a cheap cyanomethyl source in the presence of tert-butyl peroxybenzoate (TBPB) under metal-free conditions. Various oxidants, bases and temperatures were screened to optimize the reaction conditions. A variety of alkynoates bearing different substituents on the aromatic (phenoxy) ring were tested. Both electron-donating and electron-withdrawing substituents were compatible. The effect of substituents present on the alkyne was also investigated. No significant electronic influence was observed with differently substituted groups. The alkynes bearing an alkyl group however remained inert under the optimized reaction conditions. They further explored the scope using acetone instead of acetonitrile and the desired products (2-oxopropyl substituted coumarins 204) were isolated in good yields. The nitrile functionality was further transformed into other functional groups under different conditions to yield the corresponding products (Scheme 50). For instance, 2-(2-oxo-4-phenyl-2H-chromen-3-yl)acetonitrile was converted into methyl 2-(2-oxo-4-phenyl-2H-chromen-3-yl)acetate 205 and 2-(2-oxo-4-phenyl-2H-chromen-3-yl)acetamide 206.
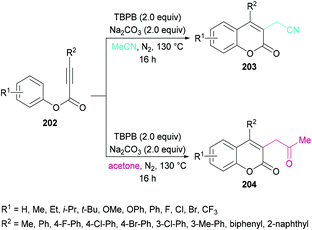 |
| | Scheme 49 Synthesis of 3-functionalized coumarins from aryl alkynoates. | |
 |
| | Scheme 50 Synthetic manipulation of the nitrile functionality in cyanomethylated coumarin 203. | |
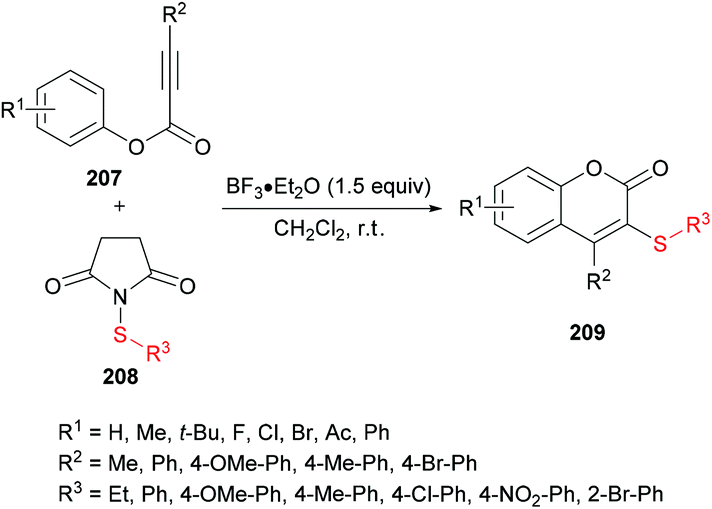
Chang and co-workers206 described a facile metal-free sulfenylation/cyclization approach involving aryl alkynoates 207 and N-sulfanylsuccinimides 208 in the presence of BF3·Et2O to produce 3-sulfenylated coumarins 209 in moderate to excellent yields (Scheme 51). Initial screening included the investigation of various parameters to identify the optimal conditions to perform the metal-free transformation. Without the addition of a Lewis acid, no corresponding product was observed, and the electrophilic cyclization furnished the coumarin product in 87% yield when the reaction was performed with 1.5 equivalents of BF3·Et2O in dichloromethane at room temperature. Various other Lewis acids (FeCl3, AlCl3) and Brønsted acids (TFA or TfOH), however, led to inferior results. The use of other solvents as well as electrophilic sulfenylating reagents led to diminished reactivity. Under the established reaction conditions, the scope and limitations of the cyclization methodology were explored. A range of substituents on the phenoxyl ring of alkynoates were tested and gave the 3-sulfenylated coumarins in good yields.
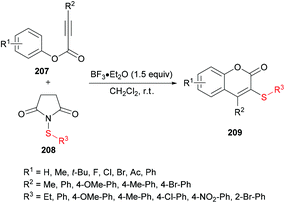 |
| | Scheme 51 BF3·Et2O-mediated synthesis of 3-sulfenylated coumarins from aryl alkynoates and N-sulfanylsuccinimides. | |
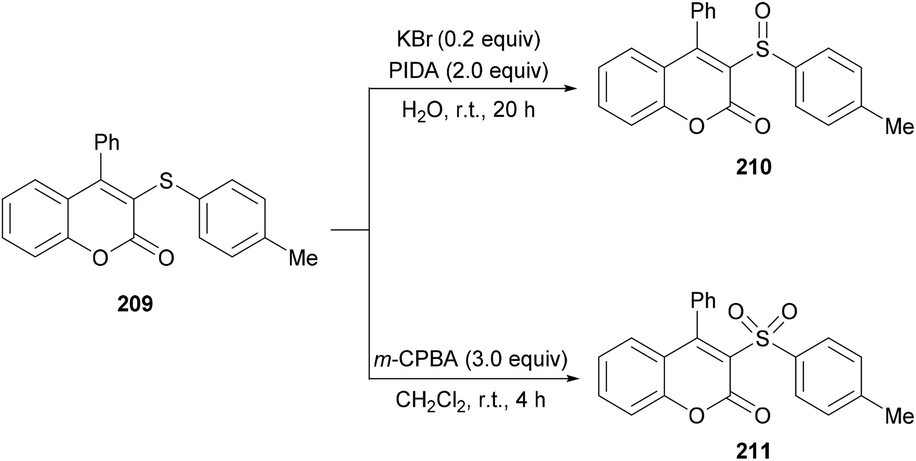
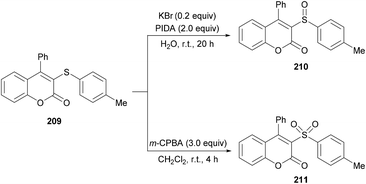
Both moderately electron-rich (methyl and tert-butyl) and electron-deficient functional groups (fluoro, chloro, bromo, acetyl) at the para-position of aryl 3-phenylpropiolates produced similar results. ortho-Substituted alkynoates also produced the desired annulated product in good to excellent yields. A sterically bulky naphthyl alkynoate also produced the coumarin product in 81% yield. The reaction scope also included the variation of substituents at the alkynyl functionality. Phenyl arylpropiolates with variable degrees of electronic character were found to be compatible in addition to alkylpropiolates. Moreover, additional sulfenylating reagents bearing different arylthiol partners were also made a part of this study and the desired products were furnished in moderate to high isolated yields. Notably, the para-methoxy substituted alkynoate led to the formation of spiro[4.5]triene-2,8-dione, generated via ipso-sulfenylcyclization. Keeping in view the applications of sulfinyl and sulfonyl functional groups in organic and medicinal chemistry,207–210 3-sulfinylated 210 and 3-sulfonated coumarins 211 were prepared under oxidative conditions using (diacetoxyiodo)benzene (PIDA) and meta-chloroperbenzoic acid (m-CPBA) as the oxidants (Scheme 52).
 |
| | Scheme 52 Synthesis of 3-sulfinylated and 3-sulfonated coumarins under oxidative conditions. | |
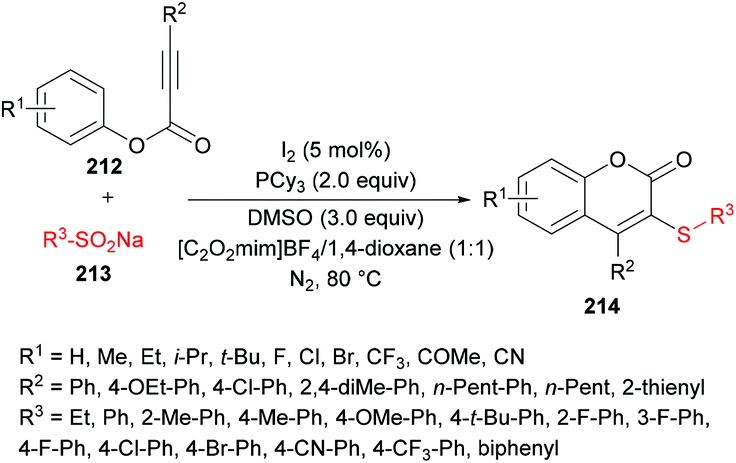
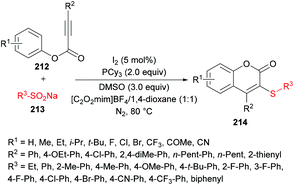
Wu, Jiang and co-workers211 reported an iodine-catalyzed electrophilic cyclization of alkynoates 212 and sodium arylsulfinates 213 to construct 3-sulfenylcoumarins 214 in moderate to good yields (Scheme 53). The optimized reaction conditions were developed using phenyl 3-phenylpropiolate and sodium benzenesulfinate as model substrates. Several oxidants including O2, H2O2, TBHP, K2S2O8, DTBP and DMSO and non-metal iodine catalysts such as I2, NIS, I2O5, ICl and TBAI were considered in a variety of solvents. After extensive screening, the optimal conditions were identified and used to explore the generality and scope of this reaction. Sodium arylsulfinates bearing electron-rich phenyl groups and halogens were compatible whereas electron-poor groups at the phenyl ring and heteroaryl and alkyl groups produced only a trace amount of the desired products. Moreover, various alkynoate substrates with diverse electronic features were tested. Aryl, alkyl, heteroaryl and extended aryl groups were well tolerated with broad functional group compatibility. Mono-, di-, and tri-substituted substrates were successfully cyclized to coumarin products in good yields.
 |
| | Scheme 53 Iodine-catalyzed synthesis of 3-sulfenylcoumarins from aryl alkynoates and sodium arylsulfinates. | |
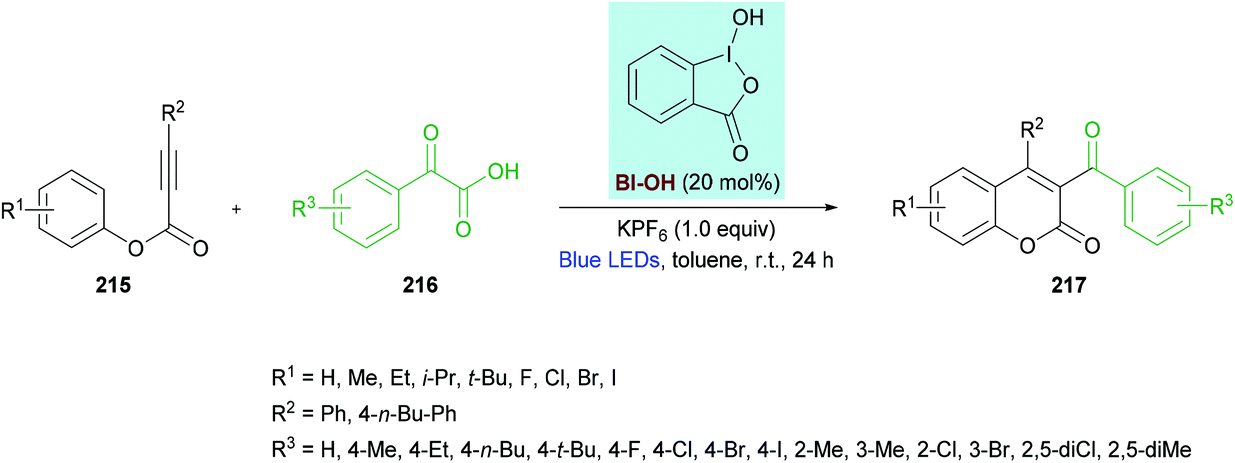
Zhang, Wang and co-workers212 unearthed a hypervalent iodine reagent (HIR)-catalyzed decarboxylative carbonylarylation of phenylpropiolates 215 with α-oxocarboxylic acids 216. The reaction proceeds under metal-free conditions generating the desired 3,4-disubstituted coumarins 217 in good yields at room temperature (Scheme 54). An extensive screening of various reaction parameters was performed to identify the optimal conditions. Blue, purple, red, green and white LEDs and sunlight were used as a light source while using eosin Y, [Ru(bpy)3]Cl2, PhI(OAc)2, [1-hydroxy-1,2-benziodoxol-3(1H)-one] (BI-OH), 1,2-benziodoxol-3(1H)-one acetate ester (BI-OAc) and BI-CC-Ph as catalysts. To enhance the reaction performance, several additives (PhSO3Na, K2CO3, Na2CO3, TsOH, PivOH, (NH4)2C2O4, KPF6) were also tested alongside a range of solvents (toluene, benzene, acetone, THF, DMSO, EtOAc, MeCN, DCE, Et3N). The optimal reaction conditions that were used to explore the scope of the reaction include 215 (0.20 mmol), 216 (0.40 mmol), BI-OH (20 mol%), and KPF6 (1.0 equiv.) in toluene (1.0 mL) under an air atmosphere. The reaction mixture was irradiated with a 1.5 W blue LED at room temperature for 24 h. Broad functional group tolerance was observed when various aryl 3-phenylpropiolates were reacted with 2-oxo-2-phenylacetic acid. A range of electron-rich substituents (methyl, ethyl, isopropyl, tert-butyl) at the para-position of the aromatic rings as well as 3,5-(Me)2 substituents produced the anticipated coumarin products in ample yields. Likewise, electron-deficient functional groups (F, Cl, Br, I) also proved to be equally tolerant to standard conditions. Similarly, the reaction scope was further extended by employing various α-oxocarboxylic acids. The electronically different substituents survived under the optimized reaction conditions; however, the ortho-substituted substrates showed slow progress and reactivity producing the target product in moderate yields. Notably, 2-(naphthalen-1-yl)-2-oxoacetic acid was successfully coupled to 3-phenyl (4-ethylphenyl)propiolate delivering the substituted coumarin in 67% isolated yield. Heteroaromatic and aliphatic substituted substrates did not produce the desired products, presumably due to the lower stability of the acyl radicals. The radical mechanism was confirmed by using TEMPO or BHT, as a radical scavenger that completely inhibited the reaction.
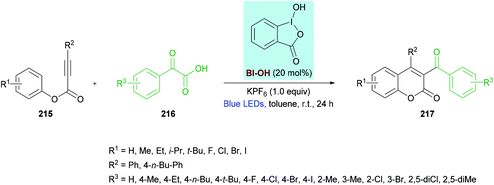 |
| | Scheme 54 Hypervalent iodine reagent (HIR)-catalyzed synthesis of 3,4-disubstituted coumarins from phenylpropiolates and α-oxocarboxylic acids. | |
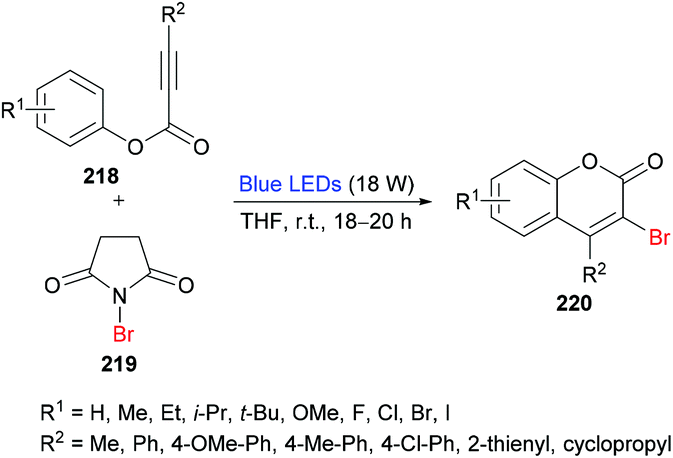
She and co-workers213 demonstrated a facile approach for the preparation of 3-bromocoumarins 220 from alkynoates 218 through visible-light-mediated radical cyclization using NBS 219 as the bromo source (Scheme 55). This strategy involves a bromo radical addition/spirocyclization/ester migration cascade sequence and can be performed under very mild reaction conditions in the absence of any catalyst or strong oxidant. Initially, various reaction conditions were screened using tolyl alkynoate and NBS with an 18 W blue LED as a visible light source. A range of solvents including MeCN, DCM, DMSO, DMF, THF, and 1,4-dioxane were employed. The best results were obtained in THF. The scope and diversity of the substrates used to prepare coumarins were investigated. Various alkyl groups (methyl, ethyl, i-propyl, tert-butyl) at the para-position of the aryl ring were suitable under the optimized reaction conditions to furnish the desired 3-bromocoumarins in 63–75% yields. In the case of the methoxy substituent, the product was isolated in 52% yield only. Substrates featuring a halogen substituent (fluoro, chloro, bromo and iodo) were also compatible, albeit producing the anticipated products in moderate yields (51–59%). ortho-Substituted alkynoate provided a complex mixture without the detection of the desired product. Different substituents at the alkyne carbon were also suitable for this transformation. Moreover, the presence of a bromo substituent at the 3-position of coumarin proved to be a viable functional handle to perform Suzuki–Miyaura cross-coupling214 and Buchwald–Hartwig amination reactions (Scheme 56). Some 3,4-biphenyl coumarin derivatives having biological significance215,216 were also synthesized using this methodology.217
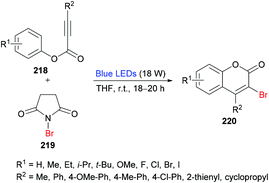 |
| | Scheme 55 Visible-light-mediated synthesis of 3-bromocoumarins from alkynoates and NBS. | |
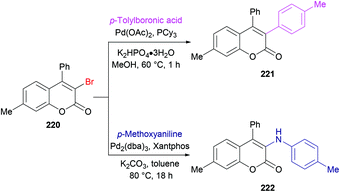 |
| | Scheme 56 Synthetic manipulation of 3-bromocoumarin 220 using Suzuki–Miyaura cross-coupling and Buchwald–Hartwig amination reactions. | |
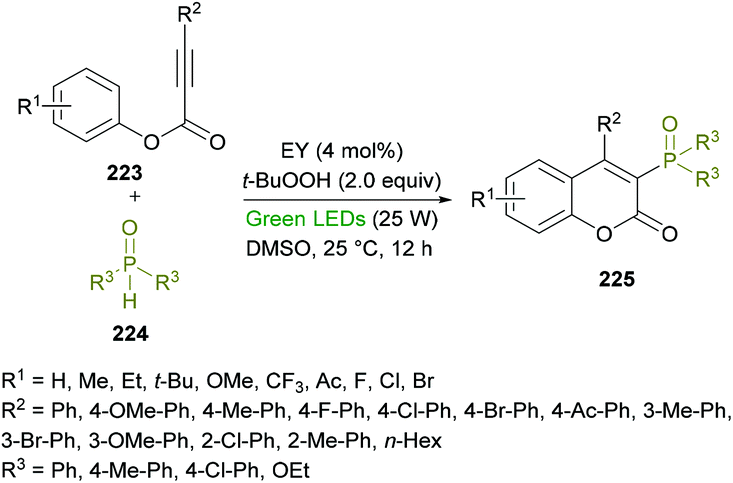
Xu and co-workers218 developed a metal-free, visible-light-promoted direct difunctionalization of alkynoates 223 using eosin Y (EY) as the photocatalyst and tert-butyl-hydroperoxide (TBHP) as the oxidant (Scheme 57). This process involves a radical tandem phosphorylation/cyclization reaction sequence to afford a range of 3-phosphorylated coumarins 225 in good yields and excellent regioselectivities. Avoiding the use of stoichiometric amounts of radical initiators or high-energy UV light,219 for the generation of P-centered radicals, visible light photoredox catalysis was employed. The reaction optimization was started with phenyl 3-phenylpropiolate, diphenylphosphine oxide (radical precursor) with TBHP as the oxidant under the influence of an organic photocatalyst and the irradiation of visible light. A survey of various photocatalysts, oxidants and solvents to produce the 3-phosphorylated coumarin was conducted with the identification of EY as the photocatalyst, TBHP as the oxidant, and DMSO as the solvent rendering the heterocyclic product in 87% yield. This set of conditions was further applied to various alkynoates and phosphoryl radical precursors.
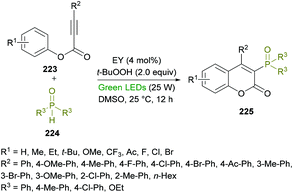 |
| | Scheme 57 Visible-light-promoted synthesis of 3-phosphorylated coumarins from alkynoates and phosphine oxides. | |
The variation of the R2 substituent at the alkyne component of the alkynoate demonstrated good functional group tolerance. Electron-donating (methyl, methoxy) and electron-withdrawing (fluoro, chloro, bromo, ketone) groups at the phenyl ring showed equal participation in this domino radical cyclization process. Alkyl group substituted alkynoates exhibited no reactivity. Although, other P-centered radical precursors proved to be good coupling partners, diethyl phosphonate furnished the corresponding product in 35% yield only. The next round of structural changes includes the variation of different substituents at the phenoxy ring of alkynoates. Again, various substituents including methyl, ethyl, tert-butyl, methoxy, trifluoromethyl, fluoro, chloro, and bromo were successfully tolerated. The extended aromatic system (2-naphthyl) also gave the desired coumarin in 55% isolated yield; however, ortho-substituted phenoxy rings were found to be sensitive to standard reaction conditions. Gram-scale synthesis was also performed with the isolation of the target product in 74% yield.
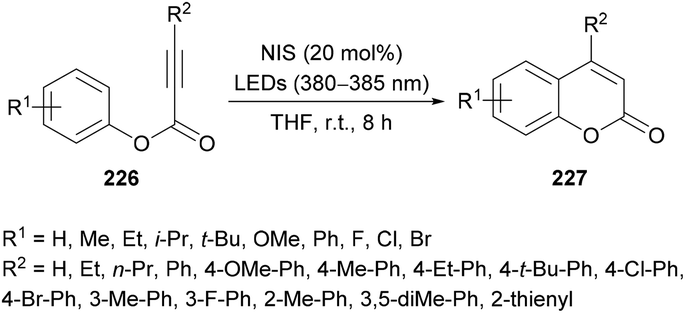
Li and co-workers220 prepared a library of coumarin derivatives 227 using N-iodosuccinimide as a free-radical initiator under photo-irradiation. This metal-free methodology uses readily available starting materials (alkynoates 226) and involves free radical intramolecular cyclization and ester rearrangement to afford coumarin products at room temperature (Scheme 58). p-Tolyl 3-phenylpropiolate served as a model substrate to optimize the reaction conditions. Various iodine sources (iodobenzene, TBAI, NIS, I2) in different solvents (PhMe, DCE, MeCN, DMF, DMSO, THF) were investigated. The best results were obtained in THF using 20 mol% of NIS delivering the coumarin product in 91% yield with a trace amount of 3-iodo-4-phenyl-coumarin. The variation of electronically and sterically different groups was introduced at the aryl component of the alkynoate to investigate the substrate scope. Electron-donating (methyl, ethyl, i-propyl, tert-butyl, methoxy, phenyl) and electron-withdrawing (fluoro, chloro, bromo) groups were successfully tolerated, However, strongly electron-deficient groups such as nitro, trifluoromethyl and formyl substituted alkynoates failed to show any reactivity under the optimized reaction conditions. Di-halogenated alkynoate also produced the desired coumarin in 77% yield. The reaction did not tolerate ortho-substituted and heterocyclic substrates. The scope was further broadened using alkyl and aryl substituents at the CC triple bond of the alkynoate. Methyl, ethyl, tert-butyl, methoxy, fluoro, chloro, and bromo at the phenyl ring were all compatible. 3-Alkyl-coumarins were successfully accessed from 3-ethyl- and 3-propylpropiolic acids in good yields; however, unsubstituted as well as heteroaryl substituted alkynoates showed no product formation. The mechanistic experiments revealed that the selection of the wavelength of light and the loading of NIS were critical in the construction of de-iodinated coumarins.
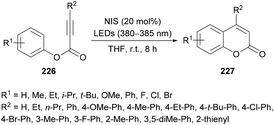 |
| | Scheme 58 Photoinduced cyclization of alkynoates to coumarins with N-iodosuccinimide as a free-radical initiator. | |
Guo and co-workers221 reported an electrochemical oxidative annulation of alkynoates 228 with diselenides or disulfides 229 to access a range of coumarins 230 (Scheme 59). In an electrochemical environment, oxidative cyclization generated new chalcogen-substituted coumarins under metal- and oxidant-free conditions. The model reaction was composed of a graphite anode and a platinum cathode to perform electrochemical oxidative cyclization of phenyl 3-phenylpropiolate, thus delivering 3-organoselenyl-2H-coumarin. A range of different electrolytes (n-Bu4NBF4, n-Bu4NPF6, n-Bu4NI, n-Bu4NBr, Et4NClO4) in various solvents (MeCN, DMF, DCE, HFIP) were screened. A variation in the constant current from 15 mA resulted in decreased reactivity. The scope of the electrochemical oxidative reaction includes the investigation of various alkynoates with diselenides or disulfides. The para-substituted phenoxy ring bearing both electron-rich (Me, Et, t-Bu) and electron-deficient (F, Cl, Br, CF3, Ac) groups demonstrated complete tolerance under the optimized conditions. In addition, the substrates incorporating alkyl (Me, Et) and aryl substituents at the alkyne moiety also showed high compatibility; however, when R2 = H, no reaction occurred. Pleasingly, the ortho-methyl substituted alkynoate produced the cyclized product in 75% yield. On the other hand, both aliphatic and aromatic diselenides were smoothly coupled to alkynoates. The reaction of 2-naphthyl alkynoate also gave the coumarin product in 75% yield. Furthermore, disulfide also exhibited good tolerance under electrochemical oxidative cyclization conditions to afford the desired products in 53%–56% yields. This green protocol could also be performed at the 5 mmol scale generating the corresponding product in 58% yield.
 |
| | Scheme 59 Electrochemical oxidative synthesis of functionalized coumarins from alkynoates and diselenides or disulfides. | |
2.2.2. Synthesis of chromones.
Liu and co-workers222 developed an organocatalytic intramolecular Stetter-type hydroacylation reaction between an aldehyde and activated alkynes to produce chromones 234, 235 (Scheme 60). A range of parameters including N-heterocyclic carbenes, bases, and solvents were tested to generate the optimized reaction conditions. After careful investigation, carbene (20 mol%), triethylamine (20 mol%) and DMF solvent were identified as the optimized conditions. Using these conditions, the scope and generality of the reaction were tested. A diverse majority of salicylaldehydes incorporating electron-rich and electron-deficient groups delivered the desired chromones in good yields. Halogens at the phenyl ring were also successfully tolerated. On the other hand, alkynes bearing a ketone functionality as the electron-deficient substituents (acetyl, trimethylacetyl, benzoyl, phenylacetyl) were efficiently used, delivering the desired products in 66–74% yields.
 |
| | Scheme 60 N-Heterocyclic carbene-catalyzed synthesis of chromones through intramolecular hydroacylation of activated alkynes. | |
The proposed mechanism for this transformation starts with the nucleophilic attack of carbene on the electrophilic carbonyl carbon of the aldehyde which on proton transfer results in an acyl anion equivalent (Breslow intermediate).223–225 The subsequent attack of a nucleophilic carbon on an activated alkyne thus generates a new carbon–carbon bond. Proton exchange followed by elimination of the catalyst produced the exocyclic kinetic product which underwent isomerization to afford the aromatized product (Scheme 61).
 |
| | Scheme 61 Proposed mechanism for the formation of chromones 234. | |
Xue and co-workers226 demonstrated the synthesis of chromones while investigating the phosphine-catalyzed α-addition reaction of 1,3-diketones 236 with terminal alkynoates 237 (Scheme 62). 1,3-Dicarbonyl substrates containing an alkyl group provide chromone derivatives 238 whereas aromatic substituents lead to the formation of vinylesters 239, important building units in the construction of nitrogen-containing heterocycles and other functionalized molecules.227–233
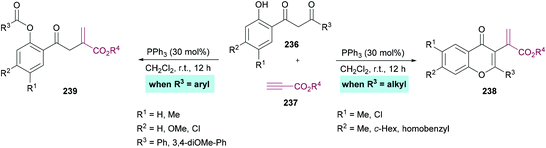 |
| | Scheme 62 PPh3-catalyzed synthesis of chromones through the α-addition reaction of 1-(o-hydroxyaryl)-1,3-diketones with terminal alkynoates. | |
Based on the literature reports,234,235 the authors proposed the reaction mechanism which starts with the nucleophilic addition of PPh3 to the electron-deficient alkyne 237 to form zwitterion 240. 1-(o-Hydroxyaryl)-1,3-diketone was deprotected to produce 241 and 242. Subsequent proton transfer in 242 produced 243 which then adds to 241 to furnish 244. A second proton transfer gave 245 which was cyclized via an intramolecular nucleophilic addition to produce 246, a key divergent intermediate for both products. In the first case, when R3 is an aryl group, a carbon–carbon bond cleavage in intermediate 246 led to 247, which on elimination of the phosphine catalyst furnished product 237. However, when R3 is an alkyl group, another proton transfer followed by the elimination of PPh3 gave 249. The corresponding heterocyclic product 238 was furnished on dehydration of 249. Notably, the divergent reactivity towards the formation of different products was possibly dependent on the stabilization of the intermediate 246 (Scheme 63).
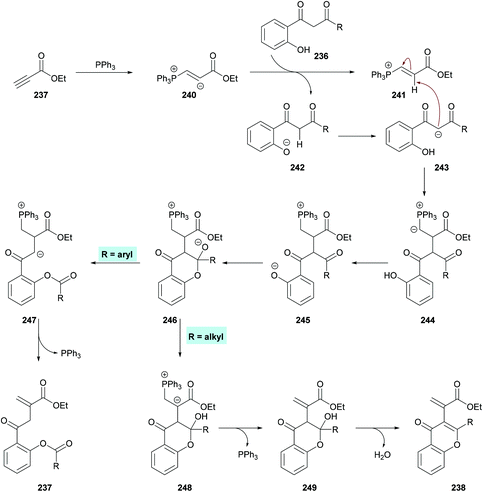 |
| | Scheme 63 Proposed mechanism for the formation of chromones 238. | |
2.2.3. Synthesis of chromanones.
Xue and co-workers236 developed a DABCO-catalyzed condensation method for the synthesis of chromanones 252 from ortho-acylphenols 250 and terminal alkynoates 251 where KOt-Bu was employed in the second step of this one-pot process for intramolecular cyclization (Scheme 64). During the course of reaction optimization, the addition of DABCO and KOt-Bu in two subsequent steps was found to be critical as a trace amount of the product was observed when both were added simultaneously. A diverse range of chromanone products were furnished using various ortho-acylphenols bearing different functional groups. Substrates endowed with electron-rich substituents on the aromatic part performed better compared to electron-deficient substituted ortho-acylphenols. Notably, the multi-substituted ortho-acylphenols were also found to be viable substrates producing the corresponding products in moderate yields.
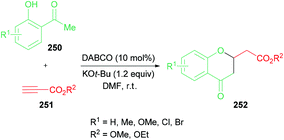 |
| | Scheme 64 DABCO-catalyzed synthesis of 2-alkyl-substituted chromanones from ortho-acylphenols and terminal alkynoates. | |
2.2.4. Synthesis of furans.
Deng and Chuang237 developed a multi-component approach involving diynedioates 253, phosphines 254 and aromatic aldehydes 255 delivering a range of multisubstituted furans 256 bearing phosphorus ylides in good yields (Scheme 65). The reaction sequence involves the nucleophilic attack of phosphine on diynedioates generating a 1,5-dipolar species which undergoes addition to aromatic aldehydes followed by 5-endo-dig cyclization. Various reaction conditions were screened, and the desired product was isolated in an optimal yield when o-dichlorobenzene was employed as a solvent at 60 °C. The scope of the reaction was examined using various phosphines, diynedioates, and aldehydes. Electron-rich phosphines performed better producing the corresponding furans in 81–85% yields compared to electron-deficient phosphines. Furan products were also isolated in comparable yields when heteroaromatic phosphines were employed. Various substituted benzaldehydes and 4-pyridinecarboxaldehyde also participated generating the anticipated heterocyclic products in remarkable yields. Moreover, di-n-butyl hexa-2,4-diynedioate, diethyl hexa-2,4-diynedioate, and dibenzyl hexa-2,4-diynedioate delivered the furans in good yields. Notably, the highly nucleophilic phosphines (PMe3 or PBu3) were unreactive under the optimized conditions in addition to electron-rich aldehydes. The furan products were further functionalized under oxidative conditions using mCPBA (2.4 equiv.) to afford α-keto esters 257.
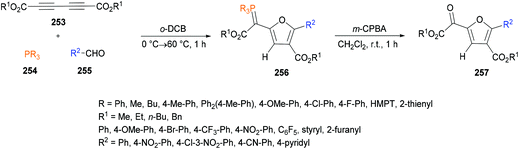 |
| | Scheme 65 Multi-component synthesis of furans containing phosphorus ylides from phosphines, diynedioates and aryl aldehydes. | |
Xu and co-workers238 highlighted a simple and straightforward synthetic approach to access a series of structurally diverse polysubstituted furans 260 from sulfur ylides 259239–245 and alkyl acetylenic carboxylates 258. This process involves a tandem sequence of Michael addition, intramolecular nucleophilic addition, 4π ring opening, intramolecular Michael addition, and elimination to afford dialkyl furan-3,4-dicarboxylates in moderate to good yields (Scheme 66).
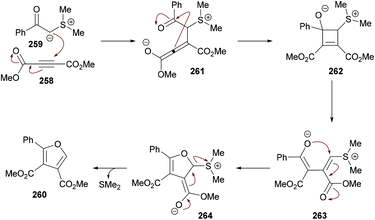 |
| | Scheme 66 Proposed mechanism for the formation of polysubstituted furans 260. | |
The model reaction was performed using dimethyl acetylenedicarboxylates and dimethylsulfonium benzoylmethylide in 1,2-dichloroethane. Further optimization of the reaction conditions included the variation of the starting material ratios in a variety of solvents (DMF, MeCN, DMSO) at different temperatures. The desired product was isolated in 79% yield when the reaction was conducted in DMSO at 80 °C under a nitrogen atmosphere (Scheme 67). Subsequent experiments were performed using these conditions to demonstrate the versatility of the protocol using various sulfonium ylides and alkynoates. Furan-3,4-dicarboxylates were isolated in moderate to good yields. A range of substituents (methyl, methoxy, fluoro, chloro, bromo, trifluoromethyl, cyano, nitro, dichloro) at the phenyl ring of sulfonium ylides were compatible. 1-Naphthyl and 2-naphthyl bearing sulfonium ylides also produced the desired products in 85% and 75% yields, respectively. Diethyl acetylenedicarboxylate was also successfully reacted with different dimethylsulfonium 2-aryl-2-oxoethylides. The scope was further extended to alkyl 2-substituted furan-3-carboxlyates using alkyl propynoates and the desired 4-trifluoromethylfuran-3-carboxylates were constructed in 44–57% yields; however, methyl propynoate remained less reactive and delivered the corresponding furan-3-carboxylate product in 15% yield. This method was also useful to deliver trialkyl furan-2,3,4-tricarboxylates when the reactions were performed at 160 °C under microwave irradiation conditions.246–251 Furthermore, dialkyl furan-2,4-dicarboxylates were prepared; however the regioselectivity was different from those reported in the literature.252
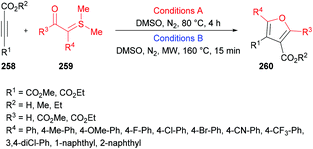 |
| | Scheme 67 Regiospecific synthesis of polysubstituted furans from sulfonium acylmethylides and acetylenic esters. | |
In view of the significant influence of the trifluoromethyl or perfluoroalkyl group on the lipophilicity, metabolic stability and permeability253–261 of biologically active molecules, trifluoromethylated or perfluoroalkylated furans have gained central importance due to their anti-HIV, antibacterial and antiparasite activities.262–264 To access these scaffolds, Zhang, Cao and co-workers265 developed a one-pot multi-component methodology using isocyanides 265, methyl perfluoroalk-2-ynoates 266, and aromatic aldehydes 267. The process operates at room temperature providing an easy access to a diverse range of 2-amino-3-perfluoroalkylfurans 268 in good to excellent yields (Scheme 68). Based on previously developed conditions for the synthesis of perfluoroalkylated cyclopentadienes, the optimization investigation was started using isocyanides, 2-isocyano-2-methylpropane, methyl 4,4,4-trifluorobut-2-ynoate and 2-nitrobenzaldehyde as model substrates in dichloromethane. Other solvents such as THF, CHCl3 and toluene produced poor results. The reaction scope survey revealed the compatibility of all the three components. Both aliphatic and aromatic aldehydes incorporating electron-donating and electron-poor groups were employed. The electronic nature of the substituents displayed significant influence on the reactivity with electron-withdrawing groups being more productive in terms of the product yields. Electron-deficient heterocyclic aldehydes (picolinaldehyde) also turned out to be good coupling units. Moreover, various perfluoroalk-2-ynoates were also included in the multi-component reaction. Alkynoates with CF3, C2F5, and n-C3F7 groups were well tolerated delivering the desired products in high yields. The scope was further expanded to isocyanides bearing tert-butyl, cyclohexyl and 2,4,6-trimethylphenyl groups displaying high reactivity and furnishing the corresponding 2-amino-3-perfluoroalkylfurans in good to excellent yields.
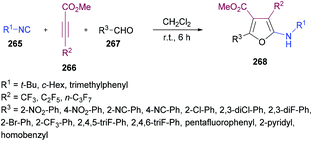 |
| | Scheme 68 Multi-component synthesis of 2-amino-3-perfluoroalkylfurans from isocyanides, methyl perfluoroalk-2-ynoates and aldehydes. | |
Mechanistically, this multi-component methodology starts with a regioselective Michael addition of isocyanide 265 to methyl 2-perfluoroalkynoate 266 generating 1,3-dipolar intermediate 269 which on nucleophilic addition to aldehyde 267 produced the zwitterionic intermediate 270. Subsequent cyclization followed by a [1,5]-H shift delivered the aminofuran product 268266–268 (Scheme 69).
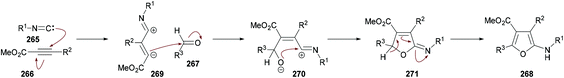 |
| | Scheme 69 Proposed mechanism for the formation of 2-amino-3-perfluoroalkylfurans 268. | |
2.2.6. Synthesis of dioxanones and benzodioxinones.
Taran and co-workers272 introduced a one-step phosphine-catalyzed method for the preparation of 1,4-dioxane-2-one 288 and 1,4-benzodioxine-2-one derivatives 289 from 1,2-diol starting materials 286, 287 (Scheme 72). A range of diols were efficiently converted to the desired products at room temperature. The methodology was further extended to the synthesis of 1,4-benzodioxin-2-ones using the phosphine-catalyzed α-O-addition and transesterification reaction. The title products were obtained as Z-isomers in acceptable yields. The reactivity of the reaction was found to be highly dependent on the pKa of the diol and/or the nucleophilicity of the corresponding catecholate. Electron-rich catechols furnished the best results compared to electron-deficient catechols.
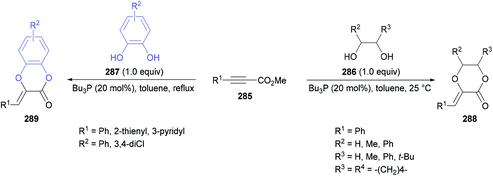 |
| | Scheme 72 Phosphine-catalyzed synthesis of 1,4-dioxane-2-ones and 1,4-benzodioxine-2-ones from 1,2-diols and alkynoates. | |
2.2.7. Synthesis of lactones.
Yu, Pu and co-workers273 discovered a convenient protocol for the asymmetric synthesis of 4-amino-2(5H)-furanones 293 involving alkynes 290, aldehydes and amines 292 (Scheme 73). The reaction can be performed at room temperature producing the heterocyclic products with high enantioselectivity (84–90% ee). The investigation is based on their previous reports where γ-hydroxy-α,β-acetylenic esters274–278 were synthesized from an enantioselective reaction of methyl propiolate and aldehydes using 1,1′-bi-2-naphthol (BINOL) in combination with ZnEt2, Ti(OiPr)4, and HMPA. In the present approach, these esters were subsequently cyclized with aliphatic amines to produce amino furanones. Various conditions were tested for this two-step process to establish the optimized protocol. Under the optimal reaction conditions, different aldehydes were initially coupled to methyl propiolate followed by the addition of benzylamine to afford 4-amino-2(5H)-furanones. Moderate to good isolated yields were obtained with high enantioselectivity. Linear and branched aliphatic aldehydes were efficiently tolerated alongside cyclic and few aromatic amines. Moreover, the use of racemic alkyl- and aryl-substituted acetylenic esters were also investigated for the addition reaction of primary and secondary amines. The corresponding products were isolated in good yields. The synthetic modification of the products illustrated that 3-brominated heterocycle 294 could be accessed when treated with Br2, although bromination and iodination of 4-amino-2(5H)-furanones using NBS279 and ICl or I(py)2BF4280–282 were previously known. Similarly, C-acylated product 296 was isolated in an excellent yield (95%) instead of an N-acylated product using trifluoroacetic anhydride at room temperature; however, N-protected product 295 was furnished in 99% yield when the reaction was performed with (Boc)2O using DMAP at room temperature (Scheme 74).
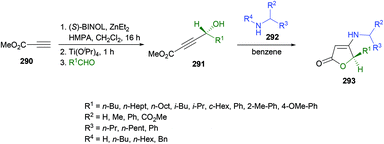 |
| | Scheme 73 Asymmetric synthesis of 4-amino-2(5H)-furanones from alkynoates, aldehydes and amines. | |
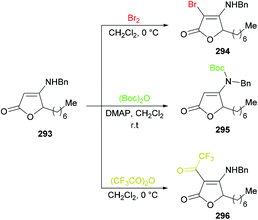 |
| | Scheme 74 Synthetic modification of 4-amino-2(5H)-furanone 293. | |
Zlotin and co-workers283 investigated the reaction of methyl 4-hydroxyalk-3-ynoates 297 and amines 298 to afford 4-aminofuran-2(5H)-ones 299 in ionic liquids (Scheme 75). The reaction rate was accelerated many fold while reducing the time and improving the product yield. Several examples were synthesized, and the data were compared with the literature showing the supremacy of the reaction conditions employing ionic liquids. Additionally, the recyclability of IL [bmim][BF4] was demonstrated in the synthesis of butenolides. Reaction rates and product yields remained unaffected over at least five reaction cycles.
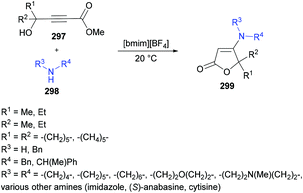 |
| | Scheme 75 Synthesis of 4-aminofuran-2(5H)-ones from methyl 4-hydroxyalk-3-ynoates and amines in ionic liquids. | |
Deng and Chuang284 reported a series of γ-lactones 203 bearing an α-phosphorus ylide functionality from hex-2-en-4-ynedioic acid dialkyl esters 200, phosphines 201 and aldehydes 202 (Scheme 76). The authors investigated various reaction conditions including solvents (THF, DCE, DCM) at different reaction times and temperatures. Chlorinated solvents were found to be less effective compared to THF. Initially, the reactivity of different phosphines was tested. Triarylphosphines produced the desired lactones in moderate to good yields (55–79%). Electron-donating aryl bearing phosphines generally performed better compared to the electron-withdrawing one whereas heteroaryl phosphine was also a smooth coupling partner, albeit producing the lactone in a lower yield (55%). Moreover, phosphine endowed with an alkyl substituent could only produce the desired product in 38% yield. On the other hand, substituted aldehydes were explored to expand the reaction scope. Electron-deficient aldehydes containing nitro, chloro and nitrile functional groups generally produced efficient results; however, attempts to use alkyl and electron-rich substituted aldehydes remained unproductive.
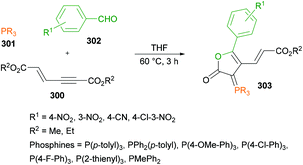 |
| | Scheme 76 Multi-component synthesis of γ-lactones featuring α-phosphorus ylides from enynes, phosphines and aldehydes. | |
Chen, Li and co-workers285 demonstrated a task-specific acidic ionic liquid-catalyzed synthesis of β-enaminolactones 306. Alkynoates 304 and β-amino alcohols 305 were successfully cyclized to the desired products in ample yields (Scheme 77). The formation of a seven-membered ring involves tandem intermolecular hydroamination and intramolecular transesterification reaction sequences. Diethyl but-2-ynedioate and 2-amino-2-phenylethanol were chosen as model substrates. Initially, various Lewis acid catalysts (AlCl3, ZnCl2, FeCl3, Yb(OTf)3, Cu(OTf)2, Zn(OTf)2) were tested and the results indicated that Cu(OTf)2 and Zn(OTf)2 exhibit good reactivity. As the role of the acid catalyst was found to be critical, the sulfonic-functionalized ionic liquid286–288 (TSFIL-1:[TMBSA]HSO4) was tested due to its many advantages289–295 and it showed an excellent reactivity and produced the desired lactone in 94% yield. Other parameters including variation of solvents and temperature exhibited inferior results. Under the established reaction conditions, the flexibility of this protocol was assessed. Both alkynoates and β-amino alcohols bearing different groups were employed. Two-point structural diversity was present in each component of this methodology. Substrates with various alkyl and aryl substituents were smoothly converted to their corresponding lactones in moderate to good yields. Primary and secondary alcohols also produced promising results. In general, this method showed a broad substrate scope with good functional group tolerance and operational simplicity.
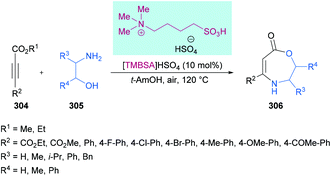 |
| | Scheme 77 Task-specific acidic ionic liquid-catalyzed synthesis of β-enaminolactones from alkynoates and β-amino alcohols. | |
Waldmann, Kumar and co-workers296 developed organocatalyzed tricyclic benzopyrones 309 involving a [4 + 2] annulation reaction of 3-formylchromones 308 with electron-deficient alkynes 307 (Scheme 78). Under the optimized reaction conditions, a varied range of substituents were endured at the chromone ring and alkynoates. Unsubstituted, alkyl- and alkoxy-substituted chromones and alkynoate esters were efficiently coupled to produce pyran-fused chromones in good yields. Moreover, acyclic oxadienes 310 were also employed as substrates with acetylene carboxylates to generate [4 + 2] annulation products that underwent a subsequent Claisen rearrangement to afford dehydropyrans 311. A range of different substituents on both starting materials were successfully tolerated. Next, the asymmetric version of this transformation was explored by employing a β-isoquinidine catalyst.
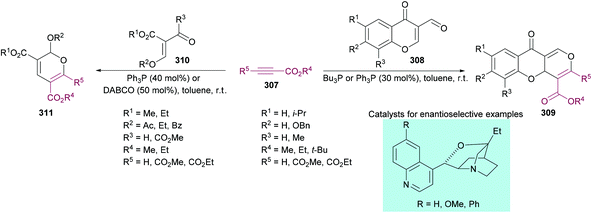 |
| | Scheme 78 Asymmetric organocatalyzed synthesis of tricyclic benzopyrones. | |
2.2.8. Synthesis of pyrones.
Jiang and co-workers297 developed a multi-component methodology involving the hydroalkylation of alkynoates 312 and activated methylenes 313 for the preparation of functionalized 2H-pyran-2-ones 314 (Scheme 79). The target products were obtained in ample yields. Various reaction parameters including temperature, alkaline additives and solvents were screened to obtain the appropriate reaction conditions. The use of 1.0 equivalent of sodium hydroxide in 1,4-dioxane solvent at 80 °C proved to be the optimal conditions producing the desired heterocycle in 89% yield; however, the further experimentation was conducted using 10 mol% of sodium hydroxide as 88% yield was observed. Using these conditions, the scope of the methodology was examined. Several active methylene compounds were successfully coupled to alkynoates to generate six-membered oxygen heterocycles in good yields. Moreover, when ethyl 3-phenylprop-2-ynoate and diethyl propanedioate and formaldehyde were coupled in one pot, diethyl 6-oxo-4-phenyl-2H-pyran-3,3(6H)-dicarboxylate 315 was obtained in 82% isolated yield. Trisubstituted 2H-pyran-2-ones were only obtained when dibenzoylmethane, 1-phenyl-1,3-butanedione, 2,4-pentanedione, and ethyl acetoacetate were used as activated methylene partners.
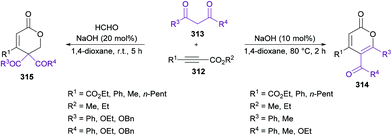 |
| | Scheme 79 Synthesis of polysubstituted 2H-pyran-2-ones from alkynoates and activated methylenes. | |
Scheme 80 illustrates the possible mechanistic pathway which starts with the activation of 1,3-dicarbonyl compound 313 to form 316. Nucleophilic addition of 316 to alkynoate 312 produced intermediate 317 which could possibly be present in different resonance structures, such as 318, 319, and 320. Nucleophilic attack on the carbonyl carbon delivered product 314. In the presence of an aldehyde, intermediate 320 was converted to intermediate 322. An intramolecular oxygen nucleophilic cyclization furnished the desired product 315.
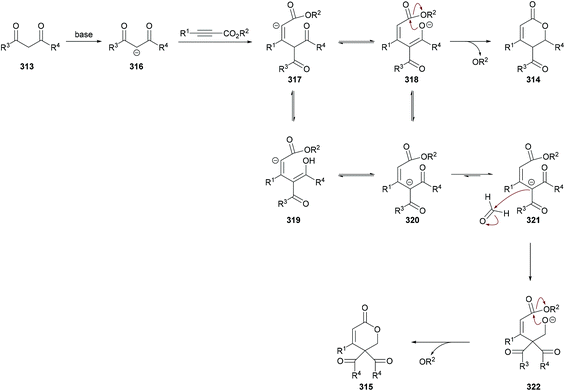 |
| | Scheme 80 Proposed mechanism for the formation of pyranones 314, 315. | |
2.3. Synthesis of saturated oxygen-containing heterocycles
Chung and Fu298 established that chiral spiro phosphepine ligand 323 can transform a variety of hydroxy-2-alkynoates 324 into various saturated oxygen heterocycles 325 (tetrahydrofurans, tetrahydropyrans, dihydrobenzopyrans) with good enantioselectivity (Scheme 81). Using the optimized reaction conditions, a variety of substrates were converted to products with high enantioselectivity and good yields.
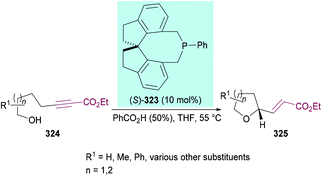 |
| | Scheme 81 Phosphine-catalyzed enantioselective synthesis of oxygen heterocycles. | |
In the same year, Pedduri and Williamson299 reported a phosphine-catalyzed reaction to access various functionalized tetrahydrofurans 328, 329 using electron-deficient propargyl alcohols 326 and Michael acceptors 327 as starting materials (Scheme 82). Several reaction conditions were investigated to identify the suitable conditions. Under the optimized conditions, alkylidene-, arylidene-, and heteroarylidene malonate/Meldrum's acid-based alkene derivatives were included to assess the diversity of the reaction. The corresponding heterocyclic products were furnished in good yields as a separable E:Z mixture.
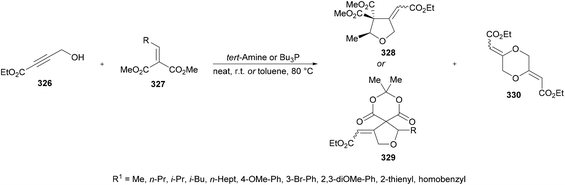 |
| | Scheme 82 Phosphine-catalyzed synthesis of functionalized tetrahydrofurans using electron-deficient propargyl alcohols and Michael acceptors. | |
Osman and Koide300 developed a new synthetic route for the synthesis of functionalized cyclic acetals 333 using 2-pyridinecarboxyaldehyde 332 and γ-hydroxy-α,β-acetylenic esters 331 (Scheme 83). The methodology operates under mild reaction conditions avoiding the need for any additive. After optimizing the suitability of various solvents, methanol was identified to produce the highest conversion and was chosen for subsequent experiments to explore the reaction scope. Various acetylenic esters bearing cyclohexyl, tert-butyl, aromatic and hetero-aromatic substituents were successfully tolerated. The post-synthetic functionalization of the products was also performed under hydrogenolysis conditions301 (Scheme 84).
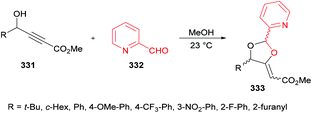 |
| | Scheme 83 Cyclic acetal formation between 2-pyridinecarboxaldehyde and γ-hydroxy-α,β-acetylenic esters. | |
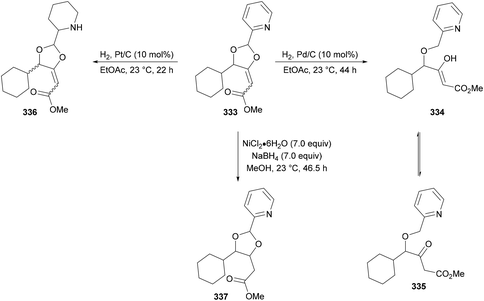 |
| | Scheme 84 Synthetic modification of cyclic acetals 333. | |
2.4. Synthesis of sulfur-containing heterocycles
Sulfur-containing heterocycles constitute the core structure of various pharmaceuticals, natural products and bioactive pharmacophores. These heterocycles also find wide applications in synthetic organic chemistry.302 Due to their widespread utility, facile and efficient synthetic methods are always required to meet the requirements in several drug discovery programs.
2.4.1. Synthesis of iminothiazolidin-4-ones and 2-thioxo-4-thiazolidinones.
Taran and co-workers303 developed an efficient and facile phosphine-catalyzed approach for the construction of sulfur heterocycles (Scheme 85). This methodology involves a tandem umpolung addition and intramolecular cyclization of bifunctional sulfur pronucleophiles on aryl propiolates 338. The method appeared to tolerate a diverse range of substrates (sulfur–oxygen or sulfur–nitrogen pronucleophiles) leading to 5- and 6-membered sulfur-containing heterocycles 339, 340. Mercapto alcohols, amines, and amides all worked well to provide the desired arylidene heterocycle Z isomers in moderate to good yields. Moreover, thioureas can also be employed as bifunctional S,N-pronucleophiles producing 2-iminothiazolidin-4-one compounds. Finally, the synthesis of rhodanine (2-thioxo-4-thiazolidinone) was accomplished using dithiocarbamates and aryl propiolates under similar reaction conditions. A variety of amines were reacted with carbon disulfide to generate dithiocarbamates in situ which were used to afford the desired cyclized product.
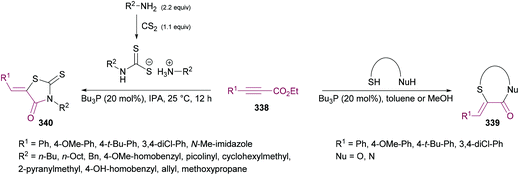 |
| | Scheme 85 Phosphine-catalyzed synthesis of sulfur heterocycles. | |
2.4.2. Synthesis of thiohydantoins.
Loreau, Taran and co-workers304 made use of arylpropiolates 341 and thioureas 342 for the synthesis of thiohydantoins 343, 344 (Scheme 86). The present methodology operates via phosphine-catalyzed tandem umpolung addition and an intramolecular cyclization process. This method is different from the classical approach which involves two-step synthesis, aiming at the preparation of the thiohydantoin moiety followed by a Knoevenagel condensation with aldehydes.305,306 This concept originates from the dual reactivity of thioureas where α-N-addition occurs at aryl propiolates under Bu3P catalysis followed by cyclization to produce thiohydantoins. With the optimized reaction conditions in hand, a range of thioureas were employed as coupling partners with aryl propiolates to afford numerous 5-arylidene-2-thiohydantoins. The regioselectivity was dependent on the nature of the substituents present in the thiourea functionality. Alkyl, aryl and hetero-aryl bearing thioureas participated efficiently. However, there is a competition in the α-N-addition step between the two nitrogen atoms of the thiourea functionality and the reactivity of the NH groups is influenced by steric hindrance, nucleophilicity and pKa. Despite some limitations (poor yield and a mixture of regioisomers in certain cases), this method presents some advantages over the reported methods.
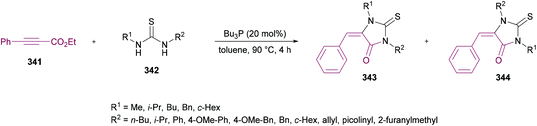 |
| | Scheme 86 Phosphine-catalyzed synthesis of thiohydantoins from thioureas and arylpropiolates. | |
2.4.3. Synthesis of thiophenes.
Liu and co-workers307 demonstrated a facile and convenient approach to access a small library of decorated thiophenes 347via the [2 + 2 + 1] cycloaddition reaction. A variety of alkynoates 345 and elemental sulfur 346 coupled together to generate the title products (Scheme 87).
 |
| | Scheme 87 Synthesis of polysubstituted thiophenes via the base-induced [2 + 2 + 1] cycloaddition reaction of alkynes and elemental sulfur. | |
2.5. Synthesis of O,N-heterocycles
2.5.1. Synthesis of oxazines.
Jiang and co-workers308 described a multi-component one-pot synthetic route for the synthesis of 3,4,5-trisubstituted-3,6-dihydro-2H-1,3-oxazines 351 from readily available starting materials (alkynoates 348, anilines 349, formaldehyde 350). The formation of a six-membered N,O-heterocyclic scaffold occurs via a Brønsted acid-catalyzed reaction sequence involving domino hydroamination/Prins reaction/cyclization/dehydration (Scheme 88). After successful identification of the optimized reaction conditions, the scope of the multi-component methodology was explored. A range of anilines and electron-deficient alkynes were employed alongside formaldehyde delivering the desired oxazines in good to excellent yields. Substituents at different positions of anilines were successfully tolerated; however, ortho-substituted anilines remained reluctant to produce the desired products.
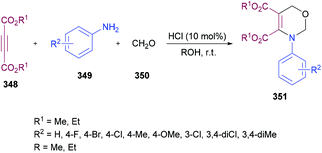 |
| | Scheme 88 Brønsted acid-promoted domino one-pot three-component synthesis of 3,4,5-trisubstituted-3,6-dihydro-2H-1,3-oxazines. | |
Curini and co-workers309 described the same methodology and produced 3,4,5-trisubstituted-3,6-dihydro-2H-1,3-oxazines 352via the ytterbium triflate promoted reaction sequence (Scheme 89). This methodology differs from the previous report in the use of both aliphatic and aromatic amines while avoiding excess formaldehyde and strong mineral acids like HCl.
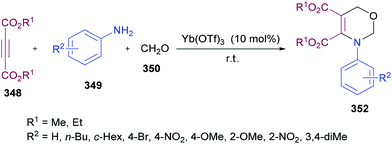 |
| | Scheme 89 Ytterbium triflate promoted one-pot three-component synthesis of 3,4,5-trisubstituted-3,6-dihydro-2H-1,3-oxazines. | |
2.5.2. Synthesis of oxazinones.
Jiang and co-workers310 developed a highly efficient three-component methodology involving but-2-ynedioic acid diethyl esters 353, amines 354 and aldehydes 355 to furnish polysubstituted ethyl 2,5-dihydro-2H-1,3-oxazine-6-one-4-carboxylates 356 in good to excellent isolated yields (Scheme 90). A range of solvents (MeCN, PhMe, Et2O, DCM, THF, EtOH, H2O) and bases (Na2CO3, Et3N, NaOH, KOH) were screened. A mixture of MeCN and H2O (10:1) was the best choice with the isolation of the heterocyclic product in 78% yield. A variety of amines and aldehydes were utilized to explore the reaction scope. In the case of aliphatic aldehydes, the corresponding products could be isolated in less than three minutes and in excellent yields. Both electron-rich and electron-deficient substituents at the aryl rings of both amines and aromatic aldehydes were also successful coupling partners.
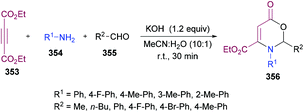 |
| | Scheme 90 Three-component synthesis of polysubstituted 2,5-dihydro-1,3-oxazin-6-ones from alkynoates, amines and aldehydes. | |
2.6. Synthesis of fused heterocycles
2.6.1. Synthesis of imidazo[1,2-a]pyridines.
Chen, Wang and co-workers311 developed a highly efficient transition-metal-free cyclization methodology for the preparation of 2,3-diarylimidazo[1,2-a]pyridines 359 involving 2-aminopyridines 357 and alkynoates 358 (Scheme 91). These starting materials were found to be compatible under the optimal conditions. The scope and limitations of the one-pot methodology were explored using a variety of both components. Electron-donating and electron-withdrawing groups were successfully tolerated on the alkynoate. In addition, the effect of different groups at various positions of the 2-aminopyridine moiety was also tested. Halogens including fluoro, chloro, and bromo substituents were also well tolerated. Alkynoates bearing a heteroaryl ring also gave the desired product in good yields; however, in certain cases, a longer reaction time was required.
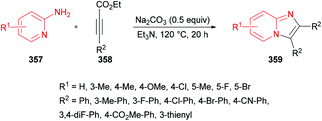 |
| | Scheme 91 Synthesis of 2,3-diarylimidazo[1,2-α]pyridines from 2-aminopyridines and alkynoates. | |
Mechanistically, 2-aminopyridine 357 was reacted with alkynoate 358 to produce intermediate 361 through 360via intermolecular Michael addition which upon an intramolecular [2 + 2] cycloaddition reaction produced intermediate 362. The desired product 359 was furnished on cleavage of the carbon–carbon bond of the cyclobutene ring via a ring-opening reaction. Diethyl but-2-enedioate byproduct 363 was detected with GC-MS (Scheme 92).
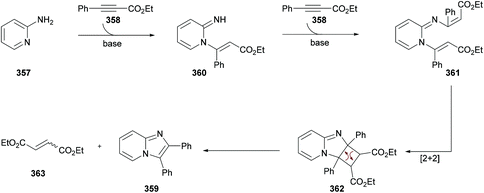 |
| | Scheme 92 Proposed mechanism for the formation of 2,3-diarylimidazo[1,2-α]pyridines 359. | |
2.6.2. Synthesis of isoindolin-1-ones.
Zhou, Lu and co-workers312 demonstrated a phosphine-catalyzed [3 + 2] annulation reaction involving electron-deficient alkynes 364 and N-hydroxyphthalimide 365 to provide a facile access to 3a-hydroxyisoxazolo[3,2-a]isoindol-8(3aH)-ones 366 (Scheme 93). The target fused products were isolated in remarkable yields. Various phosphines, solvents and temperatures were screened. Isoindolin-1-one products were isolated in excellent yields using triphenylphosphine (20 mol%) in DMF at room temperature. Under the optimized conditions, the scope of the reaction was explored delivering a variety of fused products in high yields. Initially, various terminal alkynoates were tested. A variety of ester substituents participated efficiently. The aromatic ring of benzyl propiolates bearing electron-donating and electron-withdrawing functional groups was found to be compatible. A long aliphatic chain (n-hexyl) substituted alkynoate showed no reactivity; however, ethyl 2-butynoate and ethyl 3-phenylpropiolate furnished the corresponding 3a-hydroxyisoxazolo[3,2-a]isoindol-8(3aH)-ones in 55% and 99% yields, respectively. These reaction conditions produce different results to those observed in the literature313 when the reaction was performed using DMAD and a stoichiometric amount of triphenylphosphine at room temperature in dichloromethane and a pyrroloisoindole compound was isolated.
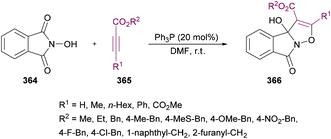 |
| | Scheme 93 Phosphine-catalyzed synthesis of 3a-hydroxyisoxazolo[3,2-a]isoindol-8(3aH)-ones via [3 + 2] annulation of electron-deficient alkynes with N-hydroxyphthalimide. | |
The authors proposed the mechanism of this transformation which was initiated by the nucleophilic addition of PPh3 to the electron-deficient CC bond of alkynoate 365 to form zwitterion 367, which was used to deprotonate N-hydroxyphthalimide 364 generating intermediates 368 and 369. Michael addition of 368 to 369 produced intermediate 370.314–316 Intramolecular nucleophilic cyclization furnished the cyclized intermediate 371 followed by proton transfer to give 372. Subsequent elimination of PPh3 produced the desired fused product 366 (Scheme 94).
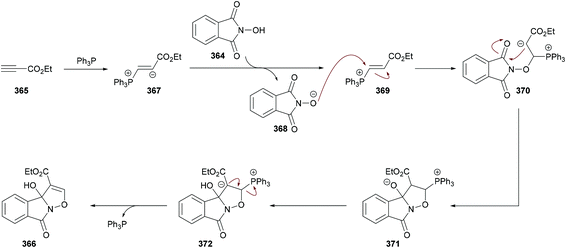 |
| | Scheme 94 Proposed mechanism for the formation of 3a-hydroxyisoxazolo[3,2-a]isoindol-8(3aH)-ones 366. | |
In a subsequent study, the same group317 reported a metal-free approach to access highly functionalized pyrazolo[5,1-a]isoindol-8(3aH)-ones 375 through a phosphine-catalyzed tandem Michael addition/intramolecular Morita–Baylis–Hillman reaction sequence involving alkynoates 374 and N-amino substituted phthalimide 373 (Scheme 95). Using the optimized reaction conditions, various alkynoates were tested producing the title products in moderate to good yields. Terminal alkynoates were compatible; however, methyl and phenyl substituted derivatives were unreactive. Various electron-rich and electron-deficient groups at the benzyl ring of propiolates appeared to be well tolerated. However, the presence of a strongly electron-withdrawing nitro group impeded the reaction, producing the corresponding product in 27% yield.
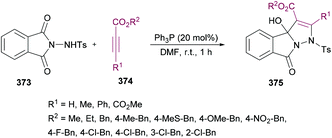 |
| | Scheme 95 Phosphine-catalyzed construction of pyrazolo[5,1-a]isoindol-8(3aH)-ones from electron-deficient alkynes and N-amino substituted phthalimide. | |
2.6.3. Synthesis of furo-thiazepines.
Ma and co-workers318 developed a three-component method involving thiazole or benzothiazole carbenes 376, substituted acetyl chloride 377 and electron-deficient alkynes 378 resulting in the formation of the polysubstituted heterocyclic scaffold furo[2,3-c]thiazepine 379. The title products were isolated in good yields using these simple and readily available starting materials (Scheme 96). Based on the previous efforts on the use of thiazole carbenes,319–325 the authors investigated a domino approach to construct a fused heterocyclic skeleton.326 For the investigation of reaction conditions, a thiazolium salt and α-methyl phenylacetyl chloride were used that generated thiazole carbene and ketene in situ. The effect of several typical bases such as NEt3, i-Pr2NEt, DBU, and NaH was also studied. i-Pr2NEt in dichloromethane provided the best result. Using the optimized reaction conditions, a diverse range of disubstituted acetyl chlorides were reacted with the thiazolium salt. The corresponding products were isolated in good to excellent yields. Substrates incorporating electron-rich and electron-deficient functional groups participated efficiently; however, the electron-poor system produced sluggish yields. The size of the alkyl group was also important as the isopropyl substituted substrate did not produce any product whereas methyl and ethyl substituents were completely productive. Dialkyl substituted substrates were also equally reactive producing the desired compounds in good yields. The reaction scope was further extended using various thiazolium salts and similar reactivities were observed in terms of the yields of the fused products. Furthermore, two different procedures were developed to investigate various electron-deficient alkynes and in most of the cases better yields were observed where ketenes were used. The authors also tested the benzothiazole carbenes and tricyclic heteroaromatic compounds were furnished in moderate yields. The synthetic utility of the selected derivatives was investigated in the Diels–Alder reaction with arynes to deliver 1,4-thiazepine-fused 7-oxanorbornadienes in excellent yields.327–329
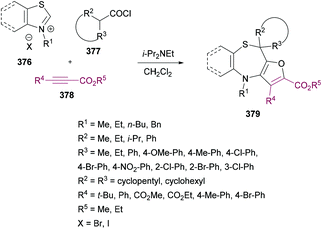 |
| | Scheme 96 Multi-component synthesis of highly substituted furan-fused 1,4-thiazepines. | |
2.6.4. Synthesis of tetrahydropyrazolopyrazolones and tetrahydropyrazolopyridazinones.
Zhong, Guo and co-workers330 unveiled a facile approach for the preparation of tetrahydropyrazolopyrazolones 382 and tetrahydropyrazolopyridazinones 383via phosphine-catalyzed [3 + 2] and [3 + 3] annulation reactions of azomethine imines 380 and ethyl 2-butynoate 381 (Scheme 97). This coupling reaction was optimized by using ethyl 2-butynoate with 1-(p-nitrobenzylidene)-3-oxopyrazolidin-1-ium-2-ide in the presence of different phosphine catalysts. Although the best results were achieved when dimethylphenylphosphine was employed with the isolation of 382 and 383 in 65% and 14% yields, respectively, the authors were interested to isolate both products due to their biological significance;331–335 therefore, the subsequent experimentation was performed with tributylphosphine as the catalyst. A range of azomethine imines were annulated with ethyl 2-butynoate in dichloromethane at room temperature. Tetrahydropyrazolopyrazolone and tetrahydropyrazolopyridazinone derivatives were obtained in moderate to good yields. Both electron-rich and electron-deficient substituents at the phenyl ring were compatible. Strongly electron-deficient groups (nitro, cyano) at the ortho- or para-position of the phenyl ring provided the target products in relatively much higher yields compared to electron-rich and weakly electron-withdrawing substituents, albeit with poor chemoselectivities toward the [3 + 2] annulation product. Moreover, azomethine imines bearing 1-naphthyl, 2-naphthyl and cyclohexyl groups also underwent a smooth reaction with 2-butynoate, producing the corresponding pyrazolidinone derivatives.
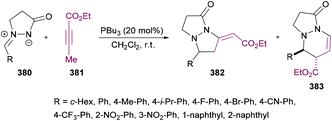 |
| | Scheme 97 Phosphine-catalyzed [3 + 2] and [3 + 3] annulations of azomethine imines with ethyl 2-butynoate. | |
2.6.5. Synthesis of spirooxindoles.
Yan and co-workers336 developed a divergent three-component approach using isatylidene malononitrile (ethyl cyanoacetate) 384 and triphenylphosphine 385 (Scheme 98). Depending upon the addition of the third component (but-2-ynedioate 386 or hex-2-en-4-ynedioate 387) in dimethoxyethane, triphenylphosphanylidene spiro[cyclopentane-1,3′-indolines] 388 and spiro[cyclopent[2]ene-1,3′-indolines] 389 were furnished in moderate to good yields. Using the optimized conditions, DMAD (1.2 mmol), isatylidene malononitrile (ethyl cyanoacetate, 1.0 mmol), and PPh3 (1.0 mmol) in DME (10 mL) at 0 °C → r.t. for 2 h, various spirocyclic compounds were accessed. Similarly, dialkyl hex-2-en-4-ynedioates were also employed to expand the substrate scope and the relevant products were isolated in good yields.
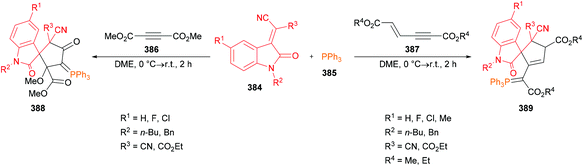 |
| | Scheme 98 Three-component synthesis of triphenylphosphanylidene spiro[cyclopentane-1,3′-indolines] and spiro[cyclopent[2]ene-1,3′-indolines]. | |
The mechanism for the formation of spiro[cyclopentane-1,3′-indolines] 388 is illustrated in Scheme 99. Intermediate 390 was furnished via a 1,4-dipolar addition reaction of PPh3385 and DMAD 386337–339 which then added to isatinylidene malononitrile 384 and delivered the intermediate 391. Subsequently, an intramolecular nucleophilic substitution reaction of the carbanion to alkoxide in the ester furnished the cyclic intermediate 392 which on 1,3-arrangement of the triphenylphosphanyl cation led to carbonium intermediate 393. The desired product was obtained by the coupling of carbonium 393 with methoxide in solution.
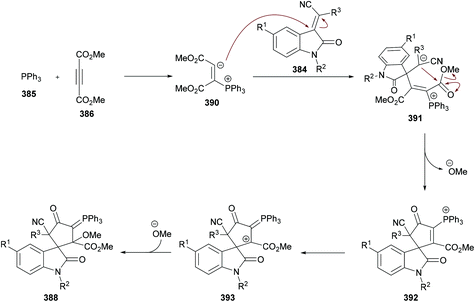 |
| | Scheme 99 Proposed mechanism for the formation of spiro[cyclopentane-1,3′-indolines] 388. | |
Scheme 100 illustrates the mechanistic pathway for the formation of product 389 in line with the literature precedents.340–343 Addition of PPh3385 to hex-2-en-4-ynedioate 387 furnished 1,3-dipolar zwitterionic intermediate 394 followed by the addition to isatinylidene malononitrile 384 leading to the formation of adduct 395. Subsequently, an intramolecular Michael addition of the carbanion to the 1,3-diene holding a stronger electron-deficient triphenylphosphanyl cation afforded the cyclized intermediate 396. Next, intermediate 397 was formed from 396 by allylic arrangement of the carbanion. Finally, the target product 389 was obtained via phosphorus ylide transfer.
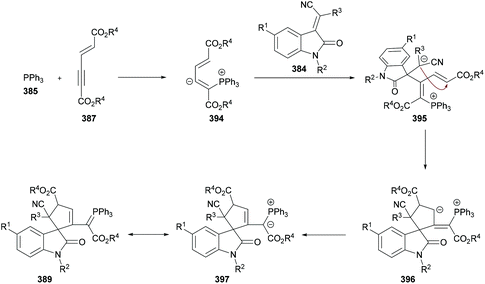 |
| | Scheme 100 Proposed mechanism for the formation of spiro[cyclopent[2]ene-1,3′-indolines] 389. | |
In a subsequent study, isatin-derived α,β-unsaturated ketones 398 were cyclized with alkynoates 399 providing access to various cyclopentene spirooxindole compounds 400 using the previously344 optimized conditions (Scheme 101). The title products were isolated in good to excellent yields with high regioselectivity and moderate to high diastereoselectivities (up to 20:1 dr). This methodology provides biologically active spiro-(cyclopentene) oxindoles featuring three contiguous stereocenters, including the quaternary stereogenic center joining the two rings. Similar to previous efforts, good functional group tolerance and structural diversity were observed.345
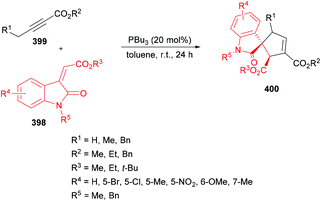 |
| | Scheme 101 Regio- and diastereoselective construction of spirocyclopenteneoxindoles through phosphine-catalyzed [3 + 2] annulation of methyleneindolinones with alkynoates. | |
2.6.6. Synthesis of spirocyclic oxindole-butenolides.
Li, Jia and co-workers346 explored a multi-component methodology to produce spirocyclic oxindole-butenolides 404via intermolecular [2 + 2 + 1] cycloaddition involving easily accessible starting materials (isocyanides 401, alkynoates 402 and isatins 403). This atom-economical approach operates through a Michael addition–nucleophilic addition–intramolecular cyclization sequence delivering the title products with good stereoselectivity (Scheme 102). After identifying the suitable conditions, the scope of the reaction with regard to all the three components was envisioned. The first point of variation includes the use of various aromatic isocyanides where sterically encumbered isocyanides were found to be less potent. Next, the scope of various isatins was examined carrying electron-neutral, electron-poor and electron-rich substituents on the aryl component. Moreover, a range of protecting groups on the indole nitrogen also proved to be viable and remained critical for the good conversion to the products. Furthermore, the coupling of [2 + 2 + 1] cycloaddition with a hydrolysis sequence led to the insertion of CO in a one-pot fashion obviating the need to achieve carbonylation under metal catalysis (product 405).
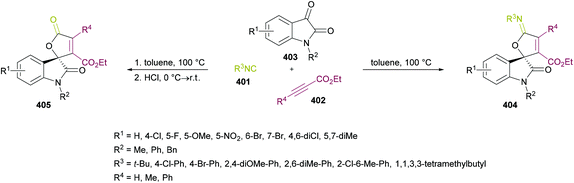 |
| | Scheme 102 Three-component synthesis of highly functionalized spirocyclic oxindole-butenolides. | |
2.6.7. Synthesis of spirorhodanines.
Miao and co-workers344 developed a diastereoselective phosphine-catalyzed [3 + 2] cycloaddition cascade reaction to generate a series of cyclopentene spirorhodanine derivatives in high yields and excellent enantioselectivities using alkynoates 406 and 5-arylidene-3-(tert-butyl)-2-thioxothiazolidin-4-ones 407 in the presence of PBu3 (Scheme 103). Ethyl 5-phenylpent-2-ynoate347 and 5-phenylidene-3-(tert-butyl)-2-thioxothiazolidin-4-one348,349 were chosen as model substrates to perform the optimization studies. The reactions were carried out using various phosphine catalysts (PPh3, PPh2Me, PBu3, DPPE, DPPP, DPPB) in different solvents (PhMe, DCM, THF, CHCl3, MeCN, MTBE) at room temperature. However, tributylphosphine (PBu3) produced the cycloaddition product cyclopentene 5-spirorhodanine with 95% yield and excellent diastereoselectivity (dr: >20:1) in toluene after 24 h. Further attention was paid to explore the scope of this spirocyclization using the optimized reaction conditions. A range of different alkynoates and 5-arylidene-3-(tert-butyl)-2-thioxothiazolidin-4-ones were coupled. The aryl component of rhodanine bearing electron-rich and electron-deficient substituents at various positions was tolerant to the optimized conditions. Disubstitution at the aryl ring was also compatible. In all cases, the desired products cyclopentene 5-spirorhodanines were isolated in good to high yields with excellent diastereoselectivities (>20:1 dr). 5-Arylidene rhodamine substrates containing naphthyl and thienyl groups also provided the corresponding products with smooth sequential cyclization in 87% and 99% yields and with excellent diastereoselectivities (>20:1 dr). However, when the benzyl group in the alkynoate was changed to a phenyl substituent, the reaction outcome was excellent in terms of the yield (81–93%) but with poor diastereoselectivities, signifying that the benzyl group in the alkynoates remains critical for achieving a high diastereoselectivity.
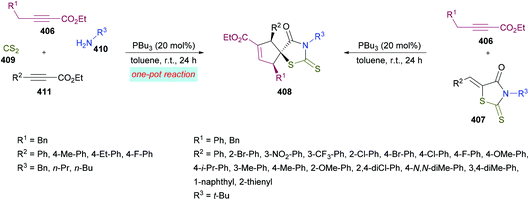 |
| | Scheme 103 Phosphine-catalyzed diastereoselective synthesis of cyclopentene spirorhodanines containing three contiguous stereocenters. | |
The scope was further extended using the one-pot multi-component methodology. Cyclopentene 5-spirorhodanine compounds 408 were accessed via phosphine-catalyzed one-pot sequential [3 + 2]/[3 + 2] cycloaddition. Ethyl 5-phenylpent-2-ynoate, phenylethylpropiolate350 benzylamine and carbon disulfide were used to optimize the reaction conditions. As the alkynoate bearing a homobenzyl group was critical for high diastereoselectivity, it was kept unchanged with carbon disulfide while changing the structural diversity on the other two components of the four-component methodology. Methyl, ethyl and fluoro substituents at the para-position were tolerated whereas benzylamine, n-butylamine and n-propylamine were employed. The corresponding products were obtained in 74–92% yields with excellent diastereoselectivities (>20:1 dr) except para-fluoro substituted spirocyclic rhodanine that was furnished with low diastereoselectivity (6:1 dr).
In line with the literature findings (Gabillet et al., 2007; Yang et al., 2013; Gabillet et al., 2014),351–353 the authors proposed the reaction mechanism for this spirocyclization methodology as shown in Scheme 104. Initially, a nucleophilic attack of the phosphine on phenylethylpropiolate produced the phosphonium salt 412 (zwitterionic intermediate) followed by the addition of an amine and carbon disulfide adduct to generate intermediate 413. Proton transfer followed by intramolecular cyclization delivered intermediate 415. Subsequent β-elimination of the catalyst furnished the phenylidene rhodanine 407 (cycle A). On the other hand, the nucleophilic attack of the regenerated catalyst PBu3 on ethyl alkynoate gave monozwitterion 416, which is in equilibrium with intermediate 417. The addition of phenylidene rhodanine 407 to 417 generated intermediate 418. Intramolecular cyclization led to phosphorane 419 which on elimination of the catalyst furnished the desired product 408 (cycle B).
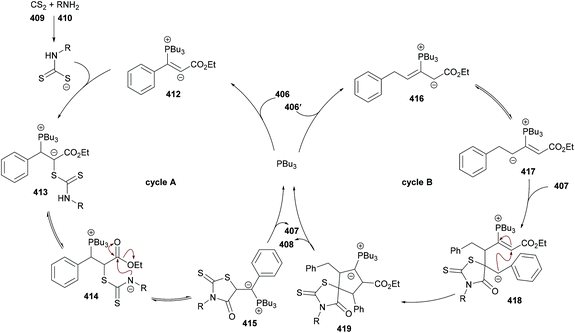 |
| | Scheme 104 Proposed mechanism for the formation of cyclopentene spirorhodanines 408. | |
2.7. Divergent reactivity of alkynoates
Mbofana and Miller354 reported a phosphine-catalyzed annulation methodology involving 2-alkynoates 420 and α-keto esters 421 to generate substituted cyclopentene products 422, 423 (Scheme 105). Initially, a range of phosphines were tested to obtain the desired products in optimal yields. Tricyclohexylphosphine was identified as the lead catalyst. The addition of 4 Å molecular sieves to the reaction mixture (to remove the methanol byproduct) led to the formation of the cyclopentene fused product in 76% yield whereas the addition of methanol after 24 h followed by stirring for 3 h generated cyclopentene exclusively in 74% yield. With two different sets of conditions, the scope of monocyclic and bicyclic products was investigated. A range of α-keto esters were employed under the optimized reaction conditions. Electron-donating (methyl, methoxy) and electron-withdrawing (chloro) groups were tolerated efficiently; however, methoxy-substituted products were isolated in low yields. Substituents at the para- and meta-positions were also cyclized successfully; however, the monosubstituted substrate remained unreactive. With a naphthyl ester, both products were isolated in 68% and 65% yields, respectively.
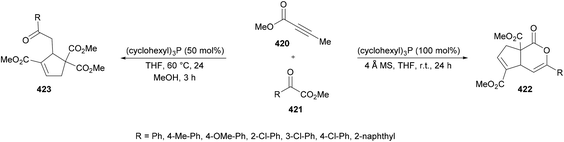 |
| | Scheme 105 Phosphine-catalyzed synthesis of cyclopentene derivatives from 2-butynoate and α-keto esters. | |
Based on the literature reports and initial mechanistic experiments, the authors proposed a catalytic cycle for this divergent methodology (Scheme 106). Phosphine coordination to methyl 2-butynoate generated zwitterionic intermediate 424 which on addition of another molecule of methyl 2-butynoate delivered intermediate 425.355 Proton transfer followed by an addition reaction with a keto ester led to intermediate 427. Subsequent isomerization and rearrangement produced intermediate 429.356–358 Another proton transfer alongside concerted ester group migration and cyclization furnished intermediate 431. Isomerization to 432 followed by deprotonation gave intermediate 433 which underwent cyclization to form intermediate 434. Proton transfer followed by catalyst elimination furnished the desired cyclopentene fused dihydropyrone heterocycle 422. Methanolysis of 422 generated the monocyclic five-membered product 423.
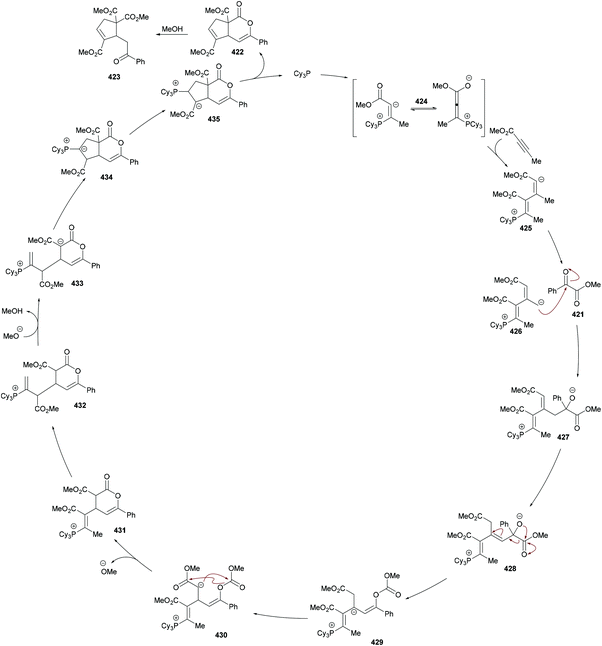 |
| | Scheme 106 Proposed mechanism for the formation of cyclopentene fused dihydropyrone 422 and monocyclic five-membered product 423. | |
Das and co-workers359 introduced a distinct multi-component approach utilizing the reactivity of nitroarenes 436, formaldehyde 437, and dialkyl acetylenedicarboxylates 438 under indium catalysis (Scheme 107). The reactions were performed in dilute aqueous HCl at room temperature. Dioxopyrrolidines 439 and tetrahydropyrimidines 440 were formed in good to high yields using 1:1:4 and 2:1:4 molar ratios of the substrates. Other metals including Sn, Zn, and Fe were also tested in aqueous HCl; however, indium produced the best results both in terms of the yield and reaction time (30–40 min). The primary point of structural variations includes the use of different nitro compounds. Various nitroarenes were employed alongside electron-deficient alkynes and formaldehyde to deliver the corresponding heterocyclic products in good to high yields. Electron-neutral, electron-rich and electron-deficient substituents were tolerated. Mechanistically, the nitroarenes were reduced to aryl amines360 in the presence of an In/aqueous HCl system and were further reacted with electron-deficient alkynes and formaldehyde, respectively.
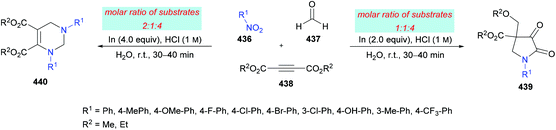 |
| | Scheme 107 Multi-component synthesis of polysubstituted pyrrolidines and tetrahydropyrimidines in water. | |
2.8. Application in the synthesis of natural products and pharmaceuticals
This section highlights the use of alkynoates in the synthesis of natural products and pharmaceuticals. Various methodologies have been employed to construct several key intermediates in the synthesis of bioactive complex molecules.
2.8.1. Total synthesis of a norsesquiterpene alkaloid.
Reddy and co-workers361 reported the first total synthesis of an anticancer norsesquiterpene alkaloid (R)-8-hydroxy-4,7,7-trimethyl-7,8-dihydrocyclopenta[e]isoindole-1,3(2H,6H)-dione 445, isolated from the solid culture of the mushroom-forming fungus Flammulina velutipes (Scheme 108). The synthesis of the key enyne intermediate 442 from D-(−)-pantolactone 441 followed by the ring-closing metathesis (RCM) using Grubbs’ 1st generation catalyst, the Diels–Alder reaction with dimethyl acetylenedicarboxylate (DMAD) and subsequent treatment with DDQ furnished aromatized compound 443 in a moderate yield (∼40%). The hydrolysis of an ester in indane 443 with aqueous KOH followed by heating with urea in ethylene glycol provided compound 444. Finally, the deprotection of the benzyl group was performed using 10% Pd/C under a hydrogen atmosphere in ethanol delivered the norsesquiterpene alkaloid 445.
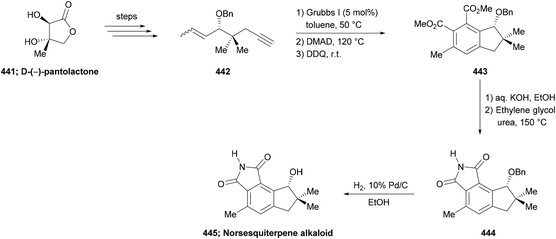 |
| | Scheme 108 Total synthesis of a norsesquiterpene alkaloid. | |
2.8.2. Synthesis of deoxypodophyllotoxin.
Andrus, Ess and co-workers362 developed a short seven-step synthesis of deoxypodophyllotoxin 453. The ISDA strategy led to the simultaneous formation of the cyclohexyl ring and lactone functionality together with a stereocenter in a single step (Scheme 109). Alcohol 447 was prepared from commercial 3,4-methylenedioxycinnamic acid 446 using Fisher esterification and DIBAL reduction procedures whereas alkyne acid 449 was obtained by the Corey–Fuchs reaction using trimethoxybenzaldehyde 448 followed by trapping with carbon dioxide. Alcohol 447 and alkyne acid 449 were reacted under Mitsunobu coupling conditions to afford the electron-rich alkyne allyl ester intermediate 450. The ISDA reaction using BHT (1.5 equiv.) in benzonitrile afforded two isomeric products 451 and 452 in 56% overall yield with 1:1.6 selectivity. Both structures were confirmed through X-ray crystallography. Finally, the reduction of 452 using Mg/MeOH conditions provided DPT with cis-stereochemistry.
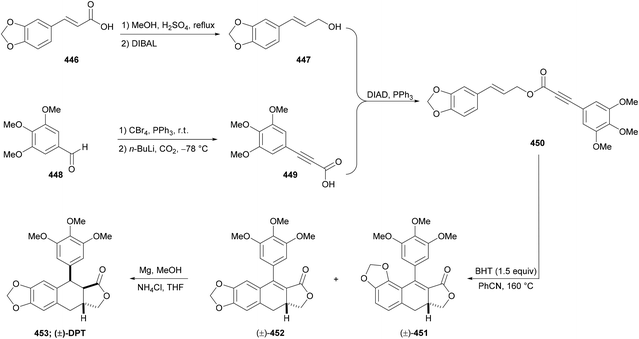 |
| | Scheme 109 Synthesis of deoxypodophyllotoxin by the ISDA strategy. | |
2.8.3. Synthesis of (−)-CJ-13,982.
Rizzacasa and co-workers363 reported an efficient and step-economical approach to produce alkyl citrate natural products from a cyclobutene diester (Scheme 110). The enantiospecific synthesis of (−)-CJ-13,982 459 was started using optically pure lactone 454364 (prepared from (S)-(+)-γ-hydroxymethyl-γ-butyrolactone) which was transformed into the silyl ketene acetal, and cycloaddition with dimethyl acetylenedicarboxylate (DMAD) afforded the major adduct cyclobutene 455. Silyl ether and acetal hydrolysis was achieved using HCl in MeOH followed by oxy-Michael addition, and cyclobutene methanolysis afforded the triester as a single stereoisomer in a good yield. Tosyl protection followed by displacement with NaI and zinc-mediated reductive elimination afforded the key alkyl citrate fragment 457. Cross metathesis (CM) with undecene followed by hydrogenation produced the (−)-CJ-13,982 trimethyl ester 458. Subsequent hydrolysis using 5.0 equivalents of 1 M aqueous NaOH in dioxane afforded the desired (−)-CJ-13,982.
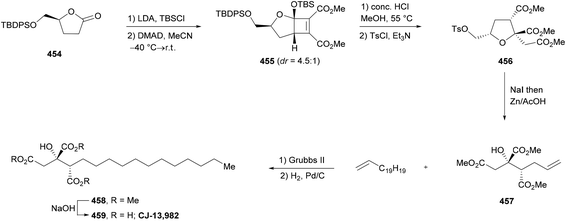 |
| | Scheme 110 Synthesis of (−)-CJ-13,982. | |
2.8.4. Synthesis of (R)-puraquinonic acid.
Elmehriki and Gleason365 developed a stereoselective method for the construction of cyclic α-quaternary stereocenters from biselectrophilic substrates and chiral bicyclic sulfonyl lactam (Scheme 111). Using this method, the total synthesis of (R)-puraquinonic acid 470 was achieved in an overall yield of 14%. Vinylation of 3-pentyn-1-ol 460 using catalytic mercury(II) acetate was performed to deliver ethyl vinyl ether 461. A one-pot sequence involving ene-yne metathesis under Grubbs II conditions forming diene, followed by Diels–Alder cycloaddition with dimethyl acetylenedicarboxylate (DMAD) and oxidation with DDQ afforded arene 462 in 42% yield (3 steps). Reduction of the ester groups using LiAlH4 followed by bromination with phosphorus tribromide produced dibromide 463 in 95% yield (2 steps). Spiroalkylation was performed using 465 to form 466 which was transformed to amide 467 after reduction/alkylation with greater than 95:5 dr. The hydrolysis of 467 using sulfuric acid in 1,4-dioxane provided carboxylic acid 468 in 90% yield which was oxidized to puraquinonic acid 470 in 84% yield using a combination of peracetic acid and acetic acid in DCM at 0 °C. On the other hand, protection of alcohol in 467 with a MOM group followed by oxidation using peracetic acid and acetic acid in chloroform at room temperature afforded quinone 469 which was transformed to puraquinonic acid 470 using the literature conditions.366
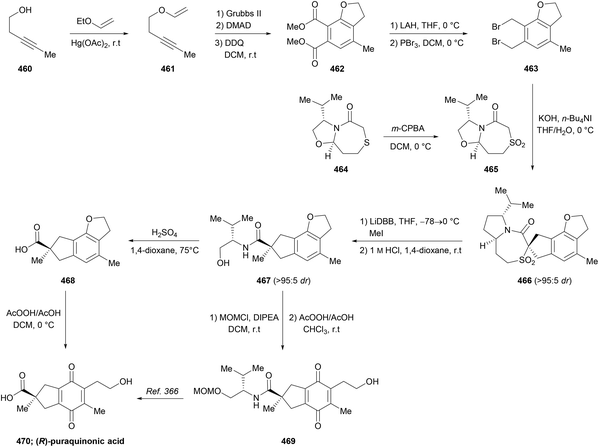 |
| | Scheme 111 Synthesis of (R)-puraquinonic acid. | |
2.8.5. Synthesis of discoipyrrole C.
Kurian and Manheri367 reported the synthesis of 1H-pyrrol-3(2H)-ones from the reaction of indolizinones with dimethyl acetylenedicarboxylate which was used in the total synthesis of the anticancer discoipyrrole C 478 (Scheme 112). Isovaleric acid 471 was converted to the corresponding Weinreb amide 472 which underwent pyridine ortho acylation followed by alkylation to provide intermediate 473. Deprotection of the TBS group followed by acetylation in the presence of triethylamine afforded 474 which on subsequent tandem amino-palladation, reductive elimination and 1,2-alkyl shift afforded indolizinone 475.368 Coupling of 475 with DMAD provided a mixture of tricyclic compound 476 and 1H-pyrrol-3(2H)-one 477 which on deacetylation delivered the discoipyrrole C 478.
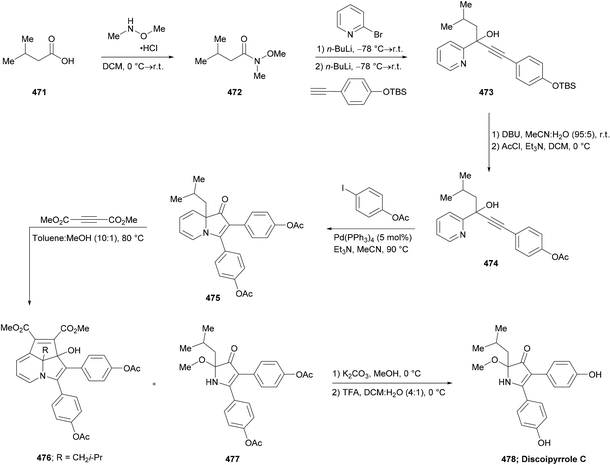 |
| | Scheme 112 Synthesis of discoipyrrole C. | |
2.8.6. Synthesis of amlodipine.
Kim and co-workers369 reported the synthesis of amlodipine 486, a calcium ion channel blocker, using the aza-Diels–Alder reaction between the Knoevenagel adduct 479, methyl butynoate 480 and benzylamine 481 as the starting point. Debenzylation of the Diels–Alder product 482 was achieved under palladium catalysis followed by a selective allylic bromination with good selectivity (>80:20, mono:di) using a buffered solution of pyridinium perbromide. An azidoethanol moiety was introduced370 which was reduced by Zn/HCl to produce the racemic amlodipine 486 (Scheme 113).
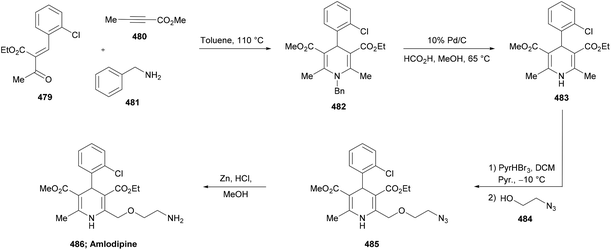 |
| | Scheme 113 Synthesis of amlodipine. | |
2.8.7. Synthesis of raltegravir.
Raltegravir (496, isentress) is an important pyrimidine-based anti-HIV drug launched by Merck in 2008.371 The structural features of this HIV-integrase inhibitor include a functionalized pyrimidone core flanked by an oxadiazole ring with a terminal para-fluorobenzyl moiety. The synthesis of the central pyrimidone ring was initiated by the amination of acetone cyanohydrin 487 followed by Cbz-protection of the resulting amine and aminolysis of the nitrile with hydroxylamine to give amidoxime 489. The reaction of 489 with dimethyl acetylenedicarboxylate (DMAD) produced pyrimidine 490 which was N-methylated to form 491. Subsequent amide formation and debenzylation produced 492. Finally, coupling of 492 with the corresponding acid chloride of oxadiazole derivative 495 (prepared via a sequence of reactions) afforded the desired raltegravir 496 (Scheme 114).
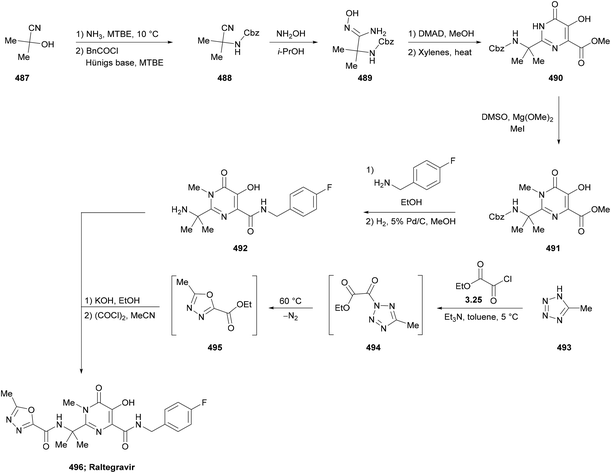 |
| | Scheme 114 Synthesis of raltegravir. | |
3. Conclusion and perspectives
Considering the high proportion of heterocycles/heteroaromatics in numerous natural products, synthetic drugs, commercial pharmaceuticals, agrochemicals and designed bioactive pharmacophores, the development of efficient chemical methodologies providing facile access to architecturally complex structures has remained a formidable challenge for both synthetic and medicinal chemists. In the present review, we summarize the remarkable developments in novel synthetic methods allowing the construction of numerous nitrogen-, oxygen-, sulfur-, and fused-heterocycles as well as hetero-spirocycles from alkynoates as simple, readily accessible and commercially available starting materials under transition-metal-free conditions. The discussion focuses on the investigation of the chemical reactivity of the triple bond directly attached to an electron-withdrawing ester group. The unique and inherent properties such as the electronic bias on the carbon–carbon triple bond posed by electron-withdrawing groups allow a range of chemical reactions (Wittig, annulations, cycloadditions) to occur, exploiting the reactivity of the zwitterions derived from activated acetylenediesters particularly with nucleophilic trivalent phosphines. In addition, the reactions of acetylenic esters in the presence of phosphines with various carbon-, nitrogen-, or sulfur-based nucleophiles were attempted to deliver heterocyclic scaffolds. Organocatalyzed, photocatalyzed, radical, and Lewis and Brønsted-acid-catalyzed reactions of alkynoates were also investigated. Moreover, the alkynoates were also employed in multi-component reactions involving the unusual reactivity of the isocyanide functionality providing ample opportunities for the generation of complex and diverse drug-like small heterocyclic compounds. The investigation of the optimized reaction conditions, viability of the substrates and coupling partners, functional group tolerance and limitations and mechanistic studies as well as post-synthetic functionalization of the heterocyclic products have been performed. Various reaction sequences such as Michael addition/azaheterocyclization, hydroamination/nucleophilic addition/amidation, hydroamination/amidation/intramolecular cyclization/imineenamine tautomerization, hydroamination/Knoevenagel condensation/Michael-type addition/intramolecular cyclization, hydroamination/Mannich-type reaction/amine-aldehyde dehydration–cyclization, nucleophilic addition/intramolecular Wittig reaction, radical addition/cyclization, radical tandem phosphorylation/cyclization, nucleophilic addition/5-endo-dig cyclization, Michael addition/intramolecular nucleophilic addition/4π ring opening/intramolecular Michael addition/elimination, intermolecular hydroamination/intramolecular transesterification, umpolung addition/intramolecular cyclization, hydroamination/Prins reaction/cyclization/dehydration, Michael addition/intramolecular Morita–Baylis–Hillman reaction and one-pot sequential [3 + 2]/[3 + 2] cycloaddition reactions were involved to deliver a range of heterocyclic products.
Despite the broad utility of alkynoates leading to various heterocycles over the past decade, there remain several challenges and unexplored opportunities in the demand to develop more general, efficient, sustainable and practical synthetic methods. We present a selection of recommendations for future work. (1) The hindered substrate scope could further be expanded under mild and green reaction conditions. (2) The effect of substitution on the alkyne component could potentially be envisioned from experimental and computational methods. (3) The installation of possibly transformable groups into impactful drugs and pharmaceuticals should be focused. (4) In light of the recent reports on electrochemical oxidative annulation of alkynoates, the field still retains considerable room for synthetic extensions and substantial improvements. (5) It is evident from the literature findings that the development of asymmetric methods to construct enantioenriched heterocycles using alkynoates needs ample attention to further explore the wider chemical space. (6) The strategic combination of efficient chemical synthesis with enzymatic methods to produce small heterocyclic molecules stands as the future goal. Collectively, there is no doubt that the enriched alkynoate chemistry will continue to grow and offer ample opportunities to both synthetic and medicinal chemists in the generation of diverse heterocyclic frameworks of pharmaceutical interest.
Abbreviations
| Ac | Acetyl |
| AQN | Anthraquinone |
| BHT | 2,6-Di-tert-butyl-4-methylphenol |
| Boc |
tert-Butoxycarbonyl |
| BINOL | 1,1′-Bi-2-naphthol |
| BI-OH | 1-Hydroxy-1,2-benziodoxol-3(1H)-one |
| BI-OAc | 1,2-Benziodoxol-3(1H)-one acetate |
| Bmim | 1-Butyl-3-methylimidazolidin |
| Bn | Benzyl |
| BPO | Benzoyl peroxide |
| bpy | 2,2′-Bipyridine |
| CAN | Cerium ammonium nitrate |
| CM | Cross metathesis |
| CPME | Cyclopentyl methyl ether |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DABSO | 1,4-Diazabicyclo[2.2.2]octane bis(sulfur dioxide) adduct |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCB | Dichlorobenzene |
| DCE | 1,2-Dichloroethane |
| DCM | Dichloromethane |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| DEAD | Diethyl acetylenedicarboxylate |
| DFT | Density functional theory |
| DIAD | Diisopropyl azodicarboxylate |
| DIBAL | Diisobutylaluminium hydride |
| DIPEA |
N,N-Diisopropylethylamine |
| DLP | Dilauroyl peroxide |
| DMF |
N,N-Dimethylformamide |
| DMAD | Dimethyl acetylenedicarboxylate |
| DMAP | Dimethylaminopyridine |
| DMSO | Dimethyl sulfoxide |
| DPP | Diphenylphosphinyl |
| Dppb | 1,4-Bis(diphenylphosphino)butane |
| Dppe | 1,2-Bis(diphenylphosphino)ethane |
| Dppp | Diphenylpropylenediphosphine |
| DPT | Deoxypodophyllotoxin |
| DTBP | Di-tert-butylperoxide |
| EDC |
N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimide |
| EDG | Electron-donating group |
| Equiv | Equivalent |
| EWG | Electron-withdrawing group |
| EY | Eosin Y |
| Hex | Hexyl |
| HFIP | Hexafluoroisopropanol |
| HIR | Hypervalent iodine reagent |
| HMPA | Hexamethylphosphoramide |
| HMPT | Hexamethylphosphorous triamide |
| ICl | Iodine monochloride |
| IL | Ionic liquid |
| IPA | Isopropyl alcohol |
| ISDA | Intramolecular styryl Diels–Alder |
| KHMDS | Potassium bis(trimethylsilyl)amide |
| LAH | Lithium aluminum hydride |
| LED | Light-emitting diode |
| LiDBB | Lithium 4,4-di-tert-butylbiphenylide |
| MCR | Multicomponent reaction |
|
mCPBA |
meta-Chloroperbenzoic acid |
| MOMCl | Chloromethyl methyl ether |
| MTBE | Methyl tert-butyl ether |
| MW | Microwave |
| NBS |
N-Bromosuccinimide |
| NIS |
N-Iodosuccinimide |
| Pent | Pentyl |
| Ph | Phenyl |
| PIDA | Phenyliodine(III) diacetate |
| PMP |
p-Methoxyphenyl |
| PPTS | Pyridinium p-toluenesulfonate |
| RCM | Ring-closing metathesis |
| R.T. | Room temperature |
| SET | Single electron transfer |
| TBAB | Tetrabutylammonium bromide |
| TBAC | Tetrabutylammonium chloride |
| TBAF | Tetrabutylammonium fluoride |
| TBAI | Tetrabutylammonium iodide |
| TBHP |
tert-Butyl hydroperoxide |
| TEMPO | 2,2,6,6-Tetramethyl-1-piperidinyloxy |
| TBPB |
tert-Butyl perbenzoate |
| TBSCl |
tert-Butyldimethylsilyl chloride |
| TFA | Trifluoroacetic acid |
| TFAA | Trifluoroacetic anhydride |
| THF | Tetrahydrofuran |
| TMS | Trimethylsilyl |
| TMBSA | Tri-methylammonium-butane sulfonate |
| UV | Ultraviolet |
Conflicts of interest
There are no conflicts to declare.
References
-
Alkaloids, ed. G. A. Cordell, Academic Press, 2010, vol. 69 Search PubMed.
-
J. A. Joule and K. Mills, in Heterocyclic chemistry, Wiley, 5th edn, 2010 Search PubMed.
- N. A. Meanwell, Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety, Chem. Res. Toxicol., 2011, 24, 1420–1456 Search PubMed.
- T. J. Ritchie, S. J. F. Macdonald, R. J. Young and S. D. Pickett, The impact of aromatic ring count on compound developability: Further insights by examining carbo- and hetero-aromatic and -aliphatic ring types, Drug Discovery Today, 2011, 16, 164–171 Search PubMed.
- B. Eftekhari-Sis, M. Zirak and A. Akbari, Arylglyoxals in synthesis of heterocyclic compounds, Chem. Rev., 2013, 113, 2958–3043 Search PubMed.
- I. Khan, S. Ali, S. Hameed, N. H. Rama, M. T. Hussain, A. Wadood, R. Uddin, Z. Ul-Haq, A. Khan, S. Ali and M. I. Choudhary, Synthesis, antioxidant activities and urease inhibition of some new 1,2,4-triazole and 1,3,4-thiadiazole derivatives, Eur. J. Med. Chem., 2010, 45, 5200–5207 Search PubMed.
- I. Khan, M. Hanif, M. T. Hussain, A. A. Khan, M. A. S. Aslam, N. H. Rama and J. Iqbal, Synthesis, acetylcholinesterase and alkaline phosphatase inhibition of some new 1,2,4-triazole and 1,3,4-thiadiazole derivatives, Aust. J. Chem., 2012, 65, 1413–1419 Search PubMed.
- M. Hanif, I. Khan, N. H. Rama, S. Noreen, M. I. Choudhary, P. G. Jones and M. Iqbal, Synthesis, crystal structure and β-glucuronidase inhibition activity of some new hydrazinecarboxamides and their 1,2,4-triazole derivatives, Med. Chem. Res., 2012, 21, 3885–3896 Search PubMed.
- A. Ibrar, I. Khan and N. Abbas, Structurally diversified heterocycles and related privileged scaffolds as potential urease inhibitors: A brief overview, Arch. Pharm. Chem. Life Sci., 2013, 346, 423–446 Search PubMed.
- I. Khan, A. Ibrar and N. Abbas, Triazolothiadiazoles and triazolothiadiazines-Biologically attractive scaffolds, Eur. J. Med. Chem., 2013, 63, 854–868 Search PubMed.
- I. Khan, A. Ibrar and N. Abbas, Oxadiazoles as privileged motifs for promising anticancer leads: Recent advances and future prospects, Arch. Pharm. Chem. Life Sci., 2014, 347, 1–20 Search PubMed.
- I. Khan, A. Ibrar, S. Zaib, S. Ahmad, N. Furtmann, S. Hameed, J. Simpson, J. Bajorath and J. Iqbal, Active compounds from a diverse library of triazolothiadiazole and triazolothiadiazine scaffolds: Synthesis, crystal structure determination, cytotoxicity, cholinesterase inhibitory activity, and binding mode analysis, Bioorg. Med. Chem., 2014, 22, 6163–6173 Search PubMed.
- I. Khan, S. Zaib, A. Ibrar, N. H. Rama, J. Simpson and J. Iqbal, Synthesis, Crystal structure and biological evaluation of some novel 1,2,4-triazolo[3,4-b,]-1,3,4-thiadiazoles and 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines, Eur. J. Med. Chem., 2014, 78, 167–177 Search PubMed.
- I. Khan, S. M. Bakht, A. Ibrar, S. Abbas, S. Hameed, J. M. White, U. A. Rana, S. Zaib, M. Shahid and J. Iqbal, Exploration of a library of triazolothiadiazole and triazolothiadiazine compounds as a highly potent and selective family of cholinesterase and monoamine oxidase inhibitors: design, synthesis, X-ray diffraction analysis and molecular docking studies, RSC Adv., 2015, 5, 21249–21267 Search PubMed.
- I. Khan, A. Ibrar, S. A. Ejaz, S. U. Khan, S. J. A. Shah, S. Hameed, J. Simpson, J. Lecka, J. Sévigny and J. Iqbal, Influence of the diversified structural variations at the imine functionality of 4-bromophenylacetic acid derived hydrazones on alkaline phosphatase inhibition: synthesis and molecular modelling studies, RSC Adv., 2015, 5, 90806–90818 Search PubMed.
- I. Khan, S. Hameed, N. A. Al-Masoudi, N. A. Abdul-Reda and J. Simpson, New triazolothiadiazole and triazolothiadiazine derivatives as kinesin Eg5 and HIV inhibitors: synthesis, QSAR and modeling studies, Z. Naturforsch., 2015, 70, 47–58 Search PubMed.
- H. Andleeb, Y. Tehseen, S. J. A. Shah, I. Khan, J. Iqbal and S. Hameed, Identification of novel pyrazole-rhodanine hybrid scaffolds as potent inhibitors of aldose reductase: design, synthesis, biological evaluation and molecular docking analysis, RSC Adv., 2016, 6, 77688–77700 Search PubMed.
- M. Kazmi, S. Zaib, S. T. Amjad, I. Khan, A. Ibrar, A. Saeed and J. Iqbal, Exploration of aroyl/heteroaroyl iminothiazolines featuring 2,4,5-trichlorophenyl moiety as a new class of potent, selective, and in vitro efficacious glucosidase inhibitors, Bioorg. Chem., 2017, 74, 134–144 Search PubMed.
- H. Andleeb, Y. Tehseen, F. Jabeen, I. Khan, J. Iqbal and S. Hameed, Exploration of thioxothiazolidinone-sulfonate conjugates as a new class of aldehyde/aldose reductase inhibitors: A synthetic and computational investigation, Bioorg. Chem., 2017, 75, 1–15 Search PubMed.
- N. Abbas, S. Zaib, S. M. Bakht, A. Ibrar, I. Khan, S. Batool, A. Saeed and J. Iqbal, Symmetrical aryl linked bis-iminothiazolidinones as new chemical entities for the inhibition of monoamine oxidases: Synthesis, in vitro biological evaluation and molecular modelling analysis, Bioorg. Chem., 2017, 70, 17–26 Search PubMed.
- M. Kazmi, S. Zaib, A. Ibrar, S. T. Amjad, Z. Shafique, S. Mehsud, A. Saeed, J. Iqbal and I. Khan, A new entry into the portfolio of α-glucosidase inhibitors as potent therapeutics for type 2 diabetes: Design, bioevaluation and one-pot multi-component synthesis of diamine-bridged coumarinyl oxadiazole conjugates, Bioorg. Chem., 2018, 77, 190–202 Search PubMed.
- N. A. Khan, I. Khan, S. M. A. Abid, S. Zaib, A. Ibrar, H. Andleeb, S. Hameed and J. Iqbal, Quinolinic carboxylic acid derivatives as potential multi-target compounds for neurodegeneration: Monoamine oxidase and cholinesterase inhibition, Med. Chem., 2018, 14, 74–85 Search PubMed.
- M. Kazmi, I. Khan, A. Khan, S. A. Halim, A. Saeed, S. Mehsudf, A. Al-Harrasi and A. Ibrar, Developing new hybrid scaffold for urease inhibition based on carbazole-chalcone conjugates: Synthesis, assessment of therapeutic potential and computational docking analysis, Bioorg. Med. Chem., 2019, 27, 115123 Search PubMed.
- A. Ibrar, M. Kazmi, A. Khan, S. A. Halim, A. Saeed, S. Mehsud, A. Al-Harrasi and I. Khan, Robust therapeutic potential of carbazole-triazine hybrids as a new class of urease inhibitors: A distinctive combination of nitrogen-containing heterocycles, Bioorg. Chem., 2020, 95, 103479 Search PubMed.
- H. Andleeb, S. Hameed, S. A. Ejaz, I. Khan, S. Zaib, J. Lecka, J. Sévigny and J. Iqbal, Probing the high potency of pyrazolyl pyrimidinetriones and thioxopyrimidinediones as selective and efficient non-nucleotide inhibitors of recombinant human ectonucleotidases, Bioorg. Chem., 2019, 88, 102893 Search PubMed.
- S. A. Shehzadi, I. Khan, A. Saeed, F. A. Larik, P. A. Channar, M. Hassan, H. Raza, Q. Abbas and S.-Y. Seo, One-pot four-component synthesis of thiazolidin-2-imines using CuI/ZnII dual catalysis: A new class of acetylcholinesterase inhibitors, Bioorg. Chem., 2019, 84, 518–528 Search PubMed.
- P. Schlack, Polymerizable lactams, Pure Appl. Chem., 1967, 15, 507–523 Search PubMed.
- J.-M. Weibel, A. Blanc and P. Pale, Ag-mediated reactions: Coupling and heterocyclization reactions, Chem. Rev., 2008, 108, 3149–3173 Search PubMed.
- G. Fanga and X. Bi, Silver-catalysed reactions of alkynes: Recent advances, Chem. Soc. Rev., 2015, 44, 8124–8173 Search PubMed.
- Z. Li, C. Brouwer and C. He, Gold-catalyzed organic transformations, Chem. Rev., 2008, 108, 3239–3265 Search PubMed.
- A. Corma, A. Leyva-Pérez and M. J. Sabater, Gold-catalyzed carbon-heteroatom bond-forming reactions, Chem. Rev., 2011, 111, 1657–1712 Search PubMed.
- N. Krause and C. Winter, Gold-catalyzed nucleophilic cyclization of functionalized allenes: A powerful access to carbo- and heterocycles, Chem. Rev., 2011, 111, 1994–2009 Search PubMed.
- A. S. K. Hashmi, Gold-catalyzed organic reactions, Chem. Rev., 2007, 107, 3180–3211 Search PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Copper in cross-coupling reactions: The post-Ullmann chemistry, Coord. Chem. Rev., 2004, 248, 2337–2364 Search PubMed.
- G. Evano, N. Blanchard and M. Toumi, Copper-mediated coupling reactions and their applications in natural products and designed biomolecules synthesis, Chem. Rev., 2008, 108, 3054–3131 Search PubMed.
- X. Tang, W. Wu, W. Zeng and H. Jiang, Copper-catalyzed oxidative carbon-carbon and/or carbon-heteroatom bond formation with O2 or internal oxidants, Acc. Chem. Res., 2018, 51, 1092–1105 Search PubMed.
- W. Hess, J. Treutwein and G. Hilt, Cobalt-catalysed carbon-carbon bond-formation reactions, Synthesis, 2008, 3537–3562 Search PubMed.
- C. Bolm, J. Legros, J. Le Paih and L. Zani, Iron-catalyzed reactions in organic synthesis, Chem. Rev., 2004, 104, 6217–6254 Search PubMed.
- D. D. Díaz, P. O. Miranda, J. I. Padrón and V. S. Martín, Recent uses of Iron(III) chloride in organic synthesis, Curr. Org. Chem., 2006, 10, 457–476 Search PubMed.
- E. B. Bauer, Recent advances in iron catalysis in organic synthesis, Curr. Org. Chem., 2008, 12, 1341–1369 Search PubMed.
- E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, C-C, C-O, C-N bond formation on sp2 Carbon by Pd(II)-catalyzed reactions involving oxidant agents, Chem. Rev., 2007, 107, 5318–5365 Search PubMed.
- W. Wu and H. Jiang, Palladium-catalyzed oxidation of unsaturated hydrocarbons using molecular oxygen, Acc. Chem. Res., 2012, 45, 1736–1748 Search PubMed.
- B. M. Trost, M. U. Frederiksen and M. T. Rudd, Ruthenium-catalyzed reactions-A treasure trove of atom-economic transformations, Angew. Chem., Int. Ed., 2005, 44, 6630–6666 Search PubMed.
- M. Arisawa, Y. Terada, C. Theeraladanon, K. Takahashi, M. Nakagawa and A. Nishida, Development of novel reactions using ruthenium carbene catalyst and its application to novel methods for preparing nitrogen-containing heterocycles, J. Organomet. Chem., 2005, 690, 5398–5406 Search PubMed.
- J. Faller and J. Parr, Lewis acid catalysis by ruthenium complexes, Curr. Org. Chem., 2006, 10, 151–163 Search PubMed.
- M. Nishizawa, H. Imagawa and H. Yamamoto, A new catalyst for organic synthesis: Mercuric triflate, Org. Biomol. Chem., 2010, 8, 511–521 Search PubMed.
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W.-L. Lam, Rare-earth metal triflates in organic synthesis, Chem. Rev., 2002, 102, 2227–2302 Search PubMed.
- M. Lautens, W. Klute and W. Tam, Transition metal-mediated cycloaddition reactions, Chem. Rev., 1996, 96, 49–92 Search PubMed.
- S. Kotha, E. Brahmachary and K. Lahiri, Transition Metal Catalyzed [2+2+2] Cycloaddition and application in organic synthesis, Eur. J. Org. Chem., 2005, 4741–4767 Search PubMed.
- P. R. Chopade and J. Louie, [2+2+2] Cycloaddition reactions catalyzed by transition metal complexes, Adv. Synth. Catal., 2006, 348, 2307–2327 Search PubMed.
- B. Heller and M. Hapke, The fascinating construction of pyridine ring systems by transition metal-catalysed [2+2+2] cycloaddition reactions, Chem. Soc. Rev., 2007, 36, 1085–1094 Search PubMed.
- M. Meldal and C. W. Tornøe, Cu-catalyzed azide-alkyne cycloaddition, Chem. Rev., 2008, 108, 2952–3015 Search PubMed.
- V. Michelet, P. Toullec and Y. J.-P. Genêt, Cycloisomerization of 1,n-enynes: Challenging metal-catalyzed rearrangements and mechanistic insights, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 Search PubMed.
- P. Belmont and E. Parker, Silver and gold catalysis for cycloisomerization reactions, Eur. J. Org. Chem., 2009, 6075–6089 Search PubMed.
- R. Chinchilla and C. Najera, The Sonogashira reaction: A booming methodology in synthetic organic chemistry, Chem. Rev., 2007, 107, 874–922 Search PubMed.
- M. M. Heravi and S. Sadjadi, Recent advances in the application of the Sonogashira method in the synthesis of heterocyclic compounds, Tetrahedron, 2009, 65, 7761–7775 Search PubMed.
- P. Thansandote and M. Lautens, Construction of nitrogen–containing heterocycles by C–H bond functionalization, Chem. – Eur. J., 2009, 15, 5874–5883 Search PubMed.
- R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Functionalization of organic molecules by transition-metal-catalyzed C(sp3)-H activation, Chem. – Eur. J., 2010, 16, 2654–2672 Search PubMed.
- E. M. Beccalli, G. Broggini, A. Fasana and M. Rigamonti, Palladium-catalyzed C–N bond formation via direct C–H bond functionalization. Recent developments in heterocyclic synthesis, J. Organomet. Chem., 2011, 696, 277–295 Search PubMed.
- A. Deiters and S. F. Martin, Synthesis of oxygen- and nitrogen-containing heterocycles by ring-closing metathesis, Chem. Rev., 2004, 104, 2199–2238 Search PubMed.
- T. J. Donohoe, A. J. Orr and M. Bingham, Ring-closing metathesis as a basis for the construction of aromatic compounds, Angew. Chem., Int. Ed., 2006, 45, 2664–2670 Search PubMed.
- T. J. Donohoe, L. P. Fishlock and P. A. Procopiou, Ring-closing metathesis: Novel routes to aromatic heterocycles, Chem. – Eur. J., 2008, 14, 5716–5726 Search PubMed.
- D. M. D'Souza and T. J. J. Muller, Multi-component syntheses of heterocycles by transition-metal catalysis, Chem. Soc. Rev., 2007, 36, 1095–1108 Search PubMed.
- V. Estévez, M. Villacampa and J. C. Menéndez, Multicomponent reactions for the synthesis of pyrroles, Chem. Soc. Rev., 2010, 39, 4402–4421 Search PubMed.
- I. Khan, A. Ibrar, N. Abbas and A. Saeed, One-pot access to a privileged library of six membered nitrogenous heterocycles through multi-component cascade approach, Res. Chem. Intermed., 2016, 42, 5147–5196 Search PubMed.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Organic azides: An exploding diversity of a unique class of compounds, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 Search PubMed.
- S. Cenini, E. Gallo, A. Caselli, F. Ragaini, S. Fantauzzi and C. Piangiolino, Coordination chemistry of organic azides and amination reactions catalyzed by transition metal complexes, Coord. Chem. Rev., 2006, 250, 1234–1253 Search PubMed.
- T. G. Driver, Recent advances in transition metal-catalyzed N-atom transfer reactions of azides, Org. Biomol. Chem., 2010, 8, 3831–3846 Search PubMed.
- B. Eftekhari-Sis and M. Zirak, Chemistry of α-oxoesters: A powerful tool for the synthesis of heterocycles, Chem. Rev., 2015, 115, 151–264 Search PubMed.
- J. Zhu, Recent developments in the isonitrile–based multicomponent synthesis of heterocycles, Eur. J. Org. Chem., 2003, 1133–1144 Search PubMed.
- A. V. Ivachtchenko, Y. A. Ivanenkov, V. M. Kysil, M. Y. Krasavin and A. P. Ilyin, Multicomponent reactions of isocyanides in the synthesis of heterocycles, Russ. Chem. Rev., 2010, 79, 787–817 Search PubMed.
- A. V. Lygin and A. de Meijere, Isocyanides in the synthesis of nitrogen heterocycles, Angew. Chem., Int. Ed., 2010, 49, 9094–9124 Search PubMed.
- A. V. Gulevich, A. G. Zhdanko, R. V. A. Orru and V. G. Nenajdenko, Isocyanoacetate derivatives: Synthesis, reactivity, and application, Chem. Rev., 2010, 110, 5235–5331 Search PubMed.
- B. Eftekhari-Sis, M. Zirak and A. Akbari, Arylglyoxals in synthesis of heterocyclic compounds, Chem. Rev., 2013, 113, 2958–3043 Search PubMed.
- A. Padwa, Rhodium(II) mediated cyclizations of diazo alkynyl ketones, J. Organomet. Chem., 2001, 617–618, 3–16 Search PubMed.
- Z. Zhang, Recent studies on the reactions of α-diazocarbonyl compounds, Tetrahedron, 2008, 64, 6577–6605 Search PubMed.
- C. Garett and K. Prasad, The art of meeting palladium specifications in active pharmaceutical ingredients produced by Pd-catalyzed reactions, Adv. Synth. Catal., 2004, 346, 889–900 Search PubMed.
- L. Ackermann, Carboxylate-assisted transition-metal-catalyzed C-H bond functionalizations: Mechanism and scope, Chem. Rev., 2011, 111, 1315–1345 Search PubMed.
- Y. Boutadla, D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Computational and synthetic studies on the cyclometallation reaction of dimethylbenzylamine with [IrCl2Cp*]2: role of the chelating base, Dalton Trans., 2009, 5887–5893 Search PubMed.
- P. J. Dunn, The importance of green chemistry in process research and development, Chem. Soc. Rev., 2012, 41, 1452–1461 Search PubMed.
- A. Ramazani, Y. Ahmadi, H. Aghahosseini and S. W. Joo, “β-Cyclodextrin nano-reactor”-catalyzed synthesis of 2H-chromene-2,3-dicarboxylates from in-situ-generated stabilized phosphorus ylides via intramolecular Wittig reaction in water, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 354–358 Search PubMed.
- C.-Z. Liu, Y. Han, Y.-Y. Zhang, J. Sun and C.-G. Yan, Nucleophilic phosphine-promoted domino reaction of dialkyl acetylenedicarboxylates and 3-arylamino-1-methyl-1H-pyrrole-2,5-diones, Synthesis, 2018, 50, 3715–3722 Search PubMed.
- A. Alizadeh, R. Mohammadi, F. Bayat and L.-G. Zhu, Metal-free regioselective construction of diazabenzo[e]acephenanthrylene-1,2-dicarboxylates via a phosphine-mediated cycloadditon, Tetrahedron, 2017, 73, 4433–4438 Search PubMed.
- A. Ramazani and A. R. Kazemizadeh, Preparation of stabilized phosphorus ylides via multicomponent reactions and their synthetic applications, Curr. Org. Chem., 2011, 15, 3986–4020 Search PubMed.
- A. Alizadeh, S. Rostamnia and M.-L. Hu, A novel four-component reaction for the synthesis of 2,5-diaminofuran derivatives, Synlett, 2006, 1592–1594 Search PubMed.
- A. Shaabani, A. Maleki, A. H. Rezayan and A. Sarvary, Recent progress of isocyanide-based multicomponent reactions in Iran, Mol. Divers., 2011, 15, 41–68 Search PubMed.
- V. Estévez, M. Villacampa and J. C. Menéndez, Multicomponent reactions for the synthesis of pyrroles, Chem. Soc. Rev., 2010, 39, 4402–4421 Search PubMed.
- J.-P. Wan and Y. Liu, Recent advances in new multicomponent synthesis of structurally diversified 1,4-dihydropyridines, RSC Adv., 2012, 2, 9763–9777 Search PubMed.
- V. Estévez, M. Villacampa and J. C. Menéndez, Recent advances in the synthesis of pyrroles by multicomponent reactions, Chem. Soc. Rev., 2014, 43, 4633–4657 Search PubMed.
- E. M. El-Sheref and A. B. Brown, Utility of acetylenedicarboxylate in organic synthesis, J. Heterocycl. Chem., 2017, 54, 825–843 Search PubMed.
- C. G. Neochoritis, T. Zarganes-Tzitzikas and J. Stephanidou-Stephanatou, Dimethyl acetylenedicarboxylate: A versatile tool in organic synthesis, Synthesis, 2014, 46, 0537–0585 Search PubMed.
- M. M. Heravi and T. Alishiri, Dimethyl acetylenedicarboxylate as a building block in heterocyclic synthesis, Adv. Heterocycl. Chem., 2014, 113, 1–66 Search PubMed.
- A. L. Odom and T. J. McDaniel, Titanium-catalyzed multicomponent couplings: Efficient one-pot syntheses of nitrogen heterocycles, Acc. Chem. Res., 2015, 48, 2822–2833 Search PubMed.
- M. Anary-Abbasinejad, K. Charkhati and H. Anaraki-Ardakani, A novel approach to the synthesis of highly functionalized pyrroles, Synlett, 2009, 1115–1117 Search PubMed.
- A. V. Lygin, O. V. Larionov, V. S. Korotkov and A. de Meijere, Oligosubstituted pyrroles directly from substituted methyl isocyanides and acetylenes, Chem. – Eur. J., 2009, 15, 227–236 Search PubMed.
- O. A. Attanasi, S. Berretta, L. De Crescentini, G. Favi, G. Giorgi and F. Mantellini, Lewis acid-catalyzed synthesis of functionalized pyrroles, Adv. Synth. Catal., 2009, 351, 715–719 Search PubMed.
- O. A. Attanasi, G. Favi, F. Mantellini, G. Moscatelli and S. Santeusanio, A novel assembly of substituted pyrroles by acid–catalyzed sequential three–component reaction of amines, alkynoates, and 1,2−diaza–1,3−dienes, Adv. Synth. Catal., 2011, 353, 1519–1524 Search PubMed.
- B. Das, N. Bhunia and M. Lingaiah, A simple and efficient metal-free synthesis of tetrasubstituted pyrroles by iodine-catalyzed four-component coupling reaction of aldehydes, amines, dialkyl acetylenedicarboxylates, and nitromethane, Synthesis, 2011, 3471–3474 Search PubMed.
- O. A. Attanasi, S. Berretta, L. De Crescentini, G. Favi, G. Giorgi, F. Mantellini and S. Nicolini, Zinc(II) triflate–catalyzed divergent synthesis of polyfunctionalized pyrroles, Adv. Synth. Catal., 2011, 353, 595–605 Search PubMed.
- S. Z. Sayyed-Alangi, Z. Hossaini and F. Rostami-Charati, Synthesis of highly functionalized pyrroles from primary amines and activated acetylenes in water, Chin. Chem. Lett., 2012, 23, 1119–1121 Search PubMed.
- Y. Han, Y. Sun, J. Sun and C.-G. Yan, Efficient synthesis of pentasubstituted pyrroles via one-pot reactions of arylamines, acetylenedicarboxylates, and 3-phenacylideneoxindoles, Tetrahedron, 2012, 68, 8256–8260 Search PubMed.
- L. Wang, Y. Zhang, H. Y. Hu, H. K. Fun and J. H. J. Xu, Photoreactions of 1-acetylisatin with alkynes: Regioselectivity in oxetene formation and easy access to 3-alkylideneoxindoles and dispiro[oxindole[3,2′]furan[3′,3′′]oxindole]s, J. Org. Chem., 2005, 70, 3850–3858 Search PubMed.
- S. Madabhushi, V. S. Vangipuram, K. K. R. Mallu, R. Jillella, S. Kurva and S. R. Pamulaparthi, Ceric (IV) ammonium nitrate (CAN) mediated synthesis of pyrrole-2,3,4,5-tetracarboxylates by reaction of dimethyl acetylenedicarboxylate with an amine, Tetrahedron Lett., 2013, 54, 6737–6739 Search PubMed.
- X. Feng, Q. Wang, W. Lin, G.-L. Dou, Z.-B. Huang and D.-Q. Shi, Highly efficient synthesis of polysubstituted pyrroles via four-component domino reaction, Org. Lett., 2013, 15, 2542–2545 Search PubMed.
- W. M. Shu, Y. Yang, D. X. Zhang, L. M. Wu, Y. P. Zhu, G. D. Yin and A. X. Wu, Highly efficient synthesis of 3a,6a-dihydrofuro[2,3-b]furans via a novel bicyclization, Org. Lett., 2013, 15, 456–459 Search PubMed.
- W. Liu, L. Tan, P. Zhou, C. Chen and Q. Zhang, Approach to trisubstituted 1H-pyrroles from alkynoates and amines mediated by tert-butyl perbenzoate, Synlett, 2013, 24, 991–994 Search PubMed.
- H. Mehrabi, M. Anary-Abbasinejad and F. Mirhashemi, An efficient and convenient synthesis of pentasubstituted pyrroles from alkyl acetoacetates, dialkyl acetylenedicarboxylates, and amines, Tetrahedron Lett., 2014, 55, 4310–4314 Search PubMed.
- Q. Zhu, H. Jiang, J. Li, S. Liu, C. Xia and M. Zhang, Concise and versatile multicomponent synthesis of multisubstituted polyfunctional dihydropyrroles, J. Comb. Chem., 2009, 11, 685–696 Search PubMed.
- N. Pinto, N. Fleury-Brégeot and A. Marinetti, Enantioselective binaphthophosphepine–promoted [3+2] annulations of N–Ts– and N–DPP–imines with allenoates and 2−butynoates, Eur. J. Org. Chem., 2009, 146–151 Search PubMed.
- H. Jiang, R. Mai, H. Cao, Q. Zhu and X. Liu, l-Proline-catalyzed synthesis of highly functionalized multisubstituted 1,4-dihydropyridines, Org. Biomol. Chem., 2009, 7, 4943–4953 Search PubMed.
- N. N. Karade, V. H. Budhewar, S. V. Shinde and W. N. Jadhav, l-Proline as an efficient organo-catalyst for the synthesis of polyhydroquinoline via multicomponent Hantzsch reaction, Lett. Org. Chem., 2007, 4, 16–19 Search PubMed.
- C.-L. Shi, D.-Q. Shi, S. H. Kim, Z.-B. Huang, S.-J. Ji and M. Ji, A novel and efficient one-pot synthesis of furo[3′,4′:5,6]pyrido[2,3-c]pyrazole derivatives using organocatalysts, Tetrahedron, 2008, 64, 2425–2432 Search PubMed.
- S. Abdolmohammadi and S. Balalaie, Novel and efficient catalysts for the one-pot synthesis of 3, 4-dihydropyrano[c]chromene derivatives in aqueous media, Tetrahedron Lett., 2007, 48, 3299–3303 Search PubMed.
- J. Sun, Q. Wu, E.-Y. Xia and C.-G. Yan, Molecular diversity of three–component reactions of aromatic aldehydes, arylamines, and acetylenedicarboxylates, Eur. J. Org. Chem., 2011, 2981–2986 Search PubMed.
- T. Glotova, M. Dvorko, I. Ushakov, N. Chipanina, O. Kazheva, A. Chekhlov, O. Dyachenko, N. Gusarova and B. Trofimov, Chemo-, regio-and stereospecific addition of amino acids to acylacetylenes: a facile synthesis of new N-acylvinyl derivatives of amino acids, Tetrahedron, 2009, 65, 9814–9818 Search PubMed.
- A. Ziyaei-Halimehjani and M. R. Saidi, Synthesis of aza-Henry products and enamines in water by Michael addition of amines or thiols to activated unsaturated compounds, Tetrahedron Lett., 2008, 49, 1244–1248 Search PubMed.
- X. Li, J. Y. Wang, W. Yu and L. M. Wu, PtCl2-catalyzed reactions of o-alkynylanilines with ethyl propiolate and dimethyl acetylenedicarboxylate, Tetrahedron, 2009, 65, 1140–1146 Search PubMed.
- I. Yavari, M. J. Bayat, M. Sirouspour and S. Souri, One-pot synthesis of highly functionalized 1,2-dihydropyridines from primary alkylamines, alkyl isocyanides, and acetylenic esters, Tetrahedron, 2010, 66, 7995–7999 Search PubMed.
- Q. H. Zhu, H. F. Jiang, J. H. Li, M. Zhang, X. J. Wang and C. R. Qi, l-Proline-catalyzed five-component domino reaction leading to multifunctionalized 1,2,3,4-tetrahydropyridines, Tetrahedron, 2010, 66, 9721–9728 Search PubMed.
- S. A. Shehzadi, M. L. Christophe, V. Velde, A. Saeed and K. A. Tehrani, Synthesis of highly functionalized 1,6-dihydropyridines via the Zn(OTf)2-catalyzed three-component cascade reaction of aldimines and two Alkynes (IA2-coupling), Org. Biomol. Chem., 2018, 16, 3241–3247 Search PubMed.
- M. Zhang, H. Jiang, H. Liu and Q. Zhu, Convenient one-pot synthesis of multisubstituted tetrahydropyrimidines via catalyst-free multicomponent reactions, Org. Lett., 2007, 9, 4111–4113 Search PubMed.
- H. Cao, X. Wang, H. Jiang, Q. Zhu, M. Zhang and H. Liu, Development, scope and mechanisms of multicomponent reactions of asymmetric electron-deficient alkynes with amines and formaldehyde, Chem. – Eur. J., 2008, 14, 11623–11633 Search PubMed.
- Q. Zhu, H. Jiang, J. Li, M. Zhang, X. Wang and C. Qi, Practical synthesis and mechanistic study of polysubstituted tetrahydropyrimidines with use of domino multicomponent reactions, Tetrahedron, 2009, 65, 4604–4613 Search PubMed.
- H. Cho, K. Shima, M. Hayashimatsu, Y. Ohnaka, A. Mizuno and Y. Takeuchi, Synthesis of novel dihydropyrimidines and tetrahydropyrimidines, J. Org. Chem., 1985, 50, 4227–4230 Search PubMed.
- S. Asghari, M. Qandalee, Z. Naderi and Z. Sobhaninia, One-pot synthesis of 4-arylquinolines from aromatic aminoketones and vinylphosphonium salts, Mol. Divers., 2010, 14, 569–574 Search PubMed.
- R. J. Lundgren, A. Wilsily, N. Marion, C. Ma, Y. K. Chung and G. C. Fu, Catalytic asymmetric C-N bond formation: Phosphine-catalyzed intra- and intermolecular γ-addition of nitrogen nucleophiles to allenoates and alkynoates, Angew. Chem., Int. Ed., 2013, 52, 2525–2528 Search PubMed.
- J. Sun, Q. Wu, E.-Y. Xia and C.-G. Yan, Molecular diversity of three–component reactions of aromatic aldehydes, arylamines, and acetylenedicarboxylates, Eur. J. Org. Chem., 2011, 2981–2986 Search PubMed.
- F. Nasiri, M. Bayzidi and A. Zolali, Reaction between enaminones and acetylenic esters in the presence of triphenylphosphine: A convenient synthesis of alkyl 2(1-benzyl-2,4-dioxo-2,3,4,5,6,7-hexahydro-1H-indol-3-yl)acetates, Mol. Divers., 2012, 16, 619–623 Search PubMed.
- J.-C. Deng and S.-C. Chuang, Three-component and nonclassical reaction of phosphines with enynes and aldehydes: Formation of γ-lactones featuring α-phosphorus ylides, Org. Lett., 2011, 13, 2248 Search PubMed.
- Y.-W. Lin, J.-C. Deng, Y.-Z. Hsieh and S.-C. Chuang, One-pot formation of fluorescent γ-lactams having an α-phosphorus ylide moiety through three-component α(δ’)-Michael reactions of phosphines with an enyne and N-tosyl aldimines, Org. Biomol. Chem., 2014, 12, 162–170 Search PubMed.
- J.-C. Deng, F.-W. Chan, C.-W. Kuo, C.-A. Cheng, C.-Y. Huang and S.-C. Chuang, Assembly of dimethyl acetylenedicarboxylate and phosphanes with aldehydes leading to γ–lactones bearing α–phosphorus ylides as Wittig reagents, Eur. J. Org. Chem., 2012, 5738–5747 Search PubMed.
- A. A. Esmaeili, H. Khoddam-Mohammadi, A. Moradi, E. Davamdar, M. Izadyar, M. Khavani and M. R. Islami, A facile access to highly functionalized triphenylphosphoranylidene succinimides through a three-component reaction and DFT investigation of the reaction mechanism, RSC Adv., 2014, 4, 37900–37907 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. A. Cheeseman, J. R. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. D. Farkas, K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09, Revision B.09, Gaussian, Inc., Pittsburgh, PA, 2009 Search PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behaviour, Phys. Rev. A: Gen. Phys., 1988, 38, 3098–3100 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 371, 785 Search PubMed.
- P. Kaur, R. Arora and N. S. Gill, Review on oxygen heterocycles, Indo Am. J. Pharm. Res., 2013, 3, 9067–9084 Search PubMed.
- M. Y. Yoon, J. H. Kim, D. S. Choi, U. S. Shin, J. Y. Lee and C. E. Song, Metal triflate–catalyzed regio– and stereoselective Friedel-Crafts alkenylation of arenes with alkynes in an ionic liquid: Scope and mechanism, Adv. Synth. Catal., 2007, 349, 1725–1737 Search PubMed.
- R. A. C. Leão, P. de F. de Moraes, M. C. B. C. Pedro and P. R. R. Costa, Synthesis of coumarins and neoflavones through zinc chloride catalyzed hydroarylation of acetylenic esters with phenols, Synthesis, 2011, 22, 3692–3696 Search PubMed.
- K. D. Kaufman and R. C. Kelly, A new synthesis of coumarins, J. Heterocycl. Chem., 1965, 2, 91–92 Search PubMed.
- X. Mi, C. Wang, M. Huang, Y. Wu and Y. Wu, Preparation of 3-acyl-4-arylcoumarins via metal-free tandem oxidative acylation/cyclization between alkynoates with aldehydes, J. Org. Chem., 2015, 80, 148–155 Search PubMed.
- D.-P. Specht, P.-A. Martic and S. Farid, Ketocoumarins: a new class of triplet sensitizers, Tetrahedron, 1982, 38, 1203–1211 Search PubMed.
- M.-M. Heravi, N. Poormohammad, Y.-S. Beheshtiha, B. Baghernejad and R. Malakooti, A new strategy for the synthesis of 3−acyl–coumarin using mesoporous molecular sieve MCM–41 as a novel and efficient catalyst, Chin. J. Chem., 2009, 27, 968–970 Search PubMed.
- M.-M. Heravi, S. Sadjadi, H.-A. Oskooie, R.-H. Shoar and F.-F. Bamoharram, The synthesis of coumarin-3-carboxylic acids and 3-acetyl-coumarin derivatives using heteropolyacids as heterogeneous and recyclable catalysts, Catal. Commun., 2008, 9, 470–474 Search PubMed.
- B.-C. Ranu and R. Jana, Ionic liquid as catalyst and reaction medium – A simple, efficient and green procedure for Knoevenagel condensation of aliphatic and aromatic carbonyl compounds using a task–specific basic ionic liquid, Eur. J. Org. Chem., 2006, 3767–3770 Search PubMed.
- X. Tang, A.-J. Blake, W. Lewis and S. Woodward, Asymmetric conjugate additions to 1,1′-diactivated cyclic enones - A comparative study, Tetrahedron: Asymmetry, 2009, 20, 1881–1891 Search PubMed.
- P. R. H. Surya and S. Sivakumar, Condensation of alpha-aroylketene dithioacetals and 2-hydroxyarylaldehydes results in facile synthesis of a combinatorial library of 3-aroylcoumarins, J. Org. Chem., 2006, 71, 8715–8723 Search PubMed.
- K. C. Majumdar, S. Samanta, I. Ansary and B. Roy, An unusual one-pot synthesis of 3-benzoylcoumarins and coumarin-3-carbaldehydes from 2-hydroxybenzaldehydes under esterification conditions, RSC Adv., 2012, 2, 2137–2143 Search PubMed.
- H. Sun, Y. Zhang, F. Guo, Y. Yan, C. Wan, Z. Zha and Z. Wang, One–pot synthesis of 3,4−disubstituted coumarins under catalysis of Mn3O4 nanoparticles, Eur. J. Org. Chem., 2012, 480–483 Search PubMed.
- W. Liu, Y. Li, K. Liu and Z. Li, Iron-catalyzed carbonylation-peroxidation of alkenes with aldehydes and hydroperoxides, J. Am. Chem. Soc., 2011, 133, 10756–10759 Search PubMed.
- J.-Y. Luo, H.-L. Hua, Z.-S. Chen, Z.-Z. Zhou, Y.-F. Yang, P.-X. Zhou, Y.-T. He, X.-Y. Liu and Y.-M. Liang, Metal-free cascade radical cyclization of 1,6-enynes with aldehydes, Chem. Commun., 2014, 50, 1564–1566 Search PubMed.
- Z. Shi and F. Glorius, Synthesis of fluorenones viaquaternary ammonium salt-promoted intramolecular dehydrogenative arylation of aldehydes, Chem. Sci., 2013, 4, 829–833 Search PubMed.
- X. Zhu, Y. Shi, H. Mao, Y. Cheng and C. Zhu, Tetraethylammonium bromide–catalyzed oxidative thioesterification of aldehydes and alcohols, Adv. Synth. Catal., 2013, 355, 3558–3562 Search PubMed.
- S.-G. Yang, J.-P. Hwang, M.-Y. Park, K. Lee and Y.-H. Kim, Highly efficient epoxidation of electron-deficient olefins with tetrabutylammonium peroxydisulfate, Tetrahedron, 2007, 63, 5184–5188 Search PubMed.
- K. Kawaai, T. Yamaguchi, E. Yamaguchi, S. Endo, N. Tada, A. Ikari and A. Itoh, Photoinduced generation of acyl radicals from simple aldehydes, access to 3-acyl-4-arylcoumarin derivatives, and evaluation of their antiandrogenic activities, J. Org. Chem., 2018, 83, 1988–1996 Search PubMed.
- Y. Shimada, K. Hattori, N. Tada, T. Miura and A. Itoh, Facile aerobic photooxidation of alcohols using 2-chloroanthraquinone under visible light irradiation, Synthesis, 2013, 45, 2684–2688 Search PubMed.
- N. Tada, T. Nobuta, S.-i. Hirashima, T. Miura, A. Itoh and Y. Ikebata, Direct aerobic photo-oxidative syntheses of aromatic methyl esters from methyl aromatics using anthraquinone-2,3-dicarboxylic acid as organophotocatalyst, Photochem. Photobiol. Sci., 2012, 11, 616–619 Search PubMed.
- N. Tada, K. Hattori, T. Nobuta, T. Miura and A. Itoh, Facile aerobic photooxidation of methyl group in the aromatic nucleus in the presence of an organocatalyst under VIS irradiation, Green Chem., 2011, 13, 1669–1671 Search PubMed.
- W. Yang, S. Yang, P. Lia and L. Wang, Visible-light initiated oxidative cyclization of phenyl propiolates with sulfinic acids to coumarin derivatives under metal-free conditions, Chem. Commun., 2015, 51, 7520–7523 Search PubMed.
- S. G. Modha, V. P. Mehta and E. Van der Eycken, Transition metal-catalyzed C-C bond formation via C-S bond cleavage: An overview, Chem. Soc. Rev., 2013, 42, 5042–5055 Search PubMed.
- Y. Xi, B. Dong, E. J. McClain, Q. Wang, T. L. Gregg, N. G. Akhmedov, J. L. Petersen and X. Shi, Gold-catalyzed intermolecular C-S bond formation: Efficient synthesis of α-substituted vinyl sulfones, Angew. Chem., Int. Ed., 2014, 53, 4657–4661 Search PubMed.
- Q. Lu, J. Zhang, G. Zhao, Y. Qi, H. Wang and A. Lei, Dioxygen-triggered oxidative radical reaction: Direct aerobic difunctionalization of terminal alkynes toward β-keto sulfones, J. Am. Chem. Soc., 2013, 135, 11481–11484 Search PubMed.
- W.-T. Wei, R.-J. Song, X.-H. Ouyang, Y. Li, H.-B. Li and J.-H. Li, Copper-catalyzed oxidative ipso-carboalkylation of activated alkynes with ethers leading to 3-etherified azaspiro[4.5]trienones, Org. Chem. Front., 2014, 1, 484 Search PubMed.
- X.-H. Ouyang, R.-J. Song, Y. Li, B. Liu and J.-H. Li, Metal-free oxidative ipso-carboacylation of alkynes: Synthesis of 3-acylspiro[4,5]trienones from N-arylpropiolamides and aldehydes, J. Org. Chem., 2014, 79, 4582–4589 Search PubMed.
- D. Zheng, J. Yu and J. Wu, Generation of sulfonyl radicals from aryldiazonium tetrafluoroborates and sulfur dioxide: The synthesis of 3-sulfonated coumarins, Angew. Chem., Int. Ed., 2016, 55, 11925–11929 Search PubMed.
- S. Ye, H. Wang, Q. Xiao, Q. Ding and J. Wu, Aminosulfonylation of arenes, sulfur dioxide, and hydrazines cocatalyzed by gold(III) chloride and palladium acetate, Adv. Synth. Catal., 2014, 356, 3225–3230 Search PubMed.
- F. Eugéne, B. Langlois and E. Laurent,
N-ethyldiisopropylamine and sulfur dioxide solutions. 2. Reactions with conjugate acceptors, J. Org. Chem., 1994, 59, 2599–2603 Search PubMed.
- S. Shaaban, A. Jolit, D. Petkova and N. Maulide, A family of low molecular-weight, organic catalysts for reductive C-C bond formation, Chem. Commun., 2015, 51, 13902–13905 Search PubMed.
-
N. S. Simpkins, Sulfones in organic synthesis, Pergamon Press, Oxford, 1993 Search PubMed.
- K. G. Petrov, Y. Zhang, M. Carter, G. S. Cockerill, S. Dickerson, C. A. Gauthier, Y. Guo, R. A. Mook, D. W. Rusnak, A. L. Walker, E. R. Wood and K. E. Lackey, Optimization and SAR for dual ErbB-1/ErbB-2 tyrosine kinase inhibition in the 6-furanylquinazoline series, Bioorg. Med. Chem. Lett., 2006, 16, 4686–4691 Search PubMed.
- M. N. Noshi, A. El-Awa, E. Torres and P. L. Fuchs, Conversion of cyclic vinyl sulfones to transposed vinyl phosphonates, J. Am. Chem. Soc., 2007, 129, 11242–11247 Search PubMed.
- J. N. Desrosiers and A. B. Charette, Catalytic enantioselective reduction of β,β-disubstituted vinyl phenyl sulfones by using bisphosphine monoxide ligands, Angew. Chem., Int. Ed., 2007, 46, 5955–5957 Search PubMed.
- S. Kotha and A. S. Chavan, Design and synthesis of benzosultine-sulfone as a o-xylylene precursor via cross-enyne metathesis and rongalite: Further expansion to polycyclics via regioselective Diels-Alder reaction, J. Org. Chem., 2010, 75, 4319–4322 Search PubMed.
- C. Cassani, L. Bernardi, F. Fini and A. Ricci, Catalytic asymmetric Mannich reactions of sulfonylacetates, Angew. Chem., Int. Ed., 2009, 48, 5694–5697 Search PubMed.
- V. Sikervar, J. C. Fleet and P. L. Fuchs, A general approach to the synthesis of enantiopure 19-nor-vitamin D3 and its C-2 phosphate analogs prepared from cyclohexadienyl sulfone, Chem. Commun., 2012, 48, 9077–9079 Search PubMed.
- V. Sikervar, J. C. Fleet and P. L. Fuchs, Fluoride-mediated elimination of allyl sulfones: application to the synthesis of a 2,4-dimethyl-A-ring vitamin D3 analogue, J. Org. Chem., 2012, 77, 5132–5138 Search PubMed.
- E. A. Rodkey, D. C. McLeod, C. R. Bethel, K. M. Smith, Y. Xu, W. Chai, T. Che, P. R. Carey, R. A. Bonomo, F. Akker and J. D. Buynak,
β-Lactamase inhibition by 7-alkylidenecephalosporin sulfones: Allylic transposition and formation of an unprecedented stabilized acyl-enzyme, J. Am. Chem. Soc., 2013, 135, 18358–18369 Search PubMed.
- E. J. Emmett, B. R. Hayter and M. C. Willis, Palladium-catalyzed three-component diaryl sulfone synthesis exploiting the sulfur dioxide surrogate DABSO, Angew. Chem., Int. Ed., 2013, 52, 12679–12683 Search PubMed.
- W. Wei, J. Wen, D. Yang, M. Guo, Y. Wang, J. You and H. Wang, Direct and metal-free arylsulfonylation of alkynes with sulfonylhydrazides for the construction of 3-sulfonated coumarins, Chem. Commun., 2015, 51, 768–771 Search PubMed.
- A. El-Shafei, A. A. Fadda, I. I. Abdel-Gawad and E. H. E. Youssif, Stereospecificity of Diels-Alder reactions validated using ab initio calculations: Synthesis of novel coumarin and phenanthridine derivatives, Synth. Commun., 2009, 39, 2954–2972 Search PubMed.
- T. A. Dias and M. F. Proença, An ecofriendly approach to the synthesis of 2-imino-and 2-oxo-3-phenylsulfonyl-2H-chromenes, Tetrahedron Lett., 2012, 53, 5235–5237 Search PubMed.
- J. K. Augustine, A. Bombrun, B. Ramappa and C. Boodappa, An efficient one-pot synthesis of coumarins mediated by propylphosphonic anhydride (T3P) via the Perkin condensation, Tetrahedron Lett., 2012, 53, 4422–4425 Search PubMed.
- J. R. Merchant and P. J. Shah, Synthesis of 3−coumaryl phenyl sulfones or sulfoxides, J. Heterocycl. Chem., 1981, 18, 441–442 Search PubMed.
- H. Yoshida, Y. Ito and J. Ohshita, Three-component coupling using arynes and DMF: Straightforward access to coumarins via ortho-quinone methides, Chem. Commun., 2011, 47, 8512–8514 Search PubMed.
- T. Liu, Q. Ding, G. Qiu and J. Wu, Tandem metal-free oxidative radical 5-exo dearomative spirocyclization and ester migration: Generation of 3-functionalized coumarins from alkynoates, Tetrahedron, 2016, 72, 279–284 Search PubMed.
- S. Ni, J. Cao, H. Mei, J. Han, S. Li and Y. Pan, Sunlight-promoted cyclization versus decarboxylation in the reaction of alkynoates with N-iodosuccinimide: easy access to 3-iodocoumarins, Green Chem., 2016, 18, 3935–3939 Search PubMed.
- D. T. Racys, C. E. Warrilow, S. L. Pimlott and A. Sutherland, Highly regioselective
iodination of arenes via iron(III)-catalyzed activation of N-iodosuccinimide, Org. Lett., 2015, 17, 4782–4785 Search PubMed.
- F. Mo, J. M. Yan, D. Qiu, F. Li, Y. Zhang and J. Wang, Gold-catalyzed halogenation of aromatics by N-halosuccinimides, Angew. Chem., Int. Ed., 2010, 49, 2028–2032 Search PubMed.
- D. T. Racys, S. A. I. Sharif, S. L. Pimlott and A. Sutherland, Silver(I)-catalyzed iodination of arenes: Tuning the Lewis acidity of N-iodosuccinimide activation, J. Org. Chem., 2016, 81, 772–780 Search PubMed.
- C. Q. O'Broin, P. Fernández, C. Martínez and K. Muñiz,
N-iodosuccinimide-promoted Hofmann-Löffler reactions of sulfonimides under visible light, Org. Lett., 2016, 18, 436–439 Search PubMed.
- S. Ni, W. Sha, L. Zhang, C. Xie, H. Mei, J. Han and Y. Pan,
N-iodosuccinimide-promoted cascade trifunctionalization of alkynoates: Access to 1,1-diiodoalkenes, Org. Lett., 2016, 18, 712–715 Search PubMed.
- S. N. Mendis and J. A. Tunge, Palladium-catalyzed stereospecific decarboxylative benzylation of alkynes, Org. Lett., 2015, 17, 5164–5167 Search PubMed.
- G. Qiu, T. Liu and Q. Ding, Tandem oxidative radical brominative addition of activated alkynes and spirocyclization: Switchable synthesis of 3-bromocoumarins and 3-bromo spiro-[4,5] trienone, Org. Chem. Front., 2016, 3, 510–515 Search PubMed.
- K.-M. Kim and I.-H. Park, Unprecedented stereospecific synthesis of a novel tetracyclic ring system, a hybrid of tetrahydropyrrolo[2,3-b]indole and tetrahydroimidazo[1,2-a]indole, via a domino reaction upon a tryptophan-derived amino nitrile, Synthesis, 2004, 2641–2644 Search PubMed.
- G. V. Ramanarayanan, V. G. Shukla and K. G. Akamanchi, A novel and one step procedure for preparation of α-bromo-α,β-unsaturated carbonyl compounds, Synlett, 2002, 2059–2061 Search PubMed.
- M. A. Kumar, C. N. Rohitha, S. J. Kulkarni and N. Narender, Bromination of aromatic compounds using ammonium bromide and Oxone®, Synthesis, 2010, 1629–1632 Search PubMed.
- C.-G. Cho, J.-S. Park, I.-H. Jung and H. Lee, One step preparation of bromo-2-pyrones via bromo-decarboxylation of 2-pyrone-carboxylic acids, Tetrahedron Lett., 2001, 42, 1065–1067 Search PubMed.
- R. M. Kelkar, U. K. Joshi and M. V. Paradkar, A novel synthesis of 4-aminomethylcoumarins, Synthesis, 1986, 214–216 Search PubMed.
- W. Yang, S. Yang, P. Li and L. Wang, Visible-light initiated oxidative cyclization of phenyl propiolates with sulfinic acids to coumarin derivatives under metal-free conditions, Chem. Commun., 2015, 51, 7520–7523 Search PubMed.
- W. Fu, M. Zhu, G. Zou, C. Xu, Z. Wang and B. Ji, Visible-light-mediated radical aryldifluoroacetylation of alkynes with ethyl bromodifluoroacetate for the synthesis of 3-difluoroacetylated coumarins, J. Org. Chem., 2015, 80, 4766–4770 Search PubMed.
- K. Yan, D. Yang, W. Wei, F. Wang, Y. Shuai, Q. Li and H. Wang, Silver-mediated radical cyclization of alkynoates and α-keto acids leading to coumarins via cascade double C–C bond formation, J. Org. Chem., 2015, 80, 1550–1556 Search PubMed.
- W. Wei, J. Wen, D. Yang, M. Guo, Y. Wang, J. You and H. Wang, Direct and metal-free arylsulfonylation of alkynes with sulfonylhydrazides for the construction of 3-sulfonated coumarins, Chem. Commun., 2015, 51, 768–771 Search PubMed.
- Y. Li, Y. Lu, G. Qiu and Q. Ding, Copper-catalyzed direct trifluoromethylation of propiolates: Construction of trifluoromethylated coumarins, Org. Lett., 2014, 16, 4240–4243 Search PubMed.
- X. Mi, C. Wang, M. Huang, J. Zhang, Y. Wu and Y. Wu, Silver-catalyzed synthesis of 3-phosphorated coumarins via radical cyclization of alkynoates and dialkyl H-phosphonates, Org. Lett., 2014, 16, 3356–3359 Search PubMed.
- C. Pan, R. Chen, W. Shao and J.-T. Yu, Metal-free radical addition/cyclization of alkynoates with xanthates towards 3-(β-carbonyl)coumarins, Org. Biomol. Chem., 2016, 14, 9033–9039 Search PubMed.
- Y. Yu, S. Zhuang, P. Liu and P. Sun, Cyanomethylation and cyclization of aryl alkynoates with acetonitrile under transition-metal-free conditions: Synthesis of 3-cyanomethylated coumarins, J. Org. Chem., 2016, 81, 11489–11495 Search PubMed.
- W.-C. Gao, T. Liu, B. Zhang, X. Li, W.-L. Wei, Q. Liu, J. Tian and H.-H. Chang, Synthesis of 3-sulfenylated coumarins: BF3•Et2O-mediated electrophilic cyclization of aryl alkynoates using N-sulfanylsuccinimides, J. Org. Chem., 2016, 81, 11297–11304 Search PubMed.
- L. Du, P. Cao, J. Xing, Y. Lou, L. Jiang, L. Li and J. Liao, Hydrogen-bond-promoted palladium catalysis: Allylic alkylation of indoles with unsymmetrical 1,3-disubstituted allyl acetates using chiral bis(sulfoxide) phosphine ligands, Angew. Chem., Int. Ed., 2013, 52, 4207–4211 Search PubMed.
- L. Melzig, C. B. Rauhut, N. Naredi-Rainer and P. Knochel, Difunctionalisation of arenes and heteroarenes by directed metallation and sulfoxide-magnesium exchange, Chem. – Eur. J., 2011, 17, 5362–5372 Search PubMed.
- A. Abramovitch and I. Marek, Zinc homologation-elimination reaction of α–sulfinyl carbanions as a new route to olefins, Eur. J. Org. Chem., 2008, 4924–4931 Search PubMed.
- J. Aziz, S. Messaoudi, M. Alami and A. Hamze, Sulfinate derivatives: Dual and versatile partners in organic synthesis, Org. Biomol. Chem., 2014, 12, 9743–9759 Search PubMed.
- W. Wu, Y. An, J. Li, S. Yang, Z. Zhu and H. Jiang, Iodine-catalyzed cascade annulation of alkynes with sodium arylsulfinates: Assembly of 3-sulfenylcoumarin and 3-sulfenylquinolinone derivatives, Org. Chem. Front., 2017, 4, 1751–1756 Search PubMed.
- S. Yang, H. Tan, W. Ji, X. Zhang, P. Li and L. Wang, Visible light–induced decarboxylative acylarylation of phenyl propiolates with α–oxocarboxylic acids to coumarins catalyzed by hypervalent iodine reagents under transition metal–free conditions, Adv. Synth. Catal., 2017, 359, 443–453 Search PubMed.
- S. Feng, J. Li, Z. Liu, H. Sun, H. Shi, X. Wang, X. Xie and X. She, Visible-light-mediated radical cascade reaction: Synthesis of 3-bromocoumarins from alkynoates, Org. Biomol. Chem., 2017, 15, 8820–8826 Search PubMed.
- L. Zhang, T. Meng, R. Fan and J. Wu, General and efficient route for the synthesis of 3,4-disubstituted coumarins via Pd-catalyzed site-selective cross-coupling reactions, J. Org. Chem., 2007, 72, 7279–7286 Search PubMed.
- T. Yamaguchi, T. Fukuda, F. Ishibashi and M. Iwao, The first total synthesis of lamellarin α, 20-sulfate, a selective inhibitor of HIV-1 integrase, Tetrahedron Lett., 2006, 47, 3755–3757 Search PubMed.
- L. Santana, E. Uriarte, H. Gonzalez-Diaz, G. Zagotto, R. Soto-Otero and E. Mendez-Alvarez, A QSAR model for in silico screening of MAO-A inhibitors. Prediction, synthesis, and biological assay of novel coumarins, J. Med. Chem., 2006, 49, 1149–1156 Search PubMed.
- H. Chen, L. Zhou, S. Li, Y. Yao, Y. Gu, C. Li, N. Li, M. Meng and X. Li, Synthesis of 3,4-biphenyl coumarin derivatives and their promoting effects on the killing activity of natural killing cell, Chin. J. Org. Chem., 2013, 33, 164–168 Search PubMed.
- D. Liu, J.-Q. Chen, X.-Z. Wang and P.-F. Xu, Metal–Free, Visible–light–promoted synthesis of 3−phosphorylated coumarins via radical C-P/C-C bond formation, Adv. Synth. Catal., 2017, 359, 2773–2777 Search PubMed.
- X.-Q. Pan, J.-P. Zou, W.-B. Yi and W. Zhang, Recent advances in sulfur-and phosphorous-centered radical reactions for the formation of S–C and P–C bonds, Tetrahedron, 2015, 71, 7481–7529 Search PubMed.
- Z. Wang, X. Li, L. Wang and P. Li, Photoinduced cyclization of alkynoates to coumarins with N-iodosuccinimide as a free-radical initiator under ambient and metal-free conditions, Tetrahedron, 2019, 75, 1044–1051 Search PubMed.
- J. Hua, Z. Fang, J. Xu, M. Bian, C. Liu, W. He, N. Zhu, Z. Yang and K. Guo, Electrochemical oxidative cyclization of activated alkynes with diselenides or disulfides: Access to functionalized coumarins or quinolinones, Green Chem., 2019, 21, 4706–4711 Search PubMed.
- S. Vedachalam, Q.-L. Wong, B. Maji, J. Zeng, J. Ma and X.-W. Liu,
N–heterocyclic carbene catalyzed intramolecular hydroacylation of activated alkynes: Synthesis of chromones, Adv. Synth. Catal., 2011, 353, 219–225 Search PubMed.
- R. Breslow and C. Schmuck, The mechanism of thiazolium catalysis, Tetrahedron Lett., 1996, 37, 8241–8242 Search PubMed.
- R. Breslow and R. Kim, The thiazolium catalyzed benzoin condensation with mild base does not involve a “dimer” intermediate, Tetrahedron Lett., 1994, 35, 699–702 Search PubMed.
- R. Breslow, On the mechanism of thiamine action. IV.1 Evidence from studies on model systems, J. Am. Chem. Soc., 1958, 80, 3719–3726 Search PubMed.
- L.-G. Meng, B. Hu, Q.-P. Wu, M. Liang and S. Xue, PPh3-catalyzed unexpected α-addition reaction of 1-(o-hydroxyaryl)-1,3-diketones to terminal alkynoates: A straightforward synthesis of multifunctional vinylesters, Chem. Commun., 2009, 6089–6091 Search PubMed.
- P. R. Krishna, E. R. Sekhar and F. Mongin, Lewis acid-and/or Lewis base-catalyzed [3+2] cycloaddition reaction: Synthesis of pyrazoles and pyrazolines, Tetrahedron Lett., 2008, 49, 6768–6772 Search PubMed.
- S. H. Kim, H. S. Lee, K. H. Kim and J. N. Kim, Indium-mediated double Barbier reaction of γ-cyanoesters derived from Baylis–Hillman adduct, Tetrahedron Lett., 2009, 50, 1696–1698 Search PubMed.
- B. Yamada, P. Zetterlund and E. Sato, Utility of propenyl groups in free radical polymerization: Effects of steric hindrance on formation and reaction behavior as versatile intermediates, Prog. Polym. Sci., 2006, 31, 835–877 Search PubMed.
- R. Takagi, Y. Inoue and K. Ohkata, Construction of the adamantane core of Plukenetione-type polycyclic polyprenylated acylphloroglucinols, J. Org. Chem., 2008, 73, 9320–9325 Search PubMed.
- B. Prabhudas and D. L. J. Clive, All-carbon intramolecular conjugate displacement reactions: An effective route to carbocycles, Angew. Chem., Int. Ed., 2007, 46, 9295–9297 Search PubMed.
- K. Aikawa, Y. Takabayashi, S. Kawauchi and K. Mikami, Axial chirality control of tropos BIPHEP-Rh complexes by chiral dienes: Synergy effect in catalytic asymmetric hydrogenation, Chem. Commun., 2008, 5095–5097 Search PubMed.
- L. D. Yadav, R. Patel and V. P. Srivastava, An easy access to functionalized allyl dithiocarbamates from Baylis–Hillman adducts in water, Tetrahedron Lett., 2009, 50, 1335–1339 Search PubMed.
- B. M. Trost and G. R. Dake, Nucleophilic α-addition to alkynoates. A synthesis of dehydroamino acids, J. Am. Chem. Soc., 1997, 119, 7595–7596 Search PubMed.
- M. Hanédanian, O. Loreau, F. Taran and C. Mioskowski,
α-Addition of activated methylenes to alkynoates. A straightforward synthesis of multifunctional compounds, Tetrahedron Lett., 2004, 45, 7035 Search PubMed.
- L.-G. Meng, H.-F. Liu, J.-L. Wei, S.-N. Gong and S. Xue, One-pot reaction of ortho-acylphenols and terminal alkynoates for synthesis of 2-alkyl-substituted chromanones, Tetrahedron Lett., 2010, 51, 1748–1750 Search PubMed.
- J.-C. Deng and S.-C. Chuang, Multicomponent reactions of phosphines, diynedioates, and aryl aldehydes generated furans appending reactive phosphorus ylides through cumulated trienoates as key intermediates: A phosphine α-addition-δ-evolvement of an anion pathway, Org. Lett., 2014, 16, 5792–5795 Search PubMed.
- J. Dong, H. Du and J. Xu, Regiospecific synthesis of polysubstituted furans with mono-to tricarboxylates from various sulfonium acylmethylides and acetylenic esters, RSC Adv., 2019, 9, 25034–25038 Search PubMed.
- L. Q. Lu, T. R. Li, Q. Wang and W. J. Xiao, Beyond sulfide-centric catalysis: Recent advances in the catalytic cyclization reactions of sulfur ylides, Chem. Soc. Rev., 2017, 46, 4135–4149 Search PubMed.
- A. C. B. Burtoloso, R. M. P. Dias and I. A. Leonarczyk, Sulfoxonium and sulfonium ylides as diazocarbonyl equivalents in metal–catalyzed insertion reactions, Eur. J. Org. Chem., 2013, 5005–5016 Search PubMed.
- V. K. Aggarwal and C. L. Winn, Catalytic, asymmetric sulfur ylide-mediated epoxidation of carbonyl compounds: Scope, selectivity, and applications in synthesis, Acc. Chem. Res., 2004, 37, 611–620 Search PubMed.
- A. H. Li, L. X. Dai and V. K. Aggarwal, Asymmetric ylide reactions: Epoxidation, cyclopropanation, aziridination, olefination, and rearrangement, Chem. Rev., 1997, 97, 2341–2372 Search PubMed.
- J. Dong and J. X. Xu, Facile synthesis of thietanes via ring expansion of thiiranes, Org. Biomol. Chem., 2017, 15, 836–844 Search PubMed.
- S. Kramer and T. Skrydstrup, Gold-catalyzed carbene transfer to alkynes: Access to 2,4-disubstituted furans, Angew. Chem., Int. Ed., 2012, 51, 4681–4684 Search PubMed.
- X. Huang, B. Peng, M. Luparia, L. F. Gomes, L. F. Veiros and N. Maulide, Gold-catalyzed synthesis of furans and furanones from sulfur ylides, Angew. Chem., Int. Ed., 2012, 51, 8886–8890 Search PubMed.
- M. Higo and T. Mukaiyama, A convenient method for the synthesis of 3,4-furandicarboxylic acid derivatives by the reactions of sulfonium ylides with dimethyl acetylenedicarboxylate, Tetrahedron Lett., 1970, 11, 2565–2568 Search PubMed.
- X. H. Li and J. X. Xu, Identification of microwave selective heating effort in an intermolecular reaction with Hammett linear relationship as a molecular level probe, Curr. Microwave Chem., 2017, 4, 339–346 Search PubMed.
- X. H. Li and J. X. Xu, Determination on temperature gradient of different polar reactants in reaction mixture under microwave irradiation with molecular probe, Tetrahedron, 2016, 72, 5515–5520 Search PubMed.
- X. H. Li and J. X. Xu, Effects of the microwave power on the microwave-assisted esterification, Curr. Microwave Chem., 2017, 4, 158–162 Search PubMed.
- L. B. Hu, Y. K. Wang, B. N. Li, D. M. Du and J. X. Xu, Diastereoselectivity in the Staudinger reaction: A useful probe for investigation of nonthermal microwave effects, Tetrahedron, 2007, 63, 9387–9392 Search PubMed.
- D. Q. Yang and J. X. Xu, Influence of the product polarity on temperature profiles in the microwave-assisted Claisen rearrangement, Curr. Microwave Chem., 2018, 5, 120–127 Search PubMed.
- Y. Hayasi, M. Kobayasi and H. Nozaki, Furans from sulphonium diacylmethylides and acetylenes, Tetrahedron, 1970, 26, 4353–4360 Search PubMed.
-
K. Uneyama, Organofluorine chemistry, Blackwell, Oxford, 2006 Search PubMed.
-
R. D. Chambers, Fluorine in organic chemistry, Blackwell, Oxford, 2004 Search PubMed.
-
P. Kirsch, Modern fluoroorganic chemistry: Synthesis, reactivity, applications, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed.
- S. Wang, J. Han, C. Zhang, H. Qin and J. Xiao, An overview of reductive trifluoromethylation reactions using electrophilic ‘+CF3’ reagents, Tetrahedron, 2015, 71, 7949–7976 Search PubMed.
- K. Muller, C. Faeh and F. Diederich, Fluorine in pharmaceuticals: Looking beyond intuition, Science, 2007, 317, 1881–1886 Search PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Fluorine in medicinal chemistry, Chem. Soc. Rev., 2008, 37, 320–330 Search PubMed.
- J. Wang, M. S. Rosello, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011), Chem. Rev., 2014, 114, 2432–2506 Search PubMed.
- H. Liu and V. A. Soloshonok, Recent advances in the trifluoromethylation methodology and new CF3-containing drugs, J. Fluorine Chem., 2014, 167, 37–54 Search PubMed.
- P. Jeschke, The Unique role of halogen substituents in the design of modern agrochemicals, Pest Manage. Sci., 2010, 66, 10–27 Search PubMed.
- T. R. Elworthy, E. R. Brill, C. C. Caires, W. Kim, L. K. Lach, J. L. Tracy and S. Chiou, Lactams as prostanoid receptor ligands. Part 4: 2-Piperidones as selective EP4 receptor agonists, Bioorg. Med. Chem., 2005, 15, 2523–2526 Search PubMed.
- P. Bendale, S. Olepu, P. K. Suryadevara, V. Bulbule, K. Rivas, L. Nallan, B. Smart, K. Yokoyama, S. Ankala, P. R. Pendyala, D. Floyd, L. J. Lombardo, D. K. Williams, F. S. Buckner, D. Chakrabarti, C. L. M. J. Verlinde, W. C. Van Voorhis and M. H. Gelb, Second generation tetrahydroquinoline-based protein farnesyltransferase inhibitors as antimalarials, J. Med. Chem., 2007, 50, 4585–4605 Search PubMed.
- J. Liddle, M. J. Allen, A. D. Borthwick, D. P. Brooks, D. E. Davies, R. M. Edwards, A. M. Exall, C. Hamlett, W. R. Irving, A. M. Mason, G. P. McCafferty, F. Nerozzi, S. Peace, J. Philp, D. Pollard, M. A. Pullen, S. S. Shabbir, S. L. Sollis, T. D. Westfall, P. M. Woollard, C. Wu and D. M. B. Hickey, The discovery of GSK221149A: A potent and selective oxytocin antagonist, Bioorg. Med. Chem., 2008, 18, 90–94 Search PubMed.
- Y. Wang, L. Tao, J. Chen, W. He, H. Deng, M. Shao, H. Zhang and W. Cao, Isocyanide-based multicomponent reactions: A concise approach to 2-amino-3-perfluoroalkylfurans using methyl perfluoroalk-2-ynoates as fluorinated building blocks, Asian J. Org. Chem., 2019, 8, 710–715 Search PubMed.
- S. Sadjadi and M. M. Heravi, Recent application of isocyanides in synthesis of heterocycles, Tetrahedron, 2011, 67, 2707–2752 Search PubMed.
- T. Kaur, P. Wadhwa, S. Bagchi and A. Sharma, Isocyanide based [4+1] cycloaddition reactions: An indispensable tool in multi-component reactions (MCRs), Chem. Commun., 2016, 52, 6958–6976 Search PubMed.
- S. Sadjadi, M. M. Heravi and N. Nazari, Isocyanide-based multicomponent reactions in the synthesis of heterocycles, RSC Adv., 2016, 6, 53203–53272 Search PubMed.
- L.-G. Meng, N.-L. Ge, M.-M. Yang and L. Wang, Cyclization reaction of 2-azido-1-(2-hydroxyphenyl)ethanones with terminal alkynoates catalyzed by 4-dimethylaminopyridine (DMAP): Synthesis of 2-aminobenzofuran-3(2H)-one derivatives, Eur. J. Org. Chem., 2011, 3403–3406 Search PubMed.
- B. Batanero, J. Escudero and F. Barba, Cathodic reduction of phenacyl azides, Org. Lett., 1999, 1, 1521–1522 Search PubMed.
- X. Fan and Y. Zhang, SmI2-mediated synthesis of 2,4-diarylpyrroles from phenacyl azides, Tetrahedron Lett., 2002, 43, 1863–1865 Search PubMed.
- S. Gabillet, D. Lecerclé, O. Loreau, S. Dézard, J.-M. Gomis and F. Taran, Phosphine-catalyzed tandem α-O-addition and transesterification of oxgen pronucleophiles on arylpropiolates: A new route to dioxygenated heterocyles, Synthesis, 2007, 515–522 Search PubMed.
- L.-H. Zhou, X.-Q. Yu and L. Pu, Reactivity of γ-Hydroxy-α,β-acetylenic esters with amines: Facile synthesis of the optically active 4-amino-2(5H)-furanones, J. Org. Chem., 2009, 74, 2013–2017 Search PubMed.
- G. Gao, Q. Wang, X.-Q. Yu, R.-G. Xie and L. Pu, Highly enantioselective synthesis of γ–hydroxy–α,β–acetylenic esters by asymmetric alkyne addition to aldehydes, Angew. Chem., Int. Ed., 2006, 45, 122–125 Search PubMed.
- A. R. Rajaram and L. Pu, Regiospecific hydration of γ-hydroxy-α,β-acetylenic esters: A novel asymmetric synthesis of tetronic acids, Org. Lett., 2006, 8, 2019–2021 Search PubMed.
- B. M. Trost, A. H. Weiss and A. J. von Wangelin, Dinuclear Zn-catalyzed asymmetric alkynylation of unsaturated aldehydes, J. Am. Chem. Soc., 2006, 128, 8–9 Search PubMed.
- F. Yang, P. Xi, L. Yang, J. Lan, R. Xie and J. S. You, Facile, mild, and highly enantioselective alkynylzinc addition to aromatic aldehydes by BINOL/N-methylimidazole dual catalysis, J. Org. Chem., 2007, 72, 5457–5460 Search PubMed.
- H. Lebel and M. Parmentier, Copper-catalyzed methylenation reaction: Total synthesis of (+)-desoxygaliellalactone, Org. Lett., 2007, 9, 3563–3566 Search PubMed.
- R. H. Schlessinger, T. R. R. Pettus, J. P. Springer and K. Hoogsteen, Diastereoselective Diels-Alder reactions using furan substituted with a nonracemic amine, J. Org. Chem., 1994, 59, 3246–3247 Search PubMed.
- K. Matsuo, S. Ishida and Y. Takuno, Reaction of enaminones with benzyltrimethylammmonium dichloroiodate, Chem. Pharm. Bull., 1994, 42, 1149–1150 Search PubMed.
- P. J. Campos, J. Arranz and M. A. Rodriguez,
α-Iodination of enaminones with bis(pyridine)iodonium(I) tetrafluoroborate, Tetrahedron Lett., 1997, 38, 8397–8400 Search PubMed.
- J. M. Kim, J. E. Na and J. N. Kim,
α-Iodination of enaminones using the modified Johnson's procedure: The use of I2 and Et3N, Tetrahedron Lett., 2003, 44, 6317–6318 Search PubMed.
- N. I. Simirskaya, N. V. Ignat'ev, M. Schulte and S. G. Zlotin, Unprecedented acceleration of the domino reaction between methyl 4-hydroxyalk-3-ynoates and amines in ionic liquids, Mendeleev Commun., 2011, 21, 94–96 Search PubMed.
- J.-C. Deng and S.-C. Chuang, Three-component and nonclassical reaction of phosphines with enynes and aldehydes: Formation of γ-lactones featuring α-phosphorus ylides, Org. Lett., 2011, 13, 2248–2251 Search PubMed.
- L. Chen, B. Chen, F. Zhao, Y. Li, B. Li and M. Zhang, Task-specific acidic ionic liquid-catalyzed efficient synthesis of β-enaminolactones from alkynoates and β-amino alcohols, RSC Adv., 2017, 7, 30376–30379 Search PubMed.
- S.-G. Lee, Functionalized imidazolium salts for task-specific ionic liquids and their applications, Chem. Commun., 2006, 1049–1063 Search PubMed.
- G. Y. Zhao, T. Jiang, H. X. Gao, B. X. Han, J. Huang and D. H. Sun, Mannich reaction using acidic ionic liquids as catalysts and solvents, Green Chem., 2004, 6, 75–77 Search PubMed.
- D. Q. Xu, J. Wu, S. P. Luo, J. Zhang, J. Y. Wu, X. H. Du and Z. Y. Xu, Fischer indole synthesis catalyzed by novel SO3H-functionalized ionic liquids in water, Green Chem., 2009, 11, 1239–1246 Search PubMed.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Ionic liquids: A pathway to environmental acceptability, Chem. Soc. Rev., 2011, 40, 1383–1403 Search PubMed.
- N. Isambert, M. M. S. Duque, J.-C. Plaquevent, Y. G'enisson, J. Rodriguez and T. Constantieux, Multicomponent reactions and ionic liquids: A perfect synergy for eco-compatible heterocyclic synthesis, Chem. Soc. Rev., 2011, 40, 1347–1357 Search PubMed.
- R. Sheldon, Catalytic reactions in ionic liquids, Chem. Commun., 2001, 23, 2399–2407 Search PubMed.
- T. Welton, Room-temperature ionic liquids. Solvents for synthesis and catalysis, Chem.
Rev., 1999, 99, 2071–2083 Search PubMed.
- J. P. Hallett and T. Welton, Room-temperature ionic liquids: Solvents for synthesis and catalysis. 2, Chem. Rev., 2011, 111, 3508–3576 Search PubMed.
- T. Welton, Ionic liquids in catalysis, Coord. Chem. Rev., 2004, 248, 2459–2477 Search PubMed.
- A. S. Amarasekara, Acidic ionic liquids, Chem. Rev., 2016, 116, 6133–6183 Search PubMed.
- H. Waldmann, V. Khedkar, H. Dückert, M. Schürmann, I. M. Oppel and K. Kumar, Asymmetric synthesis of natural product inspired tricyclic benzopyrones by an organocatalyzed annulation reaction, Angew. Chem., Int. Ed., 2008, 47, 6869–6872 Search PubMed.
- W.-B. Liu, H.-F. Jiang and C.-L. Qiao, Hydroalkylation leading to heterocyclic compounds. Part 1: New strategies for the synthesis of polysubstituted 2H-pyran-2-ones, Tetrahedron, 2009, 65, 2110–2115 Search PubMed.
- Y. K. Chung and G. C. Fu, Phosphine-catalyzed enantioselective synthesis of oxygen heterocycles, Angew. Chem., Int. Ed., 2009, 48, 2225–2227 Search PubMed.
- Y. Pedduri and J. S. Williamson, One-pot synthesis of highly substituted tetrahydrofurans from activated propargyl alcohols using Bu3P, Tetrahedron Lett., 2008, 49, 6009–6012 Search PubMed.
- S. Osman and K. Koide, Cyclic acetal formation between 2-pyridinecarboxaldehyde and γ-hydroxy-α,β-acetylenic esters, Tetrahedron Lett., 2008, 49, 6550–6552 Search PubMed.
- T. W. Russell, D. M. Duncan and S. C. Hansen, Catalytic reduction. 4. Hydrogenation of aldehydes over borohydride reduced nickel and palladium, J. Org. Chem., 1977, 42, 551–552 Search PubMed.
- Y. S. Kim, S. H. Kwak and Y. D. Gong, Application of thio-Ugi adducts for the preparation of benzo[b]thiophene and S-heterocycle library via copper catalyzed intramolecular C-S bond formation, ACS Comb. Sci., 2015, 17, 365–373 Search PubMed.
- S. Gabillet, D. Lecerclé, O. Loreau, M. Carboni, S. Dézard, J.-M. Gomis and F. Taran, Phosphine-catalyzed construction of sulfur heterocycles, Org. Lett., 2007, 9, 3925–3927 Search PubMed.
- S. Carboni, J.-M. Gomis, O. Loreau and F. Taran, Synthesis of thiohydantoins by phosphine-catalyzed reaction of thioureas with arylpropiolates, Synthesis, 2008, 417–424 Search PubMed.
- B. T. Gregg, W. G. Earley, K. C. Golden, J. F. Quinn, D. A. Razzano and W. M. Rennells, First example of Lewis acid catalyzed 3-substituted 5-arylidene-1-methyl-2-thiohydantoin formation, Synthesis, 2006, 4200–4204 Search PubMed.
- B. T. Gregg, K. C. Golden, J. F. Quinn, D. O. Tumoshenko, W. G. Early, D. A. Maynard, D. A. Razzano, W. M. Rennells and J. Butcher, Expedient Lewis acid catalyzed synthesis of a 3-substituted 5-arylidene-1-methyl-2-thiohydantoin library, J. Comb. Chem., 2007, 9, 1036–1040 Search PubMed.
- W. Liu, C. Chen and H. Liu, Synthesis of polysubstituted thiophenes via base–induced [2+2+1] cycloaddition reaction of alkynes and elemental sulfur, Adv. Synth. Catal., 2015, 357, 4050–4054 Search PubMed.
- H. Cao, H.-F. Jiang, C.-R. Qi, W.-J. Yao and H.-J. Chen, Brønsted acid-promoted domino reactions: A novel one-pot three-component synthesis of 3,4,5-trisubstituted-3,6-dihydro-2H-1,3-oxazines, Tetrahedron Lett., 2009, 50, 1209–1214 Search PubMed.
- F. Epifano, C. Pelucchini, O. Rosati, S. Genovese and M. Curini, Ytterbium triflate promoted one-pot three component synthesis of 3,4,5-trisubstituted-3,6-dihydro-2H-1,3-oxazines, Catal. Lett., 2011, 141, 844–849 Search PubMed.
- M. Zhang, H. Jiang and A. Wang, Highly Efficient three-component synthesis of polysubstituted 2,5-dihydro-1,3-oxazin-6-ones under mild conditions, Synlett, 2007, 3214–3218 Search PubMed.
- Z. Chen, Y. Wen, G. Luo, M. Ye and Q. Wang, Transition-metal-free C–C bond cleavage and formation: Efficient synthesis of 2,3-diarylimidazo[1,2-α]pyridines from 2-aminopyridines and alkynoates, RSC Adv., 2016, 6, 86464–86467 Search PubMed.
- Q.-F. Zhou, X.-P. Chu, F.-F. Ge, Y. Wang and T. Lu, Phosphine–catalyzed [3+2] annulation of electron–deficient alkynes with N–hydroxyphthalimide: Synthesis of 3a–hydroxyisoxazolo[3,2−a]isoindol–8(3aH)–ones, Adv. Synth. Catal., 2013, 355, 2787–2792 Search PubMed.
- I. Yavari and M. R. Islami, Vinyltriphenylphosphonium salt mediated serendipitous synthesis of a functionalized pyrroloisoindole derivative. New synthesis of trimethyl 5-arylpyrrole-2,3,4-tricarboxylates, J. Chem. Res., 1998, 166–167 Search PubMed.
- J. Inanaga, Y. Baba and T. Hanamoto, Organic synthesis with trialkylphosphine catalysts. Conjugate addition of alcohols to α,β-unsaturated alkynic acid esters, Chem. Lett., 1993, 22, 241–244 Search PubMed.
- I. C. Stewart, R. G. Bergman and F. D. Toste, Phosphine-catalyzed hydration and hydroalkoxylation of activated olefins: Use of a strong nucleophile to generate a strong base, J. Am. Chem. Soc., 2003, 125, 8696–8697 Search PubMed.
- F. Zhong, X. Dou, X. Han, W. Yao, Q. Zhu, Y. Meng and Y. Lu, Chiral phosphine catalyzed asymmetric Michael addition of oxindoles, Angew. Chem., Int. Ed., 2013, 52, 943–947 Search PubMed.
- Q.-F. Zhou, F.-F. Ge, Q.-Q. Chen and T. Lu, Construction of pyrazolo[5,1-a]isoindol-8(3aH)-one derivatives via phosphine-catalyzed cyclization of electron-deficient alkynes and N-amino substituted phthalimide, RSC Adv., 2016, 6, 1395–1402 Search PubMed.
- H. Ding, Y. Zhang, M. Bian, W. Yao and C. Ma, Concise assembly of highly substituted furan-fused 1,4-thiazepines and their Diels-Alder reactions with benzynes, J. Org. Chem., 2008, 73, 578–584 Search PubMed.
- Y. Cheng and O. Meth-Cohn, Heterocycles derived from heteroatom-substituted carbenes, Chem. Rev., 2004, 104, 2507–2530 Search PubMed.
- V. Nair, S. Bindu and V. Sreekumar,
N-heterocyclic carbenes: Reagents, not just ligands!, Angew. Chem., Int. Ed., 2004, 43, 5130–5135 Search PubMed.
- V. Nair, C. Rajesh, A. U. Vinod, S. Bindu, A. R. Sreekanth, J. S. Mathen and L. Balagopal, Strategies for heterocyclic construction via novel multicomponent reactions based on isocyanides and nucleophilic carbenes, Acc. Chem. Res., 2003, 36, 899–907 Search PubMed.
- N. Marion, S. Díez-González and S. P. Nolan,
N-heterocyclic carbenes as organocatalysts, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 Search PubMed.
- R. Breslow, On the mechanism of thiamine Action. IV.1 Evidence from studies on model systems, J. Am. Chem. Soc., 1958, 80, 3719–3726 Search PubMed.
- R. Breslow and C. Schmuck, The mechanism of thiazolium catalysis, Tetrahedron Lett., 1996, 37, 8241–8242 Search PubMed.
- C. Ma and Y. Yang, Thiazolium-mediated multicomponent reactions: A facile synthesis of 3-aminofuran derivatives, Org. Lett., 2005, 7, 1343–1345 Search PubMed.
- C. Ma, H. Ding, Y. Zhang and M. Bian, A ring–expansion methodology involving multicomponent reactions: Highly efficient access to polysubstituted furan–fused 1,4−thiazepine derivatives, Angew. Chem., Int. Ed., 2006, 45, 7793–7797 Search PubMed.
- E. J. Corey, Catalytic enantioselective Diels–Alder reactions: Methods, mechanistic fundamentals, pathways, and applications, Angew. Chem., Int. Ed., 2002, 41, 1650–1667 Search PubMed.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogian-nakis, The Diels–Alder reaction in total synthesis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 Search PubMed.
- H. Pellissier and M. Santelli, The use of arynes in organic synthesis, Tetrahedron, 2003, 59, 701–730 Search PubMed.
- J. Liu, H. Liu, R. Na, G. Wang, Z. Li, H. Yu, M. Wang, J. Zhong and H. Guo, Phosphine-catalyzed [3+2] and [3+3] annulations of azomethine imines with ethyl 2-butynoate, Chem. Lett., 2012, 41, 218–220 Search PubMed.
-
T. Eicher and S. Hauptmann, The chemistry of heterocycles: Structures, reactions, synthesis, and applications, Wiley-VCH, Weinheim, 2nd edn, 2003 Search PubMed.
-
J. Elguero, in Five-membered rings with two heteroatoms and fused carbocyclic derivatives in comprehensive heterocyclic chemistry II, ed. A. R. Katritzky and C. W. Rees and E. F. V. Scriven, Elsevier, Oxford, 1996 Search PubMed.
-
J. Marchand-Brynaert and L. Ghosez, in Recent progress in the chemical synthesis of antibiotics, ed. G. Lukacs and M. Ohno, Springer, Berlin, 1990 Search PubMed.
- S. Hanessian, G. McNaughton-Smith, H.-G. Lombart and W. D. Lubell, Design and synthesis of conformationally constrained amino acids as versatile scaffolds and peptide mimetics, Tetrahedron, 1997, 53, 12789–12854 Search PubMed.
- M. I. Konaklieva and B. J. Plotkin, Bioisosters of β-lactams as anti-infectives, Curr. Med. Chem.: Anti-Infect. Agents, 2003, 2, 287–302 Search PubMed.
- Y. Han, Y.-J. Sheng and C.-G. Yan, Convenient synthesis of triphenylphosphanylidene spiro[cyclopentane-1,3′-indolines] and spiro[cyclopent[2]ene-1,3′-indolines] via three-component reactions, Org. Lett., 2014, 16, 2654–2657 Search PubMed.
- N. Kielland and R. Lavilla, Recent developments in Reissert-type multicomponent reactions, Top. Heterocycl. Chem., 2010, 25, 127–168 Search PubMed.
- G. Zhu, Z. Chen, Q. Jiang, D. Xiao, P. Cao and X. Zhang, Asymmetric [3+2] cycloaddition of 2,3-butadienoates with electron-deficient olefins catalyzed by novel chiral 2, 5-dialkyl-7-phenyl-7-phosphabicyclo [2.2.1] heptanes, J. Am. Chem. Soc., 1997, 119, 3836–3837 Search PubMed.
- D. B. Ramachary, C. Venkaah and P. M. Krishan, Stereoselective synthesis of five-membered spirooxindoles through Tomita zipper cyclization, Org. Lett., 2013, 15, 4714–4717 Search PubMed.
- J. C. Deng and S. C. Chuang, Three-component and nonclassical reaction of phosphines with enynes and aldehydes: Formation of γ-lactones featuring α-phosphorus ylides, Org. Lett., 2011, 13, 2248–2251 Search PubMed.
- S. C. Chuang, J. C. Deng, F. W. Chan, S. Y. Chen, W. J. Huang, L. H. Lai and V. Rajeshkumar, [3+2] Cycloaddition of dialkyl (E)–hex–2−en–4−ynedioates to [60]fullerene by phosphane–promoted tandem α(δ′)–Michael additions, Eur. J. Org. Chem., 2012, 2606–2613 Search PubMed.
- J. C. Deng, F. W. Chan, C. W. Kuo, C. A. Cheng, C. Y. Huang and S. C. Chuang, Assembly of dimethyl acetylenedicarboxylate and phosphanes with aldehydes leading to γ–lactones bearing α–phosphorus ylides as Wittig reagents, Eur. J. Org. Chem., 2012, 5738–5747 Search PubMed.
- Y. W. Lin, J. C. Deng, Y. Z. Hsieh and S. C. Chuang, One-pot formation of fluorescent γ-lactams having an α-phosphorus ylide moiety through three-component α(δ′)-Michael reactions of phosphines with an enyne and N-tosyl aldimines, Org. Biomol. Chem., 2014, 12, 162–170 Search PubMed.
- J. Zhang, M. Zhang, Y. Li, S. Liu and Z. Miao, Diastereoselective synthesis of cyclopentene spiro-rhodanines containing three contiguous stereocenters via phosphine-catalyzed [3+2] cycloaddition or one-pot sequential [3+2]/[3+2] cycloaddition, RSC Adv., 2016, 6, 107984–107993 Search PubMed.
- J. Zhang, C. Cheng, D. Wang and Z. Miao, Regio- and diastereoselective construction of spirocyclopenteneoxindoles through phosphine-catalyzed [3+2] annulation of methyleneindolinone with alkynoate derivatives, J. Org. Chem., 2017, 82, 10121–10128 Search PubMed.
- J. Li, Y. Liu, C. Li, H. Jie and X. Jia, Atom-economical synthesis of the functionalized spirocyclic oxindole-butenolide via three-component [2+2+1] cycloaddition strategy, Green Chem., 2012, 14, 1314–1321 Search PubMed.
- G. Henrion, T. E. J. Chavas, X. Le Goff and F. Gagosz, Biarylphosphonite gold(I) complexes as superior catalysts for oxidative cyclization of propynyl arenes into indan–2−ones, Angew. Chem., Int. Ed., 2013, 52, 6277–6282 Search PubMed.
- S. Shah and B. Singh, Urea/thiourea catalyzed, solvent-free synthesis of 5-arylidenethiazolidine-2,4-diones and 5-arylidene-2-thioxothiazolidin-4-ones, Bioorg. Med. Chem. Lett., 2012, 22, 5388–5391 Search PubMed.
- A. Urbanaitė, M. Jonušis, R. Bukšnaitienė, S. Balkaitis and I. Čikotienė, Electrophile–mediated reactions of functionalized propargylic substrates, Eur. J. Org. Chem., 2015, 7091–7113 Search PubMed.
- H. Gao and J. Zhang, Cationic rhodium(I)-catalyzed regioselective tandem heterocyclization/[3+2] cycloaddition of 2-(1-alkynyl)-2-alken-1-ones with alkynes, Chem. – Eur. J., 2012, 18, 2777–2782 Search PubMed.
- L.-J. Yang, W. Wang, J. Nie, S. Li and J.-A. Ma, Bisphosphine-triggered one-pot sequential [3+2]/[3+2] annulation of allenoates with cyclic ketimines, Org. Lett., 2013, 15, 5214–5217 Search PubMed.
- S. Gabillet, D. Lecercle, O. Loreau, M. Carboni, S. Dezard, J. M. Gomis and F. Taran, Phosphine-catalyzed construction of sulfur heterocycles, Org. Lett., 2007, 9, 3925–3927 Search PubMed.
- S. Gabillet, O. Loreau, S. Specklin, E. Rasalofonjatovo and F. Taran, A phosphine-catalyzed preparation of 4-arylidene-5-imidazolones, J. Org. Chem., 2014, 79, 9894–9898 Search PubMed.
- C. T. Mbofana and S. J. Miller, Phosphine-catalyzed annulation reactions of 2-butynoate and α-keto esters: Synthesis of cyclopentene derivatives, ACS Catal., 2014, 4, 3671–3674 Search PubMed.
- W. Meng, H.-T. Zhao, J. Nie, Y. Zheng, A. Fu and J.-A. Ma, Allenoate-derived 1,5-, 1,7-, and 1,9-zwitterions as highly versatile coupling-partners for phosphine-triggered cycloaddition reactions, Chem. Sci., 2012, 3, 3053–3057 Search PubMed.
- A. N. Pillai, C. H. Suresh and V. Nair, Pyridine-catalyzed stereoselective addition of acyclic 1,2-diones to acetylenic esters: Synthetic and theoretical studies of an unprecedented rearrangement, Chem. – Eur. J., 2008, 14, 5851–5860 Search PubMed.
- H. Kwart and M. M. Baevsky, The cyanide ion catalyzed cleavage of aromatic α-diketones, J. Am. Chem. Soc., 1958, 80, 580–588 Search PubMed.
- J. C. Trisler and J. L. Frye, Reaction of benzil with cyanide ion in dimethyl sulfoxide, J. Org. Chem., 1965, 30, 306–307 Search PubMed.
- B. Das, D. B. Shinde, B. S. Kanth and G. Satyalakshmi, An efficient multicomponent synthesis of polysubstituted pyrrolidines and tetrahydropyrimidines starting directly from nitro compounds in water, Synthesis, 2010, 2823–2827 Search PubMed.
- J. C. Lee, K. I. I. Choi, H. Y. Koh, Y. Kang, Y. Kuin, Y. Kang and Y. S. Cho, Indium mediated reduction of nitro and azide groups in the presence of HCl in aqueous media, Synthesis, 2001, 81–84 Search PubMed.
- K. Kashinath, P. D. Jadhav and D. S. Reddy, Total synthesis of an anticancer norsesquiterpene alkaloid isolated from the fungus Flammulina velutipes, Org. Biomol. Chem., 2014, 12, 4098–4103 Search PubMed.
- D. I. Saavedra, B. D. Rencher, D.-H. Kwon, S. J. Smith, D. H. Ess and M. B. Andrus, Synthesis and computational studies demonstrate the utility of an intramolecular styryl Diels-Alder reaction and di–t–butylhydroxytoluene assisted [1,3]-shift to construct anticancer dl-deoxypodophyllotoxin, J. Org. Chem., 2018, 83, 2018–2026 Search PubMed.
- L. Atkin, Z. Chen, A. Robertson, D. Sturgess, J. M. White and M. A. Rizzacasa, Synthesis of alkyl citrates (-)-CJ-13,981, (-)-CJ-13,982, and (-)-l-731,120 via a cyclobutene diester, Org. Lett., 2018, 20, 4255–4258 Search PubMed.
- S. Hanessian and P. J. Murray, Stereochemical control of nature's biosynthetic pathways: A general strategy for the synthesis of polypropionate-derived structural units from a single chiral progenitor, Tetrahedron, 1987, 43, 5055–5072 Search PubMed.
- A. A. H. Elmehriki and J. L. Gleason, A spiroalkylation method for the stereoselective construction
of α–quaternary carbons and its application to the total synthesis of (R)–puraquinonic acid, Org. Lett., 2019, 21, 9729–9733 Search PubMed.
- E. A. Tiong, D. Rivalti, B. M. Williams and J. L. Gleason, A concise total synthesis of (R)-puraquinonic acid, Angew. Chem., Int. Ed., 2013, 52, 3442–3445 Search PubMed.
- J. Kurian and M. K. Manheri, Synthesis of new cyclazines and 4,5-diaryl-1H-pyrrol-3(2H)-one units in discoipyrroles from indolizinone-DMAD cycloadducts, Org. Biomol. Chem., 2019, 17, 8832–8848 Search PubMed.
- H. Cho and I. Kim, Palladium-catalyzed construction of polysubstituted indolizinones, Tetrahedron, 2012, 68, 5464–5480 Search PubMed.
- S.-C. Kim, K.-M. Choi and C.-S. Cheong, Synthesis of amlodipine using aza-Diels-Alder reaction, Bull. Korean Chem. Soc., 2002, 23, 143–144 Search PubMed.
- I. Sircar, K. R. Anderson and L. Bonadies, An improved and efficient synthesis of 2-substituted 1,4-dihydropyridine derivatives via regiospecific bromination, Tetrahedron Lett., 1988, 29, 6835–6838 Search PubMed.
- V. Summa, A. Petrocchi, F. Bonelli, B. Crescenzi, M. Donghi, M. Ferrara, F. Fiore, C. Gardelli, O. G. Paz, D. J. Hazuda, P. Jones, O. Kinzel, R. Laufer, E. Monteagudo, E. Muraglia, E. Nizi, F. Orvieto, P. Pace, G. Pescatore, R. Scarpelli, M. V. Witmer and M. Rowley, Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection, J. Med. Chem., 2008, 51, 5843–5855 Search PubMed.
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Sumera
Zaib
*ab,
Sumera
Zaib