
Self-assembly of Au nanoparticles on graphene sheets as a catalyst with controlled grafting density and high reusability†
Haiqing Yaoab,
Tsao-Cheng Huangab and
Hung-Jue Sue*abc
aDepartment of Materials Science and Engineering, Texas A&M University, College Station, TX 77843, USA. E-mail: hjsue@tamu.edu
bPolymer Technology Center, Texas A&M University, College Station, TX 77843, USA
cDepartment of Mechanical Engineering, Texas A&M University, College Station, TX 77843, USA
First published on 11th November 2014
Abstract
In the present work, we have developed a method to achieve self-assembly of Au nanoparticles (NPs) on graphene sheets through chemical functionalization. Transmission electron microscopy and UV-Vis spectroscopy results indicate that the grafting density of Au NPs on graphene sheets can be controlled by changing the types of functional groups. The ability to control the density of Au NPs cannot be overestimated, because it provides tunable catalytic activity. These graphene/Au composites display good stability against agglomeration and show excellent catalytic activity in a model reduction reaction of 4-nitrophenol into 4-aminophenol. This study demonstrates a general approach to the preparation of tunable and reusable catalysts containing densely loaded and well dispersed Au NPs.
1 Introduction
Gold nanoparticles (Au NPs) have been extensively studied in catalysts for petroleum processing, energy conversion, and pollutant removal.1–3 The use of NPs results in large contact areas between the active catalyst and the surrounding environment.4 This ensures that the Au NPs are used effectively as a catalytic material. However, one of the challenges associated with the use of Au NPs as catalysts is that the high surface area reduces the colloidal stability, drives the aggregation of the NPs, and lowers the catalytic activity.5 Another challenge is post-reaction separation and the reusability of the tiny particles.6 To overcome these challenges, heterogeneous catalysts based on supported Au NPs have been developed to improve the colloidal stability. Various materials have been reported to support Au NPs for the heterogeneous catalysis. These materials include polymers, carbon materials, silica, and metal oxides.6–14 Although these methods have realized engineered local environments to stabilize Au NPs, they lack the control over the grafting density of Au NPs.15 Controlling the particle density is crucial to understand reaction kinetics and to control reaction rate, which are scientific challenges related to the catalytic application of Au NPs.Graphene, a two-dimensional sheet consisting of sp2 hybridized carbon atoms, has emerged as one of the most promising support materials for Au NPs due to its high surface area and chemical stability.16–20 One of the methods to prepare graphene sheets is the chemical exfoliation of graphite in aqueous medium, which produces oxygen functionalized graphene sheets – graphene oxide (GO). The hybridization of GO with the Au NPs synergistically enhances their functionalities and expands their catalytic applications.21–23 Efforts have been made to synthesize GO/Au nanocomposites by mixing and in situ approaches.24–28 Goncalves et al.27 reported a method to prepare GO/Au composites by directly mixing Au(III) ions with GO in water, and the oxygen groups present at GO surface serve as nucleation sites for Au NPs. However, the resulting GO/Au composites showed a low density of Au NPs on the graphene sheet, i.e. approximately 8%. In order to improve the grafting density, organic spacers, e.g. octadecylamine,29 protein,26 and 1-pyrene butyric acid,15 were used to anchor Au NPs on graphene sheets. An additional spacer was reported in our previous work,21 where we used a silylating agent (N1-(3-trimethoxysilylpropyl) diethylenetriamine) to anchor Au NPs on GO surface. The silylating agent forms covalent bonding with GO and simultaneously serves as a reductant to reduce GO. This method provides effective anchoring sites for Au NPs, leading to a density of 24%.
To fully exploit the use of GO sheets as a support for Au NPs, it is desirable to decorate the GO surface with appropriate functional groups that generate anchoring sites for Au NPs with controlled density. The present work intends to extend the self-assembly approach in our previous work21 to develop GO/Au composites with tunable particle densities. One of the consequences of using the self-assembly method is large areas of densely packed particles on a support. This becomes prominent in expanding the GO/Au composite family and exploring additional applications of the composites. In the present work, GO is chemically functionalized by three different silane coupling agents, which vary at their end groups, i.e. mono-, bi- and tri-amine. These different functional groups enable manipulating the grafting density of Au NPs and tailoring their catalytic applications. Various spectroscopies and microscopies are used to characterize the resulting composites and study the catalytic activities in the reduction reaction of 4-nitrophenol (4-NPh) to 4-aminophenol (4-APh).
2 Experimental section
2.1 Materials
Graphite (SP-1 graphite, average particle sizes of 45 μm) was obtained from Bay Carbon Inc, USA. The following reagents were purchased from Sigma Aldrich and were used as received: (3-aminopropyl) trimethoxysilane (1N), N-[3-(trimethoxysilyl) propyl] ethylenediamine (2N), N1-(3-trimethoxysilylpropyl) diethylene triamine (3N), hydrazine, NaNO3, KMnO4, H2SO4, and H2O2.2.2 Synthesis of GO and organosilane-functionalized GO
GO was synthesized by the Hummers' method.30 Graphite powder (0.5 g) was firstly treated with a solution containing concentrated H2SO4 (50 mL) and NaNO3 (0.5 g) below 5 °C. KMnO4 (3 g) was added gradually, and the mixture was stirred continuously for 2 h. The mixture was then kept at 35 °C for another 2 h. The mixture was diluted with deionized (DI) water (23 mL) and stirred for 15 min. Additional DI water (71 mL) and 30% H2O2 (10 mL) were added to the mixture, which changed the mixture color to brilliant yellow. Finally, the mixture was washed with water several times and HCl once to remove metallic ions.GO (100 mg) was dispersed in ethanol, followed by addition of organosilane (5.4 mM). The mixture was stirred at 90 °C for 24 h. The resulting suspension was washed thoroughly with ethanol and acetone to remove unreacted silylating agents. The final samples were dried at 60 °C in a vacuum oven for 24 h.
2.3 Preparation of GO/Au nanocomposites
The synthesis of Au NPs was carried out following a similar procedure reported in the literature.31 HAuCl4 aqueous solution (1 mM, 100 mL) was refluxed and vigorously stirred. Rapid addition of sodium citrate aqueous solution (38.8 mM, 10 mL) to the boiling solution resulted in a color change from pale yellow to wine red. After stirring for 10 min, the heat source was removed. Stirring was continued for an additional 15 min till the solution reached room temperature. The synthesized Au NPs have an average particle size of 10 nm according to the measurements in TEM image. To prepare GO/Au nanocomposites, Au NP solution (20 mL) was mixed with organosilane-functionalized GO (1 mg) at room temperature and the mixture was incubated at 35 °C for 20 h. The final GO/Au composites were collected by centrifugation and washed with DI water twice.2.4 Characterization
Fourier transform infrared spectroscopy-attenuated total reflectance (FTIR-ATR) spectra of the samples were acquired using Nicolet 380 (Thermo Fisher Scientific) in conjunction with ATR accessory (AVATAR OMNI Sampler, Germanium crystal) under ambient condition. X-ray photoelectron spectroscopy (XPS) data were obtained with a Kratos Axis Ultra using a non-monochromatic MgKα photon source (1486 eV). Data were fitted using XPSPEAK 4.1 by applying a linear-type background correction. The section height of organosilane-functionalized GO was identified by an atomic force microscope (AFM, Bruker Dimension Icon AFM). Transmission electron microscopy (TEM) was performed using a JEOL 2010 high-resolution TEM operated at 200 kV. UV-Vis absorption spectra were acquired with a UV-Vis-NIR spectrophotometer (Shimadzu, UV-3600).3 Results and discussion
3.1 Preparation of organosilane-functionalized GO
Fig. 1 illustrates a one-step chemical functionalization of GO through silane agents and the self-assembly of Au NPs on the functionalized GO. Three silylating reagents with different amine groups were used to react with GO to form GO-xN (x = 1, 2, and 3): (3-aminopropyl) trimethoxysilane (GO-1N), N-[3-(trimethoxysilyl) propyl] ethylenediamine (GO-2N), and N1-(3-trimethoxysilylpropyl) diethylene triamine (GO-3N). In general, all silane coupling agents have one organic substituent and three hydrolyzable groups (–OCH3). The alkoxysilane groups react with hydroxyl groups on the GO surface, followed by the formation of a monomolecular layer on the GO surface.21,32 Note that the brown colored dispersion of GO turned into black immediately after the addition of silane agents, which is similar to the phenomenon of the hydrazine reduced GO. It suggests that the π–π conjugation network of GO was partially restored due to the amine functionality of silane agents during the reaction.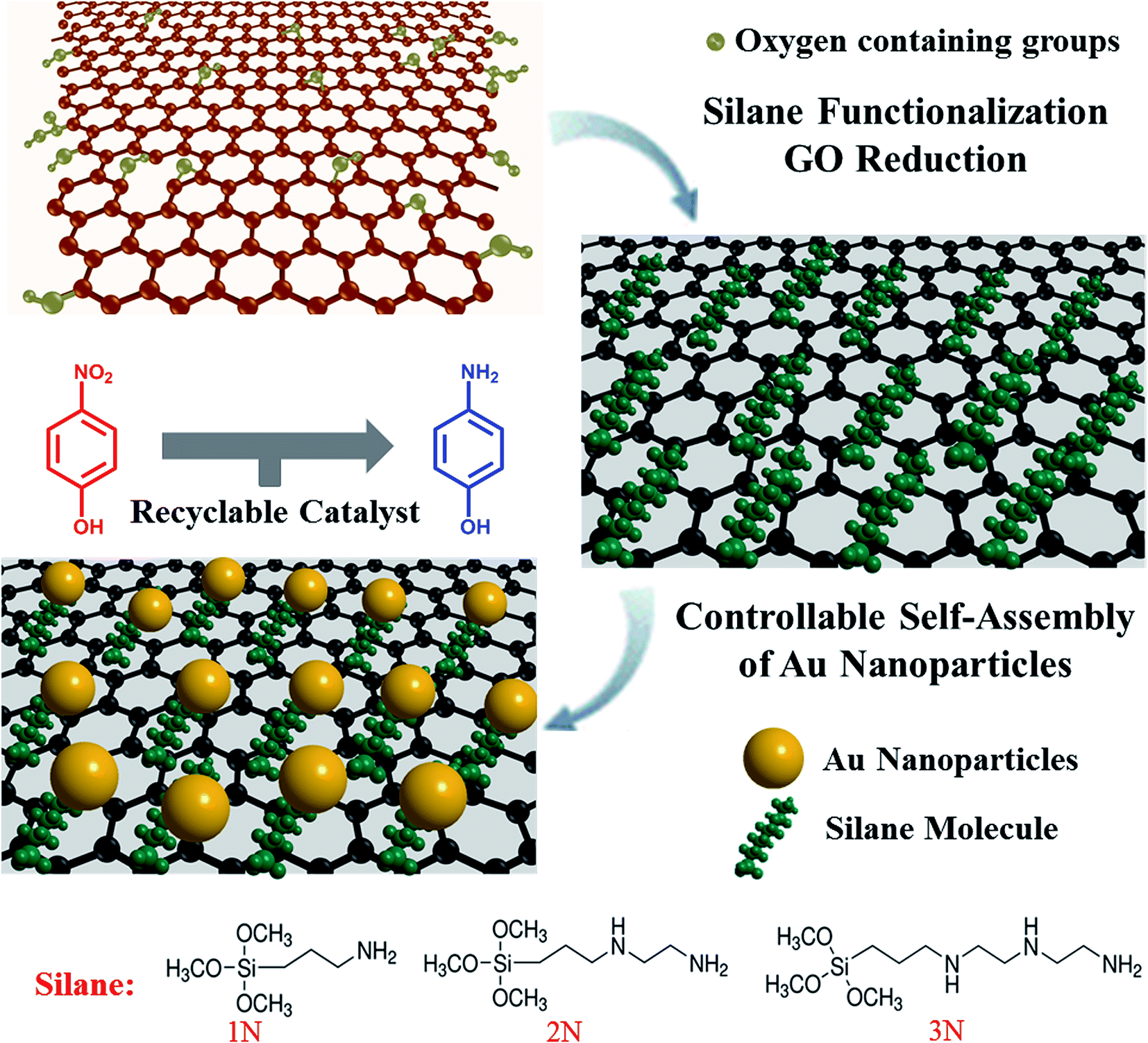 | ||
| Fig. 1 Schematic illustration of the preparation of effective catalysts based on Au nanoparticles and chemically functionalized GO. | ||
The reaction between GO and silane can be confirmed by analyzing the core-level Binding Energy (BE) obtained from XPS (Fig. 2). The XPS spectrum of C1s on GO (Fig. 2a) shows three main components, which correspond to different oxidation states of the C atoms. These states include the non-oxygenated C (284.8 eV), the C in C–O bonds (286.2 eV), and the C in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (287.8 eV).33 As compared to GO, the peak intensities of C–O and CO in GO-1N (Fig. 2b) significantly decreased. GO-2N and GO-3N show similar phenomenon (not shown). The carbon-to-oxygen (C/O) atomic ratio was calculated by the decomposition of C1s showing in the XPS spectrum. As shown in Table 1, GO has a C/O ratio of 1.9. In contrast, the C/O ratio of GO-1N increases to 2.7, and GO-2N and GO-3N have even higher C/O ratios of 4.0 and 4.5, respectively. This suggests that GO reduction has occurred and some of the oxygen-containing groups have been removed during the silylating reaction.21 In particular, the silane agent with triamine end groups reduces GO most effectively. Additionally, significant amounts of N1s and Si2p from functionalized GO were detected (Fig. 2c and d), indicating that the GO surface has also been successfully functionalized with silane molecules.
O bonds (287.8 eV).33 As compared to GO, the peak intensities of C–O and CO in GO-1N (Fig. 2b) significantly decreased. GO-2N and GO-3N show similar phenomenon (not shown). The carbon-to-oxygen (C/O) atomic ratio was calculated by the decomposition of C1s showing in the XPS spectrum. As shown in Table 1, GO has a C/O ratio of 1.9. In contrast, the C/O ratio of GO-1N increases to 2.7, and GO-2N and GO-3N have even higher C/O ratios of 4.0 and 4.5, respectively. This suggests that GO reduction has occurred and some of the oxygen-containing groups have been removed during the silylating reaction.21 In particular, the silane agent with triamine end groups reduces GO most effectively. Additionally, significant amounts of N1s and Si2p from functionalized GO were detected (Fig. 2c and d), indicating that the GO surface has also been successfully functionalized with silane molecules.
 | ||
| Fig. 2 XPS spectra of (a) C1s of GO, (b) C1s of GO-1N, (c) N1s of GO-1N, and (d) Si2p of GO-1N. | ||
| C/O ratio | Residual weight after 900 °C (wt%) | –NH group concentration (mmol g−1) | |
|---|---|---|---|
| GO | 1.9 | 0 | 0 |
| GO-1N | 2.7 | 34 | 5.6 |
| GO-2N | 4.0 | 25.5 | 8.4 |
| GO-3N | 4.5 | 20.8 | 10.2 |
To further confirm the presence of the functional groups on the GO surface, GO and organosilane-functionalized GO were characterized by FTIR (Fig. 3). GO shows a broad and tense peak at 3500–3300 cm−1, corresponding to the stretching vibrations of the O–H groups on graphene sheet. The CO stretching at 1728 cm−1 and C–O stretching at 1200–1100 cm−1 indicate the presence of carboxylic and epoxide groups on GO sheets. Compared with GO, functionalized GO shows several new peaks in the ranges of 2700–3000 cm−1 and 1700–1000 cm−1. The new peaks at 2925 and 2850 cm−1 are associated with the stretching vibration of –CH2– groups from silane molecules, and the sharp peak at 1577 cm−1 is assigned to the bending vibration of –NH–.34 The stretching vibration of Si–O–C at 1107 cm−1 strongly indicates that the silylation of GO has occurred. The 1728 cm−1 (CO asymmetric stretching) peak of GO spectrum has disappeared after functionalization, implying the removal of –COOH from GO sheets after the reaction.
 | ||
| Fig. 3 FTIR spectra of GO and organosilane-functionalized GO. | ||
The compositions of organosilane-functionalized GO were measured by TGA. Fig. 4 shows the TGA curves of GO-1N, GO-2N, and GO-3N, as well as the pristine GO. GO in air exhibits two weight losses at 200 °C and 500 °C, respectively, attributing to the decomposition of oxygen-containing groups and carbon combustion. The organic components in silylating agents decompose at a temperature range from 200–400 °C.34 Assuming that all the residual weight after 900 °C for GO-xN was SiO2, the compositions of GO-xN, especially the concentration of amine groups, were calculated and summarized in Table 1. GO-1N (5.6 mmol g−1) has the lowest amine group concentration. GO-2N (8.4 mmol g−1) and GO-3N (10.4 mmol g−1) have much larger concentrations, which are 1.5 times and 1.8 times larger than that of GO-1N. The ability to control the concentration of amine groups cannot be overestimated, because the amine group is the key for further tailoring the grafting density of Au NPs.
 | ||
| Fig. 4 TGA curves of GO and organosilane-functionalized GO. | ||
Introducing surface functionalization for Au NPs can improve their colloidal stability, but the strong binding of the surface capping ligands often results in “poisoning” – deactivation of the nanocatalysts. To obtain catalytically active GO/Au composites, Au NPs are required to immobilize solely on the graphene surface, but not embedded within the organosliane layer. To that end, a monomolecular layer of silane on the graphene surface is critical to avoid the embedding of Au NPs. Direct evidence concerning the molecular morphology of GO-xN sheets comes from AFM studies. The dispersion of GO-xN was drop-dried on a mica substrate, and the thicknesses of the GO-xN are measured in the range of 2.2–2.8 nm (a typical AFM image is shown in Fig. 5 for the case of GO-2N). It is well known that GO has a thickness of 0.8 nm. The average thickness of the silane agents is in the range of 1–2 nm, which adds up to a total thickness of GO-xN in the range of 1.8–2.8 nm.35 This is a compelling evidence that silane indeed forms a monomolecular layer on the graphene surface.
 | ||
| Fig. 5 (a) AFM image of GO-2N coated onto a freshly cleaved mica surface showing single or multilayered sheets, (b) and (c) height profiles of GO-2N as labeled in (a). | ||
3.2 Preparation of GO/Au nanocomposites
Our protocol for fabricating GO/Au nanocomposites exploits the simplicity of self-assembly of Au NPs from solution and their affinity for amine functional groups. As discussed above, silane molecules form a stable self-assembled monolayer on GO surface through covalent bond. Terminated amine groups point outwards away from the graphene surface and serve as anchoring sites for Au NPs. Immersion of the functionalized graphene into a solution of Au NPs causes Au NPs to anchor on graphene surface through Au–N covalent bond formation. The advantage of the colloidal self-assembly approach is that the covalent bond between nanoparticles and the substrate reduces the surface mobility of the nanoparticles and prevents spontaneous coalescence of nanoparticles.The self-assembly of Au NPs on functionalized GO was achieved through immersion of GO-xN in pre-synthesized Au NPs aqueous solutions at 35 °C for 20 hours. After washing off excess Au NPs, the solution of GO/Au exhibited a reddish color (inset in Fig. 6a). The binding of Au NPs to organosilane-coated substrates develops in two stages: (1) reaction rate-controlled Au NPs deposition on GO-xN, and (2) saturation coverage of Au NPs controlled by repulsive interaction between the neighboring particles.21
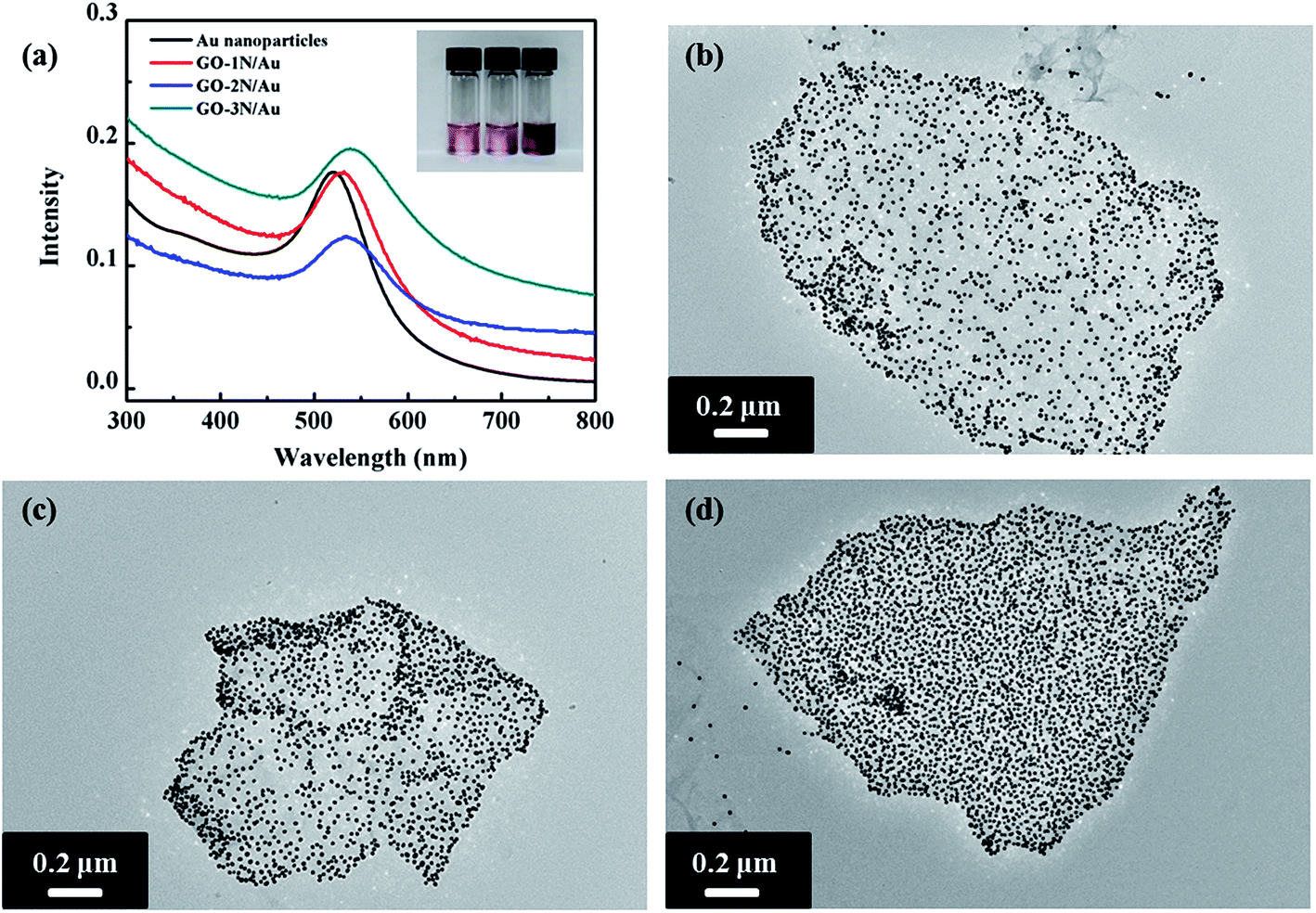 | ||
| Fig. 6 (a) UV-Vis spectra of Au, GO-1N/Au, GO-2N/Au, and GO-3N/Au. Inset: photographs from left to right of dispersions of GO-1N/Au, GO-2N/Au, and GO-3N/Au. TEM images of (b) GO-1N/Au, (c) GO-2N/Au, and (d) GO-3N/Au. | ||
UV-Vis spectroscopy was used to characterize the GO/Au nanocomposites. Pristine Au NPs with 10 nm size have a surface plasmon resonance peak around 520 nm in the visible spectrum.36 The surface plasmon resonance peaks of the assembled GO/Au nanocomposites become much broader and have redshifts due to the strong inter-particle coupling of closely packed particles (Fig. 6a). Au NPs are well known for their strong interactions with visible light, i.e. resonant excitations of the collective oscillations of the conductive electrons within the particles.37 The surface plasmon resonance energy of Au NPs depends on their size, shape, and surrounding environment.38 When Au NPs are closely packed, the coupling effect between particles becomes eminent, and the surface plasmon resonance wavelength of the two coupled particles will shift to a high wavelength.36,39 Among these three nanocomposites, GO-3N/Au has the largest redshift (18 nm), whereas GO-1N/Au has the smallest redshift (10 nm). This indicates that the inter-particle spacing between Au NPs could be altered by varying functional groups on the graphene surface. More amine groups on the graphene surface lead to higher grafting density of NPs and smaller inter-particle distance.
TEM observation reveals the same trend toward the grafting density of Au NPs as confirmed from UV-Vis results. TEM images of GO-1N/Au and GO-2N/Au show relatively loose-packed NPs, but GO-3N/Au displays closely packed NPs on the graphene surface. The Au NP density on the graphene sheets was determined using ImageJ (freeware from the National Institutes of Health). The densities of Au NPs on GO-3N/Au, GO-2N/Au and GO-1N/Au nanocomposites are 7200, 6000 and 4800 μm−2, respectively. Compared with the other two silane-functionalized graphene, GO-3N has the highest Au NP grafting density. As discussed above, there are two stages for Au NP binding to organosilane-coated substrates: initial Au NP deposition on GO substrates, which increases with time and late saturated Au NP coverage due to repulsive interaction among Au NPs. During the initial stage, terminated amine groups on graphene surface serve as anchoring sites for Au NPs. Immersion of the functionalized graphene into a solution of Au NPs causes Au NPs to anchor on graphene surface through Au–N covalent bond formation. In addition to the time factor, the concentration of amine groups is one factor to determine the binding rate. As confirmed by the TGA results, GO-3N has the highest amine group concentration, followed by GO-2N and GO-1N. Because of the high concentration of amine groups, GO-3N has the highest chance to attract and graft Au NPs among these three GO/Au nanocomposites. This conclusion agrees with the results observed from TEM images. Immobilization of Au NPs with high density on the graphene surface can maximize the active surface area exposed to reaction media, which is crucial for a consistent, highly efficient catalyst performance.
3.3 Catalytic properties of GO/Au nanocomposites
The effectiveness of the catalytic activity of the GO/Au nanocomposites is validated through a typical catalytic reaction, i.e. the hydrogenation of 4-nitrophenol (4-NPh) to 4-aminophenol (4-APh) using NaBH4 as the hydrogen source. 4-NPh is a harmful and hazardous chemical that is typically found in industrial products and agricultural waste.40 The hydrogenation of 4-NPh to 4-APh is one of the necessary processes for manufacturing fine and industrial chemicals. However, this hydrogenation process could not be realized, unless a catalyst (e.g. Au NPs) is used.6 This process involves three steps: (1) hydrogen generation from NaBH4, (2) absorption of hydrogen on Au NPs, and (3) reduction of 4-NPh to 4-APh through the Au-absorbed hydrogen.22,41 The third step allows a real-time monitoring of the reaction process by UV-Vis spectroscopy. 4-NPh forms 4-nitrophenolate anions in alkaline reaction media with a strong absorption peak at 400 nm, and 4-APh has an absorption peak at 295 nm. When the GO/Au nanocomposite was introduced into the reaction mixture, the hydrogenation reaction proceeded simultaneously with an increase in the absorption intensity at 295 nm (indicator of the product) and a decrease at 400 nm (indicator of the reactant) (Fig. 7a). The solution color changes from yellow to colorless when all 4-NPh is consumed (inset in Fig. 7a). | ||
| Fig. 7 (a) Time-dependent UV-Vis spectra of the reaction mixture of catalytic reduction of 4-NPh with GO-1N/Au, inset: photograph of aqueous solutions of 4-NPh before and after reduction. (b) Dependence of ln(A/A0) on reaction time for the reactions catalyzed by GO-N/Au catalysts (R2 for linear fitting = 0.93 (GO-1N/Au), 0.99 (GO-2N/Au), 0.99 (GO-3N/Au)). All catalysts are used at the same molar ratio based on graphene and 40 equiv. of NaBH4 for the reaction. | ||
All three GO/Au nanocomposites show high efficiency in catalyzing the hydrogenation reaction, finishing the reaction within 16 min. The reaction kinetics is studied to better understand the relationship between the grafting density of Au NPs and their catalytic performance. The pseudo-first-order reaction kinetics is applicable to analyze the reaction rate constant, considering that the concentration of NaBH4 is in large excess relative to that of 4-NPh (Fig. 7b).6 The highest efficiency was obtained from GO-3N/Au, followed by GO-2N/Au and GO-1N/Au, having a rate constant of 0.15 min−1, 0.13 min−1, 0.11 min−1, respectively. As expected, the catalytic efficiency becomes higher as the particle grafting density increases. The catalytic rate constant for GO/Au nanocomposites is proportional to the Au NP density. According to the particle density calculation described above, the Au NP densities of GO-2N/Au and GO-3N/Au are 1.2 and 1.5 times larger than that of GO-1N/Au. The corresponding rate constants for GO-2N/Au and GO-3N/Au are expected as 0.13 min−1 and 0.16 min−1, which are in agreement with the experimental findings.
It has been demonstrated up to this point that the GO/Au nanocomposite allows tailoring the catalytic reaction rate. The section below demonstrates that these nanocomposites could be easily separated from the reaction system and reused in a new catalytic reaction. To check the reproducibility and stability of the catalyst, repeated hydrogenation reactions of 4-NPh to 4-APh were carried out using GO-3N/Au. Between each repeated reaction, the GO-3N/Au was removed from the reaction and rinsed with water. Our results show that there is no detectable loss of catalytic activity after 8 successive reactions (Fig. 8a), which is the highest cycle number ever reported.6,25 The stability of the GO-3N/Au nanocomposite was confirmed by UV-Vis spectroscopy (Fig. 8b). The absorption peak of GO-3N/Au does not shift after 8 successive reactions. Additionally, a TEM image of GO-3N/Au after successive reactions was taken (Fig. S1†). There is no discernable difference in structures before and after 8 times of reuses. The results from UV-Vis spectra and TEM image indicate that (1) Au NPs does not change their inter-particle spacing after repeated catalytic reactions, and (2) the GO-3N/Au nanocomposite survived after catalytic cycles without showing signs of detachment or aggregation. In addition, the turnover frequency (TOF) of the GO-3N/Au is 35 h−1 (Table S1†). Although the TOF of the GO-3N/Au is not the highest value reported, this catalyst is found to be as efficient as other catalysts with similar sizes and supporting materials. Note that the catalytic performance of Au NPs is appreciably affected by both the size of Au NPs and the supporting materials.12 Despite the comparable TOF values, silane coupling agents with different amine end groups lead to GO/Au nanocomposites with controlled Au NP grafting density, which help better understand how the Au packing density influences catalytic activity. Therefore, it is clearly demonstrated that silane-functionalized GO plays a vital role in preventing Au NPs from aggregation and maintaining full catalytic activity after successive cycles of reaction.
 | ||
| Fig. 8 (a) Catalytic conversion efficiency of 4-NPh in 8 continuous reaction cycles, and (b) UV-Vis spectra of GO-3N/Au before (black line) and after 8 continuous reaction cycles (red line). | ||
4 Conclusions
We have developed a method to fabricate highly active, tunable, and reusable catalysts through self-assembly of Au NPs on organosilane-functionalized GO. Silane coupling agents with different amine end groups lead to GO/Au nanocomposites with controlled Au NP grafting density, which help better understand how the Au packing density influences catalytic activity. This approach realizes tailored morphology, grafting density, and chemical configuration, which could not be otherwise obtained by other methods. This realization is crucial toward the development and optimization of heterogeneous nanocatalysts.Acknowledgements
The authors thank Kaneka for their financial support. Special thanks are also given to the Microscopy Image Center in Texas A&M University for access of the TEM facility and the Materials Characterization Facility in Texas A&M University for the help of XPS Spectroscopy.References
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- R. A. Sperling, P. R. Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896 RSC.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC.
- B. Hvolbaek, T. V. W. Janssens, B. S. Clausen, H. Falsig, C. H. Christensen and J. K. Norskov, Nano Today, 2007, 2, 14 CrossRef.
- R. Long, K. Mao, X. Ye, W. Yan, Y. Huang, J. Wang, Y. Fu, X. Wang, X. Wu, Y. Xie and Y. Xiong, J. Am. Chem. Soc., 2013, 135, 3200 CrossRef CAS PubMed.
- Z. Jiajing, D. Bo, F. Zheng, S. Jibin, W. Chenxu, P. B. Messersmith and D. Hongwei, Adv. Mater., 2014, 26, 701 CrossRef PubMed.
- T. Ji, L. Li, M. Wang, Z. Yang and X. Lu, RSC Adv., 2014, 4, 29591 RSC.
- C. T. Campbell, J. C. Sharp, Y. X. Yao, E. M. Karp and T. L. Silbaugh, Faraday Discuss., 2011, 152, 227 RSC.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981 CrossRef CAS PubMed.
- H. J. Yin, H. J. Tang, D. Wang, Y. Gao and Z. Y. Tang, ACS Nano, 2012, 6, 8288 CrossRef CAS PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed.
- J. Hu, Y.-L. Dong, X.-J. Chen, H.-J. Zhang, J.-M. Zheng, Q. Wang and X.-G. Chen, Chem. Eng. J., 2014, 236, 1 CrossRef CAS PubMed.
- E. Lam, S. Hrapovic, E. Majid, J. H. Chong and J. H. T. Luong, Nanoscale, 2012, 4, 997 RSC.
- Y. Wang, G. Wei, W. Zhang, X. Jiang, P. Zheng, L. Shi and A. Dong, J. Mol. Catal. A: Chem., 2007, 266, 233 CrossRef CAS PubMed.
- W. Hong, H. Bai, Y. Xu, Z. Yao, Z. Gu and G. Shi, J. Phys. Chem. C, 2010, 114, 1822 CAS.
- J. Zhu, M. Chen, Q. He, L. Shao, S. Wei and Z. Guo, RSC Adv., 2013, 3, 22790 RSC.
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288 RSC.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906 CrossRef CAS PubMed.
- H. Q. Yao, L. Jin, H. J. Sue, Y. Sumi and R. Nishimura, J. Mater. Chem. A, 2013, 1, 10783 CAS.
- K. Nguyen Tri, S. W. Kim, D.-H. Yoo, E. J. Kim and S. H. Hahn, Appl. Catal., A, 2014, 469, 159 CrossRef PubMed.
- J. Li, C.-Y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426 RSC.
- X. Xie, J. Long, J. Xu, L. Chen, Y. Wang, Z. Zhang and X. Wang, RSC Adv., 2012, 2, 12438 RSC.
- W. Liu, D. Sun, J. Fu, R. Yuan and Z. Li, RSC Adv., 2014, 4, 11003 RSC.
- J. Liu, S. Fu, B. Yuan, Y. Li and Z. Deng, J. Am. Chem. Soc., 2010, 132, 7279 CrossRef CAS PubMed.
- G. Goncalves, P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Gracio, Chem. Mater., 2009, 21, 4796 CrossRef CAS.
- M. Mahyari, A. Shaabani and Y. Bide, RSC Adv., 2013, 3, 22509 RSC.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263 CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939 CrossRef CAS PubMed.
- J. Sagiv, J. Am. Chem. Soc., 1980, 102, 92 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS PubMed.
- Y. Matsuo, Y. Nishino, T. Fukutsuka and Y. Sugie, Carbon, 2007, 45, 1384 CrossRef CAS PubMed.
- E. Metwalli, D. Haines, O. Becker, S. Conzone and C. G. Pantano, J. Colloid Interface Sci., 2006, 298, 825 CrossRef CAS PubMed.
- Q. H. Wei, K. H. Su, S. Durant and X. Zhang, Nano Lett., 2004, 4, 1067 CrossRef CAS.
- U. Kreibig and M. Vollme, Optical properties of metal clusters, Springer, Berlin, New York, 1995 Search PubMed.
- L. L. Zhao, K. L. Kelly and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 7343 CrossRef CAS.
- K. H. Su, Q. H. Wei, X. Zhang, J. J. Mock, D. R. Smith and S. Schultz, Nano Lett., 2003, 3, 1087 CrossRef CAS.
- Y. Choi, H. S. Bae, E. Seo, S. Jang, K. H. Park and B.-S. Kim, J. Mater. Chem., 2011, 21, 15431 RSC.
- T. Fujitani, I. Nakamura, T. Akita, M. Okumura and M. Haruta, Angew. Chem., Int. Ed., 2009, 48, 9515 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11231h |
| This journal is © The Royal Society of Chemistry 2014 |