
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring borinane-based multi-ammonium salts for epoxide (co)polymerization: insights into the structure–activity relationship†
Vamshi K.
Chidara
 ,
Yves
Gnanou
* and
Xiaoshuang
Feng
*
,
Yves
Gnanou
* and
Xiaoshuang
Feng
*
Physical Sciences and Engineering (PSE) Division, King Abdullah University of Science and Technology (KAUST), Thuwal-23955, Saudi Arabia. E-mail: yves.gnanou@kaust.edu.sa; fxs101@gmail.com; vamshi.chidara@kaust.edu.sa
First published on 31st May 2024
Abstract
The reactivity of bifunctional borinane-based mono-ammonium salts has previously been demonstrated in various polymerization processes, including ring-opening polymerization (ROP) of epoxides and copolymerization with CO2 or anhydrides. In this study, three bifunctional borinane-based multi-ammonium salts (N+/B) B, C and D (B8N3(C6)Br3; B8N3(C2)Br3; B10N4(C2)Br4) were synthesized with varying B/N ratios and linker lengths between two ammoniums, along with a monoammonium bifunctional salt, catalyst A, B3NBr, used as a reference. The polymerization activities of these catalysts which were essentially used as initiators were evaluated in ROP of propylene oxide (PO), epichlorohydrin (ECH), and glycidyl azide (GA), and ROCOP of PO and ECH with CO2. Specifically, this work focused on the ROP of ECH, which exhibited temperature-dependent reactivity. Lower temperatures favored chain propagation and resulted in well-controlled polymerization behavior, while higher temperatures favored chain transfer reactions to the monomer resulting in low molar mass polymers. Catalysts A and B demonstrated comparable reactivities across all polymerizations, indicating that catalyst B, with a higher B/N ratio and spatially arranged ammoniums, may be an ideal candidate. Overall, the general trend of catalytic activity was observed to be A ≅ B > C > D. This study provides valuable insights into the design and synthesis of forthcoming bifunctional N+/B catalysts.
1. Introduction
Since the first report on the organocatalytic copolymerization of CO2/epoxides by binary systems associating triethylborane (TEB) with onium salts,1 much effort has been devoted to their application to other oxygenated monomers and to the search for more efficient boron-based catalysts.2,3 These binary systems based on TEB/onium salts promote CO2/epoxide copolymerization and give rise to polycarbonates with a high carbonate content and exceptional selectivity.1 Detailed density functional theory (DFT) calculations have shed light on the mechanism, wherein one TEB serves to make an ate complex with the growing oxyanions preventing the formation of cyclic carbonates by backbiting, while another TEB activates the epoxides and thus reduces the energy barrier associated with the alternating enchainment of CO2 and epoxide.4 Subsequent to this work, Wu and co-workers designed and reported bifunctional boron-based catalysts following an approach described earlier for organometallic systems.5 The activity of their bifunctional catalysts which link the ammonium cation to 9-borabicyclo(3.3.1)nonane (9-BBN) was enhanced compared to that of boron-based binary complexes.6–8 Such bifunctional catalytic systems feature proximity between the propagating active species and 9-BBN, thus catalyzing more efficiently CO2/epoxide copolymerization at low loadings.9,10Inspired by Wu's work, Cheng and Liu designed and synthesized a series of bifunctional hexanuclear organoboron catalysts including six 9-BBN centers and two quaternary ammonium bromides linked by o-, m-, or p-benzenedimethylenyl groups.11 Their catalytic performances were investigated in the copolymerization of CO2 with epoxides. They discovered that the polymerization activities were dependent on the structure of the disubstituted linker, with bifunctional catalysts with an ortho-disubstituted linker exhibiting the highest activity in CHO/CO2 copolymerization. Instead of using hindered 9-BBN, our research group reported a series of bifunctional catalysts with ammonium cations linked to six-membered cyclic borinane centers.12–14 The results obtained for the copolymerization of epoxides with CO2 or ring opening polymerization of epoxides showed superior activities in comparison to those reported for 9-BBN-based bifunctional catalysts, indicating that the structure of the boron centers has a great influence on the performance of the catalysts. Several other key factors, such as the B/N ratio within the catalyst and the B–N distance in the catalyst, as well as the electronic and steric environment around the central ammonium core also play a pivotal role in the efficiency and performance of the bifunctional catalysts in various polymerization processes. Inspired by the contributions of Cheng and Liu and of Wu, we present in this study boron-based bifunctional catalysts in which multiple quaternary ammonium bromide centers are connected to borinane moieties through short alkyl chain linkers. It is now well established that bifunctional catalysts exhibit higher activities than binary systems, but little is known about multi-ammonium-bromide based bifunctional catalysts fitted with borinane centers when used to homopolymerize epoxides and copolymerize them with CO2.
In this study, we take advantage of the availability of various polyamines to synthesize multi-ammonium bromide salts fitted with borinane moieties and use them to catalyze the homopolymerization of propylene oxide, epichlorohydrin and glycidyl azide and their copolymerization with CO2. One of the motivations behind this research was to investigate how these borinane-based multi-ammonium bromide salts perform compared to mono-ammonium borinane catalysts with similar B/N ratios and to 9-BBN-based bifunctional systems.
2. Results and discussion
In previous reports, we described the synthesis of bifunctional organoboron catalysts including a unique quaternary ammonium bromide salt to which was linked several borinane centers (Fig. 1A); we then demonstrated their quite remarkable efficiency at catalyzing the homopolymerization of PO and its copolymerization with CO2.12,13 In this study we wished to investigate whether multi-quaternary ammonium bromides to which are linked several borinane centers with approximately the same B/N ratio but various distances between ammonium cations show any synergetic effect when used to catalyze the homopolymerization of epoxides and their copolymerization with CO2.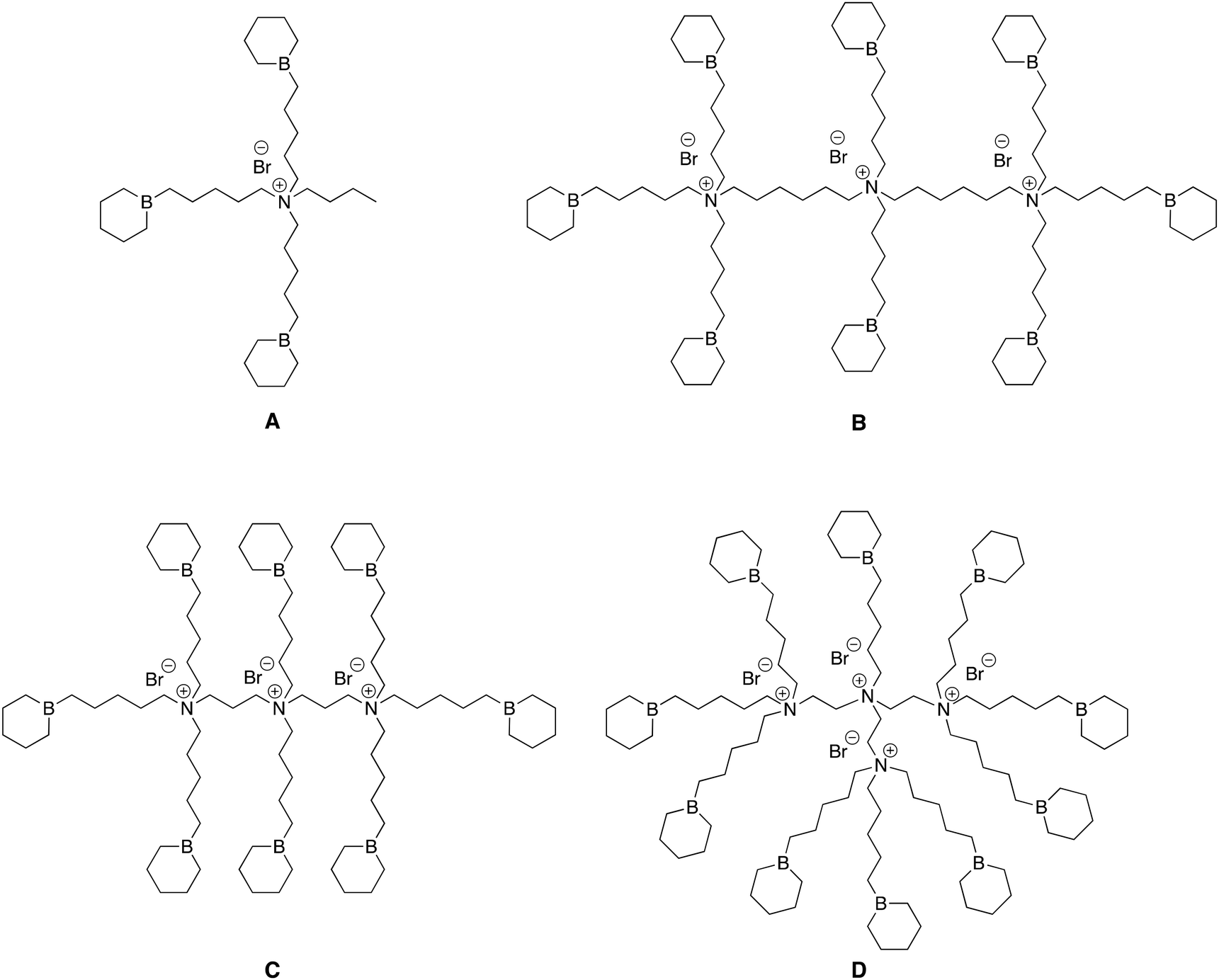 | ||
| Fig. 1 Structures of representative catalysts A, B, C and D. B3NBr; B8N3(C6)Br3; B8N3(C2)Br3; B10N4(C2)Br4. | ||
For this, we selected three different commercial pluriamines, namely bis(hexamethylene)triamine (BHMT), bis(3-aminopropyl)amine (BAPA) and tris(2-aminoethyl)amine (TREN), that subsequently served as precursors for the synthesis of the catalysts B, C and D (Fig. 1), respectively. As shown in the ESI,† these pluriamines containing both primary and secondary amines were first reacted with stoichiometric amounts of 5-bromo-1-pentene and further quaternized by Menshutkin reaction to form quaternary ammonium bromide salts fitted with 8 or 10 terminal double bonds, respectively (Schemes S1–S3 in the ESI†).
In detail, the primary and secondary amine functional groups present in BHMT, BAPA, and TREN were each reacted with 5-bromo-1-pentene for 8 hours at 70 °C. This led to the conversion of primary and secondary amines to tertiary amines terminated by double bonds. Complete transformation was confirmed by 1H NMR spectroscopy, where characteristic peaks appeared at 2.38 ppm corresponding to –CH2N of a tertiary amine, and two peaks at 4.99 ppm (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) and 5.82 ppm (–CHCH2) corresponding to the terminal double bonds of the tertiary amines. Subsequently, these tertiary amines were reacted with stoichiometric amounts of 5-bromo-1-pentene to form the respective quaternary ammonium bromide salts fitted with terminal double bonds. The complete quaternization of the tertiary amines was confirmed by 1H NMR spectroscopy, where the characteristic peak corresponding to –CH2N shifted downfield from 2.38 ppm (as observed in the case of the tertiary amine) to a broad range of 3.00–3.75 ppm after the quaternization reaction, confirming the formation of quaternary ammonium bromide salts or pre-catalysts B, C, and D (Fig. S1–S4 in the ESI†).
CH2) and 5.82 ppm (–CHCH2) corresponding to the terminal double bonds of the tertiary amines. Subsequently, these tertiary amines were reacted with stoichiometric amounts of 5-bromo-1-pentene to form the respective quaternary ammonium bromide salts fitted with terminal double bonds. The complete quaternization of the tertiary amines was confirmed by 1H NMR spectroscopy, where the characteristic peak corresponding to –CH2N shifted downfield from 2.38 ppm (as observed in the case of the tertiary amine) to a broad range of 3.00–3.75 ppm after the quaternization reaction, confirming the formation of quaternary ammonium bromide salts or pre-catalysts B, C, and D (Fig. S1–S4 in the ESI†).
In the next step, the terminal double bonds present in the pre-catalysts B, C, and D underwent a hydroboration reaction with the six-membered cyclic borinane to form the respective quaternary ammonium bromide salts fitted with terminal borinanes, specifically catalysts B, C, and D. The formation of these structures was confirmed by 1H NMR and 11B NMR spectroscopies; the peaks corresponding to terminal double bond protons at 4.99 ppm (–CHCH2) and 5.82 ppm (–CHCH2) in the 1H NMR disappeared, confirming the efficient hydroboration reaction. For all multi-ammonium bromide catalysts B, C, and D, the 11B NMR spectra showed two peaks: one at 86 ppm expected for a 3-coordinated boron center, representing the borinane, and a minor uncharacterized peak at 55 ppm (Fig. S2–S4 in the ESI†).
In this way, quaternary ammonium bromide salt catalysts B, C, and D fitted with borinane centers were obtained with B/N ratios between 2.5 and 3; namely, B8N3(C6)Br3, B8N3(C2)Br3, and B10N4(C2)Br4, where Bn, Nn, and Brn represent the numbers of borinane moieties, ammoniums and bromides contained in the catalysts, and Cn represents the carbon distance between two ammonium cations. Catalysts B (B8N3(C6)Br3) and C (B8N3(C2)Br3) both have the same B/N ratio of 2.67 but differ in the number of carbons (six for B and two for C) between their ammonium cations; catalyst D (B10N4(C2)Br4) has the lowest B/N ratio of 2.50, two carbons between its ammonium cations and carries 4 ammonium and 10 borinane moieties. For comparison, the monoammonium catalyst A (B3NBr) that has a B/N ratio of 3.00 was synthesized as a reference following our previous report (Fig. 1).13 In these borinane-carrying ammonium bromide salts, the distance between B and N atoms was kept constant at 5 carbons. Of particular interest was the study of the influence of parameters such as the B/N ratio – or the ratio of borinanes to ammoniums – and the distance between two ammonium cations on the overall reactivity of these three catalysts when used to catalyze the homopolymerization of epoxides and their copolymerization.
I. Homopolymerization of epoxides using catalysts A, B, C and D
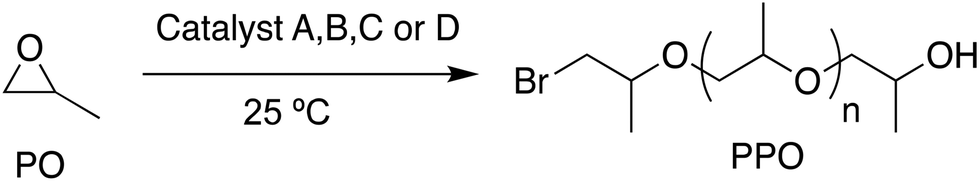
The B, C, and D (B8N3(C6)Br3; B8N3(C2)Br3; B10N4(C2)Br4) bifunctional catalysts are three different quaternary ammonium bromides fitted with borinane centers. Catalyst A (B3NBr) which was described in an earlier work is a quaternary ammonium bromide salt carrying borinane centers with a B/N ratio of 3. It serves as a benchmark reference for evaluating the relative efficiencies of the above three catalysts B, C and D. All four catalysts A, B, C and D demonstrated strong activity when used in the ring-opening polymerization of various epoxides including propylene oxide (PO), epichlorohydrin (ECH) and glycidyl azide (GA) leading to the formation of the corresponding polyethers, which is detailed in the following sections a, b and c.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Type of catalyst | Type of epoxide | [Br−]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[epoxide]c :[epoxide]c |
Temp (°C) | Time (h) | Conv.d (%) | M n, Theoretical (kg mol−1) |
M
n, GPCe (kg mole) |
Đ |
| a Almost instantaneous reactions. b Polymerizations were run under neat conditions. c Molar ratio of one Br− initiator present in the catalyst to PO. d Conversion determined by 1H NMR. e Determined by GPC in THF using narrow polystyrene standards as calibrants. | |||||||||
| 1 | A | PO | 1:1000 |
25 | 100 | 58.0 | 57.6 | 1.05 | |
| 2 | B | PO | 1:1000 |
25 | 100 | 58.0 | 57.2 | 1.10 | |
| 3 | C | PO | 1:1000 |
25 | 100 | 58.0 | 54.7 | 1.63 | |
| 4 | D | PO | 1:1000 |
25 | 100 | 58.0 | 53.9 | 1.20 | |
 000 g mol−1 through a monomer-activated mechanism, albeit requiring a five-fold excess of tri-isobutyl aluminum relative to the onium initiator.21
000 g mol−1 through a monomer-activated mechanism, albeit requiring a five-fold excess of tri-isobutyl aluminum relative to the onium initiator.21
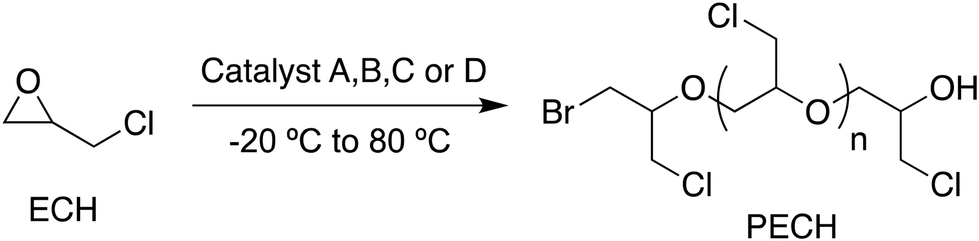
In our study, ECH polymerizations conducted at −20 °C in the presence of catalysts A and B resulted in the formation of well-controlled PECH samples with molar masses in the range of 20000 g mol−1 (entries 1 and 2, Table 2). The performances of catalysts C and D in the polymerization of ECH were mixed, exhibiting lower activity compared to catalysts A and B. After 3 hours of reaction at −20 °C, only 70% and 30% conversions were achieved for a targeted molar mass of 23000 g mol−1 with catalysts C and D, respectively (entries 3 and 4, Table 2). The short distance between ammonium cations in catalysts C and D might well be the reason for the observed slower rate of propagation. Above −20 °C, particularly at temperatures ranging from 25 °C to 80 °C, transfer reactions became significant compared to chain propagation, leading to significantly lower molar masses than those expected.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Type of catalyst | Type of epoxide | [Br−]:[epoxide]b |
Temp (°C) | Time (h) | Conv.c (%) |
M
n, Theoreticalc (kg mol−1) |
M
n, GPCd (kg mol−1) |
Đ |
| a Polymerizations were run under neat conditions at specified temperatures for specified times. b Molar ratio of one Br− initiator present in the catalyst to ECH. c Conversion determined by 1H NMR. d Determined by GPC in THF using narrow polystyrene standards a calibrants. | |||||||||
| 1 | A | ECH | 1:250 |
−20 | 3 | 100 | 23.2 | 21.4 | 1.35 |
| 2 | B | ECH | 1:250 |
−20 | 3 | 100 | 23.1 | 20.1 | 1.36 |
| 3 | C | ECH | 1:250 |
−20 | 3 | 70 | 16.2 | 15.9 | 1.38 |
| 4 | D | ECH | 1:250 |
−20 | 3 | 35 | 8.1 | 6.1 | 1.44 |
| 5 | B | ECH | 1:250 |
25 | 3 | >99 | 23.1 | 16.7 | 1.44 |
| 6 | B | ECH | 1:250 |
80 | 3 | >99 | 23.1 | 9.0 | 1.40 |
| 7 | B | ECH | 1:2500 |
−20 | 48 | 53 | 122.6 | 53.4 | 1.74 |
At 25 °C with catalyst B, the transfer to the monomer resulted in the formation of PECH macromonomers, PECH chains end-capped with an epoxide ring moiety. The resulting PECH samples exhibited lower molar masses than those expected in the absence of transfer (entry 5, Table 2; Fig. 2). The 1H NMR spectrum of the resulting crude sample, dissolved in CDCl3, revealed the formation of PECH, indicated by the presence of polyether peaks spanning from 3.50 to 3.80 ppm, representing all five protons of the PECH repeating unit. Additionally, the 1H NMR spectrum of this sample exhibited three distinct sets of peaks at 2.69 ppm, 2.88 ppm, and 3.24 ppm, corresponding to the protons of the oxirane ring. These peaks differed from those of the unreacted oxirane ring of ECH, which displayed peaks at 2.63 ppm, 2.79 ppm, and 3.16 ppm (refer to Fig. S6† for the 1H NMR of PECH formed at −20 °C from entry 2 and Fig. S7† for that in entry 5 of Table 2). The appearance of this new set of peaks at 2.69 ppm, 2.88 ppm, and 3.24 ppm was thus attributed to the chains end-capped with an oxirane moiety resulting from the chain transfer to the ECH monomer, as represented by II in Fig. 2. Due to the distinct pendant group compared to the alkyl chloride of ECH, these new peaks were observed slightly downfield in the 1H NMR spectrum, indicative of the chain transfer to the ECH monomer and end-capping of the PECH chain. It was observed that the oxirane ring at the PECH chain-end is stable even for several days at 25 °C. A new chloride anion is produced as a result of such a transfer event, which in turn can initiate the polymerization of ECH to form a new PECH chain, thereby increasing the number of chains in the reaction medium and consequently lowering the molar masses of the PECH produced.
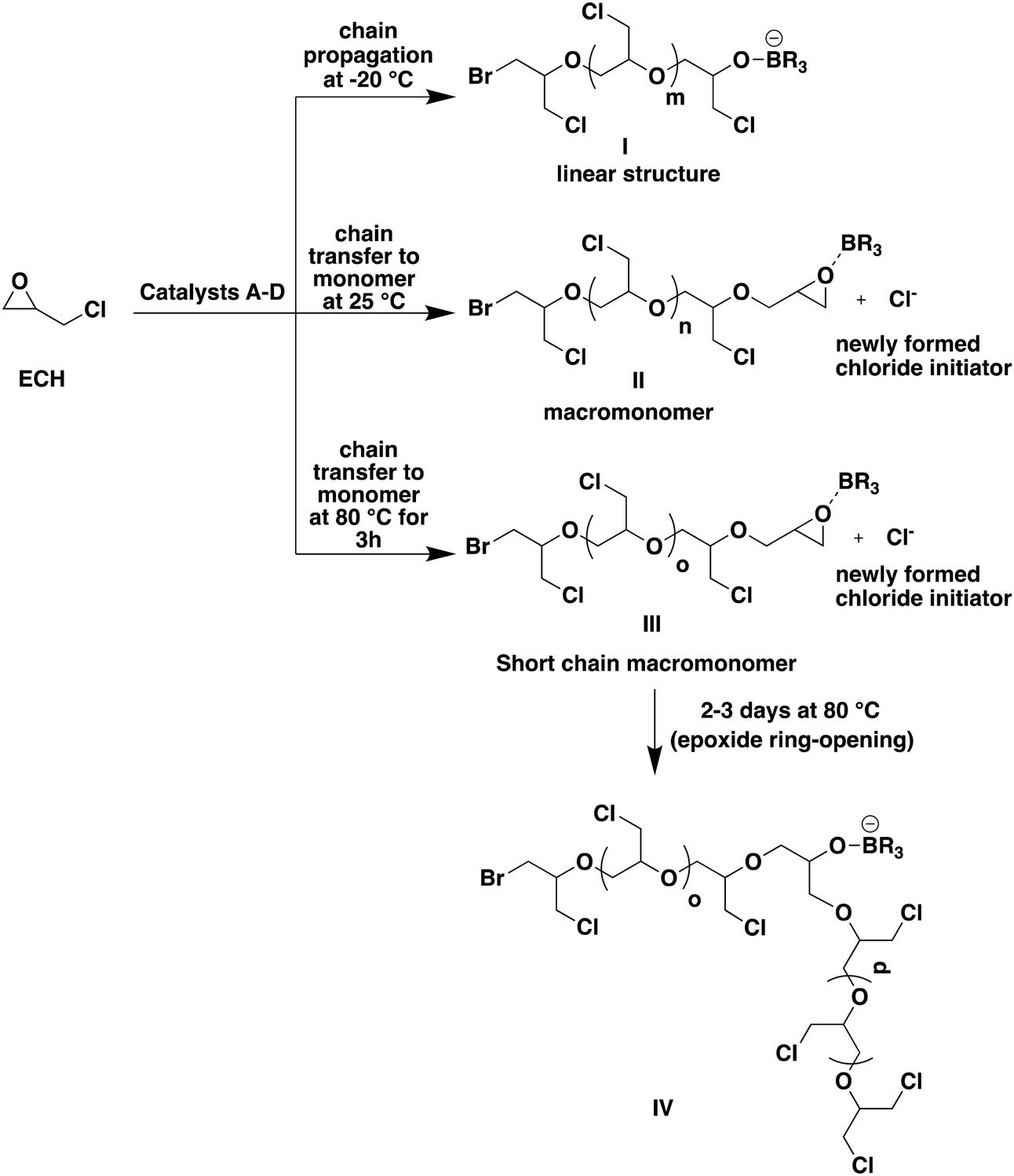 | ||
| Fig. 2 Schematic representing different modes of ECH polymerization under different reaction conditions. BR3 is used only as a simplified graphical representation and it represents one of the multiple borinanes fitted to ammonium salts in catalysts A, B, C and D. | ||
The reaction of ECH with catalyst B at 80 °C also resulted in the formation of PECH with a terminal oxirane moiety and a much lower molar mass of only 9.0 kg mol−1 compared to the expected molar mass of 23.1 kg mol−1 (entry 6, Table 2). At 80 °C, the rate of chain transfer is increased compared to that at 25 °C, leading to the formation of PECH with lower molar masses as represented by III in Fig. 2. The 1H NMR spectrum of the resulting PECH exhibited a similar peak pattern to the one described in the previous case, indicating the presence of an oxirane moiety at the chain end (Fig. S8 in the ESI†). However, this terminal oxirane ring disappeared when the reaction medium was maintained at 80 °C for 2 to 3 days as it underwent a ring-opening by the anionic active species present in the medium (IV in Fig. 2; Fig. S9 in the ESI†).
The PECHs formed at various temperatures were further investigated by 13C NMR spectroscopy. It was confirmed that the low temperature of −20 °C resulted in linear PECH, while higher temperatures (25 °C and 80 °C) led to low molar mass PECHs due to enhanced chain transfer reactions compared to chain propagation reactions. For example, in the case of the PECH formed at −20 °C in entry 2, Table 2, the 13C NMR spectrum showed three prominent peaks a, b, and c corresponding to chain propagation, and three minor peaks a′, b′, and c′ corresponding to chain initiations at the terminal (Fig. 3). In more detail, the peaks at 79 ppm correspond to –CH– (peak a), those at 69.5 ppm represent –CH2– (peak b), and that at 43.6 ppm is the α-carbon of the methylene chloride substituent (–CH2Cl) (peak c) of the repeating unit, [–CH2–CH(CH2Cl)–O–] of PECH. Similarly, the peaks at 79.6 ppm correspond to –CH– (peak a′), whereas the peak at 43.2 ppm represents Br–CH2– or Cl–CH2– (peaks b′ and c′) of the initiating unit [X–CH2–CH(CH2Cl)–O–], where X may be either bromide or chloride.
