
Polydopamine-functionalized polymer particles as templates for mineralization of hydroxyapatite: biomimetic and in vitro bioactivity†
Junli Cui‡
a,
Chao Ma‡b,
Zhenni Lia,
Longyun Wub,
Wei Weic,
Min Chen*b,
Bo Peng*d and
Ziwei Deng*a
aSchool of Materials Science and Engineering, Shaanxi Normal University, Xi'an, 710062, China. E-mail: zwdeng@snnu.edu.cn; Tel: +86-29-81530804
bThe Affiliated Drum Tower Hospital of Nanjing University Medical School, Nanjing, 210008, China. E-mail: croweminchan@aliyun.com
cDepartment of Gastrointestinal Surgery, The Second Affiliated Hospital of Nanjing Medical University, Nanjing, 210011, China
dDepartment of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QZ, UK. E-mail: pengbo006@gmail.com; Tel: +44 (0)1865285417
First published on 12th January 2016
Abstract
The use of polymer particles as organic templates for producing inorganic materials is an intriguing approach, because it offers the synthetic feasibility of organic–inorganic hybrid materials which may promise a variety of interesting applications. Here, inspired by mussel-adhesion phenomena in nature, we combined the use of the active surface of polydopamine (PDA) with biomimetic mineralization of hydroxyapatite (HAP) to synthesize a novel HAP-based, organic–inorganic hybrid material. Monodisperse micro-sized polystyrene (PS) particles were first prepared as inert polymer templates via dispersion polymerization, which, subsequently, were coated with PDA through the self-polymerization of dopamine in a weakly alkaline aqueous environment (pH = 8.5). This chemical modification activated the inert surface of PS particles to become bioactive. Thus, these synthesized PS/PDA composite particles were ideal templates for the biomimetic mineralization of HAP when they were incubated in a simulated body fluid (SBF) solution in aid of the biocompatible PDA layer. During the PDA-assisted biomimetic mineralization, the PDA layer interacted with the mineral ions introduced, and anchored the mineralization of HAP exclusively onto the surfaces of the composite particles. The detailed structures and morphologies of HAP-based hybrid materials (PS/PDA/HAP hybrid materials) were characterized. These HAP-based hybrid materials did not show a significant in vitro cytotoxicity against HeLa cells, indicating their potential biocapacity for drug delivery, protein and gene transfer, and as coating materials for bioimplants or as the injectable scaffold materials in bone tissue engineering for defect filling and regeneration.
Introduction
In nature, biological organisms employ a whole range of organic–inorganic hybrid materials with complex structures to fulfill various biological functions, such as mechanical support, navigation, storage, protection and defense.1,2 Intrigued by these features, the mimicking of natural mineralization processes for advanced organic–inorganic hybrid materials under ambient environments has received significant attention in recent years.3,4 Among them, natural bone, one of the most widely studied hybrid biomaterials, originates from the organized mineralization of HAP (Ca10(PO4)6(OH)2) which spatially aligns the growth between collagen fibrils,5 exhibiting numerous unique chemical and mechanical properties for living beings. Recently, the understanding and ultimately mimicking of such a hierarchical organization of bone tissue, has provided new routes for the fabrication of specialized organic–inorganic hybrid materials that possess compositional and partly structural analogies to natural bones.6,7 Due to the feasibility to control polymorphism, crystal structure and orientation, and crystal organization, biomimetic mineralization has been extensively explored to synthesize a series of bone-like HAP minerals.7 To obtain the artificial bone-like materials, a variety of organic materials (such as collagen, biopolymers, biodegradable synthetic polymers, organoapatites, and supramolecular assemblies) have been employed as organic matrixes for the biomimetic mineralization of HAP.7–10 Moreover, extensive studies have been performed in search of an optimal environment for the synthesis of biologically relevant inorganic materials via a controllable assembly. In general, the compatibility or adhesion at the interfaces between inorganic components and organic matrixes is crucial for a successful formation of organic–inorganic hybrid materials.11 Unlike the natural organic materials, such as collagen, osteonectin and sialoprotein, which can nucleate and align to HAP crystals, thus forming overall shapes with cell-adhesive moieties,12 many current synthetic materials are bio-inertness and lack of interfacial bioactivity, which are the significant hurdles for the subsequent fabrication of organic–inorganic hybrid biomaterials. Therefore, the development of a simple and universal way towards the construct of biointerfaces is eagerly required to integrate the natural inorganic crystals (e.g. HAP) into those synthetic materials, which may finally lead to a new generation of hybrid biomaterials.Mussels, which are underwater fouling organisms, enable to achieve a long-lasting adhesion in a wet environment by attaching to any natural or synthetic substrates based on the proteins secreted in their byssus.13 Inspired by the mussel-adhesion phenomena in nature, a universal modification of a surface has been reported, in which the self-polymerization of dopamine produces a thin surface-adherent PDA coating onto various surfaces. Thanks to their rich amount of active catechol and amine groups, these PDA coatings can be served as a universal adherent platform for various secondary surface-mediated reactions.14,15 Up to now, this mussel-inspired PDA-based surface modification has been widely applied to various fields, such as biomimetic mineralization, surface-initiated polymerization, surface decoration and functionalization of nanomaterials.16,17 More importantly, due to their excellent adhesive versatility with organic and inorganic materials, PDA coatings provide a simple, yet universal way for the construction of biointerfaces, through which natural inorganic components (e.g. HAP,18,19 silica,20 and metal nanoparticles21,22) can be readily integrated into diverse synthetic materials. Hence, this PDA-assisted biomimetic mineralization approach presents a new paradigm to create novel organic–inorganic hybrid materials.
