
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical synthesis of (hetero)aryl Au(I) complexes†
Fredric J. L.
Ingner
 a,
Zachary X.
Giustra
a,
Sebastian
Novosedlik
a,
Andreas
Orthaber
b,
Paul J.
Gates
c,
Christine
Dyrager
a and
Lukasz T.
Pilarski
*a
a,
Zachary X.
Giustra
a,
Sebastian
Novosedlik
a,
Andreas
Orthaber
b,
Paul J.
Gates
c,
Christine
Dyrager
a and
Lukasz T.
Pilarski
*a
aDepartment of Chemistry—BMC, Uppsala University, BOX 576, 75-123, Uppsala, Sweden. E-mail: lukasz.pilarski@kemi.uu.se
bDepartment of Chemistry—Ångström, Uppsala University, BOX 523, 75-120, Uppsala, Sweden
cSchool of Chemistry, University of Bristol, Cantock's Close, Clifton, Bristol BS8 1TS, UK
First published on 11th August 2020
Abstract
Growing demand for sustainable chemical syntheses casts mechanochemistry in a new light as an environmentally benign alternative to traditional solvent-based methods. Given recent interest in Au(I) complexes for catalytic, materials, and medicinal applications, we developed a mechanochemical protocol to prepare (hetero)aryl Au(I) complexes under green conditions. The procedure reported here uses C–H or C–B activation to afford the corresponding Au(I) complexes in high yields. Our approach bypasses external heating, long reaction times and the use of toxic solvents. We demonstrate that mechanochemical C–H auration can be used on highly functionalised bioactive substrates. Mechanistic aspects of the C–H auration are discussed.
Introduction
Organogold compounds have demonstrated usefulness in a diverse range of research areas including synthetic,1 physical,2 and medicinal chemistry.3 An important subset of this class of compounds comprises neutral (hetero)aryl-ligated Au(I) complexes, which have attracted interest as mechanistic probes in organometallic catalysis,4 as luminescent5 and often mechanochromic6 materials for optical devices and sensors, and as biologically active molecules for anticancer and antibacterial treatments.7Despite the considerable level of attention garnered by these complexes, their preparation traditionally relies on the addition of reactive, unstable, or otherwise inconvenient-to-handle aryl organometallic reagents (e.g., Grignards, lithiates, zincates) to a Au(I) source,1i,n,5c,6a,8 or transmetalation from toxic arylstannanes.5e,9 More recent reports describe the synthesis of aryl Au(I) complexes via transmetalation from various organoboron reagents5b,f,5g,7a,10 and organosilanes11 or by direct C(sp2)–H activation.12 While a handful of these methods use so-called green solvents13 (e.g., H2O, EtOH),10e,f they still suffer from the need for extended reaction times or elevated temperatures. More generally, the problem of solvent waste remains an ongoing challenge for the greening of chemical synthesis, including that of organometallic species.14
We sought to address these issues by developing a method to synthesize (hetero)aryl Au(I) complexes using a mechanochemical ball milling approach.15 Ball milling uses the physical impact of ball bearings against the interior walls of reaction vessels to provide reagent mixing and activation energy. The generation of organometallic compounds under milling conditions has been reported previously for a number of transition metals.16 With respect to gold specifically, however, only a very small number of complexes, virtually all of them NHC-ligated, have hitherto been prepared mechanochemically,16q,17 the majority by manual grinding with a mortar and pestle.17a,b The reproducibility of mortar-and-pestle techniques is conspicuously problematic due, in part, to variability in energy inputs delivered by different operators.16a
Herein, we develop a standardized approach to the synthesis of a broad range of (hetero)aryl Au(I) complexes, via either transmetalation from organoboronic acids or by C–H auration (Fig. 1). In particular, our use of an automated mixer mill allows for the precise application of consistent mechanochemical force. We note that our method is applicable to the chemoselective auration of highly functionalized bioactive substrates. We also demonstrate direct C(sp2)–H auration to be reversible under milling conditions and investigate the implications this has for achieving chemoselectivity between different aryl moieties.
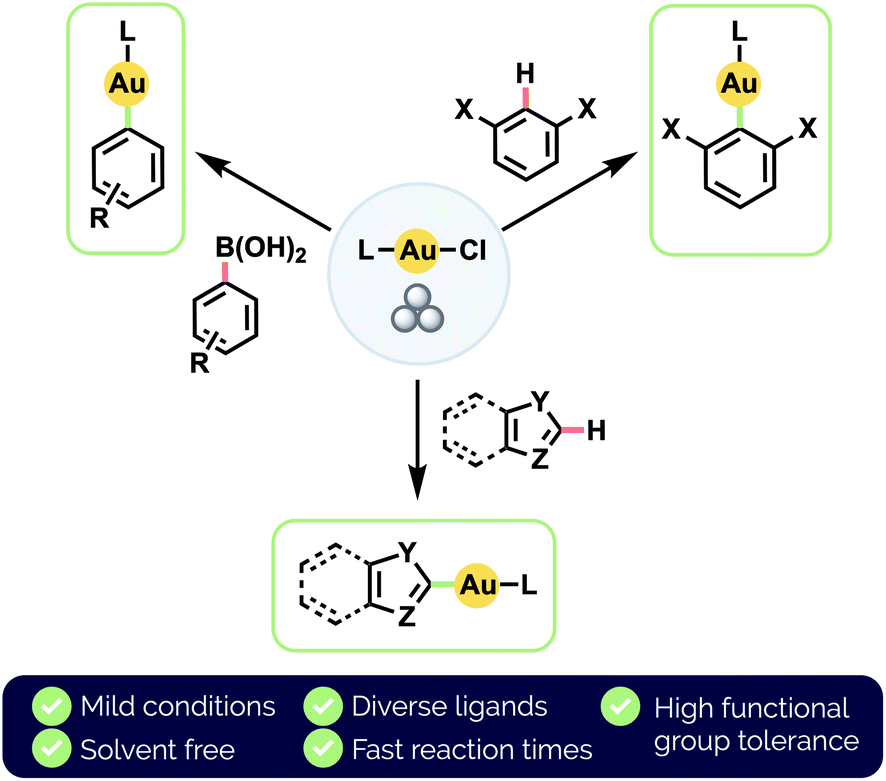 | ||
| Fig. 1 This work: mechanochemical synthesis of diverse (hetero)aryl Au(I) complexes by ball milling. | ||
Results and discussion
We initially investigated the possibility of using boronic acids as aryl sources, given that such compounds have been demonstrated to be competent reagents in several solvent-based methods.5g,10a–f All reactions were conducted in a Teflon™ vessel loaded with one stainless steel ball bearing. Using 3-tolylboronic acid (1a) with (Ph3P)AuCl as a model Au(I) precursor, we observed clean formation of the desired product, 2a, by 31P NMR spectroscopy after milling for 1 h at 30 Hz in the presence of either 1.0 equiv. KOH or CsF (Table 1, entries 1 and 2).18 No product was observed without inclusion of a base (entry 3). Accordingly, the addition of a small volume of water to enable liquid-assisted grinding (LAG)19 was only effective when combined with a base additive (compare entries 4 and 5). In particular, product formation became effectively quantitative with a modest excess of KOH (entry 6). Substituting the separate boronic acid/KOH pairing for a pre-formed trialkoxyborate led to diminished yield (entry 7).|
|
||
|---|---|---|
| Entry | Additives | Yield a (%) |
| a Determined by 31P NMR spectroscopy for reactions conducted on 0.1 mmol scale in a 14 mL Teflon™ vessel with one stainless steel ball (15 mm, 13.6 g). b Using potassium 2-hydroxymethyl-2-methylpropane-1,3-diol borate in place of boronic acid. | ||
| 1 | KOH (1.0 equiv.) | 75 |
| 2 | CsF (1.0 equiv.) | 52 |
| 3 | None | 0 |
| 4 | H2O (0.1 mL) | 0 |
| 5 | KOH (1.0 equiv.) + H2O (0.1 mL) | 94 |
| 6 | KOH (1.2 equiv.) + H2O (0.1 mL) | >99 |
| 7b | H2O (0.1 mL) | 84 |
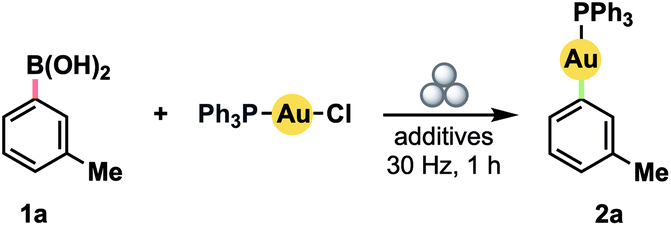
Having established a set of optimised conditions, we screened boronic acids to generate a variety of aryl Au(I) phosphine complexes (Scheme 1). Significantly, most of the product complexes could be isolated from the reaction mixture by extraction with EtOAc and a Celite filtration, which speaks to the overall ease of their synthesis using our procedure. The reaction did not appear significantly sensitive to the steric demands of ortho-substituted aryl groups; these transmetalated efficiently even when paired with a bulky20 tricyclohexyl phosphine ligand (2c). In general, transmetalation of either electron-rich or electron-neutral aryl groups proceeded in excellent to quantitative yield (2a–h). Greater substituent dependence was observed, however, for electron-poor arylboronic acids: while 4-(trifluoromethyl)phenyl complex 2i was obtained in very good isolated yield (84%), 3-nitrophenyl complex 2j was observed to form in only 23% spectroscopic yield. A similar trend was found for heteroarene substrates: pleasingly, electron-rich moieties such as thiophene (2k) and 9-phenyl-9H-carbazole (2l) – both motifs of significant interest in organic materials applications21 – transmetalated smoothly. In contrast, electron-poor 3-pyridineboronic acid afforded only trace amounts of product (2m). Lastly, although p-vinyl complex 2h was obtained in very good yield (88%), the generation of an α-aurated styrenyl complex22 (2n) proved less efficient.
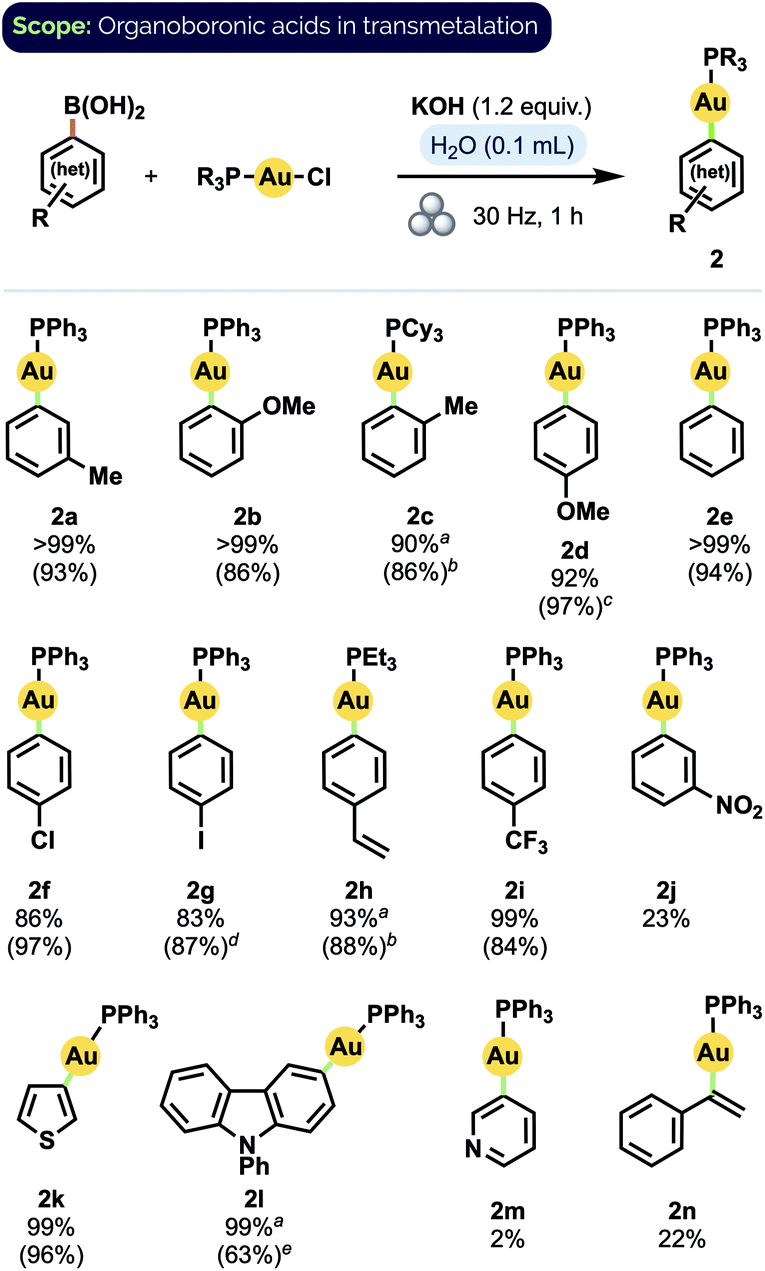 | ||
Scheme 1 Substrate scope for aryl boronic acid transmetalation to Au(I) phosphines. Yields determined by 31P NMR spectroscopy; values in parentheses are isolated yields using 1.2 equiv. Ar-B(OH)2 and 1.5 equiv. KOH(aq) (1.25 M) on 0.1 mmol scale. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2.0 equiv. KOH(s)b3.0 equiv. KOH(aq) (2.50 M), 25 Hz c1.4 equiv. Ar-B(OH)2d2.5 equiv. KOH(aq) (2.50 M), 2 h, 25 Hz e3.0 equiv. KOH(aq) (2.50 M), 36 Hz. 2.0 equiv. KOH(s)b3.0 equiv. KOH(aq) (2.50 M), 25 Hz c1.4 equiv. Ar-B(OH)2d2.5 equiv. KOH(aq) (2.50 M), 2 h, 25 Hz e3.0 equiv. KOH(aq) (2.50 M), 36 Hz. | ||
Our success with mechanically induced transmetalations from (hetero)arylboronic acids prompted us to attempt the more challenging23 synthesis of (hetero)aryl Au(I) complexes directly via arene C–H activation. Specifically, we aimed to develop a general mechanochemical protocol for the generation of Au(I) complexes of significantly greater scope than the handful of polyfluoroarene derivatives generated manually so far.17b With 1,3-dichlorobenzene as a model substrate, we determined that increasing the equivalents of arene relative to (Et3P)AuCl led to steadily higher yields (Table 2, entries 1–4). Even at 4.0 equiv. of arene, however, product yield did not exceed 50%. In contrast to the boronic acids case, the addition of water to facilitate LAG proved detrimental (entry 5). Accordingly, by substituting the hydroxide base with KOtBu – and thus establishing anhydrous conditions – the yield improved dramatically (entry 6).
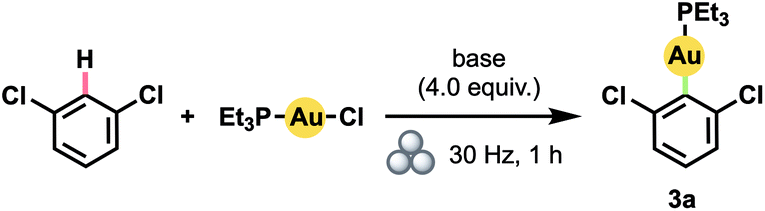
Using these optimized conditions, we were able to prepare a series of polyhaloaryl Au(I) phosphine complexes in very good to excellent yields (Scheme 2). The general effectiveness of our mechanochemical approach notably extended in this instance to the synthesis of pyridyl complexes both with (3c) and without (3f) steric hindrance about the nitrogen center. The required di-ortho-halide substitution pattern of the aryl group likewise did not appear to encumber the reaction and various combinations of F, Cl, and Br substituents were all well tolerated. Products also formed readily with bulky PtBu3 ligands. The formation of complex 3d, for which solution-phase syntheses had previously proven unsuccessful,12d proceeded in 91% isolated yield under our milling conditions. Moreover, the preparation of pentafluorophenyl complex 3e could be scaled up by a factor of 5 without significant decrease in yield. The structure of 3f was confirmed by X-ray crystallographic analysis.24
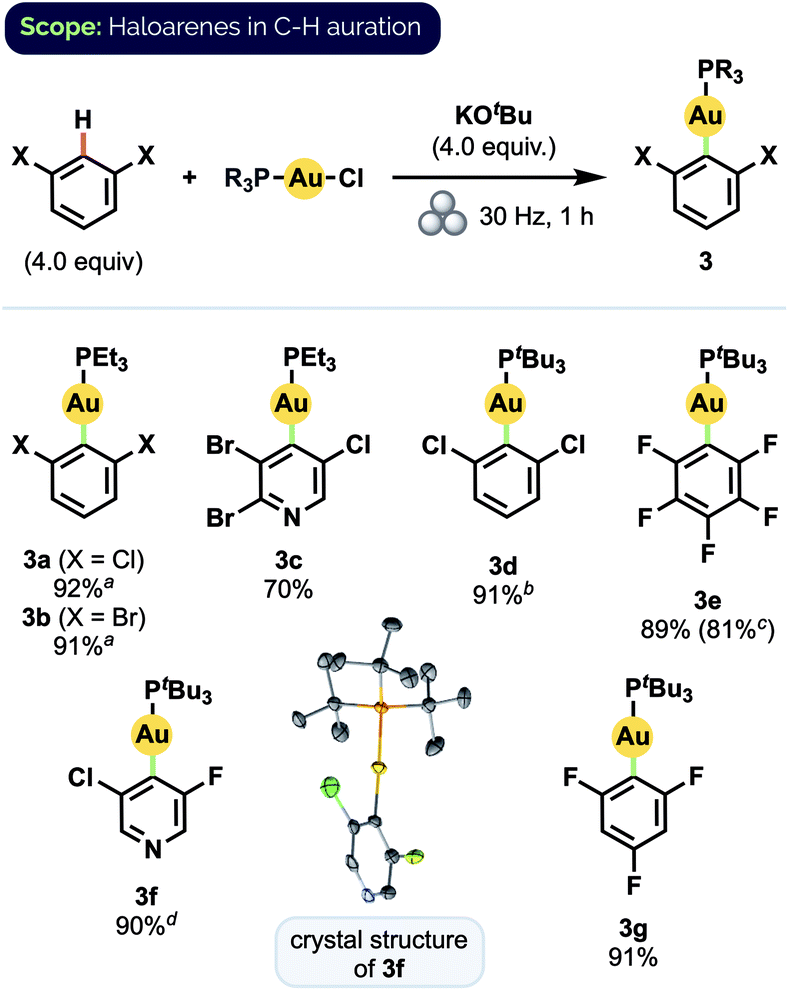 | ||
| Scheme 2 Substrate scope for haloarene C–H auration. Isolated yields on 0.1 mmol scale. a1.5 h b2 h, 36 Hz c0.5 mmol scale d4.0 equiv. KOH. For 3f the crystal structure has ellipsoids set at 50% probability. | ||
By a simple switch of base from KOtBu to KOH, we could readily expand our C–H auration protocol to include a series of heteroarenes (Scheme 3).25 Notably, benzothiazole complex 4a was obtained in 85% isolated yield after milling for 1 h, compared to 55% yield obtained after stirring in 1,4-dioxane at 35 °C for 15 h.12d (Temperature measurements of the interior of our reaction vessels and the balls using a laser thermometer immediately after the cessation of milling never exceeded 32 °C, even after milling for 60 min at 30 Hz.) Despite bearing a comparatively more acidic proton at C2,26 benzoxazole (pKa(DMSO) = 24.8) exhibited lower reactivity than benzothiazole (pKa(DMSO) = 27.3), suggesting that C–H bond acidity may not be the sole determinant of the relative ease of auration in these cases. Specifically, we propose that initial S⋯Au interactions in the case of benzothiazole may pre-organise the reactants to better facilitate subsequent C–H auration to produce 4a. Quantitative formation of benzoxazole complex 4b could nevertheless be achieved by simply increasing the oscillation frequency of the mill to 36 Hz. High yield, and complete regioselectivity for C2 functionalization, were likewise observed for unsubstituted thiophene (pKa(DMSO) = 33.5), in stark contrast to the reported difficulty of forming complex 4c in solution.12d 2-Bromo-substituted thiophene also underwent C–H auration to afford complex 4d in high isolated yield.
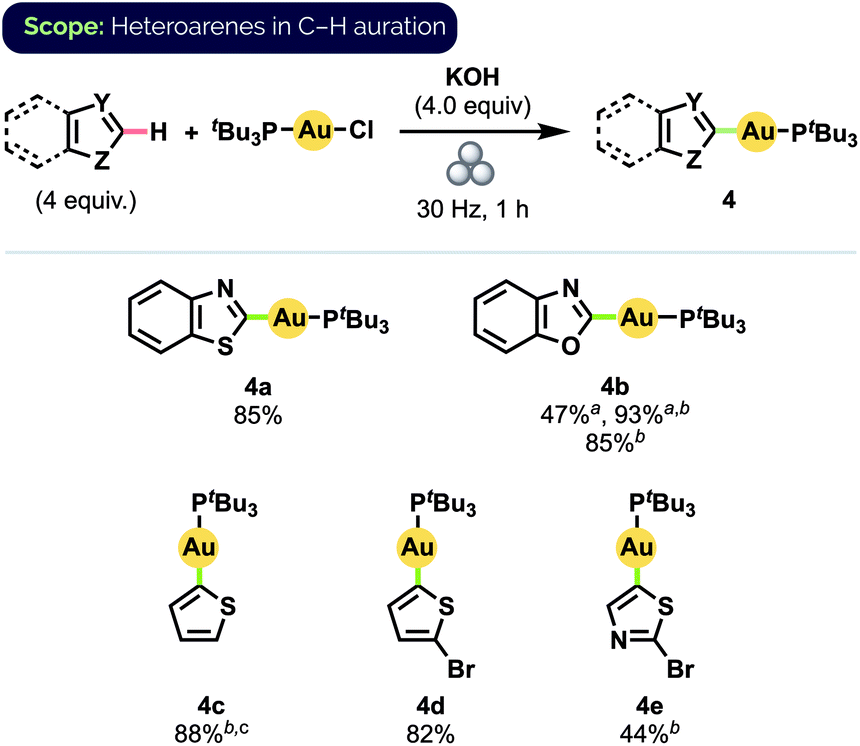 | ||
| Scheme 3 Substrate scope for heteroarene C–H auration. Isolated yields on 0.1 mmol scale. aYield determined by 31P NMR spectroscopy b36 Hz c4.0 equiv. KOtBu. | ||
Gratifyingly, our optimized mechanochemical conditions for preparing Au(I) phosphine complexes translated well to the complementary synthesis of NHC-ligated compounds (Scheme 4). Of particular note was that 2,4,6-trifluorophenyl complex 5a was obtained in 82% isolated yield, whereas earlier attempts with mortar-and-pestle grinding produced only trace amounts.17b Additionally, complex 5b, isolated in 93% yield, has recently been demonstrated by Nolan and co-workers to serve as a versatile precursor to many other NHC-Au(I) species.10f
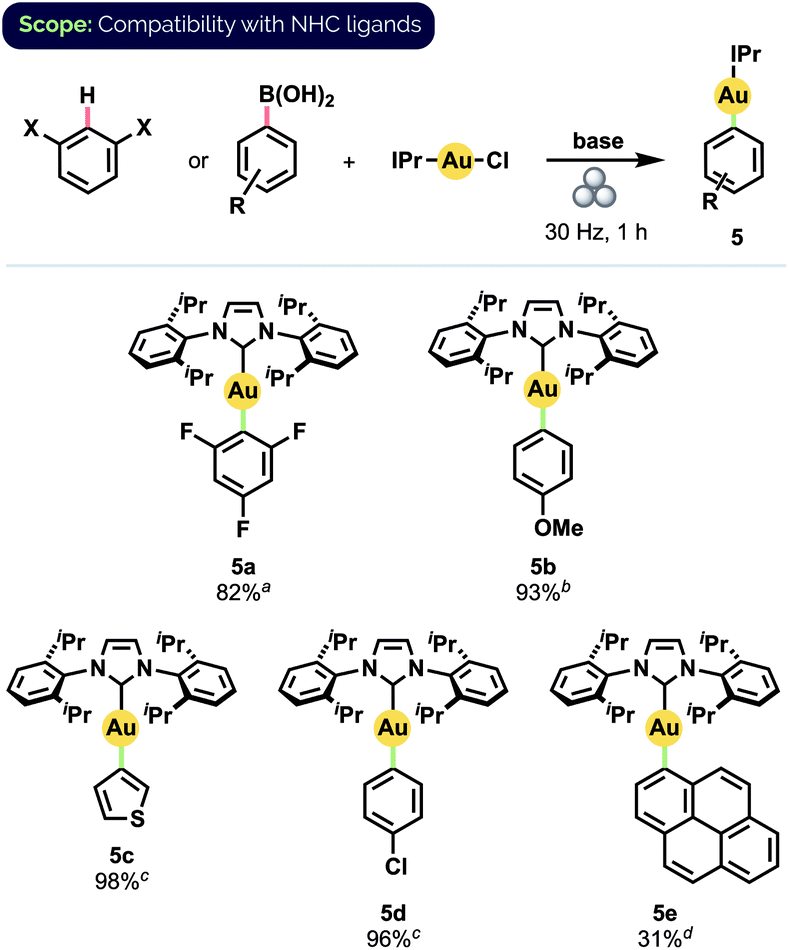 | ||
| Scheme 4 Synthesis of NHC-ligated complexes. Isolated yields on 0.1 mmol scale. a4.0 equiv. arene, 4.0 equiv. KOtBu b1.4 equiv. Ar-B(OH)2, 1.5 equiv. KOH(aq) (1.25 M) c1.5 equiv. KOH(aq) (1.25 M) d1.0 equiv. Ar-B(OH)2, 2.0 equiv. KOH(s). | ||
To further demonstrate the general applicability of our milling conditions to the preparation of aryl Au(I) complexes, we attempted the C–H auration of highly functionalized, biologically active substrates (Fig. 2). Such late-stage derivatisation under mechanochemical conditions has not been demonstrated previously. The C2–H auration of caffeine proceeded in 67% yield after milling for 1 h at 30 Hz (cf. 76% after 65 h stirring in 1,4-dioxane12d). Interestingly, Diflufenican, a commercialized herbicide,27 was selectively aurated on the difluorophenyl group. This is complementary to analogous metalations with palladium and cobalt directed by the pyridyl moiety to the trifluoromethylated phenyl ring instead.28 Functionalization of the commercial antifungal agent Econazole29 also occurred exclusively on the 1,3-dichlorophenyl ring, despite the presence of a pendant imidazole group.
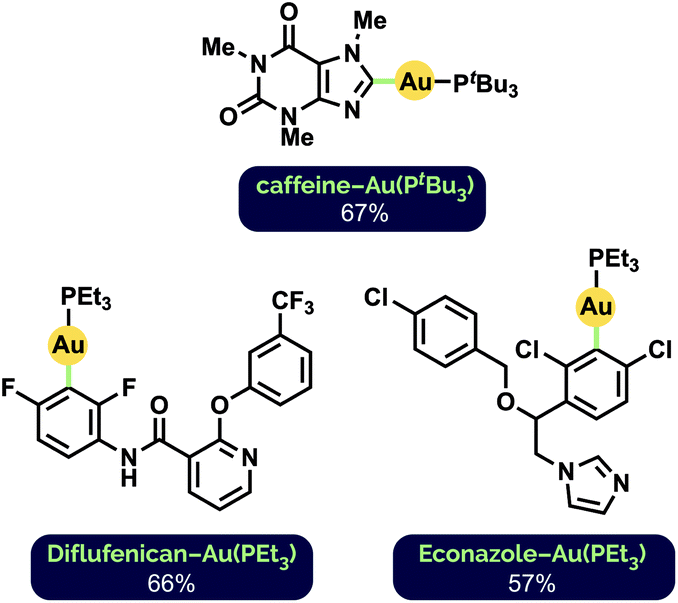 | ||
| Fig. 2 Synthesized Au(I) phosphine complexes of biologically active compounds. See ESI† for reaction details. | ||
Given the prevalence of biologically active compounds bearing both halo- and heteroaryl motifs (vide supra), we were interested to investigate the degree to which our mechanochemical C–H activation conditions would afford chemoselectivity between auration of these two groups. To that end, a series of competition experiments was conducted using equimolar amounts of haloarene and heteroarene substrates with (tBu3P)AuCl and excess KOtBu.30 As shown in Scheme 5A, combining pentafluorobenzene (C6F5H) and benzothiazole resulted in the formation of complexes 3e and 4a in an 11:1 ratio under conditions analogous to those for their separate synthesis. Lower ratios were observed with C6F5H and benzoxazole (Scheme 5B, 3e/4b = 4:1) and trifluorobenzene and benzothiazole (Scheme 5C, 3g/4a = 1:1). In all cases, analysis by 31P NMR spectroscopy indicated complete consumption of the (tBu3P)AuCl starting material after 1 h. Effectively the same product ratios were also obtained in each case upon milling for a longer period (2 h).
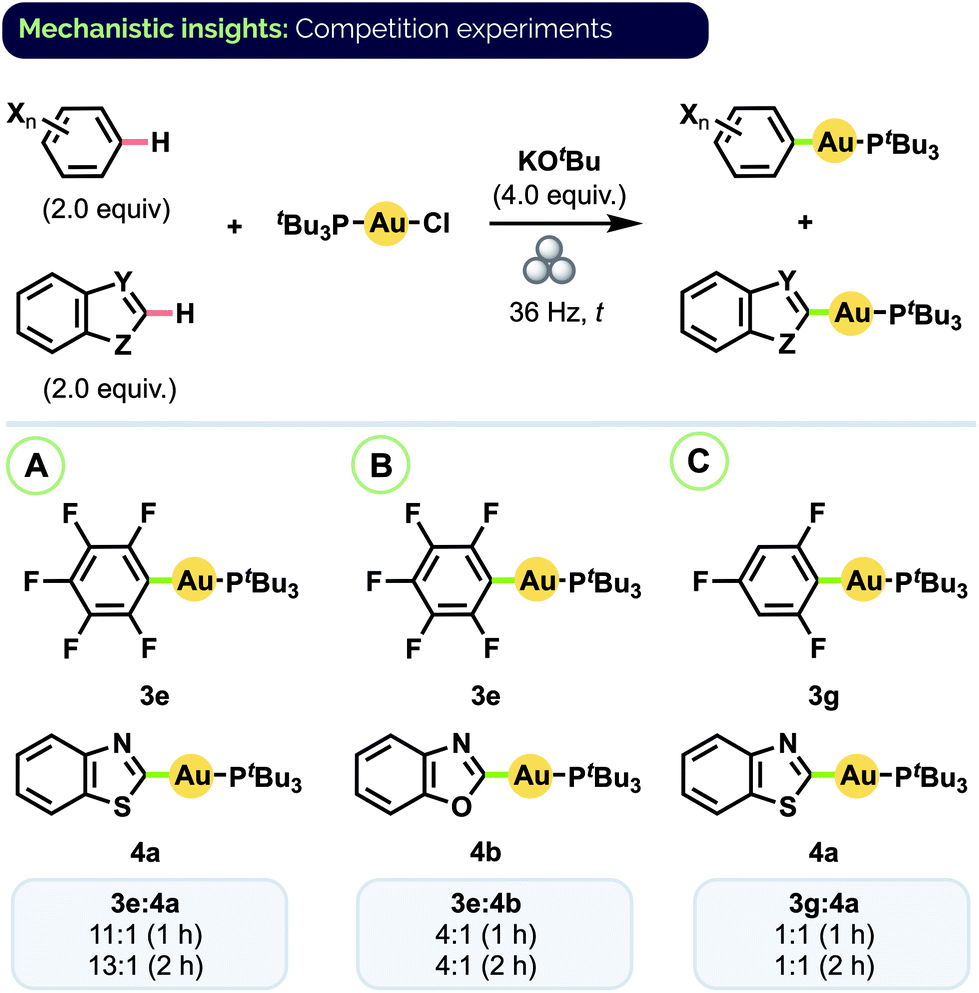 | ||
| Scheme 5 Haloarene/heteroarene C–H auration competition experiments. Product ratios determined by 31P NMR spectroscopy. | ||
To determine if the origin of the product ratios was thermodynamic or kinetic in nature, we carried out additional experiments in which a pre-formed complex was subjected to milling in the presence of another arene substrate, with or without base present (Scheme 6). (An equivalent of free arene corresponding to the aryl group of the complex was also added so that the total number of equivalents of each aryl group present would be the same as in the prior competition experiments.) When complex 3e was milled together with benzothiazole and excess KOtBu, only partial conversion to benzothiazole complex 4a was observed and the ratio of product complexes was 3e/4a = 13:1 (Scheme 6A).31 Starting instead from pure 4a and free C6F5H also favoured 3e (3e/4a = 5:1; Scheme 6B). These results indicate the C–H auration is reversible and that the product ratios from the competition experiments reflect an overall equilibrium between different aryl Au(I) complexes. DFT (PBE0/TZVP/SDD) calculations also point to the major species in the mixtures from the competition experiments as being the more thermodynamically stable complex (see ESI† for details).
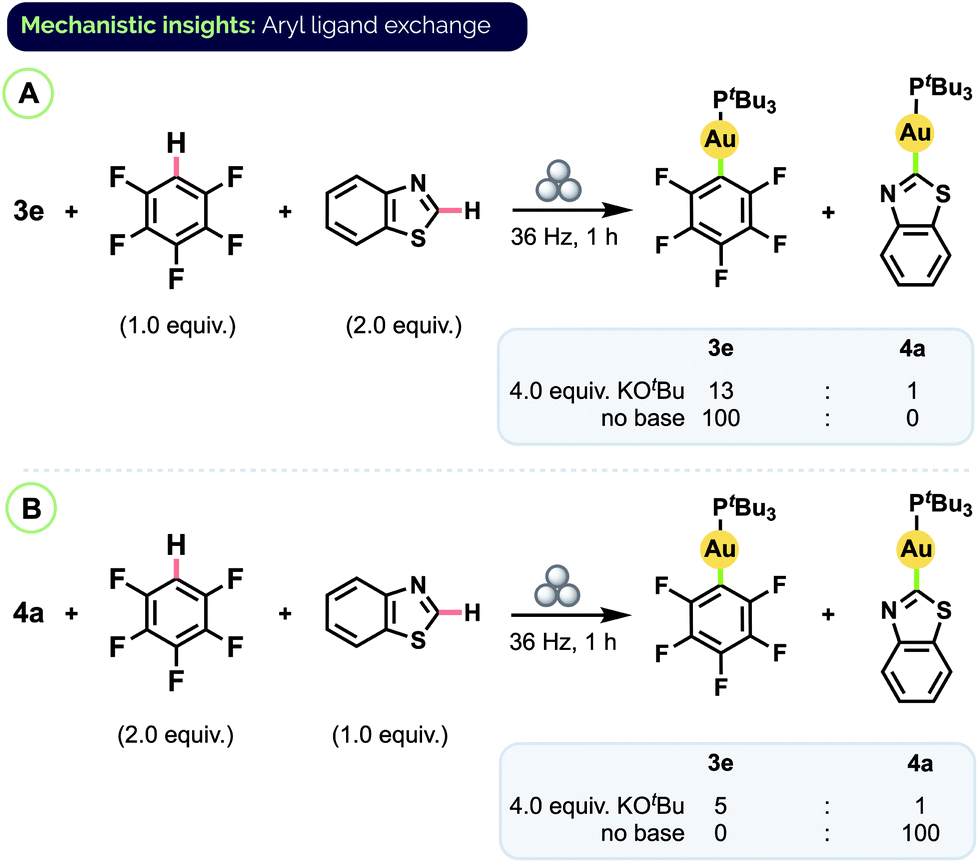 | ||
| Scheme 6 Aryl group exchange experiments. Final complex ratios determined by combined 31P and 19F NMR analysis, see ESI† for details. | ||
The actual mechanism of the observed aryl exchange remains to be elucidated fully. The complete lack of reactivity of 3e and 4a with benzothiazole and C6F5H, respectively, in the absence of KOtBu rules out the possibility of a direct metathesis-type process. Larrosa and co-workers have proposed that aryl C–H auration is mediated in solution by a gold alkoxide species when KOtBu is used as base.12d Indeed, upon milling (tBu3P)AuCl together with KOtBu, we observed complete conversion to a single species that we assigned as (tBu3P)AuOtBu (6) on the basis of 1H, 13C, 31P, DOSY NMR spectroscopy and HRMS analysis (Scheme 7).32 Significantly, this material demonstrated competency equal to that of (tBu3P)AuCl in forming 3e and 4a under standard milling conditions. If formed in catalytic amounts in situ, 6 could thus also potentially facilitate the observed aryl exchange between these complexes. At this time, however, we cannot discount the possibility of an exchange proceeding directly through the reaction with a deprotonated aryl species.
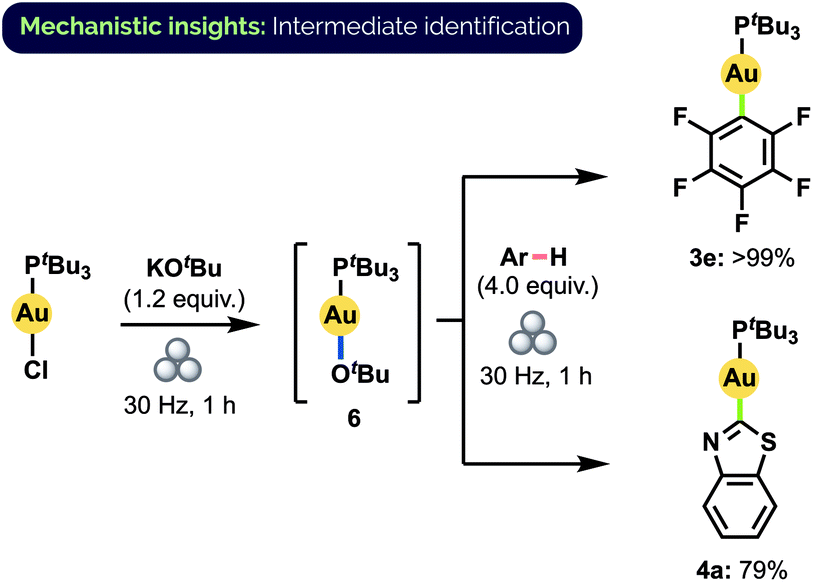 | ||
| Scheme 7 Synthesis of Au(I) alkoxide complex 6 and subsequent aryl C–H activation. Yields determined by 31P NMR spectroscopy. | ||
Conclusions
We have developed a green protocol for the preparation of (hetero)aryl Au(I) complexes by leveraging the practical advantages of automated mechanochemical synthesis. Specifically, the method reported herein generates products with greater overall expediency than previous syntheses while avoiding the need for prolonged heating or environmentally hazardous solvents. Under our mechanochemical regime, Au(I) complexes can be formed by either transmetalation from readily available aryl boronic acids or direct C–H activation of various halo- and heteroarenes. The method allows the synthesis of several previously challenging compounds and demonstrates, for the first time, the viability of a mechanochemical approach to the late-stage C–H auration of biologically active substrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Swedish Research Council (Vetenskapsrådet) and the Carl Trygger Foundation for financial support. We also thank Dr. Johanna Larsson and Prof. Graham Budd for help with proof-reading the manuscript.Notes and references
- For reviews of various facets of modern organogold homogeneous catalysis, see: (a) A. Nijamudheen and A. Datta, Chem. – Eur. J., 2020, 26, 1442–1487 CrossRef CAS PubMed; (b) X. Zhao, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2019, 55, 12127–12135 RSC; (c) V. Pirovano, Eur. J. Org. Chem., 2018, 2018, 1925–1945 CrossRef CAS; (d) S. Kramer, Chem. – Eur. J., 2016, 22, 15584–15598 CrossRef CAS PubMed; (e) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed; (f) W. Zi and F. D. Toste, Chem. Soc. Rev., 2016, 45, 4567–4589 RSC; (g) B. Ranieri, I. Escofet and A. M. Echavarren, Org. Biomol. Chem., 2015, 13, 7103–7118 RSC; (h) M. Bandini, Chem. Soc. Rev., 2011, 40, 1358–1367 RSC. For examples of organometallic gold complexes used as stoichiometric reagents, see: (i) M. Contel, M. Stol, M. A. Casado, G. P. M. van Klink, D. D. Ellis, A. L. Spek and G. van Koten, Organometallics, 2002, 21, 4556–4559 CrossRef CAS; (j) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Rudolph, T. D. Ramamurthi and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8243–8246 CrossRef CAS PubMed; (k) S. Yili, S. D. Ramgren and S. A. Blum, Organometallics, 2009, 28, 1275–1277 CrossRef; (l) M. Peña-López, M. Ayán-Varela, L. A. Sarandeses and J. P. Sestelo, Chem. – Eur. J., 2010, 16, 9905–9909 CrossRef PubMed; (m) J. J. Hirner and S. A. Blum, Organometallics, 2011, 30, 1299–1302 CrossRef CAS; (n) M. Hejda, L. Dostál, R. Jambor, A. Růžička, R. Jirásko and J. Holeček, Eur. J. Inorg. Chem., 2012, 2012, 2578–2587 CrossRef CAS; (o) M. Peña-Lõpez, L. A. Sarandeses and J. P. Sestelo, Eur. J. Org. Chem., 2013, 2013, 2545–2554 CrossRef.
- (a) M. Enomoto, A. Kishimura and T. Aida, J. Am. Chem. Soc., 2001, 123, 5608–5609 CrossRef CAS PubMed; (b) A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179–183 CrossRef CAS PubMed; (c) M.-C. Tang, D. P.-K. Tsang, M. M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 446–449 CrossRef CAS PubMed; (d) W.-X. Ni, M. Li, J. Zheng, S.-Z. Zhan, Y.-M. Qiu, S. W. Ng and D. Li, Angew. Chem., Int. Ed., 2013, 52, 13472–13476 CrossRef CAS PubMed; (e) M.-C. Tang, D. P.-K. Tsang, Y.-C. Wong, M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 17861–17868 CrossRef CAS PubMed; (f) S. T. Madrahimov, T. A. Atesin, O. Karagiaridi, A. A. Sarjeant, O. K. Farha, J. T. Hupp and S. T. Nguyen, Cryst. Growth Des., 2014, 14, 6320–6324 CrossRef CAS; (g) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed; (h) E. Y.-H. Hong, C.-T. Poon and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 6368–6371 CrossRef CAS PubMed; (i) N. B. Jayaratna, M. M. Olmstead, B. I. Kharisov and H. V. R. Dias, Inorg. Chem., 2016, 55, 8277–8280 CrossRef CAS PubMed; (j) J. Cored, O. Crespo, J. L. Serrano, A. Elduque and R. Giménez, Inorg. Chem., 2018, 57, 12632–12640 CrossRef CAS PubMed; (k) M. B. E. Griffiths, Z. S. Dubrawski, G. Bačić, A. Japahuge, J. D. Masuda, T. Zeng and S. T. Barry, Eur. J. Inorg. Chem., 2019, 2019, 4927–4938 CrossRef CAS; (l) L.-K. Li, M.-C. Tang, S.-L. Lai, M. Ng, W.-K. Kwok, M.-Y. Chan and V. W.-W. Yam, Nat. Photonics, 2019, 13, 185–191 CrossRef CAS.
- For general reviews, see: (a) M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462 RSC; (b) T. Zou, C. T. Lum, C.-N. Lok, J.-J. Zhang and C.-M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC; (c) B. Đ. Glišić and M. I. Djuran, Dalton Trans., 2014, 43, 5950–5969 RSC; (d) C. F. Shaw, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS PubMed. For selected recent studies: (e) J. Weaver, S. Gaillard, C. Toye, S. Macpherson, S. P. Nolan and A. Riches, Chem. – Eur. J., 2011, 17, 6620–6624 CrossRef CAS PubMed; (f) E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS PubMed; (g) H. Bjelosevic, I. A. Guzei, L. C. Spencer, T. Persson, F. H. Kriel, R. Hewer, M. J. Nell, J. Gut, C. E. J. van Rensburg, P. J. Rosenthal, J. Coates, J. Darkwa and S. K. C. Elmroth, J. Organomet. Chem., 2012, 720, 52–59 CrossRef CAS; (h) A. Gutiérrez, I. Marzo, C. Cativiela, A. Laguna and M. C. Gimeno, Chem. – Eur. J., 2015, 21, 11088–11095 CrossRef PubMed; (i) S. Vanicek, H. Kopacka, K. Wurst, S. Vergeiner, S. Kankowski, J. Schur, B. Bildstein and I. Ott, Dalton Trans., 2016, 45, 1345–1348 RSC; (j) A. Luengo, V. Fernández-Moreira, I. Marzo and M. C. Gimeno, Inorg. Chem., 2017, 56, 15159–15170 CrossRef CAS PubMed; (k) V. G. Reddy, T. S. Reddy, S. H. Privér, Y. Bai, S. Mishra, D. Wlodkowic, N. Mirzadeh and S. Bhargava, Inorg. Chem., 2019, 58, 5988–5999 CrossRef PubMed; (l) J. Oberkofler, B. Aikman, R. Bonsignore, A. Pöthig, J. Platts, A. Casini and F. E. Kühn, Eur. J. Inorg. Chem., 2020, 2020, 1040–1051 CrossRef CAS; (m) A. P. A. Oliveira, J. T. J. Freitas, R. Diniz, C. Pessoa, S. S. Maranhão, J. M. Ribeiro, E. M. Souza-Fagundes and H. Beraldo, ACS Omega, 2020, 5, 2939–2946 CrossRef CAS.
- (a) K. E. Roth and S. A. Blum, Organometallics, 2010, 29, 1712–1716 CrossRef CAS; (b) D. Weber and M. R. Gagné, Chem. Commun., 2011, 47, 5172–5174 RSC; (c) M. T. Johnson, J. M. J. van Rensburg, M. Axelsson, M. S. G. Ahlquist and O. F. Wendt, Chem. Sci., 2011, 2, 2373–2377 RSC; (d) K. E. Roth and S. A. Blum, Organometallics, 2011, 30, 4811–4813 CrossRef CAS; (e) M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 7777–7782 CrossRef CAS PubMed; (f) J. J. Hirner and S. A. Blum, Tetrahedron, 2015, 71, 4445–4449 CrossRef CAS PubMed; (g) S. Taschinski, R. Döpp, M. Ackermann, F. Rominger, F. de Vries, M. F. S. J. Menger, M. Rudolph, A. S. K. Hashmi and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2019, 58, 16988–16993 CrossRef CAS PubMed.
- (a) R. Bayón, S. Coco and P. Espinet, Chem. – Eur. J., 2005, 11, 1079–1085 CrossRef PubMed; (b) D. V. Partyka, A. J. Esswein, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2007, 26, 3279–3282 CrossRef CAS; (c) C. R. Wade, A. A. Yakovenko and F. P. Gabbaï, New J. Chem., 2010, 34, 1646–1651 RSC; (d) Y. Ogino, T. Kusamoto, Y. Hattori, M. Shimada, M. Tsuchiya, Y. Yamanoi, E. Nishibori, K. Sugimoto and H. Nishihara, Inorg. Chem., 2017, 56, 3909–3915 CrossRef CAS PubMed; (e) T. G. Do, E. Hupf, E. Lork, J. F. Kögel, F. Mohr, A. Brown, R. Toyoda, R. Sakamoto, H. Nishihara, S. Mebs and J. Beckmann, Eur. J. Inorg. Chem., 2019, 2019, 647–659 CrossRef CAS; (f) J. J. Mihaly, D. J. Stewart, T. A. Grusenmeyer, A. T. Phillips, J. E. Haley, M. Zeller and T. G. Gray, Dalton Trans., 2019, 48, 15917–15927 RSC; (g) T. Li, D. S. M. Ravinson, R. Haiges, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2020, 142, 6158–6172 CrossRef PubMed. For a recent review, see: (h) J. M. López-de-Luzuriaga, M. Monge and M. E. Olmos, Dalton Trans., 2017, 46, 2046–2067 RSC; (i) T. Seki, C. Feng, K. Kashiyama, S. Sakamoto, Y. Takasaki, T. Sasaki, S. Takamizawa and H. Ito, Angew. Chem., Int. Ed., 2020, 59, 8839–8843 CrossRef CAS PubMed.
- (a) T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 CrossRef CAS PubMed; (b) T. Seki, M. Jin and H. Ito, Inorg. Chem., 2016, 55, 12309–12320 CrossRef CAS PubMed; (c) K. Sakurada, T. Seki and H. Ito, CrystEngComm, 2016, 18, 7217–7220 RSC; (d) T. Seki, N. Tokodai, S. Omagari, T. Nakanishi, Y. Hasegawa, T. Iwasa, T. Taketsugu and H. Ito, J. Am. Chem. Soc., 2017, 139, 6514–6517 CrossRef CAS PubMed; (e) T. Seki, K. Kashiyama, S. Yagai and H. Ito, Chem. Lett., 2017, 46, 1415–1418 CrossRef CAS; (f) T. Seki, K. Kobayashi, T. Mashimo and H. Ito, Chem. Commun., 2018, 54, 11136–11139 RSC; (g) T. Seki, K. Ida, H. Sato, S. Aono, S. Sakaki and H. Ito, Chem. – Eur. J., 2020, 26, 735–744 CrossRef CAS PubMed.
- (a) S. Craig, L. Gao, I. Lee, T. Gray and A. J. Berdis, J. Med. Chem., 2012, 55, 2437–2451 CrossRef CAS PubMed; (b) M. Frik, J. Jiménez, I. Gracia, L. R. Falvello, S. Abi-Habib, K. Suriel, T. R. Muth and M. Contel, Chem. – Eur. J., 2012, 18, 3659–3674 CrossRef CAS PubMed; (c) S. K. Verma, S. N. Ansari, P. Kumari and S. M. Mobin, Organometallics, 2019, 38, 2591–2596 CrossRef CAS.
- C. Croix, A. Balland-Longeau, H. Allouchi, M. Giorgi, A. Duchêne and J. Thibonnet, J. Organomet. Chem., 2005, 690, 4835–4843 CrossRef CAS.
- (a) N. Meyer, C. W. Lehmann, T. K.-M. Lee, J. Rust, V. W.-W. Yam and F. Mohr, Organometallics, 2009, 28, 2931–2934 CrossRef CAS; (b) N. Meyer, S. Sivanathan and F. Mohr, J. Organomet. Chem., 2011, 696, 1244–1247 CrossRef CAS.
- (a) D. V. Partyka, M. Zeller, A. D. Hunter and T. G. Gray, Angew. Chem., Int. Ed., 2006, 45, 8188–8191 CrossRef CAS PubMed; (b) D. V. Partyka, J. B. Updegraff III, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2009, 28, 1666–1674 CrossRef CAS; (c) S. Dupuy, L. Crawford, M. Bühl, A. M. Z. Slawin and S. P. Nolan, Adv. Synth. Catal., 2012, 354, 2380–2386 CrossRef CAS; (d) H. K. Lenker, T. G. Gray and R. A. Stockland Jr., Dalton Trans., 2012, 41, 13274–13276 RSC; (e) F. J. L. Ingner, A. C. Schmitt, A. Orthaber, P. J. Gates and L. T. Pilarski, ChemSusChem, 2020, 13, 2032–2037 CrossRef CAS PubMed; (f) N. V. Tzouras, M. Saab, W. Janssens, T. Cauwenbergh, K. Van Hecke, F. Nahra and S. P. Nolan, Chem. – Eur. J., 2020, 26, 5541–5551 CrossRef CAS PubMed.
- S. Dupuy, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2012, 18, 14923–14928 CrossRef CAS PubMed.
- (a) P. Lu, T. C. Boorman, A. M. Z. Slawin and I. Larrosa, J. Am. Chem. Soc., 2010, 132, 5580–5581 CrossRef CAS PubMed; (b) S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC; (c) S. R. Patrick, I. I. F. Boogaerts, S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2011, 7, 892–896 CrossRef CAS PubMed; (d) N. Ahlsten, G. J. P. Perry, X. C. Cambeiro, T. C. Boorman and I. Larrosa, Catal. Sci. Technol., 2013, 3, 2892–2897 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- (a) C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed; (b) K. L. Wilson, J. Murray, H. F. Sneddon, K. M. P. Wheelhouse and A. J. B. Watson, Chem, 2017, 3, 365–368 CrossRef CAS; (c) C. P. Ashcroft, P. J. Dunn, J. D. Hayler and A. S. Wells, Org. Process Res. Dev., 2015, 19, 740–747 CrossRef CAS.
- (a) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC; (b) D. Tan and T. Friščić, Eur. J. Org. Chem., 2018, 2018, 18–33 CrossRef CAS; (c) I. N. Egorov, S. Santra, D. S. Kopchuk, I. S. Kovalev, G. V. Zyryanov, A. Majee, B. C. Ranu, V. L. Rusinov and O. N. Chupakhin, Green Chem., 2020, 22, 302–315 RSC; (d) J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC; (e) J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed; (f) M. Leonardi, M. Villacampa and J. C. Menéndez, Chem. Sci., 2018, 9, 2042–2064 RSC; (g) D. Tan and F. García, Chem. Sci., 2019, 48, 2274–2292 CAS; (h) J. M. Andersen and J. Mack, Chem. Sci., 2017, 8, 5447–5453 RSC.
- For representative examples, see: (a) V. D. Makhaev, A. P. Borisov and L. A. Petrova, J. Organomet. Chem., 1999, 590, 222–226 CrossRef CAS; (b) V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, Chem. Commun., 2002, 2, 1606–1607 RSC; (c) D. Braga, S. L. Giaffreda, F. Grepioni, A. Pettersen, L. Maini, M. Curzi and M. Polito, Dalton Trans., 2006, 60, 1249–1263 RSC; (d) J. G. Hernández, I. S. Butler and T. Friščić, Chem. Sci., 2014, 5, 3576–3582 RSC; (e) J. G. Hernández, N. A. J. Macdonald, C. Mottillo, I. S. Butler and T. Friščić, Green Chem., 2014, 16, 1087–1092 RSC; (f) M. Ferguson, N. Giri, X. Huang, D. Apperley and S. L. James, Green Chem., 2014, 16, 1374–1382 RSC; (g) M. Juribašić, K. Užarević, D. Gracin and M. Ćurić, Chem. Commun., 2014, 50, 10287–10290 RSC; (h) N. R. Rightmire, T. P. Hanusa and A. L. Rheingold, Organometallics, 2014, 33, 5952–5955 CrossRef CAS; (i) C. S. B. Gomes, P. T. Gomes and M. T. Duarte, J. Organomet. Chem., 2014, 760, 101–107 CrossRef CAS; (j) D. W. Peters and R. G. Blair, Faraday Discuss., 2014, 170, 83–91 RSC; (k) C. J. Adams, M. Lusi, E. M. Mutambi and A. G. Orpen, Chem. Commun., 2015, 51, 9632–9635 RSC; (l) J. G. Hernández and C. Bolm, Chem. Commun., 2015, 51, 12582–12584 RSC; (m) J. Wang, R. Ganguly, L. Yongxin, J. Díaz, H. S. Soo and F. García, Dalton Trans., 2016, 45, 7941–7946 RSC; (n) D. H. Woen, C. M. Kotyk, T. J. Mueller, J. W. Ziller and W. J. Evans, Organometallics, 2017, 36, 4558–4563 CrossRef CAS; (o) D. V. Aleksanyan, S. G. Churusova, R. R. Aysin, Z. S. Klemenkova, Y. V. Nelyubina and V. A. Kozlov, Inorg. Chem. Commun., 2017, 76, 33–35 CrossRef CAS; (p) A. Beillard, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, New J. Chem., 2017, 41, 1057–1063 RSC; (q) H. Cheng, J. G. Hernández and C. Bolm, Adv. Synth. Catal., 2018, 360, 1800–1804 CrossRef CAS; (r) T. E. Shaw, L. R. Shultz, L. R. Garayeva, R. G. Blair, B. C. Noll and T. Jurca, Dalton Trans., 2018, 47, 16876–16884 RSC; (s) K. Kubota, R. Takahashi and H. Ito, Chem. Sci., 2019, 10, 5837–5842 RSC. For a review, see: (t) N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC.
- (a) A. Zhdanko, M. Ströbele and M. E. Maier, Chem. – Eur. J., 2012, 18, 14732–14744 CrossRef CAS PubMed; (b) J. D. Egbert, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 2271–2274 CrossRef CAS; (c) A. Beillard, E. Golliard, V. Gillet, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, Chem. – Eur. J., 2015, 21, 17614–17617 CrossRef CAS PubMed; (d) J.-L. Do, D. Tan and T. Friščić, Angew. Chem., Int. Ed., 2018, 57, 2667–2671 CrossRef CAS PubMed; (e) G. N. Hermann, M. T. Unruh, S.-H. Jung, M. Krings and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 10723–10727 CrossRef CAS PubMed.
- CsF-mediated reactivity is surprising here given that, in the analogous boron-to-palladium transmetalation, fluoride coordination to boron has been demonstrated to have an inhibitory effect on the process: (a) C. Amatore, A. Jutand and G. Le Duc, Angew. Chem., Int. Ed., 2012, 51, 1379–1382 CrossRef CAS PubMed . Also, the intermediacy of a Au-F complex in boron-to-gold transmetalation has, to our knowledge, only been reported for Au(III) species: (b) J. Miró and C. del Pozo, Chem. Rev., 2016, 116, 11924–11966 CrossRef PubMed.
- (a) N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2, 2372–2373 RSC; (b) F. C. Strobridge, N. Juda and T. Friščić, CrystEngComm, 2010, 12, 2409–2418 RSC; (c) E. A. Losev and E. V. Boldyreva, CrystEngComm, 2014, 16, 3857–3866 RSC.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- (a) R. Kroon, D. A. Mengistie, D. Kiefer, J. Hynynen, J. D. Ryan, L. Yu and C. Müller, Chem. Soc. Rev., 2016, 45, 6147–6164 RSC; (b) C. Rodríguez-Seco, L. Cabau, A. Vidal-Ferran and E. Palomares, Acc. Chem. Res., 2018, 51, 869–880 CrossRef PubMed.
- To our knowledge, only three examples of this type of compound have been isolated previously: (a) J. A. Akana, K. X. Bhattacharyya, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 7736–7737 CrossRef CAS PubMed; (b) M. Joost, P. Gualco, S. Mallet-Ladeira, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2013, 52, 7160–7163 CrossRef CAS PubMed; (c) T. Seki, T. Mashimo and H. Ito, Chem. Sci., 2019, 10, 4185–4191 RSC. For isolations of the corresponding β-aurated species, see: ref. 1k and. (d) A. S. K. Hashmi, T. D. Ramamurthi and F. Rominger, J. Organomet. Chem., 2009, 694, 592–597 CrossRef CAS; (e) T. P. Cornell, Y. Shi and S. A. Blum, Organometallics, 2012, 31, 5990–5993 CrossRef CAS.
- These protocols require extended reaction times and/or the use of hazardous solvents, i.e., DMF or 1,4-dioxane (ref. 12a and d).
- See ESI† for details.
- As with haloarenes, existing solution-phase methods to prepare these complexes require long reaction times and hazardous solvents (ref. 12d).
- K. Shen, Y. Fu, J.-N. Li and Q.-X. Guo, Tetrahedron, 2007, 63, 1568–1576 CrossRef CAS.
- M. C. Cramp, J. Gilmour, L. R. Hatton, R. H. Hewett, C. J. Nolan and E. W. Parnell, Pestic. Sci., 1987, 18, 15–28 CrossRef CAS.
- (a) S.-J. Lou, Q. Chen, Y.-F. Wang, D.-Q. Xu, X.-H. Du, J.-Q. He, Y.-J. Mao and Z.-Y. Xu, ACS Catal., 2015, 5, 2846–2849 CrossRef CAS; (b) Q. Gou, X. Tan, M. Zhang, M. Ran, T. Yuan, S. He, L. Zhou, T. Cao and F. Luo, Org. Lett., 2020, 22, 1966–1971 CrossRef CAS PubMed.
- R. C. Heel, R. N. Brogden, T. M. Speight and G. S. Avery, Drugs, 1978, 16, 177–201 CrossRef CAS PubMed.
- Although KOH was ultimately found to be the optimal base for heteroarene C–H auration, comparable activity was also observed with KOtBu.
- Similar reactivity was also observed in solution; see ESI† for details.
- Despite extensive efforts, we were unable fully to isolate this species from remaining KOtBu. For reported preparation and characterization of related (R3P)Au(I) alkoxide complexes, see: (a) B. R. Sutherland, K. Folting, W. E. Streib, D. M. Ho, J. C. Huffman and K. G. Caulton, J. Am. Chem. Soc., 1987, 109, 3489–3490 CrossRef CAS; (b) S. Komiya, M. Iwata, T. Sone and A. Fukuoka, J. Chem. Soc., Chem. Commun., 1992, 1109–1110 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral characterization of new compounds, NMR spectra. CCDC 2007939 (3d) and 2007940 (3f). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0gc02263b |
| This journal is © The Royal Society of Chemistry 2020 |