
Nano-capsules of amphiphilic poly(ethylene glycol)-block-poly(bisphenol A carbonate) copolymers via thermodynamic entrapment†
A. Sutti,
T. Chaffraix,
A. S. Voda,
A. Taylor and
K. Magniez*
Institute for Frontier Materials, Deakin University, Geelong, Victoria 3217, Australia. E-mail: kevin.magniez@research.deakin.edu.au; Tel: +61-5227-1305
First published on 8th January 2016
Abstract
The synthesis of amphiphilic poly(ethylene glycol)-block-poly(bisphenol A carbonate) (PEG-b-PC) block copolymer is presented here using a simple bio-chemistry coupling reaction between poly(bisphenol A carbonate) (PC) with a monomethylether poly(ethylene glycol) (mPEG-OH) block, mediated by dicyclohexylcarbodiimide/4-dimethylaminopyridine. This method inherently allows great flexibility in the choice of starting materials as well as easy product purification only requiring phase separation and water washing. Collective data from Fourier transform infrared spectroscopy (FTIR), nuclear magnetic resonance spectroscopy (NMR) and modulated dynamic scanning calorimetry (MDSC) confirmed the successful attachment of the poly(ethylene glycol) (mPEG-OH) and poly(bisphenol A carbonate) (PC) blocks. The preparation of nano-capsules was carried out by sudden addition of water to PEG-b-PC copolymers dispersed in THF, resulting in the controlled precipitation (i.e. thermodynamic entrapment) of the copolymer. Nano-capsules as small as 85 nm ± 30 nm were produced using this simple and fast methodology. We also demonstrate that encapsulating a water-insoluble bisphenol A diglycidyl ether (DGEBA) epoxy resin is possible highlighting the potential use of these capsules as a chemical delivery system.
Introduction
Micro and nano-capsules of high performance polymers appeal to industry due to a wide range of fields of application including sensors, chemical delivery, textiles, composites and food science.1–3 Their preparation commonly involves any of the following, widely accepted, techniques: block copolymer self-assembly,4,5 seeded polymerization/dynamic swelling method,6,7 interfacial polymerisation of mini-emulsions8–11 and layer-by-layer (LbL).12,13 Although well established in the literature, these methods can be time consuming, require a number of complex steps or achieve low yield. Additionally, the processes might be very starting-material-specific.The use of amphiphilic block copolymers to form nano-particulate structures, including nanospheres, nanocapsules and polymersomes has been described elsewhere in the literature.14 Amphiphilic block copolymers are typically a macromolecule composed of two regions, one hydrophilic and one hydrophobic. Particles composed of amphiphilic block copolymers present advantages over other nano-engineered materials. The competitive hydrophilic/hydrophobic characteristics of each respective blocks can be exploited in a synergistic fashion. For example, in the case of self-assembled polymersomes the water-affinity can be retained in the inner layer of the capsule, whereas the wall of the polymersome will display a lipophilic character. The hydrophobic/hydrophilic balance of an amphiphilic copolymer, determined by both the block nature and block length ratio, has a significant effect on the overall self-assembly behaviour of the copolymer in water or organic solvents.15 Hence, achieving control over the block length ratio in amphiphilic block copolymers is critical. This is usually attained through the controlled polymerisation of a homo-polymer block (e.g. hydrophobic) onto a pre-existing block (e.g. hydrophilic). Ring opening,16 atom transfer radical,17 living cationic18 and RAFT19 polymerisation have been reported for the synthesis of amphiphilic block copolymers of controlled length. Although these approaches are widely established, the synthesis of block copolymers having a certain desired block length ratio is not easily achieved in a single polymerization step and will often require a combination of synthetic steps.20 This can be considered a major drawback and hinders the range of possible amphiphilic copolymers.
The use of carbodiimides derivatives such as dicyclohexylcarbodiimide (DCC) is very prevalent in bio-chemistry to promote amide bond formation and peptide coupling.21 This is because they display excellent reaction yield, are relatively easy to handle, possess good solubility and the urea by-products can be easily isolated.22 The conversion of an alcohol via esterification with a carboxylic is also possible by combining DCC with 4-dimethylaminopyridine (DMAP) as catalyst. This reaction (often referred to as Steglich esterification) and its mechanism has been discussed earlier.23 This manuscript draws on such established and simple bio-chemistry methodology to synthesise an amphiphilic poly(ethylene glycol)-block-poly(bisphenol A carbonate) (PEG-b-PC) block copolymer. The method inherently allows great flexibility in the choice of homopolymer blocks starting materials of pre-determined molecular weights and very easy product purification, only requiring phase separation and water washing. Poly(ethylene glycol) (PEG) was used in this work as the hydrophilic block component due to its neutral-charge hydrophilic character whereas poly(bisphenol A carbonate) (PC) was chosen as the more hydrophobic block due to its appealing engineering properties suitable for a range of higher end applications.
Additionally, this manuscript demonstrates the fast production of PEG-b-PC polymer nano-capsules of controllable diameters through sudden water addition, which induces instantaneous precipitation and the thermodynamic locking of the copolymer chains into formed nano-capsules.14,24,25 The obtained nano-capsules were colloidally stable in water over weeks. The copolymer was also demonstrated to be capable of encapsulating a water-insoluble bisphenol-A diglycidyl ether (DGEBA) epoxy resin. The degradable nature of the poly(ethylene glycol) block demonstrates the capsules' potential for use as a chemical delivery system.
Methodology
Materials and methods
All materials were obtained from Sigma Aldrich and used as received without any further purification. The chemicals employed were poly(ethylene glycol) monomethyl ether (mPEG-OH, Mw ∼ 5000 Da), poly(bisphenol A carbonate) (PC, Mw ∼ 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da), tetrahydrofuran (THF) and the coupling agents 4-dimethylaminopyridine (DMAP) and dicyclohexylcarbodiimide (DCC).
000 Da), tetrahydrofuran (THF) and the coupling agents 4-dimethylaminopyridine (DMAP) and dicyclohexylcarbodiimide (DCC).
Synthesis of poly(ethylene glycol)-block-poly(bisphenol A carbonate) (PEG-b-PC)
All syntheses were carried out using a Schlenk oxygen exclusion method in the presence of argon (Ar) using a dual chamber manifold. Reaction vessels, attachments and stirring equipment were flame dried under vacuum prior to synthesis. Each reaction vessel was purged with argon three times prior to the introduction of reagents. Following the addition of all reagents the system was purged with a steady stream of argon for 3 to 5 minutes prior to commencing heating.PEG-b-PC was prepared by reacting 0.75 g of mPEG-OH (0.15 mmol) with 3.0 g (0.07 mmol) of PC in the presence of 0.1 g (1 mmol) of DMAP and 0.2 g of DCC (1 mmol) as a coupling agent. The PC and mPEG-OH starting materials were added to a three neck round bottom flask blanketed with nitrogen at room temperature. The materials were suspended in 150 ml of anhydrous THF until complete dissolution. Dry DMAP and DCC were added to the solution while stirring. The reaction was allowed to occur at room temperature overnight under nitrogen. The reaction mixture was poured into a 2 fold volume of MilliQ water. The milky phase which immediately formed was collected by filtration and washed several times with MilliQ water, followed by drying in vacuo at 65 °C.
Equipment and testing
Results and discussion
Synthesis and characterization of PBocLTrp-b-PEG-b-PBocLTrp
The collective data from FTIR, NMR, and MDSC presented below confirmed the successful attachment of the poly(ethylene glycol) (mPEG-OH) and poly(bisphenol A carbonate) (PC) blocks (Scheme 1). | ||
| Scheme 1 Synthetic route to poly(ethylene glycol)-block-poly(bisphenol A carbonate) (PEG-b-PC) copolymer. | ||
It is first important to point out that a large excess of water soluble mPEG-OH starting material was used in order to minimise the presence of residual poly(bisphenol A carbonate) (PC) after reaction and to simplify the purification process (as all by-products are water-soluble). After quenching the reaction with MilliQ water, a suspension containing a white milky product was formed. The milky product was filtered carefully and washed several times with MilliQ water to remove any trace of unreacted mPEG-OH. The infrared spectrum of the isolated product is shown in Fig. 1 along with the spectra of the mPEG-OH and PC starting materials.
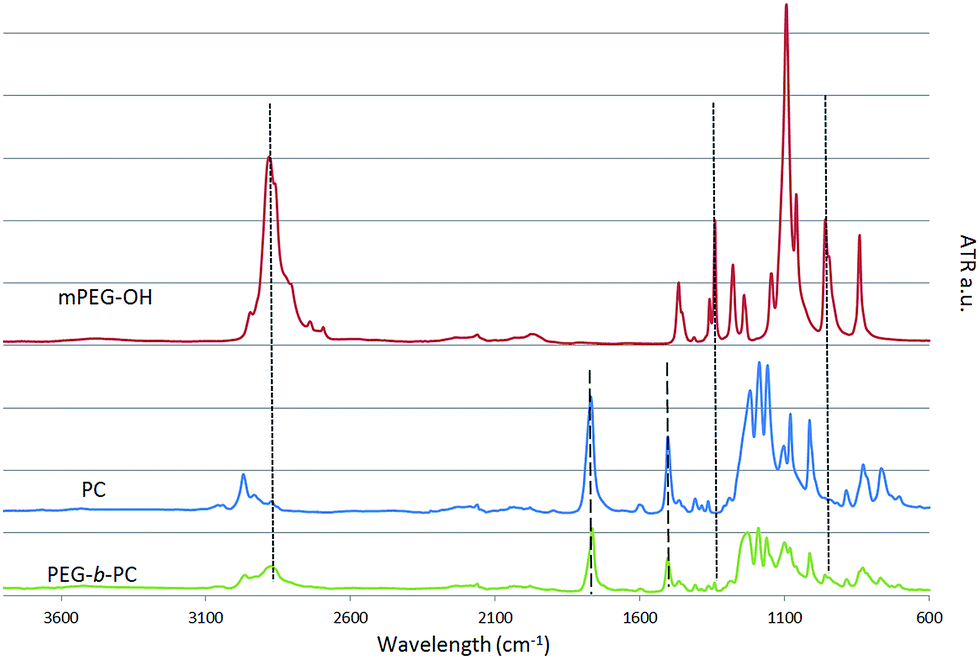 | ||
| Fig. 1 ATR spectrum of the materials. | ||
The spectra of the reaction product displays the characteristic vibration peaks of both the mPEG-OH and PC starting materials, providing an indication that both blocks are present in the product. The stretching bands νs and νas corresponding to the methyl CH2 group of the polyethylene glycol block are visible at 2880 and 1340 cm−1 whilst their out-of-plane bending ω is visible at 960 cm−1.26
The sharp characteristic stretching band νs of the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1770 cm−1 and the aromatic stretching at 1500 cm−1 of the PC are also discernible.27 It seems reasonable to assume that the presence of PEG in the isolated insoluble fraction proves the formation of block copolymers; if no reactions had occurred between PC and mPEG-OH, the water soluble mPEG-OH would have only been found in the aqueous/THF fractions. Due to the attachment of the hydrophobic PC block to the hydrophilic mPEG-OH block, the PEG-b-PC copolymers displayed a macroscopic level of hydrophilicity with a measured surface contact angle θ of ∼60° (ESI Fig. S1†). The surface contact angle θ of the PC starting material was ∼90°, consistent with Tang et al.,28 whereas the surface contact angle of the mPEG-OH could not be measured as expected (i.e. too hydrophilic).
O at 1770 cm−1 and the aromatic stretching at 1500 cm−1 of the PC are also discernible.27 It seems reasonable to assume that the presence of PEG in the isolated insoluble fraction proves the formation of block copolymers; if no reactions had occurred between PC and mPEG-OH, the water soluble mPEG-OH would have only been found in the aqueous/THF fractions. Due to the attachment of the hydrophobic PC block to the hydrophilic mPEG-OH block, the PEG-b-PC copolymers displayed a macroscopic level of hydrophilicity with a measured surface contact angle θ of ∼60° (ESI Fig. S1†). The surface contact angle θ of the PC starting material was ∼90°, consistent with Tang et al.,28 whereas the surface contact angle of the mPEG-OH could not be measured as expected (i.e. too hydrophilic).
The 1H NMR spectra of the materials in CDCl3 are shown in Fig. 2. The peak at 3.58 ppm, attributed to the protons (protons positions 1 and 2) in the methylene (–O–CH2–CH2–) group of the PEG starting material29 was found to shift slightly to 3.63 ppm after attachment of the PC block. Conversely, the peak resonating at 1.69 ppm of the protons (protons positions 1) in the methyl (–CH3–) group of the PC methyl peak shifted downfield to 1.66 ppm. In the aromatic region (7.0 to 7.5 ppm), the protons in the phenyl group (protons positions 2 and 3) of the PC block27,30 were observed to shift upfield with the quartet centred at 7.22 ppm in the starting material having a resonance centred at 7.19 ppm in the block copolymer. Heteronuclear multiple-bond correlation spectroscopy (2D-HMBC) NMR did not provide any further elucidation regarding the point of attachment between the two blocks. The large molecular weight of the copolymer associated with inadequate resolution made it impossible to provide a meaningful interpretation (ESI Fig. S2 and S3†).
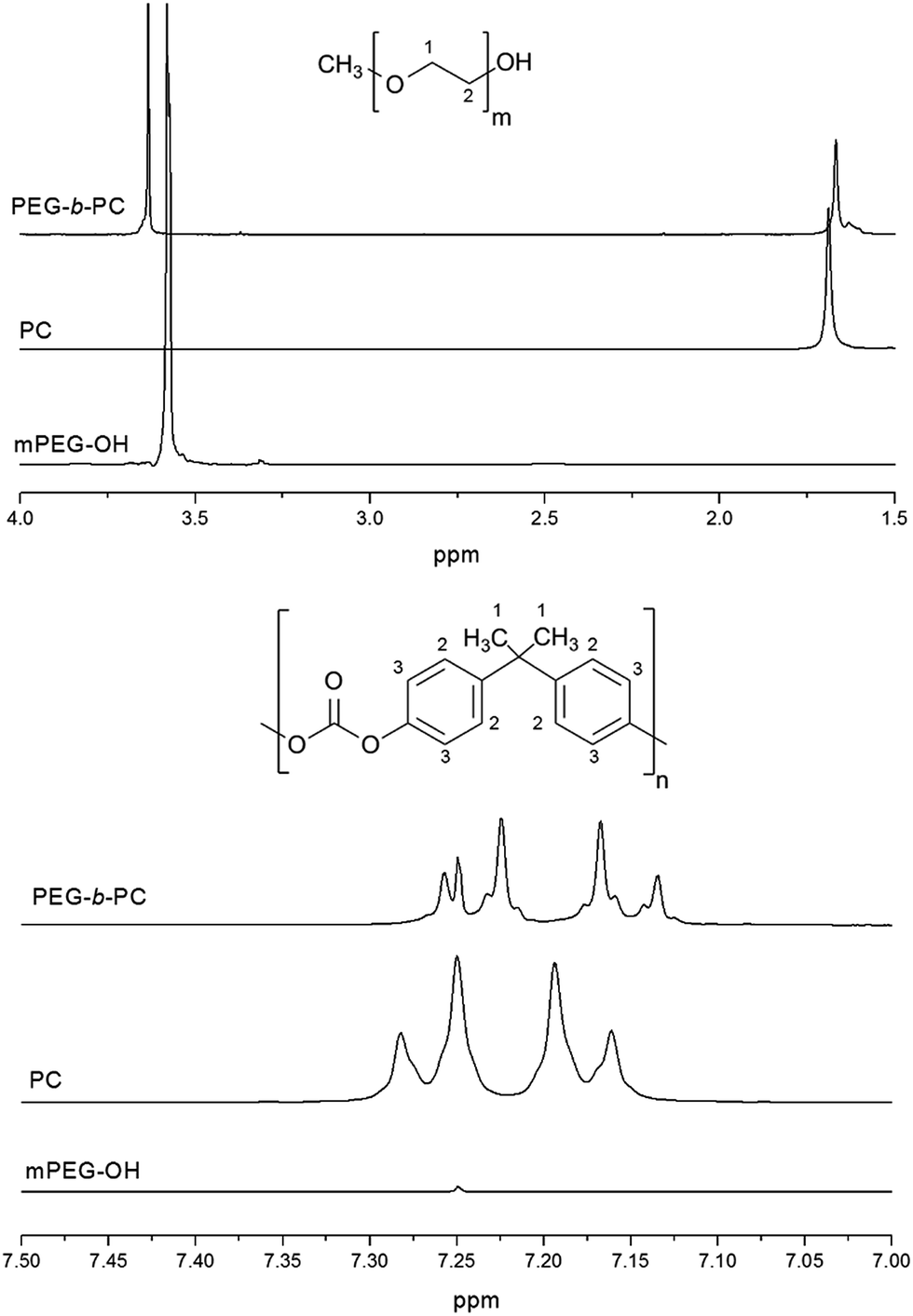 | ||
| Fig. 2 1H NMR for the mPEG-OH and PC starting materials and the synthesised PEG-b-PC block copolymer in CDCl3. The chemical structure of the mPEG-OH and PC starting materials are shown. | ||
The thermal properties of the starting and synthesised materials were analysed by differential scanning calorimetry (DSC) using both conventional and modulated modes. Thermal analysis of the starting homo-polymer materials showed that the glass transition of the PC and the melting of the PEG blocks could be detected in conventional mode (see ESI Fig. S4 and S5†). However the glass transition of the PEG block could only be captured in modulated mode (see ESI Fig. S6†), which is not surprising as the difficulties in determining the glass transition of PEG using conventional DSC mode have been highlighted in the literature.31
In the MDSC endotherm of the synthesised PEG-b-PC copolymers (Fig. 3), a melting peak at around 53 °C was observed which corresponds to the melting of the polyethylene glycol block. The melting of the PEG blocks however is noticeably depressed in comparison to the mPEG-OH homo-polymer which melts at 63.5 °C (Fig. S5†). Depression in melting peak upon attachment of another crystalline and non-crystalline block to poly(ethylene glycol) unit has been explained from the kinetically induced chain folding effects derived from the end interfacial theory.32
 | ||
| Fig. 3 MDSC endotherm of the PEG-b-PC block copolymer showing the melting and the various glass transitions. | ||
The glass transition respective to the PEG block was observed at −6.5 °C along with an additional glass transition at a higher temperature of −8.5 °C, which possibly relates to relaxation of the entangled PEG chains (of reduced molecular mobility) at the miscible interphase. A large glass transition with respect to the PC block was observed between 140 and 160 °C.
The behaviour of the amphiphilic PEG-b-PC copolymers was investigated using dynamic light scattering (DLS) analysis in THF. A z-average diameter of 15.4 nm with narrow size distribution (PDI of 0.308, see ESI, Fig. S7†) was measured, however over an extended period of time (t > 2 h) the z-average diameter was found to increase to 200 nm. The corresponding DLS histogram was also found to be skewed and the distribution was bimodal, centred at 15 and 200 nm (see ESI, Fig. S7†). Bimodal distributions are commonly observed for amphiphilic polymers33 and the appearance of larger particles after coalescence may be ascribed to the relative high amount of the more hydrophobic block in the amphiphilic block copolymer aggregating as a result of intermolecular association.34,35 After coalescence and evaporation of the THF, the morphology of the particles was analysed by scanning electron microscopy (SEM). We observed a wide distribution of smooth nano-capsules with diameters varying between approximately 150 nm and just under 1 micron (Fig. 4). The morphology of the nano-capsules (constituted of a polymer shell and an empty core) was revealed as some of the capsules had ruptured under vacuum.
 | ||
| Fig. 4 SEM images of the coalesced amphiphilic PEG-b-PC copolymer after evaporation of the THF. Inset (A) shows a magnified image of a nano-capsule. | ||
In order to better control the size of the PEG-b-PC nano-capsules the thermodynamic trapping method25 was employed by adding water (a miscible non-solvent) to the PEG-b-PC copolymer/THF mixture. The study first started by analysing the effect of the solvent ratio on the size of the nano-capsules at a fixed PEG-b-PC concentration of 1 mg per ml of solvent mixture. The THF to water volume ratio was varied from 4 to 1 and 1 to 4 (see Experimental section), resulting in visually homogeneous, but phase separated systems in all cases. The milky suspensions, formed immediately upon quickly mixing the two liquids in the appropriate ratios, were colloidally stable over a number of days, aside from reversible density-driven settling. The dry collected product showed a spherical morphology associated with a core–shell nano-morphology (showed in the SEM images in Fig. 5). The spherical-capsule morphology is indicative of two phenomena: the presence of liquid–liquid phase separation induced by interfacial tension mismatch (spherical) and the surface-driven precipitation of the polymer, in the dispersed polymer-rich phase. Although THF and water are miscible, it is not uncommon that mixing polymer solutions in THF with water may lead to temporary thermodynamic instability, and the formation of two phases, as the solvent quality is reduced. Considering the copolymer's higher solubility in THF than in water, the likely polymer-rich phase is dispersed within the aqueous-rich but polymer-poor phase. The phase separation and precipitation of the copolymer can be considered non-solvent induced, and related to a fine balance between thermodynamically favoured states, regulated by kinetically-favoured transitions. We hypothesise a transient liquid–liquid separation stage, followed by the precipitation and consolidation of the polymer shell. It can be expected that larger amounts of non-solvent (i.e. water) may shift the balance of forces, affecting the emulsion droplet size and the kinetics of polymer precipitation, thus affecting capsule diameter distributions.
 | ||
| Fig. 5 SEM images of the PEG-b-PC capsules prepared from (A) 1 to 4; (B) 2 to 3; (C) 3 to 2 and (D) 4 to 1 THF to water ratio mixtures (1 mg per ml PEG-b-PC). The size distribution (nm) of the capsules at each ratio is shown. | ||
The size distribution of the nano-capsules was analysed statistically. It was found that when the THF was volumetrically dominant, the size distribution of the nano-capsules was skewed with large variations in sizes (Fig. 5 images C and D). The capsules prepared from a 4 to 1 solution ratio presented a mean diameter of 350 nm with a standard deviation of ±150 nm and some large particles of up to 2000 nm were observed. Increased water content reduced the size of the nano-capsules and tightened the size distribution. In the particular case of the 1 to 4 THF to water ratio, the average size of the nano-capsules was found to be 115 nm ± 75 nm (Fig. 5 image A).
Following that, the effect of PEG-b-PC copolymer concentration (0.2, 0.5, 1 and 2 mg ml−1) on the size of the nano-capsules was analysed, using a 1 to 4 THF to water ratio mixture. The SEM images, presented in Fig. 6, show that the size distribution of the nano-capsules was tight at the lowest concentration of 0.2 mg ml−1 with a mean average diameter of 85 nm ± 30 nm. As the concentration of the PEG-b-PC in solution increased to 2 mg ml−1 much larger capsules were formed (>1 microns) as expected. This is in agreement with previous works from Eisenberg who investigated the changes in polystyrene-b-poly(acrylic acid) vesicle size upon changes in the acrylic acid block length, water content in dioxane/water mixtures and polymer concentration.36,37 Eisenberg reported that the size and concentration of the vesicles increased with the polymer concentration. On the other hand, in response to water addition, an increase in the interfacial tension was observed causing an increase in the vesicle size and a decrease in the total number of vesicles. In more recent time, a similar trend was reported by Marsden who investigated the water addition/solvent evaporation method (referred to as the WASE) to rapidly assemble amphiphilic block copolymer into stable polymersomes.24 Sanson et al.38 also used a solvent-injection method to precipitate PTMC24-b-PGA12 copolymer vesicles of sizes varying between 50 and 450 nm depending on the nature of organic phase (i.e. THF, DMSO or MeOH), injection time and pH.
 | ||
| Fig. 6 SEM images of the PEG-b-PC capsules prepared in a 1 to 4 THF to water ratio mixture at (A) 0.2 mg ml−1; (B) 0.5 mg ml−1; (C) 1 mg ml−1 and 2 mg ml−1 (D). The size distribution (nm) of the capsules at each ratio is shown. | ||
Here we found that by increasing water content the size of the nano-capsules was reduced, and even though our results may seem in disagreement with the research work of Marsden and Eisenberg discussed just above, it is to be noted that our process is sensibly different. The response to water addition was extensively studied by Eisenberg39 where it was noted that the sequential addition of small amount e.g. 5 to 10 wt% of water to amphiphilic polystyrene-b-poly(acrylic acid) block copolymers dissolved in DMF induced transitions in morphologies (spheres, rods, worms and bilayers). The type of observed morphology was also found to be dependent on the water content used, the copolymer type and concentration in the organic solvent. The morphological transitions indicate that the shells of the polymersomes were highly flexible in nature, thanks to a combination of chain mobility and dynamic equilibrium between unimers and aggregates.14 In the same work, it was mentioned that dynamic equilibrium was preserved, providing the water content was not too high, otherwise a freezing of the morphology was observed.39 The formed nano-capsules presented in our work are the result of a ‘frozen’ kinetically favoured structure, resulting in a solid-like shell. In the interest of showing that water addition induces instantaneous precipitation and kinetic locking of the PEG-b-PC copolymer chains, we conducted a parallel set of experiment where water was added sequentially to a solution consisting of 0.5 mg PEG-b-PC per ml of THF so that the THF to water ratio would vary from 4 to 1, to 1 to 4 (similar to what we did in the previous set of experiments). With the 4 to 1 THF to water ratio, we found that the size of the capsules was ∼400 nm. Subsequent addition of water to this solution however did not induce any noticeable change in size as shown from the DLS analysis results (ESI Fig. S8†). This experiment somewhat proved that the formed nano-structure are locked into a configuration at the moment of their formation, for the relative concentrations used. This further indicates the solid nature of the capsules' shells produced here, rather than a flexible polymersome-like nature. The ability to tune particle size through the controlled initial addition of water is a great advantage from a processing perspective, especially when coupled with the inherent increased speed of the process.
The possibility of encapsulating a water-insoluble bisphenol-A diglycidyl ether (DGEBA) resin (which has a very similar chemical structure to that of the PC block) was investigated. We dissolved approximately 5 mg of PEG-b-PC copolymer with approximately 20 mg of epoxy resin, bisphenol A diglycidyl ether (DGEBA) with 2 ml of THF. To this clear and transparent solution, approximately 3 ml of distilled water was added. The liquid mixture became cloudy and milky in appearance (Fig. 7, image A). The vial was vigorously stirred and left to rest to let large particles settle. Phase separated large micro-spheres were collected at the bottom of the vial (Fig. 7, image B). To test the performance of the supposed epoxy-filled capsules, the THF was evaporated at room temperature, and the remaining water was evaporated in a conventional oven at 80 °C for approximately 1 hour. The micro-capsules collected at the bottom of the vial (Fig. 7, image C) seemed intact. To test whether the micro-capsules contained any epoxy, a syringe needle was used under a microscope to induce shell rupture. Fig. 7, image D, illustrates the effect of puncture on the capsules, with DGEBA leaching out of the capsules after rupture.
 | ||
| Fig. 7 Photo of the epoxy diglycidyl ether of bisphenol A (DGEBA)/PEG-g-PC mixture in water/THF solvent (20/80 ratio) after vigorous stirring (A) and after coalescence (∼5 minutes, B). After evaporation of the water/THF, the DGEBA filled PEG-b-PC micro-capsules were visible at the bottom of the vial (C). Rupture of the capsules using a needle tip was achieved, with epoxy DGEBA leaching out of the capsules (D). | ||
Conclusions
The collective findings of the research work presented in this manuscript is two-fold. Firstly, we presented a simple way to prepare amphiphilic poly(ethylene glycol)-block-poly(bisphenol A carbonate) block copolymers starting from the poly(bisphenol A carbonate) (PC) and monomethyl ether poly(ethylene glycol) (mPEG-OH) homo-polymer blocks of pre-determined block length. The synthesis was achieved using a simple bio-chemistry coupling methodology associated with an easy product purification. Although the results reported here are confined to one type of amphiphilic copolymer, this methodology can be adapted to a wide library of pre-existing homo-polymers having similar chemical functionalities in order to tune the amphiphilicity and the type of building blocks.Secondly, we demonstrated that the thermodynamic locking of the copolymer chains could be utilised for the fast formation (∼a few seconds) of nano-capsules of controllable diameters. This simple methodology, only requiring water addition into solubilised copolymer in THF, offers a much better alternative to other chemical methods presented in the literature for the preparation of amphiphilic copolymer nano-capsules typically requiring long times and strict conditions.
Collectively we believe that the ability to tune copolymer block ratio from a wide library of pre-existing polymers whilst being able to speed up both the copolymer synthesis and capsule formation process are all advantageous from a technological stand point.
Notes and references
- F. Caruso, Colloids and colloid assemblies: synthesis, modification, organization and utilization of colloid particles, John Wiley & Sons, 2006 Search PubMed.
- H. Möhwald, E. Donath and G. Sukhorukov, Smart capsules, Wiley-VCH, Weinheim, 2003 Search PubMed.
- Y. Wang, A. S. Angelatos and F. Caruso, Chem. Mater., 2008, 20, 848–858 CrossRef CAS.
- D. G. Shchukin and G. B. Sukhorukov, Adv. Mater., 2004, 16, 671–682 CrossRef CAS.
- J. Rodriguez-Hernandez, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS.
- M. Okubo, M. Shiozaki, M. Tsujihiro and Y. Tsukuda, Colloid Polym. Sci., 1991, 269, 222–226 CAS.
- M. Okubo and T. Nakagawa, Colloid Polym. Sci., 1992, 270, 853–858 CAS.
- K. Bouchemal, S. Briançon, E. Perrier, H. Fessi, I. Bonnet and N. Zydowicz, Int. J. Pharm., 2004, 269, 89–100 CrossRef CAS PubMed.
- K. Bouchemal, S. Briançon, H. Fessi, Y. Chevalier, I. Bonnet and E. Perrier, Mater. Sci. Eng. C, 2006, 26, 472–480 CrossRef CAS.
- L. Janssen and K. Te Nijenhuis, J. Membr. Sci., 1992, 65, 59–68 CrossRef CAS.
- L. Janssen and K. Te Nijenhuis, J. Membr. Sci., 1992, 65, 69–75 CrossRef CAS.
- Dekker Encyclopaedia of Nanoscience and Nanotechnology, ed. J. Schwartz, C. Contescu and K. Putyera, Marcel Dekker, Inc, New York, 2004, vol. 5 Search PubMed.
- A. P. Johnston, C. Cortez, A. S. Angelatos and F. Caruso, Curr. Opin. Colloid Interface Sci., 2006, 11, 203–209 CrossRef CAS.
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65, 259–269 CrossRef CAS PubMed.
- Y. S. Lee, Self-assembly and nanotechnology: a force balance approach, John Wiley & Sons, 2008 Search PubMed.
- V. Heroguez, Y. Gnanou and M. Fontanille, Macromolecules, 1997, 30, 4791–4798 CrossRef CAS.
- M. M. Ali and H. D. Stöver, Macromolecules, 2004, 37, 5219–5227 CrossRef CAS.
- S. Aoshima, S. Sugihara, M. Shibayama and S. Kanaoka, Synthesis and Self-Association of Stimuli-Responsive Diblock Copolymers by Living Cationic Polymerization, Macromol. Symp., 2004, 215(1), 151–164 CrossRef CAS.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS PubMed.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- C. A. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- G. T. Hermanson, Bioconjugate techniques, Academic press, 2013 Search PubMed.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- H. R. Marsden, L. Gabrielli and A. Kros, Polym. Chem., 2010, 1, 1512–1518 RSC.
- D. Lensen, D. M. Vriezema and J. C. M. van Hest, Macromol. Biosci., 2008, 8, 991–1005 CrossRef CAS PubMed.
- A. K. Gupta and S. Wells, IEEE Transactions on NanoBioscience, 2004, 3, 66–73 CrossRef PubMed.
- J. Devaux, P. Godard, J. Mercier, R. Touillaux and J.-M. Dereppe, J. Polym. Sci., Polym. Phys. Ed., 1982, 20, 1881–1894 CrossRef CAS.
- L. Tang and N. Y. Lee, Lab Chip, 2010, 10, 1274–1280 RSC.
- K. Jankova, X. Chen, J. Kops and W. Batsberg, Macromolecules, 1998, 31, 538–541 CrossRef CAS.
- P. M. Henrichs, J. Tribone, D. J. Massa and J. M. Hewitt, Macromolecules, 1988, 21, 1282–1291 CrossRef CAS.
- H. Petersen, P. M. Fechner, D. Fischer and T. Kissel, Macromolecules, 2002, 35, 6867–6874 CrossRef CAS.
- I. Hamley, in Interfaces Crystallization Viscoelasticity, Springer, 1999, pp. 113–137 Search PubMed.
- Y. Wang, Y. Tan, X. Huang, Y. Che and X. Du, J. Appl. Polym. Sci., 2009, 112, 1425–1435 CrossRef CAS.
- N. A. B. Vieira, J. R. Neto and M. J. Tiera, Colloids Surf., A, 2005, 262, 251–259 CrossRef CAS.
- S. Verbrugghe, A. Laukkanen, V. Aseyev, H. Tenhu, F. M. Winnik and F. E. Du Prez, Polymer, 2003, 44, 6807–6814 CrossRef CAS.
- A. A. Choucair, A. H. Kycia and A. Eisenberg, Langmuir, 2003, 19, 1001–1008 CrossRef CAS.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- C. Sanson, C. Schatz, J.-F. le Meins, A. Brûlet, A. Soum and S. Lecommandoux, Langmuir, 2010, 26, 2751–2760 CrossRef CAS PubMed.
- L. Zhang and A. Eisenberg, Macromolecules, 1999, 32, 2239–2249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra23555c |
| This journal is © The Royal Society of Chemistry 2016 |