
Synthesis, characterization and biological evaluation of cationic hydrazone copper complexes with diverse diimine co-ligands†
Shin Thung Chewa,
Kong Mun Lo*a,
Saravana Kumar Sinniahb,
Kae Shin Simb and
Kong Wai Tan*a
aDepartment of Chemistry, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: kmlo@um.edu.my; kongwai@um.edu.my
bInstitute of Biological Sciences, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia
First published on 10th November 2014
Abstract
Four new copper(II) complexes containing triphenylphosphonium conjugated salicylaldehyde-(4-fluorobenzhydrazone), (L) with the formulation [CuL]Cl(1), [Cu(phen)L]Cl(2), [Cu(bpy)L]Cl(3), [Cu(dmbpy)L]Cl(4), (where L = doubly deprotonated hydrazone; phen = 1,10′-phenanthroline; bpy = 2,2′-bipyridine; dmbpy = 5,5′-dimethyl-2,2′-bipyridine) have been synthesized. The compounds were characterized by spectroscopic methods and, in the case of crystalline products, by X-ray crystallography. The topoisomerase I (topo I) inhibition, DNA binding and cleavage activities and cytotoxicity of the compounds were studied. A DNA relaxation study demonstrated that all the copper complexes successfully inhibit topo I enzyme by binding to topo I as the preferred pathway. Complex 1 is the most active with starting inhibitory concentration ≈20 μM. The planarity of the tridentate hydrazone Schiff base ligand and the diimine co-ligands increase the binding affinity to DNA. The presence of the 1,10′-phenanthroline co-ligand in complex 2 induces plasmid DNA (pBR322) cleavage without exogenous agents. It is noteworthy that the addition of diimine co-ligands to the copper(II) complex enhanced the cytotoxicity of the compounds, especially against the human prostate adenocarcinoma cell line (PC-3). Complex 2 exhibits the highest activity against PC-3 with the IC50 value of 2.47 ± 0.37 μM. Annexin V/propidium iodide analysis showed that compound 1 induces apoptotic and necrotic cell death, whereas compound 2–4 work mainly through apoptosis.
1. Introduction
The great successes achieved with platinum-based antitumor agents,1,2 such as cisplatin, carboplatin, and oxaliplatin, have promoted the development of metal-based drugs.3,4 Copper complexes are regarded as the most promising alternatives to cisplatin as anticancer drugs.5 A large body of evidence indicates that copper chelation is an effective method to inhibit tumor growth, and copper chelators have become promising agents in the treatment of cancer.6–8 Schiff base derivatives of hydrazones exhibit various bioactivities and play vital role in certain pharmaceutical functions such as DNA binding and cleavage,9 antimicrobial,10 antifungal, antibacterial,11,12 antimitotic,13 anticancer14 and antioxidant behavior.15 It is pertinent to mention here that a recent article revealed that two copper(II) hydrazone complexes coordinated via NNO atoms are found to interact with CT-DNA through intercalation mode and exhibited excellent cytotoxicity and selectivity.16 The use of heterocyclic diimine as the co-ligand in ternary complexes is of considerable interest because some of the diimine containing copper complexes exhibit interesting biological as well as pharmacological properties.17 The pivotal role of diimine ligands in antitumour activities have been clarified by numerous literatures earlier.18,19 Overall, it has been demonstrated that physicochemical features, such as planarity, hydrophobicity, and size of the diimine, the nature of the co-ligand, and the coordination geometry of the metal complex, all played important roles in determining the binding mode of copper complexes to DNA. Cellular processing of DNA damage drives the cell to the activation of apoptosis signal transduction pathways.20 In view of the diversified roles of Schiff base copper(II) complexes, the prospect of designing copper based complexes as effective anticancer drugs is rather tempting. The encouraging results from our cationic topoisomerase I inhibitor complexes has provoke our appetency in designing better compounds.21,22 Our current modification is inspired by the advent of 5-fluorouracil, a highly cytotoxic drug derived from the fluorination of the non-cytotoxic uracil. It has become evident that organofluorine compounds hold a special place in therapeutic chemistry.23–28 In fact, approximately 20% of all currently approved drugs contain at least one fluorine atom. An increasing number of fluorinated antitumor agents have now become available for cancer treatment.29–31 In light of the increased antitumour activities from numerous related literatures, we attempt to develop a new series of copper(II) complexes containing 5-(triphenylphosphoniummethyl)-salicylaldehyde 4-fluorobenzoylhydrazone] chloride (L) with general formula [CuL]Cl (1), [Cu(phen)L]Cl (2), [Cu(bpy)L]Cl (3), [Cu(dmbpy)L]Cl (4), (where L = doubly deprotonated benzhydrazone ligands with different substituents; phen = 1,10′-phenanthroline; bpy = 2,2′-bipyridine; dmbpy = 5,5′-dimethyl-2,2′-bipyridine) as copper based anticancer agents. As part of our attempt to unravel the possible mode of action of the copper complexes, their interaction with DNA and topoisomerase are reported herein.2. Synthesis and characterization
The synthetic route for the ligands and corresponding copper(II) complexes is shown in Scheme 1. The hydrazone Schiff base ligand L was synthesized from the condensation reaction of (3-formyl-4-hydroxybenzyl)triphenylphosphonium chloride with 4-fluorobenzoic hydrazide in ethanol. The copper(II) complexes 1–4 were prepared by the reaction of the corresponding ligand and/or diimine ligands in ethanol with copper acetate in ethanol, as indicated in Scheme 1. All the complexes are highly soluble in MeOH, DMF and DMSO. The new Cu(II) complexes were obtained in good yields and characterized by elemental analysis, IR and UV-visible spectroscopy. All compounds gave satisfactory elemental analysis. Their exact structures were finally confirmed by single crystal X-ray diffraction studies. Hydrazone ligand L is yellow in colour while all the copper complexes 1–4 are green.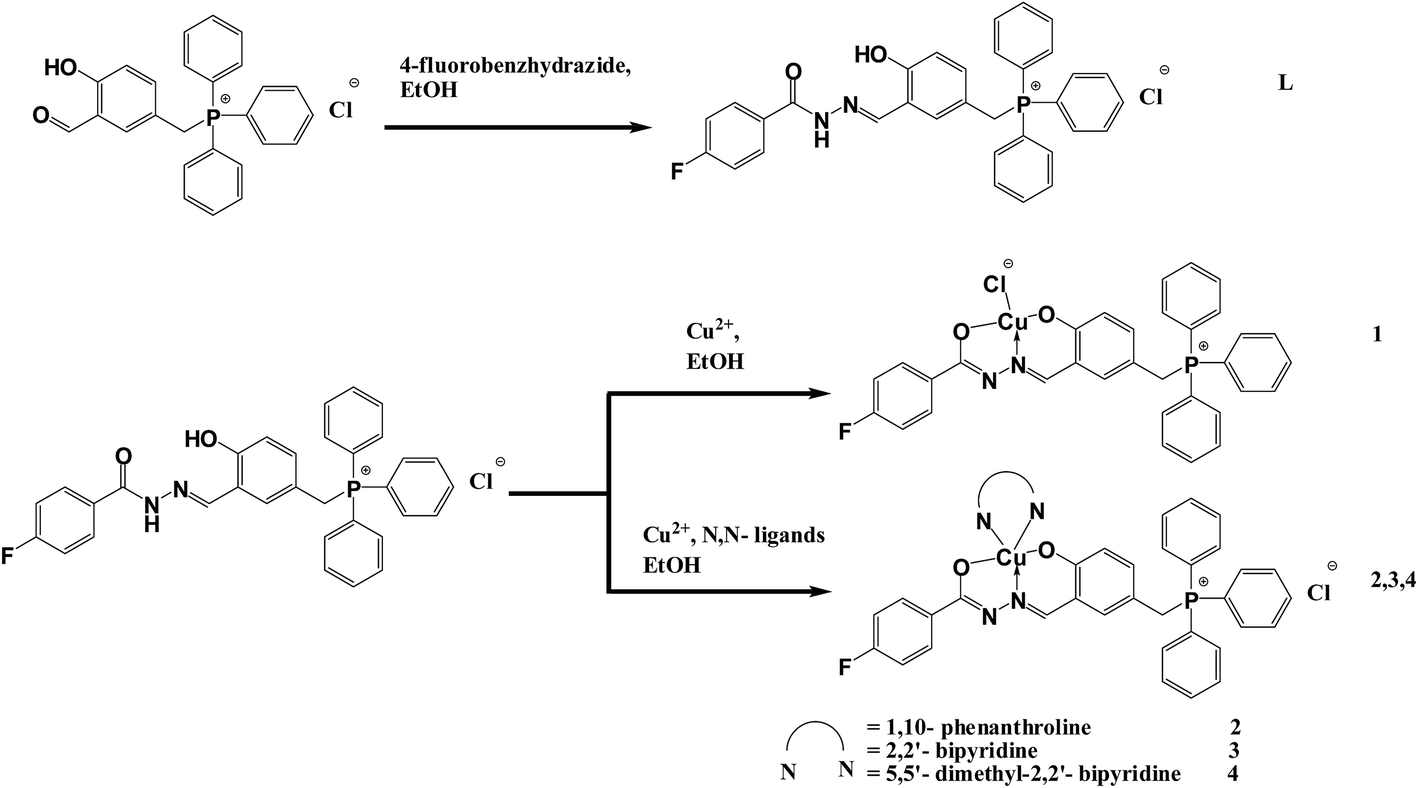 | ||
| Scheme 1 Reaction scheme and proposed structures. | ||
2.1. Infrared spectra
The IR spectra of the complexes in the region 4000–400 cm−1 were analyzed. In comparison with the free ligand, the data gave evidence of coordination of ligand L to the copper metal ion via phenolate oxygen, azomethine nitrogen and enolate oxygen. In the IR spectrum of L, a strong band at 1661 cm−1 has disappeared in all the IR spectra of complexes which is assigned to the stretching vibration of carbonyl [v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)].32,33 The infrared spectra of 1–4 display IR absorption bands at 1614, 1612, 1611, and 1610 cm−1 respectively which are ascribed to the v(CN) stretching frequencies of the complexes, whereas for the free ligand the same band was observed at 1627 cm−1. The shift of this band on complexation towards lower wavenumbers indicates the coordination of azomethine nitrogen v(CN) to the copper centre.9,34,35 The appearance of the bands in the 1499–1501 cm−1 range in the complexes are due to the asymmetric stretching vibration of the newly formed NC bond as a result of the enolization of ligand (L).9,36 The infrared spectrum of L showed a band at 1232 cm−1 assigned to the stretching of phenolic v(C–O). However, the band shifted to the lower region (1214–1219 cm−1) for the complexes.37 In addition, the characteristic v(N–N) stretching of the free ligand L (observed at 1014 cm−1) undergoes a positive shift to a higher wavenumber upon complexation which is attributed to the diminished repulsion between the lone pairs of adjacent nitrogen atoms.38–40 The formation of complexes 1–4 have been confirmed by the presence of bands around ∼492 and ∼518 cm−1 corresponding to v(Cu–N) and v(Cu–O) respectively.32,33,36 The sharp and relatively strong v(C–P) band at 1438 cm−1 remains practically unchanged after the formation of ligands and complexes.41,42
