
Water–chromophore electron transfer determines the photochemistry of cytosine and cytidine†

Abstract
Many of the UV-induced phenomena observed experimentally for aqueous cytidine were lacking the mechanistic interpretation for decades. These processes include the substantial population of the puzzling long-lived dark state, photohydration, cytidine to uridine conversion and oxazolidinone formation. Here, we present quantum-chemical simulations of excited-state spectra and potential energy surfaces of N1-methylcytosine clustered with two water molecules using the second-order approximate coupled cluster (CC2), complete active space with second-order perturbation theory (CASPT2), and multireference configuration interaction with single and double excitation (MR-CISD) methods. We argue that the assignment of the long-lived dark state to a singlet nπ* excitation involving water–chromophore electron transfer might serve as an explanation for the numerous experimental observations. While our simulated spectra for the 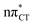 state are in excellent agreement with experimentally acquired data, the electron-driven proton transfer process occurring on the
state are in excellent agreement with experimentally acquired data, the electron-driven proton transfer process occurring on the 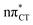 surface may initiate the subsequent damage in the vibrationally hot ground state of the chromophore.
surface may initiate the subsequent damage in the vibrationally hot ground state of the chromophore.

 Please wait while we load your content...
Please wait while we load your content...