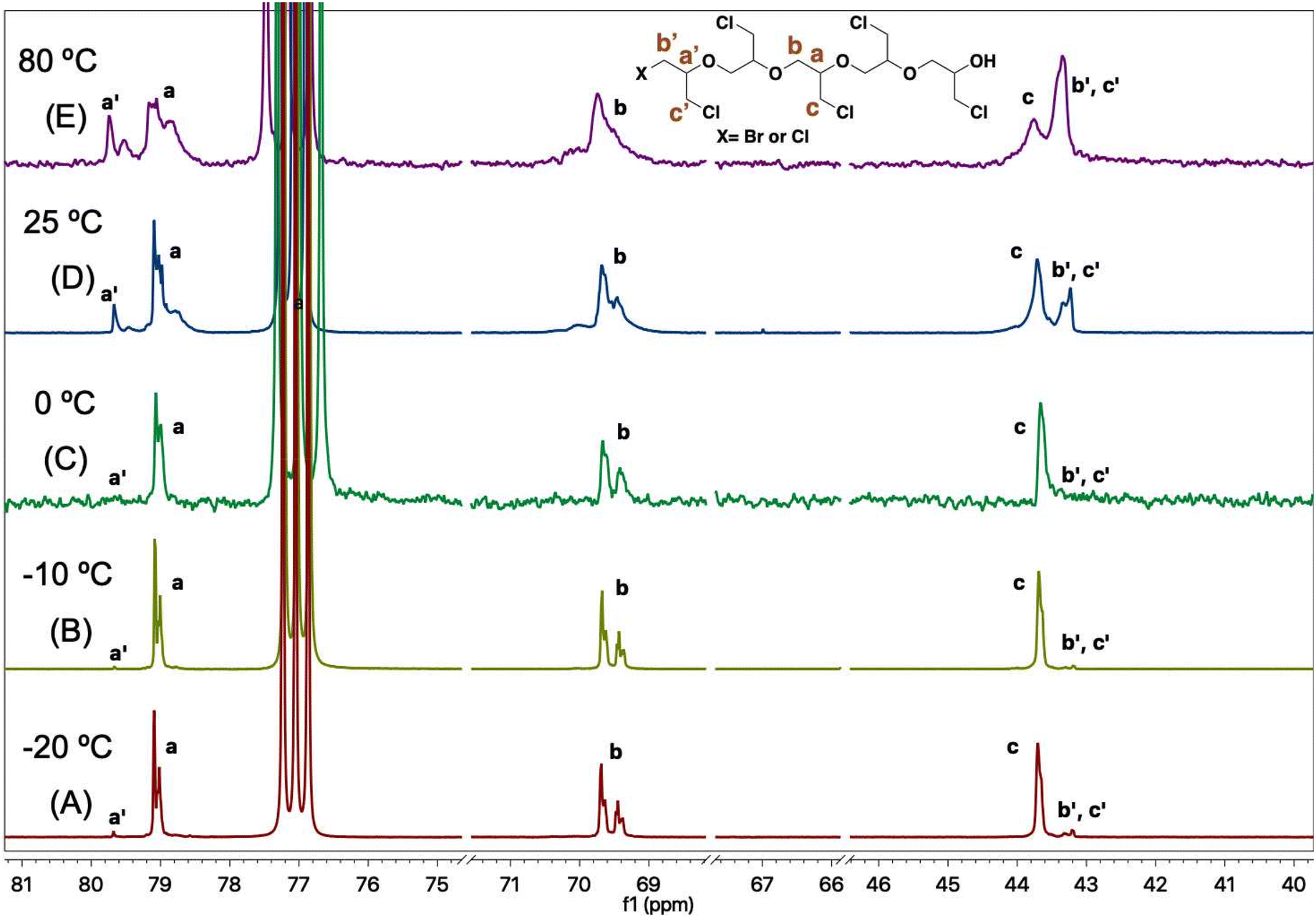 | ||
| Fig. 3 13C NMR spectra of poly(epichlorohydrin) PECH at temperatures (A) −20 °C, (B) −10 °C, (C) 0 °C, (D) 25 °C, and (E) 80 °C, respectively, from bottom to top. | ||
During ECH polymerization, if chain propagation dominates over chain transfer reactions, peaks a, b, and c are expected to be the major peaks, while a′, b′, and c′ would be minor sets of peaks. Conversely, if chain transfer dominates over chain propagation reactions, peaks a′, b′, and c′ become equally prominent or even the major set of peaks entirely, depending on the chain length of the formed PECH. It is observed from the 13C NMR spectra in Fig. 3 that the PECH formed at −20 °C exhibits peaks a, b, and c as the major set of peaks. However, in the PECH formed at 80 °C, the intensity of peaks corresponding to a′, b′, and c′ becomes considerable, demonstrating increased chain-transfer reactions at higher temperatures.
Our findings reveal that the polymerization behavior of ECH is highly influenced by temperature. At very low temperatures, such as −20 °C, chain propagation predominates, facilitating the formation of well-controlled linear PECH through successive ring-opening reactions of ECH. However, at higher temperatures (25 °C and 80 °C), chain propagation and chain transfer reactions compete. At 25 °C chain transfer to the monomer occurs, leading to the formation of PECH macromonomers; at 80 °C shorter PECH macromonomers are produced due to stronger transfer to the monomer; they subsequently undergo ring-opening of their terminal oxirane ring as confirmed by the disappearance of the peaks at 2.69 ppm, 2.88 ppm, and 3.24 ppm. In both cases, higher temperatures resulted in a loss of molar mass control and the production of shorter chains (Fig. 2).
On the other hand when targeting very high molar masses (more than 230000 g mol−1), transfer reactions dominate propagation, producing shorter PECH chains than expected (entry 7, Table 2; see Fig. S10 in the ESI† for rate equations determining chain transfer reactions over chain propagation reactions).
Coupling of PECHs at higher temperatures. The results presented in Table 3 pertain to three sets of reactions, each varying in their monomer loading. Entries 1a, 1b, and 1c depict the outcomes of a reaction utilizing a 1
:250 bromide ion to ECH ratio. In entry 1a of Table 3 corresponding to entry 1 of Table 2, a linear PECH with a molar mass of 21400 g mol−1 is formed as confirmed via1H NMR, matching the theoretical value. Subsequently, the same reaction flask free of any unpolymerized monomer was then heated to 80 °C and stirred for 3 hours; the characterization by GPC of the sample isolated after this period at 80 °C showed an increase of molar mass from 21400 g mol−1 to 50150 g mol−1, nearly doubling its initial value (entry 1b, Table 3; Fig. S11 in the ESI†). Their doubled molar masses along with the same unimodal distributions as the precursors indicate that coupling occurred efficiently but only once and not repetitively as shown in Fig. 4, through reaction of the oxyanion carried by the chain end of I with the carbon in the α-position to the terminal Br atom of II. However, further stirring of the reaction mixture for longer durations did not yield any significant increase in molar mass (entry 1c, Table 3). Entries 2a–2c in Table 3 represent similar outcomes of a reaction employing a 1:125 ECH ratio. Initially, the reaction mixture was reacted at −20 °C for 3 hours resulting in 100% conversion of ECH (entry 2a, Table 3). Using a PECH sample of lower molar mass prepared at −20 °C under similar conditions to those described above, molar masses again double to 29500 g mol−1 upon heating to 80 °C (entries 2b and 2c, Table 3; Fig. S12 in the ESI†). Chain end coupling was responsible for such doubling of molar mass. With a sample of even lower mass, 6104 g mol−1, the molar mass tripled to 19200 g mol−1 after increasing the temperature of the reaction medium to 80 °C and maintaining it for 3 h (entries 3a and 3b, Table 3). In the three examples corresponding to samples of various molar masses, the carbon atom in the α-position to the terminal bromide atom is more electrophilic than the dangling alkyl chloride which then resulted in the above-mentioned couplings (Fig. 4). Further, the glass-transition temperature (Tg) of PECH samples before and after coupling was tested to evaluate the effect of coupling on the Tg. Specifically, the PECH samples from entry 3a (before coupling) and entry 3b (after coupling) in Table 3 were selected. The differential scanning calorimetry (DSC) analysis indicated that the value of −32 °C for the Tg of the PECH sample before coupling slightly changed to −32.4 °C after coupling. This suggests no significant variation in the glass-transition temperature due to the coupling process (Fig. S13 and S14†).
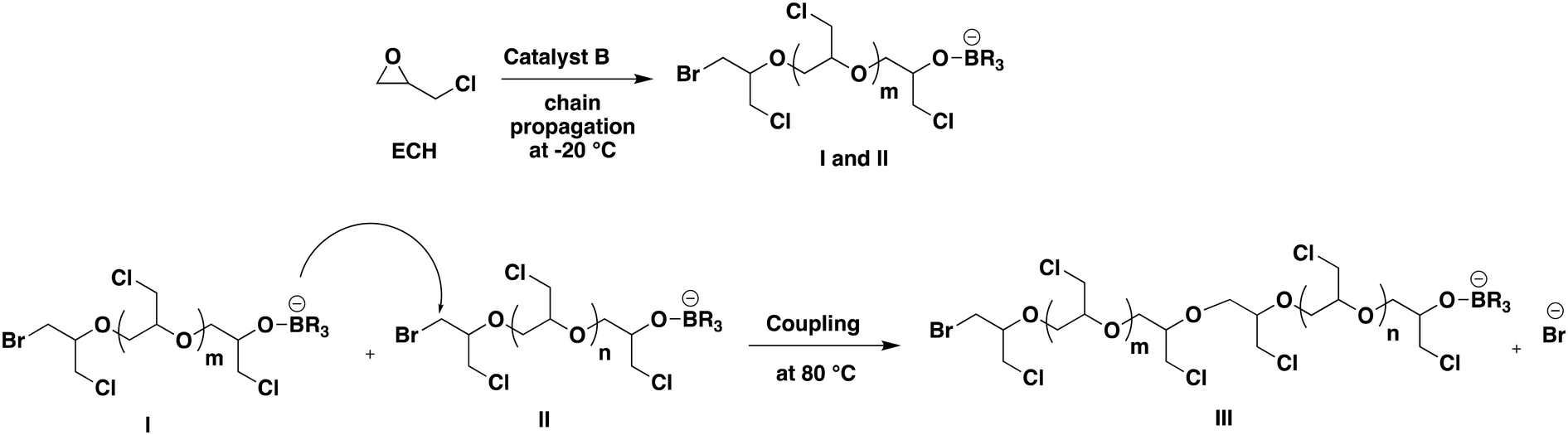 | ||
| Fig. 4 Coupling of PECH chains at 80 °C. | ||
| Entry | Type of catalyst | [Br−]:[ECH]b |
Conv.c (%) | Temp (°C) | Time |
M
n, Theoreticalc (kg mol−1) |
M n, GPC (kg mol−1) | Đ |
|---|---|---|---|---|---|---|---|---|
| a Polymerizations were run under neat conditions at specified temperatures for specified times. b Molar ratio of one Br− initiator present in the catalyst to ECH. c Conversion determined by 1H NMR. d Determined by GPC in THF using narrow polystyrene standards as calibrants. | ||||||||
| 1a | B | 1:250 |
100 | −20 | 3 h | 23.13 | 21.4 | 1.36 |
| 1b | — | 80 | 3 h | 23.13 | 50.15 | 1.38 | ||
| 1c | — | 80 | 48 h | 23.13 | 50.15 | 1.38 | ||
| 2a | B | 1:125 |
100 | −20 | 3 h | 11.56 | 12.09 | 1.36 |
| 2b | — | 80 | 3 h | 11.56 | 29.50 | 1.35 | ||
| 2c | — | 100 | 12 h | 11.56 | 29.50 | 1.35 | ||
| 3a | B | 1:64 |
100 | −20 | 3 h | 5.90 | 6.10 | 1.38 |
| 3b | — | 80 | 3 h | 5.90 | 19.20 | 1.35 | ||
| 3c | — | 100 | 12 h | 5.90 | 19.20 | 1.35 | ||
:250 ratio of bromide ion initiator to GA in the presence of catalyst B at 60 °C resulted in 100% monomer conversion in 18 h but the molar mass obtained was only 6015 g mol−1 compared to the theoretical value of 24770 g mol−1 (entry 1, Table 4). Reducing the temperature to 25 °C resulted in 100% monomer conversion but only 7910 g mol−1 molar mass (entry 2, Table 4; Fig. S15 in the ESI†). Performing the polymerization at an even lower temperature of −20 °C mitigated to some extent chain transfer to the monomer, resulting in 54% conversion in 72 h and a molar mass of 9600 g mol−1 (entry 3, Table 4).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Type of catalyst | Type of epoxide | [Br−]:[epoxide]b |
Temp (°C) | Time (h) | Conv.c (%) |
M
n, Theoreticalc (kg mol−1) |
M
n, GPC
(kg mol−1) |
Đ |
| a Polymerizations were run under neat conditions at specified temperatures for specified times. b Molar ratio of one Br− initiator present in the catalyst to GA. c Conversion determined by 1H NMR. d Determined by GPC in THF using narrow polystyrene standards as calibrants. e Phenyl glycidyl ether (PGE) was used as a representative glycidyl ether monomer. | |||||||||
| 1 | B | GA | 1:250 |
60 | 18 | 100 | 24.8 | 6.0 | 1.10 |
| 2 | B | GA | 1:250 |
25 | 48 | 100 | 24.8 | 7.9 | 1.15 |
| 3 | B | GA | 1:250 |
−20 | 72 | 54 | 13.4 | 9.6 | 1.11 |
| 4 | A | GA | 1:250 |
−20 | 72 | 63 | 15.6 | 10.4 | 1.12 |
| 5 | C | GA | 1:250 |
−20 | 72 | 29 | 7.2 | 6.2 | 1.12 |
| 6 | D | GA | 1:250 |
−20 | 72 | 11 | 5.0 | 2.0 | 1.13 |
| 7e | B | PGE | 1:250 |
25 | 0.5 | 100 | 37.5 | 22.9 | 1.15 |
The bulkiness of the azide group might be the reason for the slow rate of propagation of GA which is obviously not immune to transfer reactions even at −20 °C. Under similar conditions, catalyst A resulted in PGA with a similar molar mass of 10.4 kg mol−1 (entry 4, Table 4; Fig. S16 in the ESI†) and catalyst C afforded only 29% monomer conversion in 72 h and a sample with 6.15 kg mol−1 molar mass (entry 5, Table 4). As for catalyst D, the least active of all four catalysts, monomer conversion reached 11% within 3 h resulting in a molar mass of 1.91 kg mol−1 for the sample isolated (entry 6, Table 4). The results for the ROP of glycidyl azide also followed the trend observed previously with catalyst A ≅ catalyst B > catalyst C > catalyst D confirming that the distance between ammonium cations plays a crucial role in the activity of these catalysts. Further, catalyst B was used to polymerize phenyl glycidyl ether (PGE) and test its actual activity. A reaction was carried out with a 1:250 ratio of [bromide ion] to [PGE] at 25 °C which resulted in 100% monomer conversion in 0.5 h, providing a PPGE sample of 22900 g mol−1; this value is lower than the theoretical one of 37500 g mol−1 due to the presence of moisture impurities (entry 7, Table 4). See the ESI, Fig. S17† for the 1H NMR spectrum and Fig. S18† for the GPC trace of poly(phenyl glycidyl ether).
II. Copolymerization of epoxides with CO2 using catalysts A, B, C and D
The ring-opening copolymerization (ROCOP) of epoxides, namely PO and ECH, with CO2 was also investigated using the above-described multi-ammonium bromide, borinane carrying catalysts. Using an initiator to PO ratio of 1:1000, 100% conversion of PO could be achieved within 12 h at 40 °C with catalysts A and B (entries 1 and 2, Table 5). Both copolymerizations occurred selectively with formation of linear polymers over cyclics. Because of the presence of several boron atoms with their Lewis acid character, back-biting reactions could be avoided to a large extent but the formation of ether linkages was unavoidable. All attempts at copolymerization of PO with CO2 produced samples containing ether linkages, thereby resulting in the formation of poly(propylene carbonate-co-propylene oxide) (PPCPO). With catalyst A the PPCPO sample formed contained 54% carbonate linkages (entry 1, Table 5; Fig. S19 in the ESI†) and with catalyst B it contained 51% carbonate content (entry 2, Table 5) showcasing the similar reactivity patterns of catalysts A and B.
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Type of catalyst | Type of epoxide | [Br]:[epoxide]b |
Temp (°C) | Time (h) | Conv.c (%) | Linear vs. cyclic carbonate selectivityc (%) | Carbonate content in polymer (mol%)c |
M
n, Theoreticalc (kg mol−1) |
M
n, GPCd (kg mol−1) |
Đ |
| a Polymerizations were run under neat conditions using 10 bar CO2 at 0 °C to 40 °C for 12 h to 72 h. b Molar ratio of one Br− initiator to epoxide present in the catalyst. c Conversion, selectivity (the molar ratio of linear carbonate to cyclic carbonate in the crude reaction mixture), and linear carbonate content (the molar ratio of linear carbonate linkages to ether linkages in the polymer chains) were determined by 1H NMR. d Determined by GPC in THF using narrow polystyrene standards as calibrants. | |||||||||||
| 1 | A | PO | 1:1000 |
40 | 12 h | 100 | >99 | 54 | 81.7 | 79.4 | 1.04 |
| 2 | B | PO | 1:1000 |
40 | 12 h | 100 | >99 | 51 | 80.4 | 74.8 | 1.15 |
| 3 | C | PO | 1:1000 |
40 | 12 h | 94 | >99 | 45 | 73.1 | 72.1 | 1.20 |
| 4 | D | PO | 1:1000 |
40 | 12 h | 38 | 97 | 78 | 33.2 | 31.8 | 1.10 |
| 5 | A | ECH | 1:500 |
40 | 12 h | 80 | 82 | 38 | 39.8 | 1.2 | 2.10 |
| 6 | A | ECH | 1:500 |
25 | 48 h | 100 | 92 | 37 | 56.7 | 4.7 | 2.32 |
| 7 | A | ECH | 1:500 |
0 | 72 h | 72 | 99 | 58 | 42.5 | 6.9 | 1.74 |
| 8 | B | ECH | 1:500 |
40 | 12 h | 75 | 89 | 43 | 39.7 | 1.0 | 1.87 |
| 9 | B | ECH | 1:500 |
25 | 48 h | 98 | 93 | 44 | 54.1 | 3.0 | 2.06 |
| 10 | B | ECH | 1:500 |
0 | 72 h | 51 | 99 | 61 | 30.4 | 4.9 | 2.02 |
Under similar conditions, catalyst C with a B/N ratio 2.67 but with shorter linkers between two ammonium cations afforded comparable 94% conversion but lower carbonate content (45%) (entry 3, Table 5; Fig. S20 in the ESI†). As for catalyst D with the lowest B/N ratio of 2.5 and even shorter linkers between ammonium cations, only 38% conversion of PO could be achieved in 12 h at 40 °C; the carbonate content increased to 78% in the latter case thus illustrating the lower activity for ROCOP of PO with CO2 (entry 4, Table 5).
The copolymerization of CO2 with epoxides such as ECH with its electron-withdrawing group is a decade old problem that is deemed very challenging as the formation of cyclic carbonates is favored over linear propagation. Over the last ten years, several solutions have been proposed to address this problem resorting either to homogeneous bifunctional metallic catalysts, or heterogenous catalysts such as DMC or zinc glutarate.22–26 Recently, Wu and coworkers reported that using a bifunctional 9-BBN-based ammonium salt CO2 could be successfully copolymerized with ECH up to 30.0 kg mol−1 molar mass.27 Catalysts A and B were tested in the ROCOP of ECH with CO2 for the formation of poly(epichlorohydrin ether carbonate) (PEEC). A ratio of 1:500 initiator to ECH was used initially at 40 °C with catalyst A and 80% conversion of ECH could be achieved in 12 h with formation of 82% linear chains and 18% cyclic carbonate (entry 5, Table 5). Because of the presence of electron withdrawing chlorides the formation of cyclic carbonates could not be avoided. The GPC traces of this sample showed a broad distribution of molar masses and a molar mass of 1200 g mol−1 against the 39800 g mol−1 expected (entry 5, Table 5). Lowering the reaction temperature to 25 °C resulted in polymers with 37% carbonate content and a molar mass of 4.7 kg mol−1 with catalyst A (entry 6, Table 5; Fig. S21 in ESI†), and 44% carbonate content with a molar mass of 3.0 kg mol−1 with catalyst B (entry 9, Table 5). Upon further lowering the temperature to 0 °C the linear versus cyclic selectivity could be raised to 99% and the linear sample isolated contained up to 58% carbonate linkages with catalyst A and 61% with catalyst B (entries 7 and 10, Table 5). However, the polymers obtained at 0 °C were characterized by unavoidable chain transfer reactions which resulted in lower molar masses compared to the targeted ones.
3. Conclusions
In the present study, we investigated multi-ammonium bromide salts fitted with borinanes for ROP of PO, ECH, and GA, and ROCOP of PO and ECH with CO2. Three variants of the bifunctional multi-ammonium bromide–borinanes, catalysts B, C, and D, were reported with various B/N ratios and linker lengths. A previously reported bifunctional catalyst A was used as a reference to compare the catalytic activities throughout our investigation. The catalysts B, C, and D could be easily synthesized from readily available precursors in three steps with quantitative yields.Using these catalysts, ROP of epoxides can be conducted at temperatures ranging from −20 °C to 80 °C. The ROP of PO at 25 °C occurred under “living” conditions with the formation of PPO chains of well-controlled molar mass with all the catalysts; in contrast ECH showed a temperature dependent reactivity. With the latter monomer, various polymeric structures could be obtained depending upon the temperature applied to the reaction medium; at −20 °C ECH polymerization occurred under well-controlled conditions affording PECH samples with expected molar masses; at 25 °C chain transfer reactions to the monomer produced well-defined PECH macromonomers end-capped with an oxirane ring; at 80 °C chain transfer to the monomer was even more vigorous, resulting in the formation of PECH macromonomers of lower molar masses. Maintaining the reaction medium at 80 °C for several hours led to the polymerization of the terminal oxirane ring as showed by the NMR characterization. As for the ROP of GA it proceeded slowly with catalysts A and D as compared with that of ECH, with unavoidable chain transfer reactions even at −20 °C.
The ROCOP of PO with CO2 followed the same reactivity order as for homopolymerization; catalyst A and catalyst B gave comparable results with 54% and 51% carbonate content and catalyst D afforded the highest carbonate content (78%) in the PPCPO with expected molar masses. In contrast the ROCOP of ECH with CO2 didn't proceed with good control with both catalysts A and B due to strong transfer reactions, resulting in low molar masses irrespective of the reaction temperature.
In conclusion, these findings underscore the multifaceted interplay between catalyst structure and catalytic activity in ROP and ROCOP reactions, though with no obvious synergistic effect. While higher B/N ratios generally correlated with enhanced performance, the results revealed that the linker length also significantly impacts the reactivity of the catalysts. Catalyst A with a B/N ratio of 3 showed a comparable reactivity to catalyst B with a B/N ratio of 2.67. Catalysts B and C with the same B/N ratio of 2.67 behave differently; catalyst B with its long linker length between two ammoniums showed greater reactivity compared to catalyst C with a shorter linker length. Catalyst D with a B/N ratio of 2.5 and a shorter linker length showed the least reactivity of all the catalysts. The general trend of catalytic activity was observed to be catalyst A ≅ catalyst B > catalyst C > catalyst D. A deeper investigation to understand the structure–performance relationship of such multi-ammonium systems will be carried out in our future work.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research work is supported by KAUST under baseline funding (BAS/1/1374-01-01).References
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- C. Zhang, X. Geng, X. Zhang, Y. Gnanou and X. Feng, Prog. Polym. Sci., 2023, 136, 101644 CrossRef CAS.
- S. Naumann, Polym. Chem., 2023, 14, 1834–1862 RSC.
- D.-D. Zhang, X. Feng, Y. Gnanou and K.-W. Huang, Macromolecules, 2018, 51, 5600–5607 CrossRef CAS.
- G.-W. Yang, Y.-Y. Zhang, R. Xie and G.-P. Wu, J. Am. Chem. Soc., 2020, 142, 12245–12255 CrossRef CAS PubMed.
- K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., 2006, 118, 7432–7435 CrossRef.
- E. K. Noh, S. J. Na, S. Sujith, S.-W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS PubMed.
- W.-M. Ren, Z.-W. Liu, Y.-Q. Wen, R. Zhang and X.-B. Lu, J. Am. Chem. Soc., 2009, 131, 11509–11518 CrossRef CAS PubMed.
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS.
- J. Schaefer, H. Zhou, E. Lee, N. S. Lambic, G. Culcu, M. W. Holtcamp, F. C. Rix and T.-P. Lin, ACS Catal., 2022, 12, 11870–11885 CrossRef CAS.
- Y. T. Tong, R. H. Cheng, H. Dong, Z. Liu, J. X. Ye and B. P. Liu, J. CO2 Util., 2022, 60, 101979 CrossRef CAS.
- C. Chen, Y. Gnanou and X. Feng, Polym. Chem., 2022, 13, 6312–6321 RSC.
- C. Chen, Y. Gnanou and X. Feng, Macromolecules, 2022, 55, 10662–10669 CrossRef CAS.
- C. Chen, Y. Gnanou and X. Feng, Macromolecules, 2023, 56, 892–898 CrossRef CAS.
- E. J. Vandenberg, J. Elastomers Plastics, 1982, 14, 243–256 CrossRef CAS.
- E. J. Vandenberg, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 1423–1431 CrossRef CAS.
- T. Biedron, P. Kubisa and S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 619–628 CrossRef CAS.
- R.-J. Wei, Y.-Y. Zhang, X.-H. Zhang, B.-Y. Du and Z.-Q. Fan, RSC Adv., 2014, 4, 21765–21771 RSC.
- C. H. Tran, L. T. T. Pham, Y. Lee, H. B. Jang, S. Kim and I. Kim, J. Catal., 2019, 372, 86–102 CrossRef CAS.
- S. Hafner, T. Keicher and T. M. Klapötke, Propellants, Explos., Pyrotech., 2018, 43, 126–135 CrossRef CAS.
- S. Carlotti, A. Labbé, V. Rejsek, S. Doutaz, M. Gervais and A. Deffieux, Macromolecules, 2008, 41, 7058–7062 CrossRef CAS.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- Z. Shen, X. Chen and Y. Zhang, Macromol. Chem. Phys., 1994, 195, 2003–2011 CrossRef CAS.
- R.-J. Wei, X.-H. Zhang, B.-Y. Du, Z.-Q. Fan and G.-R. Qi, Polymer, 2013, 54, 6357–6362 CrossRef CAS.
- P. Sudakar, D. Sivanesan and S. Yoon, Macromol. Rapid Commun., 2016, 37, 788–793 CrossRef CAS PubMed.
- G.-P. Wu, S.-H. Wei, W.-M. Ren, X.-B. Lu, T.-Q. Xu and D. J. Darensbourg, J. Am. Chem. Soc., 2011, 133, 15191–15199 CrossRef CAS PubMed.
- G.-W. Yang, C.-K. Xu, R. Xie, Y.-Y. Zhang, X.-F. Zhu and G.-P. Wu, J. Am. Chem. Soc., 2021, 143, 3455–3465 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; materials information; characterization information; general procedures for synthesis of catalysts A–D; general procedures for ROP of PO, ECH, and GA, and ROCOP of PO and CO2; 1H NMR and 11B NMR spectra; and GPC traces. See DOI: https://doi.org/10.1039/d4py00435c |
| This journal is © The Royal Society of Chemistry 2024 |