Fluorescent small-molecule agonists as follicle-stimulating hormone receptor imaging tools†
Received
14th May 2020
, Accepted 20th July 2020
First published on 3rd August 2020
Abstract
Fluorescent cell surface receptor agonists allow visualization of processes that are set in motion by receptor activation. This study describes the synthesis of two fluorescent, low molecular weight ligands for the follicle-stimulating hormone receptor (FSHR), based on a dihydropyridine (DHP) agonist. We show that both BODIPY- and Cy5-conjugated DHP (m-DHP-BDP and m-DHP-Cy5) are potent FSHR agonists, able to activate receptor signalling with nanomolar potencies and to effect receptor internalisation at higher concentrations. FSHR-dependent uptake of m-DHP-Cy5 is in stark contrast to the cellular uptake of m-DHP-BDP which was efficiently internalised also in the absence of FSHR. Our results comprise a first-in-class fluorescent low molecular weight ligand for in situ FSHR imaging and pertain the potential means for targeted delivery of drugs into the endolysosomal pathway of FSHR-expressing cells.
Introduction
The glycoprotein hormone receptors belong to the rhodopsin-like family of G protein-coupled receptors (GPCRs) and are characterized by a large N-terminal hormone-binding ectodomain.1 They bind and are activated by one of the glycoprotein hormones: luteinizing hormone (LH), follicle-stimulating hormone (FSH), thyroid-stimulating hormone (TSH) or human chorionic gonadotropin (hCG). The glycoprotein hormones all share a common alpha subunit and have a unique beta-subunit that confers glycoprotein hormone receptor specificity.2 The luteinizing hormone/choriogonadotropin receptor (LH/CGR, recognizing both LH and hCG) and the follicle-stimulating hormone receptor (FSHR) are key regulators of human reproduction,3–5 whereas the thyroid-stimulating hormone receptor (TSHR) is important for thyroid function.6
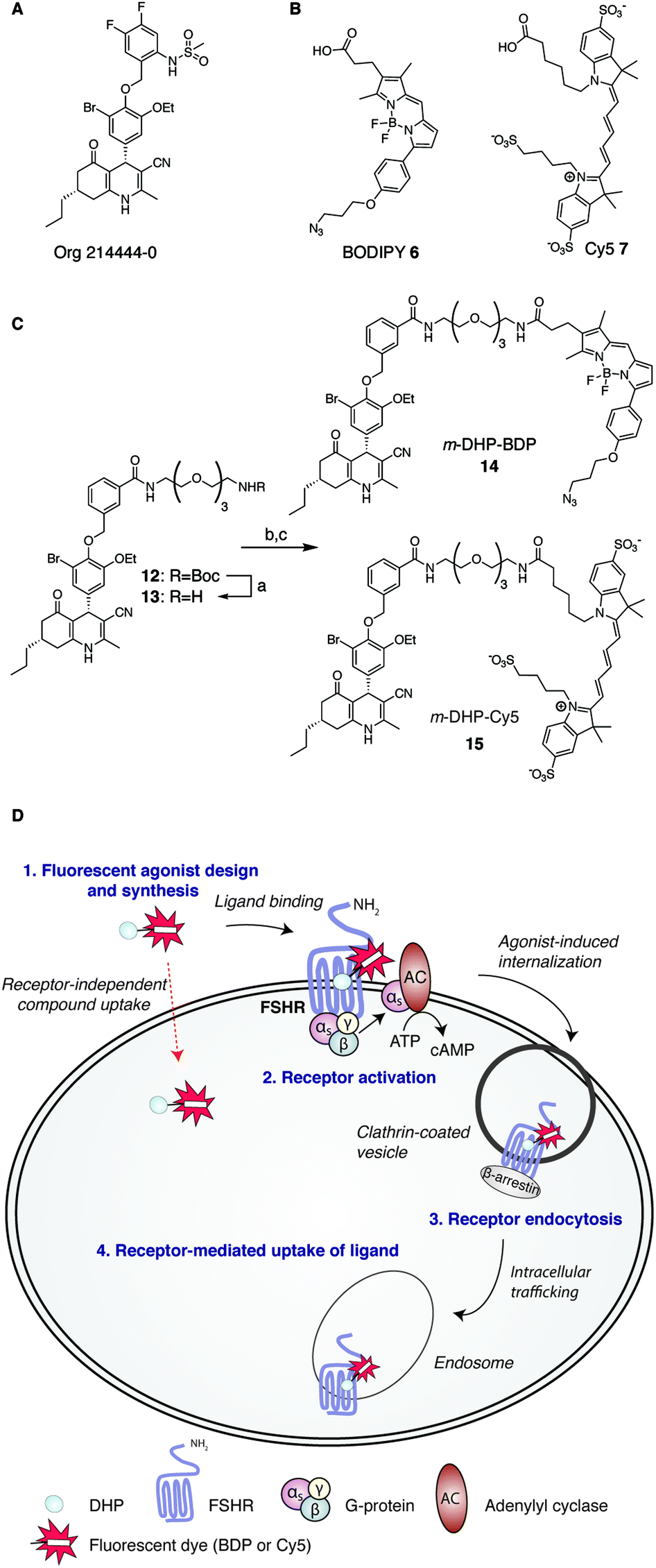
Consistent with its role in human reproduction, the FSHR is mainly present on granulosa cells in the ovaries and on Sertoli cells in the testes. Interestingly, newly formed blood vessels of a variety of tumours also express the FSHR. This finding suggests an additional role for this receptor in angiogenesis and indicates that the FSHR may be a relevant oncological drug target.7–10 Currently, the most effective clinical strategy to induce or enhance female fertility is to administer recombinant human FSH (rFSH). Though often effective, this therapy suffers from heterogeneity in the protein preparation in terms of glycosylation (predominantly in sialylation pattern), the instability of the protein at higher temperatures and, perhaps most important from a practical point of view, that rFSH cannot be administered orally. In addition, the long half-life of rFSH in circulation may lead to overstimulation (ovarian hyperstimulation syndrome). In recent years and with the aim to overcome these limitations, low molecular weight (LMW) FSHR ligands as modulators of human fertility were developed. LMW FSHR agonists include compounds from the thienopyrimidine class (also active on LHR),11 the thiazolidinone class,12–14 diketopiperazine class15,16 and the dihydropyridine class (DHPs).17 In a previous study by van Koppen et al., the dihydropyridine, Org214444-0 (Fig. 1A) proved to be a nanomolar potent FSHR agonist, with selectivity for the FSHR over both the LHR (>200-fold) and the TSHR (>1000-fold). In contrast to other LMW FSHR agonists, Org214444-0 is an allosteric FSHR agonist that does not compete for FSH binding. Org214444-0-mediated FSHR activation leads to increased cyclic AMP (cAMP) levels as well as estradiol production in human granulosa cells. In addition, stimulation of U2OS cells expressing an eGFP-tagged FSHR with this small molecule leads to the translocation of the FSHR from the plasma membrane to intracellular vesicles, albeit with an EC50 value ∼2 orders of magnitude higher than that for the stimulation of cAMP (as it is also the case with the endogenous receptor ligand, FSH). In vivo studies in rats showed that this compound has similar action as FSH in supporting the follicular phase.18
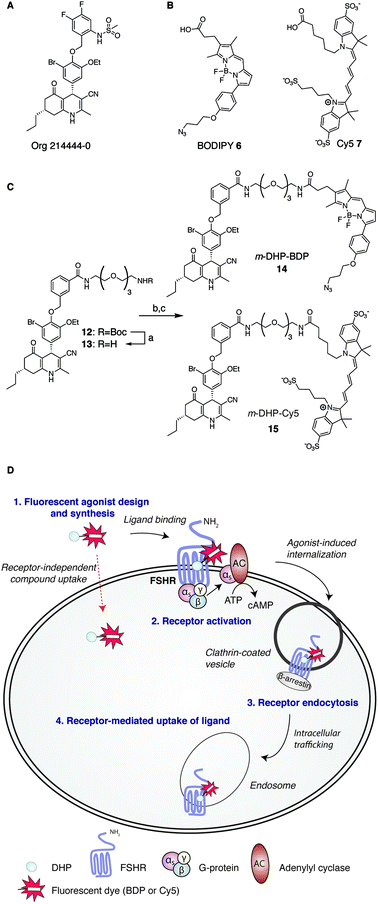 |
| | Fig. 1 Development of fluorescent DHP analogues for FSH receptor imaging. (A) Structure of previously published Org 214444-0.18 (B) Structures of BODIPY (6) and Cy5 (7) dyes. (C) Synthesis of fluorescent FSHR agonists. Reagents and conditions: [a] 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 TFA/DCM; [b] azido-BODIPY acid 6, EDC·HCl, HOBt, TEA, DCM, 48%; [c] Cy5-acid 7, PyBOP, DiPEA, DMF, 55%. (D) Schematic representation of receptor-mediated, agonist-induced uptake of the receptor-compound complex. :1 TFA/DCM; [b] azido-BODIPY acid 6, EDC·HCl, HOBt, TEA, DCM, 48%; [c] Cy5-acid 7, PyBOP, DiPEA, DMF, 55%. (D) Schematic representation of receptor-mediated, agonist-induced uptake of the receptor-compound complex. | |
Fluorescent ligands are attractive tools to study cell surface receptor-related processes. Fluorescent GPCR probes are often based on peptide ligands, both for practical (synthesis) reasons and because the attachment of a fluorophore to a peptide is a relatively minor increase in bulk and molecular weight compared to appending a dye to a LMW ligand.19,20 Indeed, both the choice of linker to separate the dye and the ligand and the nature of the fluorophore can have a profound influence on the pharmacological properties of a probe.21,22 Considering the key role of the FSHR in human reproduction and the increasing body of literature supporting its importance in cancer, fluorescent FSHR ligands could both be instrumental to study fundamental FSHR-related processes as well as function as diagnostic tumour markers. To date, only a few examples of fluorescent ligands for the FSHR have appeared in the literature, and all are based on peptide ligands that are poorly characterized in terms of potency and uptake selectivity.23,24 The aim of the present study was to develop and evaluate fluorescent FSHR ligands that are based on a small-molecule ligand rather than a FSH-derived peptide ligand. For this, we synthesised different fluorescent analogues of the very potent DHP pharmacophore. Since the physicochemical properties of the fluorophore can have pronounced effects on the potency, binding, and uptake of the final construct, we grafted fluorophores from different dye classes (BODIPY 6 and cyanine 7) onto the DHP agonist (Fig. 1B and C). As schematically depicted in Fig. 1D, treatment of FSHR-expressing cells with (potent) agonists, leads to agonists-induced receptor internalisation. An ideal ligand would then be internalised together with the receptor so that intracellular events can also be studied. To function as a diagnostic tool, compound uptake should furthermore be dependent on receptor expression levels. Agonistic potency of our fluorescent probes was thoroughly investigated by essaying their ability to activate the receptor in a cAMP luciferase reporter assay. We then investigated whether these ligands can be used to stain receptor-expressing cells using flow cytometry. We finally used confocal fluorescence microscopy to assess the ability of the compounds to induce receptor-mediated internalisation as well as the receptor-dependency of compound uptake. We show that LMW FSHR agonists form an attractive starting point for the development of FSHR imaging tools. We identify m-DHP-Cy5 15 (Fig. 1C) as a nanomolar potent FSHR agonist suitable for in situ FSHR imaging. Flow cytometry and confocal microscopy analysis revealed that m-DHP-Cy5 15 is capable of inducing FSHR endocytosis and is selectively taken up in eGFP-FSHR expressing cells via FSHR-mediated endocytosis. We thus demonstrate that DHP-based FSHR agonists are a useful addition to the existing FSHR imaging tools to study receptor expression and moreover that the DHP core structure may be recruited for the development of targeted drug strategies aimed to deliver cytotoxic agents to FSHR-expressing cells, including those involved in angiogenesis.
Experimental
Cell culture conditions
All cells were grown in a humidified atmosphere in 5% CO2 at 37 °C. CHO cells stably expressing the human FSH receptor together with a CRE-inducible luciferase reporter gene (CHOhFSHR_luc cells) and control cells without receptor but with luciferase gene (CHO_luc cells) were provided by MSD (Organon), Oss, The Netherlands. Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) and Ham's F12 medium (1:1) with penicillin/streptomycin sulphate (100 U mL−1/0.1 mg mL−1) and 5% fetal bovine serum (FBS). U2OS cells stably expressing the rat FSHR fused to the N-terminus of eGFP (U2OS-FSHR cells) were obtained from MSD (Organon), Oss, The Netherlands (FSHR redistribution assay, BioImage). Cells were grown in DMEM (high glucose (4.5 g L−1) and with stable glutamine, Invitrogen), penicillin/streptomycin sulphate (100 U mL−1/0.1 mg mL−1), G418 (0.5 mg mL−1) and FBS (10%). Wild-type U2OS cells were grown under the same conditions, without G418.
Measurement of CRE-induced luciferase expression
CHO_luc or CHO-hFSHR_luc cells were cultured to ∼80% confluency before use in the assay. On the day of the experiment, cells were harvested using enzyme-free dissociation solution (Millipore) and resuspended in assay medium (DMEM–F12 (1:1), without phenol-red, with penicillin/streptomycin (100 U mL−1/0.1 mg mL−1) supplied with 1 μg mL−1 bovine insulin (Tebu-Bio) and 5 μg mL−1 human apo-transferrin (Sigma)) to a concentration of 7.5 × 105 cells per mL. Experiments were conducted in 96-wells white Optiplates (PerkinElmer) and each well contained 30 μL of test compound, rFSH (200 pM final concentration, positive control) or assay medium (negative control), 30 μL of assay medium and 30 μL cell suspension. Final concentration of DMSO was 1% for all compounds, including controls. After 4 h of stimulation, 50 μL Neolite (PerkinElmer) was added to each well and luminescence signal was detected on a Microbeta Trilux 1450 Luminescence Counter (PerkinElmer). To control for non-FSHR mediated effects on intracellular cAMP levels, highest compound concentrations used in the dose–response experiments (typically 1 μM) were tested on CHO_luc cells. Data was analysed using GraphPad Prism 5 (GraphPad Software, La Jolla, USA), and values were normalized to the maximal effect obtained for rFSH (200 pM, Org32489, gift from MSD, Oss, The Netherlands). Each experiment was performed on duplicate plates and mean ± SEM values of at least three independent experiments are given.
Flow cytometry
U2OS or U2OS-FSHR cells were cultured as described under “Cell culture”. Cells were seeded onto 6-well plates (4 × 105 cells per well) and cultured for 48 h, before start of the experiment. Compounds (as 2× stock solution in culture medium) 14 or 15 were added to the cells (1 μM final concentration) and the cells were incubated for 2 h (37 °C, 5% CO2). Cells were washed twice with ice-cold phosphate-buffered saline and harvested using enzyme-free dissociation solution. After centrifugation (5 min, 1200 rpm), the cell pellet was resuspended in PBS to obtain a single-cell suspension of ∼1 × 106 cells per mL. Cells were transferred to a 96-wells plate and samples measured using a BD FACSCanto II flow cytometer. The following laser/filter settings were used: GFP (FSHR): λex: 488 nm, λem: 530/30 nm; BODIPY (14): λex: 488 nm, λem: 585/42 nm; Cy5 (15): λex: 633 nm, λem: >670 nm and values were compensated using the instrumentation software and mono-stained controls. Data was analysed using FCS Express 4 Flow Research Edition (De Novo Software, LA, USA). All samples were measured twice and values were calculated from three independent experiments.
Uptake and FSHR internalisation microscopy experiments
U2OS-FSHR or U2OS cells were maintained as described under “Cell culture”. Forty-eight hours before the experiment, cells were harvested (Trypsin–EDTA), counted and seeded onto sterile Labtek II 4- or 8-chamber borosilicate coverglass systems (Fisher Emergo) at a density of 25–50 × 104 cells per well. On the day of the experiment, cells were incubated with the indicated concentration of compound (10 or 1 μM) or rFSH (12 nM) for 2 h at 37 or 4 °C, before being washed with PBS (2×), fixed (4% formaldehyde in PBS), washed again (PBS) and nuclei stained with Draq5 (Thermo Scientific) (optional, only when the compound fluorescence is detected in the dsRed channel). Cells were imaged on a Leica TCS SPE confocal microscope, using GFP (FSHR), dsRed (BODIPY) or Cy5 (Cy5, Draq5) filter settings (λex 488, 532 or 635 nm) with optimised detection range to exclude bleed-through of dye signal in different channels. For competition experiments, cells were pre-incubated for 1 h with THQ 16 (10 μM) or chlorpromazine (10 or 20 μM), before addition of m-DHP-Cy5 15 (1 μM) to the medium. Images were taken under the same settings (laser power, gain, offset, pinhole) and were analysed using ImageJ. Global fluorescence was determined either for the whole picture or for a specific region of interest (ROI), which only included regions with cells present, and corrected for the selected area. Intensities were normalized to an average of ∼1 for untreated or rFSH-treated U2OS-FSHR cells (GFP signal), m-DHP-BODIPY 14-treated U2OS-FSHR cells (BODIPY signal), or m-DHP-Cy5 15-treated U2OS-FSHR cells (Cy5 signal), since these can be regarded as positive controls. The percentage of GFP fluorescence signal on the membranes compared to that in the cytoplasm was determined by selection of membranes (typically 5–7 cells per image that were inside the focal plane) with the brush tool (ROI:’membranes’), followed by the creation of a ROI that included the selected cells (ROI:’cells’). Two greyscale images were then thresholded according to Yen,25 followed by selection of the’membranes’ ROI in each picture which was then either cleared (giving’cytoplasm’) or cleared outside (giving’membranes’). Fluorescence intensities of cells/membranes within the ROI were measured and calculated as a fraction of total GFP signal (membrane + cytoplasm = 100%). For representative images showing the selected ROIs, see Fig. 4B. Colocalization was calculated on background-corrected images using the intensity correlation analysis and colocalization threshold plugins.
Synthesis
Full synthetic procedures of all new compounds are available online in the ESI.†
Results and discussion
Synthesis of fluorescent FSHR ligands
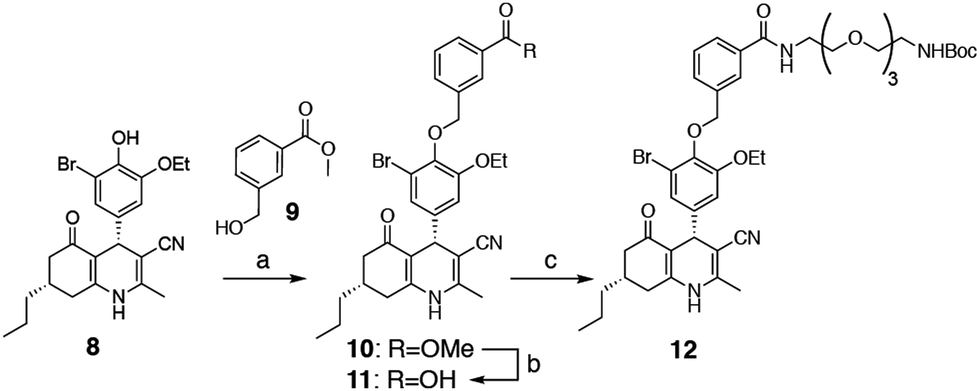
We selected two fluorescent dyes based on their different physicochemical properties: an uncharged, lipophilic BODIPY dye and a charged, hydrophilic Cy5 dye for modification of the DHP core (Fig. 1B). The synthesis of bifunctional BODIPY dye 6 was previously reported by us and optimised.26 (Scheme S1; see for synthetic details the ESI†). Cy5 derivative 7 equipped with a carboxylate for conjugation was prepared according to the literature procedure.27 Amino-m-DHP 13 was synthesised from core DHP 8 (Scheme 1 and Fig. 1C).17 DHP 8 was coupled with meta-hydroxymethyl benzoate ester 9 under Mitsunobu conditions, and ensuing saponification yielded carboxylic acid 11. The free acid 11 was condensed with mono-Boc protected diaminotetra-ethyleneglycol using EEDQ as the activating agent. After Boc removal, amino-m-DHP 13 was condensed with BODIPY dye 6 or Cy5 dye 7, resulting in “m-DHP-BDP” 14 and “m-DHP-Cy5” 15, respectively (Fig. 1C).
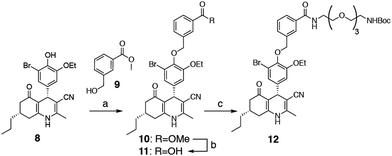 |
| | Scheme 1 Synthesis of meta-DHP 12. Reagents and conditions: [a] hydroxymethylbenzoate 9, PPh3, DIAD, THF; [b] MeOH, NaOH, 40 °C, 65% (2 steps); [c] 1-Boc-1,11-diamino-tetraethylene glycol spacer, EEDQ, DCM, 90%. | |
Fluorescent m-DHP analogues are potent FSHR agonists
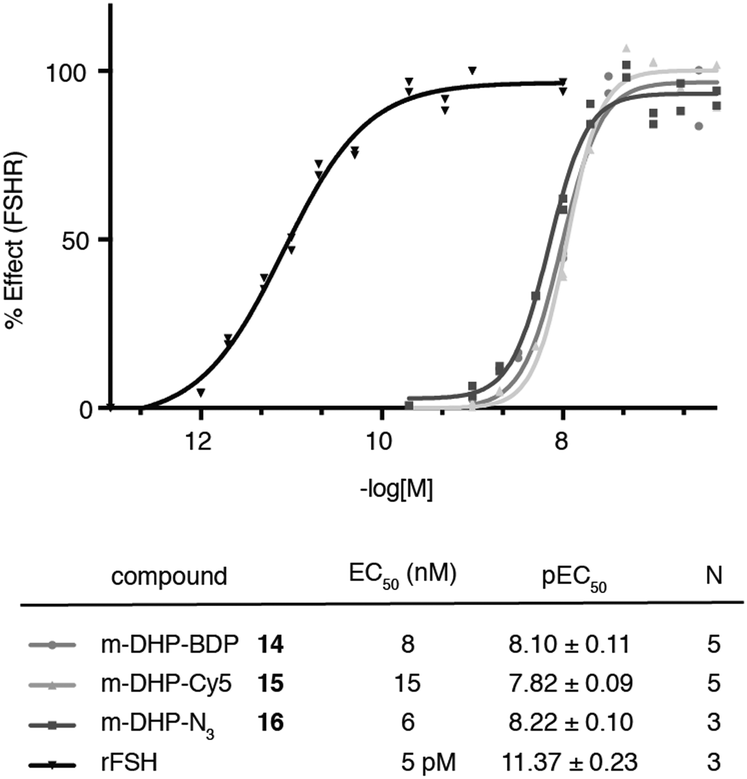
We evaluated the fluorescent ligands m-DHP-BDP 14 and m-DHP-Cy5 15 for their agonistic activity towards the FSHR in a CRE-luciferase assay. As a control, we included m-DHP-N316 (Scheme S2; see for synthetic details the ESI†), that lacks the bulk of the fluorophore, and the endogenous ligand rFSH. CHO cells stably expressing human FSHR and a CRE-luciferase reporter gene were cultured for 4 h in the presence of increasing probe concentrations. Cells were lysed and cAMP-driven luciferase expression detected by the addition of luciferase substrate. Emerging luminescence signals were normalized against the maximum response as obtained after treatment with rFSH (200 pM). CHO cells not expressing human FSHR but with the CRE-inducible luciferase (reporter) cDNA were used to control for non-receptor-mediated increases in cAMP levels. No response above background was detected in the control CHO cells for all compounds tested at the highest concentration. Representative dose–response curves are shown in Fig. 2. All compounds proved to be full FSHR agonists with low nanomolar potencies (m-DHP-BDP 14:EC50 8 nM, m-DHP-Cy5 15:EC50 15 nM; m-DHP-N316:EC50 6 nM). Based on this, we conclude that the nature and the bulk of the attached dye has little influence on FSHR agonistic activity of the probes.
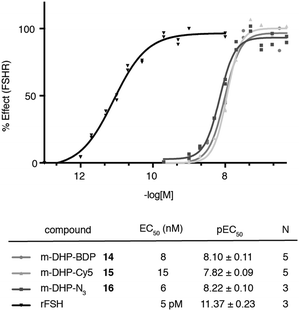 |
| | Fig. 2 Agonistic luciferase dose–response curves of fluorescent ligands m-DHP-BDP 14 and m-DHP-Cy5 15, control compound m-DHP-N316, and rFSH on CHO cells stably expressing the human FSHR. The response was normalized against the maximal effect obtained with rFSH (200 pM). The mean agonistic potency (EC50) values are calculated from the pEC50 (mean ± SEM) values from N independent experiments performed in duplicate. Representative curves are shown. | |
Binding and uptake of m-DHP-Cy5 is restricted to FSHR-expressing cells
With potent agonistic fluorescent ligands in hand, we investigated whether these compounds could be used to stain FSHR-expressing cells in a receptor-dependent manner.
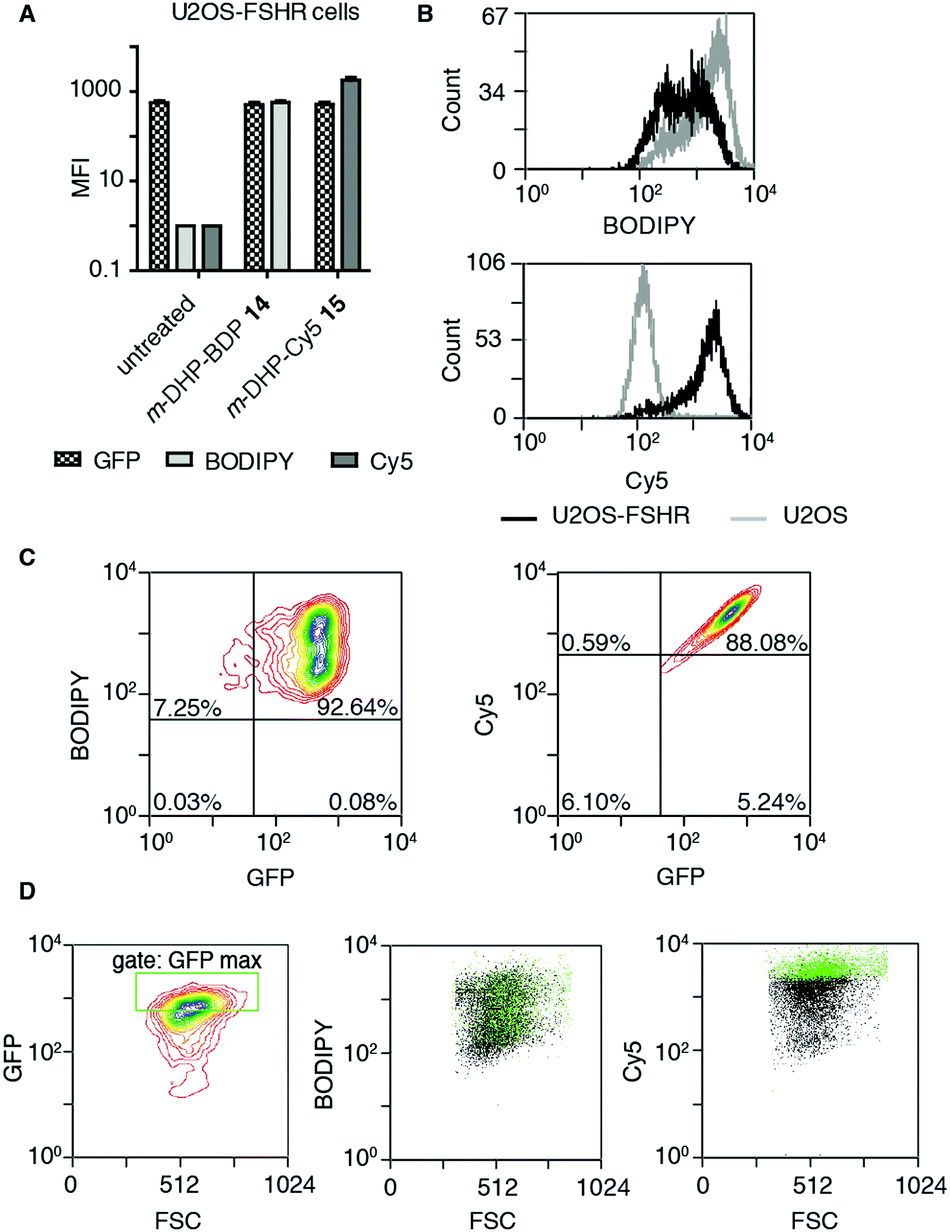
The binding/uptake of the fluorescent agonists in U2OS cells stably expressing rat FSHR fused to the N-terminus of enhanced green fluorescent protein (eGFP) (U2OS-FSHR cells) was determined using flow cytometry. As a biological control, wild-type U2OS cells lacking the FSHR were employed.28 We used agonists m-DHP-BDP 14 and m-DHP-Cy5 15 in a flow cytometry experiment to quantitatively correlate receptor expression levels (GFP signal) with compound binding/uptake (Fig. 3 and Table S1, ESI†). For this, we incubated U2OS-FSHR or U2OS cells for 2 h with 1 μM m-DHP-BDP 14 or m-DHP-Cy5 15. Cells were thoroughly washed and a single-cell suspension in PBS was subjected to flow cytometry analysis. The median fluorescence intensities (MFI) of untreated versus compound-treated U2OS-FSHR cells are shown in Fig. 3A. No influence of compound treatment on total FSHR expression levels was observed as the GFP fluorescence remained constant under all experimental conditions. BODIPY or Cy5 fluorescence increased ∼1000-fold upon treatment of U2OS-FSHR cells with compound m-DHP-BDP 14 or m-DHP-Cy5 15, respectively. Comparison of cells with and without receptor led to interesting observations. Overlaying the histograms of U2OS-FSHR vs. U2OS cells (Fig. 3B) revealed similar BODIPY fluorescence in both cell types after treatment with m-DHP-BDP 14, indicating that this compound was taken up or binding to the cells, irrespective of the presence of receptor. U2OS-FSHR cells treated with fluorescent agonist m-DHP-Cy5 15 on the other hand, were on average >10-fold more fluorescent than compound-treated control cells. Quadrant analysis of 2D ligand-fluorescence versus GFP-fluorescence intensity plots (Fig. 3C and Table S1, ESI†) corroborated this finding. Only for m-DHP-Cy5 15 did we observe a linear relationship between receptor expression and compound fluorescence (Fig. 3C). When the brightest population of cells in the GFP channel (i.e. cells that express the highest levels of FSHR) was selected and marked in the cell populations in the respective dye fluorescent channels, as shown in Fig. 3D, a clear correlation was again found between receptor expression levels and fluorescent agonist signal for m-DHP-Cy5 15, but not for m-DHP-BDP 14. This data thus strongly supported receptor-dependent cell staining for agonist m-DHP-Cy5 15, be it by binding of the ligand to cell-surface receptors or through stimulating receptor internalisation and concurrent ligand uptake. To distinguish between these possibilities, we turned to fluorescence microscopy and investigated the fluorescence localization of both the receptor and the ligand.
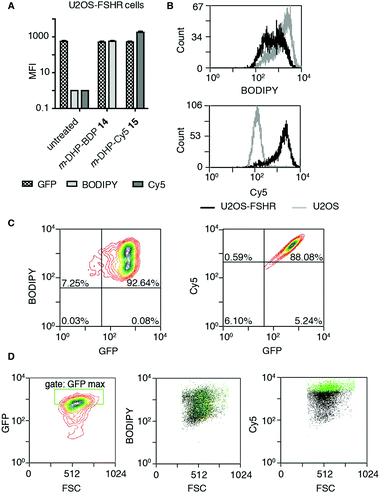 |
| | Fig. 3 Probe m-DHP-Cy5 15 selectively stains FSHR-expressing cells. U2OS-FSHR (GFP channel) and U2OS cells were treated with 1 μM m-DHP-BDP 14 (BODIPY channel) or m-DHP-Cy5 15 (Cy5 channel). After treatment (2 h, 37 °C, 5% CO2), cells were washed and analysed by flow cytometry. Representative plots are shown from three independent experiments measured in duplo. (A) Compound treatment stains U2OS-FSHR cells (increase in MFI between untreated and compound 14 or 15 treated cells), but does not alter FSHR expression levels (no change in MFI for GFP in untreated vs. treated) (B) m-DHP-BDP 14 stains both U2OS-FSHR (black histogram) and control U2OS (grey histogram) cells, while m-DHP-Cy5 15 selectively stains U2OS-FSHR cells. (C and D) Receptor expression levels correlate with m-DHP-Cy5 15 staining intensity. (C) Representative contour plots of dye fluorescence vs. GFP of treated U2OS-FSHR cells, including the quadrants that were used for the data shown in Table S1 (ESI†), show no correlation between GFP and BODIPY signals and a linear relationship between GFP and Cy5 signals. (D) The correlation between the intensity of GFP versus dye fluorescence was further visualized by selection of an additional gate “GFP max” that corresponds to cells with the highest levels of receptor expression. Cells within this gate are marked green in the population (black) of each dye in its corresponding channel. Only for m-DHP-Cy5 15 it was found that cells with maximum FSHR-eGFP levels correspond to those cells that have maximum dye fluorescence. | |
m-DHP-BDP 14 induces FSHR internalisation and is internalised in a non-receptor dependent fashion
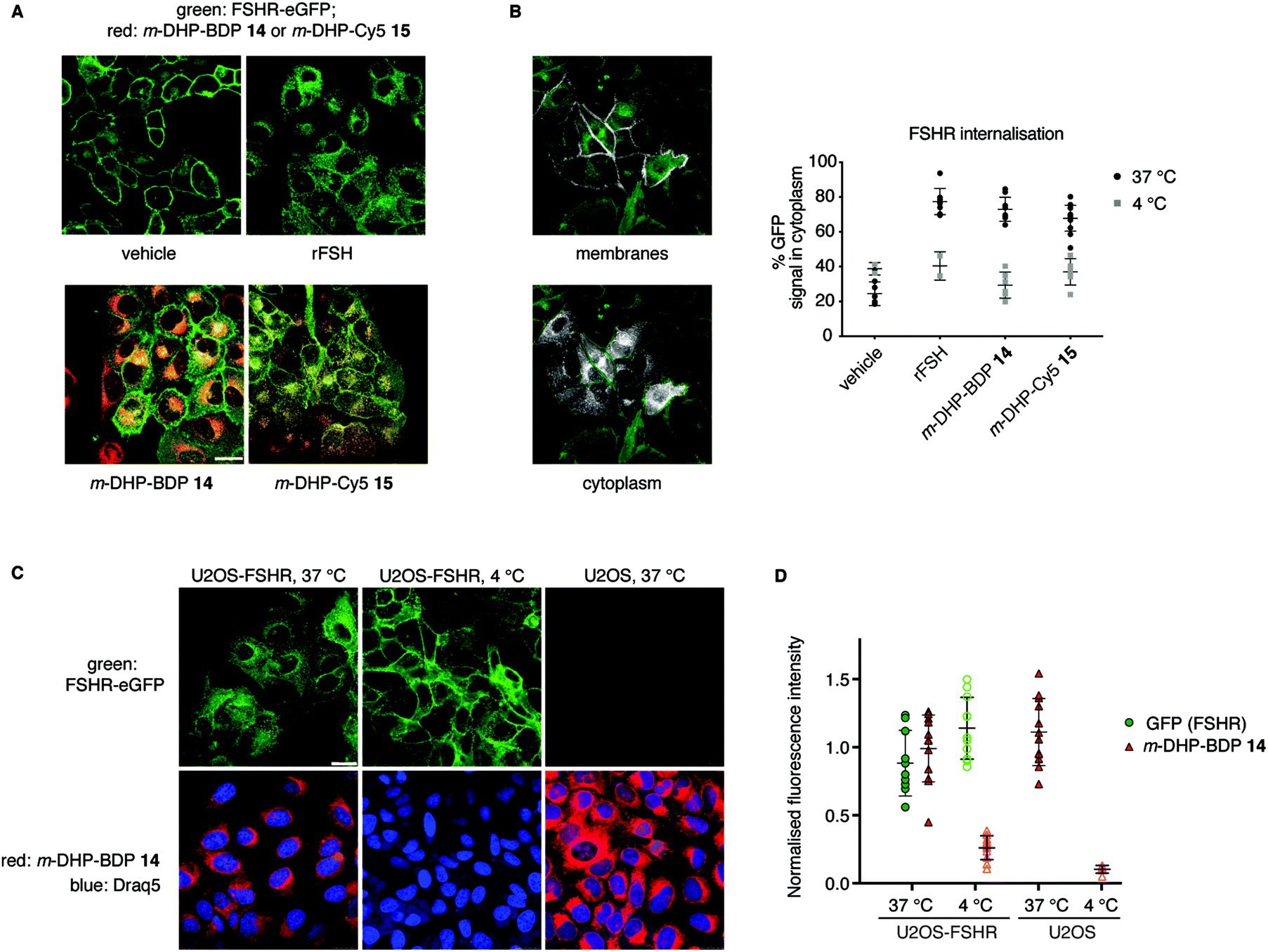
From our flow cytometry experiments it was clear that the fluorescent ligands stained FSHR-expressing cells and we next set out to establish, using confocal fluorescence microscopy, whether m-DHP-BDP 14 and m-DHP-Cy5 15 are internalised and, if so, whether internalisation coincides with FSHR internalisation. U2OS-FSHR cells were incubated with either m-DHP-BDP 14 or m-DHP-Cy5 15 for 2 h and compared to mock-treated and recombinant FSH-treated cells. As shown in Fig. 4A and B vehicle-treated cells mainly show membrane GFP fluorescence, whereas treatment with the endogenous ligand recombinant human FSH (rFSH, 12 nM) led to the translocation of the receptor from the membrane to bright intracellular vesicles, indicative of endocytosis. We observed no receptor internalisation at 50 nM of probe (Fig. S1, ESI†), but clear intracellular receptor fluorescence was observed when using 1 μM of m-DHP-BDP 14 or m-DHP-Cy5 15, in agreement with previous observations that concentrations of about two orders of magnitude higher than the EC50 found in the luciferase assay are needed to induce ∼50% receptor internalisation by FSH and low molecular weight FSHR agonists.18,29m-DHP-BDP 14 induced receptor internalisation to a similar extent as rFSH (Fig. 4A and B), confirming that m-DHP-BDP 14 is a potent, functional FSHR agonist able to induce both FSHR-induced cAMP signalling and receptor internalisation. BODIPY fluorescence was observed in the perinuclear region in membrane-like structures (Fig. 4C), most likely corresponding to the membranes of the endoplasmic reticulum (ER) or Golgi apparatus, rather than intracellular vesicles indicative of endocytosis.30,31 Analysis of BODIPY fluorescence and GFP (receptor) fluorescence as shown in Fig. 4C, D, Fig. S2 and Table S2 (ESI†) show that indeed only moderate colocalization exists between the BODIPY dye and the GFP signal.32 Moreover, we observed the same intracellular localization pattern when control U2OS cells were treated with m-DHP-BDP 14 at 37 °C (Fig. 4C, right panel). No difference in fluorescence levels was found between FSHR expressing cells and wild type cells (Fig. 4D), confirming the flow cytometry data. When U2OS-FSHR cells were treated with m-DHP-BDP 14 at 4 °C, both receptor internalisation and compound uptake were inhibited (Fig. 4C, middle panel and Fig. 4D). Receptor internalisation is an active process, and the rate of internalisation is dependent on the temperature.33 Our observation that also compound uptake was abolished at low temperature indicates that the compound was not (only) taken up by passive diffusion but that other, active, processes played a role. Although diffusion through the cell membrane could be expected to be slowed down at lower temperatures as well, this would not explain the complete lack of compound fluorescence at 4 °C. The absence of differential uptake of m-DHP-BDP 14 between wild-type U2OS cells and FSHR-U2OS cells at 37 °C makes it however very unlikely that the internalisation is FSHR-mediated. Further experiments are needed to elucidate the uptake mechanism for m-DHP-BDP 14.
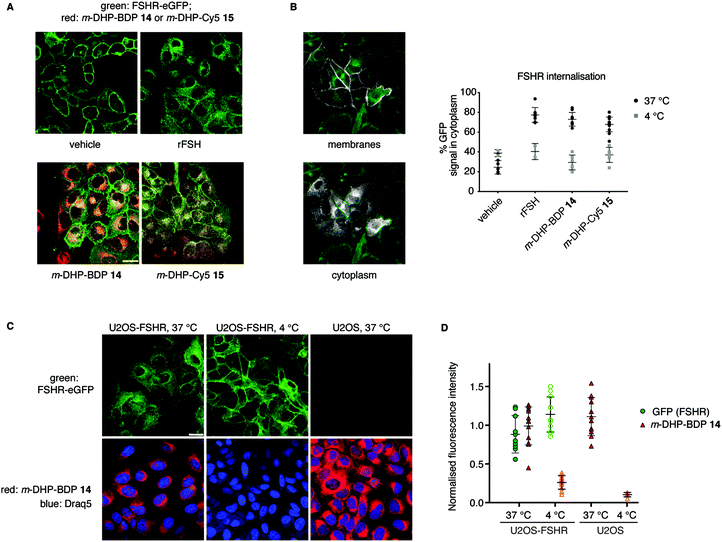 |
| | Fig. 4 Compound m-DHP-BDP 14 induces FSHR internalisation but stains cells in an FSHR-independent manner. Scale bar: 25 μm each data point represents an analysed image from three independent experiments. (A) Representative micrographs of U2OS-FSHR cells that were treated with LMW agonists or rFSH at 37 °C, but not DMSO vehicle, show receptor internalisation (green). (B) FSHR internalisation was quantified using the ratio between GFP signal on the membrane and in the cytoplasm. Only at 37 °C FSHR-internalisation was observed, when treating the cells with an agonist. Left: Images showing the different ROIs (white) that were set to define membranes or cytoplasm, see the Experimental section for more details on image analysis. (C and D) Treatment of U2OS-FSHR cells with m-DHP-BDP 14 (1 μM) at 4 °C, prevented receptor internalisation (quantification in (B)) as well as the uptake of the compound. Control U2OS cells at 37 °C became brightly red fluorescent when treated with m-DHP-BDP 14, indicating that the uptake process is temperature-dependent, but not FSHR selective. Representative images are shown in (C) and the integrated fluorescence intensity values for GFP and BDP are given in (D) to exclude effects of m-DHP-BDP 14 on total receptor levels (GFP signal) and to quantify the amount of non-specific compound uptake via active (37 °C, U2OS cells) or passive (4 °C) processes. | |
m-DHP-Cy5 15 induces FSHR internalisation and is selectively taken up in FSHR-expressing cells
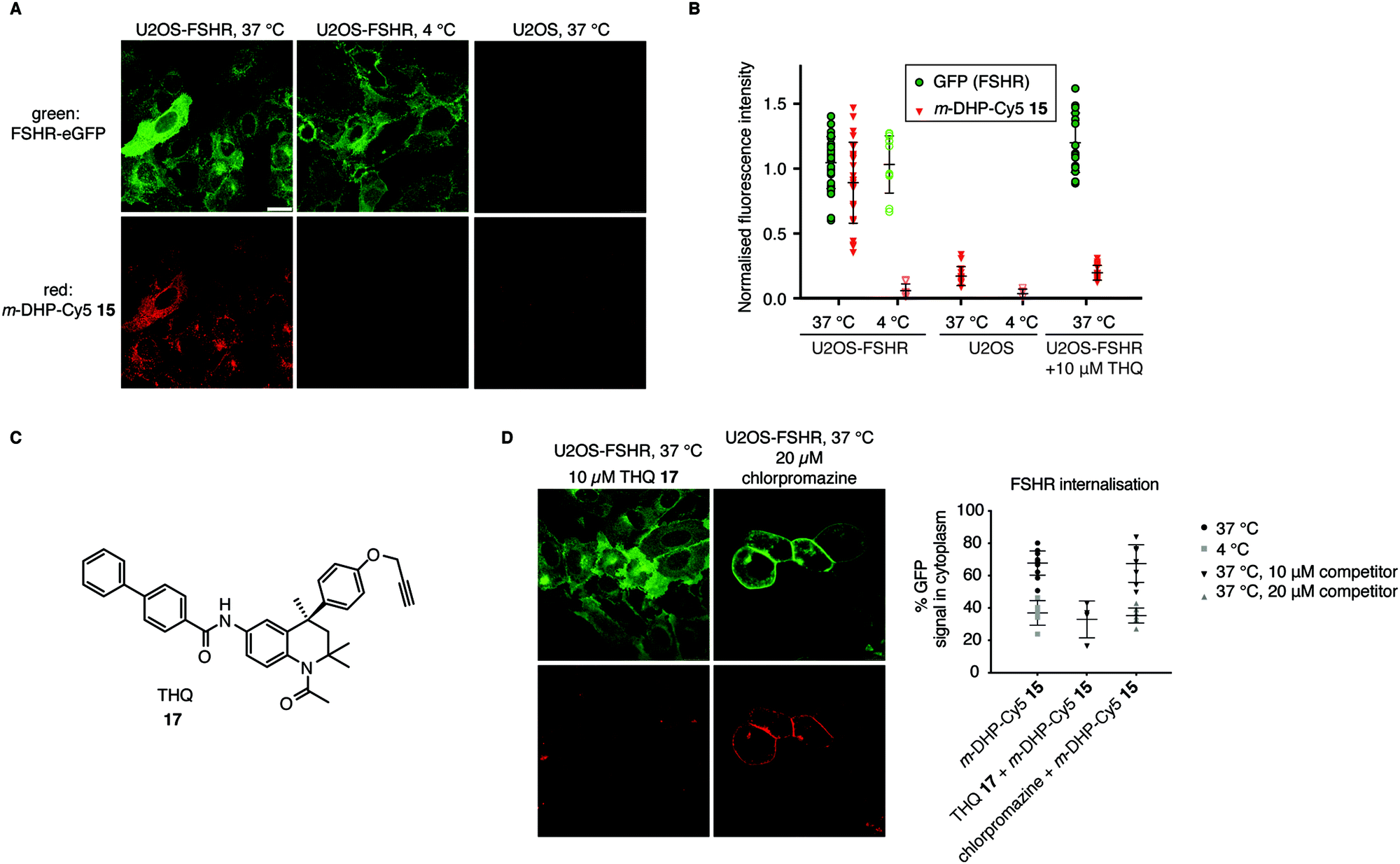
From the flow cytometry analysis, it was already apparent that m-DHP-Cy5 15 is the better candidate for selective uptake by the FSHR. Indeed, we found that m-DHP-Cy5 15 is able to effect FSHR internalisation to comparable levels as m-DHP-BDP 14 and rFSH at 37 °C (Fig. 4A and B). In stark contrast to the fluorescence pattern we observed for m-DHP-BDP 14, m-DHP-Cy5 15 fluorescence is limited to intracellular vesicles (Fig. 5Avs.Fig. 4C). A strong colocalization of the receptor and m-DHP-Cy5 15 (Fig. S2 and Table S2, ESI†),32 as well as a higher intensity correlation quotient (ICQ) was observed compared to m-DHP-BDP 14 (0.2852 vs. 0.1994, dependent staining: 0 ≤ x ≤ 0.5).34 Again, treatment at 4 °C did not lead to FSHR internalisation, whereas GFP fluorescence levels remained unchanged (Fig. 4B and 5B). No Cy5 fluorescence was observed under these conditions either (Fig. 5A and B), which indicates that the compound was taken up in an active manner. We did find it surprising that there was no or very little membrane fluorescence of m-DHP-Cy5 15 bound to the receptor detectable at 4 °C. However, binding of FSH to the FSHR is reported to become increasingly reversible at low temperatures. Also, the free energy of binding is calculated to be endothermic at temperatures below 12.5 °C.35 Similar effects might be affecting LMW ligand binding as well. This, in combination with the washing protocols might thus result in complete dissociation of possibly low levels of bound ligand to the membrane. Control U2OS cells treated with 1 μM of m-DHP-Cy5 15 at 37 °C did not show any intracellular Cy5 fluorescence (Fig. 5A right panel, Fig. 5B), in accordance with the flow cytometry data. These data strongly support the notion of FSHR-mediated uptake of m-DHP-Cy5 15 in the endolysosomal pathway of cells expressing this receptor.36
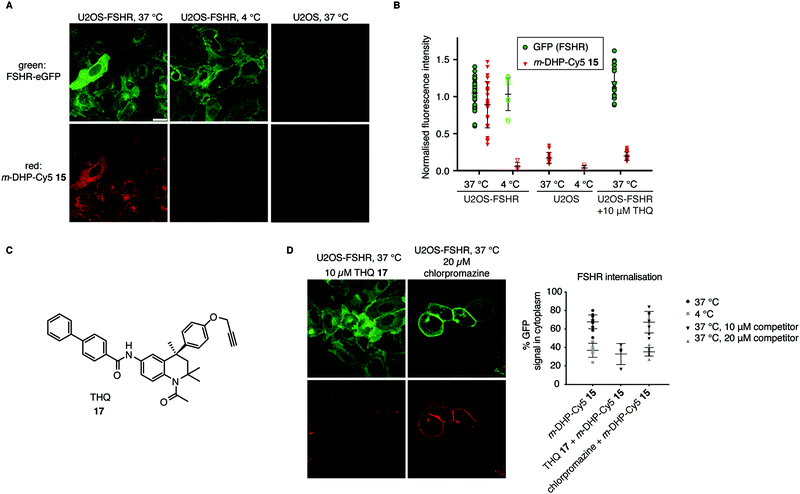 |
| | Fig. 5 Compound m-DHP-Cy5 15 induces FSHR internalisation and stains cells in an FSHR-independent manner. Scale bar: 25 μm. Each data point represents an analysed image from three independent experiments. (A and B) U2OS-FSHR and control U2OS cells were treated with m-DHP-BDP 15 (1 μM) at 37 °C or 4 °C, showing compound-induced internalisation of the receptor as well as compound uptake exclusively at 37 °C in U2OS-FSHR cells. Representative micrographs are shown in (A) and the fluorescence intensity for GFP and Cy5 is quantified in (B). (C) Chemical structure of the previously reported small molecule FSHR antagonist, THQ 17. (D) Chemical inhibition of receptor internalisation either by pre-treatment with FSHR antagonist THQ 17 or endocytosis blocker chlorpromazine prevents receptor internalisation by m-DHP-Cy5 15. (Data for m-DHP-Cy5 15 from Fig. 4B is replicated for clarity). Treatment with THQ 17 also blocks m-DHP-Cy5 15 binding and uptake (quantified in (B)). | |
Chemical inhibition of FSHR internalisation prevents m-DHP-Cy5 uptake in FSHR-expressing cells
To further confirm that m-DHP-Cy5 15 is endocytosed in an FSHR-dependent fashion, we conducted control experiments with the Cy5 dye 7 alone or in combination with rFSH. No intracellular Cy5 fluorescence was observed in these experimental settings, proving that ligand binding to the receptor is a requirement for its uptake (Fig. S3, ESI†). As additional proof, we applied a LMW FSHR ligand of a compound class distinct of the DHPs, namely a tetrahydroquinoline (THQ) antagonist previously reported by us (Fig. 5C).37,38 Pre-incubation of U2OS-FSHR cells with 10 μM final concentration of THQ 1737 for 1 h, 37 °C, followed by incubation with m-DHP-Cy5 15 (1 μM, 2 h, 37 °C) to a large extent blocked receptor internalisation and reduced m-DHP-Cy5 15 uptake to background levels (Fig. 5D). We used the relatively high dose of THQ 17 (10 μM) with respect to m-DHP-Cy5 15 (1 μM) to compensate for its lower potency (IC50: 39 nM) compared to that of m-DHP-Cy5 15 (EC50: 15 nM) (note that THQ 17 is an antagonist, whereas DHP 15 is an agonist). Colocalization analysis (Fig. S2 and Table S2, ESI†) confirmed that the low levels of remaining Cy5 fluorescence were due to non-specific background. We thus show that simultaneous incubation of U2OS-FSHR with THQ 17 and m-DHP-Cy5 15 prevents FSHR internalisation and m-DHP-Cy5 15 uptake. THQ is an allosteric antagonist of the FSHR38 and one explanation for this would be that the compound competes for the same binding pocket as m-DHP on the receptor, thereby preventing binding of m-DHP-Cy5 15 and subsequent activation and internalisation of the receptor. Alternatively, binding of the antagonist might result in a conformational shift of the receptor for which the agonist has reduced affinity. Further research is needed to elucidate the effect of THQ on DHP binding.
In a final experiment, we blocked receptor internalisation by pre-incubation with chlorpromazine, a cationic amphiphilic drug that interferes with clathrin-mediated endocytosis.39 Inhibition is concentration-dependent and also varies with the cell line used.39–42 In our initial experiments we used 10 μM of chlorpromazine, but this did not prevent agonist-induced receptor endocytosis (Fig. 5D). We then increased the concentration to 20 μM, which inhibited receptor internalisation, but was clearly also toxic to the cells as seen by profound changes in morphology. Although receptor internalisation could be blocked under these conditions, m-DHP-Cy5 15 was still able to bind to the FSHR as shown by strong, colocalized, fluorescence on the cell membrane (Fig. 5D and Fig. S2, Table S2, ESI†). Taken together, these results show that m-DHP-Cy5 15 induced FSHR internalisation in a clathrin-mediated fashion, which can be inhibited by pre-blocking FSHR (signalling) with the antagonist THQ 17, by disrupting clathrin-coated pit formation, as well as by lowering the temperature.
Conclusions
Whereas the biological role of the FSHR in human reproduction is well established, the involvement of this receptor in cancer, and specifically in angiogenesis, is a relatively young field of active research, with many open questions about the physiological relevance. The few reported peptide-based fluorescent FSHR ligands are poorly characterized and therefore not ideally suited to probe receptor function. In this study we present an alternative approach to create fluorescent FSHR ligands as valuable tools to visualize the receptor, to study receptor-mediated processes, and ultimately, to visualize whether targeting of this receptor is a viable method for selective delivery of drugs to FSHR-expressing tumours. Instead of employing a peptide-based ligand, we synthesised fluorescent ligands for the FSHR based on the class of DHP small-molecule agonists. We found these fluorescent ligands to be highly potent agonists for the FSHR. Both fluorescent agonists were able to induce receptor endocytosis as seen by receptor fluorescence using a GFP-FSHR cell line, but only one of the compounds, m-DHP-Cy5 15, was selectively internalised together with the receptor. This illustrates that even when agonistic potency is unaffected by the physicochemical nature of the dye, other pharmacological parameters can be greatly altered. m-DHP-Cy5 15 was not taken up by wild-type U2OS cells without FSHR or at low temperature in eGFP-FSHR-expressing U2OS cells. Furthermore, internalisation of both ligand and receptor could be inhibited by pharmacological intervention with either a low molecular weight FSHR antagonist or with chlorpromazine, an inhibitor of clathrin-mediated endocytosis, confirming the selective uptake of this compound via FSHR-binding, activation, and subsequent endocytosis. This opens up the exciting possibility of selective drug targeting via agonist-induced FSHR internalisation. The here reported small molecule m-DHP-NH213 forms the ideal basis for this, as we show that even modification with bulky fluorophores does not result in big changes in potency. Furthermore, through careful tuning of the properties of the attached cargo (fluorophore and/or drug) selective uptake in FSHR-expressing cells can be accomplished.
Conflicts of interest
CJvK and CMT were employees of MSD (Organon).
Acknowledgements
We thank Dr Laura Heitman for assistance with the luciferase assays and C. Erkelens, F. Lefeber and K. B. S. S. Gupta for assistance with NMR measurements. This research was supported by the Netherlands Organisation for Scientific Research (NWO).
Notes and references
- G. Vassart, L. Pardo and S. Costagliola, A molecular dissection of the glycoprotein hormone receptors, Trends Biochem. Sci., 2004, 29, 119–126 CrossRef CAS PubMed.
- J. G. Pierce and T. F. Parsons, Glycoprotein Hormones: Structure and Function, Annu. Rev. Biochem., 1981, 50, 465–495 CrossRef CAS PubMed.
- A. Dierich, M. R. Sairam, L. Monaco, G. M. Fimia, A. Gansmuller, M. LeMeur and P. Sassone-Corsi, Impairing follicle-stimulating hormone (FSH) signaling in vivo: Targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13612–13617 CrossRef CAS PubMed.
- Z. M. Lei, S. Mishra, W. Zou, B. Xu, M. Foltz, X. Li and Ch. V. Rao, Targeted Disruption of Luteinizing Hormone/Human Chorionic Gonadotropin Receptor Gene, Mol. Endocrinol., 2001, 15, 184–200 CrossRef CAS PubMed.
- A. P. N. Themmen and I. T. Huhtaniemi, Mutations of Gonadotropins and Gonadotropin Receptors: Elucidating the Physiology and Pathophysiology of Pituitary-Gonadal Function, Endocr. Rev., 2000, 21, 551–583 CrossRef CAS PubMed.
- M. W. Szkudlinski, V. Fremont, C. Ronin and B. D. Weintraub, Thyroid-Stimulating Hormone and Thyroid-Stimulating Hormone Receptor Structure-Function Relationships, Physiol. Rev., 2002, 82, 473–502 CrossRef CAS PubMed.
- A. Radu, C. Pichon, P. Camparo, M. Antoine, Y. Allory, A. Couvelard, G. Fromont, M. T. V. Hai and N. Ghinea, Expression of Follicle-Stimulating Hormone Receptor in Tumor Blood Vessels, N. Engl. J. Med., 2010, 363, 1621–1630 CrossRef CAS PubMed.
- M. Simoni, J. Gromoll and E. Nieschlag, The Follicle-Stimulating Hormone Receptor: Biochemistry, Molecular Biology, Physiology, and Pathophysiology, Endocr. Rev., 1997, 18, 739–773 CAS.
- A. Siraj, V. Desestret, M. Antoine, G. Fromont, M. Huerre, M. Sanson, P. Camparo, C. Pichon, F. Planeix, J. Gonin, A. Radu and N. Ghinea, Expression of follicle-stimulating hormone receptor by the vascular endothelium in tumor metastases, BMC Cancer, 2013, 13, 246 CrossRef CAS PubMed.
- F. Planeix, M.-A. Siraj, F.-C. Bidard, B. Robin, C. Pichon, X. Sastre-Garau, M. Antoine and N. Ghinea, Endothelial follicle-stimulating hormone receptor expression in invasive breast cancer and vascular remodeling at tumor periphery, J. Exp. Clin. Cancer Res., 2015, 34, 12 CrossRef PubMed.
-
R. G. J. M. Hanssen and C. M. Timmers, Thieno[2,3-d]pyrimidines with combined lh and fsh agonistic activity, WO2003020726A1, 2003 Search PubMed.
- D. Maclean, F. Holden, A. M. Davis, R. A. Scheuerman, S. Yanofsky, C. P. Holmes, W. L. Fitch, K. Tsutsui, R. W. Barrett and M. A. Gallop, Agonists of the Follicle Stimulating Hormone Receptor from an Encoded Thiazolidinone Library, J. Comb. Chem., 2004, 6, 196–206 CrossRef CAS PubMed.
- J. C. Pelletier, J. Rogers, J. Wrobel, M. C. Perez and E. S. Shen, Preparation of highly substituted γ-lactam follicle stimulating hormone receptor agonists, Bioorg. Med. Chem., 2005, 13, 5986–5995 CrossRef CAS PubMed.
- J. Wrobel, J. Jetter, W. Kao, J. Rogers, L. Di, J. Chi, M. C. Peréz, G.-C. Chen and E. S. Shen, 5-Alkylated thiazolidinones as follicle-stimulating hormone (FSH) receptor agonists, Bioorg. Med. Chem., 2006, 14, 5729–5741 CrossRef CAS PubMed.
- T. Guo, A. E. P. Adang, G. Dong, D. Fitzpatrick, P. Geng, K.-K. Ho, C. H. Jibilian, S. G. Kultgen, R. Liu, E. McDonald, K. W. Saionz, K. J. Valenzano, N. C. R. van Straten, D. Xie and M. L. Webb, Small molecule biaryl FSH receptor agonists. Part 2: Lead optimization via parallel synthesis, Bioorg. Med. Chem. Lett., 2004, 14, 1717–1720 CrossRef CAS PubMed.
- T. Guo, A. E. P. Adang, R. E. Dolle, G. Dong, D. Fitzpatrick, P. Geng, K.-K. Ho, S. G. Kultgen, R. Liu, E. McDonald, B. F. McGuinness, K. W. Saionz, K. J. Valenzano, N. C. R. van Straten, D. Xie and M. L. Webb, Small molecule biaryl FSH receptor agonists. Part 1: Lead discovery via encoded combinatorial synthesis, Bioorg. Med. Chem. Lett., 2004, 14, 1713–1716 CrossRef CAS PubMed.
-
P. M. G. Poveda, W. F. J. Karstens and C. M. Timmers, 4-phenyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline derivatives the treatment of infertility, WO2006117368A1, 2006 Search PubMed.
- C. J. van Koppen, P. M. Verbost, R. van de Lagemaat, W.-J. F. Karstens, H. J. J. Loozen, T. A. E. van Achterberg, M. G. A. van Amstel, J. H. G. M. Brands, E. J. P. van Doornmalen, J. Wat, S. J. Mulder, B. C. Raafs, S. Verkaik, R. G. J. M. Hanssen and C. M. Timmers, Signaling of an allosteric, nanomolar potent, low molecular weight agonist for the follicle-stimulating hormone receptor, Biochem. Pharmacol., 2013, 85, 1162–1170 CrossRef CAS PubMed.
- C. J. Daly and J. C. McGrath, Fluorescent ligands, antibodies, and proteins for the study of receptors, Pharmacol. Ther., 2003, 100, 101–118 CrossRef CAS PubMed.
- R. J. Middleton and B. Kellam, Fluorophore-tagged GPCR ligands, Curr. Opin. Chem. Biol., 2005, 9, 517–525 CrossRef CAS PubMed.
- J. G. Baker, R. Middleton, L. Adams, L. T. May, S. J. Briddon, B. Kellam and S. J. Hill, Influence of fluorophore and linker composition on the pharmacology of fluorescent adenosine A1 receptor ligands, Br. J. Pharmacol., 2010, 159, 772–786 CrossRef CAS PubMed.
- A. J. Vernall, S. J. Hill and B. Kellam, The evolving small-molecule fluorescent-conjugate toolbox for Class A GPCRs, Br. J. Pharmacol., 2014, 171, 1073–1084 CrossRef CAS PubMed.
- C.-W. Lee, L. Guo, D. Matei and K. Stantz, Development of Follicle-Stimulating Hormone Receptor Binding Probes to Image Ovarian Xenografts, J. Biotechnol. Biomater., 2015, 5(3), 198 Search PubMed.
- X. Zhang, J. Chen, Y. Zheng, X. Gao, Y. Kang, J. Liu, M. Cheng, H. Sun and C. Xu, Follicle-Stimulating Hormone Peptide Can Facilitate Paclitaxel Nanoparticles to Target Ovarian Carcinoma In vivo, Cancer Res., 2009, 69, 6506–6514 CrossRef CAS PubMed.
- Jui-Cheng Yen, Fu-Juay Chang and Shyang Chang, A new criterion for automatic multilevel thresholding, IEEE Trans. Image Process., 1995, 4, 370–378 CrossRef CAS PubMed.
- M. Verdoes, B. I. Florea, U. Hillaert, L. I. Willems, W. A. van der Linden, M. Sae-Heng, D. V. Filippov, A. F. Kisselev, G. A. van der Marel and H. S. Overkleeft, Azido-BODIPY Acid Reveals Quantitative Staudinger–Bertozzi Ligation in Two-Step Activity-Based Proteasome Profiling, ChemBioChem, 2008, 9, 1735–1738 CrossRef CAS PubMed.
- R. B. Mujumdar, L. A. Ernst, S. R. Mujumdar, C. J. Lewis and A. S. Waggoner, Cyanine dye labeling reagents: Sulfoindocyanine succinimidyl esters, Bioconjugate Chem., 1993, 4, 105–111 CrossRef CAS PubMed.
- P. J. Thul, L. Åkesson, M. Wiking, D. Mahdessian, A. Geladaki, H. A. Blal, T. Alm, A. Asplund, L. Björk, L. M. Breckels, A. Bäckström, F. Danielsson, L. Fagerberg, J. Fall, L. Gatto, C. Gnann, S. Hober, M. Hjelmare, F. Johansson, S. Lee, C. Lindskog, J. Mulder, C. M. Mulvey, P. Nilsson, P. Oksvold, J. Rockberg, R. Schutten, J. M. Schwenk, Å. Sivertsson, E. Sjöstedt, M. Skogs, C. Stadler, D. P. Sullivan, H. Tegel, C. Winsnes, C. Zhang, M. Zwahlen, A. Mardinoglu, F. Pontén, K. Feilitzen, K. S. von, Lilley, M. Uhlén and E. Lundberg, A subcellular map of the human proteome, Science, 2017, 356, eaal3321 CrossRef PubMed.
- H. Kishi, H. Krishnamurthy, C. Galet, R. S. Bhaskaran and M. Ascoli, Identification of a Short Linear Sequence Present in the C-terminal Tail of the Rat Follitropin Receptor That Modulates Arrestin-3 Binding in a Phosphorylation-independent Fashion, J. Biol. Chem., 2002, 277, 21939–21946 CrossRef CAS PubMed.
- M.-H. Teiten, L. Bezdetnaya, P. Morlière, R. Santus and F. Guillemin, Endoplasmic reticulum and Golgi apparatus are the preferential sites of Foscan® localisation in cultured tumour cells, Br. J. Cancer, 2003, 88, 146–152 CrossRef PubMed.
- M. Terasaki, J. Song, J. R. Wong, M. J. Weiss and L. B. Chen, Localization of endoplasmic reticulum in living and glutaraldehyde-fixed cells with fluorescent dyes, Cell, 1984, 38, 101–108 CrossRef CAS PubMed.
- V. Zinchuk, Y. Wu and O. Grossenbacher-Zinchuk, Bridging the gap between qualitative and quantitative colocalization results in fluorescence microscopy studies, Sci. Rep., 2013, 3, 1365 CrossRef CAS PubMed.
- P. W. Fletcher and L. E. Reichert, Cellular processing of follicle-stimulating hormone by Sertoli cells in serum-free culture, Mol. Cell. Endocrinol., 1984, 34, 39–49 CrossRef CAS PubMed.
- Q. Li, A. Lau, T. J. Morris, L. Guo, C. B. Fordyce and E. F. Stanley, A Syntaxin 1, Gαo, and N-Type Calcium Channel Complex at a Presynaptic Nerve Terminal: Analysis by Quantitative Immunocolocalization, J. Neurosci., 2004, 24, 4070–4081 CrossRef CAS PubMed.
- T. T. Andersen, L. M. Curatolo and L. E. Reichert Jr, Follitropin binding to receptors in testis: studies on the reversibility and thermodynamics of the reaction, Mol. Cell. Endocrinol., 1983, 33, 37–52 CrossRef CAS PubMed.
- N. Sayers and A. C. Hanyaloglu, Intracellular Follicle-Stimulating Hormone Receptor Trafficking and Signaling, Front. Endocrinol., 2018, 9, 653 CrossRef PubMed.
- K. M. Bonger, S. Hoogendoorn, C. J. van Koppen, C. M. Timmers, H. S. Overkleeft and G. A. van der Marel, Synthesis and Pharmacological Evaluation of Dimeric Follicle-Stimulating Hormone Receptor Antagonists, ChemMedChem, 2009, 4, 2098–2102 CrossRef CAS PubMed.
- N. C. R. van Straten, T. H. J. van Berkel, D. R. Demont, W.-J. F. Karstens, R. Merkx, J. Oosterom, J. Schulz, R. G. van Someren, C. M. Timmers and P. M. van Zandvoort, Identification of Substituted 6-Amino-4-phenyltetrahydroquinoline Derivatives: Potent Antagonists for the Follicle-Stimulating Hormone Receptor, J. Med. Chem., 2005, 48, 1697–1700 CrossRef CAS PubMed.
- L. H. Wang, K. G. Rothberg and R. G. Anderson, Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation, J. Cell Biol., 1993, 123, 1107–1117 CrossRef CAS PubMed.
- M. Okuyama, H. Laman, S. R. Kingsbury, C. Visintin, E. Leo, K. L. Eward, K. Stoeber, C. Boshoff, G. H. Williams and D. L. Selwood, Small-molecule mimics of an α-helix for efficient transport of proteins into cells, Nat. Methods, 2007, 4, 153 CrossRef CAS PubMed.
- X. Sun, V. K. Yau, B. J. Briggs and G. R. Whittaker, Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells, Virology, 2005, 338, 53–60 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00068j |
| ‡ Current address: Department of Organic Chemistry, University of Geneva, Quai Ernest-Ansermet30, 1211 Geneve, Switzerland. |
|
| This journal is © The Royal Society of Chemistry 2020 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Gijs H. M.
van Puijvelde
b,
Gijs A.
van der Marel
*a,
Gijs H. M.
van Puijvelde
b,
Gijs A.
van der Marel