
Enhanced visible light and photocatalytic performance of TiO2 nanotubes by hydrogenation at lower temperature
Lijuan Hana,
Zheng Maa,
Zhihe Luob,
Gang Liua,
Jiantai Mac and
Xingcai An*a
aNatural Energy Institute, Gansu Academy of Sciences, Lanzhou 730046, People's Republic of China. E-mail: anxingcai@unido-isec.org; Tel: +86-931-8386630
bNorthwest Yongxin Coatings Company Limited, Lanzhou 730046, People's Republic of China
cState Key Laboratory of Applied Organic Chemistry, The Key Laboratory of Catalytic Engineering of Gansu Province and Chemical Engineering, Lanzhou University, Lanzhou 730000, People's Republic of China
First published on 20th October 2015
Abstract
Protonated titanate nanotubes were chosen as a precursor in a hydrogenation process. Owing to the high capacity for molecular hydrogen storage of nanotubes, TiO2 nanotubes can be hydrogenated through thermal treatment under N2 and H2 mixed flow at lower temperature. A series of hydrogenated TiO2 nanotubes and nanobelts were synthesized and characterized by XRD, UV-vis, TEM, EPR and XPS. The results showed that the hydrogenated TiO2 nanotubes possess tiny and uniform diameters of 8–10 nm and walls thicknesses of 2–3 nm, and were mainly anatase. The anatase TiO2 nanotubes transformed to TiO2-B nanobelts when the hydrothermal temperature was higher than 150 °C. The light absorption of hydrogenated TiO2 nanotubes was expanded to visible light. However, air-TiO2 and hydrogenated TiO2 nanobelts only absorbed ultraviolet light. According to XPS and EPR analysis, hydrogenated TiO2 nanotubes displayed stable core–shell structures, in which the surface was mainly stoichiometric TiO2 and the core was non-stoichiometric TiO2 with Ti3+ and oxygen vacancies. The adsorption and photocatalytic performance were evaluated by the removal rate of phenol. Based on a pseudo-first order kinetic model, the degradation rate constant was obtained with the regression analysis. The highest degradation rate constant of hydrogenated TiO2 nanotubes was 5.2 times higher than air-TiO2. In comparison, the degradation rate constants of hydrogenated TiO2 nanobelts were much lower than air-TiO2. The results showed that the precursor with nanotube structure can be hydrogenated easily at lower temperature compared with nanobelts, resulting in the photocatalytic activity of hydrogenated TiO2 nanotubes being enhanced drastically.
1. Introduction
Titanium dioxide (TiO2) is regarded as one of the most ideal photocatalysts. It has promising applications in many areas, owing to its nontoxicity, excellent chemical stability, and low cost.1,2 However, the band gap (Eg) of pristine TiO2 is too wide, which results in that pristine TiO2 only absorbs UV light, limiting its practical applications. So far, many approaches involving adding nonmetals3–6 or metals7–9 have been used to expand the visible light response range and enhance the photocatalytic activity of TiO2. However, the introduction of dopants, acting as charge carrier recombination centers, easily increases the recombination possibility of photo-generated electron–hole pairs, resulting in a decline in photocatalytic performance.10Hydrogenated black TiO2 has been proved to enhance visible light absorption, due to the introduction of electronic state band below the conduction band by oxygen vacancy or Ti3+ doping and the disorder surface of TiO2.11,12 The light absorption of black TiO2 can even be extended to infrared region,11 which has aroused great interest of researchers. Recently, black TiO2 through various methods have been reported,11–21 such as high pressure in H2 atmosphere,11 plasma assisted hydrogenation13, high temperature in ordinary pressure,12,14 hydrothermal method.15,19,21 Among these methods, the method of high temperature (>500 °C) in ordinary pressure is easy and simple. However, it is difficult to control the crystallite size of TiO2 and its photocatalytic activity at high temperature. The lower temperature hydrogenation is helpful to control the crystallite size of TiO2, but the lower temperature generally leads to incomplete hydrogenation of TiO2. Herein, we have developed a facile method of hydrogenated TiO2 at lower temperature (400 °C) in ordinary pressure. In the method, the tiny and hollow tubular structure protonated titanate was chosen as precursors. Due to the high capacity for molecular hydrogen storage of protonated titanate nanotubes,22–26 it is possible that the protonated titanate nanotubes are in the hydrogen-rich environment at hydrogenation procedure. The hydrogen-rich environment of nanotubes could reduce the hydrogenated temperature of TiO2 in ordinary pressure, resulting in that the hydrogenated TiO2 was generated easily at lower temperature (400 °C) under ordinary pressure compared with nanoparticles or nanobelts.
The research results show that the protonated titanate nanotubes were hydrogenated at lower temperature (400 °C) in ordinary pressure. And, the protonated titanate nanotubes transformed to black hydrogenated anatase TiO2 nanotubes in hydrogenation process, which exhibit good visible light response and photocatalytic performance.
2. Experimental section
2.1. Photocatalyst preparation
All chemicals were of analytical grade and used without further purification. TiO2 precursors (protonated titanate) were prepared by alkaline hydrothermal method and ion exchange.22 Briefly, a certain amount of anatase titania powder prepared according to sol–gel method27 were mixed with 60 mL of 10 mol L−1 NaOH solution in a Teflon-lined autoclave at appropriate temperature for 24 hours. The hydrothermal temperature affects significantly the morphology and crystal form of material.22–26 When the hydrothermal temperature is too high, the nanotubes structure of material would transform to nanobelts or nanorods.22,25 So, we controlled the tubular shape of material carefully by hydrothermal temperature. The precipitate obtained after alkaline hydrothermal treatment was washed firstly with deionized water until the pH reached about 7, and then immersed in 0.1 mol L−1 HCl solution overnight with the ion exchange. After that, the precipitate was washed by deionized water again until the pH was about 7 and dried in 100 °C for 10 hours. The dried powder was calcinated at 400 °C for 4 hours in N2 and H2 mixed flow. After that, the sample was cooled fast in inert environment. Finally, the black TiO2 obtained. The samples prepared at 110 °C, 130 °C, 150 °C, 170 °C, 190 °C, 210 °C were marked as H-TiO2(110), H-TiO2(130), H-TiO2(150), H-TiO2(170), H-TiO2(190), H-TiO2(210) respectively. Protonated titanate prepared at 130 °C was calcinated at 400 °C for 4 hours in air atmosphere to produce TiO2 without hydrogenation, which was used as reference sample and marked as air-TiO2.2.2. Characterizations
The crystal phases of the samples prepared were characterized by the X-ray diffraction (XRD) (D/Max-2400 powder diffractometer) with Cu Kα X-ray source (λ = 0.154056 nm) at room temperature. The morphology of the sample was observed on a JEM-2100 transmission electron microscopy (TEM) with an accelerating voltage of 200 kV. The chemical states of the samples were characterized by ESCALAB210 X-ray photoelectron spectroscopy (XPS) with an Mg Kα X-ray source. UV-vis diffuse reflection spectra of the samples were obtained by UV-vis spectrophotometer (UV-2550) to know ultraviolet and visible light absorption from 200 nm to 800 nm. Electron paramagnetic resonance (EPR) spectra were recorded on a Bruker EMX-10/12 spectrometer at room temperature.2.3. Adsorption and photocatalytic performance measurement
The adsorption and photocatalytic performance of samples were measured as follow procedure. Herein, phenol was selected as a model pollutant to evaluate the adsorption and photocatalytic activity in visible light. The visible light source was a 300 W Xe lamp (Beijing Bofeilai Co., Ltd, Microsolar300) equipped with an AM 1.5 glass filter and a UV-cut-420 nm optical filter. Firstly, 50 mg photocatalyst was dispersed in 250 mL of 10 mg L−1 aqueous solution of phenol in cylindrical quartz vessel. Then, the suspension above was magnetically stirred in the dark for 30 minutes to reach adsorption–desorption equilibrium. Then, 5 mL dispersion was withdrawn and centrifuged to test the adsorption activity according to the concentration of phenol. After the adsorption reached equilibrium, the suspension was illuminated by Xe lamp. At 1 hour intervals, 5 mL suspension was withdrawn, centrifuged, and filtered by a 0.22 μm membrane filter to remove the remaining particles. The concentration of phenol at different time interval was measured at λ = 270 nm by UV2500 spectrophotometer (Shimadzu Corporation). Three replicate experiments were performed for every sample.According to the former works,28–30 the photocatalytic degradation kinetics of phenol can be described using a pseudo-first order kinetic model based on the concentration of phenol:
 | (1) |
3. Results and discussion
3.1. Structure and morphology characterization
The crystal structure of hydrogenated TiO2 prepared at different hydrothermal temperatures (110–210 °C) and air-TiO2 were analyzed by XRD, as shown in Fig. 1. From Fig. 1, it can be seen that hydrothermal temperature significantly influences the formation of crystal structure. When the hydrothermal temperature was from 110 °C to 150 °C, hydrogenated TiO2 exhibited the phase of pure anatase16. Comparing with air-TiO2, the diffraction peaks of hydrogenated TiO2 had not obvious shift, and the intensity of hydrogenated TiO2 became weaker, which is accordance with the other hydrogenation TiO2.11,31 The decrement of the peak intensity can be ascribed to the increase of defect density in the crystal structure. However, when the hydrothermal temperature was higher than 170 °C, several new broad peaks at 2θ = 14.0°, 24.7°, 28.8°, 44.7° appeared, which were in accordance with the patterns of TiO2-B.32–34 This means that the anatase had not transformed to rutile but transformed to TiO2-B at higher hydrothermal temperature.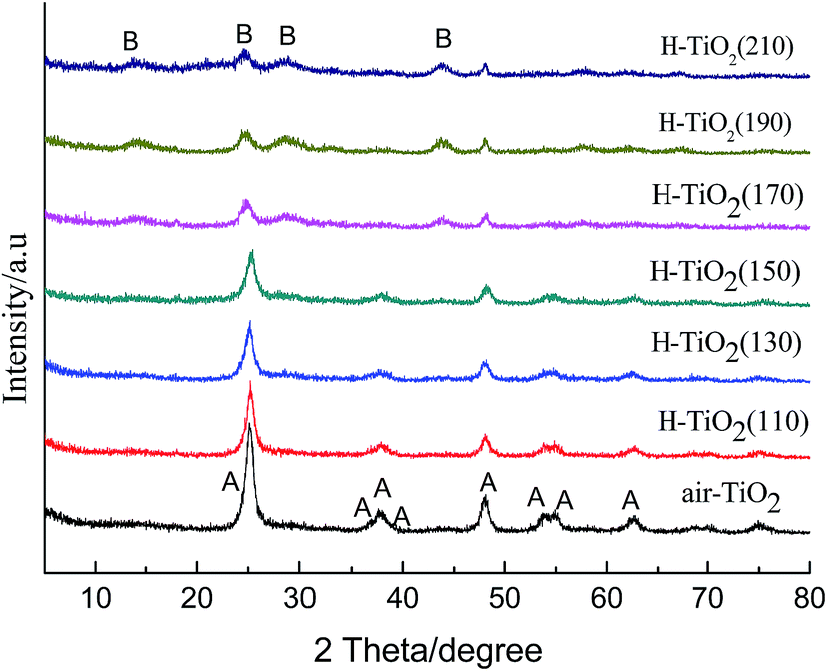 | ||
| Fig. 1 XRD patterns of H-TiO2(110), H-TiO2(130), H-TiO2(150), H-TiO2(170), H-TiO2(190), H-TiO2(210) and air-TiO2; (A): anatase TiO2, (B): TiO2-B. | ||
In order to further probe the morphology of hydrogenated TiO2 prepared at different hydrothermal temperatures, transmission electron microscopy (TEM) images were obtained, as shown in Fig. 2. Fig. 2a is the TEM micrograph of hydrogenated TiO2 prepared at 110 °C. It shows that most of the material is tubular structure, but a few nanosheets still exist. When the hydrothermal temperature was increased to 130 °C, it can be seen clearly from Fig. 2b that the nanotubes with a nearly uniform diameters (8–10 nm) are dispersed homogeneously, and the lengths of these nanotubes are 50 nm–300 nm. The inset in Fig. 2b displays the electron diffraction pattern of these nanotubes. Two diffraction rings in inset is clear, which can be identified as (200) and (101) of the anatase TiO2 crystal. The other strong diffractions of anatase TiO2 crystal are barely identified, which indicates that the shells should consist of a quasi-two-dimensional lattice.24 Fig. 2c is the TEM micrograph of H-TiO2(130) at high magnification. The right inset in Fig. 2c is an enlarged picture of the nanotube walls as marked by the white circle. Through measurement, the lattice fringes spacing are of around 0.19 nm, corresponding to the (200) plane of the anatase phase, which implied that the axis of TiO2 nanotubes grew along (200) plane of the anatase phase. With the hydrothermal temperature increased to 150 °C, these nanotubes (Fig. 2d) grew longer and were intertwined each other. It is possible that the long intertwined nanotubes may not store enough molecular hydrogen. Fig. 2e is the TEM micrograph of H-TiO2(170), which reveals the sample prepared at 170 °C is the mixture of nanobelts and nanotubes. When the hydrothermal temperature was increased to 210 °C, the nanotubes had entirely transformed to TiO2 nanobelts. From Fig. 2f, it can be seen that the width of TiO2 nanobelts was about 50 nm and many TiO2 nanobelts overlapped each other. The inset in Fig. 2f displays the electron diffraction pattern of these nanobelts. The electron diffraction pattern is accordance with the other report about TiO2-B,32,34 which implied that the crystal phase had changed to TiO2-B at higher hydrothermal temperature. The result is consistent with analysis of XRD.
 | ||
| Fig. 2 TEM images of hydrogenated TiO2 (a): TEM image of H-TiO2(110); (b): TEM image of H-TiO2(130) at low magnification, the inset is electron diffraction pattern; (c): TEM image of H-TiO2(130) at high magnification, the inset is the enlarged picture of nanotube wall; (d): TEM image of H-TiO2(150); (e): TEM image of H-TiO2(170); (f): TEM image of H-TiO2(210), the inset is electron diffraction pattern. | ||
The UV-vis diffuse reflectance spectra of hydrogenated TiO2 nanotubes prepared in different hydrothermal temperatures and air-TiO2 are shown in Fig. 3. It can be seen that air-TiO2 only responds to ultraviolet light. The hydrogenated TiO2 nanotubes (H-TiO2(110), H-TiO2(130), H-TiO2(150)) had absorption tail in the visible light regions. And, the onset of the optical absorption of hydrogenated TiO2 nanotubes had not exhibited red shift comparing with air-TiO2, which implies the valence band or conduction band had not shift. As reported previously,26 oxygen vacancies and Ti3+ in hydrogenated TiO2 nanotubes would be formed as isolated states in the band gap of TiO2 to induce visible light absorption, rather than a shift in the position of either band edges. However, the light response of hydrogenated TiO2(H-TiO2(170), H-TiO2(190), H-TiO2(210)) had not been expanded to visible light but had a shift to ultraviolet light, which may be due to band structure of TiO2-B itself on the one hand, on the other hand, the nanobelts possess low capacity for molecular hydrogen storage, resulting in incomplete hydrogenation. So, the visible light response of these samples (H-TiO2(170), H-TiO2(190), H-TiO2(210)) are lower obviously than hydrogenated TiO2 nanotubes (H-TiO2(110), H-TiO2(130), H-TiO2(150)), even air-TiO2.
 | ||
| Fig. 3 UV-visible diffuse reflectance spectra of H-TiO2(110), H-TiO2(130), H-TiO2(150), H-TiO2(170), H-TiO2(190), H-TiO2(210) and air-TiO2. | ||
To further examine the effect of hydrogenation on the chemical composition of TiO2 nanotubes surfaces, XPS spectra of the hydrogenated TiO2 nanotubes prepared in 130 °C and air-TiO2 were investigated. In order to avoid the effect of carbon caused in hydrogenation, the samples were sputtered by Au before measurement. Fig. 4a presents the Ti 2p XPS spectra of air-TiO2 and H-TiO2(130). From Fig. 4a, it can be seen that both of H-TiO2(130) and air-TiO2 had two symmetric peaks at ∼464.6, ∼458.8 eV, respectively, which are the characteristic Ti 2p1/2 and Ti 2p3/2 peaks of Ti4+.16,19,25 The Ti 2p XPS spectrum of hydrogenated TiO2 nanotubes did not give any Ti3+ or Ti2+ signals, which indicated that no titanium with lower oxidation states were detected in the surface of samples. So, we deduce that the surface of hydrogenated TiO2 nanotubes is made of stoichiometric TiO2.
 | ||
| Fig. 4 (a) XPS spectra of hydrogenated TiO2 nanotubes H-TiO2(130) and air-TiO2. (b) EPR spectra of H-TiO2(110), H-TiO2(130), H-TiO2(150), H-TiO2(170), and air-TiO2. | ||
Since Ti3+ was not detected by XPS analysis, EPR of hydrogenated TiO2 nanotubes (H-TiO2(110), H-TiO2(130), H-TiO2(150), H-TiO2(170)) and air-TiO2 were conducted to further identify the presence of oxygen vacancy and Ti3+. The EPR spectra are shown in Fig. 4b. According to Fig. 4b, all the hydrogenated TiO2 samples gave rise to a signal at g = 2.002, which is the typical g value for paramagnetic Ti3+ centers due to the oxygen depletion.35,36 However, the hydrogenated TiO2 samples revealed the different intensity, which implied the different Ti3+ concentration. The EPR intensity of hydrogenated TiO2 nanotubes, such as H-TiO2(110), H-TiO2(130) and H-TiO2(150), were very strong. The intensity of hydrogenated TiO2 nanobelts (H-TiO2(170)) was lower than the others, which implied the low concentration of Ti3+ of hydrogenated TiO2 nanobelts and the high concentration of Ti3+ of hydrogenated TiO2 nanotubes. Because the concentration of Ti3+ is related to the degree of hydrogenation, the different concentrations of Ti3+ of hydrogenated TiO2 imply that the nanotubes are easily hydrogenated than nanobelts, which is accordance with our speculation. As reported, when a part of Ti3+ located in the surface of TiO2 nanotubes, the surface Ti3+ can efficiently trap oxygen molecules on defect sites to form ˙O2−, which can generate another EPR signal. This signal has not been found in all the EPR spectra, which imply that Ti3+ is absent from the surface of the hydrogenated TiO2. The result is in good agreement with XPS analysis. In contrast, air-TiO2 had not shown any signals at the same measurement condition, which reveals that there was not any Ti3+ in the bulk or the surface for air-TiO2. EPR result reveals that a high concentration of Ti3+ was located in the bulk of TiO2 nanotubes, resulting in defective and non-stoichiometric TiO2. In addition, the surface of hydrogenated TiO2 nanotubes is composed of stoichiometric TiO2. The stable stoichiometric TiO2 can protect Ti3+ in the bulk of TiO2 nanotubes from being oxidized while non-stoichiometric TiO2 in the bulk promote visible light response and photocatalytic activity. The structure is similar to the core–shell structure of hydrogenated TiO2 nanoparticles prepared by the other methods,12,37 which is considered stable and high photocatalytic activity.
3.2. Adsorption and photocatalytic performance of the samples
The relative concentrations plotted over time for the adsorption and photocatalytic performance of as-prepared samples are presented in Fig. 5. The result shows that all samples have scarcely any adsorption activity for phenol. So, the effect of adsorption activities of samples compared with their photocatalytic performance can be ignored. For the photocatalytic performance of the samples, phenol can be degraded in different extent under visible light irradiation. Among these samples, the hydrogenated TiO2 nanotubes prepared at 110 °C, 130 °C, 150 °C exhibited high photocatalytic activity for phenol. However, the hydrogenated TiO2 prepared at 170 °C, 190 °C, 210 °C exhibited very poor photocatalytic activity for phenol, even badly than air-TiO2.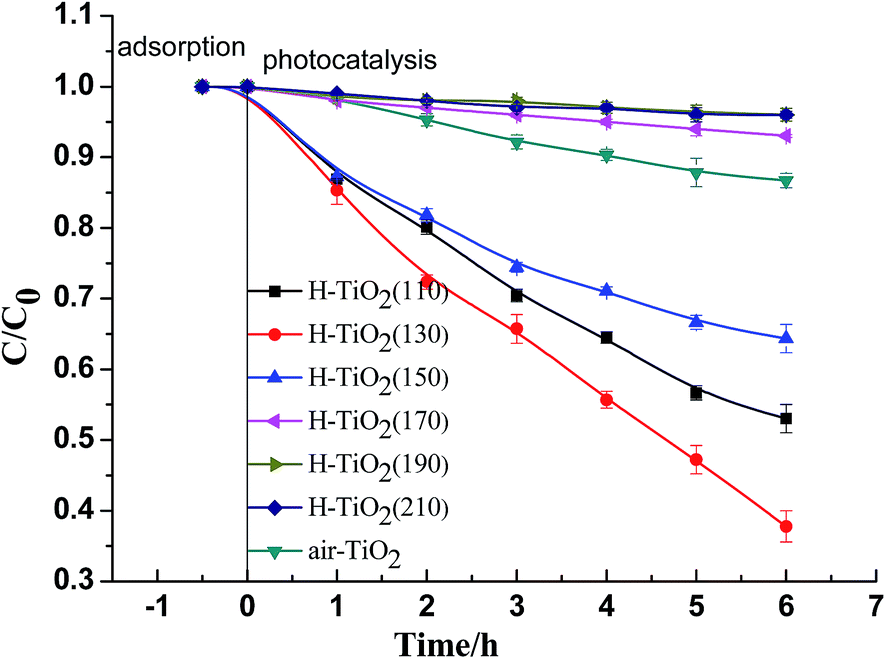 | ||
| Fig. 5 Adsorption and photocatalytic performance for degradation phenol of H-TiO2(110), H-TiO2(130), H-TiO2(150), H-TiO2(170), H-TiO2(190), H-TiO2(210) and air-TiO2. | ||
The photocatalytic degradation of phenol follows an apparent first-order reaction. Based on pseudo-first order kinetic model (eqn (1)),28–30 the degradation rate constant (kp) is obtained with the regression analysis based on the minimization of the squared errors, and the correlation coefficient of the line is also presented (R2). The results of the regression analysis for these samples are given in Table 1. It is known that an R2 closer to 1.0 indicates that the regression line perfectly fits the data.29 From Table 1, it can be seen that the R2 values of the most of samples were higher than 0.96, and only the R2 values of H-TiO2(190) and H-TiO2(210) were about 0.94, which mean the regression analysis is credible.
| Samples | kp | R2 |
|---|---|---|
| H-TiO2(110) | 0.1036 ± 0.002 | 0.9947 |
| H-TiO2(130) | 0.1559 ± 0.005 | 0.9918 |
| H-TiO2(150) | 0.0715 ± 0.003 | 0.9654 |
| H-TiO2(170) | 0.0115 ± 0.001 | 0.9869 |
| H-TiO2(190) | 0.0069 ± 0.001 | 0.9418 |
| H-TiO2(210) | 0.0063 ± 0.001 | 0.9469 |
| Air-TiO2 | 0.0253 ± 0.002 | 0.9896 |
In order to probe whether the hydrogenation is beneficial to the photocatalytic performance of TiO2 or not, the degradation rate constants of the samples are compared, as shown in Fig. 6. It is obviously that the hydrogenation has not always enhanced the photocatalytic performance. When the structure of TiO2 is nanotubes (hydrothermal temperature ≤ 150 °C), all the degradation rate constants of hydrogenated TiO2 nanotubes are much higher than air-TiO2. The highest degradation rate constant of hydrogenated TiO2 nanotubes (H-TiO2(130)) reaches to 0.1559 h−1, which is 5.2 times higher than air-TiO2. Furthermore, the degradation rate constant of hydrogenated TiO2 nanotubes (H-TiO2(150)), which is the lowest among the hydrogenated TiO2 nanotubes, is higher than air-TiO2. However, when the structure of TiO2 is nanobelts (hydrothermal temperature ≥ 170 °C), the degradation rate constants are lower than air-TiO2. It shows that the structure of precursor and crystal phase effect significantly the photocatalytic performance. The hydrogenated anatase TiO2 nanotubes with high concentration of oxygen vacancies and Ti3+ exhibit high photocatalytic activity while the hydrogenated TiO2-B nanobelts exhibit low photocatalytic activity.
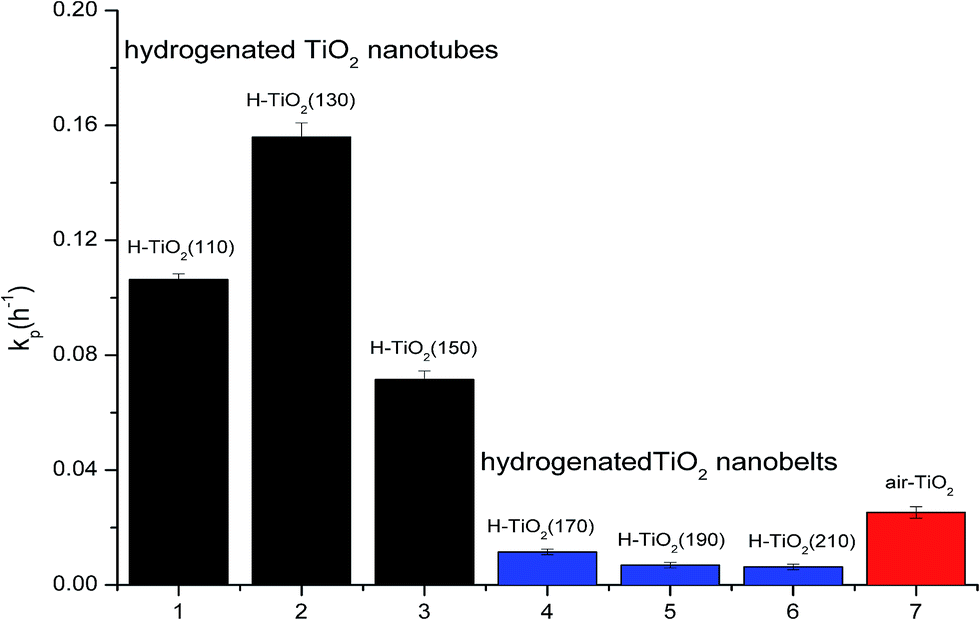 | ||
| Fig. 6 Apparent first-order degradation constants kp (h−1) for photocatalysis of H-TiO2(110), H-TiO2(130), H-TiO2(150), H-TiO2(170), H-TiO2(190), H-TiO2(210) and air-TiO2. | ||
The high concentration of oxygen vacancies and Ti3+ species presented in hydrogenated anatase TiO2 nanotubes would form some isolate defect energy levels in the band gap, which not only enhance the visible light absorption of photocatalysts but also suppress the recombination of electron–hole pairs to promote the photocatalytic activity. In contrast, the concentration of oxygen vacancies and Ti3+ of hydrogenated TiO2-B nanobelts is lower than hydrogenated anatase TiO2 nanotubes, which may not enough to increase the photocatalytic performance. More importantly, the photocatalytic activity of TiO2-B crystal is usually lower than anatase crystal, caused the poor photocatalytic performance of hydrogenated TiO2-B nanobelts.
Among the photocatalysts of H-TiO2(110), H-TiO2(130), H-TiO2(150), H-TiO2(130) had the highest photocatalytic activity. Compared with H-TiO2(110), H-TiO2(150), H-TiO2(130) is composed of the short and thin nanotubes which can adsorb a large number of molecular hydrogen causing the most effective hydrogenation. Owing to the effective hydrogenation, more oxygen vacancies and Ti3+ were generated, which is confirmed by EPR data. So, the visible light photocatalytic activity of H-TiO2(130) was the best due to the higher visible light response and electron–hole separation efficiency.
3.3. The mechanisms of formation of hydrogenated nanotubes
The mechanism of hydrogenated nanotubes was illustrated by Fig. 7. Owing to the protonated titanate nanotubes possess a high capacity for molecular hydrogen storage and many protons on their walls,14 the walls of protonated titanate nanotubes adsorb a large number of hydrogen molecules. According to TEM analysis, TiO2 nanotubes grow along (200) plane of the anatase phase. The (200) plane of nanotube is exposed with mainly unsaturated O and Ti modes. These molecular hydrogens are easily adsorbed on the unsaturated O and Ti sites favorably under the H2 atmosphere.14,38 With the temperature increasing (I), OH bands are formed in the first stage of annealing on the surface of TiO2 nanotubes. Meanwhile, it is possible that Ti–H bonds on the surface of nanotubes are formed, which make the surface Ti recovered octahedral coordination. Upon increasing the annealing temperature (II), Ti–OH and Ti–H become unstable and produced H2O molecules, which result in formation of more oxygen vacancies.39 With the formation of oxygen vacancies, low valence titanium (Ti3+) are formed. Furthermore, hydrogen molecules pass into the bulk of TiO2 nanotubes forming more oxygen vacancies and Ti3+ in its lattice. Then, through fast cooling in inert environment, the active surface is freezed and Ti3+ in its lattice are protected.12 Oxygen vacancies and Ti3+ form located states between conduction band and valence band, which can enhance the efficiency separation of the photogenerated electron–hole and extend the visible light absorption. And then, the photocatalytic activity of hydrogenated TiO2 nanotubes in visible light was enhanced remarkably. | ||
| Fig. 7 Schematic illustration of the formation process of hydrogenated nanotubes. | ||
4. Conclusions
In summary, nanotubes and nanobelts protonated titanate were prepared by alkaline hydrothermal method. Through calcinating protonated titanate in N2 and H2 mixed flow at lower temperature (400 °C), hydrogenated nanotubes anatase and hydrogenated nanobelts TiO2-B obtained. Hydrogenated nanotubes have stable core–shell structure and exhibit excellent photocatalytic activity while hydrogenated nanobelts TiO2-B exhibit poor photocatalytic activity. Due to high capacity for molecular hydrogen storage of nanotubes, the protonated titanate nanotubes can be easily hydrogenated at mild condition. With the effective hydrogenation, oxygen vacancies and Ti3+ were generated, which not only enhance the separation efficiency of the photogenerated electron–hole but also extend the visible light absorption. The highest degradation rate constant of hydrogenated TiO2 nanotubes in visible light is 5.2 times higher than air-TiO2. It reveals that the photocatalytic activity of hydrogenated TiO2 nanotubes in visible light was enhanced remarkably.Acknowledgements
The authors are grateful for financial aid from Key Science and Technology Foundation of Gansu Province (143GKDA013), Youth Science and Technology Fund of Gansu Province (1308RJYA051), and Youth Science and Technology Innovation Fund of Gansu Academy of Sciences (2013QN-16).References
- N. Venkatachalam, M. Palanichamy and V. Murugesan, Mater. Chem. Phys., 2007, 104, 454–459 CrossRef.
- M. Hussain, R. Ceccarelli, D. Marchisio, D. Fino, N. Russo and F. Geobaldo, Chem. Eng. J., 2010, 157, 45–51 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- J. Ananpattarachai, P. Kajitvichyanukul and S. Seraphin, J. Hazard. Mater., 2009, 168, 253–261 CrossRef CAS PubMed.
- S. Liu and X. Chen, J. Hazard. Mater., 2008, 152, 48–55 CrossRef CAS PubMed.
- H. Li, D. Wang, H. Fan, P. Wang, T. Jiang and T. Xie, J. Colloid Interface Sci., 2011, 354, 175–180 CrossRef CAS PubMed.
- C. Jeffrey and C. Chen, J. Photochem. Photobiol., A, 2004, 163, 509–515 CrossRef.
- C. Lazau, P. Sfirloaga, C. Orha, C. Ratiu and L. Grozescu, Mater. Lett., 2011, 65, 337–339 CrossRef CAS.
- J. Liu, R. Han, Y. Zhao, H. Wang, W. Lu, T. Yu and Y. Zhang, J. Phys. Chem. C, 2011, 115, 4507–4515 CAS.
- K. Tan, H. Zhang, C. Xie, H. Zheng, Y. Gu and W. Zhang, Catal. Commun., 2010, 11, 331–335 CrossRef CAS.
- X. Chen, L. Liu, P. Yu and S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. Bianchi, R. Psaro and V. Santo, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef CAS PubMed.
- L. Pranevicius, D. Milcius, S. Tuckkute and K. Gedvilas, Appl. Surf. Sci., 2012, 258, 8619–8622 CrossRef CAS.
- Z. Zheng, B. Huang, J. Lu, Z. Wang, X. Qin, X. Zhang, Y. Dai and M. Whanqbo, Chem. Commun., 2012, 48, 5733–5735 RSC.
- Z. Zhao, H. Tan, H. Zhao, Y. Lv, L. Zhou, Y. Song and Z. Sun, Chem. Commun., 2014, 50, 2755–2757 RSC.
- T. Xia and X. Chen, J. Mater. Chem. A, 2013, 1, 2983–2989 CAS.
- T. Leshuk, R. Parviz, P. Everett, H. Krishnakumar, R. Varin and F. Gu, ACS Appl. Mater. Interfaces, 2013, 5, 1892–1895 CAS.
- Z. Zheng, B. Huang, X. Meng, J. Wang, S. Wang, Z. Lou, Z. Wang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2013, 49, 868–870 RSC.
- X. Liu, S. Gao, H. Xu, Z. Lou, W. Wang, B. Huang and Y. Dai, Nanoscale, 2013, 5, 1870–1875 RSC.
- H. Tan, Z. Zhao, M. Niu, C. Mao, D. Cao, D. Cheng, P. Feng and Z. Sun, Nanoscale, 2014, 6, 10216–10223 RSC.
- W. Wang, Y. Ni, C. Lu and Z. Xu, RSC Adv., 2012, 2, 8286–8288 RSC.
- B. Zhao, F. Chen, Y. Jiao and J. Zhang, J. Mater. Chem., 2010, 20, 7990–7997 RSC.
- Y. Wang, G. Hu, X. Duan, H. Sun and Q. Xue, Chem. Phys. Lett., 2002, 365, 427–431 CrossRef CAS.
- B. Yao, Y. Chan, X. Zhang, W. Zhang, Z. Yang and N. Wang, Appl. Phys. Lett., 2003, 82, 281–283 CrossRef CAS.
- W. Liu, J. Gao, F. Zhang and G. Zhang, Mater. Trans., 2007, 48, 2464–2466 CrossRef CAS.
- S. Putdee, O. Mekasuwandumrong, A. Soottitantawat and J. Panpranot, Ceram. Int., 2014, 40, 2323–2329 CrossRef CAS.
- X. An, L. Han, Z. Chen, G. Liu and W. Xi, Acta Energ. Sol. Sin., 2012, 33, 874–877 CAS.
- X. van Doorslaer, K. Demeestere, P. M. Heynderickx, H. van Langenhove and J. Dewulf, Appl. Catal., B, 2011, 101(3), 540–547 CrossRef CAS.
- X. Huang, Y. Feng, C. Hu, X. Xiao, D. Yu and X. Zou, Chemosphere, 2015, 138, 183–189 CrossRef CAS PubMed.
- M. A. Abdullah and F. K. Chong, Chem. Eng. J., 2010, 158(3), 418–425 CrossRef.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. Fitzmorris, C. Wang, J. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- S. Pavasupree, Y. Suzuki, S. Yoshikawa and R. Kawhata, J. Solid State Chem., 2005, 178, 3110–3116 CrossRef CAS.
- Y. Suzuki, S. Pavasupree, S. Yoshikawa and R. Kawhata, J. Mater. Res., 2005, 20, 1063–1070 CrossRef CAS.
- B. Zhao, F. Chen, W. Qu and J. Zhang, J. Solid State Chem., 2009, 182, 2225–2230 CrossRef CAS.
- J. Huo, Y. Hu, H. Jiang and C. Li, Nanoscale, 2014, 6(15), 9078–9084 RSC.
- F. Napoli, M. Chiesa, S. Livraghi, E. Giamello, S. Agnoli, G. Granozzi and C. Di Valentin, Chem. Phys. Lett., 2009, 477(1), 135–138 CrossRef CAS.
- Q. Zhu, Y. Peng, L. Lin, C. Fan, G. Gao, R. Wang and A. Xu, J. Mater. Chem. A, 2014, 2, 4429–4437 CAS.
- F. Tian, Y. Zhang, J. Zhang and C. Pan, J. Phys. Chem. C, 2012, 116, 7515–7519 CAS.
- W. Wang, C. Lu, Y. Ni, M. Su and Z. Xu, Appl. Catal., B, 2012, 127, 28–35 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |