
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A zinc–porphyrin–peptide conjugate via “click-chemistry”: synthesis and amyloid-β interaction†
Rita
Tosto
,
Stefania
Zimbone
,
Giuseppe
Di Natale
,
Maria Laura
Giuffrida
 ,
Tiziana
Campagna
,
Giuseppe
Pappalardo
* and
Giuseppina
Sabatino
*
,
Tiziana
Campagna
,
Giuseppe
Pappalardo
* and
Giuseppina
Sabatino
*
Istituto di Cristallografia, Consiglio Nazionale delle Ricerche, Via Paolo Gaifami 9, 95126, Catania, Italy. E-mail: giuseppina.sabatino@cnr.it
First published on 8th July 2024
Abstract
The discovery of systems capable of recognizing amyloid-β protein (Aβ) oligomeric species with high sensitivity and specificity, also producing detectable signals, could represent an attractive approach for the early diagnosis of Alzheimer's disease (AD). In this regard, peptide-based inhibitors of Aβ aggregation have been extensively studied with particular attention to those derived from original amyloid sequences, such as the hydrophobic Aβ16–20 core (KLVFF). In this study we combined the antifibrillogenic action of the KLVFF peptide motif with the spectroscopic features of the porphyrin macrocycle. Specifically, we describe the synthesis of a new water-soluble zinc metallated porphyrin–peptide conjugate, in which the porphyrin macrocycle is linked via 1,2,3-triazole linkage to the hydrophobic Aβ16–20 sequence. The zinc–porphyrin–peptide conjugate was obtained by copper-catalyzed azide–alkyne cycloaddition (CuAAC) in the presence of Cu(I) as a catalyst. The “click” reaction was carried out between the azido-KLVFF peptide and the alkyne-porphyrin. The ability of the porphyrin–peptide conjugate to interact with Aβ was investigated. The zinc-metallated porphyrin–peptide conjugate was studied by biophysical techniques, including UV-vis, circular dichroism (CD), and Bis-Ans fluorescence. Finally, cell viability studies were performed on differentiated neuroblastoma cells.
1. Introduction
Alzheimer's disease (AD) is a multifactorial pathological condition that is mainly characterized by the extracellular accumulation of amyloid-β protein (Aβ), and intracellular aggregation of hyperphosphorylated tau protein.1–3 It causes severe impairment to memory and cognitive function, leading to behavioural and psychological symptoms such as depression, stress, anxiety, and mood disturbances with time.4 Currently AD is incurable, and the major risk factor of AD spreading is associated with aged population. The Aβ peptide exists in multiple assembly states such as monomers, oligomers, protofibrils, and fibrils.5 Decreased Aβ monomer levels coupled with increased Aβ oligomer (AβO) concentrations are considered one of the major neuropathologic biomarkers for AD. Current research efforts are aimed to develop therapeutic strategies to reduce Aβ burden via detection and inhibition of Aβ production to explore potential therapeutics against AD. Timely detection of different aggregation state of Aβ may be an useful diagnostic approach for preventive pharmacological intervention aimed at slowing down the disease at the early stage.We previously described6 the synthesis of a novel meso-substituted tricationic zinc-metalled porphyrin–peptide conjugate in which the carboxyphenyl group at mesoposition of the porphyrin core is linked via amide bond to the N-terminal amino function of the well-known hydrophobic pentapeptide Aβ16–20 (Lys-Leu-Val-Phe-Phe, named KLVFF).7 This zinc–porphyrin–peptide conjugate showed anti-aggregating properties and the ability to inhibit Aβ oligomer cytotoxicity. The porphyrin scaffold8,9 improved the water solubility of the peptide and enhanced the antifibrillogenic action of KLVFF.10–12 Using an experimental approach based on ion mobility mass spectrometry coupled with a multivariate statistical analysis we demonstrated the inhibition of a Zn–porphyrin–peptide conjugate in the early self-assembly of Aβ40 peptide.13
Herein we describe a new zinc-metallated porphyrin-peptide conjugate 5 (Fig. 1) in which the porphyrin macrocycle is linked via 1,2,3-triazole bond to the peptide Aβ16–20. The porphyrin–peptide conjugate 5 was obtained by the copper catalysed azide–alkyne cycloaddition (CuAAC),14,15 the selective “click” reaction between an alkyne and an azide group in the presence of Cu(I) as a catalyst.16
 | ||
| Fig. 1 Zinc–porphyrin-KPGKLVFF conjugate 5. | ||
The mild condition of this approach is compatible with the peptides chemistry, both on resin17,18 and in solution,19 and offers the possibility to develop peptide-based drugs, which could be potentially useful therapeutically.20,21 On the other hand, 1,2,3-triazoles are stable under oxidative and reductive conditions and hydrolysis, which makes this moiety more resistant to metabolism in living cells, compared to amides.22
Our aim was to generate, via “click-chemistry” a hybrid metallated porphyrin–peptide system capable of interacting with Aβ and at the same time to act for the detection of Aβ's aggregated species.8,23 We coupled the well-known ability of the KLVFF peptide to interact with the homologous sequence of full-length Aβ42 with the peculiar physical, optical, and electronic properties of the porphyrin macrocycle.24 Indeed, porphyrin possess attractive property making them versatile molecular platforms in a wide range of biomedical purposes.9,25 In the present study we demonstrate, by means of CD spectroscopy, that the presence of the porphyrin chromophore can be advantageously exploited as a molecular tool capable of evoking an induced dichroic signal in the presence of Aβ42.8,26
2. Experimental section
2.1. Materials
Peptide-synthesis grade N,N-dimethylformamide (DMF) and HPLC-grade CH3CN were purchased from Sigma-Aldrich. Rink Amide AM resin, Fmoc-protected amino acids (Fmoc-Gly-OH, Fmoc-Pro-OH, Fmoc-Lys(Boc)-OH, Fmoc-Phe-OH, Fmoc-Val-OH, Fmoc-Leu-OH) and the activators N,N′-diisopropylcarbodiimide (DIC) and Oxyma pure were purchased from Iris Biotech. Fmoc-Lys(N3)-OH was purchased from Sigma-Aldrich (Milan). Propargylamine was purchased from Chem Impex. 5-(4-carboxyphenyl)-10,15,20-(tri-N-methyl-4-pyridyl)porphyrin trichloride was purchased from Porphychem Company (France). Trifluoacetic acid (TFA) and scavengers for the cleavage of peptides from resin, triisopropyl silane (TIS), were purchased from Sigma Aldrich (Milano, Italy). Diisopropyl ether (iPr2O), click reagent copper (II) sulfate pentahydrate (CuSO4·5H2O), and sodium L-ascorbate were purchased from Sigma Aldrich (Milano, Italy). PyBOP, DIPEA, tert-butanol (tBuOH), tetrahydrofuran (THF), hexafluoroisopropanol (HFIP) were from Sigma-Aldrich (France). 4,4′-Dianilino-1,1′-binaphthyl-5,5′-disulfonic acid, dipotassium salt (Bis-Ans) was purchased from ChemCruz.Microwave assisted solid phase peptide synthesis (MW-SPPS) was performed by using an automatic peptide synthesizer Liberty Blue 2.0 (CEM Corporation, Matthews, NC, USA). Peptide was lyophilized using a Labconco FreeZone lyophilizer.
Analytical and preparative RP-HPLC were performed using a SHIMADZU LC-20A chromatography system equipped with a SPD-M20A photodiode array detector. Detection at 222 or 254 nm (absorption wavelength of peptide bond) and 400 nm (absorption wavelength of porphyrin). HPLC eluents were A: 0.1% TFA/H2O and B: 0.1% TFA/CH3CN.
MALDI-MS were recorded on the SCIEX TOF/TOF™ 5800 instrument using α-cyano-4-hydroxycinnamic acid (α-CHCA) as a matrix with thin layer deposition method. 0.1 mg of lyophilized samples were dissolved in 100 μL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.01 CH3CN/H2O/TFA. α-CHCA was prepared dissolving 4 mg/vial of matrices 1 mL of 30% acetonitrile in 0.3% TFA.
:1:0.01 CH3CN/H2O/TFA. α-CHCA was prepared dissolving 4 mg/vial of matrices 1 mL of 30% acetonitrile in 0.3% TFA.
High-resolution (HR) electrospray mass spectrometry (ESI-MS) were recorded using Q Exactive (Orbitrap) mass spectrometer (Thermo Fisher scientific instruments). The experimental conditions for spectra acquired in the positive ion mode were: spray voltage = 3.5 kV, capillary temperature 250°C; m/z range 200–2000, S-lens RF level 60 V, Sheath gas 5.
2.2. Synthesis of the mono-substituted N-(2-propynyl)benzamide porphyrin (2)
To a stirred solution of 5-(4-carboxyphenyl)-10,15,20-(tri-N-methyl-4-pyridyl)porphyrin trichloride (1) (50 mg, 0.06 mmol) and propargylamine (33.7 mg, 0.6 mmol, 10 eq.) in DMF (2 mL), the following coupling mixture: PyBOP (103 mg, 0.2 mmol, 3.3 eq.), HOBt (30.3 mg, 0.2 mmol, 3.3 eq.) and DIEA (69 μL, 0.39 mmol, 6.6 eq.) in DMF (2 mL) was added. The mixture was stirred at room temperature for 3 h. The crude porphyrin-derivative 2 was concentrated under vacuum and directly purified via preparative RP-HPLC on a Jupiter C4 250 × 21.2 mm (300 A pore size, AXIA Packet) column at a flow rate 10 mL min−1 using the following method: isocratic 10% B in 5 min, gradient 10–40% B in 15 min, isocratic 40% B in 10 min. Yield 55%. Compound 2 was characterized by analytical HPLC on a Jupiter C12 250 × 4.6 mm (Proteo 90 A pore size, AXIA Packet) column using the following method: isocratic 10% B in 5 min, gradient 10–80% B in 15 min, isocratic 80% B in 5 min, Rt 15.8. MALDI-MS [obsd: m/z (M + H)+ 743.19; calcd for C48H39N8O: 743.87] (Fig. S1, ESI†).2.3. Metallation of the mono-substituted N-(2-propynyl)benzamide porphyrin (2) with Zn
A solution of mono-substituted N-(2-propynyl)benzamide porphyrin (2) (30 mg, 0.04 mmol) in THF/acetic acid 1:1 (4 mL) was treated with Zn(CH3COO)2 (73.2 mg, 0.4 mmol, 10 eq.). The zinc(II) ion incorporation reaction into 2 was carried out, at room temperature, overnight. The crude metallated porphyrin derivative 3 was concentrated under vacuum and directly purified via preparative RP-HPLC on a Jupiter C12 250 × 21.2 mm (Proteo 90 A pore size, AXIA Packet) column, at a flow rate 10 mL min−1 using the following method: isocratic 0% B in 5 min, gradient 0–60% B in 20 min, isocratic 60% B in 10 min. Yield 60%. Compound 3 was characterized by analytical HPLC on a Jupiter C12 250 × 4.6 mm (Proteo 90 A pore size, AXIA Packet) column using the following method: isocratic 10% B in 5 min, gradient 10–80% B in 15 min, isocratic 80% B in 5 min, Rt 15.7. ESI-MS [obsd: m/z [M + H]+ 806.4, [M + H]2+ 403.68; calcd for C47H34ZnN9O: 805.4] (Fig. S2, ESI†).
