
Amino acid acrylamide mimics: creation of a consistent monomer library and characterization of their polymerization behaviour†
Dries
Wyers
 ,
Toon
Goris
,
Yana
De Smet
and
Tanja
Junkers
*
,
Toon
Goris
,
Yana
De Smet
and
Tanja
Junkers
*
Polymer Reaction Design Group, School of Chemistry, Monash University, 19 Rainforest Walk, Clayton, VIC 3800, Australia. E-mail: tanja.junkers@monash.edu
First published on 10th August 2021
Abstract
Many attempts to mimic the structure of biopolymers via precision polymer synthesis with reversible deactivation radical polymerization (RDRP) techniques have been made. For peptides and proteins and their building blocks in particular, a broad variety of mimics have previously been suggested. However, general consistency in the design of these materials has been lacking. In this work, the foundations of a consistent acrylamide monomer library that mimics essential amino acids has been laid, defining mimics as the reaction product of acryloyl chloride and the decarboxylized respective amino acid. Five of these monomers were then selected as model monomers, each representing one of the the five major groups of amino acids. The model monomers were synthesized in flow procedures directly from acrylic acid and subjected to reversible addition fragmentation chain transfer (RAFT) polymerizations. 2-Hydroxyethyl acrylamide, N-isobutylacrylamide, N-methylacrylamide and isopropyl 4 – acrylamidobutanoate (mimicking serine, valine, glycine and glutamic acid) were polymerized via a general thermal RAFT approach resulting in high monomer conversions and good molecular weight control after short reaction times of 5 to 15 minutes. For the mimic of arginine, 1,3-di-boc-guanidinobutyl acrylamide, photoRAFT at room temperature was required to reach consistent polymerization conditions, then also resulting in well-defined materials.
Introduction
In the eyes of a polymer chemist, proteins can be regarded as the ultimate perfect polymer structures as they exhibit high molecular weights, uniformity in chain length and perfect control over the monomer sequence.1 The combination of these factors define the unique properties and characteristics that these macromolecules display. DNA and proteins, the two most well-known examples of nature's precisely engineered macromolecules, are largely responsible for the diversity and complexity of designer materials that can be found in nature. Over the years, with a better understanding of their structures, also research towards the design of modified or completely artificial analogues of these molecules has gained significant momentum. For the synthesis of artificial peptides, in particular, the development of solid phase peptide synthesis (SPPS) by Merrifield and co-workers in 1963 marked the first major breakthrough.2 Various peptide mimics have since been developed with β-peptides,3 peptoids4 and peptide/polymer conjugates as best-known examples. However, while β-peptides and peptoids have proven to be valid candidates to mimic the action of peptides and proteins, their practical use is sometimes limited due to their sensitivity towards temperature, pH, organic solvents or degradation.5 Peptide/protein–polymer conjugates, on the other hand, benefit from the advantages of biomolecules whilst tuning their limitations by employing suitable synthetic polymers in addition.5,6 They find use in applications such as gas separation, optoelectronics7 and catalysis.8 However, biomedicine and pharmacology are the fields that thrive the most by using such hybrid compounds.Although the use of peptide/protein–polymer conjugates has resulted in promising applications, their use is still dependent on and limited by the physiological environment they are used in. Therefore, the ability to produce synthetic polymers without adding the original biomolecule moiety has gained significant attention in recent years. In principle, the specificity and superiority in properties of peptides and proteins can be mimicked by synthetic compounds if a similar level of complexity can be reached. The use of fully synthetic polymers offers the great advantage of featuring fully synthetic backbones. These structures can be designed to be chemically more robust compared to their biological counterparts. Thus, synthetic analogues are likely to display increased stability while it can be expected that they exhibit at least some similarity in function.9 Recent advances in polymerization techniques and especially since the development of reversible deactivation radical polymerization (RDRP) led the field of polymer chemistry into a rapid evolution and have made the precise synthesis of well-defined polymeric structures possible.10–12 Polymers from RDRP exhibit controlled molecular weights, narrow dispersities and high end-group fidelity. Moreover, such polymers can display a very specific topology (linear, branched, star-shaped, etc.) or a very specific sequence (sequence-controlled, sequence-defined) resulting in very unique physiochemical properties.13 As a result, synthetic materials that could replicate the precision and functionality of the materials found in nature, are by these routes in principle possible, even if sequence lengths and overall dispersities of RDRP-made polymers are still far off from the precision of proteins. However, the broad array of monomers that can be used in these polymerization reactions make them nonetheless appealing.
The synthesis of fully synthetic peptide mimics via RDRP reactions has gathered much attention in recent years and fully synthetic peptide mimicking polymers have been synthesized by using reversible addition fragmentation chain transfer (RAFT) polymerization,14,15 nitroxide-mediated polymerization (NMP)16,17 and atom transfer radical polymerization (ATRP).18 In general, the design of the vinyl monomers used in these reactions can be divided in two main classes: (i) vinyl monomers that display the full structure of a natural amino acid (or peptide sequence) as their functional group; and (ii) monomers that only copy the main functionality of amino acids in their respective structure. The work of Endo and coworkers, as well as Koga and coworkers greatly defined the standard for the first class of monomers. In their work, straightforward procedures were developed for the synthesis of vinyl monomers that carry amino acid residues in there side chain trough the reaction of an α-amino acid and either acryloyl or methacryloyl chloride, resulting in respective acrylamide or methacrylamide monomers that display full amino acid moieties as their functional groups.9,19,20 Polymeric materials made thereof, mainly focused on applications like stimuli-responsive materials,18,21,22 self-assembly20,23 and optoelectronics.14,24
Regarding the second class of monomer mimics, various reports display the use of a broad array of monomers that carry functional groups characteristic of amino acids. Polymer structures composed of positively charged amino acid residues probably are the best-known examples of this second class of monomers. They have been widely explored for their use as antibacterial agents as cationic amino acids are highly common in so-called antimicrobial peptide (AMP) sequences. Monomers mimicking the structure of lysine (Lys) have been extensively investigated for these applications.25–29 Arginine-like mimics, on the other hand, have been studied for cell penetration and drug-delivery applications.30–32 Different monomer species have been used to design these functional molecules. Locock et al. reported the use of methacrylates to mimic the structures of arginine, lysine and tryptophan.33 Wong and co-workers, described the design and use of single-chain polymeric nanoparticles comprised of acrylate mimics of lysine and phenyl alanine for the effective destruction of planktonic and biofilm bacteria.34
As can be concluded from previous examples, various monomers of the second monomer class have been synthesized and used in production of different materials. The main focus in the monomer design, however, was placed on the incorporation of the exact functional groups that are displayed by natural amino acids whereas little to none attention has been given to the overall consistency of the monomer structures. Moreover, limited attention has been given to the influence of the length of the spacer separating the core functionality and the vinyl group of the monomer. This latter part, however, can play a vital part in the action of the final polymeric structure. Recently, Maron et al. designed a synthetic peptide mimicking oligomer sequence, comprised of amino acid mimicking acrylamide monomers via RAFT single unit insertion (SUMI) reactions, for hydrophobic drug solubilization applications.15 The design of synthesized sequence was based on a known peptide sequence that was able to solubilize meta-tetra(hydroxyphenyl)-chlorin (m-THPC), a photosensitizer for photodynamic cancer therapy. This model pentamer consisted of four phenyl alanine and one leucine unit. The translation of the amino acids phenyl alanine and leucine, resulted in N-phenylmethyl acrylamide and N-isobutyl acrylamide as mimics. The importance of the monomer sequence was proven by switching the leucine mimic to a valine mimic, demonstrating its change in properties. This minimal change of only one carbon atom, resulted in a significant decrease in drug solubility. Thus, indicating the major importance in side group and general monomer design consistency.
In this manuscript, the foundations towards a general design principle for amino acid analogues are proposed (Fig. 1). A uniform and consistent library containing functional monomers is developed based on the amide formation of acryloyl chloride with primary amines carrying the functionalities displayed on natural amino acids. The amines are thereby derived from decarboxylation of the respective amino acid compound. From this library, five different model monomers, each representing a distinct group of amino acids, were studied in detail towards their RAFT polymerization characteristics in order to determine ideal reaction parameters for each specific group of monomers.
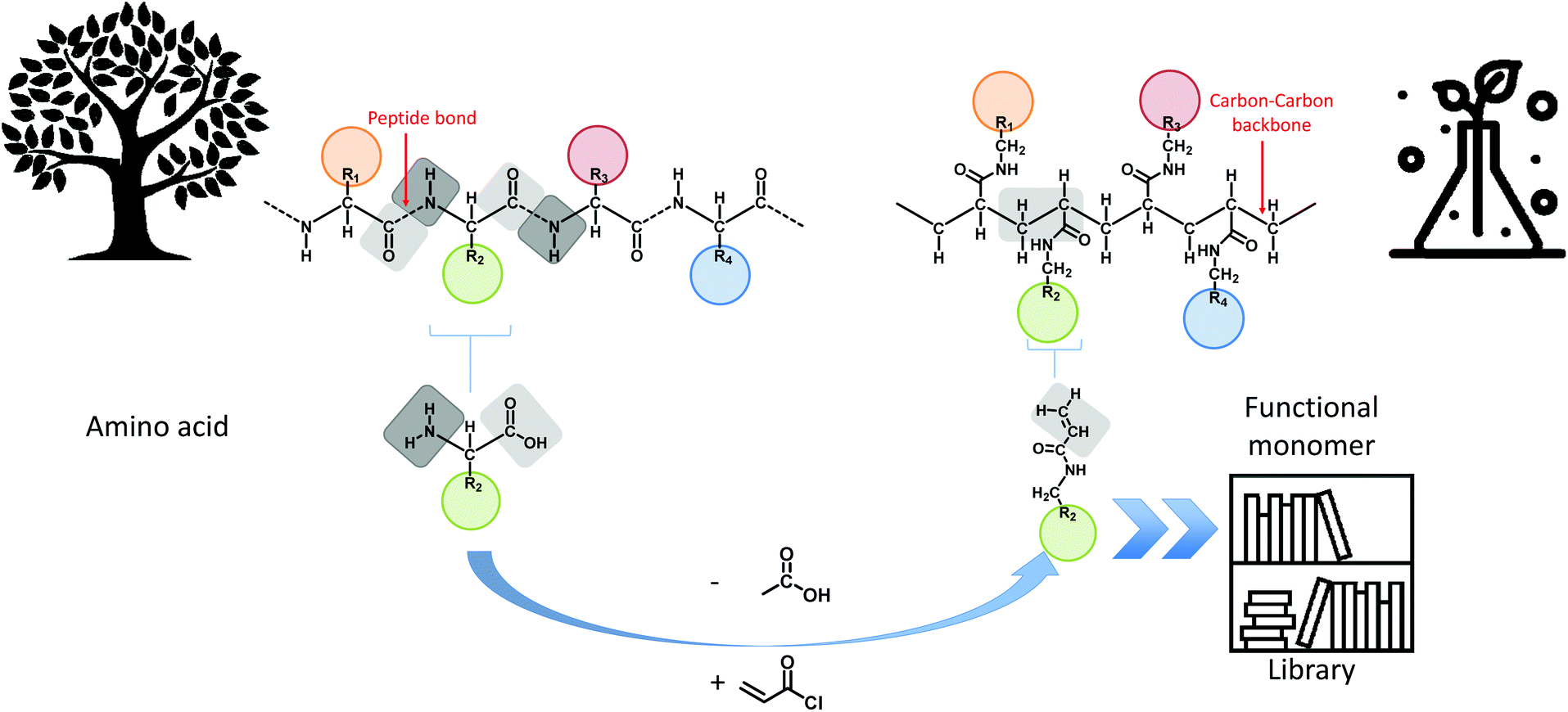 | ||
| Fig. 1 Overview of the main concept of translating amino acid structures into acrylamides. A comparison between the structure of a peptide sequence and a synthetic oligomer sequence is made. | ||
Experimental
Materials
Acrylic acid (Merck), thionyl chloride (97%, Sigma) and N,N-dimethylformamide (DMF, Merck) were used as received. Tetrahydrofuran (THF, Chem-Supply) and dichloromethane (DCM, Merck) were dried over molecular sieves. Triethylamine (Merck), isobutylamine (Merck), methylamine (2 M solution in THF, Sigma), phenethylamine (Merck), isopentylamine (≥98%, Sigma), amino-2-propanol (93%, Sigma), β-alanine (99%, Sigma), 4-aminobutyric acid (>98%, Chem-Supply), S-methylisothiourea hemisulfate salt (98%, Sigma), di-tert-butyl dicarbonate (di-boc, 99+%, AK Scientific), 1,4-diaminobutane (>98%, Chem-Supply), tetrapropyl ammonium bromide (98%, Sigma), ethanethiol (99+%, Fisher Scientific), 2-bromopropionic acid (99+%, Fisher Scientific) and 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride were used without further purification.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 or 7:6 v/v) were added in a glass vial and sealed with a rubber septum. The solution was degassed for 15 min by argon purging. Next, the prepared solution was loaded into a 1 mL gastight syringe and placed in the holder of a syringe pump (chemyx). The syringe pump delivered the reaction solution with the correct flowrate into a 25 μL tubular reactor (Idex, Peek tube Yellow, 1/32′′ OD × 0.007′′ ID) submerged in an oil bath of 90 °C. Various reaction times were screened and analysed by 1H NMR and SEC GPC.
:6 or 7:6 v/v) were added in a glass vial and sealed with a rubber septum. The solution was degassed for 15 min by argon purging. Next, the prepared solution was loaded into a 1 mL gastight syringe and placed in the holder of a syringe pump (chemyx). The syringe pump delivered the reaction solution with the correct flowrate into a 25 μL tubular reactor (Idex, Peek tube Yellow, 1/32′′ OD × 0.007′′ ID) submerged in an oil bath of 90 °C. Various reaction times were screened and analysed by 1H NMR and SEC GPC.
Results and discussion
Design of a consistent monomer library
As previously stated, numerous reports have displayed the use of amino acid mimicking monomers for the synthesis of functional polymer materials, however, consistent monomer design principles have been lacking. On first glance this might seem a minor obstacle, yet, once one tries to translate the whole series of essential amino acids, one inevitably crosses problems that need to be solved. Thus, our first goal was to establish the foundations of a consistent monomer library that comprises acrylamide monomers that mimic amino acid structures as closely as possible.Initially, we targeted acrylate monomers rather than acrylamides as the monomer class of choice as the use of these molecules would result in an easier purification both after synthesis of the monomers and certainly when precision monodisperse materials are targeted. However, it soon became clear that the use of acrylates would not be ideal as the availability of the required alcohols would be a very limiting factor in synthesis. Moreover, some acrylate monomers proofed to be unstable upon synthesis (prone to thermal elimination at low temperatures) and would require longer aliphatic spacers between its desired functionality and the acrylate ester group, resulting in an inconsistent library. We solved this issue by switching to acrylamides, which by nature are more stable. Also, by using amines as source materials that theoretically (or practically) are derived directly from the respective amino acid, an extra carbon (the stereogenic centre of the original amino acid) is consistently added as natural spacer. Such amines are available from decarboxylation of natural amino acids and have been the subject of research for several years. Decarboxylation pathways involving the use of bacterial strains,35–37 enzymatic reactions,35 radical approaches38–40 and organocatalytic reactions41 have been investigated. Polyzos and coworkers recently developed a straightforward flow procedure for the decarboxylation of amino acids thus generating a fast and easy method to access biological amines in high purified yields.42 The respective amines for the synthesis of the acrylamide library are hence readily available. Moreover, acrylamides are preferred over acrylates since they are in principle more water soluble compared to acrylates or methacrylates, a very important factor for the development of polymeric structures that are envisaged to be used in biological systems. Last but not least, while not containing the stereoinformation present in natural peptides, acrylamides are nonetheless capable to form hydrogen bonds, which may serve as a useful addition. Fig. 2 gives an overview over the whole amino acid mimic library as resulting from the above design principles.
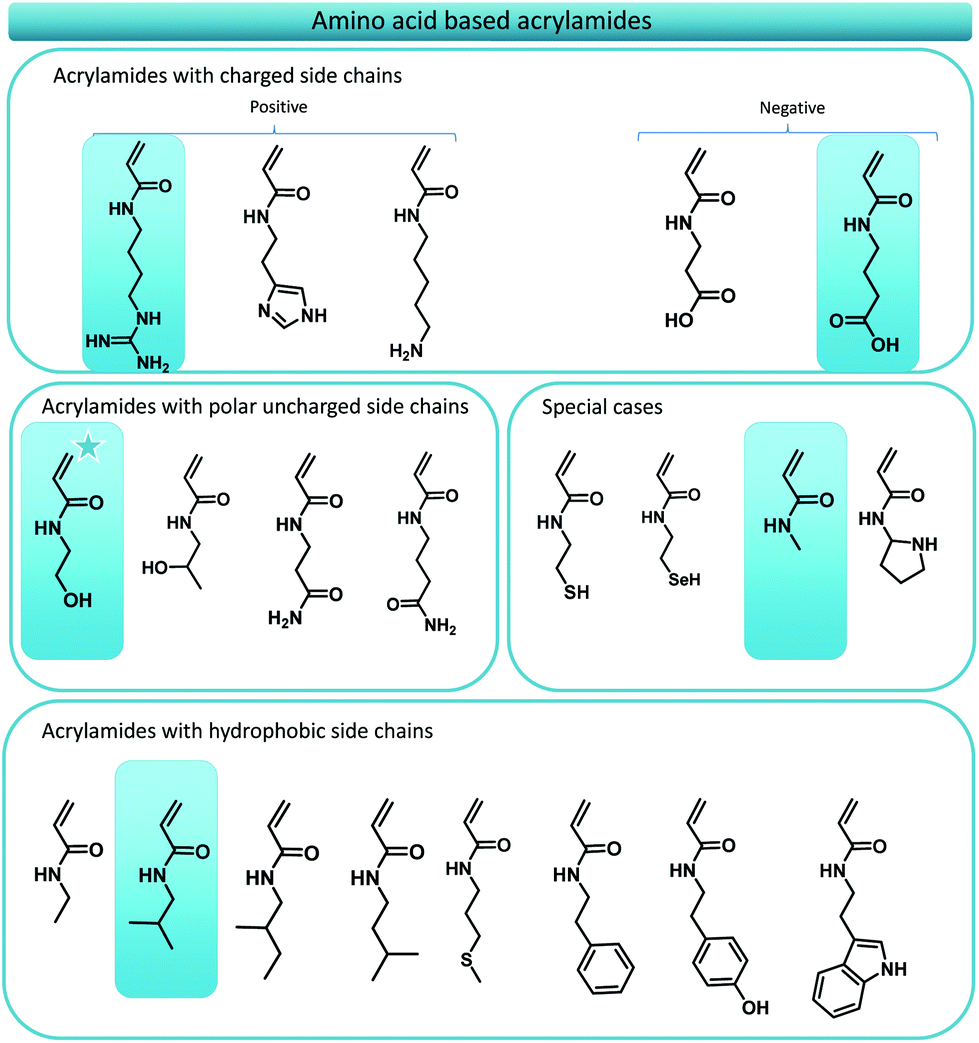 | ||
| Fig. 2 Targeted library of amino acid mimicking acrylamides. Each acrylamide displays a functional group that can be found in the 21 natural amino acids. The structure marked with a star is commercially available. The structures indicated with in the blue boxes are the five model monomers investigated in this manuscript. | ||
Of the total of 21 natural occurring amino acids, only one, namely the serine mimic, is commercially available (2-hydroxyethyl acrylamide). This structure is marked with a star in Fig. 2. Most mimic structures are fairly straightforward. Some examples, however, stand out, namely glycine and proline. Proline is an amino acid which displays a side group that is directly attached to the amino functionality. A feature which cannot be directly translated to an acrylamide. To circumvent this problem, pyrrolidine is suggested as functional mimic, taking the shortcomings of the design change into account. Glycine on the other hand, is accompanied with the problem that in principle it does not contain any functional R-group. The formal acrylamide equivalent of glycine would hence be acrylamide. This would, however, not be an apolar spacer as glycine is in peptides. Therefore, N-methyl acrylamide is used knowing that it still presents a considerable polarity. Yet, this approach is consistent of using the theoretically decarboxylated amino acid as parent amine for the acrylamide.
Next to the design of the monomer library, it is of course interesting to know how well the respective monomers would polymerize. The presence of the various functional groups can complicate polymerization behaviour, and reactions might not always be straightforward. Thus, in here we selected five model monomers, representing each an amino acid group. Four of these monomers, the mimics for valine, glycine, glutamic acid and arginine were synthesized from the respective amines and for all five the radical polymerization behaviour was studied. 2-Hydroxyethyl acrylamide (the serine mimic) was the only commercially available monomer, representing monomers with hydrophilic side groups. N-Isobutylacrylamide (valine mimic), displaying a simple apolar alkyl sidechain, was chosen as the representative of the amino acids with hydrophobic sidechains. N-Methylacrylamide was used as the equivalent of glycine. Isopropyl 4-acrylamidobutanoate is designed to mimic glutamic acid, an anionic amino acid, which is deprotonated under physiological conditions. The synthesis of this monomer started from 4-aminobutyric acid, a γ-amino acid, resulting in limited solubility in organic solvents. Therefore, we chose to protect the carboxylic acid functionality with an isopropyl protection group as this allows for the solubilisation of 4-aminobutyric acid in organic solvents as well as the straightforward purification of the residual monomer. The isopropyl protection group can be easily removed post-polymerization via saponification.
The final monomer that was used as a model was 1,3-di-boc-guanidinobutyl acrylamide, a mimic of the natural amino acid arginine. Arginine mimicking monomers, in particular, have been widely used for the design of functional polymeric materials due to their inherent antibacterial and cell penetrating properties. Several publications on the polymerization and use of arginine like monomers have been published over the years but kinetic data on their polymerizations have been scarce. Acrylate,26,43,44 methacrylate,30,31,45,46 acrylamides32,47 and methacrylamides48–52 displaying various spacer lengths and either protected or unprotected guanidinium functionalities have been described. The key functionality that needs to be introduced to mimic arginine is the guanidine functionality. Two general synthesis strategies exist to this end: (i) the synthesis of a pre-monomer featuring an amine terminal group, which can be modified post-polymerization to guadinine,31 or (ii) the synthesis of a monomer directly bearing a guanidinium functionality. The use of amine functional monomers generally requires the post-polymerization modification from amine to guanidine, as well as the prior protection of the amine during radical polymerization. With respect to RAFT polymerization, this approach is tedious, and will lead to aminolysis of end groups during the amine deprotection. Thus, in this work a protected guanidinium functionalized acrylamide monomer design was employed, carrying a butyl spacer in order to stay in line with our uniform monomer design approach. Perrier and coworkers reported a similar synthetic pathway before.32,53 In their work, an arginine mimicking acrylamide was synthesized carrying a protected guanidinium functionality, however connected with an ethyl spacer. The protection of the guanidinium functionality is required to use the monomer in conventional RAFT polymerization reactions without causing unwanted side reactions.
Monomer synthesis
All model monomers were synthesized through a simple amidation reaction from the primary amine and acryloyl chloride, and were conducted in a two-step coupled flow-batch procedure as depicted in Fig. 3. Acryloyl chloride was synthesized in situ from acrylic acid and thionyl chloride in a continuous flow procedure based on a previously developed approach by our group (Fig. 3).54,55 The resulting acryloyl chloride was then directly subjected to amidation without intermediate purification. Exact reaction parameters for the synthesis of acryloyl chloride as well as for each monomer can be found in ESI S3.† The synthesis of acid chlorides in continuous flow systems are a safer and greener approach compared to conventional batch reactions. This method greatly benefits from the increased surface-to-volume ratio and fast mass and heat transfer that flow reactors offer. As a result, enhanced control of the reaction parameters, high selectivity, and a high efficiency are obtained.56 In principle also the amidation itself could be carried out in flow. Here it was, however, sufficient to directly drip the acid chloride into a solution containing the amine. The isolated yields for the monomers synthesized through this method varied between 28–66% (see ESI S3†). Full optimization of the monomer synthesis procedure in order to increase the final yield was beyond the scope of this manuscript and no further optimization was thus attempted.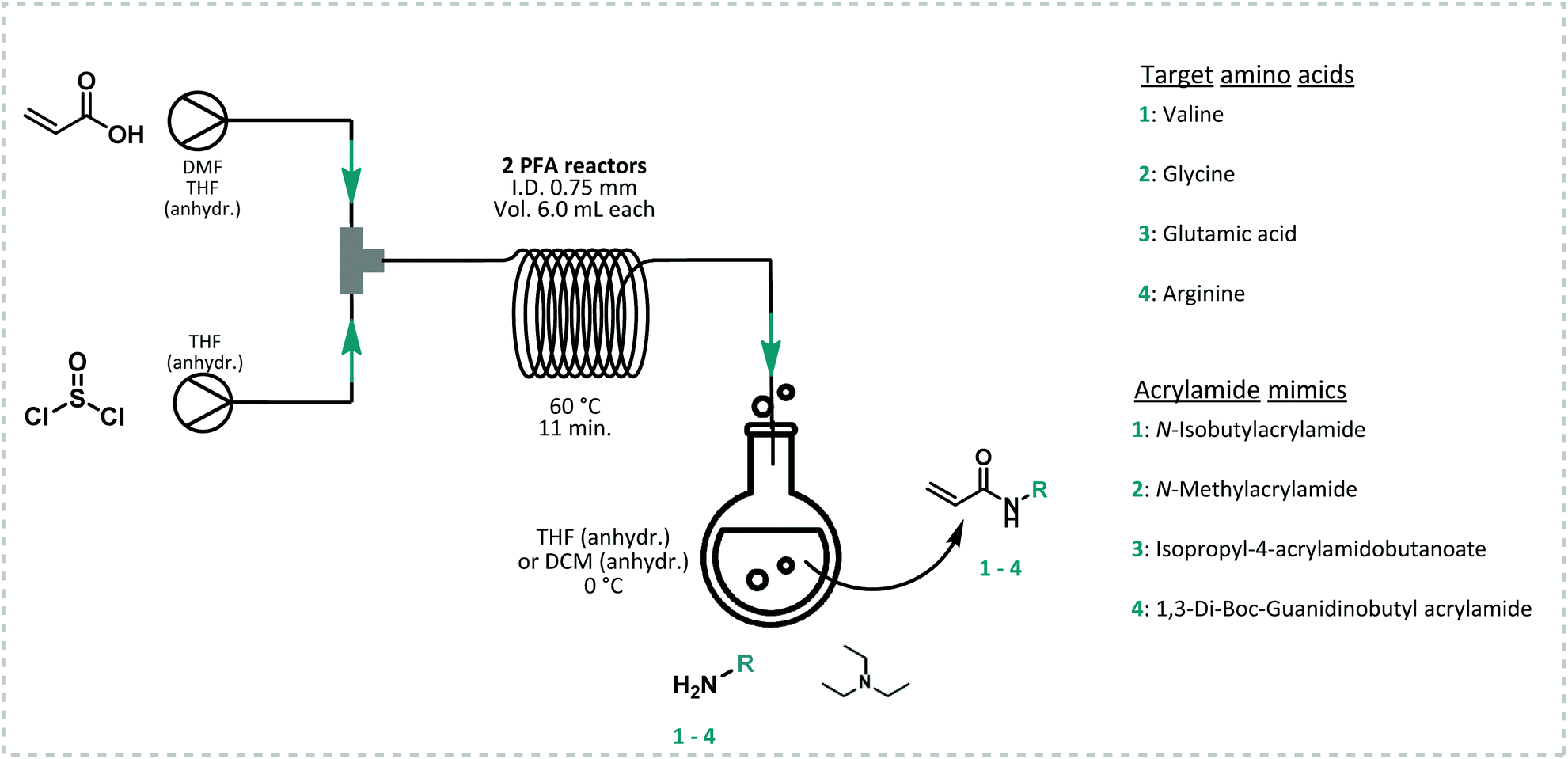 | ||
| Fig. 3 Schematic representation of the coupled flow-batch procedure to synthesize four different model acrylamide monomers. In a first step, acryloyl chloride is synthesized in two PFA reactors and without intermediate purification used in the synthesis of a functional acrylamide via a simple nucleophilic addition/elimination reaction with a primary amine in presence of triethylamine (TEA). | ||
RAFT polymerization screening
After synthesizing the four non-commercially available monomers, the radical polymerization behaviour of all five model monomers was investigated. The use of acrylamide monomers in reversible deactivation radical polymerization (RDRP) reactions is well known.57–59 Thermal RAFT polymerization was chosen as the standard method of choice, as this reaction mode is the most used method in RDRP with high functional group tolerance.58,60 Small scale screening reactions were conducted in a simple tubular flow reactor with a total volume of 25 μL (ESI Fig. S3†). Flow chemistry allows here to establish well reproducible reaction conditions, and to use minimal amounts of monomers during the screening itself. As reaction solvent, a mixture of 1,4-dioxane and MilliQ water in varying ratios depending on the monomer polarity was selected. The use of water as a (co-)solvent is known to enhance the polymerization rate of acrylamide monomers through formation of hydrogen bonds.61,62 2-(Propionic acid)ylethyl trithiocarbonate (PAETC) was used as chain transfer agent (CTA) as it is generally found to be a good RAFT agent for the polymerization of acrylamide monomers (Scheme 1).63 The reaction temperature was set to 90 °C in order to further enable fast polymerization. By increasing the temperature and employing a thermal radical initiator that decomposes rapidly at this temperature, a high radical flux is generated. As the rate of polymerization (Rp) is directly related to the concentration of propagating radicals and the propagation rate coefficient of the monomer, the process can thus be drastically accelerated.58 2,2′-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) was selected as thermal initiator. It features a half-life of 10 hours at 44 °C (3.5 min at 90 °C). A [CTA]0/[I]0 ratio of 40 was employed for all targeted chain lengths in order to ensure good control of the polymerization reaction and high livingness of the final product. A full summary of the exact reaction conditions and reactant concentrations for each monomer species can be found in ESI.† The initial monomer concentration was kept constant (1.66 M) and different degrees of polymerization (DP) were targeted by varying both the CTA and initiator concentrations. For each monomer, a total reaction time of 5 to 15 minutes was applied and samples were taken at various points in time. The results of the kinetic screening for the first four monomers are shown in Fig. 4. We assume that the kinetics of each model monomer is a reasonable representative of its group of amino acid mimics. To cover a broad range of chain lengths, DP 10, 25, 50 and 100 were targeted for each monomer.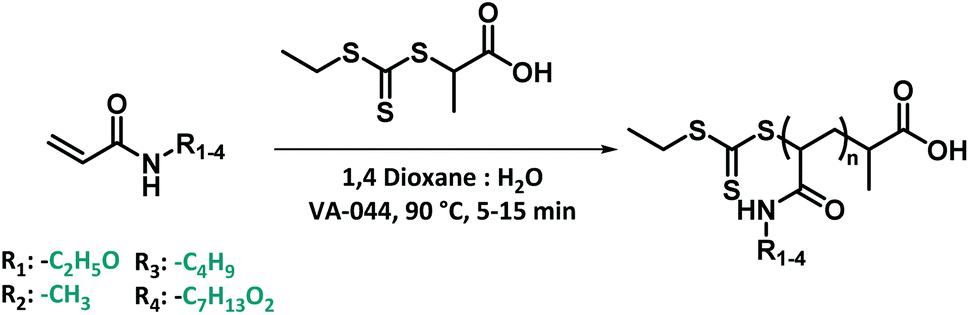 | ||
| Scheme 1 Thermal RAFT polymerization for 4 model monomers using PAETC as chain transfer agent and VA-044 as radical initiator. | ||
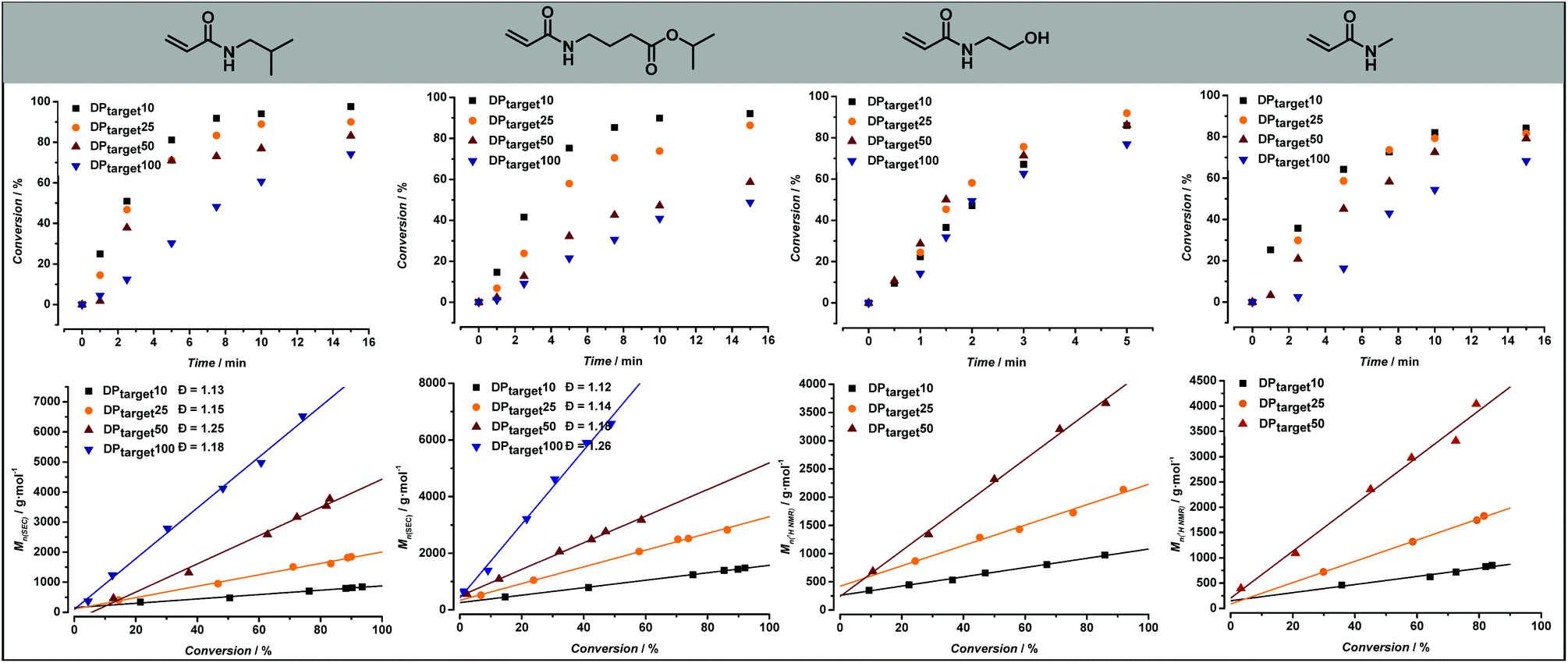 | ||
| Fig. 4 Summary of the results obtained from the RAFT polymerization screening reactions of four model monomers. Evolution of monomer conversion over time is given for each monomer in the upper panel of the figure. Number-average molecular weight evolution with conversion under variation of [RAFT]/[monomer] ratio is given for each monomer in the lower panel of the figure. The best linear fit of the data points is displayed. | ||
2-Hydroxyethyl acrylamide was the first model monomer to be studied. As it is a hydrophilic monomer, a solvent mixture of 60:40 water:1,4-dioxane was used. A minimum amount of 40% 1,4-dioxane was required to ensure the full solubility of the CTA. By using a temperature of 90 °C and an initial monomer concentration of 1.6 M, high conversion values (>90%) could be achieved in 5 minutes reaction time. It should be noted that since the reaction was carried out in flow, evaporation of water posed no issue.
Different chain lengths were targeted by keeping the monomer concentration constant and adjusting the CTA and initiator concentration accordingly. In this way, one would expect the reaction targeting the lowest DP (and thus featuring the highest concentration of CTA and radical initiator) to be the fastest, as the rate of polymerization is directly proportional to the initial radical concentration. However, as can be seen from Fig. 4, the reaction for DP10 is slower than both the reactions for DP25 and DP50. This effect is only visible for 2-hydroxyethyl acrylamide, and not in the reactions of other model monomers, which follow expectations better. The exact cause for this difference is unclear and has yet to be determined, but it can be assumed that the specific RAFT pre-equilibrium is the cause for this effect. Determination of the exact molecular weights for these polymers using GPC-SEC is not straightforward due to solubility issues and lack of appropriate Mark–Houwink parameters. Therefore, the experimental number average molecular weight (Mn) of these polymers was determined from 1H NMR by end group integration (SEC results are shown in ESI Fig. S6†). The full molecular weight evolution for all monomers and target DPs is shown in Fig. 4 alongside the kinetic data; the full lines give represent best linear fits of the data. For all polymerizations, linear progression of the Mn with increasing monomer conversion is observed. The linearity of the data depicted in Fig. 4 is indicative of a well-controlled RAFT polymerization.
Similar reaction conditions were used for the screening of N-methylacrylamide. As it also is a rather hydrophilic monomer, a 60:40 H2O:dioxane solvent mixture was likewise employed. After 5 minutes reaction time, targeting a DP of 10, however, a monomer conversion percentage of only 64% was obtained. As a result, reaction times were increased to 15 minutes. The exact cause for the slower polymerization rate in comparison with the polymerization of N-hydroxyethyl acrylamide is not clear. However, high monomer conversions were achieved after 15 minutes, which is still a reasonably fast polymerization. Molecular weight determination for the poly(N-methylacrylamide) samples was also conducted via1H NMR, resulting in a clear linear correlation between Mn and conversion.
The remaining three model monomers, were less polar and thus slightly different solvent ratios had to employed. A maximal amount of 30 v/v% H2O could be employed for the polymerization of both N-isobutylacrylamide and isopropyl-4-acrylamidobutanoate. Similarly, as was the case with N-methylacrylamide, reaction times up to 15 minutes were required to reach high monomer conversion values. In these cases, since solubility was less an issue for these polymers, molecular weight analysis was conducted via SEC resulting in a clear linear relationship between Mn and monomer conversion, again underpinning the well-controlled polymerization behaviour. Moreover, narrow molecular weight distributions, with dispersity values between 1.16 and 1.26 for poly(N-isobutyl acrylamide) and between 1.12 and 1.26 for poly(isopropyl-4-acrylamidobutanoate) samples, were obtained in good agreement with all previous observations.
The final model monomer that was investigated, was 1,3-di-boc-guanidinobutyl acrylamide, the acrylamide equivalent of the cationic amino acid arginine carrying a protected guanidinium functional group. Initially, a thermal RAFT procedure, similar to the method employed for the other monomers, was tested, using PAETC as CTA, VA-044 as initiator and a temperature of 90 °C. A lower monomer concentration (0.45 M) and a slightly different solvent ratio (20:80 v/v% H2O:1,4-dioxane) was used based on literature procedures of a similar monomer.32,53 The lower monomer concentration was also chosen to ensure proper solubility of the monomer in the solvent mixture. After an initial test screening reaction, targeting a DP of 10, only 26% of monomer conversion could be observed through 1H NMR analysis after a reaction time of 15 minutes (ESI Fig. S19†). This amount of monomer conversion is low for the reaction conditions used. Longer reaction times did not result in higher monomer conversions. 1H NMR analysis of the crude reaction mixture also indicated the presence of a prominent singlet at 1.11 ppm. This peak is characteristic for the tert-butyl protons in tert-butanol. tert-Butanol is indicative of in situ deprotection of the boc protection groups during the attempted polymerization. Boc protection groups are very sensitive to acids and often selective deprotection is possible under very acidic conditions. During boc deprotection reactions, both CO2 and tert-butyl cations are generated. These cations will either readily react with water to form tert-butanol or undergo deprotonation and form isobutylene.64,65 Additionally, new peaks became visible in the vinyl proton region further indicating deprotection rather than polymerization.
Classically, boc protection groups are removed with strong acids like HCl or TFA. In order to test if the pH of the reaction solution had an influence, a different CTA was used. PAETC features a carboxylic acid resulting in a reaction solution with a slightly acidic pH. Changing the CTA to 2-cyano-2-propyl dodecyl trithiocarbonate (CPD-TTC) resulted to an increase of the reaction solution to pH 6. However, undesired deprotection of the monomer was still occurring during polymerization. Moreover, literature reports indicate the possible influence of water in boc group removal as it acts as a selective dual acid/base catalyst at elevated temperatures.66 To test the influence of water, the reaction was repeated without the presence of H2O as co-solvent. 1H NMR still indicated a significant singlet peak at 1.11 ppm, indicative of tert-butanol formation, and the absence of polymerization after a reaction time of 48 hours at 46 °C (ESI Fig. S20†). One would expect the absence of water as reaction solvent leading to the formation of isobutylene rather then tert-butanol. However, as the solvent used in the polymerization reaction was not anhydrous, traces of water are present, and this amount is sufficient to form predominantly tert-butanol.
Finally, the influence of temperature was tested by subjecting three NMR tubes containing a small amount of monomer dissolved in DMSO-d6 to different temperatures (90 °C, 70 °C and 46 °C) for 15 minutes. The destructive influence of temperature was clearly indicated as the characteristic singlet peak was clearly present for the 90 °C sample after 15 minutes (ESI Fig. S21†). The samples that were subjected to 70 °C and 46 °C, resulted in lower amounts of deprotection after 15 minutes, leading to the conclusion that temperature is the main cause for the deprotection and consequently inhibition of polymerization. A quantitative analysis of the thermal deprotection of the monomer was conducted through in situ NMR. A solution of 0.045 M of 1,3-di-boc-guandinobutyl acrylamide in DMSO-d6 was prepared and heated at 70 °C for 80 minutes and NMR spectra were taken at five minute intervals (ESI Fig. S22–S24†). A clear increase of tert-butanol formation with increasing exposure times could be observed with 0.020 M of tert-butanol present after 80 minutes exposure time. Besides the formation of tert-butanol, characteristic peak signals of isobutylene (4.66 ppm ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 and 1.70 ppm –CH3, 1:3 integration ratio) were observed as well, although in smaller quantities, and only later than tert-butanol formation. Thermogravimetric analysis (TGA) of the bulk monomer under aerobic conditions revealed first signs of decomposition of 1,3-di-boc-guanidinobutyl acrylamide start at 125 °C (ESI Fig. S26†).
CH2 and 1.70 ppm –CH3, 1:3 integration ratio) were observed as well, although in smaller quantities, and only later than tert-butanol formation. Thermogravimetric analysis (TGA) of the bulk monomer under aerobic conditions revealed first signs of decomposition of 1,3-di-boc-guanidinobutyl acrylamide start at 125 °C (ESI Fig. S26†).
The presence of thermal decomposition of the monomer could clarify the effects mentioned in earlier literature reports published by Perrier and coworkers regarding the polymerization of a similar monomer. In their work, 1,3-di-boc-guanidinoethyl acrylamide was synthesized and polymerized via a thermal RAFT approach. They demonstrated successful polymerization of the monomer at 46 °C but indicated the loss of molecular mass control at higher temperatures.53 However, polymerization reactions of our monomer at 46 °C for longer amounts of time (24–48 hours) did not result in any formation of polymer and deprotection was clearly occurring, even though at slower rates. Increasing the initiator concentration from 0.0045 M to 0.045 M, when employing lower reaction temperatures, did not resolve the issue.
As a result, we switched from thermal to room-temperature photo-induced RAFT polymerization (photoRAFT). In this way, the thermal deprotection of the guanidinium functionality was prevented. Initial test polymerizations, targeting oligomeric materials with a DP of 10, were conducted in a 36 W Coscelia Nail Dryer UV LED (365 + 405 nm) lamp. In order to negate a possible influence of the acidity of the reaction solution on deprotection of the monomer, 2-cyano-2-propyl dodecyl trithiocarbonate was chosen as CTA. Benzoin was selected as supplemental initiator. A slightly higher [CTA]/[initiator] ratio of 20 was used to ensure proper initiation of the reaction. Samples were allowed to react at room temperature for 24 hours resulting in 98% monomer conversion with minor deprotection of the guanidinium functionality as confirmed by the presence of a minor peak at 1.11 ppm on 1H NMR analysis (ESI Fig. S25†). When using the identical reaction solution, i.e. 1,4-dioxane:H2O (80:20 v:v%), as were previously used in thermal RAFT reactions, phase separation of the reaction solution and precipitation of the oligomeric material could be observed at higher monomer conversions. This effect is not visible when 1,4-dioxane is used as sole reaction solvent.
After these initial test reactions, a more in-depth screening of the polymerization kinetics was attempted. For this, a TL-D 15 W BLB 1SL/25 UV lamp (peak emission at 365 nm) was used (ESI Fig. S4†) as a more sophisticated UV light source. By switching to a lamp with a narrower emission spectrum, the reaction could be accelerated to completion in 10 hours. However, a full kinetic characterization of the polymerization reaction could not be obtained as all attempted reactions showed varying inhibition periods, making conversion vs. time plots rather meaningless. However, full monomer conversion could be achieved consistently after a reaction time of 10 hours using CPD-TTC as CTA and benzoin as initiator resulting in oligomer materials with molecular weights around 3800 g mol−1 with narrow dispersity values of 1.10–1.18. The presence of a brief inhibition period is often observed in RAFT polymerization and the exact causes for this are still not entirely understood.67 For photoactivation, the exact causes are likely even more complex than for classical thermal RAFT polymerization. Nevertheless, polymerization could be carried out successfully and consistently. The various attempts and identification of suitable polymerization conditions for the monomer are summarized in Fig. 5.
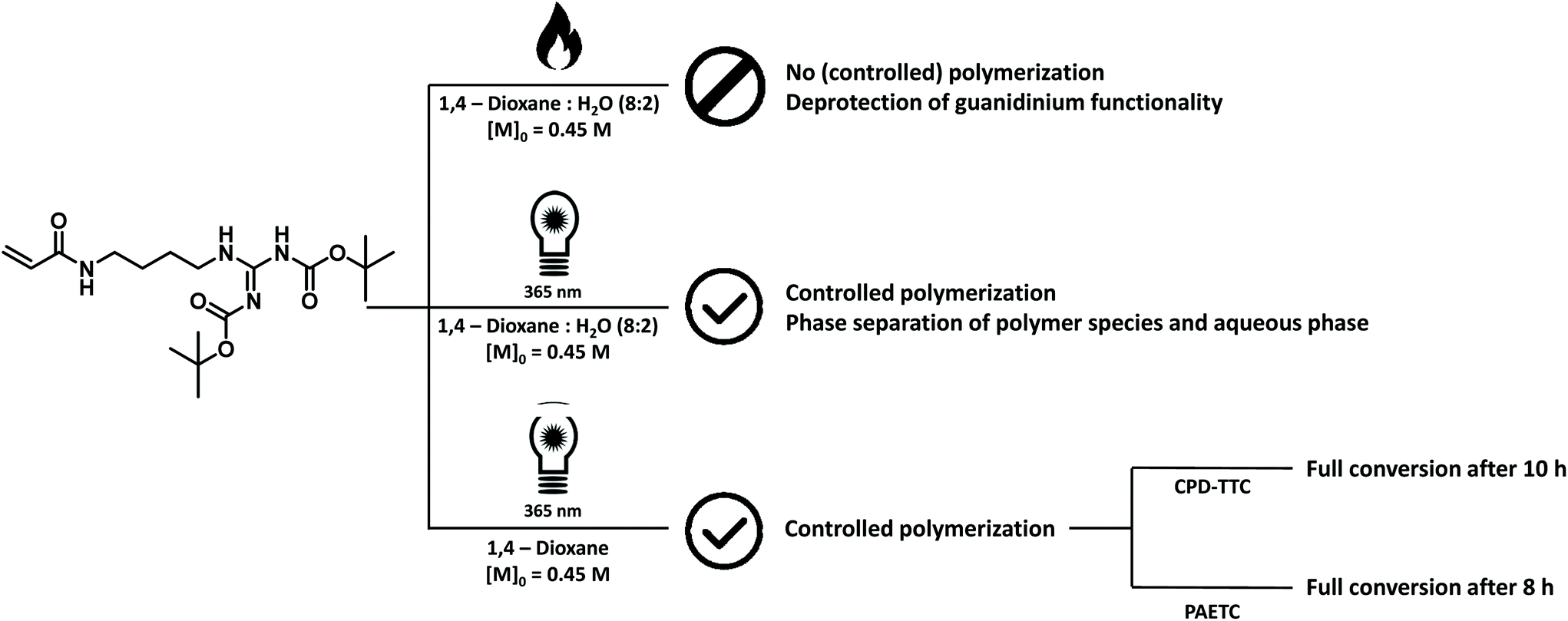 | ||
| Fig. 5 Summarizing figure of reaction conditions tested for the polymerization of 1,3-di-boc-guanidinobutyl acrylamide. Thermal initiated RAFT polymerization attempts lead to the deprotection of the guanidinium functionality of the monomer and no polymerization occurred. Photo initiated RAFT polymerization lead to well-defined polymer materials with the final polymer crashing out when a mixture of 1,4-dioxane and water was employed. | ||
By substituting CPD-TTC back to PAETC, the overall reaction time could be consistently shortened to 8 hours. Yet, also here a clear kinetic screening failed for the same reason of seemingly random inhibition periods. However, good control over molecular weight was demonstrated by targeting different molecular weights of 4000, 10000, 20000 and 40000 g mol−1 corresponding to polymers with 10, 25, 50 and 100 monomer insertions. After a reaction time of 8 hours, materials with apparent molecular weights of 3800, 6900, 12400 and 13620 g mol−1 were obtained (see Fig. 6). For the low molecular weight products, there is a good match between theoretical and experimental Mn (considering the SEC was based on polystyrene standard calibration). Narrow distributions were obtained, displaying dispersity values of 1.10 for DP 10 and slightly increased values of 1.22 for DP 100 at full conversion. Polymerization is thus also for this monomer proceeding with good control. However, for the target DP 100 a mismatch in molecular weight is clearly observed, indicating that at the lowered RAFT agent concentration still deprotection may negatively influence the polymerization, and that photoRAFT limits the maximum achievable chain length.
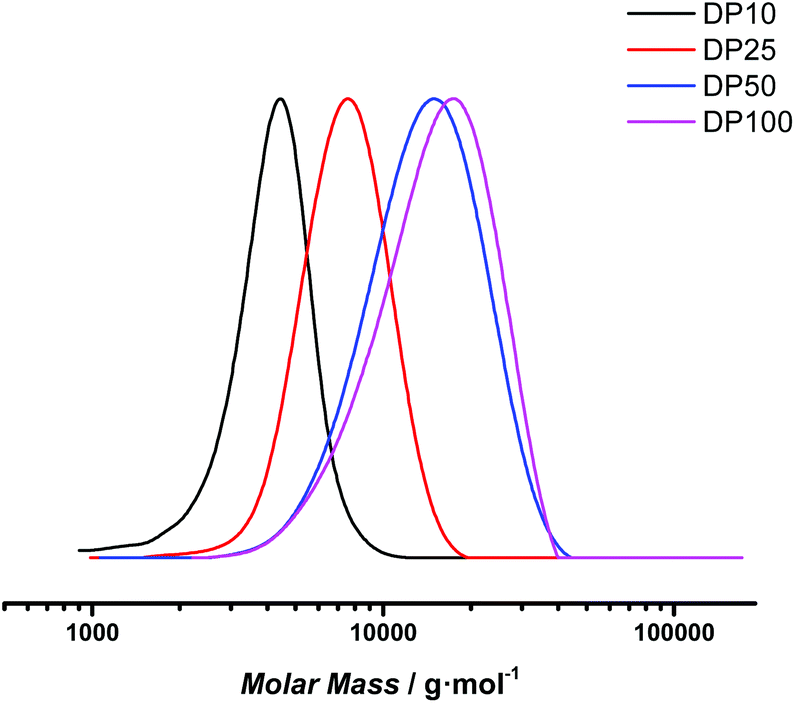 | ||
| Fig. 6 SEC traces of poly(1,3-di-boc-guanidinobutyl acrylamide) samples targeting different degrees of polymerization after 8 hours reaction time. | ||
Conclusions
The first fundaments towards an acrylamide monomer library that mimics natural amino acids were laid. Five model monomers, each representing one of the five main groups of amino acids, were selected and four of them were synthesized directly from acrylic acid in a flow procedure. Next, the RAFT polymerization of these monomers was studied via thermal RAFT polymerization procedures, indicating good control over molecular weights and dispersities for four out of five model monomers after short reaction times of 5 to 15 minutes.Polymerization of 1,3-di-boc-guanidinobutyl acrylamide (a mimic for arginine) was more difficult to polymerize due to thermal deprotection occurring, leading to inhibition of polymerization. However, also this monomer could successfully be polymerized by applying photoRAFT conditions at room temperature, resulting in narrowly dispersed polymers (Đ = 1.10–1.22) after 8 hours reaction time.
Conflicts of interest
No conflicts of interests to declare.Acknowledgements
Funding from Monash University is kindly acknowledged. We acknowledge Dr. Joris Haven, Dr. Camille Bakkali-Hassani and Dr. Jordan Hooker for scientific discussions and Dr. Alasdair McKay for the help with the in situ NMR experiments.References
- A. Bruce, Molecular biology of the cell, Garland Pub., [1989] ©1989, New York, 2nd edn, 1989 Search PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- M. Overhand, F. N. M. Kühnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 913–941 Search PubMed.
- S. D. Ganesh, N. Saha, O. Zandraa, R. N. Zuckermann and P. Sáha, Polym. Bull. (Berlin), 2017, 74, 3455–3466 CrossRef CAS.
- J. Y. Shu, B. Panganiban and T. Xu, Annu. Rev. Phys. Chem., 2013, 64, 631–657 CrossRef CAS.
- N. ten Brummelhuis, P. Wilke and H. G. Borner, Macromol. Rapid Commun., 2017, 38, 1700632 CrossRef PubMed.
- J. Wigenius, P. Björk, M. Hamedi and D. Aili, Macromol. Biosci., 2010, 10, 836–841 CrossRef CAS PubMed.
- G. Delaittre, I. C. Reynhout, J. J. Cornelissen and R. J. Nolte, Chem. – Eur. J., 2009, 15, 12600–12603 CrossRef CAS PubMed.
- F. Sanda and T. Endo, Macromol. Chem. Phys., 1999, 200, 2651–2661 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- D. H. Solomon, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5748–5764 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- J. J. Haven and T. Junkers, Polym. Chem., 2019, 10, 679–682 RSC.
- H. Mori and T. Endo, Macromol. Rapid Commun., 2012, 33, 1090–1107 CrossRef CAS PubMed.
- E. Maron, J. H. Swisher, J. J. Haven, T. Y. Meyer, T. Junkers and H. G. Börner, Angew. Chem., Int. Ed., 2019, 58, 10747–10751 CrossRef CAS.
- S.-n. Nishimura, N. Higashi and T. Koga, Polym. Chem., 2019, 10, 71–76 RSC.
- S.-n. Nishimura, N. Higashi and T. Koga, Chem. Commun., 2019, 55, 1498–1501 RSC.
- N. Higashi, A. Hirata, S.-n. Nishimura and T. Koga, Colloids Surf., B, 2017, 159, 39–46 CrossRef CAS PubMed.
- F. Sanda, T. Kurokawa and T. Endo, Polym. J. (Tokyo, Jpn.), 1999, 31, 353–358 CrossRef CAS.
- F. Sanda, M. Nakamura, T. Endo, T. Takata and H. Handa, Macromolecules, 1994, 27, 7928–7929 CrossRef CAS.
- N. Higashi, R. Sonoda and T. Koga, RSC Adv., 2015, 5, 67652–67657 RSC.
- T. Yamano, N. Higashi and T. Koga, Macromol. Rapid Commun., 2020, 41, 1900550 CrossRef CAS PubMed.
- F. Sanda, M. Nakamura and T. Endo, Macromolecules, 1996, 29, 8064–8068 CrossRef CAS.
- H. Mori, S. Nakano and T. Endo, Macromolecules, 2005, 38, 8192–8201 CrossRef CAS.
- C. Ergene, K. Yasuhara and E. F. Palermo, Polym. Chem., 2018, 9, 2407–2427 RSC.
- J. L. Grace, A. G. Elliott, J. X. Huang, E. K. Schneider, N. P. Truong, M. A. Cooper, J. Li, T. P. Davis, J. F. Quinn, T. Velkov and M. R. Whittaker, J. Mater. Chem. B, 2017, 5, 531–536 RSC.
- J. L. Grace, J. X. Huang, S. E. Cheah, N. P. Truong, M. A. Cooper, J. Li, T. P. Davis, J. F. Quinn, T. Velkov and M. R. Whittaker, RSC Adv., 2016, 6, 15469–15477 RSC.
- K. Kuroda, G. A. Caputo and W. F. DeGrado, Chem. – Eur. J., 2009, 15, 1123–1133 CrossRef CAS PubMed.
- T. D. Michl, K. E. S. Locock, N. E. Stevens, J. D. Hayball, K. Vasilev, A. Postma, Y. Qu, A. Traven, M. Haeussler, L. Meagher and H. J. Griesser, Polym. Chem., 2014, 5, 5813–5822 RSC.
- A. M. Funhoff, C. F. van Nostrum, M. C. Lok, M. M. Fretz, D. J. A. Crommelin and W. E. Hennink, Bioconjugate Chem., 2004, 15, 1212–1220 CrossRef CAS PubMed.
- K. E. S. Locock, T. D. Michl, J. D. P. Valentin, K. Vasilev, J. D. Hayball, Y. Qu, A. Traven, H. J. Griesser, L. Meagher and M. Haeussler, Biomacromolecules, 2013, 14, 4021–4031 CrossRef CAS PubMed.
- L. Martin, R. Peltier, A. Kuroki, J. S. Town and S. Perrier, Biomacromolecules, 2018, 19, 3190–3200 CrossRef PubMed.
- K. E. S. Locock, T. D. Michl, N. Stevens, J. D. Hayball, K. Vasilev, A. Postma, H. J. Griesser, L. Meagher and M. Haeussler, ACS Macro Lett., 2014, 3, 319–323 CrossRef CAS.
- T.-K. Nguyen, S. J. Lam, K. K. K. Ho, N. Kumar, G. G. Qiao, S. Egan, C. Boyer and E. H. H. Wong, ACS Infect. Dis., 2017, 3, 237–248 CrossRef CAS PubMed.
- B. Belleau and J. Burba, J. Am. Chem. Soc., 1960, 82, 5751–5752 CrossRef CAS.
- E. F. Gale, Biochem. J., 1940, 34, 392 CrossRef CAS PubMed.
- E. F. Gale, Biochem. J., 1941, 35, 66 CrossRef CAS PubMed.
- D. H. Barton, Y. Hervé, P. Potier and J. Thierry, J. Chem. Soc., Chem. Commun., 1984, 1298–1299 RSC.
- J. Moenig, R. Chapman and K. D. Asmus, J. Phys. Chem., 1985, 89, 3139–3144 CrossRef CAS.
- L. K. Steffen, R. S. Glass, M. Sabahi, G. S. Wilson, C. Schoeneich, S. Mahling and K. D. Asmus, J. Am. Chem. Soc., 1991, 113, 2141–2145 CrossRef CAS.
- L. Claes, M. Janssen and D. E. De Vos, ChemCatChem, 2019, 11, 4297–4306 CrossRef CAS.
- R. L. Pilkington, M. A. Dallaston, G. P. Savage, C. M. Williams and A. Polyzos, React. Chem. Eng., 2021, 6, 486–493 RSC.
- L. Paltrinieri, E. Huerta, T. Puts, W. van Baak, A. B. Verver, E. J. R. Sudhölter and L. C. P. M. de Smet, Environ. Sci. Technol., 2019, 53, 2396–2404 CrossRef CAS PubMed.
- T. N. Tran, A. Nourry, G. Brotons and P. Pasetto, Prog. Org. Coat., 2019, 128, 196–209 CrossRef CAS.
- F. Mohammed, W. Ke, J. F. Mukerabigwi, A. A.-W. M. M. Japir, A. Ibrahim, Y. Wang, Z. Zha, N. Lu, M. Zhou and Z. Ge, ACS Appl. Mater. Interfaces, 2019, 11, 31681–31692 CrossRef CAS PubMed.
- J. M. Sarapas, C. M. Backlund, B. M. deRonde, L. M. Minter and G. N. Tew, Chem. – Eur. J., 2017, 23, 6858–6863 CrossRef CAS PubMed.
- A. Malfanti, F. Mastrotto, Y. Han, P. Král, A. Balasso, A. Scomparin, S. Pozzi, R. Satchi-Fainaro, S. Salmaso and P. Caliceti, Mol. Pharm., 2019, 16, 1678–1693 CrossRef CAS PubMed.
- Q. Zhao, Q. Liu, C. Li, L. Cao, L. Ma, X. Wang and Y. Cai, Chem. Commun., 2020, 56, 4954–4957 RSC.
- Z. Tan, Y. K. Dhande and T. M. Reineke, Bioconjugate Chem., 2017, 28, 2985–2997 CrossRef CAS PubMed.
- F. Wang, W. Sun, L. Li, L. Li, Y. Liu, Z.-r. Zhang and Y. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 27563–27574 CrossRef CAS PubMed.
- J. Li, S. Xiao, Y. Xu, S. Zuo, Z. Zha, W. Ke, C. He and Z. Ge, ACS Appl. Mater. Interfaces, 2017, 9, 17727–17735 CrossRef CAS PubMed.
- B. Chou, P. Mirau, T. Jiang, S.-W. Wang and K. J. Shea, Biomacromolecules, 2016, 17, 1860–1868 CrossRef CAS PubMed.
- A. Kuroki, A. Kengmo Tchoupa, M. Hartlieb, R. Peltier, K. E. S. Locock, M. Unnikrishnan and S. Perrier, Biomaterials, 2019, 217, 119249 CrossRef CAS PubMed.
- J. Salaklang, E. Mertens, V. Maes, R. Dams, W. Dermaut and T. Junkers, J. Flow Chem., 2020, 10, 673–679 CrossRef.
- J. Salaklang, V. Maes, M. Conradi, R. Dams and T. Junkers, React. Chem. Eng., 2018, 3, 41–47 RSC.
- M. Movsisyan, T. S. A. Heugebaert, R. Dams and C. V. Stevens, ChemSusChem, 2016, 9, 1945–1952 CrossRef CAS PubMed.
- R. W. Lewis, R. A. Evans, N. Malic, K. Saito and N. R. Cameron, Polym. Chem., 2018, 9, 60–68 RSC.
- G. Gody, R. Barbey, M. Danial and S. Perrier, Polym. Chem., 2015, 6, 1502–1511 RSC.
- G. R. Jones, Z. Li, A. Anastasaki, D. J. Lloyd, P. Wilson, Q. Zhang and D. M. Haddleton, Macromolecules, 2016, 49, 483–489 CrossRef CAS.
- J. Tanaka, P. Gurnani, A. B. Cook, S. Häkkinen, J. Zhang, J. Yang, A. Kerr, D. M. Haddleton, S. Perrier and P. Wilson, Polym. Chem., 2019, 10, 1186–1191 RSC.
- B. De Sterck, R. Vaneerdeweg, F. Du Prez, M. Waroquier and V. Van Speybroeck, Macromolecules, 2010, 43, 827–836 CrossRef CAS.
- A. Valdebenito and M. V. Encinas, Polym. Int., 2010, 59, 1246–1251 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- F. S. Gibson, S. C. Bergmeier and H. Rapoport, J. Org. Chem., 1994, 59, 3216–3218 CrossRef CAS.
- I. W. Ashworth, B. G. Cox and B. Meyrick, J. Org. Chem., 2010, 75, 8117–8125 CrossRef CAS PubMed.
- J. Wang, Y.-L. Liang and J. Qu, Chem. Commun., 2009, 5144–5146, 10.1039/B910239F.
- G. Moad, Macromol. Chem. Phys., 2014, 215, 9–26 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00735a |
| This journal is © The Royal Society of Chemistry 2021 |