O)].32,33 The infrared spectra of 1–4 display IR absorption bands at 1614, 1612, 1611, and 1610 cm−1 respectively which are ascribed to the v(CN) stretching frequencies of the complexes, whereas for the free ligand the same band was observed at 1627 cm−1. The shift of this band on complexation towards lower wavenumbers indicates the coordination of azomethine nitrogen v(CN) to the copper centre.9,34,35 The appearance of the bands in the 1499–1501 cm−1 range in the complexes are due to the asymmetric stretching vibration of the newly formed NC bond as a result of the enolization of ligand (L).9,36 The infrared spectrum of L showed a band at 1232 cm−1 assigned to the stretching of phenolic v(C–O). However, the band shifted to the lower region (1214–1219 cm−1) for the complexes.37 In addition, the characteristic v(N–N) stretching of the free ligand L (observed at 1014 cm−1) undergoes a positive shift to a higher wavenumber upon complexation which is attributed to the diminished repulsion between the lone pairs of adjacent nitrogen atoms.38–40 The formation of complexes 1–4 have been confirmed by the presence of bands around ∼492 and ∼518 cm−1 corresponding to v(Cu–N) and v(Cu–O) respectively.32,33,36 The sharp and relatively strong v(C–P) band at 1438 cm−1 remains practically unchanged after the formation of ligands and complexes.41,42
2.2. Electronic spectra
The significant electronic absorption bands in the spectra of the hydrazone ligand L and all the complexes recorded in methanol solution are presented in Table 1. The bands in the region of 312–327 nm and 227–276 nm of complexes 1–4 are due to the n → π* and π → π* transitions of the hydrazone ligands and the coordinated diimine ligands.32,35 The n → π* transition, which appears in the region of 288–299 nm in the spectrum of the uncomplexed hydrazone ligand was slightly shifted to a higher wavelength upon complexation. This is an indication of the enolization followed by the deprotonation of the ligand during complexation.36 The broad bands observed at approximately 400 nm are attributed to the ligand-to-metal-charge transfer (LMCT) transition.9,35,40 Their broadness may be due to the overlapping of the LMCT transitions of O → Cu and N → Cu. The complexes with N,N donor ligands (2–4) displayed d–d bands in the 648–655 nm range, which tend to be indicative of a distorted square pyramidal geometry32 while complex 1 that displays a square planar coordination, absorbs at 637 nm.43| Compounds | d–d | LMCT | n → π* | π → π* |
|---|---|---|---|---|
| L | — | — | 288 (42226.4), 299 (38476.8) | 277 (38105.6) |
| 1 | 637 (168.33) | 392 (20439.2) | 312 (23340), 327 (21943.2) | 227 (49990.4), 269 (33652) |
| 2 | 655 (150.63) | 391 (17470.4) | 312 (22656), 327 (20615.2) | 228 (79638.4), 266 (54267.2) |
| 3 | 648 (149.66) | 391 (18281.6) | 312 (22324), 327 (21142.4) | 228 (56865.6), 276 (39990.4) |
| 4 | 655 (161.13) | 391 (19775.2) | 313 (26191.2), 327 (21796.8) | 229 (55390.4), 276 (42432) |
2.3. 1H- and 13C-NMR
Additional structural information can be deduced from the 1H NMR and 13C NMR spectra. Since copper(II) complexes are paramagnetic in nature, its NMR spectrum could not be obtained. In the 1H NMR spectrum of ligand L, the chemical shift for the hydroxyl proton (–OH) proton and azomethine proton (NCH) appear at 11.40 ppm and 8.58 ppm respectively. The formation of ligand L is further corroborated by the presence of –NH proton at 12.44 ppm. The multiplets that appear at the region of 7.66–7.92 ppm are ascribed to the aromatic protons from triphenylphosphine, whereas the aromatic protons of the 4-fluoro-benzhydrazide appear in the region of 7.33–7.38 ppm and 8.07–8.10 ppm. The sharp doublets signal around 5.00 ppm is assigned to the methylene proton.
From 13C NMR spectrum of ligand L, the chemical shift at 157.21 ppm and 146.91 ppm is due to the hydroxyl carbon (C–OH) and azomethine carbon (CN) respectively. Additionally, the signal at 163.01 ppm is ascribed to the carbonyl carbon from the 4-fluoro-benzhydrazide.
2.4. X-ray crystal structures
The crystalline compounds were structurally characterized by single crystal X-ray crystallography. Selected crystallographic data are summarized in Table 2. Fig. 1–4 show the ORTEP plots of compounds. Selected bond lengths and angles and hydrogen bonding interactions are given in Table 3 and Table 4, respectively. Single crystals of 1 were obtained from slow evaporation of the methanolic solution at room temperature. The compound crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. As depicted in Fig. 1, the copper ion is coordinated to the tridentate hydrazone ligand, through the phenolate oxygen (O1), azomethine nitrogen (N1) and enolate oxygen (O2) and with a fourth bond formed between the copper metal and a chloride ion. This gives rise to a zwitterion species with a positive charge at the phosphorus centre. The diagonal angles, N1–Cu1–Cl1 [173.12(9)°] and O1–Cu1–O2 [172.25(11)°] deviate significantly from 180° (ref. 43) and the sum (Σ) of the six inter-bond angles for complex 1 is 705.44°, which deviates from the ideal angle of 720°, suggesting that the copper centre adopts a distorted square planar geometry.44 The other bond distances involving the Cu atom such as Cu1–O1, Cu1–O2 and Cu1–N1 [1.889(2) Å, 1.930(2) Å and 1.932(3) Å, respectively] are within the range found in similar copper complexes. Also, the cis angles in the basal plane involving the phenolate oxygen and azomethine nitrogen [O1–Cu1–N1, 93.15(11)°] and that involving azomethine nitrogen and enolate oxygen [O2–Cu1–N1, 81.03(11)°] deviate from the ideal value of 90°, suggesting that the square planar coordination geometry in 1 is slightly distorted due to the chelate effect. In the molecular packing of the molecules are found to be hydrogen bonded between the water molecules and imino N2 and the Cl1 counterion forming a one-dimensional polymeric structure extending along the b axis.
space group. As depicted in Fig. 1, the copper ion is coordinated to the tridentate hydrazone ligand, through the phenolate oxygen (O1), azomethine nitrogen (N1) and enolate oxygen (O2) and with a fourth bond formed between the copper metal and a chloride ion. This gives rise to a zwitterion species with a positive charge at the phosphorus centre. The diagonal angles, N1–Cu1–Cl1 [173.12(9)°] and O1–Cu1–O2 [172.25(11)°] deviate significantly from 180° (ref. 43) and the sum (Σ) of the six inter-bond angles for complex 1 is 705.44°, which deviates from the ideal angle of 720°, suggesting that the copper centre adopts a distorted square planar geometry.44 The other bond distances involving the Cu atom such as Cu1–O1, Cu1–O2 and Cu1–N1 [1.889(2) Å, 1.930(2) Å and 1.932(3) Å, respectively] are within the range found in similar copper complexes. Also, the cis angles in the basal plane involving the phenolate oxygen and azomethine nitrogen [O1–Cu1–N1, 93.15(11)°] and that involving azomethine nitrogen and enolate oxygen [O2–Cu1–N1, 81.03(11)°] deviate from the ideal value of 90°, suggesting that the square planar coordination geometry in 1 is slightly distorted due to the chelate effect. In the molecular packing of the molecules are found to be hydrogen bonded between the water molecules and imino N2 and the Cl1 counterion forming a one-dimensional polymeric structure extending along the b axis.
| Compounds | 1 | 2 | 4 |
|---|---|---|---|
| Chemical formula | C33H26ClCuFN2O3P | C47H39ClCuFN4O4P | C45H39ClCuFN4O4P |
| Mr | 647.52 | 872.78 | 848.76 |
| Crystal system | Triclinic, P |
Triclinic, P |
Triclinic, P |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Unit cell dimension | |||
| a (Å) | 9.8737 (2) | 10.3225 (8) | 10.7354 (4) |
| b (Å) | 11.0307 (2) | 14.0607 (11) | 13.2515 (5) |
| c (Å) | 14.8193 (3) | 14.8610 (11) | 14.9015 (6) |
| α (°) | 81.3800 (10) | 107.7460 (10) | 80.534 (3) |
| β (°) | 78.7180 (10) | 98.8120 (10) | 69.109 (3) |
| γ (°) | 72.2110 (10) | 91.6650 (10) | 81.430 (3) |
| V (Å3) | 1500.13 (5) | 2023.5 (3) | 1944.15 (13) |
| Z | 2 | 2 | 2 |
| F(000) | 664 | 902 | 878 |
| Dx (Mg m−3) | 1.434 | 1.432 | 1.450 |
| Melting point (K) | 521–522 | 423–424 | 473 |
| θ (°) | 2.3–23.7 | 2.4–25.5 | 3.0–25.1 |
| μ (mm−1) | 0.91 | 0.70 | 0.73 |
| T (K) | 100 (2) | 100 (2) | 100 (2) |
| Crystal colour, habit | Block, green | Block, green | Block, green |
| Crystal size (mm) | 0.25 × 0.16 × 0.05 | 0.2 × 0.15 × 0.05 | 0.16 × 0.10 × 0.05 |
| Tmin | 0.804 | 0.670 | 0.893 |
| Tmax | 0.956 | 0.746 | 0.965 |
| Observed data [I > 2σ(I)] | 4273 | 5461 | 5431 |
| Rint | 0.037 | 0.051 | 0.039 |
| θmax (°) | 26.0 | 25.5 | 25.5 |
| θmin (°) | 1.4 | 1.5 | 1.5 |
| h | −12 12 | −12 12 | −12 12 |
| k | −13 13 | −17 17 | −16 15 |
| l | −18 17 | −17 18 | −18 18 |
| R [F2 > 2σ(F2)] | 0.047 | 0.054 | 0.045 |
| wR(F2) | 0.137 | 0.155 | 0.146 |
| S | 1.04 | 1.04 | 1.05 |
| Δρmax (e Å−3) | 0.61 | 1.04 | 0.69 |
| Δρmin (e Å−3) | −0.37 | −0.94 | −0.70 |
| Complex 1 | |||
|---|---|---|---|
| Cu1–O1 | 1.889 (2) | O1–Cu1–O2 | 172.25 (11) |
| Cu1–O2 | 1.930 (2) | O1–Cu1–N1 | 93.15 (11) |
| Cu1–N1 | 1.932 (3) | O2–Cu1–N1 | 81.03 (11) |
| Cu1–Cl1 | 2.252 (1) | O1–Cu1–Cl1 | 93.71 (8) |
| O1–C23 | 1.315 (4) | O2–Cu1–Cl1 | 92.18 (8) |
| F1–C31 | 1.364 (5) | N1–Cu1–Cl1 | 173.12 (9) |
| O2–C27 | 1.294 (4) | ||
| N1–C26 | 1.277 (4) | ||
| N1–N2 | 1.397 (4) | ||
| N2–C27 | 1.313 (5) | ||
| Complex 2 | |||
|---|---|---|---|
| Cu1–O1 | 1.914 (3) | O1–Cu1–N1 | 92.85 (12) |
| Cu1–N1 | 1.926 (3) | O1–Cu1–O2 | 157.55 (12) |
| Cu1–O2 | 1.932 (3) | N1–Cu1–O2 | 81.00 (13) |
| Cu1–N4 | 2.009 (3) | O1–Cu1–N4 | 94.86 (12) |
| Cu1–N3 | 2.274 (3) | N1–Cu1–N4 | 170.05 (13) |
| O1–C23 | 1.324 (5) | O2–Cu1–N4 | 94.09 (12) |
| O2–C27 | 1.288 (5) | O1–Cu1–N3 | 97.70 (12) |
| O3–C46 | 1.354 (8) | N1–Cu1–N3 | 93.73 (12) |
| N1–C26 | 1.294 (5) | O2–Cu1–N3 | 104.20 (12) |
| N1–N2 | 1.400 (4) | N4–Cu1–N3 | 79.01 (12) |
| N2–C27 | 1.326 (5) | ||
| N3–C34 | 1.321 (5) | ||
| N3–C45 | 1.357 (5) | ||
| N4–C43 | 1.328 (5) | ||
| N4–C44 | 1.363 (5) | ||
| Complex 4 | |||
|---|---|---|---|
| Cu1–O1 | 1.917 (2) | O1–Cu1–N1 | 93.75 (11) |
| Cu1–N1 | 1.932 (3) | O1–Cu1–O2 | 160.69 (10) |
| Cu1–O2 | 1.956 (2) | N1–Cu1–O2 | 81.29 (11) |
| Cu1–N4 | 2.011 (3) | O1–Cu1–N4 | 91.55 (11) |
| Cu1–N3 | 2.246 (3) | N1–Cu1–N4 | 174.64 (12) |
| F1–C31 | 1.365 (4) | O2–Cu1–N4 | 93.96 (11) |
| O1–C23 | 1.312 (4) | O1–Cu1–N3 | 96.73 (11) |
| O2–C27 | 1.295 (4) | N1–Cu1–N3 | 101.48 (11) |
| N1–C26 | 1.287 (4) | O2–Cu1–N3 | 102.53 (11) |
| N1–N2 | 1.397 (4) | N4–Cu1–N3 | 77.02 (11) |
| N2–C27 | 1.329 (4) | ||
| N3–C34 | 1.333 (5) | ||
| N3–C38 | 1.347 (5) | ||
| N4–C43 | 1.345 (4) | ||
| N4–C39 | 1.352 (4) | ||
| D–H⋯A | d(D–H) (Å) | d(H⋯A) (Å) | d(D⋯A) (Å) | d(D–H⋯A) (°) |
|---|---|---|---|---|
| Complex 1 | ||||
| O3–H3B⋯O4i | 0.87 | 1.62 | 2.460 (1) | 163 |
| O3–H3A⋯O3ii | 0.87 | 2.07 | 2.860 (1) | 151 |
| Symmetry codes: (i) x + 1, y, z; (ii) −x + 2, −y, −z + 1 | ||||
|
||||
| Complex 2 | ||||
| O3–H3A⋯O1 | 0.84 | 2.02 | 2.815 (5) | 158 |
| O4–H4A⋯Cl1 | 0.87 | 2.11 | 2.974 (6) | 173 |
|
||||
| Complex 4 | ||||
| O3–H3A⋯Cl1i | 0.85 | 2.34 | 3.167 (5) | 165 |
| Symmetry codes: (i) x, y, z + 1 | ||||
 | ||
| Fig. 1 An ORTEP view of 1 showing the atom labeling scheme (50% probability thermal ellipsoids). | ||
In contrast, the molecule of 2 (Fig. 2) adopts a distorted square pyramidal geometry at the copper centre and the five coordination at Cu is formed by the dianionic tridentate ONO-donor ligand and the N4 atom of phen, while the axial position is occupied by N3 of phen. All the bond lengths and bond angles are well in agreement with the previously reported square pyramidal copper(II) Schiff base complexes.21 It is anticipated that the axial Cu1–N3 bond length is slightly longer than the equatorial Cu1–N4 bond distance as a consequence of pseudo Jahn–Teller effect. It has long been proven that Jahn–Teller effect is operative in the case of d9 transition metal complexes. Thus, one trans pair of coordination bonds are elongated while the remaining four are shortened.9 The molecular structure of 2 was found to contain water and an ethanol solvent molecule, which form hydrogen bond with the Cl1 and the phenolate O3, respectively.
 | ||
| Fig. 2 An ORTEP view of 2 showing the atom labeling scheme (50% probability thermal ellipsoids). | ||
The complex 4, which was obtained by replacing the phenanthroline with 5,5′-dimethyl-2,2′-bipyridine (dmbpy) also gives a five-coordinated copper complex (Fig. 3), with a distorted square pyramidal geometry. In the five coordinated structure, the ONO atoms of the Schiff base occupied the basal plane and dmbpy displays axial (N3)-equatorial (N4) coordination. Similarly, there is a typical pseudo Jahn–Teller effect influence copper(II) resulted in an elongated square pyramidal environment for 4.45 The Cu1–N3 distance [2.246(3) Å] is significantly longer than the equatorial Cu1–N4 distance [2.011(3) Å ]46 forming a distorted square pyramid with a parameter τ of 0.23 The trigonality index, τ is calculated by the equation τ = (β − α)/60,47 where β and α are the largest angles in the coordination sphere (for perfect square pyramidal and trigonal bipyramidal geometries the values of τ are zero and unity, respectively).15,36 A similar trigonality index was also evidenced for the square pyramidal phen containing complex (3).
 | ||
| Fig. 3 An ORTEP view of 4 showing the atom labeling scheme (50% probability thermal ellipsoids). | ||
3. Biological evaluation
3.1. Topoisomerase I inhibition
DNA topoisomerase I (topo I), an essential enzyme in metazoans has been identified as a target among the most widely used clinical drugs for treatment of cancer.48–50 Topoisomerases have pivotal roles in DNA replication and transcription.33,51 The enzymatic activity of topo I, like that of other topoisomerases, breaks the DNA backbone reversibly by replacing a DNA phosphodiester bond with a bond between a phosphate at one end of the DNA break and a tyrosine residue of the topo I protein.51–54 In the case of topo I, the tyrosine links to the phosphate at the 3′-hydroxyl end of the DNA break. DNA relaxation takes place by rotation of the free end of the broken DNA around the intact DNA strand.55This assay provides a direct means of determination whether the drugs affect the unwinding of a supercoiled duplex DNA to a nicked open circular and relaxed DNA.33 One unit of topo I could fully convert the supercoiled pBR322 DNA to fully relaxed topoisomers. After the incubation of DNA with topo I, the appearance of the slowest moving bands of more relaxed DNA can be seen on the gel image (Fig. 4(A)–(E), Lane 5) The disappearance or the decrease in the intensity of the slowest moving band upon incubation of DNA, topo I and the complexes is the indication of topo I inhibition. The degree of inhibition of topo I increases with increasing concentration (5–160 μM) of the complexes (Fig. 4(B)–(E)). But, the function of topo I is not fully inhibited for some complexes even up to 160 μM.
 | ||
| Fig. 4 Human topoisomerase I inhibition assay by gel electrophoresis. The gel images from (A–E) are for compounds L, 1, 2, 3 and 4, respectively. Electrophoresis results of incubating human topoisomerase I (0.25 unit/20 μL) with pBR322 in the absence or presence of 5–160 μM of compound: Lane 1 and 13, GeneRulerTM 1 kb DNA ladder; Lane 2, DNA alone (control); Lane 3, DNA + 160 μM compound (control); Lane 5, DNA + 1 unit human topoisomerase I (control); Lane 7–12, DNA + 1 unit human topoisomerase I + varying concentration of compound; Lane 7, 5 μM; Lane 8, 10 μM; Lane 9, 20 μM; Lane 10, 40 μM; Lane 11, 80 μM; Lane 12, 160 μM. | ||
It is evident from the gel image that the Schiff base ligand L does not exhibit any topo I inhibition ability. Surprisingly, the incorporation of the compound L towards copper metal centre has conspicuously provoked the topo I inhibition activity of the complexes. By comparing all the compounds, complex 1 is the most active with starting inhibitory concentration of 20 μM. This might be due to the electron withdrawing fluoro substituent of benzhydrazone that improve the interaction of the molecule with topoisomerase I.56 Surprisingly, the addition of various diimine co-ligand into the copper complex exacerbated the topo I inhibitory ability. This is reflected in Fig. 4, where all the ternary complexes 2, 3 and 4 demonstrate a starting inhibitory concentration of 40 μM (Fig. 4(C)–(E), Lane 10) regardless of the variation of diimine ligand phen, bpy or dmbpy. These distinctive results are conflicting with our previous research where diimines lead to enhancement of topo I inhibition. Further investigations are required to identify the underlying mechanism between complex and the enzyme.
Of primary concern is the inevitable interaction between all the compounds with DNA without addition of topo I that would lead to the decline in rate of DNA migration (Fig. 4(A)–(E), Lane 3). To this end, it is logical to assume that the cationic complexes bind to DNA at high concentration and cause the DNA to travel slower because of the formation of DNA and metal complex aggregate with higher molecular weight or the decrease in the negative charge of the DNA due to the binding with the cationic complexes.57–60 Therefore, we could not determine the concentration of compounds that lead to full inhibition of topo I.
In an attempt to obtain more insight into the inhibition activity of the topo I in this work, we used three variations of mixing the DNA, topo I and compounds [1 (15 μM), 2 (30 μM), 3 (30 μM), 4 (30 μM)] for topo I inhibition assay. It is possible to predict a generalized mechanism of action of topo I inhibition based on the three variations of mixing. When the three components are mixed simultaneously, there is slight inhibition of topo I as can seen by the presence of the fastest moving band with low intensity (Form I) which consists of supercoiled DNA and poorly relaxed DNA (Fig. 5, Lane 5). Similar band can be observed by incubating DNA and compound 1 before the addition of topo I (Fig. 5, Lane 6). Interestingly, when complex 1 is incubated with topo I before the addition of DNA (Fig. 5, Lane 7), the intensity of the fastest moving bands are the highest (Form I). However, the slowest moving band still remains with lower intensity (Form II). These observations suggest that the mechanism of action for topo I inhibition is based on two pathways, one involving the binding of complex to DNA and the other comprise the binding of complex to topoisomerase. In light of these results, we can deduce that binding of complex to topo I is a preferred inhibition pathway regardless of the structures of the complexes (Fig. 5 and S1(A)–(C)† Lane 7). Nevertheless, continued research on this will hopefully lead to a better assessment of the usefulness of these compounds as anticancer drugs.
 | ||
| Fig. 5 Effect of sequence of mixing for the human topoisomerase I inhibition assay of complex 1. Electrophoresis results of incubating human topoisomerase I (0.25 unit/20 μL) with pBR322: Lane 1, 4 and 8, GeneRulerTM 1 kb DNA ladder; Lane 2, DNA alone (control); Lane 3, DNA + 1 unit human topoisomerase I (control); Lane 5, DNA + 15 μM 1 + 1 unit human topoisomerase I, all components mixed at the same time; Lane 6, DNA + 15 μM 1 + 1 unit human topoisomerase I, DNA + complex 1 incubated for 30 min first before the addition of topo I; Lane 7, DNA + 15 μM 1 + 1 unit human topoisomerase I, complex 1 + topo I incubated for 30 min first before DNA is added. | ||
3.2. DNA binding study – absorption titration
As the primary pharmacological target of many antitumor drugs, DNA and DNA binding activities of metal complexes have been a clue of paramount importance for the development of effective metal based chemotherapeutic drugs.61,62 DNA binding is the critical step for chemical nuclease activities of metal complexes in most cases. Monitoring the changes in absorption spectrum of the metal complexes upon addition of increasing amounts of DNA is one of the most widely used methods for determining overall binding constants.63,64The potential binding ability of hydrazone ligand L and its copper complexes (1–4) towards CT-DNA were studied by UV-vis spectroscopy in the range of 200–500 nm. The typical absorption titration curves of the compounds at constant concentration (20 μM) in the presence of different concentrations of CT-DNA are shown representatively in Fig. 6 and the UV absorption titration data were listed in Table 5. In the presence of DNA, the absorption bands of the ligands L exhibited hypochromism of about 19% at 288 nm. However, complex 1 exhibited marked hypochromism of about 23% at 267 nm. Upon the incremental addition of CT-DNA to the complexes with various diimine co-ligands, the absorption spectrum exhibit similar changes to the ligand with hypochromicity of 22–33 % in the range of 267–276 nm. These absorption bands are attributed to the π–π* transition of the ligands and its respective complexes. The intercalative mode of binding usually results in hypochromism along with or without a small red or blue shift due to the strong stacking interaction between an aromatic chromophore and the base pairs of DNA.16 The observed hypochromic effect may suggest tight binding to CT-DNA probably by intercalation.
 | ||
| Fig. 6 Absorption titration curves by UV-vis spectroscopy. The gel images from (A–E) are for compounds L, 1, 2, 3 and 4, respectively. DNA binding affinity of compounds (20 μM) towards CT-DNA in TN buffer. Inset: plot of [DNA]/[εb − εf] vs. [DNA] for titration of CT-DNA of the aforementioned compounds. | ||
| Compound | λmax (nm) | Change in absorbance | Hypochromicity (%) | Kb ×105 (M−1) |
|---|---|---|---|---|
| L | 288 | Hypochromism | 19 | 3.9532 |
| 1 | 267 | Hypochromism | 23 | 2.9365 |
| 2 | 268 | Hypochromism | 26 | 2.5241 |
| 3 | 276 | Hypochromism | 22 | 1.4958 |
| 4 | 268 | Hypochromism | 33 | 1.6994 |
In order to compare quantitatively the binding strength of the compounds, their intrinsic binding constants (Kb) with CT-DNA were determined from the following equation:65
| [DNA]/[εa − εf] = [DNA]/[εb − εf] + 1/Kb [εb − εf] |
The magnitudes of intrinsic binding constants (Kb) were calculated as 3.95 × 105 M −1 and 2.94 × 105 M −1 for the ligand and complex 1, respectively (Table 5). However, the incorporation of diimines in complex 1 does not give rise to a higher Kb values. Complex 2, 3 and 4 revealed strong binding strength with Kb values of 2.52 × 105 M −1, 1.50 × 105 M −1 and 1.70 × 105 M −1, respectively. The Kb values of most compounds are similar or higher than that of the classical intercalator EB (Kb = 1.23 × 105 M −1).66,67 The promising results obtained from the DNA binding studies gave a root to think on the interaction of the compounds with DNA where the binding takes place via intercalative interaction.
3.3. DNA cleavage studies
There has been considerable interest in DNA endonucleolytic cleavage reactions that are activated by metal ions.68,69 The chemical nuclease activities of the selected hydrazone copper complexes have been studied using supercoiled pBR322 plasmid DNA as a substrate in a medium of 50 mM Tris–HCl/NaCl buffer (pH = 7.2) in the absence and presence of external exogenous agents (500 μM of hydrogen peroxide, H2O2 and 30 μM of ascorbate acid) under physiological conditions. When circular plasmid DNA is conducted by electrophoresis, the fastest migration will be observed for the supercoiled form (Form I). If one strand is cleaved, the supercoils will relax to produce a slower-moving nicked circular form (Form II). If both strands are cleaved, a linear form (Form III) will be generated which migrates in between Form I and Form II. Control experiments revealed that no measurable DNA cleavage occurred when supercoiled pBR322 plasmid DNA was incubated with CuCl2 (Fig. 7, Lane 3) clearly indicating that the observed cleavage was solely due to the Cu(II) complexes. | ||
| Fig. 7 Cleavage in the absence of exogenous agents by gel electrophoresis. The gel images from (A–C) are compound 2 with incubation time of 24, 48 and 72 h, respectively. Electrophoresis results of incubating pBR322 plasmid DNA in the absence or presence of 5–160 μM of compound in TN buffer: Lane 1 and 11, GeneRulerTM 1 kb DNA ladder; Lane 2, DNA alone (control); Lane 3, DNA + CuCl2 (control); Lane 5–10, DNA + varying concentration of compound; Lane 5, 5 μM; Lane 6, 10 μM; Lane 7, 20 μM; Lane 8, 40 μM; Lane 9, 80 μM; Lane 10, 160 μM. | ||
 | ||
| Fig. 8 Oxidative cleavage assay by gel electrophoresis. The gel images of A and B are the incubation of 10 μM complex 2 with external exogenous agents (hydrogen peroxide, H2O2 (A) or ascorbate acid (B)). Electrophoresis results of incubating complex 2 with pBR322 plasmid DNA in the absence or presence of 10–500 μM H2O2 (A) or 5–30 μM ascorbate acid (B) in TN buffer: Lane 1 and 10, GeneRulerTM 1 kb DNA ladder; Lane 2, DNA alone (control); Lane 3, DNA + H2O2 (A) or ascorbate acid (B) (control); Lane 5–9, DNA + complex 2 + varying concentration of H2O2 (A) or ascorbate acid (B); Lane 5, 10 μM H2O2 (A) or 5 μM ascorbate acid (B); Lane 6, 30 μM H2O2 (A) or 10 μM ascorbate acid (B); Lane 7, 100 μM H2O2 (A) or 15 μM ascorbate acid (B); Lane 8, 300 μM H2O2 (A) or 20 μM ascorbate acid (B); Lane 9, 500 μM H2O2 (A) or 30 μM ascorbate acid (B). | ||
For nuclease activity in the absence of co-reagents, only copper complexes with diimine co-ligand phen (2) is able to cleave DNA while other tested complexes do not cause any DNA damage. This could be due to hydrolytic,70 oxidative71,72 or combination of both pathways.73 To assess the DNA cleaving propensity of the hydrazone copper complex 2, supercoiled (SC) plasmid pBR322 DNA was incubated with varying concentrations of complexes (5–160 μM). Surprisingly, it was observed that 10 μM of 2 (Fig. 7(A), Lane 6) is capable of causing single strand DNA breaks, as indicated by the nicked circular formation and the disappearance of supercoiled DNA, even in the absence of exogenous agents. When incubation time increases, the same concentration of compound (Fig. 7(B), Lane 6) presented higher activity with a mixture of nicked and linear DNA form. As can be seen from Fig. 7(C) (Lane5), when 5 μM of 2 is incubated with supercoiled DNA for 72 hours, significant conversion of nicked DNA form was observed. It is to be noted that, upon incremental increase of the complex concentration, there is a remarkable reverse in nuclease sensitivities from 80 μM onwards (Fig. 7(A)–(C), Lane 9–10). This observed trend in activity was unexpected since nuclease activity normally increases with increasing complex concentration. The experiment of increasing concentration of external agents was also carried out for complex 2. In this respect it is worth mentioning that, with the same concentration of complex, addition of H2O2 and ascorbate acid cause the conversion of supercoiled DNA to nicked circular form in a shorter period, which is 2 hours time. However, the nuclease activity of 2 in the presence of exogenous agents is elevated in a great deal (Fig. 8(A) and (B)). These proves that both H2O2 and ascorbate acid are effective activating agents that play a crucial role in DNA cleavage.
 | ||
| Fig. 9 Oxidative cleavage assay by gel electrophoresis. The gel images from (A–D) are for compounds L, 1, 3 and 4, respectively. Electrophoresis results of incubating pBR322 plasmid DNA and external exogenous agents (500 μM hydrogen peroxide, H2O2) in the absence or presence of 5–160 μM of compound in TN buffer: Lane 1 and 11, GeneRulerTM 1 kb DNA ladder; Lane 2, DNA alone (control); Lane 3, DNA + H2O2 (control); Lane 5–10, DNA + H2O2 + varying concentration of compound; Lane 5, 5 μM; Lane 6, 10 μM; Lane 7, 20 μM; Lane 8, 40 μM; Lane 9, 80 μM; Lane 10, 160 μM. | ||
The oxidative cleavage of the remaining complexes in the presence of external exogenous agents (hydrogen peroxide, H2O2 and ascorbate acid) were also investigated. All the copper complexes were found to exhibit pronounced nuclease activity on treatment with the activators (Fig. 9 and 10(B)–(D)). The results unambiguously establish the concentration dependent DNA cleavage activity of the complexes. This clearly indicates that the observed DNA cleavage by the copper(II) complexes in the presence of H2O2 is due to diffusible hydroxyl radicals or molecular oxygen which are capable of damaging DNA by Fenton type chemistry.74 Nevertheless, a peculiar behavior is observed when it comes to higher concentration (>80 μM). The cleaving potencies appeared to be contrary to the concentration of the complexes. It is undeniable that the hydrazone copper complexes with different diimine co-ligands illustrate appreciable DNA cleavage that is more significant than the copper complex without the diimine moiety. It is obvious that the free hydrazone ligands do not play any important role in DNA cleavage while the complexation with copper(II) metal center lead to nuclease activity. At this juncture, we also can conclude that the DNA nuclease activity of copper complex bearing 5,5′-dimethyl-2,2′-bipyridine co-ligand (complex 4) is better than the one with 2,2′-bipyridine (complex 3). This study clearly demonstrated that the DNA cleavage activity follow the trend of 2 > 4 > 3 > 1.
 | ||
| Fig. 10 Oxidative cleavage assay by gel electrophoresis. The gel images from (A–D) are for compounds L, 1, 3 and 4, respectively. Electrophoresis results of incubating pBR322 plasmid DNA and external exogenous agents (30 μM ascorbate acid) in the absence or presence of 5–160 μM of compound in TN buffer: Lane 1 and 11, GeneRulerTM 1 kb DNA ladder; Lane 2, DNA alone (control); Lane 3, DNA + ascorbate acid (control); Lane 5–10, DNA + ascorbate acid + varying concentration of compound; Lane 5, 5 μM; Lane 6, 10 μM; Lane 7, 20 μM; Lane 8, 40 μM; Lane 9, 80 μM; Lane 10, 160 μM. | ||
Several authors have studied the influence of different activators on the cleavage of DNA by copper(II) complexes.75,76 Sigman et al.75 have found that MPA to be superior to ascorbic acid as a activating agent on the nuclease activity of a bis(o-phenanthroline)copper(II) complex because it produces less background cleavage. Chiou et al.77 have showed that ascorbate is more effective in inducing DNA cleavage than other reducing agents such as MPA and dithiothreitol due to its ability to generate hydrogen peroxide in the presence of oxygen and metal ions, whereas other reducing agents are known to produce superoxide, which rapidly undergoes dismutation in aqueous solution. Detmer et al.76 have indicated that a mixture of H2O2 and ascorbate is a more effective activating agent than ascorbate alone. In our work, we found that both hydrogen peroxide and ascorbate are good activators for DNA cleavage.78
3.4. In vitro cytotoxicity assay on selected tumour cell lines
The in vitro cytotoxicity of L and its copper(II) complexes were evaluated by MTT assay on selected human tumour cell lines involving human lung carcinoma cell line (A549), human prostate adenocarcinoma cell line (PC-3), non-cancer human fibroblast cell line (MRC-5), and normal human prostate epithelial cell line (RWPE-1). Cell viability was determined by MTT assay, which serves as an index of cell viability by detecting the reduction of tetrazolium salt to blue formazan by mitochondrial enzyme activity of succinate dehydrogenase in living cells after 72 hours of treatment with increasing concentration of the compounds. The compounds were dissolved in DMSO and blanks containing same concentration of DMSO were taken as controls to identify the activity of solvents in this MTT assay. The results were analyzed by mean of cell viability expressed as IC50 values. As shown in Table 6, complexes 1–4 exhibit various in vitro cytotoxicities against cancerous and non-cancerous cell lines. In contrast to free ligand L, generally, copper coordinated compounds exhibit enhanced cytotoxicities. Complex 1 showed lower IC50 values compared to free ligand L which are 21.26 ± 7.03 μM and 12.90 ± 1.40 μM against PC-3 and RWPE-1 cell lines respectively. It is remarkable that complex 2 (IC50 = 2.47 ± 0.37 μM) is the most cytotoxic among the compounds against the aforementioned cell lines. In general, the cytotoxicity of copper complexes which contain N,N-ligands (2, 3 and 4) are more effective than the one without any diimine (1) and free ligand (L). The cytotoxicity of the ternary complexes (2–4) decreases by the following order: 2 > 4 > 3. The observed order of decrease reflects the important role of extended aromatic ring and hydrophobicity of the diimines in improving cytotoxicity.| Compounds | Cytotoxicity IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| A549 | PC-3 | MRC-5 | RWPE-1 | Selectivity index | ||
| (SI1) | (SI2) | |||||
| a SI1 = IC50 of RWPE-1/IC50 of PC-3, SI2 = IC50 of MRC-5/IC50 of A549. | ||||||
| L | >30 | 28.40 ± 5.73 | >30 | 19.49 ± 0.44 | 0.69 | — |
| 1 | >30 | 21.26 ± 7.03 | >30 | 12.90 ± 1.40 | 0.61 | — |
| 2 | 4.12 ± 0.97 | 2.47 ± 0.37 | 2.35 ± 0.47 | 3.77 ± 0.03 | 1.53 | 0.57 |
| 3 | 23.32 ± 4.56 | 5.90 ± 0.13 | 15.63 ± 2.83 | 10.64 ± 0.27 | 1.80 | 0.67 |
| 4 | 10.10 ± 0.39 | 5.31 ± 0.17 | 8.74 ± 0.11 | 10.01 ± 0.35 | 1.89 | 0.87 |
In order to determine the selectivity of the compounds, the cytotoxicity activities of the compounds were tested on RWPE-1 and MRC-5 non-cancerous cell lines. It is notable that although free ligand L and complex 1 are not selectively cytotoxic towards PC-3 cancerous cell lines than RWPE-1 normal cell lines, complexes 2–4 which bear extra diimines moiety somehow show higher selectivity. Therefore, complexation of L by copper and diimine co-ligands not only leads to enhancement in cytotoxicity but cytoselectivity with selectivity index (SI) of 1.53, 1.80 and 1.89 for complexes 2, 3 and 4 respectively. The highest cytotoxicity exhibited by complex 2 is consistent with the strong binding constant of the complex through deeper intercalation of the phen co-ligand in between the base pair of DNA and its higher ability to cleave DNA in the absence of reductant is responsible for its potency to induce cell death through different modes.79 By comparing both tumour cell lines, all the compounds seems to be more toxic against PC-3 cell line than A549 cell line. Researchers point out that there is an increase of 2- to 10-fold of topo I enzyme in prostate tumours, compared to benign hyperplastic prostate tissue from the same patients.51 Therefore, prostate carcinoma cells are good models to study compounds that are designed to target topo I.
3.5. Annexin V-FITC/PI double staining study using flow cytometry
Annexin V is a Ca2+ dependent phospholipid-binding protein with high affinity for phosphatidylserine. One of the early characteristic of apoptosis is phospotidylserine externalization from the inner side to the outer layer.80–82 Therefore Annexin V can be used as a sensitive probe for phospotidylserine externalization which indicates early stage of apoptosis. Another dye used in this experiment is propidium iodide which enters the damaged membranes of the cells and stains the DNA.83The results for Annexin V(AV)/Propidium iodide (PI) double staining study by flow cytometry are shown is Fig. 11 and Fig. 12. After 72 hours incubation, all the compounds (20 μM) showed a great increment of PC-3 cells in the lower right quadrant (Q4: AV+/PI−), which can be assumed that the treated cells undergo early apoptosis. However, the early apoptotic cells are found to be higher upon complexation of L (49.4%); with complexes 1, 2, 3 and 4 showing 53.4%, 72.9%, 58.9% and 73.5% respectively. Viable cells (Q3: AV−/PI−) drastically reduced by 60.5–67.4 % compared to the negative control (91.4%). It is to be noted that compound L and 1 with poorer nuclease property have more cells in the upper right quadrant (Q2: AV+/PI+) which is suggestive of late apoptotic or necrotic cell death.
 | ||
| Fig. 11 Annexin V/PI against PC-3 cells treated with 20 μM of compounds L, 1, 2, 3 and 4 for 72 hours. Results were analyzed by flow cytometry. The images from (A–F) are for negative control, ligand L and complexes 1, 2, 3, and 4. At least 10000 cells were counted in each sample. | ||
 | ||
| Fig. 12 Results for Annexin V/PI against PC-3 cells treated with 20 μM of compounds L, 1, 2, 3 and 4 for 72 hours. Results were analyzed by flow cytometry. At least 10000 cells were counted in each sample. | ||
4. Conclusion
All the complexes exhibit cytotoxicity through their ability to bind and cleave DNA and inhibit topo I. In brief, the diimine ligands are vital in promoting the selectivity and cytotoxicity of the compounds. Compound 2 with phen is the most cytotoxic compound. This could be due to its highest DNA cleavage efficiency and the ability to cleave DNA in the absence of exogenous agents. Even though the complex without N,N-ligand is the best topo I inhibitor, however, it is less selective, less cytotoxic, and the weakest DNA cleavage agent. This underscores the importance of nuclease activity in dictating the final cytotoxicity of this class of compound.5. Experimental
5.1. Materials
Paraformaldehyde (BDH Limited poole England), salicylaldehyde (Merck), triphenylphosphine (Merck), 4-fluoro benzoic hydrazide (Sigma-Aldrich), 1,10′-phenanthroline (Acros), 2,2′-bipyridine, 5,5′-dimethyl-2,2′-bipyridine and copper(II) acetate monohydrate (Fluka) were used as received without further purification. All the solvents such as ethanol, methanol, dimethylformamide were of reagent grade. The pBR322, GeneRulerTM 1 kb DNA ladder, 6× loading buffer and Tris-(hydroxymethyl)aminomethane (Tris) were procured from BioSyn Tech (Fermentas). Analytical grade agarose powder was obtained from Promega. Sodium chloride, human DNA topoisomerase I and ethidium bromide were purchased from Sigma Chemical Co. (USA). MTT (methylthiazolyldiphenyl-tetrazolium bromide), RPMI 1640 medium, EMEM (Eagle's Minimum Essential Medium), sodium bicarbonate, cis-platin, carboxymethyl cellulose, EDTA, DMSO were purchased from Sigma-Aldrich company. Foetal bovine serum, penicillin/streptomycin (100×), amphotericin B (250 μg ml−1) and sodium pyruvate (100 mM) were from PAA Laboratories.5.2. Physical measurements
The infrared spectra were recorded in the region 400–4000 cm−1 as KBr pellets with a Perkin-Elmer Spectrum 2000 FT-IR spectrophotometer and a Perkin-Elmer Spectrum RX1 FT-IR spectrophotometer. Elemental analyses for all the Schiff base ligands and copper(II) complexes were performed on a Perkin-Elmer EA2400 CHNS elemental analyzer. 1H and 13C NMR spectra of the ligands were recorded in deuterated DMSO-d6 on a JEOL JNM GX-270 FT NMR System Spectrometer. UV-Vis spectroscopic measurements of compounds in methanol were carried out on a Perkin-Elmer Lambda 40 spectrophotometer in the wavelength range of 200–800 nm. The X-ray crystallographic unit cell parameters and the intensity data were collected on a Bruker SMART APEX CCD diffractometer, equipped with a Mo Kα X-ray source (λ = 0.71073 Å). The APEX2 software was used for data collection and the SAINT software for cell refinement and data reduction. Absorption corrections on the data were made using SADABS. Molecular graphics were drawn by using XSEED.84 The structure were solved by direct-methods and refined by a full-matrix least-squares procedure on F2 with anisotropic displacement parameters for non-hydrogen atoms.5.3. Preparation of ligand and copper complexes
Synthesis of [5-(triphenylphosphoniomethyl)-salicylaldehyde 4-fluorobenzoylhydrazone] chloride monohydrate (L)The ligand was synthesized by condensing 4-fluorobenzhydrazide (0.166 g, 1 mmol) with 5-(triphenylphosphoniomethyl)-salicylaldehyde (0.433 g, 1 mmol) in ethanol (30 ml) for 4 hours. Slow evaporation of the solvent yielded yellow powder. The product were filtered, washed with cold ethanol and air-dried.
Yield: 83%, yellow solid, m.p.: 290–291 °C. Anal. calc. for C33H27ClFN2O2P: C, 69.66; H, 4.78; N, 4.92. Found: C, 69.25; H, 4.66; N, 4.88%. IR (KBr disc, cm−1): v(CO): 1661, v(CN): 1627, v(C–O): 1232, v(NN): 1014.
Characteristic 1H NMR (DMSO-d6, TMS, ppm 400 MHz, s, singlet; d, duplet; t, triplet; m, multiplet): 12.44 (s, 1H, NH); 11.40 (s,1H, OH); 8.58 (s, 1H, CHN); 8.07–8.10 (m, 2H, aromatic CH); 7.66–7.92 (m, 15H, aromatic CH);, 7.33–7.38 (t, 2H, aromatic CH); 7.21 (s, 1H, aromatic CH); 6.78–6.88 (m, 2H, aromatic CH); 5.11–5.15 (d, 2H, CH2). 13C NMR (DMSO-d6, TMS, ppm 100 MHz): 163.01 (CO); 157.21 (C–OH); 146.91 (CN); 115.35–135.06, 161.74 (Ar); 27.25, 27.72 (CH2).
5.4. Synthesis of chlorido[N-5-(triphenylphosphoniomethyl)-salicylaldehyde 4-fluoro benzoylhydrazonato] copper(II) monohydrate (1)
Copper(II) acetate monohydrate (0.200 g, 1 mmol) and L (0.569 g, 1 mmol) were refluxed in ethanol (30 ml) for 3 hours. The green complex formed was filtered off and rinsed with cold ethanol. Crystals were formed by recrystallization in methanol.Yield: 73%, green solid, m.p.: 248–249 °C. Anal. calc. for C33H27ClFCuN2O2P·H2O: C, 60.92; H, 4.49; N, 4.31. Found: C, 60.52; H, 4.31; N, 4.28%. IR (KBr disc, cm−1): v(CN): 1614, v(NC): 1499, v(C–O): 1214, v(NN): 1043.
5.5. Synthesis of (1,10′-phenanthroline)[N-5-(triphenylphosphoniomethyl)-salicylaldehyde 4-fluorobenzoylhydrazonato] copper(II) monohydrate (2)
Copper(II) acetate monohydrate (0.200 g, 1 mmol) and 1,10′-phenanthroline (0.180 g, 1 mmol) were heated in ethanol (20 ml) for 2 hours follow by addition of L (0.569 g, 1 mmol) in ethanol (20 ml) and the mixture was refluxed for another 2 hours. The green complex formed was extracted with hexane and purified by recrystallization from methanol–ethanol mixture.Yield: 63%, green solid, m.p.: 150–152 °C. Anal. calc. for C45H37ClFCuN4O2P·H2O: C, 64.90; H, 4.72; N, 6.73. Found: C, 64.90; H, 4.41; N, 6.46%. IR (KBr disc, cm−1): v(CN): 1612, v(NC): 1501, v(C–O): 1218, v(NN): 1047.
5.6. Synthesis of (2,2′-bipyridine)[N-5-(triphenylphosphoniomethyl)-salicylaldehyde 4-fluorobenzoylhydrazonato] copper(II) (3)
Same as complex 2 but using 2,2′-bipyridine instead of 1,10′-phenanthroline. The green complex formed was extracted with hexane.Yield: 67%, green solid, m.p.: 119–120 °C. Anal. calc. for C43H37ClFCuN4O2P: C, 65.31; H, 4.72; N, 7.09. Found: C, 65.11; H, 4.41; N, 6.57%. IR (KBr disc, cm−1): v(CN): 1611, v(NC): 1501, v(C–O): 1219, v(NN): 1044.
5.7. Synthesis of (5,5′-dimethyl-2,2′-bipyridine)[N-5-(triphenylphosphoniomethyl)-salicylaldehyde 4-fluorobenzoylhydrazonato] copper(II) trihydrate (4)
Same as complex 2 but using 5,5′-dimethyl-2,2′-bipyridine instead of 1,10′-phenanthroline. Slow evaporation of the solvent yielded green crystals. The products were filtered, washed with water and air-dried.Yield: 62%, green solid, m.p.: 200–201 °C. Anal. calc. for C45H41ClFCuN4O2P·H2O: C, 64.74; H, 4.95; N, 6.71. Found: C, 64.41; H, 5.02; N, 6.54%. IR (KBr disc, cm−1): v(CN): 1610, v(NC): 1501, v(C–O): 1216, v(NN): 1047.
5.8. E. coli Topoisomerase I inhibition assay
The human DNA topoisomerase I inhibition activity was determined by measuring the relaxation of supercoiled plasmid DNA pBR322. For measurement of human topoisomerase I activity, the reaction mixtures were comprised of 10 mM Tris–HCl, pH 7.5, 100 nM NaCl, 1 mM phenylmethylsulfonyl fluoride, α-toluenesulfonyl fluoride, PMSF, and 1 mM 2-mercatoethanol, 0.25 μg plasmid DNA pBR322, 1 unit of human topoisomerase I, and the test compound with final concentration of 160 μM. All reactions were conducted at a final volume of 20 μl and were prepared on ice. Upon enzyme addition, reaction mixtures were incubated at 37 °C for 30 min. The reactions were terminated by addition of 2 μl of 10% SDS, followed by 3 μl of dye solution comprising 0.02% bromophenol blue and 50% glycerol. SDS is required to observed a linear DNA fragment and to denature topoisomerase I, preventing further functional enzymatic activity. The mixtures were applied to 1.25% agarose gel and electrophoresed for 2 hours at 80 V with running buffer of Tris-acetate EDTA (TAE) at pH 8.1. The gel was stained, destained, and photographed under UV light using a Syngene Bio Imaging system and the digital image was viewed with Gene Flash software.In human topoisomerase I inhibition condition study, the same protocol was applied. This study is designed to deduce the mode of action of complexes in the human DNA topoisomerase I inhibition study. The sequence of addition of the main components (human DNA topoisomerase I, plasmid DNA pBR322, and metal complexes) was varied. For the first condition, human DNA topoisomerase I with the metal complex was incubated at 37 °C for 30 min before the addition of DNA. The mixture was incubated for another 30 min at the same temperature after the addition of DNA. As for the second condition, the metal complex and DNA was incubated for 30 min at 37 °C first, followed by the addition of topoisomerase I. This mixture was incubated for another 30 min at 37 °C after the addition of topoisomerase I.
5.9. DNA binding
The DNA-binding experiments were performed at room temperature. Using the electronic absorption spectral method, the relative bindings of the hydrazone copper complexes to CT-DNA were studied in 5 mM Tris–HCl/NaCl buffer (pH = 7.2). The solution of CT-DNA gave a ratio of UV absorbance at 260 nm and 280 nm, A260/A280, of 1.8–1.9, indicating that the DNA was sufficiently free of protein.45 The CT-DNA stock solutions were prepared in 5 mM Tris–HCl/50 mM NaCl buffer, pH = 7.2 (stored at 4 °C and used within 4 days after their preparation). The concentration of CT-DNA per nucleotide phosphate was calculated from the absorbance at 260 nm by using molar extinction coefficient, ε = 6400 M−1 cm−1. Concentrated stock solutions of metal complexes were prepared by dissolving them in 1% methanol and diluting them with 99% of corresponding buffer solution. The absorption spectra were recorded on a Perkin-Elmer Lambda 40 spectrophotometer by using cuvettes of 1 cm path length. In a typical experiment, 2 ml solution of the complex (20 μM) was transferred into a cuvette. Absorbance titration was conducted by adding concentrated stock solutions of CT-DNA directly to the cuvette and the absorption spectra were recorded in the range of 200–500 nm about 5 min after each addition of DNA solution. Absorption spectral titration experiments were performed by maintaining a constant concentration of the complex (20 μM) and varying the nucleic acid concentration until [compound]/[DNA] ≈ 1.5.10. DNA cleavage
Agarose gel electrophoresis experiments were carried out on supercoiled plasmid DNA pBR322 (4.4 kb) using a horizontal gel system. For the cleavage studies, each 20 μl sample consisted of the complex dissolved in buffer, DNA, and the required volume of additional buffer. All samples were incubated in the dark in an incubator at a temperature of 37 °C. The reaction mixtures were prepared as follows: 0.5 μl of compounds or salt with increasing concentration were added to the mixture of 0.5 μl of supercoiled plasmid DNA pBR322 (0.25 μg/μl) and Tris–NaCl buffer pH 7.5 was added to give a total volume of 20 μl. The reactions were performed after incubating the reaction mixtures at 37 °C for 24, 48 or 72 h. Three microliter of 6× loading buffer was added to 20 μl of the reaction mixtures and electrophoresis was performed at 80 V for 90 min in Tris-acetate-EDTA (TAE) buffer, pH 8.1, using 1.5% agarose gel. After electrophoresis, the agarose gel was stained with ethidium bromide solution (0.5 μg/ml). For the oxidative cleavage studies, incubation of the samples was similarly carried out with the addition of exogenous agents such as hydrogen peroxide (500 μM) and ascorbate acid (30 μM). The DNA cleavage profile was analyzed using 1.5% agarose gel in a horizontal gel tank set with a running time of 90 min, at a constant voltage of 80 V. Each reaction mixture consisted of 0.5 μg μl−1 DNA and Tris–NaCl buffer pH 7.5 unless otherwise mentioned. The resultant DNA bands after the electrophoresis step for each set of experiments were stained with ethidium bromide before being photographed under UV light using a Syngene Bio Imaging system and the digital image was viewed with Gene Flash software.5.11. Cell lines and culture medium
Human lung carcinoma (A549), human prostate adenocarcinoma (PC-3) and human non-cancer fibroblast (MRC-5) cell lines were purchased from American Type Culture Collection (ATCC, USA). A549 and PC-3 cells were maintained in RPMI 1640 medium while MRC-5 cells were maintained in EMEM medium, supplemented with 10% foetal bovine serum, 2% penicillin/streptomycin (100×) and 1% amphotericin B. The cells were cultured at 37 °C in humidified atmosphere in a CO2 incubator (Shel Lab water-jacketed, USA).5.12. MTT cytotoxicity assay
The MTT cytotoxicity assay was carried out as previously described by Mosmann.85 All samples were dissolved in DMSO to form stock solution before cytotoxicity testing. The final concentration of DMSO in each well was 0.5%. The cytotoxicity of each sample was expressed as IC50 value, which is the concentration of sample that reduced the viability of cells by 50% compared to the control (cells treated with 0.5% DMSO). All the samples were assayed in triplicate. The IC50 values for cytotoxicity screening were obtained by non-linear regression using GraphPad Prism statistical software.5.13. Annexin V-FITC/PI double staining using flow cytometry
The selected compounds were evaluated for their ability to induce cell death on PC-3 cell line by Annexin V-FITC fluorescein isothiocyanate apoptosis detection kit (BD Biosciences, Pharmingen San Diego, CA, USA). The cells were seeded in 35 mm culture dish (4 × 105 cells) for sample treatment and three 35 mm culture dish for control (unstained, annexin only and Propidium iodide only) which are used to set up compensation and quadrants. The cells were treated with the concentration of 20 μM. The cells were incubated for 72 hours at 37 °C. Flow cytometric analysis of apoptosis was carried out according to the instruction of the manufacture of FITC Annexin V Apoptosis Detection Kit I (BD Biosciences, Pharmingen San Diego, CA, USA). After incubation the supernatant was collected and the adherent cells were detached with accutase from each dish. Both floating apoptosis cells and accutase cells were pooled into 15 ml tube. The collected cells were washed twice in PBS and then resuspended in 1× binding buffer at a concentration of 1 × 106 cells per ml. 100 μl f the solution (1 × 105 cells) were transferred to a 5 ml round bottom tube. Next 5 μl of FITC Annexin V and 5 μl of propidium iodide were added into the tube. The tubes are gently tapped and incubated for 15 minutes at room temperature in the dark. A volume of 400 μl of 1× Binding buffer was added to each tube and quantified by flow cytometry within an hour on a BD FACSCanto II system (Becton Dickinson, Mountain View, CA). At least 10000 cells were counted in each sample.
List of abbreviations
| cisplatin | cis-Diamminedichloroplatinum(II) |
| MTT | (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| DNA | Deoxyribonucleic acid |
| phen | 1,10′-phenanthroline |
| bpy | 2,2′-Bipyridine |
| dmbpy | 5,5′-Dimethyl-2,2′-bipyridine |
| DMSO | Dimethylsulfoxide |
| DMF | Dimethylformamide |
| Tris | Tris-(hydroxymethyl)aminomethane |
| EMEM | Eagle's minimum essential medium |
| EDTA | Ethylenediaminetetraacetic acid |
| IR | Infrared |
| KBr | Potassium bromide |
| NMR | Nuclear%20magnetic%20resonance |
| LMCT | Ligand-to-metal-charge transfer |
| TAE | Tris-acetate EDTA |
| SI | Selectivity index |
| AV | Annexin V |
| PI | Propidium iodide |
Acknowledgements
The authors would like to thank University of Malaya and MOHE (FRGS-FP016-2013A) for funding. SKS and STC would like to thank UM for fellowship and funding (PV015-2012A and PV089/2012A). We would like to thank Prof. Wong C.S. and Prof. Norhanom for the use of their facilities.Notes and references
- A. Zamora, V. Rodríguez, N. Cutillas, G. S. Yellol, A. Espinosa, K. G. Samper, M. Capdevila, Ò. Palacios and J. Ruiz, J. Inorg. Biochem., 2013, 128, 48–56 CrossRef CAS PubMed.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617–3630 RSC.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anti-Cancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS.
- G. J. Brewer, Exp. Biol. Med., 2001, 226, 665–673 CAS.
- S. A. Lowndes and A. L. Harris, J. Mammary Gland Biol., 2005, 10, 299–310 CrossRef PubMed.
- S. Munira Haidad Ali, Y.-K. Yan, P. P. F. Lee, K. Z. X. Khong, M. Alam Sk, K. H. Lim, B. Klejevskaja and R. Vilar, Dalton Trans., 2014, 1449–1459 RSC.
- P. Krishnamoorthy, P. Sathyadevi, A. H. Cowley, R. R. Butorac and N. Dharmaraj, Eur. J. Med. Chem., 2011, 46, 3376–3387 CrossRef CAS PubMed.
- M. Carcelli, P. Mazza, C. Pelizzi and F. Zani, J. Inorg. Biochem., 1995, 57, 43–62 CrossRef CAS.
- M. V. Angelusiu, S. F. Barbuceanu, C. Draghici and G. L. Almajan, Eur. J. Med. Chem., 2010, 45, 2055–2062 CrossRef CAS PubMed.
- M. B. Ferrari, F. Bisceglie, G. Pelosi, P. Tarasconi, R. Albertini, P. P. Dall'Aglio, S. Pinelli, A. Bergamo and G. Sava, J. Inorg. Biochem., 2004, 98, 301–312 CrossRef CAS PubMed.
- S. Vogel, D. Kaufmann, M. Pojarová, C. Müller, T. Pfaller, S. Kühne, P. J. Bednarski and E. V. Angerer, Bioorg. Med. Chem., 2008, 16, 6436–6447 CrossRef CAS PubMed.
- M. C. Rodrìguez-Argüelles, M. B. Ferrari, F. Bisceglie, C. Pelizzi, G. Pelosi, S. Pinelli and M. Sassi, J. Inorg. Biochem., 2004, 98, 313–321 CrossRef PubMed.
- D. Senthil Raja, N. S. P. Bhuvanesh and K. Natarajan, Eur. J. Med. Chem., 2012, 47, 73–85 CrossRef CAS PubMed.
- M. Alagesan, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2013, 7210–7223 RSC.
- S. Kashanian, M. M. Khodaei, H. Roshanfekr, N. Shahabadi and G. Mansouri, Spectrochim. Acta, Part A, 2012, 86, 351–359 CrossRef CAS PubMed.
- A. N. Wein, A. T. Stockhausen, K. I. Hardcastle, M. R. Saadein, S. Peng, D. Wang, D. M. Shin, Z. Chen and J. F. Eichler, J. Inorg. Biochem., 2011, 105, 663–668 CrossRef CAS PubMed.
- S. Ramakrishnan, V. Rajendiran, M. Palaniandavar, V. S. Periasamy, B. S. Srinag, H. Krishnamurthy and M. A. Akbarsha, Inorg. Chem., 2009, 48, 1309–1322 CrossRef CAS PubMed.
- V. Gandin, M. Porchia, F. Tisato, A. Zanella, E. Severin, A. Dolmella and C. Marzano, J. Med. Chem., 2013, 56, 7416–7430 CrossRef CAS PubMed.
- S. T. Chew, K. M. Lo, S. K. Lee, M. P. Heng, W. Y. Teoh, K. S. Sim and K. W. Tan, Eur. J. Med. Chem., 2014, 76, 397–407 CrossRef CAS PubMed.
- S. K. Lee, K. W. Tan, S. W. Ng, K. K. Ooi, K. P. Ang and M. A. Abdah, Spectrochim. Acta, Part A, 2014, 121, 101–108 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013–1029 CrossRef CAS PubMed.
- H. J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed.
- P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, 2013 Search PubMed.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- I. Ojima, Fluorine in Med. Chem. and Chem. Biol, John Wiley & Sons, Ltd, 2009 Search PubMed.
- P. Cozzi, N. Mongelli and A. Suarato, Curr. Med. Chem.: Anti-Cancer Agents, 2004, 4, 93–121 CrossRef CAS.
- C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS PubMed.
- J. P. Bégué and D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992–1012 CrossRef PubMed.
- P. R. Reddy, A. Shilpa, N. Raju and P. Raghavaiah, J. Inorg. Biochem., 2011, 105, 1603–1612 CrossRef CAS PubMed.
- S. Tabassum, W. M. Al-Asbahy, M. Afzal, F. Arjmand and V. Bagchi, Dalton Trans., 2012, 4955–4964 RSC.
- S. Banerjee, S. Mondal, S. Sen, S. Das, D. L. Hughes, C. Rizzoli, C. Desplanches, C. Mandal and S. Mitra, Dalton Trans., 2009, 6849–6860 RSC.
- P. B. Sreeja, M. R. P. Kurup, A. Kishore and C. Jasmin, Polyhedron, 2004, 23, 575–581 CrossRef CAS PubMed.
- U. L. Kala, S. Suma, M. R. P. Kurup, S. Krishnan and R. P. John, Polyhedron, 2007, 26, 1427–1435 CrossRef CAS PubMed.
- M. S. Refat, I. M. El-Deen, H. K. Ibrahim and S. El-Ghool, Spectrochim. Acta, Part A, 2006, 65, 1208–1220 CrossRef PubMed.
- M. R. Maurya, S. Khurana, C. Schulzke and D. Rehder, Eur. J. Inorg. Chem., 2001, 779–788 CrossRef CAS.
- T. Ghosh, S. Bhattacharya, A. Das, G. Mukherjee and M. G. B. Drew, Inorg. Chim. Acta, 2005, 358, 989–996 CrossRef CAS PubMed.
- P. Sathyadevi, P. Krishnamoorthy, M. Alagesan, K. Thanigaimani, P. Thomas Muthiah and N. Dharmaraj, Polyhedron, 2012, 31, 294–306 CrossRef CAS PubMed.
- I. D. Brčeski, V. M. Leovac, G. A. Bogdanović, S. P. Sovilj and M. Revenco, Inorg. Chem. Commun., 2004, 7, 253–256 CrossRef PubMed.
- H. A. Patel, R. S. Somani, H. C. Bajaj and R. V. Jasra, Appl. Clay Sci., 2007, 35, 194–200 CrossRef CAS PubMed.
- P. Talukder, A. Datta, S. Mitra, G. Rosair, M. Salah El Fallah and J. Ribas, Dalton Trans., 2004, 4161–4167 RSC.
- C. Rajarajeswari, R. Loganathan, M. Palaniandavar, E. Suresh, A. Riyasdeen and M. A. Akbarsha, Dalton Trans., 2013, 8347–8363 RSC.
- D. D. Li, F. P. Huang, G. J. Chen, C. Y. Gao, J. L. Tian, W. Gu, X. Liu and S. P. Yan, J. Inorg. Biochem., 2010, 104, 431–441 CrossRef CAS PubMed.
- S. Roy, P. Mitra and A. K. Patra, Inorg. Chim. Acta, 2011, 370, 247–253 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- S. Sunami, T. Nishimura, I. Nishimura, S. Ito, H. Arakawa and M. Ohkubo, J. Med. Chem., 2009, 52, 3225–3237 CrossRef CAS PubMed.
- Y. Pommier, Nat. Rev. Cancer, 2006, 6, 789–802 CrossRef CAS PubMed.
- B. A. Teicher, Biochem. Pharmacol., 2008, 75, 1262–1271 CrossRef CAS PubMed.
- I. Husain, J. L. Mohler, H. F. Seigler and J. M. Besterman, Cancer Res., 1994, 54, 539–546 CAS.
- J. C. Wang, Annu. Rev. Biochem., 1996, 65, 635–692 CrossRef CAS PubMed.
- J. C. Wang, Nat. Rev. Mol. Cell Biol., 2002, 3, 430–440 CrossRef CAS PubMed.
- W. Feng, M. Satyanarayana, Y. C. Tsai, A. A. Liu, L. F. Liu and E. J. LaVoie, Eur. J. Med. Chem., 2009, 44, 3433–3438 CrossRef CAS PubMed.
- C. Marchand, S. Antony, K. W. Kohn, M. Cushman, A. Ioanoviciu, B. L. Staker, A. B. Burgin, L. Stewart and Y. Pommier, Mol. Cancer Ther., 2006, 5, 287–295 CrossRef CAS PubMed.
- D.-H. Kang, J.-S. Kim, M.-J. Jung, E.-S. Lee, Y. Jahng, Y. Kwon and Y. Na, Bioorg. Med. Chem. Lett., 2008, 18, 1520–1524 CrossRef CAS PubMed.
- M. S. Mohamed, A. A. Shoukry and A. G. Ali, Spectrochim. Acta, Part A, 2012, 86, 562–570 CrossRef CAS PubMed.
- R. L. Williams, H. N. Toft, B. Winkel and K. J. Brewer, Inorg. Chem., 2003, 42, 4394–4400 CrossRef CAS PubMed.
- K. Y. Lee, I. C. Kwon, Y. H. Kim, W. H. Jo and S. Y. Jeong, J. Controlled Release, 1998, 51, 213–220 CrossRef CAS.
- J. Annaraj, S. Srinivasan, K. M. Ponvel and P. R. Athappan, J. Inorg. Biochem., 2005, 99, 669–676 CrossRef CAS PubMed.
- A. Barve, A. Kumbhar, M. Bhat, B. Joshi, R. Butcher, U. Sonawane and R. Joshi, Inorg. Chem., 2009, 48, 9120–9132 CrossRef CAS PubMed.
- E. K. Efthimiadou, H. Thomadaki, Y. Sanakis, C. P. Raptopoulou, N. Katsaros, A. Scorilas, A. Karaliota and G. Psomas, J. Inorg. Biochem., 2007, 101, 64–73 CrossRef CAS PubMed.
- J.-G. Liu, B.-H. Ye, H. Li, Q.-X. Zhen, L.-N. Ji and Y.-H. Fu, J. Inorg. Biochem., 1999, 76, 265–271 CrossRef CAS.
- M. Chauhan, K. Banerjee and F. Arjmand, Inorg. Chem., 2007, 46, 3072–3082 CrossRef CAS PubMed.
- A. G. Quiroga, J. M. Pérez, I. López-Solera, E. I. Montero, J. R. Masaguer, C. Alonso and C. Navarro-Ranninger, J. Inorg. Biochem., 1998, 69, 275–281 CrossRef CAS.
- A. Dimitrakopoulou, C. Dendrinou-Samara, A. A. Pantazaki, M. Alexiou, E. Nordlander and D. P. Kessissoglou, J. Inorg. Biochem., 2008, 102, 618–628 CrossRef CAS PubMed.
- A. Zianna, G. Psomas, A. Hatzidimitriou, E. Coutouli-Argyropoulou and M. Lalia-Kantouri, J. Inorg. Biochem., 2013, 127, 116–126 CrossRef CAS PubMed.
- L. Ronconi and P. J. Sadler, Coord. Chem. Rev., 2007, 251, 1633–1648 CrossRef CAS PubMed.
- G. Y. Li, K. J. Du, J. Q. Wang, J. W. Liang, J. F. Kou, X. J. Hou, L. N. Ji and H. Chao, J. Inorg. Biochem., 2013, 119, 43–53 CrossRef CAS PubMed.
- P. A. N. Reddy, M. Nethaji and A. R. Chakravarty, Eur. J. Inorg. Chem., 2004, 1440–1446 CrossRef CAS.
- S. Zhang, Y. Zhu, C. Tu, H. Wei, Z. Yang, L. Lin, J. Ding, J. Zhang and Z. Guo, J. Inorg. Biochem., 2004, 98, 2099–2106 CrossRef CAS PubMed.
- W. Y. Lee, Y. K. Yan, P. P. F. Lee, S. J. Tan and K. H. Lim, Metallomics, 2012, 4, 188–196 RSC.
- L. Tjioe, T. Joshi, J. Brugger, B. Graham and L. Spiccia, Inorg. Chem., 2011, 50, 621–635 CrossRef CAS PubMed.
- A. Y. Louie, T. J. Meade and S. J. Lippard, Chem. Rev., 1999, 99, 2711–2734 CrossRef CAS PubMed.
- A. Mazumder, C. L. Sutton and D. S. Sigman, Inorg. Chem., 1993, 32, 3516–3520 CrossRef CAS.
- C. A. Detmer, F. V. Pamatong and J. R. Bocarsly, Inorg. Chem., 1996, 35, 6292–6298 CrossRef CAS.
- S. H. Chiou, N. Ohtsu and K. G. Bensch, Biological and Inorganic Copper Chemistry, 1985 Search PubMed.
- H. L. Seng, K. W. Tan, M. J. Maah, W. T. Tan, H. Hamada, M. Chikira and C. H. Ng, Polyhedron, 2009, 28, 2219–2227 CrossRef CAS PubMed.
- P. Jaividhya, R. Dhivya, M. A. Akbarsha and M. Palaniandavar, J. Inorg. Biochem., 2012, 114, 94–105 CrossRef CAS PubMed.
- G. Koopman, C. P. M. Reutelingsperger, G. A. M. Kuijten, R. M. J. Keehnen, S. T. Pals and M. H. J. Van Oers, Blood, 1994, 84, 1415–1420 CAS.
- S. J. Martin, C. P. M. Reutelingsperger, A. J. McGahon, J. A. Rader, R. C. A. A. Van Schie, D. M. LaFace and D. R. Green, J. Exp. Med., 1995, 182, 1545–1556 CrossRef CAS.
- M. Van Engeland, L. J. W. Nieland, F. C. S. Ramaekers, B. Schutte and C. P. M. Reutelingsperger, Cytometry, 1998, 31, 1–9 CrossRef CAS.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutelingsperger, J. Immunol. Methods, 1995, 184, 39–51 CrossRef CAS.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1012657–1012659. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra11716f |
| This journal is © The Royal Society of Chemistry 2014 |