2.4. Synthesis of the azido-peptide Ac-K(N3)PGKLVFF-NH2 (4)
The peptide was synthesized by a fully automated microwave-assisted solid phase peptide synthesis (MW-SPPS) following the Fmoc/tBu strategy on 0.1 mmol of Rink amide AM resin (0.32 mmol g−1, 100–200 Mesh). After resin swelling in DMF, Fmoc-amino acids (Fmoc-Phe-OH, Fmoc-Val-OH, Fmoc-Leu-OH, Fmoc-Lys(Boc)-OH, Fmoc-Gly-OH, Fmoc-Pro-OH and Fmoc-Lys(N3)-OH) were introduced through the following protocol: (1) Fmoc-deprotection by 20% piperidine in DMF; (2) washes (3×) with DMF; (3) couplings with Fmoc-amino acid (5 eq., 0.2 M in DMF), oxyma pure (5 eq., 1 M in DMF) and DIC (10 eq., 1 M in DMF) prepared in separated bottles; (4) washes (3×) with DMF. Both deprotection and coupling reactions were performed in a Teflon vessel with microwave energy and nitrogen bubbling. Reaction temperatures were monitored by an internal fiberoptic sensor.The resin was exposed to the microwave-assisted cycle described in Table 1.
| Step | Temperature (°C) | Power (W) | Time (s) |
|---|---|---|---|
| Deprotection | 75 | 175 | 15 |
| 90 | 37 | 50 | |
| 65 | 220 | 30 | |
| Coupling | 75 | 175 | 15 |
| 90 | 55 | 110 |
2.5. Cleavage of the peptide 4 from the resin.
Final cleavage of the peptide from the Rink Amide AM resin, with concomitant lysine side-chain deprotection, was achieved by treatment of resin-bound peptide with a TFA/TIS/H2O solution (95:5:5, 1 mL mixture/100 mg of resin). The cleavage was carried out for approximately 3 h at room temperature. The resin was filtered and rinsed with TFA (2 × 1 mL). The peptide solution was added to the washes and the product was precipitated by addition of ice-cold Diisopropyl ether (iPr2O) (40 mL). The crude peptide was washed with ice-cold iPr2O (3 × 30 mL), dried under vacuum, dissolved in H2O (10 mL) and lyophilized. The resulting crude peptide 4 was purified by preparative RP-HPLC on a Jupiter C12 250 × 21.2 mm (Proteo 90 A pore size, AXIA Packet) column, at a flow rate 10 mL min−1 by the following method: isocratic 10% B in 5, gradient 10–80% B in 15 min, isocratic 80% B in 10 min. Yield 65%. The product was characterized by analytical HPLC on a Jupiter C12 250 × 4.6 mm (Proteo 90 A pore size, AXIA Packet) column using the following method: isocratic 10% B in 5 min, gradient 15–55% B in 15 min, isocratic 55% B in 5 min, Rt 18.3. ESI-MS [obsd: m/z (M + H)+ 1002.73; (M + Na)+ 1024.80; calcd for C50H75N13O9:1002.21] (Fig. S3, ESI†).
2.6. Synthesis of zinc–porphyrin–peptide conjugate 5via “click-chemistry”.
The metallated porphyrin derivative 3 (20 mg, 0.02 mmol) was conjugated with the azido peptide Ac-K(N3)PGKLVFF-NH2 (4) (40 mg, 0.04, 2 eq.) using CuSO4 (1.6 mg, 0.01 mmol, 0.5 eq.) reduced to Cu+ with ascorbic acid (5 mg, 0.028, 1.4 eq.). The click reaction was carried out in THF/1-But-ol/H2O (10 mL) 2/1/0.5 at 50 °C, overnight and was monitored by analytical HPLC on a Jupiter C12 250 × 4.6 mm (Proteo 90 A pore size, AXIA Packet) column, at a flow rate 1.25 mL min−1 by the following method: isocratic 10% B in 5 min, gradient 10–55% B in 15 min, isocratic 55% in 10 min, Rt 16.8 min (Fig. S4, ESI†).After completion of the reaction, the solvent was removed under vacuum and the crude product 5 was purified by preparative HPLC on a Jupiter C4 250 × 21.2 mm (300 A pore size, AXIA Packet) column, at a flow rate 10 mL min−1 by the following method: isocratic 10% B in 5 min, gradient 10–60% B in 10 min, isocratic 60% B in 10 min. Yield 25%. MALDI-MS [obsd: m/z (M + H)+ 1807.8; calcd for C98H110N21O10Zn: 1807.5] (Fig. S5, ESI†).
2.7. Monomerization: general procedure
The peptide was dissolved in TFA (1 mg mL−1) and sonicated for 10 min. Then the TFA was evaporated under a gentle stream of nitrogen until the peptide is reduced to a thin film adhering to the vial. Subsequently 1 mL of HFIP was added to the peptide. HFIP is well known for its ability to stabilize local hydrogen bonds between residues close in the amino-acid sequence, particularly those forming α-helices, it can disrupt the β-conformation by destabilizing hydrophobic interactions.After 1 h incubation at 37 °C, the peptide solution was dried under a stream of azote, the peptide film was dissolved in 2 mL HFIP, dried under azote stream to remove remaining trace of TFA, again dissolved in 1 mL HFIP and frozen at −80 °C for 4 or 5 hours, then lyophilized overnight.
2.8 Circular dicroism (CD) spectroscopy
CD were recorded on a J-810 spectrometer (Jasco, Japan) under a constant flow of N2 at room temperature. The CD measurements were performed in the UV regions (190–260 nm and 190–500 nm) using a 1 cm path length quartz cell. All aggregation kinetics were carried out in 120 hours, by incubation at 37 °C and there were collected 10 scans spectra with a scan interval of 24 h. The CD spectra were recorded for Aβ42 (5 μM and 20 μM) monomer in the absence or in the presence of porphyrin–peptide (5 μM and 20 μM for the 1:1 molar ratios). Monomerized samples of Aβ42 (5 μM and 20 μM) were solubilized in 50 μL of NaOH 20 mM (2.5%) and then diluted with 1950 μL of 10 mM phosphate buffer at pH 7.4 whether or not containing the conjugate 5.
2.9. UV-vis spectroscopy
UV-vis spectra were recorded on Jasco V-670 spectrophotometer. All measurements were performed in the UV region 300–600 nm using a 1 cm path length quartz cell and by incubation at 37 °C over time.2.10. Fluorescence spectroscoy
Fluorescence spectra were recorded on a Horiba Fluoromax-4 spectrofluorometer from 440 to 800 nm in 1-nm steps, at an excitation wavelength λexc = 425 nm, at room temperature. The final fluorescence spectra represent averages of at least three measurements. Bandwidths 5nm for both excitation and emission were used.
2.11. Dynamic light scattering (DLS)
DLS measurements were carried out on a ZetaSizer NanoZS90 Malvern instrument (UK), equipped with a 633 nm laser at a scattering angle of 90°and 25 °C temperature. The size of peptide-conjugate 5 was determined at 5 μM and 20 μM in 10 mM phosphate buffer at pH 7.4. Each measurement was performed three times.2.12. 4,4′-Dianilino-1,1′-binaphthyl-5,5′-disulfonic acid (Bis-Ans) fluorescence assay
Bis-Ans fluorescence kinetics were measured on a Flash Thermo Varioskan spectrofluorometer with excitation and emission at 385 and 510 nm, respectively.Aβ42 alone and in the presence of the porphyrin–peptide conjugate 5 in 1:1 molar ratio was dissolved in 10 mM aq. NaOH (30 μL). The samples were diluted (to 150 μL) with 60 μM Bis-Ans solution in 10 mM phosphate buffer at pH 7.4 to obtain a final Aβ42 concentration of 20 μM. The samples were incubated a 37 °C in a 96-well plate. To minimize evaporation effects the multiwall plate was sealed with a transparent heat-resistant plastic film. Readings were taken every 10 min, after weak shaking for 10 s. The fluorescence intensity was monitored for 65 h. The measurements were performed in triplicate.
2.13. Cell cultures and MTT assay
The neuroblastoma cell line, SH-SY5Y, was maintained in DMEM-F12 (Gibco, ThermoFisher) supplemented with 10% heat inactivated (HI) fetal calf serum (Gibco, ThermoFisher), 100 mg mL−1 penicillin and streptomycin (Gibco, ThermoFisher), and 2 mM L-glutamine at 37 °C, 5% CO2. Two weeks before experiments, 5 × 103 cells were plated on 96-well plates in DMEM-F12 with 5% HI fetal calf serum. The percentage of serum was gradually decreased until it was 1% of the total. All-trans-retinoic acid (RA) (Sigma), 5 μM, was used to promote neuronal differentiation, and medium-containing RA was changed every 3 days.The lyophilized sample 5 was dissolved in water at a 1 mM concentration stock solution. Fully differentiated SH-SY5Y cells were treated with increasing concentrations (0.2 μM, 2 μM, 10 μM and 20 μM) of the porphyrin–peptide conjugate 5 and the relative controls, compound 1 and KLVFF, dissolved respectively in water and dimethyl sulfoxide (DMSO) at a 1 mM concentration stock solution. After 48 h treatment, cultures were incubated with MTT (5 mg mL−1 stock solution) for 2 h at 37 °C and then lysed with DMSO, and the formazan production was evaluated in the multiplate reader Victor Nivo (Milan, Italy) through the absorbance at 570 nm.
3. Results and discussion
3.1. Synthesis of the Zn–porphyrin–peptide conjugate
The Zn–porphyrin–peptide conjugate 5 was obtained by an hybrid solid/solution phase approach (Fig. 2), through appropriate building blocks for the CuAAC click reaction: the mono-substituted N-(2-propynyl)benzamide porphyrin 2 and the azido-peptide Ac-K(N3)PGKLVFF-NH2 (4). The alkyne group was incorporated to the 5-(4-carboxyphenyl)-10,15,20-(tri-N-methyl-4-pyridyl)porphyrin (1) by coupling of the propargylamine with PyBOP/HOBt and DIEA in DMF. The intermediate N-(2-propynyl)benzamide-containing porphyrin (2) was metallated with Zn using a strong excess of Zn(CH3COO)2 in THF/acetic acid to obtain the zinc–porphyrin derivative 3. Regarding the peptide, the N-terminal GPG motif, previously inserted on the KLVFF,6 was substituted into the azido sequence Ac-K(N3)PG. The azido peptide Ac-K(N3)PGKLVFF-NH2 (4) with N-terminal acetylation and C-terminal amidation, was synthesized by microwave-assisted solid phase peptide synthesis (MW-SPPS) based on the 9-fluorenylmethoxycarbonyl (Fmoc)/tBu strategy and DIC (N,N′-diisopropylcarbodiimide)/oxyma pure as coupling reagents.27 This coupling system is safe and cheap, and does not require tertiary bases unlike the onium coupling methods (i.e. HBTU). | ||
| Fig. 2 Reagents and conditions for the preparation of the zinc–porphyrin–peptide conjugate 5. (a) Propargylamine (10 eq.)/PyBOP (3.3 eq.)/HOBt (3.3 eq.) and DIEA (6.6 eq.) in DMF, rt, 3 h. (b) Zn(CH3COO)2 (10 eq.) in THF/acetic acid 1:1, r.t., O/N. (c) MW-SPPS by Fmoc-aa/DIC/Oxyma (5/10/5) and cleavage by TFA/H2O/TIS (95/2.2/2.5). (d) THF/1-But-ol/H2O 2/1/0.5 CuSO4. | ||
Additionally, DIC is suitable in terms of time and solvent consuming since, it is stable at 90 °C (temperature reached in the microwave conditions) and requires lower DMF washing volumes because of its higher solubility compared to more classic coupling reagents.28,29 Moreover, the Oxyma pure is a good non-explosive substitute of HOBt.
Crude Aβ16–20-azido-peptide, Ac-K(N3)PGKLVFF-NH2 (4), was obtained by treating the peptidyl-rink amide AM resin with TFA and triisopropysilane/water as scavengers. After purification by preparative RP-HPLC, the peptide 4 was conjugated with the N-(2-propynyl)benzamide-containing porphyrin (3) by the click reaction, carried out in THF/1-But-ol/H2O 2/1/0.5 using CuSO4 reduced to Cu+ with ascorbic acid at 50 °C overnight, to obtain the Zn–porphyrin–peptide conjugate 5 (Fig. 2).
3.2. Characterization by UV-vis spectroscopy of the Zn–porphyrin–peptide conjugate 5 and the porphyrin derivatives 1–3
The peptide derivatives, including the Aβ42, were subjected to a monomerization protocol to remove pre-aggregated seeds just before performing any experiments. This ensures sample solutions containing monomeric peptide species to be obtained.30The UV-visible absorption of compound 1 (Fig. 3a, blue colour line) showed a characteristic intense band at approximately 422 nm corresponding at the S2 → S0 transition (the Soret band), followed by three weaker absorption bands (Q bands) from 450 to 600 nm (see inset Fig. 3a). Incorporation of the alkyne group to the porphyrin derivative 2 (Fig. 3a, orange colour line) slightly shifts the absorption maximum of Soret band toward higher wavelengths (427 nm) whereas a change of the Q bands profile was observed. After the metallation by zinc (compound 3) (Fig. 3a, grey colour line) the absorption maximum of Soret band is almost unaffected (426 nm) while the UV profile in the Q bands region revealed reasonable changes. The absorption intensity of the porphyrin intermediates 1–3 remains nearly unchanged, while it increases slightly after the conjugation of the porphyrin to the peptide (compound 5, Fig. 3b).
 | ||
| Fig. 3 UV-vis spectra of the porphyrin intermediates 1–3 at 5 μM (inset: zoom-in 490–600 nm) (a) and of the Zn–porphyrin–peptide conjugate 5 at 5 μM recorded 0, 24, 48 and 120 hours (b). | ||
To evaluate the self-assembly capacity of the KLVFF-porphyrin peptide conjugate 5 over time, UV-vis spectra were also acquired at 0, 24, 48 and 120 hours at 5 μM (Fig. 3b). The UV-vis spectra of conjugate 5 showed a Soret band with a λmax near 424 nm, almost unchanged over time, so we can say that the self-aggregation at 5 μM can be overlooked. The spectra also revealed a clear modification of the Q bands, compared to Fig. 3a, due to the conjugation of porphyrin to the peptide.
3.3. Characterization by fluorescence spectroscopy of the Zn–porphyrin–peptide conjugate 5
Considering potential interaction of compound 5 with cells, the fluorescence spectra of the Zn–porphyrin–peptide conjugate 5 at 5 μM were recorded at 0 and 24 h (Fig. 4b) and compared with the fluorescence spectra acquired for the porphyrin 1 (Fig. 4a). The fluorescence intensities of the compound 1 is an order of magnitude larger than the fluorescence intensities of the peptide conjugate 5.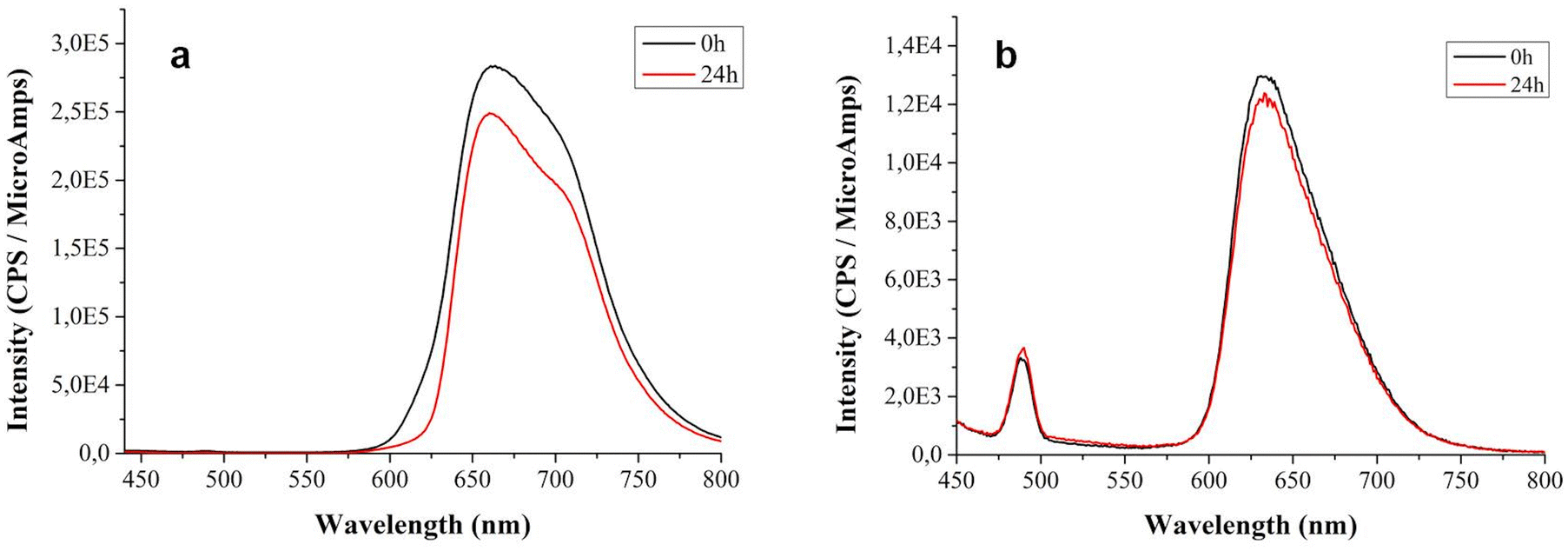 | ||
| Fig. 4 Fluorescence spectra of the compound 1 (a) and 5 (b) (excitation wavelength at 425 ± 5 nm) at 5 μM collected in the time interval 0–24 h. | ||
The signal observed in the spectrum of the compound 1, centered at 660 nm, exhibits a shoulder peak that is no longer visible in the spectrum of conjugate 5, where a slight, blue-shifted band centered at 630–631 nm was observed. After 24 h, the fluorescence intensity of the compound 1 slightly decrease while is almost the same for the Zn–porphyrin–peptide conjugate 5, so we can say that the peptide functionalization of the porphyrins result in the reduction of its known aggregation propensity. This result is in accordance with the UV-vis data reported above suggesting that no self-aggregation phenomena are evident for the compound 5.
3.4. Dynamic light scattering (DLS)
To determine the presence of aggregates in solution and to monitor their growth over time, DLS measurements at 5 μM and 20 μM, were collected at t = 0 and 24.In Fig. 5 the size distribution of scattering objects by intensity indicates that at 5 μM the freshly prepared compound 5 (t = 0), forms aggregates with an average size of 171 nm. After 24 h the DLS profile indicated the presence of larger aggregates (362 nm). Data collected at 20 μM indicated the increase of the dimension aggregates at the initial stage (531 nm) along with a very low percentage of bigger aggregates with size in the micrometric range (5560 nm). After 24 h the presence of sedimentation was observed and the majority of the scattering particles displayed a lower diameter size of around 356 nm, and the same bigger aggregates with size in the micrometric range.
 | ||
| Fig. 5 DLS size distributions by intensity (%) for Zn-peptide conjugate 5 at t = 0 and after 24 h in phosphate buffer 10 mM. | ||
We performed additional DLS experiment by decreasing the concentration of the peptide 5 to verify whether the already observed aggregates disappear/dissolve but the low concentration of the samples provided DLS measurements whose reliability must be taken with caution. However, we cannot totally exclude that at a certain diluted concentration the conjugate 5 would not present aggregated forms in solution.
3.5. Studying the interaction of the zinc–porphyrin–peptide conjugate 5 with Aβ42 by UV-vis, CD, and Bis-Ans fluorescence measurements
:1). The UV-vis spectra, recorded at 0, 24, 48 and 120 h, showed that the absorption intensity of conjugate 5 decreased gradually over time (see inset Fig. 6), and the presence of a precipitate was evident after 120 h.
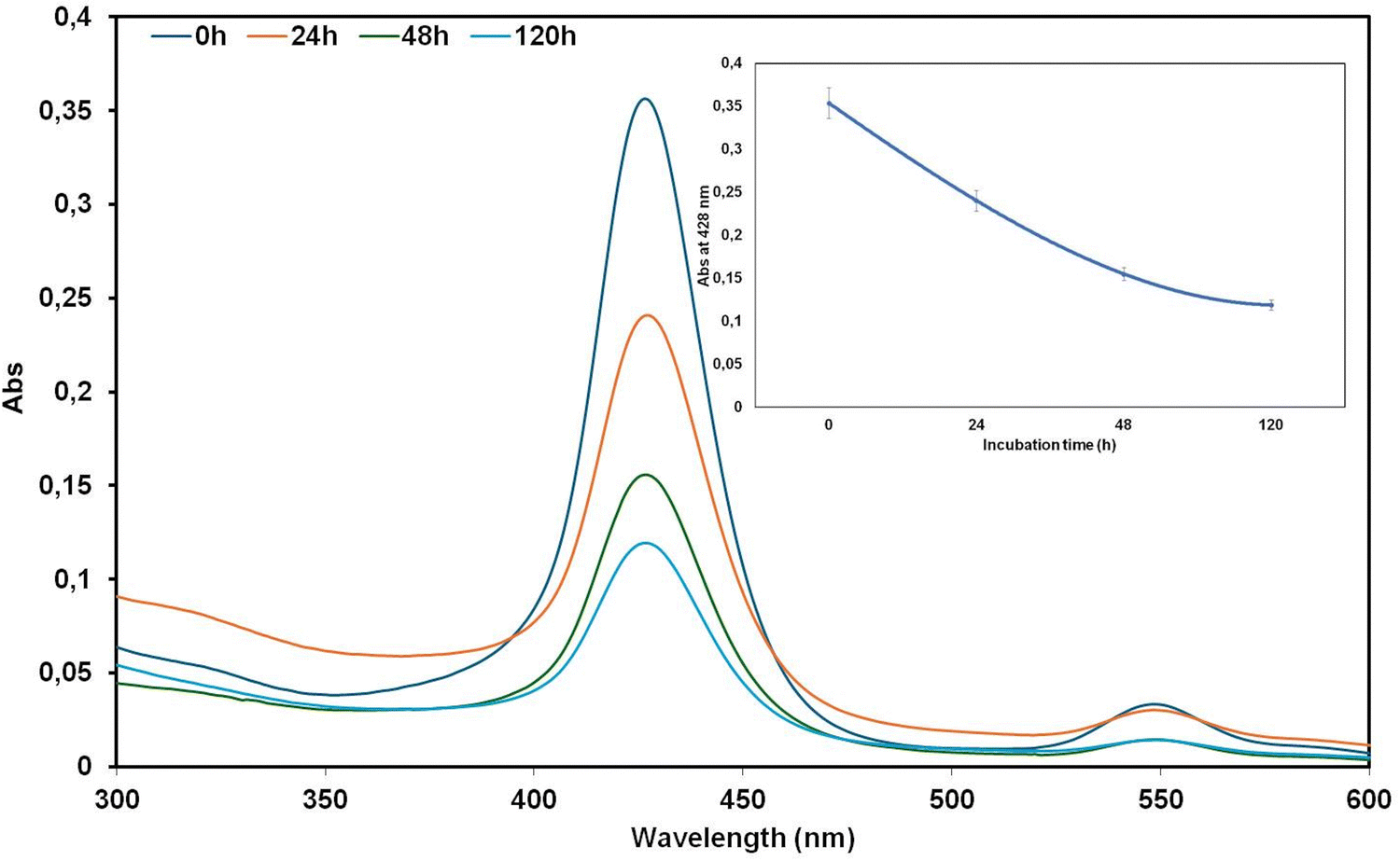 | ||
| Fig. 6 UV-vis spectra of the binary system Aβ42/5 (1:1) at 5 μM recorded at 0, 24, 48 and 120 h (inset: absorbance at 428 nm vs. incubation time). | ||
As the UV-vis spectra of the peptide-conjugate 5 at 5 μM alone (see Fig. 3b above) showed a stability over the time, from Fig. 6 we can suppose a co-aggregation Aβ42/5.
 | ||
| Fig. 7 CD Spectra recorded at different times (0, 24, 48. 120 h) of Aβ42 (a) and peptide-conjugate 5 at 5 μM (b). | ||
The CD spectra of the zinc–porphyrin–peptide conjugate 5 acquired at 5 μM (Fig. 7b), revealed an inherent propensity to the transition from random-coil to helix-type structures over time.
In order to assess the capability of the peptide conjugate 5 to affect the Aβ's aggregation, CD analyses of Aβ42 (at 5 μM) in the far UV region were recorded in the presence of porphyrin–peptide conjugate 5 at 1:1 molar ratio (Fig. 8). The resultant CD profiles were obtained by subtracting the contribute of the CD activity of the porphyrin–peptide conjugate 5. It turned out that the porphyrin–peptide conjugate 5 can interfere with the aggregation process of Aβ42 and stabilize the Aβ42's random coil conformation at least for 24 h. After this time, compound 5 is no longer able to prevent the conformational change of Aβ42 toward the β-sheet structure.
 | ||
| Fig. 8 CD profiles obtained by subtracting the contribute of the CD activity of the porphyrin–peptide conjugate 5 from the curve observed from the Aβ42/5 binary system at 5 μM, recorded at different times (0, 24, 48. 120 h). | ||
To accelerate the formation of aggregated species at the expenses of monomers in solution, we carried out CD experiments using 20 μM Aβ42 samples. The curve profiles obtained in the 195–350 nm wavelength region gave evidence of this: at t 0 h the CD spectrum revealed a strong negative ellipticity around 200 nm typical of a mixing between random coil and structured peptide chain (Fig. 9a).
 | ||
| Fig. 9 CD spectra in the UV region (195–350 nm) of Aβ42 at 20 μM (a) and of Aβ42/5 binary system (b) at 20 μM, recorded at different times (0, 24, 48. 120 h). *CD curve at 0 h was recorded in the interval 200–350 nm due to the distorted signal below 200 nm. | ||
As the incubation progressed the CD profile rapidly turned into the distinctive curve of the β-sheet structure, with a single minimum at 216 nm, an x-axis intercept at 202 nm and a maximum shift at a wavelength near 195 nm.31
Moreover, the CD spectra of Aβ42, acquired in the presence of the porphyrin–peptide conjugate 5 (Fig. 9b), confirmed the ability of 5 to stabilize the Aβ42 random coil conformation up to 48 h. The reduction of the negative ellipticity at 200 nm as the incubation time proceeds, indicated the transition of Aβ42 towards the β-sheet conformation that generally preludes the oligomerization process of Aβ42.
We also performed CD experiments of the Aβ42/5 mixture in the 350–500 nm visible region to explore any ability of the porphyrin–peptide conjugate 5 and to reveal early events of Aβ42 aggregation (Fig. 10a). We observed that the Aβ42 aggregation process was also associated to the appearance of an induced negative dichroic signal, that increase over time, nearly to the porphyrin's Soret absorption band (λmax 440 nm) (Fig. 10a).
 | ||
| Fig. 10 CD spectra in the vis region (350–500 nm) of Aβ42/5 binary system at 20 μM (a) and of zinc–porphyrin–peptide conjugate 5 at 20 μM (b). | ||
Noteworthy, the CD spectra of the porphyrin–peptide conjugate 5 alone in the 350–500 nm acquired at 20 μM did not exhibit any dichroic signal in the visible region (Fig. 10b). Such evidence strongly suggests that the growth of the dichroic band relates to the interaction of 5 with the aggregated species of Aβ42. These results points out the possibility to use porphyrin–peptide conjugate 5 as a molecular probe for detection of Aβ aggregate forms in AD.
:1 molar ratio. The kinetic profile of 5 was also recorded for comparison.
Fig. 11 shows the bar graphic of the Bis-Ans fluorescence intensities collected at the different interval of time. It is clear from Fig. 10 that, in the presence of compound 5 the aggregation process of Aβ42 is slightly affected, practically reproducing a similar trend as in Aβ. This result is in keeping with CD data where the conjugate 5 revealed its ability to interfere with the fibrillogenic process of Aβ42 however without preventing the conformational transition towards the β-sheet structure. Moreover, in accord to CD spectra (described above), a propensity of the zinc–porphyrin–peptide conjugate 5 at 20 μM to self-aggregation is evident.
 | ||
| Fig. 11 Bis-Ans Fluorescence intensities of 5 in the presence or absence of Aβ42 compared with the value of Aβ42 alone at the same time. | ||
3.6. Cytotoxicity study
To exclude any potential toxic effect of porphyrin–peptide conjugates 5, we evaluated the cell viability by using MTT assay. We chose the human neuroblastoma cell line, SH-SY5Y, fully differentiated by all-trans retinoic acid to obtain a widely used neuronal-like model. Interestingly, no toxicity was observed for the porphyrin–peptide conjugate 5 and its relative controls, compound 1 and KLVFF (Fig. S8, ESI†) at the concentrations used (0.2 μM, 2 μM, 10 μM, 20 μM) after 48 h exposure (Fig. 12). The molecular integrity of the porphyrin–peptide conjugates 5 after 48 h at 37 °C in presence of 1% FCS was confirmed by HPLC (Fig. S9, ESI†). | ||
| Fig. 12 MTT assay of fully differentiated SH-SY5Y treated for 48 hours with increasing concentrations of compound 5 (0.2, 2, 10, 20 μM). Bars represent means ±SEM of three independent experiments with n = 3 each. | ||
4. Conclusions
The aggregation of Aβ42 is believed to play a key role in the onset and development of AD. It is widely accepted that small soluble Aβ42 aggregates are toxic, and their detection might represent a great strategy for early diagnosis and therapy of AD. In the present study a new conjugate was obtained by click-reaction between an alkyne-porphyrin and the azido-peptide of KLVFF 5. The data obtained showed that the conjugate 5 differently interacts with Aβ and can affect the amyloid aggregation although it cannot totally prevent the β-sheet transition. The spectroscopic feature of porphyrin-moiety endows this peptide conjugate with diagnostic properties, potentially enabling the detection of soluble Aβ aggregates in biological fluids. The CD spectra recorded in the visible region demonstrated that the zinc-containing porphyrin–peptide conjugate 5 can generate an induced CD signal upon of β-sheet structure formation of Aβ42. This result along with the lack of toxicity on neuronal cells further support the potential use of this new zinc–porphyrin–peptide conjugate as an “Aβ sensor” in AD, laying the groundwork for future and more in-depth investigations.Author contributions
Conceptualization, G. P.; methodology and data curation, G. S., R. T., S. Z., G. D. N., M. L. G., T. C.; writing – original draft preparation, G. S., G. D. N., S. Z.; writing – review and editing, G. S. G. P. G. D. N.; visualization, G. S., G. D. N.; supervision, G. P. and G. S.; funding acquisition, G. P. All authors have read and agreed to the published version of the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or its ESI†].Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of the Sicilian MicronanoTech Research and Innovation Center - SAMOTHRACE (Ecosistema dell’Innovazione - ECS00000022) is gratefully acknowledged. We are grateful to Dr M.G.L. Consoli for her support in the DLS experiments.References
- G. G. Glenner and C. W. Wong, Biochem. Biophys. Res. Commun., 1984, 120, 885–890 CrossRef CAS PubMed.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- M. A. Busche and B. T. Hyman, Nat. Neurosci., 2020, 23, 1183–1193 CrossRef CAS PubMed.
- C. L. Masters, R. Bateman, K. Blennow, C. C. Rowe, R. A. Sperling and J. L. Cummings, Nat. Rev. Dis. Primer, 2015, 1, 1–18 Search PubMed.
- G. Chen, T. Xu, Y. Yan, Y. Zhou, Y. Jiang, K. Melcher and H. E. Xu, Acta Pharmacol. Sin., 2017, 38, 1205–1235 CrossRef CAS PubMed.
- V. Villari, R. Tosto, G. D. Natale, A. Sinopoli, M. F. Tomasello, S. Lazzaro, N. Micali and G. Pappalardo, ChemistrySelect, 2017, 2, 9122–9129 CrossRef CAS.
- L. O. Tjernberg, D. J. E. Callaway, A. Tjernberg, S. Hahne, C. Lilliehöök, L. Terenius, J. Thyberg and C. Nordstedt, J. Biol. Chem., 1999, 274, 12619–12625 CrossRef CAS PubMed.
- B. I. Lee, S. Lee, Y. S. Suh, J. S. Lee, A. Kim, O.-Y. Kwon, K. Yu and C. B. Park, Angew. Chem., Int. Ed., 2015, 54, 11472–11476 CrossRef CAS PubMed.
- N. Tsolekile, S. Nelana and O. S. Oluwafemi, Molecules, 2019, 24, 2669 CrossRef CAS PubMed.
- T. L. Lowe, A. Strzelec, L. L. Kiessling and R. M. Murphy, Biochemistry, 2001, 40, 7882–7889 CrossRef CAS.
- R. Kino, T. Araya, T. Arai, Y. Sohma and M. Kanai, Bioorg. Med. Chem. Lett., 2015, 25, 2972–2975 CrossRef CAS PubMed.
- T. Arai, D. Sasaki, T. Araya, T. Sato, Y. Sohma and M. Kanai, Chembiochem Eur. J. Chem. Biol., 2014, 15, 2577–2583 CrossRef CAS PubMed.
- S. Lazzaro, N. Ogrinc, L. Lamont, G. Vecchio, G. Pappalardo and R. M. A. Heeren, Anal. Bioanal. Chem., 2019, 411, 6353–6363 CrossRef CAS PubMed.
- K. Ladomenou, V. Nikolaou, G. Charalambidis and A. G. Coutsolelos, Coord. Chem. Rev., 2016, 306, 1–42 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- A. Stefanucci, W. Lei, S. Pieretti, E. Novellino, M. P. Dimmito, F. Marzoli, J. M. Streicher and A. Mollica, Sci. Rep., 2019, 9, 5771 CrossRef PubMed.
- A. D’Ercole, G. Sabatino, L. Pacini, E. Impresari, I. Capecchi, A. M. Papini and P. Rovero, Pept. Sci., 220, 112(4), e24159 CrossRef.
- W. Tang and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013–7039 RSC.
- H. Li, R. Aneja and I. Chaiken, Molecules, 2013, 18, 9797–9817 CrossRef CAS PubMed.
- S. M. Kondengadan, S. Bansal, C. Yang, D. Liu, Z. Fultz and B. Wang, Acta Pharm. Sin. B, 2023, 13, 1990–2016 CrossRef CAS PubMed.
- C. D. Hein, X.-M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216–2230 CrossRef CAS.
- Y. Fan, D. Wu, X. Yi, H. Tang, L. Wu, Y. Xia, Z. Wang, Q. Liu, Z. Zhou and J. Wang, ACS Omega, 2017, 2, 4188–4195 CrossRef CAS PubMed.
- F. Biscaglia and M. Gobbo, Pept. Sci., 2018, 110, e24038 CrossRef.
- R. Boscencu, N. Radulea, G. Manda, I. F. Machado, R. P. Socoteanu, D. Lupuliasa, A. M. Burloiu, D. P. Mihai and L. F. V. Ferreira, Molecules, 2023, 28, 1149 CrossRef CAS.
- A. Hirabayashi, Y. Shindo, K. Oka, D. Takahashi and K. Toshima, Chem. Commun., 2014, 50, 9543–9546 RSC.
- S. R. Manne, A. Sharma, A. Sazonovas, A. El-Faham, B. G. de la Torre and F. Albericio, ACS Omega, 2022, 7, 6007–6023 CrossRef CAS PubMed.
- A. D’Ercole, L. Pacini, G. Sabatino, M. Zini, L. Milli, F. Nuti, A. Ribecai, A. Paio, P. Rovero and A. M. Papini, Org. Process Res. Dev., 2021, 25(3), 552–563 CrossRef.
- G. Sabatino, A. D’Ercole, L. Pacini, M. Zini, A. Ribecai, A. Paio, P. Rovero and A. M. Papini, Org. Process Res. Dev., 2021, 25, 552–563 CrossRef CAS.
- M. L. Giuffrida, F. Caraci, B. Pignataro, S. Cataldo, P. De Bona, V. Bruno, G. Molinaro, G. Pappalardo, A. Messina, A. Palmigiano, D. Garozzo, F. Nicoletti, E. Rizzarelli and A. Copani, J. Neurosci., 2009, 29, 10582–10587 CrossRef CAS.
- G. D. Fasman, Circular Dichroism and the Conformational Analysis of Biomolecules, Springer Science & Business Media, 2013 Search PubMed.
- N. D. Younan and J. H. Viles, Biochemistry, 2015, 54, 4297–4306 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj02162b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |