
A fluorescence turn-on probe for selective detection of nitrogen dioxide†
Biplab Mondal* and
Vikash Kumar
Department of Chemistry, Indian Institute of Technology Guwahati, Assam 781039, India. E-mail: biplab@iitg.ernet.in; Fax: +91-361-258-2339; Tel: +91-361-258-2317
First published on 6th November 2014
Abstract
Two copper(II) complexes, 1 and 2, of two ligands, L1 and L2 [L1 = 2-{[anthracen-9-ylmethyl-(2-dimethylamino-ethyl)-amino]-methyl}-4,6-di-tert-butyl-phenol; L2 = 5-dimethylamino-naphthalene-1-sulfonic acid (3,5-di-tert-butyl-2-hydroxy-benzyl)-(2-dimethylamino-ethyl)-amide, were synthesized and characterized. In methanol solution, the quenched fluorescence intensity of the ligands in complexes 1 and 2 was found to be restored upon exposure to nitrogen dioxide. This is attributed to the reduction of the paramagnetic Cu(II) centre by nitrogen dioxide to diamagnetic Cu(I). The reduction was accompanied by simultaneous nitration in the phenol ring of the ligands.
Nitric oxide (NO) is known to play important roles in physiological functions such as vasodilation and neurotransmission. It is also known that NO could participate as a cytotoxic effector and/or a pathogenic mediator when produced at high concentration.1 NO mediated pathogenicity mostly depends on the formation of secondary intermediates like peroxynitrite (ONOO−) and nitrogen dioxide (NO2) that are more reactive and toxic than NO itself.2 The formation of these reactive nitrogen species requires the presence of oxidants such as superoxide radicals, hydrogen peroxide, and transition metal centres. Nitrogen dioxide can also be formed in hydrophobic environments from the reactions of NO with molecular oxygen.3,4 These reactive nitrogen species are known to induce nitration of protein tyrosine residues to 3-nitrotyrosine.5 This modification has attracted considerable interest to biomedical research as it can alter protein function and is associated to disease states. Though the capacity of peroxynitrite to cause protein tyrosine nitration in vitro and vivo has been demonstrated, the role of peroxynitrite as a central species in biological nitration is not yet confirmed beyond doubt.6–10 An alternative pathway involving on the formation of NO2 from nitrite (NO2−; the main metabolism product from NO) by the action of hemeperoxidases and/or transition metal complexes has been proposed recently.11–15
Thus, a probe which could selectively identify the NO2, would be of immense importance to establish the tyrosine nitration mechanism. There have not been many examples of sensor which could detect NO2 selectively.16
Recently, we have demonstrated that the reduction of Cu(II) centre by NO2 gas in the complexes of the 2,4-di-tert-butyl-6-(((2-dimethylamino)ethyl)(isopropyl)amino)methyl-phenol (DDMEP) and 6,6′-(((2-(dimethylamino)ethyl)azanediyl)bis(methylene))bis(2,4-di-tert-butylphenol) (DMABP) ligands.17 This instigate us to develop selective fluorescent NO2 sensors based on Cu(II) complexes by attaching fluorophore to the same ligand frameworks. Herein we report the example of copper(II) complexes of two such fluorophore ligands as selective sensors for NO2 (Fig. 1).
 | ||
| Fig. 1 Fluorophore ligands used for the present study. | ||
Ligand L1‡ has been synthesized in a three step procedure: (i) the condensation of N,N-dimethylethylenediamine with anthracene aldehyde; (ii) reduction of the imine compound by NaBH4 to get the corresponding amine and (iii) subsequent Mannich reaction with 2,4-di-tert-butyl phenol in presence of formaldehyde (Experimental section, ESI†). L2 has been prepared by the reaction of corresponding N,N-dimethylethylenediamine with dansyl chloride to get the corresponding sulphonamide. In the next step, the sulphonamide was treated with 2,4-di-tert-butyl-6-chloromethylphenol (Experimental section, ESI†). The ligands were characterized by various spectral analyse and micro analyses (Experimental section, ESI†). L1 has been further characterized by its X-ray single crystal structure determination. The perspective ORTEP view of L1 is shown in Fig. 2. The crystal data, bond angles and lengths are listed in the ESI.†
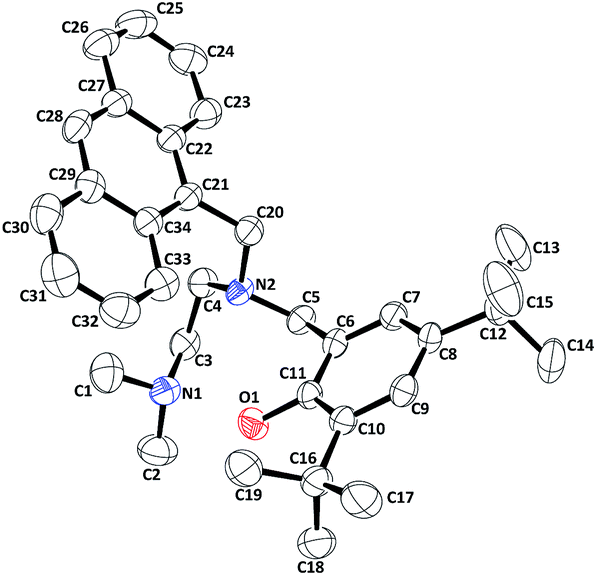 | ||
| Fig. 2 ORTEP diagram of L1 (50% thermal ellipsoid plot). | ||
The complexes 1§ and 2 were synthesized by the reaction of copper(II) acetate dihydrate with equivalent amount of the respective ligands. The complexes were characterized by various analytic techniques and elemental analyses (Experimental section; ESI†). Complex 1 has been further characterized by single crystal X-ray structure determination. The ORTEP view is shown in Fig. 3. Even after several attempts, we could not grow the X-ray quality crystals of the complex 2. It should be noted that the structure of the copper(II) complex with similar framework but isopropyl substitution at Namine position is reported earlier from our laboratory.17
 | ||
| Fig. 3 ORTEP diagram of complex 1 (50% thermal ellipsoid plot. H-atoms are omitted for clarity). | ||
In methanol solution, the Cu(II) centre in the complexes was found to undergo rapid reduction in presence of nitrogen dioxide (Fig. 4). Addition of nitric oxide to the methanol solution did not result in the reduction of the Cu(II) centre (ESI†) in the complexes. Similar observation was observed with copper(II) complexes of DDMEP and DMABP ligands.17
 | ||
| Fig. 4 UV-visible spectra of complex 1 before (black) and after (blue) addition of NO2 in methanol. | ||
Fluorescence studies of the ligands at 298 K in methanol solution exhibited a significant quenching of the intensity in presence of equivalent amount of Cu(II) ion (ESI†). This is attributed to the paramagnetic effect of Cu(II) centres.18 While nitric oxide is purged to the degassed methanol solution of the complexes, no change in fluorescence behaviour was observed (ESI†). This is in accord with the observation that the Cu(II) centre in these complexes does not undergo reduction by nitric oxide unlike other reported ones (ESI†).19 However, addition of nitrogen dioxide to the degassed methanol solution of complexes 1 and 2 restored the quenched fluorescence intensity of the ligands (Fig. 5). The restored emission intensity was found to be more in case of dansyl fluorophore (L2) compared to L1. In case of complex 1, the restored intensity was 3 ± 0.2 fold and in case complex 2, it was 5 ± 0.2 fold. The detection was observed even at 10 nmol concentration. Though addition of nitric oxide or O2 alone in the degassed methanol solution of complexes did not make any difference, when NO was added in presence of O2, the quenched fluorescence intensity of the respective ligands was restored (ESI†). This is because of the fact that NO reacts first with O2 to yield NO2 and thus formed NO2 reacts with the copper(II) centre of the complexes. Addition of H2O2, KO2 also found to be not reactive.
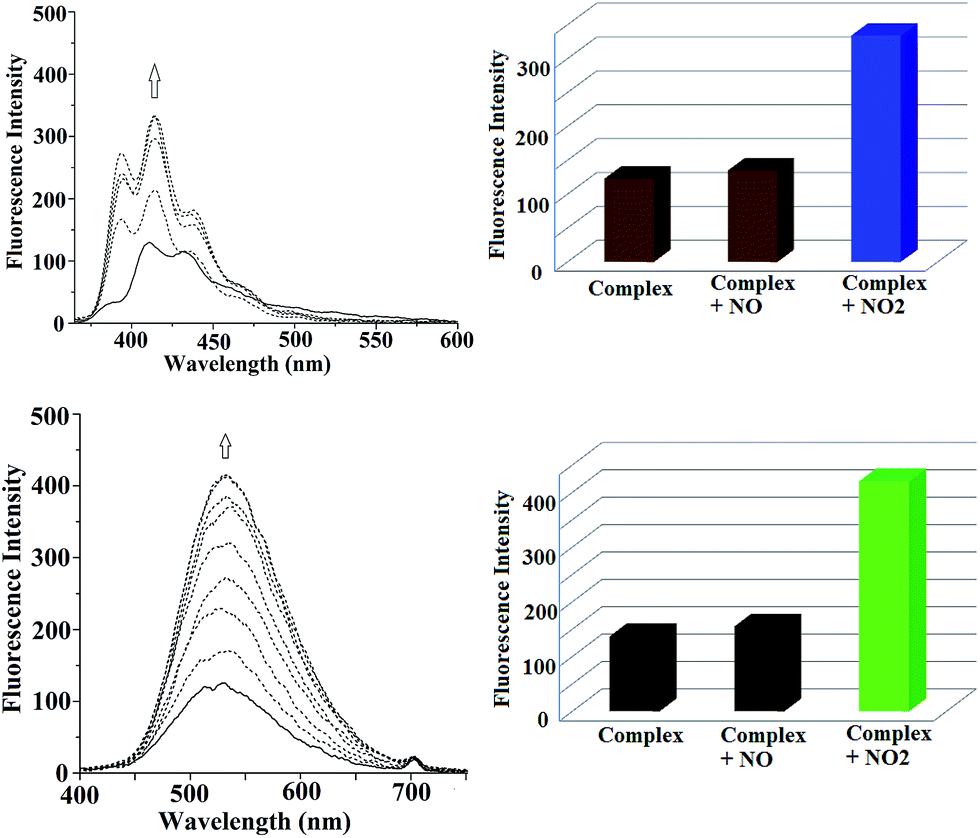 | ||
| Fig. 5 Fluorescence response of complexes 1 (Top) and 2 (Bottom) in methanol solution after addition of NO2. The respective bar-diagram shows the fluorescence responses of the complexes, after addition of NO and NO2, respectively. | ||
As such, there is only one fluorescence sensor for nitrogen dioxide is known which is based on Ni(II) complex of dithiocarbamate ligand derived from sulforhodamine B fluorophore.16 The quenching of ligand fluorescence is attributed to the photoinduced electron transfer from Ni(II) to the sulforhodamine B excited state. The reaction of NO2 with the complex results in oxidation and decomplexation of the ligand from Ni(II). This leads to dimerization of the dithiocarbamate ligand to yield strong fluorescence.16
In the present case, the turn-on of fluorescence intensity in presence of nitrogen dioxide is attributed to the reduction of Cu(II) centre to diamagnetic Cu(I). The reduction has been authenticated by UV-visible, X-band EPR and 1H-NMR spectroscopic studies. In UV-visible spectrum, complex 1 in methanol solution displays the d–d transition band at 665 nm. Upon addition of nitrogen dioxide, this disappears suggesting the reduction of the Cu(II) centre to Cu(I) (Fig. 4).
In X-band EPR, the characteristic four line signal of Cu(II) of the complexes was disappeared upon addition of nitrogen dioxide in the solution (ESI†). Similar spectral changes were observed in case of complex 2 also suggesting the occurrence of same chemistry (ESI†).
The reduction of Cu(II) centres in complexes by NO2 resulted in simultaneous nitration of the phenol ring present in the ligand frameworks (Scheme 1). The nitration products of L1 and L2 were isolated in a good yield (∼70%) and characterized by various spectral techniques in certainty (ESI,† Experimental section).
 | ||
| Scheme 1 | ||
The phenol ring nitration takes place through an electrophilic substitution where tert-butyl group is being substituted by nitronium ion (NO2+). The NO2+ formed during the reduction of Cu(II) by NO2, attacks at the 4-position of the phenol ring resulting in the corresponding 2-tert-butyl-4-nitrophenol.
The release of tert-butyl cation during the reaction is confirmed from the presence of tert-butanol in GC mass spectrum of the reaction mixture (ESI†).
In cases of earlier reported copper(II) complexes of DDMEP and DMABP ligands, the similar nitration was observed. The nitration was reported to proceed through a NO2+ species which is formed in the reduction of Cu(II) to Cu(I) by NO2.17
Conclusions
The present set of complexes demonstrate the example where NO2 induces the reduction of Cu(II) centre and thus, turns on the quenched fluorescence intensity of the fluorophore ligands. The reaction results in the simultaneous nitration of the phenol ring present in the ligand framework. The reaction was not observed with NO alone. This demonstrates the first example where the reduction of Cu(II) centre by nitrogen dioxide is utilized to develop the selective sensor for nitrogen dioxide. The in vivo applications of these complexes are warranted.Acknowledgements
VK gratefully acknowledges CSIR, India for providing the fellowship. BM would like to thank Department of Science and Technology (DST), India for financial support (SR/S1/IC-38/2010); DST-FIST program for X-ray diffraction facility.Notes and references
- L. J. Ignarro, Annu. Rev. Pharmacol. Toxicol., 1990, 30, 535–560 CrossRef CAS PubMed.
- R. Radi, A. Denicola, B. Alvarez, G. Ferrer-Sueta and H. Rubbo, in Nitric Oxide, ed. L. Ignarro, Academic, San Diego, 2000, pp. 57–82 Search PubMed.
- X. Liu, M. J. Miller, M. S. Joshi, D. D. Thomas and J. R. Lancaster Jr, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2175–2179 CrossRef CAS.
- A. Denicola, C. Batthyany, E. Lissi, B. A. Freeman, H. Rubbo and R. Radi, J. Biol. Chem., 2002, 277, 932–936 CrossRef CAS PubMed.
- M. Sokolovsky, J. F. Riordan and B. L. Vallee, Biochemistry, 1966, 5, 3582–3589 CrossRef CAS; R. Radi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4003–4008 CrossRef PubMed.
- J. S. Beckman, H. Ischiropoulos, L. Zhu, M. van der Woerd, C. Smith, J. Chen, J. Harrison, J. C. Martin and M. Tsai, Arch. Biochem. Biophys., 1992, 298, 438–445 CrossRef CAS.
- H. Ischiropoulos, L. Zhu, J. Chen, M. Tsai, J. C. Martin, C. D. Smith and J. S. Beckman, Arch. Biochem. Biophys., 1992, 298, 431–437 CrossRef CAS.
- H. Ischiropoulos, L. Zhu and J. S. Beckman, Arch. Biochem. Biophys., 1992, 298, 446–451 CrossRef CAS.
- J. S. Beckmann, Y. Z. Ye, P. G. Anderson, J. Chen, M. A. Accavitti, M. M. Tarpey and C. R. White, Biol. Chem. Hoppe-Seyler, 1994, 375, 81–88 CrossRef CAS.
- H. Ohshima, M. Friesen, I. Brouet and H. Bartsch, Food Chem. Toxicol., 1990, 28, 647–652 CrossRef CAS.
- S. Pfeiffer, K. Schmidt and B. Mayer, J. Biol. Chem., 2000, 275, 6346–6352 CrossRef CAS PubMed.
- D. D. Thomas, M. G. Espey, M. P. Vitek, K. M. Miranda and D. A. Wink, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12691–12696 CrossRef CAS PubMed.
- J. P. Eiserich, M. Hristova, C. E. Cross, A. D. Jones, B. A. Freeman, B. Halliwell and A. van der Vliet, Nature, 1998, 391, 393–397 CrossRef CAS PubMed.
- K. Bian, Z. Gao, N. Weisbrodt and F. Murad, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5712–5717 CrossRef CAS PubMed.
- M. L. Brennan, W. Wu, X. Fu, Z. Shen, W. Song, H. Frost, C. Vadseth, L. Narine, E. Lenkiewicz, M. T. Borchers, A. J. Lusis, J. J. Lee, N. A. Lee, H. M. Abu-Soud, H. Ischiropoulos and S. L. Hazen, J. Biol. Chem., 2002, 277, 17415–17427 CrossRef CAS PubMed.
- Y. Yan, S. Krishnakumar, H. Yu, S. Ramishetti, L. W. Deng, S. Wang, L. Huang and D. Huang, J. Am. Chem. Soc., 2013, 135, 5312–5315 CrossRef CAS PubMed.
- V. Kumar, A. Kalita and B. Mondal, Dalton Trans., 2013, 16264–16267 RSC.
- M. H. Lim, B. A. Wong, W. H. Pitcock Jr, D. Mokshagundam, M.-H. Baik and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 14364–14373 CrossRef CAS PubMed.
- P. Kumar, A. Kalita and B. Mondal, Dalton Trans., 2013, 5731–5739 RSC and references therein.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1009929 and 1013006. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra10426a |
| ‡ Crystal data for ligand, L1: CCDC no. 1009929, C34H44N2O, M = 496.71, monoclinic (P2(1)/c), a = 11.4955(11), b = 10.9564(13), c = 23.216(2) Å, α = 90°, β = 95.880(9)°, γ = 90°, V = 2908.7(5) Å3, Z = 4, Dc = 1.134 g cm−3, μ = 0.067 mm−1, T = 293(2) K, 5110 reflections, 2500 independent, R(F) = 0.0701 [I > 2δ(I)], wR(F2) = 0.1488 (all data), GOF = 1.174. |
| § Crystal data for complex 1: CCDC no. 1013006, C36H46N2O3Cu, M = 618.29, monoclinic C2/c, a = 28.2038(9), b = 12.6258(4), c = 22.7579(8) Å, α = 90°, β = 128.0360(10)°, γ = 90°, V = 6382.9(4) Å3, Z = 8, Dc = 1.287 g cm−3, μ = 0.722 mm−1, T = 296(2) K, 5622 reflections, 4006 independent, R(F) = 0.0410 [I > 2δ(I)], wR(F2) = 0.0596 (all data), GOF = 1.473. |
| This journal is © The Royal Society of Chemistry 2014 |