Herein, we combined mussel-inspired surface chemistry with active biomimetic mineralization to explore a simple and efficient approach for the synthesis of HAP-based, novel organic–inorganic hybrid materials, which could be promising for biomedical applications. In this approach, monodisperse micron-sized PS particles were first prepared via dispersion polymerization. PS particles were chosen as the synthetic polymer templates mainly because of their unique advantageous features, such as, their easily tunable size, shape and monodispersity, diverse and controllable surface group, inert and stable in aqueous phase, and commercial availability.23–25 Moreover, they hold a great promise for biomedical applications, for instance, cell culture, biomarker, and tissue engineering.26–31 Considering the bio-inertness and lack of interfacial bioactivity of the pristine PS particles made from dispersion polymerization, their surface functionality is vital for the HAP mineralization. It has been shown that mussel-inspired PDA coatings were excellent bio-platform which can easily be decorated onto the surface of pristine PS particles upon the self-polymerization of dopamine in a weakly alkaline aqueous environment.20,21 After decoration with a thin PDA coating, the surface of PS/PDA composite particles offered a good site for in situ mineralization of HAP in a physiological condition (simulated body fluid SBF solution), forming PS/PDA/HAP hybrid materials. Thanks for mussel-inspired PDA-based surface modification, these PDA coatings can also construct the bioactive interfaces on a wide variety of synthetic and bio-inertness materials, not only PS particles.14 Thus, this PDA-assisted biomimetic mineralization can readily integrate HAP into diverse synthetic materials, which presents a powerful approach to fabricate HAP-based, novel organic–inorganic hybrid materials regardless of type and shape of substrate materials.18 In addition, the biocompatibility of these HAP-based hybrid materials was evaluated using the Cell Counting Kit-8 assay. They did not show a significant in vitro cytotoxicity against HeLa cells. Therefore, these PS/PDA/HAP hybrid materials may potentially useful in drug delivery, protein and gene transfer, and as coating materials for bioimplants or as injectable scaffold materials in bone tissue engineering for defect filling and regeneration.
Experimental section
Materials
3-Hydroxytyramine hydrochloride (dopamine hydrochloride), Tris(hydroxymethyl) aminomethane (Trizma®base, ≥99.8%), and polyvinylpyrrolidone (PVP, MW = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) were purchased from Sigma-Aldrich and used as received. 2,2′-Azobisisobutyronitrile (AIBN) was purchased from Shanghai Chemical Reagent Co., Ltd. (China) and purified by recrystallization in ethanol prior to use. Styrene (St) was also purchased from Shanghai Chemical Reagent Co., Ltd. (China) and was distilled in vacuum to remove the inhibitor, and then, was stored at 4 °C until use. Other chemical reagents and solvents used in this work were all of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd (China) and used without further purification. Ultrapure water (>17 MΩ cm−1) obtained from a GZY-P10 water system was used throughout the experiments.
000 g mol−1) were purchased from Sigma-Aldrich and used as received. 2,2′-Azobisisobutyronitrile (AIBN) was purchased from Shanghai Chemical Reagent Co., Ltd. (China) and purified by recrystallization in ethanol prior to use. Styrene (St) was also purchased from Shanghai Chemical Reagent Co., Ltd. (China) and was distilled in vacuum to remove the inhibitor, and then, was stored at 4 °C until use. Other chemical reagents and solvents used in this work were all of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd (China) and used without further purification. Ultrapure water (>17 MΩ cm−1) obtained from a GZY-P10 water system was used throughout the experiments.
Preparation of monodisperse PS/PDA composite particles
First, monodisperse PS particles were used as polymeric templates for the fabrication of composite particles, which were prepared via dispersion polymerization according to previous works.20,21,32 In detail, all of St (20.0 g), PVP (1.8 g), AIBN (0.2 g), ethanol (60.4 g) and water (7.6 g) were added into a 250 mL four-necked round-bottomed flask equipped with a mechanical stirrer, a thermometer with a temperature controller, a N2 inlet, an Allihn condenser and a heating mantle. The reaction solution was deoxygenated by bubbling nitrogen at room temperature for ca. 1 h. Then, the reaction solution was heated to 70 °C, and allowed the dispersion polymerization to carry out for 24 h under a constant stirring at a rate of 100 rpm. After the completion of the reaction, the dispersion was centrifuged and washed three times with ethanol. Finally, PS particles were dried in a vacuum oven at room temperature for 24 h, and stored in air for further use.The process for the preparation of PS/PDA composite particles was described as follows:20,21 first, dopamine solution (2 mg mL−1) was prepared by dissolving dopamine (200 mg) in the Tris–HCl buffer (100 mL, 10 mM and pH = 8.5). Then, the dried PS powder (0.1 g) was dispersed into as-prepared dopamine solution aided by sonication. The obtained suspension was kept magnetically stirring at room temperature for 36 h to allow the coating complete. The obtained PS/PDA composite particles were separated with a centrifuge, and rinsed three times with deionized water. These core–shell PS/PDA composite particles were dried in a vacuum oven 24 h at room temperature.
Biomimetic mineralization of HAP on PS/PDA composite particles
In this study, the simulated body fluid (SBF) solution was prepared according to Kokubo's work.33 To accelerate the HAP-coating process, the solution with an ionic concentration of 1.5 times higher than SBF was used in the formation of HAP.18,34 The composition of 1.5 × SBF was listed as follows: Na+, 213.0 mM; K+, 7.5 mM; Mg2+, 2.25 mM; Ca2+, 3.75 mM; Cl−, 221.7 mM; HCO3−, 6.3 mM; HPO42−, 1.5 mM; SO42−, 0.75 mM. For the biomimetic mineralization of HAP, the as-synthesized PS/PDA composite particles (0.1 g) were immersed into 100 mL of 1.5 × SBF solution in a polyethylene beaker, and the mixture was incubated at 37 °C for 3, 7 and 10 days. The 1.5 × SBF solution was replaced daily to ensure adequate ions present for the mineral growth. After each soaking period, the PS/PDA/HAP hybrid materials were collected from the 1.5 × SBF solution and rinsed thoroughly with pure water for three times, and were then dried in the vacuum oven at room temperature for 24 h.In addition, the pristine PS particles incubated in a 1.5 × SBF solution at 37 °C over pre-determined time periods were used as controls.
Cell culture
HeLa cells (human cervix adenocarcinoma cell line, bought from ATCC) were chosen as test cells, and cultured in Dulbecco's Modified Eagle Medium (Biological Industries, Co. Ltd.) supplemented with 10% fetal bovine serum (Biological Industries, Co. Ltd.) and penicillin–streptomycin solution (100 U mL−1, 100 μg mL−1, Gibco) at 37 °C in a humidified atmosphere of 5% CO2.27In vitro cytotoxicity and cell viability evaluation
Cell viability was determined by using the Cell Counting Kit-8 assay (CCK-8, Dojindo, Japan).35 HeLa cells were seeded in 96-well plates with a density of 2 × 103 cells per well in a standard growth medium and cultured at 37 °C for 24 h in a humidified atmosphere with 5% CO2 prior to the exposure to above materials. Subsequently, HeLa cells were incubated in the growth medium containing different concentrations (50, 100, 150 and 200 μg mL−1) of PS/PDA/HAP hybrid materials for various incubation time. Meanwhile, the wells only containing cell medium were prepared as the controls (Mock groups). At the end of the treatment, CCK-8 dye was added to each well and the plates were incubated for another 1 h at 37 °C. Subsequently, the absorbance of each well was measured through single wavelength spectrophotometry at 450 nm with a microplate reader. The experimental results were averaged by three measurements.Cell morphology evaluation
For cell morphology evaluation, HeLa cells with different treatments were seeded in six-well plates with a density of 2 × 105 cells per well, and were cultured for different incubation time in the aforementioned environment, respectively. The plates were observed with an inverted phase contrast microscope (Zeiss Axiovert 10, Carl Zeiss MicroImaging Co. Ltd).Statistical analysis
All experiments were repeated at least three times, thus, data were reported as the mean ± SD. Statistical analyses were performed by using one-way ANOVA (*P < 0.05; **P < 0.01). All statistical evaluations were performed with SPSS 11.0 software (SPSS Inc., Chicago, IL, USA).Characterization
The surface morphologies of PS particles, PS/PDA composite particles and PS/PDA/HAP hybrid materials were characterized by scanning electron microscopy (SEM, Hitachi S-4800, Hitachi, Ltd. Japan). All sample dispersions were first diluted with ethanol and then dried on silica wafers at room temperature before observation. The average diameters of PS particles and PS/PDA composite particles can be calculated based on the measurement of more than 100 particles from SEM images. The polydispersity (PD) was calculated based on the standard deviation expressed as follows:36,37
 | (1) |
 | (2) |
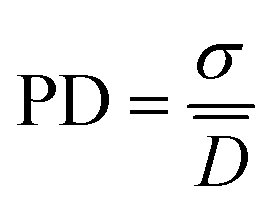 | (3) |
![[D with combining macron]](https://www.rsc.org/images/entities/i_char_0044_0304.gif) represents the number-average diameter of the particles, N the number of the particles, and Ni the number of the particle has diameter Di, respectively. PD is calculated based on the standard deviation (σ) and of the system.
represents the number-average diameter of the particles, N the number of the particles, and Ni the number of the particle has diameter Di, respectively. PD is calculated based on the standard deviation (σ) and of the system.
Energy-dispersive X-ray spectroscopy (EDX) was conducted on a Hitachi S-4800 SEM to determine the surface composition of HAP minerals on PS/PDA composite particles.
Transmission electron microscopy (TEM, JEOL JEM-2100, Japan) was used to observe the morphologies and internal structures of obtained samples (PS particles, PS/PDA composite particles and PS/PDA/HAP hybrid materials). All samples were diluted with ethanol and then ultrasonicated at 25 °C for 10 min. The particle dispersions were dried onto the carbon-coated copper grids prior to examination.
Fourier-transform infrared spectra (FTIR) of PS particles and PS/PDA composite particles were recorded on a transform infrared spectroscope (EQUINX55, Brucher Crop, Germany). All samples were centrifuged and washed with absolute ethanol, and then, dried in a vacuum oven for 24 h. The sample powders were pressed into KBr pellets for the measurement. The spectra were taken from 4000 to 500 cm−1.
Powder X-ray diffraction (XRD) was performed on a DX-2700 X-ray diffractometer equipped with a Cu tube and a diffracted beam curved graphite monochromator operating at 40 kV and 30 mA. Crystal structure identification was carried out by scanning the PS/PDA/HAP powders deposited on a glass substrate with a scanning rate of 0.02° (2θ) per second in the range of 10° and 80° (2θ).
X-ray photoelectron spectroscopy (XPS) measurement was carried out on an AXIS Ultra X-ray photoelectron spectrometer (Kratos Analytical Ltd. U.K.) equipped with a monochromatized Al Kα X-ray source (1486.6 eV). All binding energies were calibrated by using containment carbon (C1s = 284.6 eV).
Thermogravimetric analysis (TGA) was performed on a SDT Q600 (TA Instruments. U.S.A.). All dried samples (PS particles, PS/PDA composite particles and PS/PDA/HAP hybrid materials) were heated from 40 to 800 °C at a heating rate of 10 °C min−1 under a nitrogen flow with a rate of 50 mL min−1. The weight percentage of HAP minerals (CHAP) deposited on PS/PDA particles can be calculated as follows:38
| CHAP [%] = (1 − CLPS/PDA/HAP/CLPS/PDA) × 100 | (4) |
Results and discussion
Surface modification of PS particles with mussel-inspired PDA coatings
During the last decade, polymeric particles have been intensively exploited as active templates for controlled crystallization of inorganic materials, leading to a series of organic–inorganic hybrid materials suitable for a broad spectrum of applications.39,40 In particular, PS particles have been well-exploited as ideal templates for the fabrication of composites or hollow spheres at a massive scale because of their unique advantageous features, such as commercially available, monodisperse, size tunable, diverse surface groups, bio-inert and etc.21,32,41,42 Thus, here, monodisperse PS particles synthesized via dispersion polymerization32 were chosen as the polymeric templates. As shown in Fig. 1a, the SEM image confirmed that the original PS particles were fairly uniform both in size and spherical shape with an average diameter of 1.48 μm and a polydispersity of 1.65% (see Fig. 1c). In addition, the surface of PS particles was smooth which provides an ideal site for the subsequent surface modification.20 | ||
| Fig. 1 (a and b) Scanning electron microscopy (SEM) images and (c and d) their corresponding size distribution histograms and theoretic fittings of (a) original PS particles and (b) PS/PDA composite particles, respectively. X-ray photoelectron spectroscopy (XPS) scans of (e) original PS particles in (a) and (f) PS/PDA composite particles in (b). | ||
Due to their surface bio-inertness, these as-synthesized PS particles are difficult to integrate inorganic components onto their surfaces, as a result to further construct novel organic–inorganic hybrid materials is difficult. Nevertheless, mussel-inspired PDA coatings can solve this problem, specifically, making surface bio-active. In a basic environment, dopamine tends to self-polymerize, gradually forming an adherent PDA coating on the surface of PS particles, which would further lead to core–shell structured PS/PDA composite particles at room temperature. The morphology of PS/PDA composite particles has been examined by SEM as shown in Fig. 1b. In comparison with pristine PS particles in Fig. 1a, the mussel-inspired PDA coatings did not have a significant influence on the spherical shape of PS particles because the PDA shells were homogeneously deposited on the surfaces, which could efficiently hinder the formation of odd structures.43 After coating with PDA, both the average size and the surface roughness of the particles were increased. The average diameter of PS/PDA composite particles was 1.65 μm (see Fig. 1d) growing from 1.48 μm of PS template particles, suggesting that a thin PDA layer with a thickness of 85 nm has successfully coated on the PS particles. Moreover, the polydispersity of PS/PDA composite particles was retained around 3%, further verifying the homogeneous PDA coatings have been formed. Moreover, it is worthwhile pointing out that the abundant active catechol and amine groups presented on the PDA coatings facilitate a versatile platform for secondary surface-mediated reactions.20,21,44
In addition, XPS is a powerful tool employed to analyze the surface chemical composition variation between PS and PS/PDA composite particles. Before coating, a strong carbon (C1s) signal as well as the weak characteristic signals of oxygen (O1s) and nitrogen (N1s) was observed in a wide scanning XPS spectrum (see Fig. 1e). The appearance of O1s and N1s peaks at a relatively low intensity was attributed to those “auxiliary” agents, such as PVP stabilizer and AIBN initiator introduced during dispersion polymerization.20,21 After coating, both the intensities of O1s and N1s peaks were greatly enhanced, as shown in Fig. 1f. It was also found that the surface O/C mole ratio of PS/PDA composite particles was higher in comparison with that of PS particles, which may be ascribed to the rich of oxygen gradient in PDA. These XPS results again confirmed that the PDA coatings had been successfully incorporated onto the surfaces of PS particles.
Further, the thin PDA coating formed on the surface of PS particles was also confirmed by Fourier-transform infrared spectra (FTIR). Before the surface modification, the typical absorption bands including the C–H stretching of the benzene ring at ca. 3025 cm−1, the C–H stretching of saturated C–H groups of PS main-chain at the region of 2923–2840 cm−1, the C–C stretching of the benzene ring at ca. 1450 cm−1 and ca. 1601 cm−1, the C–H bending of the benzene ring at ca. 755 cm−1 and the benzene ring folding appearing at ca. 698 cm−1, have been clearly detected (see Fig. 2a).20 After coating, a new absorbance band at 1290 cm−1 appeared which was ascribed to the phenolic C–OH stretching vibration of the PDA (see Fig. 2b).20,21 Combined with previous SEM and XPS results, the formation of core–shell structured PS/PDA composite particles can be certainly concluded. These formed PDA coatings can serve as an active layer, endowing PS particles with a good surface functionality and interfacial bioactivity through their abundant active catechol and amine groups, which also facilitate PS/PDA composite particles as an active substrate for the following biomimetic mineralization of HAP under physiological conditions.
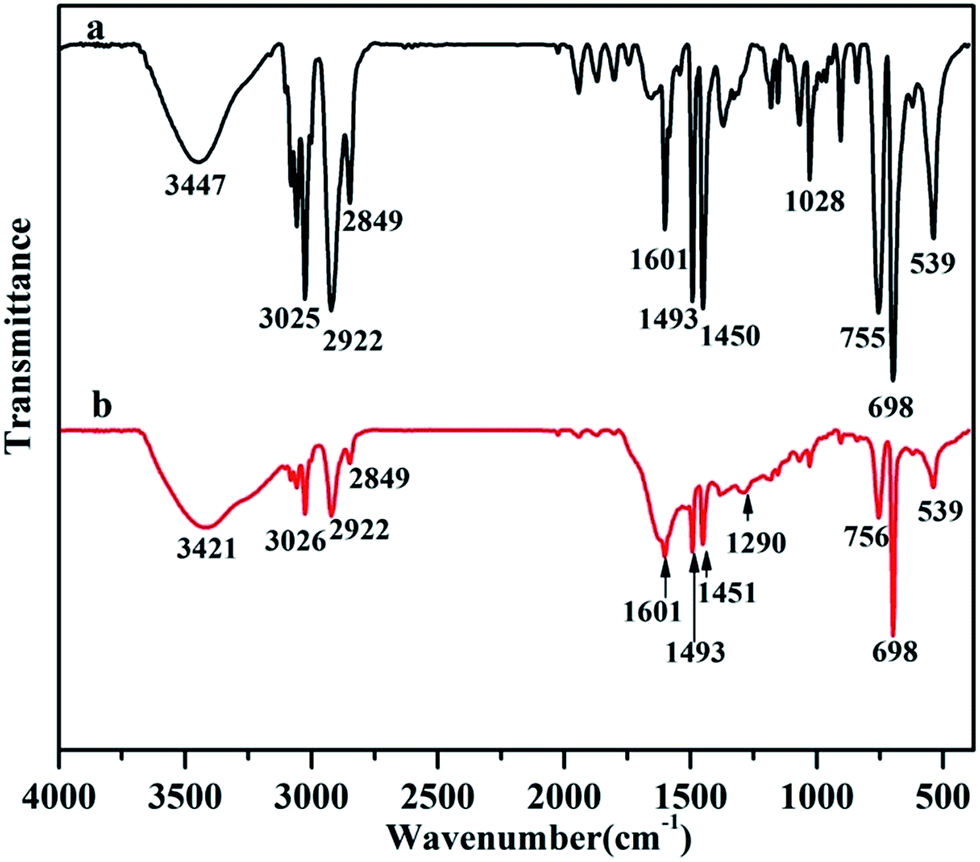 | ||
| Fig. 2 Fourier-transform IR (FTIR) spectra of (a) PS particles and (b) PS/PDA composite particles with their characteristic absorption peaks. | ||
Biomimetic mineralization of HAP on PS/PDA composite particles
Biomimetic mineralization of HAP was induced by soaking PS/PDA composite particles into a simulated body fluid (SBF) solution designed with an ionic concentration approximate to that of human blood plasma. The detailed preparation process of the PS/PDA/HAP hybrid materials has been illustrated in Scheme 1. As illustrated, PDA-coated PS particles were prepared by immersing PS particles into a weakly alkaline aqueous solution of dopamine, and subsequently, as-formed PS/PDA composite particles were transferred and incubated in a 1.5 × SBF solution for biomimetic mineralization. During mineralization, those catecholamine moieties that were abundant in PDA were responsible for the nucleation of HAP by concentrating Ca2+ ions at the interface through electrostatic interactions. The HAP was formed by co-precipitation of calcium and phosphate ions.18,45 Ultimately, the organic–inorganic PS/PDA/HAP hybrid materials were obtained through this so-called PDA-assisted biomimetic mineralization.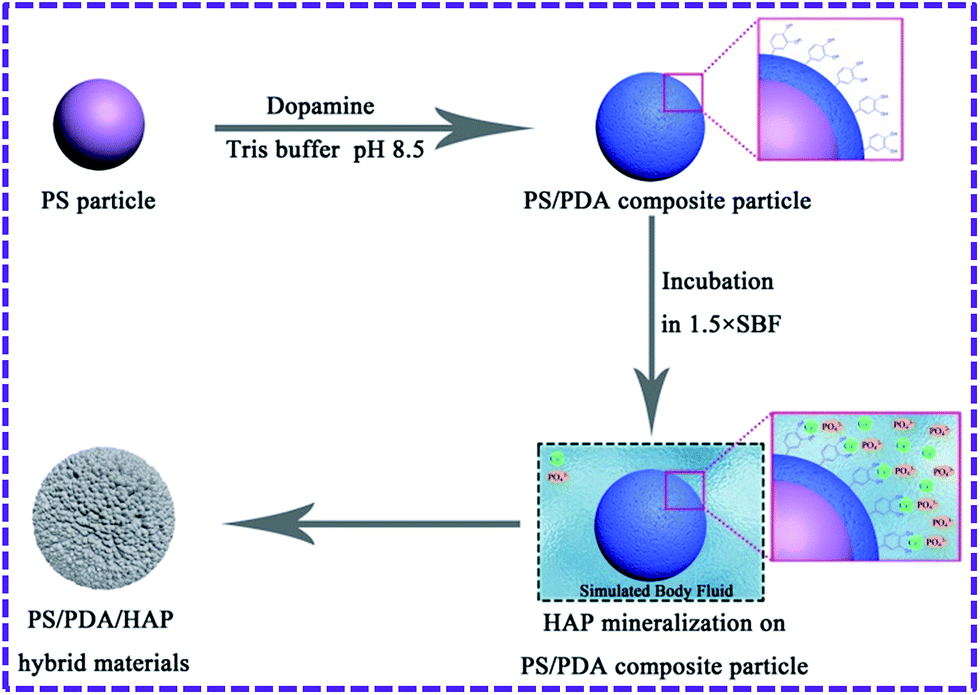 | ||
| Scheme 1 Schematic diagram illustrating formation of PS/PDA/HAP hybrid materials through PDA-assisted biomimetic mineralization. | ||
In the TEM images, the great changes on the surface morphology of composite particles were observed after the incubation of PS/PDA composite particles in the 1.5 × SBF solution (see Fig. 3). After 3 days incubation, some inorganic mineralized clusters partly covering on the surfaces of PS/PDA composite particles were illustrated as shown in Fig. 3b, indicating that PDA coatings could assist inorganic HAP minerals binding onto the surfaces of polymeric template particles. To firmly confirm the key role of PDA coatings during the biomimetic mineralization, the incubation of pristine PS particles in the 1.5 × SBF solution was also carried out. After 3 days incubation, no inorganic mineralized clusters coating was observed, solely leaving PS particles in the 1.5 × SBF solution (see Fig. S1 in ESI†). It is evidenced that pristine PS particles are surface inert and difficult to induce HAP mineralization without the assistance from PDA coatings. Moreover, as the incubation time increased to 7 days, it was observed that the surface of PS/PDA composite particles was completely covered with the inorganic mineralized clusters (see Fig. 3c). A further increase of incubation to 10 days led to a thicker layer of inorganic minerals, and a more aggregated system, as shown in Fig. 3d. In the magnified TEM images, they clearly show that the inorganic mineralized clusters compose of a stack of small plate-like materials, which were similar to the dominant morphology of nature bones (see Fig. S2 in ESI†). The morphology evolution of the particles was also monitored by SEM as shown in Fig. S3,† which are consistent with the TEM results in Fig. 3. These results suggested that PDA layers played a vital role in this biomimetic mineralization.
 | ||
| Fig. 3 Transmission electron microscopy (TEM) images of PS/PDA composite particles (a) and the resulting PS/PDA/HAP hybrid biocomposites after the incubation of PS/PDA composite particles in the 1.5 × SBF solutions for (b) 3; (c) 7; and (d) 10 days, respectively. | ||
We also applied multiple analytical tools to characterize the structure variation of biocomposites. Scanning electron microscopy (SEM) analysis further indicates that the coverage degree of the inorganic mineralized clusters on the surfaces of PS/PDA composite particles was increased with increasing the incubation time (see Fig. 4a, d and g). As revealed by Fig. 4b, e and h, the inorganic mineralized clusters formed have a plate-like structure which is a typical form of HAP crystals. By using energy dispersive X-ray (EDX), the elemental composition of the obtained materials can be investigated. Before the mineralization, the strong C and Si signal peaks accompanying with the weak characteristic signals of O and N peaks were observed in the EDX data of PS/PDA composite particles. The appearance of Si peak was attributed to silica wafers used as the substrates (see Fig. S4 in ESI†). After the mineralization, the significant Ca and P peaks were observed, which were contributed from the inorganic HAP minerals (see Fig. 4c, f and i). Moreover, the calcium-to-phosphorus (Ca/P) ratio of inorganic mineralized clusters was shown between 1.43 and 1.56, a slightly lower than the theoretical ratio of HAP (Ca/P = 1.67).34 According to literatures,46,47 human biological apatite crystals are always made of the calcium-deficient HAP nanoparticles, which usually contain a few percent of carbonate, acid phosphate, sodium, and magnesium ions because of the continuous contact with a flow of trace ions.34,48 In this regard, the HAP minerals formed with PS/PDA composite particles have a lower Ca/P ratio than pure HAP, and the chemical composition is close to human biological apatite, thus, they are probably appropriate for bioimplants and bone tissue regeneration applications.
 | ||
| Fig. 4 Scanning electron microscopy (SEM) images and the corresponding energy dispersive X-ray (EDX) spectra of the resulting PS/PDA/HAP hybrid biocomposites after incubation of PS/PDA composite particles in 1.5 × SBF solution for (a–c) 3; (d–f) 7; and (g–i) 10 days, respectively. | ||
Similarly, the surface chemical information of the as-produced PS/PDA/HAP hybrid materials was examined with XPS as well. As discussed previously, the XPS spectrum of PS/PDA composite particles only exhibited C1s, N1s, and O1s peaks (see Fig. 1f) from PDA layer, while, after mineralization, some new peaks emerged (see Fig. 5a), which were the characteristic patterns of calcium element (Ca1s, Ca2s, Ca3s, Ca2p and Ca3p) and phosphorus element (P2s and P2p). It was also found that the atomic concentrations of both calcium and phosphorus increased with increasing the incubation time. Evidently, increasing mineralization time induced more HAP minerals crystallization on the surface of PS/PDA composite particles, which was in good agreement with the TEM and SEM results.
 | ||
| Fig. 5 (a) X-ray photoelectron spectroscopy (XPS) scans and (b) X-ray diffraction (XRD) patterns of the as-produced PS/PDA/HAP hybrid biocomposites after incubation of PS/PDA composite particles in 1.5 × SBF solution for different time: 3, 7 and 10 days. | ||
Fig. 5b displayed the XRD patterns of the as-synthesized PS/PDA/HAP hybrid materials. The characteristic peaks at 25.98°, 28.90°, 31.80°, 39.88°, 46.88° and 49.54° correspond to the reflections of (002), (210), (211), (310), (222) and (213) of HAP crystals, respectively. Moreover, the XRD spectra also indicated that the peak intensities of HAP minerals increased with the increasing mineralization time. It should be noted that the broadness of the peaks in the XRD spectra of PS/PDA/HAP hybrid materials was due to the combined effect of the nanocrystalline nature of HAP and the amorphous phase of PDA.34 The as-synthesized HAP was the bonelike HAP minerals rather than octacalcium phosphate, which was structurally similar to HAP.18
To quantitatively determine how much HAP minerals have been deposited onto the surfaces of PS/PDA composite particles, thermogravimetric analysis (TGA) was used. As the results shown in Fig. 6a, the pure PS particles started to loss their weight at around 300 °C and the complete decomposition took place at the temperatures above 430 °C. On the contrary, it was found that those PS/PDA composite particles started to decompose up to 350 °C, and 36.81% of the residue was still preserved after the temperature increased to 450 °C. This was attributed to the protection of PDA coatings effectively against the pyrolysis of PS cores. When the calcination temperature was finally increased to 800 °C, still 23.71% of the residue was left over, which demonstrated that the thermal stability of polymeric template particles had been significantly improved after PDA coating. After coating with HAP, the weight percent of the residue was increased as the results shown in Fig. 6c–e. It was also found that the weight percentage of residue increased with increasing the incubation time in 1.5 × SBF solution. The residual content increased from 40.43 to 60.43 and further to 67.83% after incubating for 3, 7, and 10 days, respectively. Thus, the calculated weight fractions of HAP minerals were 21.92, 48.13 and 57.83%, corresponding to the incubation increased from 3, 7 to 10 days.
 | ||
| Fig. 6 Thermogravimetric analysis (TGA) curves of (a) PS particles, (b) PS/PDA composite particles and (c–e) the as-produced PS/PDA/HAP hybrid materials after incubation of PS/PDA composite particles in 1.5 × SBF solution for (c) 3; (d) 7; and (e) 10 days, respectively. | ||
Based on the results and discussion above, we are able to conclude that PDA coatings can efficiently assist the biomimetic mineralization of HAP under physiological conditions, leading to HAP-based, organic–inorganic hybrid materials. In principle, the chemical activation of material surfaces via PDA coatings is a convenient and environmental friendly approach, which also introduce the abundance active catechol and amine groups onto the surfaces.13,14,16 Therefore, PDA coatings may offer a versatile platform, through which a variety of natural inorganic components (e.g. HAP, silica, and metal nanoparticles) can be readily integrated into diverse synthetic materials for developing the advanced biomaterials.
In vitro cytotoxicity and cell viability evaluation
To consider the PS/PDA/HAP hybrid materials as potential biomaterials for future biomedical applications, the biocompatibility of the produced HAP-based materials should be evaluated first, which is one of the primary concerns in the use of these HAP-based materials in biological and medical fields.49,50 To investigate the biocompatibility, HeLa cells (human cervix adenocarcinoma cell line) were used as test cells in the presence of PS/PDA/HAP hybrid materials. The PS/PDA/HAP hybrid materials produced after incubation of PS/PDA composite particles in 1.5 × SBF solution for 10 days were chosen as a typical sample for the following in vitro cytotoxicity and cell viability evaluation.Fig. 7a–e and 8a–e showed the cellular morphologies of HeLa cells in culture media as a function of PS/PDA/HAP hybrid materials concentration and culture time. In comparison with the untreated controls (mock groups, Fig. 7a and 8a), the morphology of HeLa cells has not been obviously affected by those PS/PDA/HAP hybrid materials, even with a high concentration of PS/PDA/HAP hybrid materials (e.g. 200 μg mL−1) over a long incubation time (e.g. 48 h). The cell viability was evaluated by using the Cell Counting Kit-8 (CCK-8) assay. We added the solutions of PS/PDA/HAP hybrid materials to the culture media at different concentrations (e.g. 50, 100, 150, and 200 μg mL−1) to investigate the effects of the amount of HAP-based hybrid materials. Comparing with control samples in Fig. 7a and 8a, the presence of PS/PDA/HAP hybrid materials proliferated the growth of HeLa cells as shown in Fig. 7 and 8, which was also confirmed by the results shown in Fig. 9a and b, indicating that the as-prepared PS/PDA/HAP hybrid materials had a reasonably good biocompatibility. Thus, it paves the possible avenues towards the in vivo biomedical application of these PS/PDA/HAP hybrid materials for bioimplants and bone tissue regeneration.
 | ||
| Fig. 7 Inverted phase contrast microscope inspection of HeLa cells in culture media containing different concentrations of PS/PDA/HAP hybrid materials after incubation of 24 h: (a) the untreated control group (mock group), (b) 50, (c) 100, (d) 150, and (e) 200 μg mL−1. | ||
 | ||
| Fig. 8 Inverted phase contrast microscope inspection of HeLa cells in culture media containing different concentrations of PS/PDA/HAP hybrid materials after incubation of 48 h: (a) the untreated control group (mock group), (b) 50, (c) 100, (d) 150, and (e) 200 μg mL−1. | ||
 | ||
| Fig. 9 In vitro cytotoxicity (CCK-8 assay) of PS/PDA/HAP hybrid materials against HeLa cells after incubation at 37 °C for (a) 24 h and (b) 48 h, respectively. Data are presented as means ± standard deviation, n = 3 (*P < 0.05 and **P < 0.01, respectively). | ||
Conclusions
In summary, we present a simple and efficient approach for the synthesis of HAP-based, novel organic–inorganic hybrid materials with the help of combining the active surface of PDA with biomimetic mineralization. In this procedure, mussel-inspired PDA coatings were responsible for the chemical functionalization of surficial inert PS particles, forming active polymer particle templates for effective biomimetic mineralization of HAP when incubated in SBF solution. Through the PDA-assisted HAP mineralization, the HAP minerals were effectively integrated onto polymer particle templates as a result of novel HAP-based hybrid materials. These HAP minerals showed a similar crystal structure to that of human biological apatite. In vitro cytotoxicity assays indicated that these HAP-based hybrid materials showed a good biocompatibility with HeLa cells. Based on these advantages, we believe these HAP-based hybrid materials are promising potential candidates in drug delivery, protein and gene transfer, and use as coating materials for bioimplants or injectable scaffold materials in bone tissue engineering for defect filling and regeneration.Acknowledgements
We acknowledge the National Natural Science Foundation of China (No. 51203087, 51473089), the Natural Science Basic Research Plan in Shaanxi Province of China (No. 2013JQ2006), the Science and Technology Program of Shaanxi Province (No. 2015KJXX-20), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, the Fundamental Research Funds for the Central Universities (No. GK201503039, GK201501002, GK201301002, GK2011 01003) and the Outstanding Youth Project of Nanjing City (No. JQX14005) for financial supports.Notes and references
- E. Bäuerlein, Biomineralization: Progress in Biology, Molecular Biology and Application, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2nd edn, 2004 Search PubMed
.
- S. Mann, Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry, Oxford University Press, Oxford, 2001 Search PubMed
- S. Mann, D. D. Archibald, J. M. Didymus, T. Douglas, B. R. Heywood, F. C. Meldrum and N. J. Reeves, Science, 1993, 261, 1286–1292 CAS
- A. W. Xu, Y. R. Ma and H. Colfen, J. Mater. Chem., 2007, 17, 415–449 RSC
- P. Fratzl, Nat. Mater., 2008, 7, 610–612 CrossRef CAS PubMed
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed
- L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Chem. Rev., 2008, 108, 4754–4783 CrossRef CAS PubMed
- G. Wei, J. Reichert, J. Bossert and K. D. Jandt, Biomacromolecules, 2008, 9, 3258–3267 CrossRef CAS PubMed
- Z. Su, J. Li, Z. Ouyang, M. M. L. Arras, G. Wei and K. D. Jandt, RSC Adv., 2014, 4, 14833–14839 RSC
- J. Wang, H. Wang, Y. Wang, J. Li, Z. Su and G. Wei, J. Mater. Chem. B, 2014, 2, 7360–7368 RSC
- A. Ethirajan, U. Ziener and K. Landfester, Chem. Mater., 2009, 21, 2218–2225 CrossRef CAS
- A. George and A. Veis, Chem. Rev., 2008, 108, 4670–4693 CrossRef CAS PubMed
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed
- Q. Ye, F. Zhou and W. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC
- J. Ryu, S. H. Ku, H. Lee and C. B. Park, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS
- H. Liu, P. Xi, G. Xie, Y. Shi, F. Hou, L. Huang, F. Chen, Z. Zeng, C. Shao and J. Wang, J. Phys. Chem. C, 2012, 116, 3334–3341 CAS
- Z. Li, C. Wu, K. Zhao, B. Peng and Z. Deng, Colloids Surf., A, 2015, 470, 80–91 CrossRef CAS
- Y. Cong, T. Xia, M. Zou, Z. Li, B. Peng, D. Guo and Z. Deng, J. Mater. Chem. B, 2014, 2, 3450–3461 RSC
- L. Guo, Q. Liu, G. Li, J. Shi, J. Liu, T. Wang and G. Jiang, Nanoscale, 2012, 4, 5864–5867 RSC
- Z. Niu, Z. Yang, Z. Hu, Y. Lu and C. C. Han, Adv. Funct. Mater., 2003, 13, 949–954 CrossRef CAS
- M. Yang, J. Ma, C. Zhang, Z. Yang and Y. Lu, Angew. Chem., Int. Ed., 2005, 44, 6727–6730 CrossRef CAS PubMed
- M. Chen, L. Wu, S. Zhou and B. You, Adv. Mater., 2006, 18, 801–806 CrossRef CAS
- L. Bacakova, V. Mares, M. G. Bottone, C. Pellicciari, V. Lisa and V. Svorcik, J. Biomed. Mater. Res., 2000, 49, 369–379 CrossRef CAS PubMed
- T. dos Santos, J. Varela, I. Lynch, A. Salvati and K. A. Dawson, PLoS One, 2011, 6, e24438, DOI:10.1371/journal.pone.0024438
- O. Lunov, T. Syrovets, C. Loos, G. U. Nienhaus, V. Mailander, K. Landfester, M. Rouis and T. Simmet, ACS Nano, 2011, 5, 9648–9657 CrossRef CAS PubMed
- L. Bacakova, V. Mares, V. Lisa and V. Svorcik, Biomaterials, 2000, 21, 1173–1179 CrossRef CAS PubMed
- M. J. Bridgett, M. C. Davies and S. P. Denyer, Biomaterials, 1992, 13, 411–416 CrossRef CAS PubMed
- A. Zeller, A. Musyanovych, M. Kappl, A. Ethirajan, M. Dass, D. Markova, M. Klapper and K. Landfester, ACS Appl. Mater. Interfaces, 2010, 2, 2421–2428 CAS
- Z. Deng, M. Chen, G. Gu and L. Wu, J. Phys. Chem. B, 2008, 112, 16–22 CrossRef CAS PubMed
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed
- M. Lee, S. H. Ku, J. Ryu and C. B. Park, J. Mater. Chem., 2010, 20, 8848–8853 RSC
- C. Ma, C. Peng, X. Lu, X. Ding, S. Zhang, X. Zou and X. Zhang, Biochem. Biophys. Res. Commun., 2015, 458, 234–239 CrossRef CAS PubMed
- B. Peng, E. van der Wee, A. Imhof and A. van Blaaderen, Langmuir, 2012, 28, 6776–6785 CrossRef CAS PubMed
- K. Zhao, J. Zhao, C. Wu, S. Zhang, Z. Deng, X. Hu, M. Chen and B. Peng, RSC Adv., 2015, 5, 69543–69554 RSC
- X. Hu, Y. Wang and B. Peng, Chem.–Asian J., 2014, 9, 319–327 CrossRef CAS PubMed
- J. Zhang, S. Xu and E. Kumacheva, J. Am. Chem. Soc., 2004, 126, 7908–7914 CrossRef CAS PubMed
- Z. Yang, Z. Niu, Y. Lu, Z. Hu and C. C. Han, Angew. Chem., 2003, 115, 1987–1989 CrossRef
- Z. Deng, M. Chen, S. Zhou, B. You and L. Wu, Langmuir, 2006, 22, 6403–6407 CrossRef CAS PubMed
- Z. Deng, Z. Zhen, X. Hu, S. Wu, Z. Xu and P. K. Chu, Biomaterials, 2011, 32, 4976–4986 CrossRef CAS PubMed
- B. Peng and A. Imhof, Soft Matter, 2015, 11, 3589–3598 RSC
- C. Wu, G. Zhang, T. Xia, Z. Li, K. Zhao, Z. Deng, D. Guo and B. Peng, Mater. Sci. Eng., C, 2015, 55, 155–165 CrossRef CAS PubMed
- J. Zhang, W. P. Zhang, T. Bao and Z. L. Chen, Analyst, 2014, 139, 242–250 RSC
- R. M. Wilson, J. C. Elliott, S. E. P. Dowker and L. M. Rodriguez-Lorenzo, Biomaterials, 2005, 26, 1317–1327 CrossRef CAS PubMed
- Y. Lee, Y. Hahm, S. Matsuya, M. Nakagawa and K. Ishikawa, J. Mater. Sci., 2007, 42, 7843–7849 CrossRef CAS
- S. A. Hutchens, R. S. Benson, B. R. Evans, H. M. O'Neill and C. J. Rawn, Biomaterials, 2006, 27, 4661–4670 CrossRef CAS PubMed
- Z. K. Hong, P. B. Zhang, C. L. He, X. Y. Qiu, A. X. Liu, L. Chen, X. S. Chen and X. B. Jing, Biomaterials, 2005, 26, 6296–6304 CrossRef CAS PubMed
- H. Wang, Y. Li, Y. Zuo, J. Li, S. Ma and L. Cheng, Biomaterials, 2007, 28, 3338–3348 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: TEM image of PS particles after incubation in 1.5 × SBF solution for 3 days; TEM, SEM and EDX characterization of PS/PDA composite particles and the produced PS/PDA/HAP hybrid materials. See DOI: 10.1039/c5ra24821c |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |